Tumor Biology and Metastasis
Cells of multicellular organisms form part of a specialized society that cooperate to promote survival of the organism. In this society, cell division, proliferation, and differentiation are strictly controlled and a balance exists between normal cell birth and the natural cell death rate.1 Derangement of these normal homeostatic mechanisms can lead to uncontrolled proliferation or loss of the ability to die, leading to a normal cell taking on a malignant phenotype.
Cancer in animals is well documented throughout history but has taken on significance over the past hundred years for a number of reasons. Studies on chicken, feline, and bovine retroviruses have made significant contributions to our overall understanding of carcinogenesis through the discovery of oncogenes and tumor suppressor genes.2 Further contributions to our understanding of viral oncogenesis have come from studies of the DNA papilloma viruses in cattle and horses, complementing research into cervical cancer in women.3-10 This complementary cancer research has paved the way for the development of programs of research in comparative medicine that has benefits for both humans and the veterinary species. In this chapter we summarize the current understanding of the molecular mechanisms of cancer development and metastasis.
Normal Cell Division
Within an animal, all cells are subject to wear and tear, making cellular reproduction a necessity for maintenance of the individual. Reproduction of the gametes occurs by the process of meiosis, whereas reproduction of somatic cells involves two sequential phases known as mitosis and cytokinesis. Mitosis is nuclear division, and cytokinesis involves the division of the cytoplasm, the two occurring in close succession. Nuclear division is preceded by a doubling of the genetic material of the cell during a period known as interphase. As well as a copying of the chromosomes, this period is characterized by marked cellular activity in terms of RNA, protein and lipid production. The alternation between mitosis and interphase in all tissues is often referred to as the cell cycle. The phases of the cell cycle are shown in Figure 2-1.
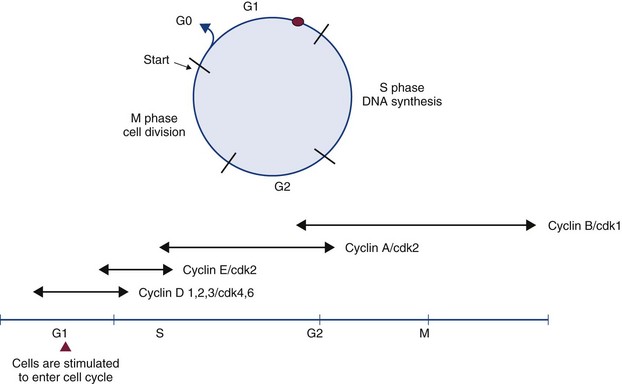
Figure 2-1 The cell cycle and its control. The cell cycle is divided into four phases (G1, S, G2, and M) and G0, which represents cycle-arrested cells. Cells are stimulated to enter the cell cycle in response to external factors, including growth factors and cell adhesion molecules. During G1, cells are responsive to mitogenic signals. Once the cell cycle has traversed the restriction point (R) in the G1 phase, the cell-cycle transitions become autonomous. Progression through the cell cycle is mediated by the sequential activation and inactivation of the cyclin-dependent kinases (CDKs). Control of CDK activity is through their interaction with specific cyclins (D, E, A, B) and with specific CDK inhibitors.
Interphase (G1, G2, and S phases) is the longest phase of the cell cycle. During interphase, the chromatin is very long and slender; however, it shortens and thickens as interphase progresses. The first phase of mitosis is referred to as prophase and sees the first appearance of the chromosomes. As the phase progresses, the chromosomes appear as two identical sister chromatids joined at the centromere. As the nuclear membrane disappears, spindle fibers form and radiate from the two centrioles, each located at opposite poles of the cell. The spindle fibers serve to pull the chromosomes to opposite sides of the cell.
During metaphase, the spindle fibers pull the centromeres of the chromosomes, which become aligned to the middle of the spindle, often referred to as the equatorial plate. During anaphase, the centromeres split and the sister chromatids are pulled apart by the contraction of the spindle fibers. The final stage of cell division is telophase, characterized by the formation of a nuclear membrane around each group of chromosomes, is followed by cytokinesis or separation of the cytoplasm to produce two identical diploid cells. Progression through the cell cycle lasts approximately 12 to 24 hours.
The Cell Cycle
The cell cycle comprises four phases (M phase, S phase, G1, and G2) (see Figure 2-1). Nonproliferating cells are usually arrested between the M (mitosis) and S (DNA synthesis) phases and are referred to as G0 cells. The majority of cells in normal tissues are in G0. Cells are stimulated to enter the cell cycle in response to external factors including growth factors and cell adhesion. During the G1 phase of the cell cycle, cells are responsive to mitogenic signals. Once the cell cycle has traversed the restriction point (R) in the G1 phase, cell cycle transitions become autonomous.
Progression through the cell cycle is mediated by the sequential activation and inactivation of a class of proteins called cyclin-dependent kinases (CDKs).11,12 CDKs consist of an inactive conserved catalytic core and are regulated at the following three levels:
1. CDK activity requires the association with regulatory subunits known as cyclins. The level of CDK remains constant throughout the cell cycle; however, the concentration of cyclins varies in a phase-specific manner during the cell cycle. The periodic synthesis and destruction of cyclins provides the primary level of cell cycle control (see Figure 2-1).
2. The activity of cyclin/CDK complexes is also regulated by phosphorylation. Activation of CDK/cyclin complexes requires phosphorylation by CDK-activating kinases (CAK); meanwhile the phosphorylation at threonine and serine residues suppresses activity.
3. CDKs are also tightly regulated by a class of inhibitory proteins known as CDK inhibitors (CDKI). CDKIs can block G1/S progression by binding CDKs/cyclin complexes and can be classified into the following two groups:
The first class of proteins induced in G1 following mitogenic stimulation of the cell cycle is the class D cyclins, which in turn activate CDK4 and CDK6. Cyclin D-CDK complexes cause phosphorylation of the substrate retinoblastoma protein (Rb), which results in dissociation of the transcription factor E2F from the Rb protein. The phosphorylation status of Rb plays a critical role in regulating G1 progression, and Rb is the molecular device that serves as the R point switch. Phosphorylated Rb releases E2F enabling transcription of numerous E2 responsive genes involved in DNA synthesis, and unphosphorylated RB remains associated with E2F, thereby inhibiting cell cycle progression. During G1 progression, activation of E2F also leads to induction of cyclin E. Cyclin E associates with CDK2 and the cyclin E-CDK2 complex maintains Rb in the phosphorylated state and is essential for cells to enter the S phase of cell cycle. At the G1/S phase transition, E2F induces cyclin A. During early S phase, cyclin D and E are degraded and cyclin A associates with CDK2 and CDK1; this kinase activity is essential for entry into S phase, completion of S phase, and entry into M phase. Mitosis is regulated by CDK1 in association with cyclins A and B causing phosphorylation of cytoskeletal proteins, including histones and lamins. This tight regulation on cell cycle progression prevents uncontrolled passage of normal cells through the cell cycle. The corollary of this is that loss of these control mechanisms can be a driving event in the development of cancer.
Cellular Responses to DNA Damage
When normal cells are subjected to stress signals, radiation, DNA damage, or oxygen depletion, the majority of cells have the ability to cause cell cycle arrest in G1, S, and G2 or enter programmed cell death (apoptosis) or both. Within cells, numerous surveillance systems called checkpoints function to recognize and respond to DNA damage. Cell cycle checkpoints occur in the G1 phase in response to DNA damage, during S phase to monitor the quality of DNA replication and the occurrence of DNA damage, and during the G2/M phase to examine the status of the spindle. The DNA damage–induced checkpoint mediated by the tumor suppressor protein p53 is the most well studied. The p53 protein plays an important role in maintaining genomic stability and forms part of a stress response pathway to various exogenous and endogenous DNA damage signals, including gamma irradiation, ultraviolet (UV) irradiation, chemicals, and oxidative stress.
p53 Functions as a Genomic Guardian
The p53 response to stress may be mediated by DNA-dependent protein kinase (DNA-PK) or by the ATM kinase and leads to phosphorylation of the N terminus of p53. In normal cells, p53 is short lived; however, phosphorylated p53 is stabilized and can then function as a transcriptional regulator binding to sequences and transactivating a number of genes, including p21.13,14 p21 has a high affinity for G1 CDK/cyclin complexes and acts as a CDKI inhibiting kinase activity, thereby arresting cells in G1. By holding cells in G1, the replication of damaged DNA is prevented, and the cell’s own DNA repair machinery has the opportunity to repair damage prior to reentering the active growth cycle (Figure 2-2).
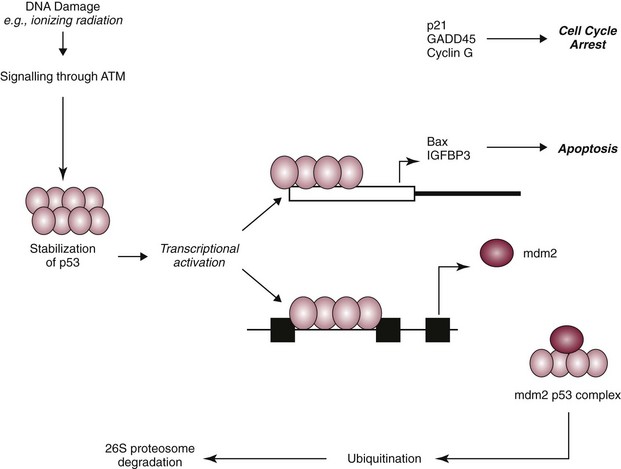
Figure 2-2 p53 is involved in cell cycle control. The p53 response to stress may be mediated by DNA-dependent protein kinase (DNA-PK) or by the ATM kinase and leads to phosphorylation of the N terminus of p53. In normal cells, p53 is short lived; however, phosphorylated p53 is stabilized and can then function as a transcriptional regulator binding to sequences and transactivating a number of genes, including p21 and Bax. Consequently, the cell cycle is arrested or the cell undergoes programmed cell death (apoptosis). This is a defense mechanism that allows for repair or death (if the cell is irreversibly damaged). The cellular levels of p53 protein are regulated by the product of another gene mouse double minute 2 oncogene (MDM2). The principal role of MDM2 is to act as a negative regulator of p53 function. One mechanism for MDM2 to downregulate p53 is to target p53 for degradation. The p53 protein is maintained in normal cells as an unstable protein, and its interaction with p53 can target p53 for degradation via a ubiquitin proteosome pathway. MDM2 can also control p53 function by suppressing p53 transcriptional activity. MDM2 is a transcriptional target of p53, and expression is induced by the binding of p53 to an internal promoter within the MDM2 gene. MDM2 can in turn bind to a domain within the amino terminus of p53, thereby inhibiting the transcriptional activity and G1 arrest function of p53 by masking access to the transcriptional machinery.
The cellular levels of p53 protein are regulated by the product of another gene MDM2 (mouse double minute 2 oncogene).15 The principal role of MDM2 is to act as a negative regulator of p53 function. One mechanism for MDM2 to downregulate p53 is to target p53 for degradation. The p53 protein is maintained in normal cells as an unstable protein, and its interaction with MDM2 can target p53 for degradation via a ubiquitin proteosome pathway. MDM2 can also control p53 function by suppressing p53 transcriptional activity. MDM2 is a transcriptional target of p53, and expression is induced by the binding of p53 to an internal promoter within the mdm2 gene. MDM2 can in turn bind to a domain within the amino terminus of p53, thereby inhibiting the transcriptional activity and G1 arrest function of p53 by masking access to the transcriptional machinery (see Figure 2-2).16
Cell Death
In contrast to necrosis, apoptosis is a distinct type of cell death most often characterized as the “programmed” self-destruction of cells that occurs in disease states, as well as part of the normal physiologic cell turnover. Whereas necrosis is characterized by swelling of the cell and lysis, in apoptosis there is cellular and nuclear shrinkage followed by fragmentation and subsequent phagocytosis. These morphologic features of apoptosis result from a number of apoptosis effectors (i.e., caspases) and regulators (particularly the Bcl-2 protein family). The molecular mechanisms involved in apoptosis are shown in Figure 2-3.17,18 Apoptosis provides a controlled mechanism for eliminating cells that are irreversibly damaged and involves an adenosine triphosphate (ATP)-dependent activation of cellular pathways, which move calcium from the endoplasmic reticulum to the cytoplasm and activation of endonucleases. As noted previously, some of these pathways are mediated through caspases. However, a wide variety of signals can initiate an apoptotic response, including Fas ligand (CD95 or FasL) and its interaction with the Fas receptor, tumor necrosis factor (TNF) and its receptor interaction, and certain oncogenes. The Fas and TNF receptors are members of the death receptor family. These transmembrane proteins with cysteine rich extracellular domains and intracellular regions share a common structure termed the “death domain.” The proapoptotic ligands for these receptors are homotrimeric peptides that are either soluble or expressed at the surface of the adjacent cell. Ligand-induced receptor clustering promotes the binding of a soluble cytosolic adapter protein called Fas-associated death domain (FADD), which itself contains a death domain as well as a caspase binding site, to the clustered death domains of the receptors. This leads to activation of caspase 8 and downstream activation of effector caspases for apoptosis.
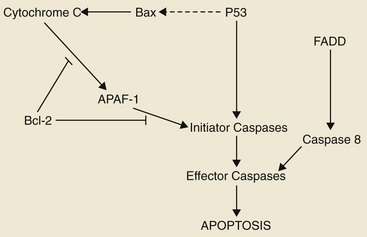
Figure 2-3 The mediators of apoptosis. A wide variety of signals can initiate an apoptotic response, including Fas ligand (CD95 or FasL) and its interaction with the Fas receptor, tumor necrosis factor (TNF) and its receptor interaction, and certain oncogenes. The Fas and TNF receptors are members of the death receptor family. These are transmembrane proteins with cysteine rich extracellular domains and intracellular regions that share a common structure termed the death domain. The proapoptotic ligands for these receptors are homotrimeric peptides that are either soluble or expressed at the surface of the adjacent cell. Ligand-induced receptor clustering promotes the binding of a soluble cytosolic adapter protein called Fas-associated death domain (FADD), which itself contains a death domain, as well as a caspase binding site, to the clustered death domains of the receptors. This leads to activation of caspase 8 and downstream activation of effector caspases for apoptosis.
Where cells are damaged and are unable to repair DNA, p53 expression can upregulate p21 and cause cell cycle arrest or can aid in directing the cell into programmed death or apoptosis through upregulation and expression of Bax (a proapoptotic Bcl-2 family protein) and also through priming of caspases. In this, the expression of p53 can also downregulate expression of the Bcl2 gene itself (a prosurvival, negative regulator of apoptosis).
Summary
The cell cycle ensures a regulated process so that each cell can complete DNA replication before cell division occurs. The cell responds to growth and environmental signals through the cell cycle. Components of the cell cycle that have a stimulatory effect include the cyclins and the CDKs. The negative influences come from a series of checkpoints that respond to external stimuli. These include tumor suppressor genes such as p53 and Rb and also genes involved in DNA repair. The process of DNA replication is also subject to the introduction of errors, and this is closely monitored by a class of enzymes called DNA repair enzymes. Consequently, there are a number of safeguards within the cell cycle to ensure that normal cells are produced during division and that the DNA is accurately replicated. The next section will describe how these systems are overcome to produce a malignant cancer cell.
From Normal Cell to Cancer Cell
It is difficult to define a cancer cell in absolute terms. Tumors are usually phenotypically recognized by the fact that their cells show abnormal growth patterns and are no longer under the control of normal homeostatic growth controlling mechanisms, including apoptosis. Although the range of mechanisms involved in the development of tumors and the spectrum of tissues from which tumors are derived is diverse, they can be classified into the following three broad types:
• Benign tumors: Broadly speaking, these tumors arise in any of the tissues of the body and grow locally. Their clinical significance is the ability to cause local pressure, cause obstruction, or form a space-occupying lesion, such as a benign brain tumor. Benign tumors do not metastasize.
• In-situ tumors: These are often small tumors that arise in the epithelium. Histologically, the lesion appears to contain cancer cells, but the tumor remains in the epithelial layer and does not invade the basement membrane or the supporting mesenchyme. A typical example of this is preinvasive squamous cell carcinoma (SCC) affecting the skin of cats, which is often referred to as Bowen’s disease.
• Cancer: This refers to a malignant tumor that has the capacity for both local invasion and distant spread by the process of metastasis.
Multistep Carcinogenesis
Cancer is the phenotypic end result of a whole series of changes that may have taken a long period of time to develop. Indeed, recent studies that have sequenced the genome of pancreatic and brain tumors have identified 63 and 60 genetic alterations on average in each cancer, respectively. From this large list of genetic alterations, there are a small number of commonly mutated genes that are “drivers” of the cancer phenotype.19,20
The application of a cancer-producing agent (carcinogen) to tissues does not lead to the immediate production of a cancer cell. After the initiation step produced by the agent, there follows a period of tumor promotion. This promotion may be caused by the same initiating agent or by other substances, such as normal growth promoters or hormones. The initiating step is a rapid step and affects the genetic material of the cell. If the cell does not repair this damage, then promoting factors may progress the cell toward a malignant phenotype. In contrast to initiation, progression may be a very slow process and may not even manifest in the lifetime of the animal. Each stage of multistep carcinogenesis reflects genetic changes in the cell with a selection advantage that drives the progression toward a highly malignant cell. The age-dependent incidence of cancer suggests a requirement for between four and seven rate-limiting, stochastic events to produce the malignant phenotype.21
These sequential events in tumor formation are a consequence of changes at the genetic level. Over the past 25 years, cancer research has generated a rich and complex body of information revealing that cancer is a disease involving dynamic changes in the genome. Seminal to our understanding of cancer biology has been the discovery of the so-called cancer genes or oncogenes and tumor suppressor genes. Mutations that produce oncogenes with dominant gain of function and tumor suppressor genes with recessive loss of function have been identified through their alteration in human and animal cancer cells and by their elicitation of cancer phenotypes in experimental models.
Oncogenes
The RNA tumor viruses (retroviruses) provided the first evidence that genetic factors play a role in the development of cancer. The initial observation came in 1910 when Rous demonstrated that a filterable agent (later classified as a retrovirus and termed avian leukosis virus) was capable of producing lymphoid tumors in chickens.22 Retroviruses have three core genes (gag, pol, and env) and an additional gene that gives the virus the ability to transform cells. Retroviral sequences that are responsible for transforming properties are called viral oncogenes (v-onc). The names of these genes are derived from the tumors in which they were first described (e.g., v-ras from rat sarcoma virus).
Viral oncogenes were subsequently shown to have cellular homologues called cellular oncogenes (c-onc). Later, the term proto-oncogene was used to describe cellular oncogenes that do not have transforming potential to form tumors in their native state but can be altered to lead to malignancy.23 Most proto-oncogenes are key genes involved in the control of cell growth and proliferation and their roles are complex. For simplicity, their sites and modes of action in the normal cell can be divided as follows (Table 2-1):
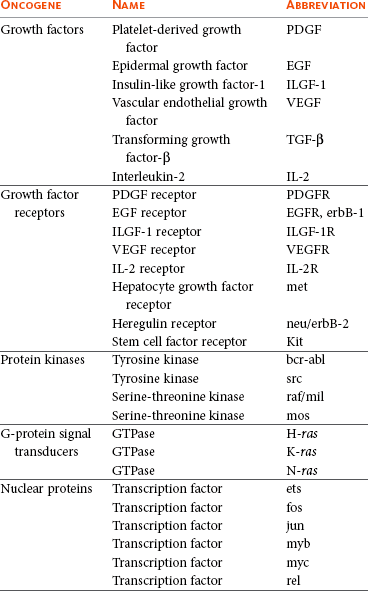
Growth Factors
Growth factors are molecules that act on the cell via cell surface receptors. Their contribution to carcinogenesis may be through excessive production of the growth factor or where a growth factor is expressed in a cell but does not normally function in that cell.
Growth Factor Receptors
Several proto-oncogene–derived proteins form a part of cell surface receptors for growth factors. The binding of ligand to receptor is the initial stage of delivery of mitogenic signals to cells. Their role in carcinogenesis may be through structural alterations in these proteins.
Protein Kinases
Protein kinases are associated with the inner surface of the plasma membrane and are involved in signal transduction following ligand-receptor binding. Structural changes in these genes and proteins lead to increased kinase activity that can have profound effects on signal transduction pathways.
Signal Transduction
The binding of an extracellular growth factor to the membrane receptor leads to a series of events by which the mitogenic signal is transduced to the nucleus of the cell. Essential to this signaling is the successive phosphorylation of signaling intermediaries or the second messengers such as guanosine triphosphate (GTP) and proteins that bind GTP (G-proteins). During signal transduction, GTP is converted to guanosine diphosphate (GDP) by the guanosine triphosphatase (GTPase) activity of G-proteins. A group of proto-oncogenes called Ras encode proteins with GTPase and GTP-binding activity and, in the normal cell, help to modulate cellular proliferation. Mutations in the Ras proto-oncogene can contribute to uncontrolled cellular proliferation.
Nuclear Proteins and Transcription Factors
Nuclear proteins and transcription factors encode proteins that control gene expression. These proto-oncogenes may have a role in cellular proliferation. Not surprisingly, changes in transcription factor activity may contribute to the development of the malignant genotype.
Mechanisms by which Oncogenes Become Activated
The advent of recombinant DNA technology has allowed scientists to unravel a number of mechanisms by which the normal products of proto-oncogenes can be disrupted to produce uncontrolled cell division. The conversion of a proto-oncogene to an oncogene is a result of somatic events in the genetic material of the target tissue. The activated allele of the oncogene dominates the wild-type allele and results in a dominant gain of function. This means that only one allele needs to be affected to obtain phenotypic change; this is in contrast to tumor suppressor genes in which both alleles have to be lost for phenotypic change. The mechanisms of oncogene activation are outlined in the following list and are shown in Figure 2-4.23-30
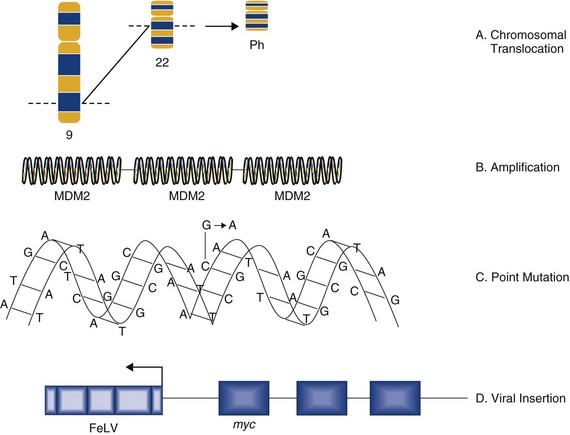
Figure 2-4 Methods of oncogene activation. Oncogenes can be activated by chromosomal rearrangements (e.g., BCR/ABL–induced leukemia), gene amplification (e.g., amplification of MDM2 in some sarcomas), point mutations (e.g., changes in nucleotide sequence that alters protein production), or by viral insertions (e.g., the insertion of feline leukemia virus [FeLV] at the myc locus in FeLV-induced lymphomas).
• Chromosomal translocation. Where proto-oncogenes are translocated within the genome (i.e., from one chromosome to another), their function can be greatly altered. The classic example in human medicine is the chromosomal breakpoint that produces the Philadelphia chromosome found in chronic myelogenous leukemia (CML). This involves the translocation of the c-abl oncogene on chromosome 9 to a gene on chromosome 22 (bcr). The point at which two genes come together is referred to as a chromosomal breakpoint (or translocation breakpoint). The BCR/ABL hybrid gene produces a novel transcript whose protein product has elevated tyrosine kinase activity and can contribute to uncontrolled cellular proliferation. Transgenic mice for this chimeric gene develop lymphoblastic leukemia and lymphoma. Since this gene product is directly linked to CML formation, it is a logical target for tyrosine kinase inhibitors in the treatment of chronic myeloid leukemia in humans (CML).
• Gene amplification. Quantitation of gene copy number is possible by a number of molecular techniques, including comparative genomic hybridization, genotyping arrays, and Southern hybridization. Amplification of oncogenes can occur in a number of tumor types and has been demonstrated in childhood neuroblastoma, where the myc proto-oncogene (nuclear transcription factor) is amplified up to 300 times. Gene amplification is possibly the most common mechanism of proto-oncogene activation. A further example is the MDM2 proto-oncogene, which has been identified in dogs and horses, and has recently shown to be amplified in a proportion of canine soft tissue sarcomas.31
• Point mutations. These are single base changes in the DNA sequence of proto-oncogenes leading to the production of abnormal proteins. Point mutations can arise through the actions of ionizing radiation, chemical carcinogens, or errors in DNA replication and repair. A mutation in a proto-oncogene or the transcriptional machinery that controls its expression may disrupt homeostasis and result in sustained proliferation signals or a failure to respond to negative feedback signals. A classical example is the Ras proto-oncogene in which point mutations are a consistent finding in a number of human tumors. K-ras mutations have also been identified in canine lung tumors. Mutations in the Erb-B (epidermal growth factor receptor) gene have been shown in a number of human cancers to lead to ligand-independent activation. Erb-2 mutations have been identified in canine mammary tumors.
• Viral insertions. The discovery of oncogenes was a direct result of studies on tumor-causing viruses. In some circumstances, proto-oncogene function can be damaged by the insertion of viral elements. Occasionally, novel retroviruses are isolated from leukemias or sarcomas in animals that have been viremic with a leukemia virus for some time. These viruses induce tumors very rapidly when inoculated into members of the species of origin and are referred to as acutely transforming oncoviruses. The prototype of the acutely transforming virus is the Rous sarcoma virus (RSV) isolated from a fowl in 1911. Subsequently, many more have been isolated from animals infected with avian, feline, murine, or simian oncoviruses. These viruses are generated by a rare recombinatorial event between the leukemia virus with which the animal was originally infected and a cellular proto-oncogene. In this, part of the viral genome is deleted and replaced with the cellular oncogene. The virus then becomes acutely transforming because this oncogene is now under the transcriptional control of very efficient viral promoters. This then allows infection of a cell and insertion of this continuously expressed oncogene into the cellular genome leading toward rapid progression and malignancy. Evidence suggests that these acutely transforming viruses are not transmitted naturally, but all events occur in the individual animal. Because the virus has itself lost some of its own genetic material, it is defective for replication. However, they are spread throughout an animal by the provision of help from the normal leukemia virus, which provides the missing proteins in co-infected cells.
In addition to acutely transforming mechanisms, retroviruses can activate cellular oncogenes by integrating adjacent to them. A good example of this is the myc gene, which is frequently activated in feline T-cell lymphomas. In one mode, the virus integrates adjacent to the oncogene and transcription initiation in the viral long terminal repeat (LTR) proceeds into the adjacent oncogene, producing a hybrid mRNA. In a second form, the enhancer of the virus overrides the regulation of the c-myc transcription from its normal promoter.
Tumor Suppressor Genes
Changes in genes can lead to either stimulatory or inhibitory effects on cell growth and proliferation. The stimulatory effects are provided by the proto-oncogenes as described. Mutations or translocations of these genes produce positive signals leading to uncontrolled growth. In contrast, tumor formation can result from a loss of inhibitory functions associated with another class of cellular genes called the tumor suppressor genes. The discovery of these genes began by observations of inherited cancer syndromes in children, in particular studies of retinoblastoma. In the early 1970s, epidemiologic studies of both retinoblastoma and Wilms’ tumor led Knudson to propose his “two hit” theory of tumorigenesis.
Retinoblastoma Forms the First Clues to the Existence of Tumor Suppressor Genes
Retinoblastoma occurs in two forms, a sporadic form and an inherited form (accounting for 40% of cases).32 In the inherited form, the mode of inheritance is autosomal dominant and about half the children are affected by the condition. Knudson’s model required the retinoblastoma tumor cells (in either sporadic or inherited forms) to acquire two separate genetic changes in the DNA before tumor development. The first or predisposing event could be inherited through the germ line (familial retinoblastoma) or it could arise de novo in somatic cells (sporadic form). The second event occurred in somatic cells. Thus, in sporadic retinoblastoma, both events arose in the retinal cells. However, in familial retinoblastoma, the individual had already inherited one mutant gene and only required a second hit in the remaining normal gene in somatic cells.
The mode of inheritance of retinoblastoma is dominant with incomplete penetrance. However, at the cellular level, loss or inactivation of both alleles is required to change the cells’ phenotype. The retinoblastoma gene codes for Rb, which was previously described as a normal cellular gene involved in control of the cell cycle. Rb is described as a tumor suppressor and, in a cell with only one normal allele, that allele usually produces enough tumor suppressor product to remain normal. Generically, mutations in tumor suppressor genes behave very differently from oncogene mutations. Whereas activating oncogene mutations are dominant to wild type (they emit their proliferating signals regardless of the wild-type gene product), suppressor mutations are recessive. Mutation in one gene copy usually has no effect, as long as a reasonable amount of wild-type protein remains. Consequently, some texts refer to tumor suppressor genes as recessive oncogenes.
More recently, Knudson’s hypothesis was confirmed when the Rb gene was cloned and characterized. The retinoblastoma tumor suppressor Rb is the principal member of a family of proteins that also encompass pRb2/p130 and p107. Rb plays a central role in regulating cell cycle progression in G1. Indeed, disruption of Rb function has been found to be a common feature of many human cancers not only retinoblastoma. Rb function can be abrogated by point mutations, deletions, or by complex formation with viral oncoproteins such as SV40 large T antigen and adenoviral E1a protein. The function of additional proteins associated with the Rb pathway are also subjected to deregulation in human cancers, including overexpression of D type cyclins, overexpression of CDK4, and downregulation of the CDKI cell cycle inhibitor p16.
Although loss of cell cycle control via the Rb pathway occurs commonly in many human tumors, little is known about the role of Rb, cyclin D, CDK4, and p16 in domesticated animal tumors.
The p53 Tumor Suppressor Gene
P53 is a gene whose product is intimately involved in cell cycle control. Its discovery by Sir David Lane in 1979 marked a major milestone in cancer research and has allowed greater understanding of molecular mechanisms of cancer and identified potential targets for therapeutic intervention.
The p53 protein has been described as the guardian of the genome by virtue of its ability to push cells into arrest or apoptosis, depending on the degree of DNA damage. Thus the p53 tumor suppressor gene plays an important role in cell cycle progression, regulation of gene expression, and the cellular response mechanisms to DNA damage. Under normal physiologic conditions, wild-type p53 can bind specific DNA sequences and regulate transcription of a number of genes involved in cell cycle progression and apoptotic pathways including p21waf1/cip1 and bax (see Figure 2-2). The p53-mediated mechanisms are responsible for tumor suppression and prevent accumulation of potentially oncogenic mutations and genomic instability. Failure by p53 to activate such cellular functions may ultimately result in abnormal uncontrolled cell growth leading to tumorigenic transformation.33-36
p53 is the most frequently inactivated gene in human neoplasia, with functional loss commonly occurring through gene mutational events, including nonsense, missense and splice site mutations, allelic loss, rearrangements, and deletions. However, p53 function can also be abrogated by several nonmutational mechanisms, including nuclear exclusion, complex formation with a number of viral proteins, and through overexpression of the cellular oncogene MDM2.
The homologs of p53 and MDM2 have both been identified in domestic animal species and a number of studies indicate that this gene also has a central role in the progression of veterinary cancers.37-41
Cancer Arises through Multiple Molecular Mechanisms
The advances in our understanding of normal cell biology and the processes that lead to malignancy have increased dramatically over the past 30 years. The last decade has shown us that transformation of a normal cell into a malignant cell requires very few molecular, biochemical, and cellular changes that can be considered as acquired capabilities.42,43 Further, despite the wide diversity of cancer types, these acquired capabilities appear to be common to all types of cancer. An optimistic view of increasing simplicity in cancer biology is further endorsed by the fact that all normal cells, irrespective of origin and phenotype, carry similar molecular machineries that regulate cell proliferation, differentiation, aging, and cell death.
We have discussed that tumorigenesis is a multistep process and that these steps reflect genetic alterations that drive the progression of a normal cell into a highly malignant cancer cell. This is supported by the finding that genomes of tumor cells are invariably altered at multiple sites. The spectrum of changes range from subtle point mutations in growth regulatory genes to obvious changes in chromosomal complement.
Cancer cells have defects in regulatory circuits that govern cellular proliferation and homeostasis and survival. A model has been proposed that suggests that the vast array of genetic abnormalities associated with cancer are a manifestation of eight alterations in cellular physiology that collectively contribute to malignant growth. First proposed in 2000 and updated in 2011, these “hallmarks” of cancer constitute an organizing principle for rationalizing the complexities of cancer and are underpinned by two overarching themes: genome instability and chronic inflammation.42,43 The eight acquired characteristics can be summarized under the following headings (see Figure 1-3):
• Insensitivity to antigrowth signals
• Evasion of programmed cell death (apoptosis)
• Limitless replicative potential
In addition, cancer exhibits another dimension of complexity in that they contain an array of “normal” cells that contribute to the acquisition and maintenance of the cancer hallmarks by creating the tumor microenvironment. The next section is an overview of these traits and the strategies by which they are acquired in cancer cells. The process of metastasis requires angiogenesis and invasion, as such the traits of sustained angiogenesis, and tissue invasion and metastasis will be collectively reviewed under a final section on metastasis.
The Hallmarks of Cancer
In the preceding section, we have described the normal cell and the role of oncogenes and tumor suppressor genes in cell cycle control and regulation. In one sense, cancer is a very common disease in animals and humans, but in fact the development of cancer is a rare event. When one considers the number of cells in the body, the proliferation and regulation of these cells and the potential for malignant transformation, then the development of a single cancer is rare. This is because of the cell’s own natural defenses against progression toward the malignant phenotype. The ability of the cell to effect DNA repair or initiate cell death is a defense mechanism against malignant transformation and serves to maintain cellular homeostasis. Each of the acquired capabilities described previously represents a breach in a cell’s homeostatic mechanisms. However, the biology of cancer cannot be understood by simply considering the phenotypic traits of the cancer cell. Rather the cancer phenotype is defined by the interactions between the cancer cell, the tumor microenvironment, and the enabling effects of fundamental genomic instability and chronic inflammation.
Self-Sufficiency in Growth Signals
Normal cells require mitogenic stimuli for growth and proliferation. These signals are transmitted to the nucleus by the binding of signaling molecules to specific receptors, the diffusion of growth factors into the cell, extracellular matrix components, or cell-to-cell adhesions or interactions. As previously discussed, many oncogenes act by mimicking normal growth signals. Tumor cells are not dependent on external mitogenic stimuli for proliferation and sustained growth but are self-sufficient. The liberation from dependency on exogenous signals severely disrupts normal cellular homeostasis. Arguably, the most fundamental trait of cancer cells is their ability to sustain chronic proliferation. By deregulating these signals, cancer cells become masters of their own destinies, promoting signaling (typically through intracellular kinase domains) to promote progression through the cell cycle, increases in cell size, increases in cell survival, and changes in energy metabolism.
The cancer cell can acquire this capability in the following ways:
• They may produce growth factor ligands themselves.
• They may induce stromal cells to produce such ligands.
• There may be an increase in receptor concentration on the cell surface that leads to receptor homodimerization or heterodimerization, making the cell hyperresponsive to ligands.
• There may be structural alterations in the receptor to support ligand-independent firing.
• Constitutive activation of the signaling pathway (downstream of the receptor). A good example of this is the constitutive activation of the PI3-AKT pathway, through mutations in the catalytic subunit of the PI3 kinase.
• Disruptions in negative-feedback mechanisms that attenuate proliferative signaling, such as the following:
• Mutations in the Ras gene compromise the Ras GTPase activity, which acts as a negative-feedback mechanism to ensure the effects of Ras are only transitory.
• PTEN is a tumor suppressor protein that counteracts PI3 signaling. PTEN loss has a similar effect to constitutive PI3 activation and promotes tumorigenesis.
Although the acquisition of growth signaling autonomy by cancer cells is conceptually satisfying, it is in fact too simplistic. One of the major problems of cancer research is to focus on the cancer cell in isolation. It is now apparent that we must also consider the contribution of the tumor microenvironment to the survival of cancer cells. Within normal tissues, paracrine and endocrine signals contribute greatly to growth and proliferation. Cell-to cell growth signaling is also likely to operate in cancer cells and may be as important as some of the autonomous mechanisms of tumor growth. It has recently been suggested that growth signals for the proliferation of carcinoma cells are derived from the tumor stromal elements (e.g., cancer or carcinoma-associated fibroblasts [CAFs]). It is therefore possible that the survival of tumor cells not only relies on the acquisition of growth signal autonomy but may also require the recruitment or modulation of stromal cells to provide these growth signals.
Insensitivity to Antigrowth Signals or Evading Growth Suppressors
Within the normal cell, multiple antiproliferative signals operate to maintain cellular quiescence and homeostasis. These signals include soluble growth inhibitors that act via cell surface receptors and immobilized inhibitors that are embedded in the extracellular matrix and on the surface of nearby cells. The signals operate to push the cell either into G0 or into a postmitotic state (usually associated with the acquisition of specific differentiation–associated characteristics) and are thus intimately associated with cell cycle control mechanisms. Cells monitor their external environment during the progression through G1 and, on the basis of external stimuli, decide whether to proliferate, become quiescent, or enter into a postmitotic state.
Most cellular programs that negatively regulate cell growth and proliferation depend on the actions of tumor suppressor genes. At the basic level, most of the antiproliferative signals are funneled through the Rb protein and its close relatives. Disruption of Rb allows cell proliferation and renders the cell insensitive to antiproliferative signals such as that provided by the well-characterized transforming growth factor-β (TGF-β).32 The Rb protein integrates signals from diverse extracellular and intracellular sources and can control cell cycle progression. The other major tumor suppressor is p53, which integrates intracellular signals and can promote either cell cycle arrest or apoptosis (depending on the degree of cellular stress or damage). However, the effects of p53 expression are highly context dependent. Loss of Rb or p53 is associated with the malignant phenotype through the cell’s ability to evade antigrowth signals.
In addition to tumor suppressor gene loss, cells can also evade antigrowth signals by the following alternative cellular programs:
• Another characteristic of the cancer cell is the evasion of contact inhibition. Cell-to-cell contact in most normal cells results in an inhibitory signal against further cell proliferation. The role of this mechanism in vivo has been thought to be to maintain tissue homeostasis. In cell culture, contact inhibition is abrogated in cancer cell monolayers, leading to their indefinite expansion. NF2 and LKB1 genes are considered tumor suppressor genes that are involved in this process and loss of these genes in vivo may promote loss of contact inhibition that contributes to the progression of cancers.
• Although TGF-β has antiproliferative effects in cancer, it is now, however, appreciated that the TGF-β pathway can be corrupted in the later stages of malignancy and can contribute to cancer progression. In this late effect, TGF-β is found to activate a cellular program termed epithelial-to-mesenchymal transition (EMT) that promotes invasion and metastasis (see later).
Evading Cell Death: The Roles of Apoptosis, Autophagy, and Necrosis
The growth of any tumor depends not only on the rate of cell division but also the rate of cellular attrition (mainly provided by apoptotic mechanisms). Basic molecular and pathologic studies of tumors have confirmed that acquired resistance toward apoptosis and other types of death is a hallmark of all types of cancer.
Cancer cells, through a variety of strategies, can acquire resistance to cell death and apoptosis. One of the most common ways is through loss of function of the tumor suppressor protein p53. Loss of p53 protein function occurs most often through mutation but can also be via sequestration or inactivation of the protein by viral proteins or by amplification of other oncogenes such as MDM2. Removal of normal p53 function leads to failure of the cells’ sensor mechanism for DNA damage. When the cell suffers an insult such as UV radiation, hypoxia, or exposure to DNA-damaging agents, signals are funneled through p53 to cause either cell cycle arrest or apoptosis. Failure of this mechanism can contribute to the progression of the cell toward malignancy and promoting the accumulation of additional genetic defects that are not corrected at defined checkpoints in cell cycle progression.35
The mechanisms involved in apoptosis are now well established, as are the strategies by which cancer cells evade its actions. However, recently there have been conceptual advances involving other forms of “programmed cell death” as a barrier to cancer development. A notable example is the emerging role that autophagy plays in cancer development. Autophagy is a normal cellular response that operates at low basal levels in cells but can be induced in states of cellular stress such as nutrient deficiency. In autophagy, there is controlled breakdown of cellular organelles that yields energy and cellular substrates that can be used for a variety of cellular functions. Recent evidence suggests that autophagy may be involved in both tumor cell survival and, paradoxically, tumor cell death, depending on the cellular state. The link between apoptosis and autophagy suggests that autophagy may represent another barrier for cells to overcome before they can attain malignancy. In contrast, it has also been shown that irradiation or cytotoxic drug treatment in late-stage tumors may promote autophagy, leading to cells attaining a state of reversible dormancy. The situation seems to suggest that autophagy is a barrier to tumor development in early disease but, in late stage disease, may allow cancer cells to survive severe cellular stress.44
In both autophagy and apoptosis, the process does not lead to the release of any “proinflammatory” signals. In contrast, the process of necrosis, observed in larger tumors, causes release of signals that support an influx of inflammatory cells. For many years, this has been considered to be a positive event, helping to expose the immune system to tumor antigens and promote immune destruction. However, recent evidence suggests that some phenotypes of inflammatory macrophages can actually support tumor growth through fostering of angiogenesis, cancer cell proliferation, and invasion. Consequently, our understanding of macrophage phenotypes in cancer progression is an area of active research.
Limitless Replicative Capacity
Over 30 years ago the pioneering observations of Hayflick established that when normal human or animal cells are grown in culture, they demonstrate a finite replicative lifespan. That is, they are capable of a finite number of cell divisions, after which they undergo what has been termed replicative senescence and are incapable of any further cell division. The mechanism underlying the replicative clock that monitors this process has been the subject of intense research. This process has further evoked considerable interest as it is also one of the mechanisms that must be overcome to establish the immortal phenotype that is characteristic of the cancer cell.45-47
In mammalian cells, the DNA is organized into chromosomes within the nucleus and these are capped by specialized DNA-protein structures known as telomeres. The major function of these structures is protection, but they are progressively eroded at each cell division because of the inability of DNA to completely replicate itself. The result is that there is progressive telomeric attrition as cell populations double. After an estimated 50 cell divisions, cells enter an irreversible (and prolonged) state of cellular senescence (sometimes referred to as mortality stage 1 [M1]). This period is characterized by arrest of proliferation without loss of biochemical function or viability. At the end of this period, cells exhibit altered morphology and chromosomal instability, a state often referred to as crisis (mortality stage 2 [M2]) (Figures 2-5 and 2-6). Thus telomeric attrition is intimately involved with the aging of cells. Cancer cells must overcome replicative senescence and take on an immortal phenotype.
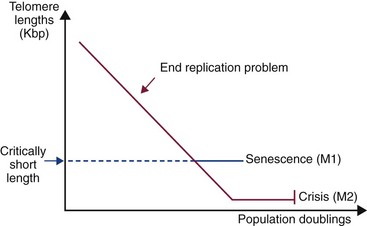
Figure 2-5 Telomeric attrition in cultured cells. In mammalian cells the DNA is organized into chromosomes within the nucleus, and these are capped by specialized DNA-protein structures known as telomeres. The major function of these structures is protection, but they are progressively eroded at each cell division because of the inability of DNA to completely replicate itself. The result is that there is progressive telomeric attrition as cell populations double. After an estimated 50 cell divisions, cells enter an irreversible (and prolonged) state of cellular senescence (sometimes referred to as mortality stage 1 [M1]). This period is characterized by arrest of proliferation without loss of biochemical function or viability. At the end of this period, cells exhibit altered morphology and chromosomal instability, a state often referred to as crisis (mortality stage 2 [M2]).
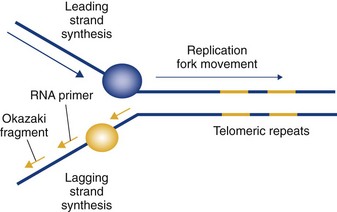
Figure 2-6 The end replication problem. Telomeric attrition arises because of the inability of chromosomes to completely replicate their extreme 5′ ends (end replication problem).
It has now been demonstrated in human tumors and, more recently, tumors of the dog, that telomere maintenance is a feature of virtually all cancer types.46-53 Tumor cells succeed in telomeric maintenance by the expression of the enzyme telomerase. From studies on cellular senescence, expression of the enzyme telomerase has emerged as a central unifying mechanism underlying the immortal phenotype of cancer cells and has thus become the most common marker of malignant cells. Telomerase is a ribonucleoprotein enzyme that maintains the protective structures at the ends of eukaryotic chromosomes, at the telomeres. In humans, telomerase expression is repressed in most somatic tissues, and telomeres shorten with each progressive cell division. In contrast, telomerase activity is a common finding in many human malignancies, resulting in stabilized telomere length. The telomerase complex consists of an RNA subunit that contains a domain complementary to the telomeric repeat sequence TTAGGG and a catalytic protein component. The catalytic protein component acts as a reverse transcriptase and can catalyze the addition of telomeric repeats onto the ends of chromosomes, using the RNA subunit as a template. It is now well documented that the level of telomerase in malignant tissue compared to normal tissue is much higher, and this differential is greater than that for classic enzymatic targets such as thymidylate synthase, dihydrofolate reductase, or topoisomerase II.
Telomerase biology is complex, and the mechanisms by which telomerase becomes reactivated in tumor cells is the subject of intense research. However, this represents an exciting opportunity for further understanding the complex biology of cancer and also the identification of completely novel targets for therapy.
Reprogramming Energy Metabolism
The sustained growth and proliferation of a cancer requires a corresponding adjustment of energy metabolism to ensure this growth can be fueled. Under normal conditions, cells respire aerobically in that they metabolize glucose to pyruvate with a net gain in energy as ATP.43 Cancer cells can undergo a “metabolic switch” so that glucose is metabolized to lactate, in the presence or absence of oxygen, causing a net energy deficit. There is a corresponding upregulation of glucose transporters (e.g., GLUT-1), which increases uptake of glucose into the cytoplasm. This process is exploited in positron-emission tomography (PET) imaging as tumors will preferentially uptake a radiolabeled analog of glucose (18F-fluorodeoxyglucose [FDG]). This metabolic switch is sometimes referred to as the Warburg effect (Figure 2-7).54
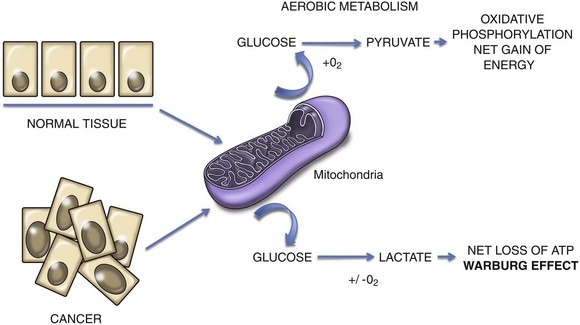
Figure 2-7 In normal tissues, cellular respiration in the presence of oxygen allows the production of a net gain in energy through metabolism of glucose to pyruvate. The Warburg effect in cancer cells refers to a metabolic switch that causes cancer cells to preferentially metabolize glucose to lactate irrespective of oxygen status, leading to a net loss of energy in the form of adenosine triphosphate (ATP).
It is difficult to appreciate the survival advantage of this mechanism. One hypothesis is that the switch allows the diversion of glycolytic intermediates into other biosynthetic pathways that support the production of new cells. This is supported by the observation that the Warburg effect can also be detected in growing cells in the embryo. An expansion of the theory of reprogramming of energy metabolism in cancer suggests that cancer cells are advantaged by a flexibility in their ability to derive ATP. This may come from a number of metabolic pathways that derive ATP from glucose metabolism under aerobic and anaerobic conditions but may also include efficiencies in metabolizing amino acids and lipids toward ATP and other biomolecule synthesis. Such metabolic flexibility may be necessary during primary tumor development but even more so during metastatic progression (see next section).
Metastasis
Metastasis is defined as the dissemination of neoplastic cells to discontinuous secondary (or higher order) sites, where they proliferate to form a macroscopic mass. Implicit in this process is the presence of a primary tumor. Metastases are not a direct extension of the primary tumor and are not dependent on the route of spread (i.e., hematogenous versus lymphatic versus peritoneal dissemination). The process of metastasis is believed to occur through the completion of a series of step-wise events. In order for this process to occur, a cancer cell must leave the site of the primary tumor, pass through the tumor basement membrane, and then through or between endothelial cells to enter the circulation (extravasation). While in the circulation, tumor cells must be able to resist anoikis (programmed cell death associated with loss of cellular contact), evade immune recognition and physical stress, and eventually arrest at distant organs. At that distant site the cell must leave the circulation and survive in the hostile microenvironment of the foreign tissue. These secondary sites are believed to be primed to receive metastatic cells through effects that are directed and mediated by the primary tumor itself (premetastatic niche). This distant site may be the eventual target organ for metastasis or may be a temporary (sanctuary) site. In either case, the cancer cell is thought to lie dormant for a variable and often protracted period of time before moving to its final location. Following a break in dormancy, cells receive signals to proliferate, create new blood vessels (angiogenesis) or co-opt existing blood vessels, and then successfully grow into a measurable metastatic lesion. It is likely that further progression is associated with the repetition of this process resulting in the development of metastases from metastases. As such, the steps outlined here continue not only after the detection of the primary tumor but also after the detection of metastases. From a therapeutic perspective, it is therefore never too late to target the biologic steps associated with metastatic progression as a means to improve outcomes for patients. The basic tenets of this model of metastasis have been intact for over 40 years; however, a greater understanding of biologic principles associated with each metastasis process is emerging.55
Metastasis-Associated Genes and Metastasis Suppressor Genes
Cancer cells are not unique in their ability to complete the individual steps required for metastasis (Figure 2-8). For example, leukocytes and neuronal cells have the ability to invade tissue planes and cross vascular barriers. Several types of leukocytes demonstrate the phenotype of intermittent adherence to vascular endothelium and are able to resist anoikis. It is also true that stem cells of various phases of differentiation are able to perform many of these steps during development and in the adult.56 What is unique about metastatic cells is that each must be able to perform all the steps required for successful metastasis. An extension of this argument is that the genetic changes that permit the metastatic process are not unique to a metastatic cancer cell; however, the metastatic cancer cell must have the appropriate set of genetic changes available to complete all the steps of the metastatic cascade.
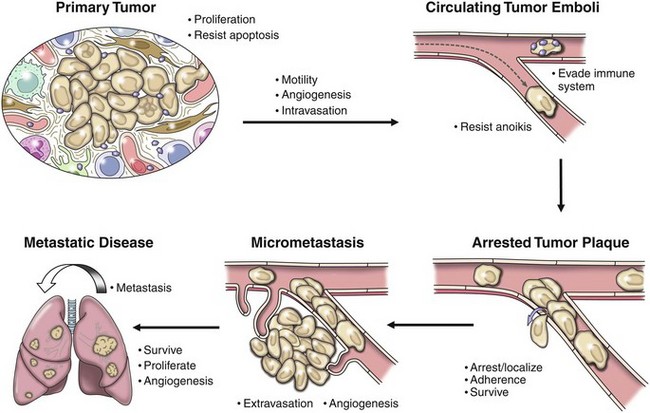
Figure 2-8 The metastatic cascade. The metastatic cascade describes a set of discrete steps that cells must move through as a part of the process of metastasis. This complexity begins with the recognized complexity associated with primary tumor development in which tumor cells must proliferate, resist apoptosis, and develop interactions with many host cells in the microenvironment. Subsequent steps then allow metastatic cancer cells to enter and survive in the circulation. Cells then arrest at distant locations, extravasate, and survive in the microenvironment of the distant locations. Arrest of cells may be at the eventual secondary organ where metastasis becomes clinically evident or cells may initially arrest at a “sanctuary site” where they may lie dormant before moving on to the eventual secondary site. The cellular programs that result in a break of dormancy are not well understood. Survival of cells at these sites of dormancy or the eventual secondary site is a significant hurdle for metastatic cells to overcome. Indeed, the majority of cells that arrive at distant locations are unable to survive in these distant locations. At secondary sites, tumor cells may proliferate and progress following the development of the angiogenic phenotype. It is likely that successful metastatic lesions at secondary sites are then the source of subsequent metastases within these secondary sites.
Literally hundreds of genes and their resultant proteins have been suggested to contribute to the development of cancers and to their eventual ability to metastasize. It is possible for a single genetic change in cancer to contribute many of the metastasis-associated processes or for several genes to work together toward a single metastasis-associated process. Metastatic cancers may achieve the metastatic phenotype through distinct constellations of genetic and “epigenetic” events that in their respective sums complete the list of necessary metastasis-associated processes needed for successful metastasis. Two classes of genes have been broadly defined as contributing to the metastatic phenotype. These include metastasis promoting genes57,58 and metastasis suppressors.59,60 These genes have functions in normal development and physiology (i.e., cell migration, tissue invasion, and angiogenesis discussed earlier) that are subverted by the cancer cell in the acquisition of the metastatic phenotype.
The use of high through-put and genome-wide investigations has uncovered many putative metastasis-associated genes in cancer. It should be noted that many metastasis-associated genes have functions that also contribute to tumor formation and progression. Several of these metastasis-associated genes have been validated in canine and feline cancers.61,62 For example, the metastasis-associated gene ezrin was identified using genomic approaches in murine and human studies of metastasis.63 Ezrin is a membrane-cytoskeleton linker protein that functionally and physically connects the actin cytoskeleton to the cell membrane. The degree of ezrin expression in the primary tumor of dogs with osteosarcoma was shown to predict a more aggressive course of disease, defined by metastasis to the lung. Furthermore, recent studies have confirmed the connection between ezrin and protein kinase C (PKC) signaling in murine, canine, and human osteosarcoma cells.64,65
Metastasis suppressor genes have been identified in several human cancers. These genes are thought to also have normal functions in the regulation of motility, invasion, and angiogenesis. The loss of these genes is not thought to be associated with the formation of tumors; however, their loss is thought to contribute to specific steps in the metastatic cascade. The characterization, biology, and clinical impact of metastasis suppressor genes have been recently reviewed elsewhere.66 The loss of reduced expression of metastasis suppressors has not been documented in canine or feline cancers at this time.
The following sections provide descriptions of the critical metastasis-associated processes (see Figure 2-8). Examples of genetic changes or resulting protein changes that contribute to each process is highlighted in each section, with particular emphasis on those with demonstrated associations with veterinary malignancies.
Intravasation
Following the successful growth of the primary tumor, intravasation of a cancer cell into the vascular or lymphatic circulation is the first step required during the metastatic cascade. The process of intravasation requires that a tumor cell be motile and able to digest, modulate, or escape the extracellular matrix.67-69 The specific mechanisms used by cancer cells to invade and intravasate include the classical model of enzymatic degradation of the extracellular matrix referred to as mesenchymal invasion (see later discussion on epithelial-mesenchymal transition). Unlike mesenchymal invasion, tumor cells may also develop so-called amoeboid invasion, in which they individually slip between fibers of the extracellular matrix without evidence or a need for enzymatic degradation.70 Finally, a distinct method termed collective invasion refers to the en masse regional extension of a tumor into surrounding tissues.71 Such collective invasion is observed clinically in dogs with oral SCC and biologically high-grade/histologically low-grade fibrosarcoma. The low rate of distant metastasis associated with collective invasion suggests a lack of functional attributes necessary for true distant metastasis. It is likely that individual metastatic tumors may utilize distinct invasion programs at distinct points during metastatic progression. Not surprisingly, studies with intravital imaging (single cell imaging of cancer cells in animals) have demonstrated that only a minority of the cells in the primary tumor develop any of these forms of invasion.72,73 Efforts are underway to define the genetic features of this minority population.74,75
In the classic mesenchymal form of invasion, matrix proteases and metalloproteases (MMPs) are believed to be necessary for the invasive phenotype. Expression of members of this enzyme family have been found in most human and several canine and feline malignancies, including canine osteosarcoma.76-79 The activity of MMPs in osteosarcoma and mast cell cancers has been correlated with grade and metastatic propensity.79,80 Similar correlative studies have been undertaken in human patients.81 The importance of MMP activity during this early step in metastasis prompted the development of pharmacological inhibitors of MMPs. Anticancer activity of these agents in preclinical animal models and in sporadic human patients was evident; however, in randomized trials, the activity of MMPs was not observed.82,83 The failure of these clinical trials may be explained at many levels and does not refute the importance of invasion as a critical step for a metastatic cell. Rather, these results suggest potential redundancy in the types of invasion (mesenchymal versus amoeboid) that may exist within a given cancer.84,85 Recent evidence also suggests that the expression of matrix-degrading enzymes and other growth factors may not be necessary in the tumor cells themselves but may be provided by inflammatory cells (i.e., macrophages) recruited by the growing tumor.86
Epithelial-Mesenchymal Transition
Observed primarily in the context of epithelial malignancies, the ability of tumor cells to engage in the early steps in the metastatic cascade has been linked to a transcriptional program referred to as epithelial-mesenchymal transition (EMT).87 This transcriptional program has been largely ascribed to the effects of a family of transcription factors, including twist, snail, and slug.88 Activation of these and other EMT transcription factors is not necessarily associated with a morphologic change in the cancer cell but may be associated with a loss of polarity (apicobasal) in epithelial cells, a greater proportion of cells losing cell-to-cell contacts similar to mesenchymal cells (observed in vitro), cell motility, and invasion. In the “mesenchymal” state, suggested by EMT, epithelial cancer cells (similar to cells involved in embryogenesis) develop the phenotypic ability to undergo invasion, migration, and intravasation.89 Interestingly, cells that are able to take on these “mesenchymal” features share signaling programs and other phenotypes with tumor-initiating cell populations.90 Opponents of the EMT hypothesis are most critical of the use of the term transition, which suggests a switch in the phenotype of individual cells from an epithelial to a mesenchymal form. It is, however, reasonable and generally agreed that the effects of EMT transcription factors contribute to the early phenotypes needed for metastatic progression. Limited data also exist that suggest a need for mesenchymal cells to take on attributes of epithelial cells (likely later during metastatic progression) for successful metastasis, the so-called mesenchymal-epithelial transition (MET). For epithelial cancers, MET is also understood to be a final step in EMT, in which following the activation an invasive phenotype and colonization of a distant site cells will revert to their original epithelial phenotypes.
The process of intravasation concludes when a cancer cell successfully enters the vascular or lymphatic circulations. Tumor cells may enter the circulation through established blood vessels, small arterioles, venules, lymphatics, or though tumor-associated (or lined) blood vessels, in a process referred to as vasculogenic mimicry.91 For larger vascular structures, the process of intravasation requires penetration of adventitial cells, including pericytes; digestion of the vascular basement membrane; and penetration between or through endothelial cells.92 Penetration of the tumor-associated vasculature may be easier than invasion through normal vessels and may only require transit from the extracellular environment between endothelial cells into the circulation.
Survival in the Circulation (Resisting Anoikis)
Frisch and Francis reported the induction of apoptosis after disruption of the interaction between epithelial cells and the extracellular matrix.93 This phenomenon was termed anoikis, from the Greek word for homelessness. In normal tissues, anoikis is a mechanism for maintaining tissue homeostasis and integrity.94 For the metastatic cancer cell, survival during dissemination requires resistance to anoikis. In normal tissues, anoikis is prevented by two systems: cell-matrix anchorage and cell-cell interactions.95 Anchorage of cells to the extracellular matrix is mediated primarily by transmembrane receptors referred to as integrins. Formation of active heterodimers trigger an intracellular cascade resulting in activation of effectors of growth and survival.96 Integrin family members have been identified in canine sarcomas and lymphomas.97-102 Cell-cell anchorage in many epithelial tissues is mediated by cadherins, a family of calcium-binding glycoproteins. Intracellularly, cadherins form complexes with members of the catenin protein family that link them to the actin cytoskeleton, as well as survival-promoting signal transduction cascades.103 Loss of either cell-cell or cell-matrix interaction in normal cells triggers the activation of the caspase proteases, the hallmark of apoptotic cell death. Metastatic cancer must resist this contact-dependent death in order to be successful and do so through two nonmutually exclusive mechanisms. The first is by maintaining cell-cell contacts with other tumor cells (homotypic interactions) or with host cells such as platelets and inflammatory cells (heterotypic interactions) during metastatic progression. Both homotypic and heterotypic interactions generate intracellular signals that prevent the initiation of anoikis. Additionally, cancer cells may overexpress proteins that directly inhibit anoikis. For example, the integrin pair αvβ3 is frequently overexpressed in malignancy, including prostate cancer and melanoma.104,105 This overexpression subverts the need for ligand binding and results in the generation of survival signals.106 Proteins reported to be involved in resistance to anoikis include Trk B, focal adhesion kinase (FAK, the immediate effector of integrin signaling), galectin-3, and TGF-β, among others. Certainly more molecular mediators of anoikis resistance have been identified and reviewed elsewhere.107 These molecules may be valuable antimetastasis targets for novel therapy.
Evasion of the Immune System
At all stages of metastatic progression, metastatic tumor cells must evade detection and destruction by the immune system. The ability of the host immune system to recognize and destroy tumor cells (immunosurveillance) was first proposed by Paul Ehrlich in 1909. Molecular support of the theory of immunosurveillance has come with studies of mice deficient in immunomodulatory and proinflammatory molecules such as interferon-γ (IFN-γ), interleukin-12 (IL-12), and perforin. Mice deficient in these molecules are known to develop tumors more readily than wild-type mice. Clinical evidence for immunosurveillance against cancer was first reported by Coley over 100 years ago. Through the administration of bacteria, Coley’s toxin, Coley was able to induce fever and tumor regression in patients with cancer. Evidence in dogs with osteosarcoma further supports the potential value of cancer immunotherapy.108 Indeed, survival times in dogs who develop bacterial infection at the site of a limb salvage surgery are significantly longer than in dogs who do not develop infection. Interestingly similar parallels may be seen in human osteosarcoma patients. In immunocompetent hosts, it is believed that immunosurveillance removes a large number of cancer cells from the primary tumor, from the circulation, and at distant metastatic sites. Cancer cells employ a wide variety of mechanisms to effect this evasion. The mechanisms for immunosurveillance and evasion from immunosurveillance by cancer are reviewed elsewhere and are summarized in Chapter 13 of this text.109
Modification of the immune system to treat cancer continues to be an attractive therapeutic strategy.110-112 Clinical trials based on this concept have been reported throughout the veterinary literature in dogs with melanoma, soft tissue sarcoma, hemangiosarcoma, osteosarcoma, and others, using a variety of immune-based therapies.113 Indeed, this principle has been the basis for the development and approval of a therapeutic vaccine directed against a melanoma antigen in dogs with melanoma.114
Arrest in Target Tissues
The arrest of circulating tumor cells at distant sites is thought to occur by two distinct but potentially overlapping mechanisms. These include size-dependent “trapping” of tumor cells within the lumen of small vessels (capillaries and veins) in the target organ, and/or receptor mediated interaction involving the tumor cell and the host vasculature.115 Data supporting the “trapping” phenomenon comes from single-cell imaging studies in which metastatic cancer cells were observed to primarily arrest at distant vascular beds as a result of size-dependent restrictions of large tumor cells in small blood vessels.116,117 This work was primarily conducted for metastasis to the liver and more recently metastasis to the brain.118 This trapping phenomenon suggests that the site for metastasis from a primary tumor is largely guided by the location of the first (small vessel) vascular bed encountered by a tumor cell. Alternatively, several groups have suggested the role of specific adhesion molecules as being necessary for initial tumor arrest in the microenvironment of the secondary metastatic sites.119-121 It is likely that both of the mechanisms play a role in the initial seeding of distant metastatic target organs. The dominant mechanism of arrest may be primarily defined by tumor type or target organ.
Early Survival at Distant Sites
Once the cancer cell or cancer embolus has arrested at its distant sites, it may immediately move out of the circulation into the target organ or may stay within the circulation.122 In either location, the cancer cell must survive in a new microenvironment. Early survival of metastatic cells at secondary sites is a significant hurdle for the cancer cell. Several studies have shown the ability of cancer cells to arrest at multiple organs in the body. Within hours, the number of remaining cells is dramatically reduced and within days, the number of viable cells may be as low as 0.1% of the original number of cells, even for the most aggressive cancer models.63,123 The organ selectivity for cancers is to a large extent defined by the organs in which a cancer cell can survive following initial arrest. The “seed and soil” hypothesis first articulated by Paget and then more recently by Fidler et al suggests that the success of a cancer, or metastasis, is defined by interactions between the seed (tumor cell) and the soil (the tumor microenvironment). Arrest of tumor cells in target organs may or may not be receptor mediated (discussed earlier); however, adhesion requires receptor-ligand interactions. Contributors to these steps include the cell adhesion molecules (CAMs). Multiple CAMs have been identified and are named based on their cell specificity (e.g., N-CAM for neural and L-CAM for liver). CD44 is a specific adhesion molecule (H-CAM for homing), initially identified as the receptor for the matrix component hyaluronic acid on hematopoietic cells. Expression of splice variants of CD44 on tumor cells has been demonstrated to correlate with poor prognosis in a wide variety of human tumors from acute myelogenous leukemia (AML) and Hodgkin’s disease to breast and colon cancer and osteosarcoma.124 Chemokines are a class of chemotactic cytokines that function in leukocyte trafficking and function. In metastasis, chemokines may also contribute to the metastatic process through enhanced metastatic cell survival.125 Chemokines may be active in the recruitment of a leukocytic infiltrate to intravasated, circulating tumor cells, therein creating an embolus that can better resist shearing stress associated with circulation or through the generation of matrix degrading proteins at the distant metastatic site.
The Premetastatic Niche and Modulation of the Microenvironment
It is increasingly clear that the sites (microenvironments) of successful secondary metastasis are modulated by the presence of a primary tumor and then by the arrival of metastatic cells at the secondary site. The concept of the premetastatic niche suggests that a primary tumor modulates the microenvironment of a secondary site before the arrival of most metastatic cells.126 The modulation of the premetastatic niche appears to be accomplished by primary tumor-induced mobilization and recruitment of specific primarily bone marrow–derived cells (bone marrow niche) to the secondary microenvironment.127 These bone marrow–derived cells are myeloid in origin and express the vascular endothelial growth factor receptor (VEGFR). Interestingly, the sites of metastatic tumor arrest appear to preferentially include sites in which these myeloid-derived cells were first recruited. Furthermore, targeting these VEGFR-positive cells using pharmacologic and genetic tools has effectively prevented metastatic development in murine models.127 Ongoing studies will likely uncover greater complexity of the populations of cells recruited to the premetastatic niche and potentially therapeutic targets for antimetastatic therapy (see Figure 1-5).
Following survival at the distant site, the tumor cell must proliferate and modulate its new environment. In most cases, it is believed that cancer cells extravasate from the circulation and then proliferate in the new organ. However, it is also possible for proliferation to occur within blood vessels, in a process referred to as intravascular metastases, and then expand out into the local tissue before further proliferation occurs.128 In both situations, modulation of the new environment is necessary for appropriate growth and progression of the metastatic lesions. It is now recognized that part of this modulation is based on tumor-induced changes in the stroma.129,130 These stromal changes may result in the production of growth factors or signals that are used by the tumor for further growth. These stromal cell–derived growth factors provide important signals for tumor cell proliferation and progression, including angiogenesis. The importance of tumor-stromal interaction has suggested that the “induced” tumor-stroma may be a credible target for novel cancer therapeutics.131
Angiogenesis
It is now well accepted that the development of new blood vessels from endothelial progenitors (vasculogenesis) or from existing blood vessels (angiogenesis) is required for cancer progression and metastasis.132,133 Endothelial cells or endothelial progenitors are activated by tumor-derived growth factors and result in new capillaries at the tumor site.134 In healthy tissue, endothelial cell proliferation is controlled by a balance between protein factors that activate endothelial cells and those that antagonize activation. Malignant tumors provide signals that result in endothelial cell survival, motility, invasion, differentiation, and organization. These steps are required to create a supportive vasculature for the tumor. In many ways, these required endothelial processes share parallel features with the processes required for the success of a metastatic cancer cell itself. The creation of new blood vessels requires the tumors to recruit circulating endothelial cells to their site, presumably through the release of growth factors such as vascular endothelial growth factor (VEGF). Circulating endothelial cells must survive at their new site with the help of survival signals (e.g., thrombospondin-I [TSP-I]) and form vascular tubes that then reorganize to sustain blood flow. The resulting vasculature of cancers is typically aberrant with often poorly organized and chaotic vascular structures that are leaky, with limited adventitial development and excessive branching.135 Once developed, this angiogenic phenotype (the result of the angiogenic switch) is associated with a diverse pattern of ongoing angiogenesis and neovascularization. This ongoing process is likely complex and involves a wide variety of tumor and microenvironmental-derived growth factors and signaling molecules. Adding to this complexity, it is likely that certain phases of cancer progression are associated with and require periods of antiangiogenesis. Indeed, these hypovascularized states may directly contribute to the progression of certain cancers.136
Many lines of evidence support the importance of angiogenesis in the biology of metastasis. The vascularity of a primary tumor (measured by microvessel density) has been correlated with metastatic behavior for most human and many veterinary tumors. The expression of angiogenesis-associated growth or survival factors and their receptors (i.e., VEGFR) in serum and in tumors, respectively, has also been correlated with outcome; more recently, functional imaging studies using magnetic resonance imaging (MRI) and other means has provided correlates of vascularity with poor outcome.137,138 The strength of this biologic argument has supported the development of a number of novel therapeutic agents with antiangiogenic activities. These agents have moved through discovery and development and are now approved drugs for cancer (see Section C in Chapter 14).
Metastasis from Metastases
For most solid tumors the appearance of a single metastatic nodule is followed shortly by the development of additional metastases within the same target organ and tertiary sites. It is unlikely that these metastatic sites all emerge from distinct and unique clones within the primary tumor and nearly simultaneously progress through the metastatic cascade to yield synchronous metastases at the distant site. It is reasonable and perhaps more likely that metastases develop from other metastatic sites. In this hypothesis a small number of successful clones colonize distant sites. As these clones move through progression to become successful metastases, the process of metastasis continues, resulting in metastasis from metastases. Although data supporting this hypothesis are limited, the implication for the treatment and management of cancer patients is substantial. If true, the process of metastasis from metastases would suggest that all steps in the metastatic cascade occur continuously, both before and after detection of metastases in patients. As such, all of the steps in the metastatic cascade may be targets for future therapeutic intervention.
Ongoing Controversies and Areas of Research in the Field of Metastasis
Does the Metastatic Propensity for Tumors Emerge Early or Late in the Biology of Cancer?
The development of the metastatic phenotype has been traditionally believed to be a process that happens late in carcinogenesis. In this model, referred to as the progression model, the genetic changes responsible for primary tumor development are in most cases distinct and precede those steps that result in the metastatic phenotype.139,140 The progression model argues that the metastatic phenotype is acquired within a small fraction of cells within the heterogeneous primary tumor. Support for the progression model came from work by Fidler and others, who demonstrated the ability to select for rodent cancers with greater metastatic potential, through the serial passage of metastatic tumor nodules back to naïve mice. This selection phenomenon suggested that a minority of tumor cell clones within a primary tumor were endowed with the metastatic phenotype and that this small proportion were enriched in the metastases compared to the primary tumor. Application of the progression model would suggest that a period of time exists between primary tumor development and acquisition of the full metastatic phenotype. This model was thought to be the basis of the improved outcome associated with early detection of cancers and the belief that effective and definitive therapy was most likely if a diagnosis was made early. Work from Ramaswamy et al provided data to support an alternative model,57 “the early oncogenic model” for metastasis. This alternative suggests that the genetic events that contribute to initial primary tumor development are the same or emerge at the same time as the events that contribute to the metastatic phenotype. As such, the early oncogenic model suggests that the biology of a cancer is defined early and may not be something that can be reduced through the early identification of a cancer. This is not to say that early detection of a cancer is not helpful for a patient, but rather that bad cancers may be “born” bad. This model may explain the phenomenon of the unknown primary tumor, wherein metastatic disease is detected without an apparent primary tumor.
To add complexity to the question of emergence of the metastatic phenotype, there is increasing evidence that host (genomic) differences can influence metastatic behavior of cancers, without necessarily influencing primary tumor growth.141,142 Using a genetically engineered mouse model of mammary cancer, Hunter et al have been able to identify specific host genes that influence metastatic behavior of tumors.141,143,144 These findings have several important implications. First, they suggest that individuals might be predisposed to aggressive metastatic progression before the development of a tumor. In addition, it suggests that there may be families in the population that are at high risk, not necessarily for tumor development, but for aggressive metastatic course once a tumor develops. Most importantly, if a significant part of a patient’s metastatic risk is encoded within the patient’s constitutional genome rather than within the mutated tumor genome, it may be feasible to identify those individuals at high risk for metastatic disease at the time of diagnosis of the primary tumor, or potentially before. This finding may be particularly relevant in the field of veterinary oncology, in which breed-associated genomic differences may explain the more aggressive course of disease seen in some dogs compared to others. Taken together, the risk for metastatic progression is in part defined by the genetics of the patient, genetic changes that develop early in the process of tumor development, and the subsequent and incremental emergence of aggressive metastatic cells.
Where Is the Inefficiency in Metastatic Inefficiency?
As devastating as the metastatic process is, it is equally inefficient. Estimates of this inefficiency in animal models suggest that less than 1% of cancer cells that successfully enter the circulation are able to survive at distant sites.145 The true metastatic inefficiency of human cancers is likely to be much lower. In most studies to date, it does not seem that entry of cancer cells into the circulation is the major barrier for successful metastases. Recent studies have identified high numbers of circulating tumor cells in cancer patients who are free of metastatic disease. The clinical importance of high circulating tumor cell numbers is not clear146; however, it appears that for some cancers, high circulating numbers of cancer cells do not necessarily correlate with risk for metastasis. Butler and Gullino estimated that between 1 to 4 × 106 cells/g of tumor enter the systemic circulation each day in human cancers.147 These data suggest that, although intravasation is necessary for metastasis, it is not sufficient nor is it process limiting. After removal of the primary tumor, these circulating cell counts drop and in many cases, no gross metastases develop. Although arrest at target organs may be a limitation for some cancers, the successful early survival of cancer cells at distant sites appears to be a major hurdle for successful metastases and therefore is a significant contributor to metastatic inefficiency. Metastatic cancer cells appear to be highly vulnerable to death (metastatic inefficiency) early after their arrival at a secondary site. The microenvironment of the secondary site is distinct from that of the primary tumor site and the initial tissues of origin of the cancer cell. These differences include changes in oxygen tension, pH, growth factor availability, and cellular binding partners. Collectively, these changes represent unique stresses to metastatic cancer cells. Successful metastatic cells must recognize, adapt, and endure these stresses in order to survive. Furthermore, appropriate modulation of the secondary microenvironment before the cancer cells arrive (premetastatic niche) and as a result of early and effective interaction with stromal and inflammatory cells within the new environment allow select populations of cells to survive and proliferate. The duration of this initial vulnerable state during metastatic progression may extend for the entire period of dormancy (see later). It is also likely that metastatic cells must successfully pass through additional vulnerable states later during metastatic progression. Targeting metastatic cancer cells during these vulnerable states may be an effective treatment strategy for cancer metastasis.148
What Is Dormancy and Where Do Dormant Cells Reside?
In spite of effective control of the primary tumor and aggressive multiagent and multimodality adjuvant therapy, the risk for metastases to distant sites remains high for several cancer histologies. Since most patients are free of gross metastases at the time of diagnosis, the development of metastases is presumed to emerge from microscopic cells that are not identifiable at the time of initial patient presentation.149 The location, size, angiogenic state, and proliferative/apoptotic state of these microscopic cells are largely unknown. Indeed, these dormant cells may exist as single cancer cells or microscopic clusters, they may exist in a quiescent (outside the cell cycle) or in a balanced state of proliferation and apoptosis, they may lie dormant in the eventual secondary metastatic tissue site (i.e., the lung) or persist in sanctuary sites (i.e., the bone marrow). Recent insights gleaned from the tumor-initiating (cancer stem cell) model may provide further support for the hypothesis that metastatic cells are capable of residence in transient sites like the bone marrow, where they may rest in a dormant state before being recruited into the metastatic cascade. The signals that induce these presumed small populations of cells to break dormancy and emerge into gross metastases likely also involve tumor cell microenvironment interaction.150,151
The Enabling Characteristics
As previously suggested, the hallmarks of cancer have been defined as functional capabilities that allow cancer cells to survive, proliferate, and disseminate. The fact that these critical hallmarks can be attained within a single cancer is explained by two key enabling characteristics of cancer: genome instability and tumor-promoting inflammation.
Genome Instability
The majority of the acquired hallmarks of cancer require changes in the genome through mutation, amplification, or chromosomal translocation. However, the process of random mutation is inefficient because of the complex and fastidious maintenance mechanisms of the normal cell that monitor DNA damage and regulate repair enzymes. As such, it is actually difficult to explain why cancers arise in animals at all because the acquisition of all traits would seem an impossible task. To explain this, it may be argued that genomes must attain increased mutability or genome instability in order to overcome the redundant homeostatic mechanisms that ordinarily prevent the emergence of a cancer cell.1,2
The following mechanisms have been suggested that may support the development of genomic instability43:
• Defects affecting various components of the DNA-maintenance machinery (caretakers of the genome), which may involve mutations in caretaker genes.
• The loss of telomeric DNA, which may cause karyotypic instability and chromosomal changes (amplification/deletion).
• Inactivation of tumor suppressor genes through genetic (mutation) or epigenetic (DNA methylation/histone modifications) mechanisms.
Tumor-Promoting Inflammation
For decades it has been recognized that tumors contain inflammatory and immune cell infiltrates that have classically been considered to be an attempt by the immune system to eradicate the tumor. However, recent evidence suggests that tumor-associated inflammation may paradoxically have a tumor-promoting effect. Inflammation can contribute to neoplastic progression through one of the following various mechanisms.43,152,153
• The supply of growth factors and growth signals to the microenvironment that promote angiogenesis, cell proliferation, and invasion.
• Induction signals that support the process of EMT.
• Fostering the progression of premalignant lesions to fully blown cancer.
• The production of reactive oxygen species that are mutagenic.
Many of the cells that contribute to this are components of the innate immune system, particularly macrophages with a specific, cancer-promoting phenotype. Specifically, they form part of the tumor microenvironment that supports the maintenance of the cancer phenotype (see Figure 1-5).
The Pathway to Cancer
The eight acquired capabilities of cancer cells and the two overarching enabling characteristics of genome instability and tumor inflammation have been outlined. It is important to stress that the pathways by which cells become malignant are highly variable. Mutations in certain oncogenes can occur early in the progression of some tumors and late in others. As a consequence, the acquisition of the essential cancer characteristics may appear at different times in the progression of different cancers. Furthermore, in certain tumors, a specific genetic event may, on its own, contribute only partially to the acquisition of a single capability, while in others it may contribute to the simultaneous acquisition of multiple capabilities. However, irrespective of the path taken, the hallmark capabilities of cancer will remain common for multiple cancer types and will help clarify mechanisms, prognosis, and the development of new treatments.
The Tumor Microenvironment
Over the past 10 years, tumors have been increasingly considered as organ systems similar to the tissues from which they derive. This is in contrast to a reductionist view of cancer biology, which considers the tumor as just a mass of tumor cells. In reality, the tumor is a complex system containing cancer cells and supporting cells, which all contribute to the maintenance of the malignant population and support ultimate dissemination and metastasis. Consequently, the major players in the cancer “organ system” are as follows:
Cancer Cells and Cancer Stem Cells
The concept that a cancer can arise from any cell in the body, along with the stochastic model of tumorigenesis, has recently been challenged.154,155 Although tumor heterogeneity is a well-established concept, a new dimension to heterogeneity has been established that suggests that a tumor may contain a hierarchical structure similar to many normal organ systems. Recent evidence suggests the existence of CSCs (sometimes termed tumor-initiating cells), which form the founder cell population for a tumor (see Figure 1-8). It was first extensively documented for leukemia and multiple myeloma that only a small subset of cancer cells are capable of extensive proliferation. For example, when mouse myeloma cells were obtained from mouse ascites, separated from normal hematopoietic cells, and put in clonal in vitro colony-forming assays, only 1 in 10,000 to 1 in 100 cancer cells were able to form colonies. Even when leukemic cells were transplanted in vivo, only 1% to 4% of cells could form spleen colonies.156-161 Because the differences in clonogenicity among the leukemia cells mirrored the differences in clonogenicity among normal hematopoietic cells, the clonogenic leukemic cells were described as leukemic stem cells. It has also been shown for solid cancers that the cells are phenotypically heterogeneous and that only a small proportion of cells are clonogenic in culture and in vivo.162-165 For example, only 1 in 1000 to 1 in 5000 lung cancer, ovarian cancer, or neuroblastoma cells were found to form colonies in soft agar. Just as in the context of leukemic stem cells, these observations led to the hypothesis that only a few cancer cells are actually tumorigenic and that these tumorigenic cells could be considered as CSCs. Although the field and concept of CSCs is a controversial one, CSCs may prove to be a common constituent of many, if not all, cancer types. Features of the CSC model also fit well with a view of metastasis in which only a small number of cells within a tumor have the ability (and plasticity) to endure the stresses of metastatic progression, survive during a dormant period, and then progress, proliferate, and differentiate into the complex heterogenous metastatic lesion.
If tumor growth and metastasis are driven by a small population of CSCs, this might explain the failure to develop therapies that are consistently able to eradicate solid tumors.154,155 Although currently available drugs can shrink metastatic tumors, these effects are usually transient and often do not appreciably extend the life of patients. One reason for the failure of these treatments is the acquisition of drug resistance by the cancer cells as they evolve; another possibility is that existing therapies fail to kill CSCs effectively.
Stem cell populations in human cancers have been identified in breast, bone, brain, colon, pancreas, liver, ovary, and skin.154,155 The actual origin of CSCs within solid tumors has not been clarified and may actually vary between tumor types. In some tumors, it may be that the resident adult or somatic stem cell may serve as the tumor-initiating cell, and in other tumors, the initiating cell may be the resident progenitor or transit-amplifying cell. A characteristic of the CSCs is that they undergo asymmetric division and self-renew, providing a continual resident population of highly resistant cancer cells.
Existing cancer therapies have been largely developed against the bulk population of tumor cells because they are often identified by their ability to shrink tumors. Because most cells with a cancer have limited proliferative potential, an ability to shrink a tumor mainly reflects an ability to kill these cells. It seems that normal stem cells from various tissues tend to be more resistant to chemotherapeutics than mature cell types from the same tissues. If the same is true of CSCs, then one would predict that these cells would be more resistant to chemotherapeutics than tumor cells with limited proliferative potential. Even therapies that cause complete regression of tumors might spare enough CSCs to allow regrowth of the tumors. Therapies that are more specifically directed against CSCs might result in much more durable responses and even cures of metastatic tumors. In veterinary oncology, putative stem cell populations have been identified for breast, bone, brain, and liver.154,155,166-168 Interestingly, stem cell populations appear to have altered DNA repair pathways, which may explain their resistance to conventional drugs. Identification of these populations, coupled with the availability of microarray and microRNA array technology, is allowing the identification of potential therapeutic targets.
Recent research has linked the acquisition of CSC characteristics with the EMT program, described previously in the section on metastasis.168 Cells that undergo EMT also take on characteristics reminiscent of the CSC phenotype. For example, they have the ability to self-renew and may support the ability of cells to colonize outside of the primary tumor. One may speculate that the signaling processes that support EMT may also serve to maintain the CSC population within a tumor and may also suggest plasticity in CSC populations.
Endothelial Cells
Much of the heterogeneity in tumors is found in the stromal compartment, and many of these cells are endothelial cells, which form the tumor-associated vasculature. VEGF and fibroblast growth factor are prominent signaling pathways in formation of these vessels. More modern sequencing techniques have also identified new pathways that may represent important signaling systems for neoangiogenesis and may represent therapeutic targets (e.g., notch signaling).
Pericytes
Pericytes are mesenchymal cells that wrap around the endothelial tubing of the blood vessels. Pericytes are considered a major cell type supporting the tumor vasculature.
Immune Inflammatory Cells
As described previously, an environment of chronic inflammation is an important enabling characteristic that supports the acquisition of cancer-related traits. Cells of the innate immune system are particularly crucial for the maintenance of the tumor environment. In particular, tumor macrophages with a specific pro-tumor phenotype can enhance cellular proliferation, invasion, and neoangiogenesis.
Cancer-Associated Fibroblasts
Cancer-associated fibroblasts comprise the supporting structure for many tumors but can also promote invasion, cell proliferation, and neoangiogenesis.43 The importance of the various cell types that make up the tumor microenvironment cannot be overstated. Although there is a complex signaling network between and within cancer cells that maintains the cancer phenotype, superimposed on top of this are the complex signaling networks between stromal components and stromal cells and cancer cells. Although we have considered earlier the potential evolution of cancer cells as they evolve from early disease to late-stage disease, it is quite possible that there is a similar evolution in the supporting structures that is dictated by the cancer itself. For example, incipient tumors may recruit stromal elements, which in turn reciprocate by promoting cell proliferation and angiogenesis. Evolution of the cancer population may then in turn feed back on the stromal population to further reprogram them to support the growing tumor. This of course could be extended to show the role of the stroma in promoting invasion and metastasis. These mechanisms underpin the complexity of understanding cancer pathogenesis as the process is highly dynamic and context dependent.
Summary and Future Directions
In this chapter we have sought to simplify as far as possible the mechanisms of carcinogenesis. It is clear that genomic instability and an environment of chronic inflammation support and provide a basis for the acquisition of the eight fundamental cancer characteristics. The identification of these pathways is providing excellent clues to the underlying mechanisms in cancer and the identification of potential therapeutic targets. However, despite our ability to make small molecules or antibodies, which aim at key targets in cancer survival, we are still a long way from a cure. The reasons for this are many but include the inherent tumor heterogeneity (in part supplied by the existence of tumor stem cells), the continual tumor evolution, and the immense and underestimated contribution of the tumor microenvironment. It is clear that multiple approaches will be required to maximize any possibility of therapeutic benefit.
References
1. McCance, KL, Roberts, LK. Cellular biology. In McCance KL, Huether SE, eds.: Pathophysiology: The biological basis of disease in adults and children, ed 3, St. Louis: Mosby, 1997.
2. Wyke, J. Viruses and cancer. In Franks LM, Teich NM, eds.: The molecular and cellular biology of cancer, ed 3, Oxford: Oxford University Press, 1997.
3. Vousden, KH. Cell transformation by human papillomaviruses. In: Minsen AC, Neil JC, McCrae MA, eds. Viruses and cancer. Cambridge: Cambridge University Press, 1994.
4. Campo, MS, O’Neil, BW, Barron, RJ, et al. Experimental reproduction of the papilloma-carcinoma complex of the alimentary canal in cattle. Carcinogenesis. 1994;15(8):1597–1601.
5. Campo, MS, Jarrett, WF, Barron, R, et al. Association of bovine papillomavirus type 2 and bracken fern with bladder cancer in cattle. Cancer Res. 1992;52(24):6898–6904.
6. Donner, P, Greiser-Wilkie, I, Moelling, K. Nuclear localization and DNA binding of the transforming gene product of avian myelocytomatosis virus. Nature. 1982;296:262–266.
7. Reid, SW, Smith, KT, Jarrett, WF. Detection, cloning and characterisation of papillomaviral DNA present in sarcoid tumours of Equus asinus. Vet Rec. 1994;135(18):430–432.
8. Gaukroger, JM, Bradley, A, Chandrachud, L, et al. Interaction between bovine papillomavirus type 4 and cocarcinogens in the production of malignant tumours. J Gen Virol. 1993;Pt 10:2275–2280.
9. Jarrett, WF. Bovine papilloma viruses. Clin Dermatol. 1985;3(4):8–19.
10. Lancaster, WD, Olson, C, Meinke, W. Bovine papillomavirus: presence of virus-specific DNA sequences in naturally occurring equine tumours. Proc Nat Acad Sci U S A. 1977;74:524–528.
11. Golias, CH, Charalabopoulos, A, Charalabopoulos, K. Cell proliferation and cell cycle control: a mini review. Int J Clin Prac. 2004;58(12):1134–1141.
12. Kong, N, Fotouhi, N, Wovkulich, PM, et al. Cell cycle inhibitors for the treatment of cancer. Drugs of the future. 2003;28(9):881–896.
13. Lane, DP. P53: Guardian of the genome. Nature. 1992;358:15–16.
14. Levine, AJ. P53, the cellular gatekeeper for growth and division. Cell. 1997;88:323–331.
15. Wu, X, Bayle, JH, Olson, D, et al. The p53-mdm2 autoregulatory loop. Genes Dev. 1993;7:1126–1132.
16. Haupt, Y, Maya, R, Kazaz, A, et al. Mdm2 promotes the rapid degradation of p53. Nature. 1997;387:296–299.
17. Wyllie, AH, Kerr, JF, Currie, AR. Cell death: the significance of apoptosis. Int Rev Cytol. 1980;68:251–306.
18. Strasser, A, Cory, S, Adams, JM. Deciphering the rules of programmed cell death to improve therapy of cancer and other diseases. EMBO J. 2011;30(18):3667–3683.
19. Jones, S, Zhang, X, Parsons, DW, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321(5897):1801–1806.
20. Parsons, DW, Jones, S, Zhang, X, et al. An integrated genomic analysis of human glioblastoma Multiforme. Science. 2008;321(5897):1807–1812.
21. McCance, KL, Roberts, LK. The biology of cancer. In McCance KL, Huether SE, eds.: Pathophysiology: The biological basis of disease in adults and children, ed 3, St. Louis: Mosby, 1997.
22. Jarrett, O, Onions, D. Leukaemogenic viruses. In Whittaker JA, ed.: Leukaemia, ed 2, Oxford: Blackwell Scientific Publications, 1992.
23. Balmain, A, Brown, K. Oncogene activation in chemical carcinogenesis. Adv Cancer Res. 1988;51:147–182.
24. Adams, GE, Cox, R. Radiation carcinogenesis. In Franks LM, Teich NM, eds.: The molecular and cellular biology of cancer, ed 3, New York: Oxford University Press, 1997.
25. Neil, JC, Hughs, D, McFarlane, R, et al. Transduction and rearrangement of the myc gene by feline leukemia virus in naturally occurring T cell leukemias. Nature. 1984;308:814–820.
26. Onions, DE, Lees, G, Forrest, D, et al. Recombinant feline viruses containing the myc gene rapidly produce clonal tumours expressing T-cell antigen receptor gene transcripts. Intern J Cancer. 1987;40:40–45.
27. Teich, NM. Oncogenes and cancer. In Franks LM, Teich NM, eds.: The molecular and cellular biology of cancer, ed 3, New York: Oxford University Press, 1997.
28. Tennent, R, Wigley, C, Balmain, A. Chemical carcinogenesis. In Franks LM, Teich NM, eds.: The molecular and cellular biology of cancer, ed 3, New York: Oxford University Press, 1997.
29. Huret, JL, Senon, S, Bernheim, A, et al. An atlas on genes and chromosomes in oncology and haematology. Cell Mol Biol. 2004;50(7):805–807.
30. Soto, AM, Sonnenschein, C. The somatic mutation theory of cancer: Growing problems with the paradigm? Bioessays. 2004;26(10):1097–1107.
31. Nasir, L, Rutteman, GR, Reid, SWJ, et al. Analysis of p53 mutational events and MM2 amplification in canine soft-tissue sarcomas. Cancer Lett. 2001;174(1):83–89.
32. Weinberg, RA. The retinoblastoma protein and cell cycle control. Cell. 1995;81:323–330.
33. Oliner, JD, Kinzler, KW, Meltzer, PS, et al. Amplification of a gene encoding a p53 associated protein in human sarcomas. Nature. 1992;358:80–83.
34. Vogelstein, B, Kinzler, KW. P53 function and dysfunction. Cell. 1992;70:525–526.
35. Sluss, HK, Jones, SN. Analysing p53 tumour suppressor functions in mice. Expert Opin Ther Targets. 2003;7(1):89–99.
36. Harris, CC. P53 tumor suppressor gene: from the basic research laboratory to the clinic—an abridged historical perspective. Carcinogenesis. 1996;17:1187–1198.
37. Nasir, L, Rutteman, GR, Reid, SW, et al. Analysis of p53 mutational events and MDM2 amplification in canine soft-tissue sarcomas. Cancer Lett. 2001;174(1):83–89.
38. Nasir, L, Burr, P, Mcfarlane, S, et al. Cloning, sequence analysis and expression of the cDNA’s encoding the canine and equine homologues of the Mouse double minute two proto-oncogene. Cancer Lett. 2000;152:9–13.
39. Nasir, L, Krasner, H, Argyle, DJ, et al. A Study of p53 tumour suppressor gene immunoreactivity in feline neoplasia. Cancer Lett. 2000;155:1–7.
40. Nasir, L, Argyle, DJ. Mutational analysis of p53 in two cases of Bull Mastiff lymphosarcoma. Vet Rec. 1999;145(1):23–24.
41. Nasir, L, Argyle, DJ, McFarlane, ST, et al. Nucleotide sequence of a highly conserved region of the canine p53 tumour suppressor gene. DNA Sequence. 1998;8(1–2):83–86.
42. Hanahan, D, Weinberg, RA. The hallmarks of cancer. Cell. 2000;100(1):57–70.
43. Hanahan, D, Weinberg, RA. Hallmarks of cancer: The next generation. Cell. 2011;144(5):646–674.
44. Kimmelman, AC. The dynamic nature of autophagy in cancer. Genes Dev. 2011;25(19):1999–2010.
45. Blasco, MA, Funk, W, Villeponteau, B, et al. Functional characterization and developmental regulation of mouse telomerase RNA. Science. 1995;269:1267–1270.
46. Blasco, MA, Lee, H-W, Hande, MP, et al. Telomere shortening and tumour formation by mouse cells lacking telomerase RNA. Cell. 1997;91:25–34.
47. Hayflick, L. Mortality and immortality at the cellular level. A review. Biochemistry. 1997;62:1180–1190.
48. Biller, BJ, Kitchel, B, Casey, D, et al. Evaluation of an assay for detecting telomerase activity in neoplastic tissues of dogs. Am J Vet Res. 1998;59(12):1526–1528.
49. McKenzie, K, Umbricht, CB, Sukumar, S. Applications of telomerase research in the fight against cancer. Mol Med Today. 1999;5(3):114–122.
50. Nasir, L, Devlin, P, Mckevitt, T, et al. Telomere lengths and telomerase activity in dog tissues: a potential model system to study human telomere and telomerase biology. Neoplasia. 2001;3(4):351–359.
51. Shay, JW, Wright, WE. Telomerase activity in human cancer. Curr Opin Oncol. 1996;8:66–71.
52. Yazawa, M, Okuda, M, Setoguchi, A, et al. Measurement of telomerase activity in dog tumours. J Vet Med Sci. 1999;61(10):1125–1129.
53. Zhu, J, Wang, H, Bishop, JM, et al. Telomerase extends the life-span of virus-transformed human cells without net telomere lengthening. Proc Natl Acad Sci U S A. 1999;96:3723–3728.
54. Shanmugam, M, McBrayer, SK, et al. Targeting the Warburg effect in hematological malignancies: from PET to therapy. Curr Opin Oncol. 2009;21(6):531–536.
55. Mendoza, M, Khanna, C. Revisiting the seed and soil in cancer metastasis. Int J Biochem Cell Biol. 2009;41(7):1452–1462.
56. Sell, S. Stem cell origin of cancer and differentiation therapy. Crit Rev Oncol Hematol. 2004;51(1):1–28.
57. Ramaswamy, S, et al. A molecular signature of metastasis in primary solid tumors. Nat Genet. 2003;33(1):49–54.
58. Clark, EA, et al. Genomic analysis of metastasis reveals an essential role for RhoC [In Process Citation]. Nature. 2000;406(6795):532–535.
59. Shevde, LA, Welch, DR. Metastasis suppressor pathways–an evolving paradigm. Cancer Lett. 2003;198(1):1–20.
60. Steeg, PS. Perspectives on classic article: metastasis suppressor genes. J Natl Cancer Inst. 2004;96(6):E4.
61. Paoloni, M, et al. Canine tumor cross-species genomics uncovers targets linked to osteosarcoma progression. BMC Genom. 2009;10:625.
62. Mayr, B, Brem, G, Reifinger, M. Absence of S100A4 (mts1) gene mutations in various canine and feline tumours. Detection of a polymorphism in feline S100A4 (mts1). J Vet Med A Physiol Pathol Clin Med. 2000;47(2):123–128.
63. Khanna, C, et al. The membrane-cytoskeleton linker ezrin is necessary for osteosarcoma metastasis. Nat Med. 2004;10(2):182–186.
64. Ren, L, et al. Dysregulation of Ezrin phosphorylation prevents metastasis and alters cellular metabolism in osteosarcoma. Cancer Res. 2012;72(4):1001–1012.
65. Hong, SH, et al. Protein kinase C regulates ezrin-radixin-moesin phosphorylation in canine osteosarcoma cells. Vet Comp Oncol. 2011;9(3):207–218.
66. Shoushtari, AN, Szmulewitz, RZ, Rinker-Schaeffer, CW. Metastasis-suppressor genes in clinical practice: lost in translation? Nat Rev Clin Oncol. 2011;8(6):333–342.
67. Liotta, LA, Kohn, EC. The microenvironment of the tumour-host interface. Nature. 2001;411(6835):375–379.
68. Friedl, P, Wolf, K, Lammerding, J. Nuclear mechanics during cell migration. Curr Opin Cell Biol. 2011;23(1):55–64.
69. Friedl, P, Wolf, K. Plasticity of cell migration: a multiscale tuning model. J Cell Biol. 2010;188(1):11–19.
70. Sabeh, F, Shimizu-Hirota, R, Weiss, SJ. Protease-dependent versus -independent cancer cell invasion programs: three-dimensional amoeboid movement revisited. J Cell Biol. 2009;185(1):11–19.
71. Scott, RW, Crighton, D, Olson, MF. Modeling and imaging 3-dimensional collective cell invasion. J Vis Exp. 58, 2011.
72. Condeelis, J, Segall, JE. Intravital imaging of cell movement in tumours. Nat Rev Cancer. 2003;3(12):921–930.
73. Condeelis, J, Singer, RH, Segall, JE. The great escape: when cancer cells hijack the genes for chemotaxis and motility. Annu Rev Cell Dev Biol. 2005;21:695–718.
74. Wang, W, et al. Single cell behavior in metastatic primary mammary tumors correlated with gene expression patterns revealed by molecular profiling. Cancer Res. 2002;62(21):6278–6288.
75. Wyckoff, JB, Segall, JE, Condeelis, JS. The collection of the motile population of cells from a living tumor. Cancer Res. 2000;60(19):5401–5404.
76. Jankowski, MK, et al. Matrix metalloproteinase activity in tumor, stromal tissue, and serum from cats with malignancies. J Vet Intern Med. 2002;16(1):105–108.
77. Loukopoulos, P, O’Brien, T, Ghoddusi, M, et al. Characterisation of three novel canine osteosarcoma cell lines producing high levels of matrix metalloproteinases. Res Vet Sci. 2004;77:131–141.
78. Hirayama, K, et al. Detection of matrix metalloproteinases in canine mammary tumours: analysis by immunohistochemistry and zymography. J Comp Pathol. 2002;127(4):249–256.
79. Lana, SE, et al. Identification of matrix metalloproteinases in canine neoplastic tissue. Am J Vet Res. 2000;61(2):111–114.
80. Leibman, NF, Lana, SE, Hansen, RA, et al. Identification of matrix metalloproteinases in canine cutaneous mast cell tumors. J Vet Intern Med. 2000;14:583–586.
81. Coussens, LM, Fingleton, B, Matrisian, LM. Matrix metalloproteinase inhibitors and cancer: trials and tribulations. Science. 2002;295:2387–2392.
82. Coussens, LM, Fingleton, B, Matrisian, LM. Matrix metalloproteinase inhibitors and cancer: trials and tribulations. Science. 2002;295(5564):2387–2392.
83. Moore, AS, et al. Doxorubicin and BAY 12–9566 for the treatment of osteosarcoma in dogs: a randomized, double-blind, placebo-controlled study. J Vet Intern Med. 2007;21(4):783–790.
84. Ramnath, N, Creaven, PJ. Matrix metalloproteinase inhibitors. Curr Oncol Rep. 2004;6(2):96–102.
85. Rucci, N, Sanita, P, Angelucci, A. Roles of metalloproteases in metastatic niche. Curr Mol Med. 2011;11(8):609–622.
86. Qian, BZ, Pollard, JW. Macrophage diversity enhances tumor progression and metastasis. Cell. 2010;141(1):39–51.
87. Foroni, C, et al. Epithelial-mesenchymal transition and breast cancer: Role, molecular mechanisms and clinical impact. Cancer Treat Rev. 2011. [Epub November 25].
88. Peinado, H, Olmeda, D, Cano, A. Snail, Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype? Nat Rev Cancer. 2007;7(6):415–428.
89. Moreno-Bueno, G, et al. The morphological and molecular features of the epithelial-to-mesenchymal transition. Nat Protoc. 2009;4(11):1591–1613.
90. Floor, S, et al. Cancer cells in epithelial-to-mesenchymal transition and tumor-propagating-cancer stem cells: distinct, overlapping or same populations. Oncogene. 2011;30(46):4609–4621.
91. Hendrix, MJ, et al. Vasculogenic mimicry and tumour-cell plasticity: lessons from melanoma. Nat Rev Cancer. 2003;3(6):411–421.
92. Kalluri, R. Basement membranes: structure, assembly and role in tumour angiogenesis. Nat Rev Cancer. 2003;3(6):422–433.
93. Frisch, SM, Francis, H. Disruption of epithelial cell-matrix interactions induces apoptosis. J Cell Biol. 1994;124(4):619–626.
94. Taddei, ML, et al. Anoikis: an emerging hallmark in health and diseases. J Pathol. 2012;226(2):380–393.
95. Grossmann, J. Molecular mechanisms of “detachment-induced apoptosis–Anoikis”. Apoptosis. 2002;7(3):247–260.
96. Guo, W, Giancotti, FG. Integrin signalling during tumour progression. Nat Rev Mol Cell Biol. 2004;5(10):816–826.
97. Fosmire, SP, Dickerson, EB, Scott, AM, et al. Canine malignant hemangiosarcoma as a model of primitive angiogenic endothelium. Lab Invest. 2004;84:562–572.
98. Akhtari, M, et al. Biology of breast cancer bone metastasis. Cancer Biol Ther. 2008;7(1):3–9.
99. Restucci, B, De Vico, G, Maiolino, P. Expression of beta 1 integrin in normal, dysplastic and neoplastic canine mammary gland. J Comp Pathol. 1995;113(2):165–173.
100. Olivry, T, et al. Investigation of epidermotropism in canine mycosis fungoides: Expression of intercellular adhesion molecule-1 (ICAM-1) and beta-2 integrins. Arch Dermatol Res. 1995;287(2):186–192.
101. Moore, PF, Rossitto, PV. Danilenko DM: Canine leukocyte integrins: characterization of a CD18 homologue. Tissue Antigens. 1990;36(5):211–220.
102. Selvarajah, GT, et al. Gene expression profiling of canine osteosarcoma reveals genes associated with short and long survival times. Mol Cancer. 2009;8:72.
103. Fukata, M, Kaibuchi, K. Rho-family GTPases in cadherin-mediated cell-cell adhesion. Nat Rev Mol Cell Biol. 2001;2(12):887–897.
104. Seftor, RE, et al. Role of the alpha v beta 3 integrin in human melanoma cell invasion. Proc Natl Acad Sci U S A. 1992;89(5):1557–1561.
105. Zheng, DQ, et al. Prostatic carcinoma cell migration via alpha(v)beta3 integrin is modulated by a focal adhesion kinase pathway. Cancer Res. 1999;59(7):1655–1664.
106. Ruoslahti, E, Reed, JC. Anchorage dependence, integrins, and apoptosis. Cell. 1994;77(4):477–478.
107. Nagaprashantha, LD, et al. The sensors and regulators of cell-matrix surveillance in anoikis resistance of tumors. Int J Cancer. 2011;128(4):743–752.
108. Lascelles, BD, et al. Improved survival associated with postoperative wound infection in dogs treated with limb-salvage surgery for osteosarcoma. Ann Surg Oncol. 2005;12(12):1073–1083.
109. Smyth, MJ, et al. New aspects of natural-killer-cell surveillance and therapy of cancer. Nat Rev Cancer. 2002;2(11):850–861.
110. Mocellin, S, et al. Colorectal cancer vaccines: principles, results, and perspectives. Gastroenterology. 2004;127(6):1821–1837.
111. Mocellin, S, Rossi, CR, Nitti, D. Cancer vaccine development: on the way to break immune tolerance to malignant cells. Exp Cell Res. 2004;299(2):267–278.
112. Rao, B, et al. Clinical outcomes of active specific immunotherapy in advanced colorectal cancer and suspected minimal residual colorectal cancer: a meta-analysis and system review. J Transl Med. 2011;9:17.
113. Bergman, PJ. Cancer immunotherapy. Vet Clin North Am Small Anim Pract. 2010;40(3):507–518.
114. Grosenbaugh, DA, et al. Safety and efficacy of a xenogeneic DNA vaccine encoding for human tyrosinase as adjunctive treatment for oral malignant melanoma in dogs following surgical excision of the primary tumor. Am J Vet Res. 2011;72(12):1631–1638.
115. Bagge, U, Skolnik, G, Ericson, LE. The arrest of circulating tumor cells in the liver microcirculation. A vital fluorescence microscopic, electron microscopic and isotope study in the rat. J Cancer Res Clin Oncol. 1983;105(2):134–140.
116. Chambers, AF, et al. Steps in tumor metastasis: new concepts from intravital videomicroscopy. Cancer Metastasis Rev. 1995;14(4):279–301.
117. Chambers, AF, et al. Critical steps in hematogenous metastasis: an overview. Surg Oncol Clin N Am. 2001;10(2):243–255. [vii].
118. Kienast, Y, et al. Real-time imaging reveals the single steps of brain metastasis formation. Nat Med. 2010;16(1):116–122.
119. Li, DM, Feng, YM. Signaling mechanism of cell adhesion molecules in breast cancer metastasis: potential therapeutic targets. Breast Cancer Res Treat. 2011;128(1):7–21.
120. Zigler, M, et al. PAR-1 and thrombin: the ties that bind the microenvironment to melanoma metastasis. Cancer Res. 2011;71(21):6561–6566.
121. Villares, GJ, Zigler, M, Bar-Eli, M. The emerging role of the thrombin receptor (PAR-1) in melanoma metastasis–a possible therapeutic target. Oncotarget. 2011;2(1–2):8–17.
122. Groom, AC, et al. Tumour metastasis to the liver, and the roles of proteinases and adhesion molecules: new concepts from in vivo videomicroscopy. Can J Gastroenterol. 1999;13(9):733–743.
123. Chambers, AF. The metastatic process: basic research and clinical implications. Oncol Res. 1999;11(4):161–168.
124. Martin, TA, et al. The role of the CD44/ezrin complex in cancer metastasis. Crit Rev Oncol Hematol. 2003;46(2):165–186.
125. Balkwill, F. Chemokine biology in cancer. Semin Immunol. 2003;15(1):49–55.
126. Kaplan, RN, Rafii, S, Lyden, D. Preparing the “soil”: the premetastatic niche. Cancer Res. 2006;66(23):11089–11093.
127. Kaplan, RN, et al. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature. 2005;438(7069):820–827.
128. Al-Mehdi, AB, et al. Intravascular origin of metastasis from the proliferation of endothelium-attached tumor cells: a new model for metastasis. Nat Med. 2000;6(1):100–102.
129. Cooper, CR, et al. Stromal factors involved in prostate carcinoma metastasis to bone. Cancer. 2003;97(suppl 3):739–747.
130. De Wever, O, Mareel, M. Role of tissue stroma in cancer cell invasion. J Pathol. 2003;200(4):429–447.
131. Engels, B, Rowley, DA, Schreiber, H. Targeting stroma to treat cancers. Semin Cancer Biol. 2011;22(1):41–49.
132. Folkman, J. Tumor angiogenesis and tissue factor. Nat Med. 1996;2:167–168.
133. Folkman, J. Angiogenesis: an organizing principle for drug discovery? Nat Rev Drug Discov. 2007;6:273–286.
134. Kerbel, RS. Tumor angiogenesis: past, present and the near future. Carcinogenesis. 2000;21(3):505–515.
135. Nagy, JA, et al. Heterogeneity of the tumor vasculature. Semin Thromb Hemost. 2010;36(3):321–331.
136. Olive, KP, et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science. 2009;324(5933):1457–1461.
137. Pircher, A, et al. Biomarkers in tumor angiogenesis and anti-angiogenic therapy. Int J Mol Sci. 2011;12(10):7077–7099.
138. Keara Boss, M, Muradyan, N, Thrall, DE. DCE-MRI: A review and applications in veterinary oncology. Vet Comp Oncol. 2011. [Epub December 8].
139. Fidler, IJ, Kripke, ML. Metastasis results from preexisting variant cells within a malignant tumor. Science. 1977;197(4306):893–895.
140. Fidler, IJ. The pathogenesis of cancer metastasis: the ‘seed and soil’ hypothesis revisited. Nat Rev Cancer. 2003;3(6):453–458.
141. Hunter, K, Welch, DR, Liu, ET. Genetic background is an important determinant of metastatic potential. Nat Genet. 2003;34(1):23–24. [author reply 25].
142. Hunter, KW. Allelic diversity in the host genetic background may be an important determinant in tumor metastatic dissemination. Cancer Lett. 2003;200(2):97–105.
143. Hunter, KW. Host genetics and tumour metastasis. Br J Cancer. 2004;90(4):752–755.
144. Khanna, C, Hunter, K. Modeling metastasis in vivo. Carcinogenesis. 2005;26(3):513–523.
145. Luzzi, KJ, et al. Multistep nature of metastatic inefficiency: Dormancy of solitary cells after successful extravasation and limited survival of early micrometastases. Am J Pathol. 1998;153(3):865–873.
146. Loberg, RD, et al. Detection and isolation of circulating tumor cells in urologic cancers: a review. Neoplasia. 2004;6(4):302–309.
147. Butler, TP, Gullino, PM. Quantitation of cell shedding into efferent blood of mammary adenocarcinoma. Cancer Res. 1975;35(3):512–516.
148. Chambers, AF, et al. Molecular biology of breast cancer metastasis. Clinical implications of experimental studies on metastatic inefficiency. Breast Cancer Res. 2000;2(6):400–407.
149. Paez, D, et al. Cancer dormancy: a model of early dissemination and late cancer recurrence. Clin Cancer Res. 2012.
150. Barkan, D, et al. Inhibition of metastatic outgrowth from single dormant tumor cells by targeting the cytoskeleton. Cancer Res. 2008;68(15):6241–6250.
151. Barkan, D, et al. Metastatic growth from dormant cells induced by a col-I-enriched fibrotic environment. Cancer Res. 2010;70(14):5706–5716.
152. Sica, A, Allavena, P, Mantovani, A. Cancer related inflammation: The macrophage connection. Cancer Lett. 2008;267(2):204–215.
153. Ben-Neriah, Y, Karin, M. Inflammation meets cancer, with NF-kappa B as the matchmaker. Nat Immunol. 2011;12(8):715–723.
154. Argyle, DJ, Blacking, T. From viruses to cancer stem cells: dissecting the pathways to malignancy. Vet J. 2008;177(3):311–323.
155. Blacking, TM, Wilson, H, Argyle, DJ. Is cancer a stem cell disease? Theory, evidence and implications. Vet Comp Oncol. 2007;5(2):76–89.
156. Park, CH, Bergsage, DE, McCulloc, EA. Mouse myeloma tumour stem cells: primary cell culture assay. J Natl Cancer Inst. 1971;46(2):411.
157. Huntly, BJ, Gilliland, DG. Leukaemia stem cells and the evolution of cancer-stem-cell research. Nat Rev Cancer. 2005;5(4):311–321.
158. Kamel-Reid, S, et al. A model of human acute lymphoblastic leukemia in immune-deficient SCID mice. Science. 1989;246(4937):1597–1600.
159. Lapidot, T, et al. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. 1994;367(6464):645–648.
160. Sirard, C, et al. Normal and leukemic SCID-repopulating cells (SRC) coexist in the bone marrow and peripheral blood from CML patients in chronic phase, whereas leukemic SRC are detected in blast crisis. Blood. 1996;87(4):1539–1548.
161. Bonnet, D, Dick, JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997;3(7):730–737.
162. Fidler, IJ, Kripke, ML. Metastasis results from preexisting variant cells within a malignant tumor. Science. 1977;197:893–895.
163. Heppner, GH. Tumor heterogeneity. Cancer Res. 1984;44:2259–2265.
164. Nowell, PC. Mechanisms of tumor progression. Cancer Res. 1986;46:2203–2207.
165. Southam, CM, Brunschwig, A. Quantitative studies of autotransplantation of human cancer. Cancer. 1961;14:971–978.
166. Pang, LY, Argyle, DJ. Using naturally occurring tumours in dogs and cats to study telomerase and cancer stem cell biology. Biochim Biophys Acta. 2009;1792(4):380–391.
167. Pang, LY, Argyle, D. Cancer stem cells and telomerase as potential biomarkers in veterinary oncology. Vet J. 2010;185(1):15–22.
168. Pang, LY, Cervantes-Arias, A, Else, RW, et al. Canine mammary cancer stem cells are radio- and chemo-resistant and exhibit an epithelial-mesenchymal transition phenotype. Cancer. 2011;3(2):1744–1762.