The Etiology of Cancer
 Section A
Section A
The Genetic Basis of Cancer
This is an exciting and rapidly evolving time in the field of cancer genetics. Although it has been clear for several decades that cancer is a disease driven by the accumulation of genetic abnormalities,1,2 new technologies are rapidly unraveling nuances in how heritable traits influence epigenetics and the tumor environment. Meticulous reductionist research done since the early 1960s helped identify hundreds of genetic abnormalities that are peculiarly associated with specific cancers, and more recent advances allowed for full sequencing of tumor genomes.3-6 The information from these experiments has reinforced current concepts and provided insight into new areas of research. This chapter will focus on contemporary information to provide context for the genetic basis of cancer and how interactions between genes and environment impact the origin, progression, and response to therapy of hematopoietic tumors. It is probably reasonable to say that the next decade will represent a new “golden age” of discovery in cancer genetics, when many of the apparent conflicts from our traditional (reductionist) experimental approaches will be resolved by integration of data from epidemiologic, molecular, and clinical studies into a more holistic understanding of the biology of cancer.
Genes and Cancer Risk
To understand cancer, one must first realize that cancer is neither a single nor a simple disease. Rather, the term cancer describes a large number of diseases whose only common feature is uncontrolled cell growth and proliferation. An important concept that is universally accepted is that cancer is a genetic disease, although it is not always heritable. Tumors arise from the accumulation of mutations that eliminate normal constraints of proliferation and genetic integrity in a somatic cell. Among other causes, mutations can arise following exposure to environmental mutagens such as cigarette smoke, ultraviolet irradiation, and others. In fact, changes in cancer incidence over the course of the twentieth century, many reflecting behavior patterns (e.g., lung cancer in smokers), infectious diseases (e.g., stomach cancer in people infected with Helicobacter pylori), or exposure to special cultural factors such as urbanization or diet (e.g., increasing breast cancer rates in the second and subsequent generations of Asian-American women), underscore the significant influence that the environment exerts on the genetic make-up of any individual. Nevertheless, it would be incorrect to assume that the environment is wholly responsible for most tumors; most associations of cancer and exposure to potential environmental carcinogens other than tobacco products and ultraviolet or gamma irradiation are relatively weak.
Rigorous experimental evidence now supports a number of “intrinsic mutagens” that interact in complex and sometimes unpredictable ways with environmental triggers to promote cancer. For example, it is clear now that oxygen free radicals that result from chronic inflammation can act as procarcinogenic mutagens. An equally important and perhaps less well-recognized “mutagen” is the inherent error rate of enzymes that control DNA replication, which introduces from 1 in 10,000,000 to 1 in 1,000,000 mutations for each base that is replicated during each round of cell division. Most mammalian genomes comprise 2 to 3 billion base pairs, so every time a cell divides, each daughter cell is likely to carry at least a few hundred mutations in its DNA. Most mutations, whether caused by extrinsic or intrinsic factors, are silent; that is, they do not hinder the cell’s ability to function. However, others can disable tumor suppressor genes or activate proto-oncogenes that respectively inhibit or promote cell division and survival. Thus it can be said that “being alive” is the single largest risk factor for cancer.
Heritable Cancer Syndromes
The existence of genetic predisposition to cancer is illustrated by well-defined heritable cancer syndromes.7 Over 200 such syndromes have now been defined.8 Even though they account for only 5% to 10% of all human cancers, studies of families with these syndromes provided many of the initial clues to understanding the genetic basis of sporadic (nonheritable) cancers. Although inheritance is recessive, these familial cancer syndromes show dominant patterns of inheritance and many have high penetrance.8 All but two of the known familial cancer syndromes are due to mutations that inactivate tumor suppressor genes. As originally proposed by Knudson in his “two-hit” hypothesis from studies in children with retinoblastoma,9 individuals at risk are obligate heterozygotes (they inherit a mutant allele and a wild-type allele). As it happens, homozygous mutations in critical growth regulatory genes usually cause embryonic lethality; however, in the case in which a single allele is affected, the mutation is present in every cell in the body. Given the rate of spontaneous mutation as described, the probability that the second, wild-type allele will be inactivated in at least one cell is extremely high, therefore facilitating tumorigenesis. This process is called loss of heterozygosity (LOH).
A curious observation worth noting is that different mutations in a single gene can predispose individuals to distinct cancer syndromes, whereas independent, single mutations of different genes can result in virtually the same disease, or at least diseases with indistinguishable phenotypes.7 This is not surprising when we consider that commonly affected genes are multifunctional and parts of complex, interactive networks or circuits10,11 (Figure 1-1). Thus a mutation may only alter gene function along one biochemical pathway, leaving its interactions with other pathways intact. Moreover, mutations that contribute to most sporadic cancers are restricted to a small subset of genes,12 many of which also are associated with heritable cancer syndromes. These observations have given rise to competing contemporary theories on the origins of cancer, which are addressed later in this chapter.
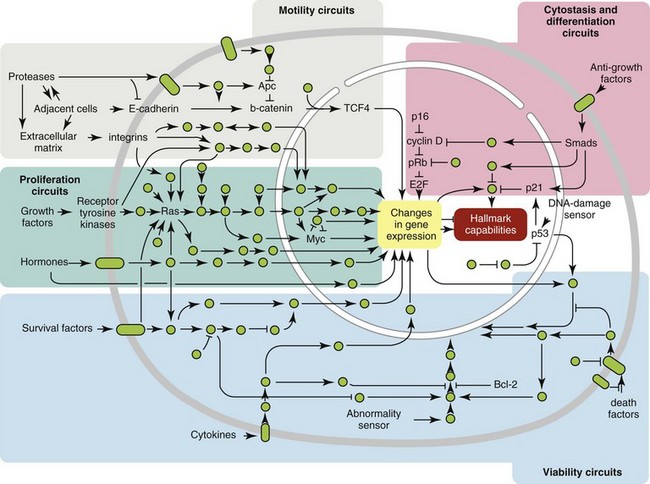
• Figure 1-1 Intracellular signaling networks regulate the operations of the cancer cell. An elaborate integrated circuit operates within normal cells and is reprogrammed to regulate hallmark capabilities within cancer cells. Separate subcircuits, depicted here in differently colored fields, are specialized to orchestrate the various capabilities. At one level, this depiction is simplistic because there is considerable crosstalk between such subcircuits. In addition, because each cancer cell is exposed to a complex mixture of signals from its microenvironment, each of these subcircuits is connected with signals originating from other cells in the tumor microenvironment. (Redrawn from Hanahan D, Weinberg RA: Hallmarks of cancer: The next generation, Cell 144:646–674, 2011, with permission.)
At least one heritable cancer syndrome (renal carcinoma and nodular dermatofibrosis [RCND] of German shepherd dogs) has been described in dogs.13 The heritable factor (or RCND gene) for this syndrome maps to dog chromosome 5 (CFA 5) and specifically to the folliculin gene, which was recently described as the heritable factor for the corresponding human disease (Birt-Hogg-Dube syndrome).14 It is probable that other syndromes comparable to those that are described in humans will eventually be identified in companion and laboratory animals, but it is unlikely that these will account for more than 5% to 10% of all cancer cases.
Genetic Influence in Sporadic Cancers
Unlike diseases due to single gene defects, cancer is a complex, multigenic disease. The “initiation, promotion, and progression” model was among the first to propose a sequential progression of mutations that could account for cancer.15,16 In this model, a genetic event would endow a somatic cell with limitless replicative potential or another growth or survival advantage from other cells in its environment (initiation). Alone, this would not be sufficient to give rise to a tumor, as the cell would remain constrained by environmental factors. A second event would further add to the cell’s ability to outcompete its neighbors in this environment, leading to its potential expansion into a recognizable tumor mass (promotion). Finally, a third event would reinforce the cell’s malignant potential (invasion, tissue destruction, and metastasis), leading to clinical disease (progression). It is important to note that an “event” is not equivalent to a single mutation but rather is more likely to represent a series of mutations that act in concert to alter the cell’s functional and morphologic phenotype. Although this model is overly simplistic and technically flawed (experimental evidence clearly shows that mutations are stochastic and do not occur in step-wise fashion),17 it is nevertheless useful to convey the events that lead to carcinogenesis and it remains the foundation for our current understanding of cancer genetics and cancer evolution.
Both the environment and the individual’s peculiar genetic background influence cancer risk and the natural history of tumors. This is especially clear in mice, in which the relative rate of spontaneous cancers and the susceptibility to chemically induced cancers differ according to the genetic background of various inbred strains.18 Similar evidence exists for humans; for example, the risk of habitual smokers to develop lung cancer was found to be tightly linked to a unique allele encoding the alpha-3 subunit of the high affinity nicotinic receptor19-21 and was later extended to this and other loci where additional nicotinic receptor subunits are encoded.22 An association between lung cancer arising from habitual tobacco use and activity of cytochrome P450 enzymes also had been observed repeatedly for many years, and there are indeed P450 alleles (e.g., CYP1B123) that also are associated with lung cancer. Together, these findings illustrate the complex relationship between genetics, environmental exposures, and probability to define cancer risk, prevention, and treatment. Specifically, alleles encoding “higher risk” nicotine receptors appear to modulate nicotine signaling, which in turn is responsible for embedding smoking behaviors and consequently exposure to dozens of potent carcinogens.22,24 Nicotine metabolism itself by P450 enzymes also influences tobacco use; however, conversion of tobacco-specific nitrosamines to mutagenic forms by highly active P450 enzymes modulates risk of transformation.25 Each component of risk is incremental and their interactions are not predictable. It is likely that similar relationships will be operative for many, if not most, common cancers of humans and domestic animals; defining these interactions will be a major emphasis of research during the coming decades.
There also is evidence to suggest that in some cases mutations are “directed” due to the presence of a “mutator phenotype,” in which the factors that control DNA replication and repair are prone to more errors than would be expected by simple random events. This leads to different rates of cancer predisposition, which would be higher than the mean in individuals bearing this “mutator phenotype,” and might explain why not all people or animals exposed to similar environmental carcinogens develop the same forms of cancer at the same rate.26 Recent information obtained from massive parallel full genome sequencing of tumor/normal pairs in diffuse large B-cell lymphoma suggests that the mutator phenotypes may be acquired and may involve genes that mediate both DNA repair and chromatin organization.5,6 It is important to note, however, that this does not exclude the possibility that mutator phenotypes also might be heritable.
In dogs and other domestic animals, the coexistence of genetic isolates in closed populations we call “breeds,” along with animals of mixed breeding, lends itself to study how a relatively homogeneous background influences cancer in out-bred populations. Preliminary data from whole genome association studies suggest there are distinct heritable traits that segregate with common cancer phenotypes in dogs.27,28 One common finding—and a pervasive obstacle for the completion of these studies—has been the observation of “fixed” risk alleles, making an association between individuals in the breed and disease challenging. Nevertheless, even though at the time of this writing none of these studies were yet published in the peer-reviewed literature, the reader should be alert for upcoming studies that will likely document specific risk genes for histiocytic sarcoma, transitional cell carcinoma, osteosarcoma, hemangiosarcoma, lymphoma, mammary cancer, and melanoma, among others, in susceptible dog breeds, and with the recent advent of the feline genome sequence,29 possibly in specific cat breeds as well.30 It remains to be seen if these traits will be shared between closely related breeds or whether they contribute to risk independently among different breeds.31
Perhaps as important, dogs are the first species in which genetic background has been shown to mold tumor genomes and tumor gene expression profiles.32-35 This knowledge, together with the demonstration that causal, pathognomonic genetic abnormalities are conserved in homologous human and canine cancers,36 opens a new area in which the precise contribution of heritable traits to sporadic cancers can be identified by using comparative systems approaches. These observations also indicate that we must assess “risk” far beyond the conventional idea of “tumor development,” as heritable traits might influence risk by modulating the probability of initiating events, the probability of promoting events, or the probability of progression through the interactions between the tumor and its microenvironment.34 A useful illustration of this concept is the occurrence of prostate cancers in men: virtually all men over 60 years of age will die with prostate cancer, but only a minority will die from prostate cancer, indicating that the major factors that influence the disease are not those that mediate transformation (at least as defined by morphologic appearance and anatomic organization), but instead those that dictate the biologic behavior of the transformed cells in the host.
Another important conceptual advance in this regard was the identification that spontaneous canine tumors had highly conserved (homologous) aberrations that had been previously characterized in human tumors. The prototypical example is a structural aberration resulting from a balanced chromosomal translocation that creates a fusion gene comprised of most of the BCR gene (located on chromosome 22 in humans and on chromosome 26 in dogs) and a truncated form of the ABL gene (coincidentally located on chromosome 9 in both humans and dogs) in chronic myelogenous leukemia (CML).36 Both translocations give rise to a derivative chromosome, the Philadelphia (Ph) chromosome in humans and the Raleigh chromosome in dogs, as illustrated by the canine form in Figure 1-2. Numeric aberrations (changes in DNA copy number) are similarly conserved among species, as illustrated by deletions of the RB1 locus, including the associated tumor-suppressing microRNAs in chronic lymphoid leukemias36 and the INK4 locus in T-cell malignancies,37 as well as copy number gains such as Runx2 amplification in osteosarcoma.38
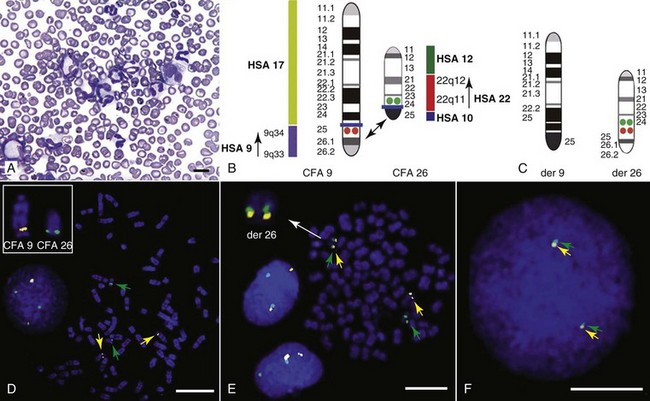
• Figure 1-2 Conserved cytogenetic rearrangement in canine chronic myelogenous leukemia (CML). A, Photomicrograph of a peripheral blood smear from a dog with CML (Diff-Quik stain; original magnification ×1600). Scale bar = 10 µm. B, Comparative ideograms showing corresponding regions for HSA 9q34 and HSA 22q11 on canine chromosomes 9 and 26. Orange and green spots indicate the location of canine ABL and BCR. A horizontal blue line on each ideogram identifies the location of the predicted breakpoints on CFA 9 and CFA 26, below which the two regions would be exchanged to form the aberrant derivative chromosomes. C, Schematic representation of the predicted derivative CFA 9 and derivative CFA 26 that would be produced by such a reciprocal translocation, with the derivative 26 (der 26) showing co-localized signals from ABL and BCR. D, Fluorescence in situ hybridization (FISH) analysis of metaphase preparations and interphase nuclei in normal canine leukocytes using canine BAC clones representing canine ABL and BCR on CFA 9 (orange spots) and CFA 26 (green spots), respectively. Inset shows enlarged, single metaphase chromosomes for CFA 9 and 26 correctly oriented. E, Hybridization of the same two BAC probes to metaphase chromosomes and interphase nuclei of a canine CML. Heterozygous co-localization of these two BAC clones to the derivative chromosome 26 is evident in the metaphase preparation and co-localization of one green and one yellow spot is also evident in the two interphase nuclei. Inset shows the derivative CFA 26 enlarged and correctly oriented (compare to predicted der 26 in C. F, Interphase nucleus from the same case of CML showing the presence of an apparent homozygous 9/26 translocation event, with both yellow and green spots showing close association. Scale bars = 8 µm. (Redrawn from Breen M, Modiano JF: Evolutionarily conserved cytogenetic changes in hematological malignancies of dogs and humans—man and his best friend share more than companionship, Chromosome Res 16:145–154, 2008, with permission.)
The development of these specific tumors from cells harboring such mutations may not be at all surprising, but why would homologous, highly conserved pathologic rearrangements, deletions, or amplifications occur in cells from organisms separated by more than 40 million years of evolution? Is it possible they are evolutionarily related on a mechanistic basis? For example, rearrangements of the immunoglobulin heavy chain locus and the MYC locus are thought to be due to recognition of MYC flanking sequences by the recombinase enzyme system.36 No such mechanism is known to be operative for other defined sites, so these other mutational events could occur stochastically, with their recurrent characterization across multiple species being the result of the selective advantage provided by the acquired gene to a cell of a highly specific lineage under highly specific conditions. This notion fits with Duesberg’s hypothesis that aneuploidy precedes genetic instability.39,40 Although it is impossible to rule out this argument, the implication would be that such mutational events are phenomenally common, and since they are not otherwise observed frequently in other cells where their occurrence was neutral makes this highly unlikely. Another possibility is that they are related to the nuclear anatomy of the cell and specifically caused by proximity of chromosomal regions, cellular stress, inappropriate DNA repair (or as mentioned previously, recombination), and DNA sequence and chromatin features.41 A third most intriguing possibility is that cellular genomes are reverting to a conformation that was found in a common ancestor (thus the high affinity and specificity between the rearranged chromosomal segments leads to the same recurrent event in many patients) but lost during the process of chromosomal reorganization in evolution, or that these sites represent targets for gene deletions or duplications that have been repeatedly advantageous to species under conditions of natural selection and so have become embedded in their contemporary descendants.
The clinical relevance of shared evolutionary and genetic origins should not be lost on physicians, veterinarians, or scientists. Shared origins mean shared biologic behaviors, thus supporting the rationale to apply the same therapies that have been developed for human patients to treat companion animals and vice versa. The best evidence for shared biologic behaviors comes from four recent studies of osteosarcoma, in which gene expression profiling documented overlapping characteristics of this disease in humans and in dogs.35,42-44 Specifically, the use of biased breed cohorts and isolated tumor explants allowed our group to filter genetic noise and stromal signatures to achieve pathologic stratification of osteosarcomas from three independent dog cohorts and five independent human cohorts into prognostically significant groups.35
The Hallmarks of Cancer
Thirty years of research culminated in the year 2000 in an insightful and thorough review paper by Douglas Hanahan and Robert Weinberg that synthesized our knowledge into six essential, acquired characteristics necessary for cellular transformation.45 These characteristics included (1) self-sufficiency in growth signals, (2) insensitivity to antigrowth signals, (3) the ability to evade apoptosis, (4) limitless replicative potential, (5) sustained angiogenesis, and (6) the capacity to invade tissues and metastasize. The importance of this paper was less in describing a list of events and more in the synthesis of these events because the concepts proposed by Hanahan and Weinberg created a paradigm shift in our understanding of cancer. Some of the important concepts that were clarified include: No single gene is universally responsible for transformation; five or six mutations are the minimum probable number required to endow the cancer phenotype (an observation that has since been confirmed experimentally3); each step is regulated by multiple interactive biochemical pathways,10 and thus mutations of different genes along a pathway can result in equivalent phenotypes and, conversely, mutations of the same gene can result in different cancers with distinct biology; tumors behave as tissues; and the interactions between the tumor and its microenvironment are major drivers of cancer behavior.
In early 2011, Hanahan and Weinberg updated the hallmarks of cancer in a new review that will likely be even more influential than the first.11 In this “next generation,” the hallmarks of cancer were refined and reassessed, and new “enabling characteristics” (genome instability and mutation and tumor-promoting inflammation) and “emerging” hallmarks (deregulating cellular energetics and avoiding immune destruction) were added to the paradigm. The impact of this unifying conceptualization of cancer genetics and this level of understanding are clearly evident when we consider how they have influenced the design, development, implementation, and success of new cancer therapies (Figure 1-3). A summary of the information with added refinements is provided later, and the reader is referred to the original manuscripts for details, since space constraints preclude an extensive review herein.
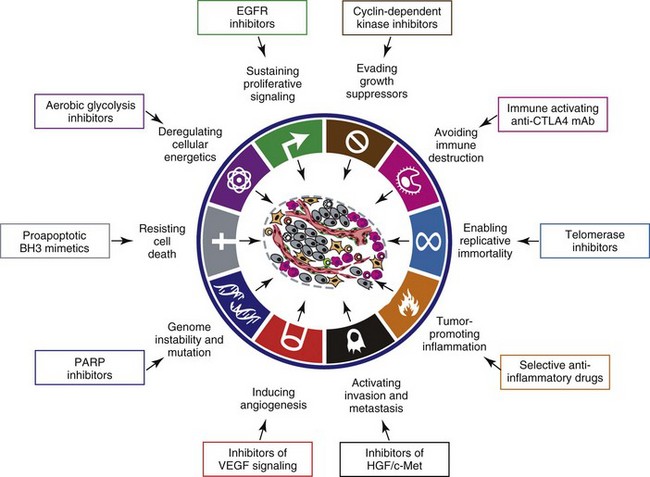
• Figure 1-3 Therapeutic targeting of the hallmarks of cancer. Drugs that interfere with each of the acquired capabilities necessary for tumor growth and progression have been developed and are in clinical trials or in some cases approved for clinical use in treating certain forms of human cancer. Additionally, investigational drugs are being developed to target each of the enabling characteristics and emerging hallmarks, which also hold promise as cancer therapeutics. The drugs listed are but illustrative examples; there is a deep pipeline of candidate drugs with different molecular targets and modes of action in development for most of these hallmarks. EGFR, Epidermal growth factor receptor; PARP, poly ADP ribose polymerase; VEGF, vascular endothelial growth factor; HGF, hepatocyte growth factor. (Redrawn from Hanahan D, Weinberg RA: Hallmarks of cancer: The next generation, Cell 144:646–674, 2011, with permission.)
Self-Sufficiency of Growth Signals
Arguably, the most important event in neoplastic transformation is the capability of cells to sustain chronic proliferation. Under normal conditions, cells communicate with each other and integrate environmental signals by sensing cues and gradients. For example, migration, metabolism, and proliferation of mature hematopoietic cells are regulated in autocrine and paracrine fashions by locally secreted cytokines. The same cytokines may travel systemically and act in an endocrine fashion. Generally, the cytokines work by binding transmembrane receptors, which in turn initiate signaling cascades that culminate in transcriptional changes that allow the cell to adapt its behavior to match the environmental signal. The activity of these cytokines, their receptors, and the corresponding signaling molecules are finely tuned. The system can be shut down when the concentration of the cytokine falls below a threshold that can stably bind the receptor, when the receptor ceases to be expressed, or when signaling molecules are downregulated or otherwise inactivated. However, mutations in even one of the molecules involved in regulating these pathways can provide sustained growth signals in the absence of the initiating cytokine. Among many examples, there is a translocation between chromosome 2 and chromosome 5 (t(2;5)) that is present in almost half of human anaplastic lymphomas. The translocation creates a fusion protein between the nucleophosmin gene (NPM1) and the anaplastic lymphoma kinase gene (ALK), which aberrantly activates the Jak2/STAT5 signaling pathway46 that is normally responsive to various interleukins (IL), including IL-2, IL-3, and IL-6. The genes that encode the normal growth-promoting proteins (such as ALK, Jak2, and STAT5) are called proto-oncogenes; the mutated versions that allow cells to gain self-sufficiency from the environmental signals are called oncogenes. It is important to note that not all growth-promoting genes have the capacity to become oncogenes and that the outcomes of oncogenic activation are most commonly senescence or apoptosis, unless there are additional events that promote stable transformation and survival.
Insensitivity to Antigrowth Signals
In addition to the hallmark capability of inducing and sustaining positively acting growth-stimulatory signals, cancer cells must also circumvent powerful programs that negatively regulate cell proliferation; many of these programs depend on the actions of tumor suppressor genes. To maintain homeostasis, cells also must integrate antigrowth signals from the environment. Quiescence in nonhematopoietic cells is enforced by signals delivered by contact inhibition.47 Hematopoietic cells, on the other hand, utilize cell-cell contacts to maintain interactions within the niche and to regulate the timing and intensity of hematopoiesis, inflammation, and immunity.48
“Stop” signals are usually delivered and integrated by the products of tumor suppressor genes, which derive their name largely from the observation that their inactivation facilitates tumor formation. Tumor suppressor genes balance the activity of growth-promoting proto-oncogenes and tend to act in tandem with these in most biochemical pathways. Loss of function of one or more tumor suppressor genes occurs in virtually every cancer, with inactivation of p53, RB1, PTEN, or CDKN2A each seen in more than 50% of all tumors. Each of these pathways may contribute to the pathogenesis of bone marrow–derived tumors in companion animals, and their dysfunction also may be predictive for outcomes.49-52
Evasion of Cell Death
Apoptosis, or programmed cell death, is the imprinted outcome for every cell in multicellular organisms. Survival requires support from extrinsic (environmental) factors, as well as precise balance of cellular energetics and metabolism. Bone marrow–derived cells normally undergo apoptosis when concentrations of survival factors (e.g., stem cell factor, IL-3, IL-7) or nutrients are limiting or when there are severe disruptions to cellular bioenergetics.53
Evasion of apoptosis is an essential acquired feature of all cancers, and it can result from loss of proapoptotic tumor suppressor genes such as p53 or PTEN or by gain of function of antiapoptotic genes, such as BCL2. Gain of function of BCL2 in humans is generally associated with indolent, follicular lymphomas that carry t(14 : 18) translocations that juxtapose BCL2 and the immunoglobulin heavy enhancer locus (IGH). These tumors are seen rarely in domestic animals, but evasion of apoptosis may be an important mechanism in the pathogenesis of indolent tumors seen more commonly in these species.
A more recent concept in the cell death field is autophagy—a process that tumor cells have efficiently coopted as a means to survive under adverse conditions.54 As part of the autophagy program, intracellular vesicles termed autophagosomes surround intracellular organelles and fuse with lysosomes. There, the organelles are broken down and then are channeled to form new molecules that support the energy-producing machinery of the cell, allowing it to survive in the stressed, nutrient-limited environment that defines most cancers.
Tumor cells also must avoid death by anoikis or loss of integral cell-to-cell or cell-to-matrix contacts.47 Absent these physiologic death pathways, the body often reacts to the anatomic and physiologic disruptions caused by cancer cells by targeting these cells for destruction through inflammatory pathways. This is but one pathway that leads to necrosis, since it appears that the process of necrosis also might be regulated genetically, providing another mechanism that favors survival of the whole (organism or tumor) over survival of the one. We are probably on the edge of an explosion of new findings that will further nuance our perception of how evasion (or inciting) of these cell death mechanisms contributes to neoplastic transformation and tumor progression.
Limitless Replicative Potential
Immortalization is another essential feature of cancer. The genetic program limits the number of times a cell is able to replicate, the so-called Hayflick limit, and when this limit is reached, replicative senescence is induced. Induction of replicative senescence does not induce death; cells maintain energetic homeostasis and remain functional, but they undergo significant genetic changes characterized by telomere erosion. Cells that are able to replicate must maintain the integrity of telomeres, which are “caps” made of repetitive DNA sequence that protect chromosomes from destruction. Solid tumors acquire immortalization predominantly by activation of the telomerase enzyme system and the consequent maintenance of telomere integrity. In hematopoietic cells, telomerase activity seems to be retained longer than in other somatic cells, so it is possible this facilitates immortalization in lymphoma and leukemia.55 The role of immortalization and the importance of telomerase (both to maintain telomere length and to maintain other biochemical functions that are essential for cell survival) are well established; however, the role of replicative senescence has recently been questioned because improved technology has allowed researchers to circumvent this process in normal cells.11 Mouse models complicate the story due to significant differences in telomere length between rodents and humans, so this is an area in which other models such as companion animals might provide clarity in the future.56
Sustained Angiogenesis
The process of angiogenesis requires the coordinated action of a variety of growth factors and cell-adhesion molecules in endothelial and stromal cells. So far, vascular endothelial growth factor-A (VEGF) and its receptors comprise the best-characterized signaling pathway in tumor angiogenesis.57 VEGF binds several receptor tyrosine kinases, including VEGF receptor-1 (VEGFR-1 [Flt-1]) and VEGFR-2 (KDR, Flk-1). VEGFR-2 is the major mediator of the angiogenic effects of VEGF. However, VEGFR-1 is expressed by some tumor cells and may mediate chemotactic signals, thus potentially having a role in cancer growth. The expression of VEGF is upregulated by hypoxia and inflammation. The transcription factor hypoxia-inducible factor-1α (HIF), which is part of a pathway that also includes regulation by the von Hippel-Lindau (VHL) tumor suppressor gene, is a major regulator of VEGF expression. Under conditions of normal oxygen tension, VHL protein targets HIF for degradation; under low oxygen conditions, HIF increases as VHL-mediated degradation is reduced, allowing for upregulation of VEGF. Other signaling molecules also contribute to angiogenesis, including platelet-derived growth factor-β (PDGF-β) and its receptor (PDGFR), and the angiopoietins. PDGF-β is required for recruitment of pericytes and maturation of new capillaries. Recent studies also document the importance of tumor-derived PDGF in recruitment of stroma that produces VEGF and other angiogenic factors (Figure 1-4).
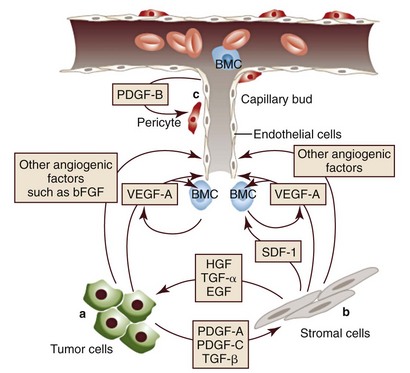
• Figure 1-4 A few of the molecular and cellular players in the tumor/microvascular microenvironment. A, Tumor cells produce VEGF-A and other angiogenic factors, such as bFGF, angiopoietins, interleukin-8 (IL-8), placenta growth factor (PlGF), and VEGF-C. These stimulate resident endothelial cells to proliferate and migrate. B, An additional source of angiogenic factors is the stroma, which is a heterogeneous compartment, comprising fibroblastic, inflammatory, and immune cells. Recent studies indicate that tumor-associated fibroblasts produce chemokines such as SDF-1, which may recruit bone marrow–derived angiogenic cells (BMC). VEGF-A or PlGF may also recruit BMC. Tumor cells may also release stromal cell-recruitment factors, such as PDGF-A, PDGF-C, or transforming growth factor (TGF-α, TGF-β). A well-established function of tumor-associated fibroblasts is the production of growth/survival factor for tumor cells such as EGFR ligands, HGF, and heregulin. C, Endothelial cells produce PDGF-β, which promotes recruitment of pericytes in the microvasculature after activation of PDGF receptor-β (PDGFR-β) factor. VEGF-A, Vascular endothelial growth factor-A; bFGF, basic fibroblast growth factor; SDF-1, stromal cell–derived factor-1; HGF, hepatocyte growth factor; PDGF, platelet-derived growth factor; EGFR, epidermal growth factor receptor. (Redrawn from Ferrara N, Kerbel RS: Angiogenesis as a therapeutic target, Nature 438:967–974, 2005, with permission.)
Folkman proposed a role for angiogenesis in cancer more than 30 years ago,58,59 but this idea took time to gain traction in the scientific community. Even after the importance of angiogenesis was recognized, the prediction was that this process would impact solid tumors but would be relatively unimportant for tumors of the blood and lymph (lymphoma, leukemia, and multiple myeloma). The first clues that this notion was mistaken came from unexpected benefits of patients with chronic lymphocytic leukemia (CLL) treated with antiangiogenic compounds,60-62 followed by similar success for some patients with multiple myeloma.63,64 More recently, a European study showed that different histologic types of human non-Hodgkin’s lymphoma show different patterns of angiogenesis, and these can predict outcomes for some of the most aggressive tumors, such as peripheral T-cell lymphomas.65 One study has shown that microvessel density is similarly correlated with the aggressive behavior of canine lymphoma,66 and similar findings have been reported for other blood-derived and solid tumors of dogs, such as mast cell cancer and mammary cancer.67,68
The concepts of how neoangiogenesis contributes to cancer also are undergoing refinement. For example, it is apparent now that clinical trials of antiangiogenic drugs have largely failed because their design was based on incomplete knowledge and thus incorrect assumptions. Perhaps the most informative example is the history of Bevacizumab (a humanized anti-VEGF antibody), which received approval to treat various cancers between 2004 and 2010 after it was shown to confer improved quality of life, albeit with modest survival benefits for patients. On December 6, 2010, the Federal Drug Administration (FDA) issued a press release (http://www.fda.gov/newsevents/newsroom/pressannouncements/ucm237172.htm) that announced the withdrawal of the indication for bevacizumab in treatment of metastatic breast cancer, stating that “… the drug has not been shown to be safe and effective for that use.” The FDA press release also stated, “The agency is making this recommendation after reviewing the results of four clinical studies of Avastin (bevacizumab) in women with breast cancer and determining that the data indicate that the drug does not prolong overall survival in breast cancer patients or provide a sufficient benefit in slowing disease progression to outweigh the significant risk to patients. These risks include severe high blood pressure; bleeding and hemorrhage; the development of perforations (or ‘holes’) in the body, including in the nose, stomach, and intestines; and heart attack or heart failure.”
Indeed, the preponderance of data suggest that antiangiogenic therapies will benefit cancer patients by promoting vessel normalization, reversing the anatomic and hemodynamic dysfunction created by the tumor microenvironment, disabling some of the intrinsic advantages that this provides for cancer cells, and allowing better penetration of drugs. Vascular normalization relies on restoring the balance among all the blood vessel–forming constituents from the bone marrow and the stroma, including pericytes, myeloid-derived cells, and endothelial progenitors, all of which contribute—and respond to—the “angiogenic switch,” whereby previously quiescent tissues trigger formation of new blood vessels associated with tumor growth.
Invasion and Metastasis
The role of genetic events in invasion and metastasis is still incompletely understood. The classic model of metastasis proposed by Fidler suggests a step-wise acquisition of assets that enables cells to leave the primary tumor site, travel through blood or lymph, invade stroma in favorable locations, and thus become reestablished at distant sites.69 More recent work suggests that most tumors possess the ability to dislodge cells that travel to distant sites, and the ability of such cells to survive in capillary beds may be the most important step in the metastatic process.70-74
Bone marrow–derived cells have intrinsic properties that allow them to travel throughout the body, traffic through all major organs, and home to areas of inflammation. Thus bone marrow–derived tumors are inherently metastatic. Nevertheless, hematopoietic tumors that are cytologically indistinguishable can have distinct and preferential tissue distribution. We do not fully understand what events make leukemic cells stay in the peripheral circulation, while cells from corresponding lymphomas (or myeloid sarcomas) with virtually identical molecular signatures stay confined to lymphoid or visceral organs.
In epithelial neoplasms that account for the majority of tumors in humans, the epithelial-to-mesenchymal transition (EMT) has received increasing attention for its role in metastasis. EMT is a developmental program that progenitor stem cells use during morphogenesis and can be physiologically reactivated during wound healing. It remains unclear whether EMT is equally (or less) important in sarcomas more commonly seen in domestic animals, in which the cells of origin seem to retain EMT capabilities to a greater extent. Similarly, there is increasing evidence of interactions between cancer cells, the “initiating” population in the tumor (cancer stem cells [CSCs]), mesenchymal stem cells (MSCs), tumor-associated fibroblasts, inflammatory cells, and angiogenic cells, which may be responsible for invasive behaviors and possibly for survival in hostile environments that exist at distant sites (Figure 1-5).
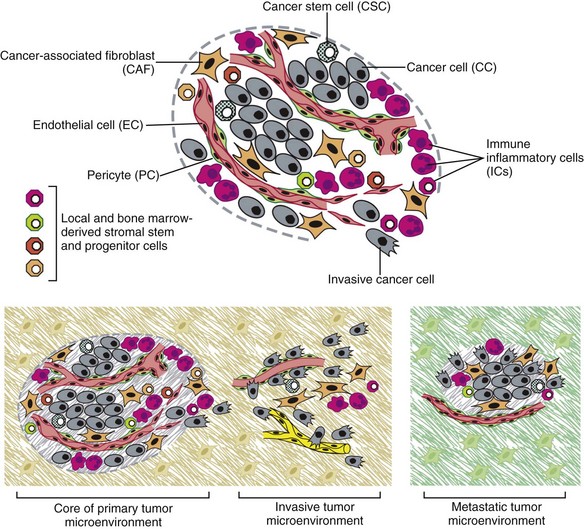
• Figure 1-5 The cells of the tumor microenvironment. Upper, An assemblage of distinct cell types constitutes most solid tumors. Both the parenchyma and stroma of tumors contain distinct cell types and subtypes that collectively enable tumor growth and progression. Notably, the immune inflammatory cells present in tumors can include both tumor-promoting, as well as tumor-killing, subclasses. Lower, The distinctive microenvironments of tumors. The multiple stromal cell types create a succession of tumor microenvironments that change as tumors invade normal tissue and thereafter seed and colonize distant tissues. The abundance, histologic organization, and phenotypic characteristics of the stromal cell types, as well as of the extracellular matrix (hatched background), evolve during progression, thereby enabling primary, invasive, and then metastatic growth. The surrounding normal cells of the primary and metastatic sites, shown only schematically, likely also affect the character of the various neoplastic microenvironments. (Not shown are the premalignant stages in tumorigenesis, which also have distinctive microenvironments that are created by the abundance and characteristics of the assembled cells.) (Redrawn from Hanahan D, Weinberg RA: Hallmarks of cancer: The next generation, Cell 144:646–674, 2011, with permission.)
Adaptive Evolution and the Tumor Microenvironment
There is a bidirectional flow of information between the tumor and the microenvironment, with each helping to mold the other into functional growing tissue that can evade or withstand attack by the host.75 Our previous reference to a “selective growth advantage” that is reminiscent of Darwinian selection is not accidental. The clonal evolution theory76 addresses the significance of sequential genetic changes providing growth and survival advantages, but to this we must add the fact that, in addition to these self-sufficient events that influence growth and survival, tumor cells must also evade “predators” (e.g., inflammation and the immune system77,78). In essence, the interaction of the tumor with its microenvironment and ultimately with the host is in fact subject to Darwinian laws of evolution, albeit in an accelerated time scale.79 This is evident in the ability of tumors to modulate stromal cells to support their own growth by providing a suitable matrix and an abundance of nutrients, while maintaining antitumor responses at bay.
As is true for other selective environments, tumors that outgrow the capability of their immediate surroundings to support their growth must alter that environment to suit their needs or identify other favorable locations where they can become established. The tumor microenvironment was recently shown to exert a significant effect on the complement of genes expressed by incipient tumor cells.80 In this case, the microenvironment was modified by gamma irradiation and the tumor was derived from orthotopic implants of chimeric Trp53-deficient mammary epithelial cells. The magnitude of change was not unlike that observed by our group when comparing the influence of breed on gene expression by canine hemangiosarcoma cells, although in our case the expression profile was maintained in a cell-autonomous fashion (i.e., ex vivo).34 Again, the behavior of carcinomas and sarcomas may differ, and for the latter, recent experiments from our group using canine hemangiosarcoma xenotransplantation models suggest that alterations in the microenvironment might favor not only the efficiency of tumor implantation but also the extent to which the microenvironment contributes to the composition of the tumor as a whole. Incipient sarcoma cells, in turn, can reside as quiescent inhabitants of distant microenvironments themselves modulating growth, morphology, and behavior of microenvironment constituents in the process of metastatic dissemination (Figure 1-6).
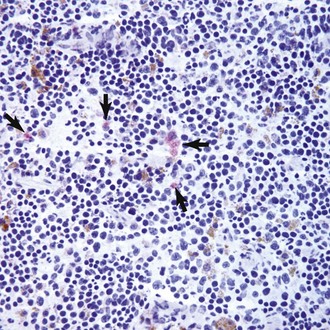
• Figure 1-6 Incipient canine hemangiosarcoma cells in the spleen of a mouse after subcutaneous tumor xenotransplantation. SB canine hemangiosarcoma cells (5 × 104) harboring a firefly luciferase gene were injected subcutaneously in the flank of immunocompromised mice. Tumors were detectable at the local site within 14 days of injection. After 48 days, the mice were humanely sacrificed and systemic tissues were examined for the presence of luciferase-expressing (tumor) cells. The photomicrograph shows immunohistochemical staining for luciferase in a frozen section of spleen from a representative mouse. Cells with red staining in the cytoplasm represent luciferase-positive canine hemangiosarcoma cells (arrows). Similar events were seen in other organs both by immunohistochemistry and by in vivo imaging. These incipient cells did not show evidence of organization into a tumor, suggesting a critical mass had not yet been reached, or possibly, that the primary subcutaneous tumor suppressed metastatic growth. In other mice, however, the incipient cells appeared to “instruct” organization of the resident spleen microenvironment into fulminant hematopoietic tumors. Magnification 200× with hematoxylin counterstain. (Experiment, staining, and image courtesy AM Frantz, EB Dickerson, TD O’Brien, and JW Wojcieszyn.)
Emerging and Enabling Hallmarks
The concept of genomic instability is not new but was incorporated as an “enabling hallmark” into the updated Hanahan and Weinberg model. Step-wise clonal evolution is satisfying because it can be correlated with discrete pathologic changes in tumor progression, especially for epithelial tumors where such progression can be seen in lesions that go through stages of hyperplasia, atypical hyperplasia (dysplasia), adenoma, carcinoma in situ, invasive carcinoma, and metastatic carcinoma (Figure 1-7). However, analysis of tumor genomes even in early stages usually shows aneuploidy (abnormal DNA copy number), as well as chaotic changes indicative of multiple numeric and structural DNA abnormalities. Similar abnormalities first noticed by Boveri more than 100 years ago in studies of sea urchin cells led him to formulate the “aneuploidy theory” of cancer.81 We know now that aneuploidy is especially evident in solid tumors; based on this, Loeb proposed the existence of the “mutator phenotype” in which cells are predisposed to undergo multiple mutations, some of which inevitably lead to cancer (see earlier).26 Some tenets of his hypothesis appear to be correct, although perhaps in different circumstances than Loeb originally envisioned, because they might relate to increased activity of polymerases with low fidelity under conditions in which the rate of DNA damage (and consequently mutations) is higher than the expected background from normal DNA replication (e.g., in lung epithelial cells from heavy smokers). However, direct measurements of mutation rates of sporadic tumors are much lower than those predicted if a “mutator phenotype” was operative in these tumors.12 Indeed, the minimum number of mutations that are required for clinical onset of cancer in solid tumors based on sequencing of solid tumor genomes is 15 to 25,4 but this may apply to tumors with chaotic karyotypes, as the number of mutations identified in a cytogenetically stable leukemia was significantly smaller.3
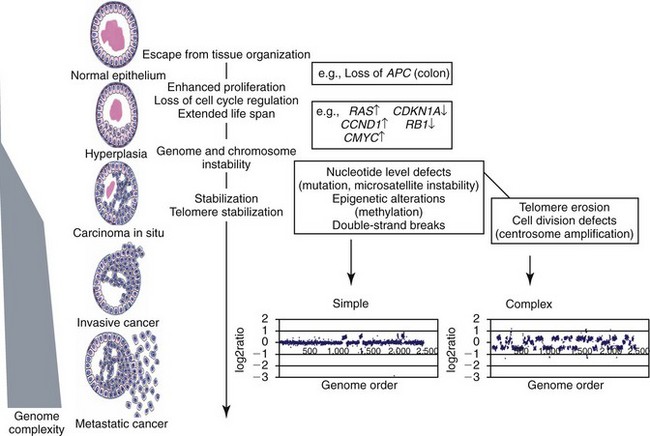
• Figure 1-7 Schematic representation of chromosomal evolution in human solid tumor progression. The stages of progression are arranged with the earlier lesions at the top. Cells may begin to proliferate excessively owing to the loss of tissue architecture, abrogation of checkpoints, and other factors. In general, relatively few aberrations occur before the development of in situ cancer. As indicated, a sharp increase in genome complexity (the number of independent chromosomal aberrations) in many (but not all) tumors coincides with the development of in situ disease. The types and range in aberration number varies markedly between tumors, probably owing to the specific failures in checkpoint or damage surveillance that are present, as illustrated by the whole-genome array comparative genomic hybridization (CGH) profiles of HCT116, a mismatch repair-defective cell line, and T47D, a mismatch repair-proficient cell line. The copy number profiles of HCT116 and T47D are labeled as “simple” and “complex,” respectively, to distinguish between tumor genomes with few or many copy number changes. The spectrum of aberrations in in situ lesions is similar to those found in more advanced malignancies. Thus an early increase in chromosomal aberration composition is followed by more modest chromosomal evolution. (Redrawn from Albertson DG, Collins C, McCormick F, et al: Chromosome aberrations in solid tumors, Nat Genet 34:369–376, 2003, with permission.)
Still, genetic instability is a hallmark of most tumors, and while it can be partly explained by increased errors in DNA replication and chromosomal segregation in cells that are rapidly dividing, other mechanisms are clearly operative, involving telomeres and telomerase.12,79,82-85 Although many of these changes are not “recurrent” and appear to be random products of instability, some may in fact contribute to proliferative crisis.86 It is possible that the initiation events for many tumors occur early in life during highly proliferative stages of tissue growth and remodeling (e.g., prior to closure of the growth plates in bone cancer), but they become evident later in life when a last series of mutations allows the transformed cell to reach this crisis stage. As we alluded earlier, hematopoietic tumors seem to avoid the chaotic chromosomal instability associated with solid tumors. We do not fully understand the reasons for this, although it may be partly due to intrinsic protective mechanisms associated with the proliferative rate of bone marrow precursor cells.
Tumor-Promoting Inflammation
The role of inflammation in cancer has received considerable attention in the past 10 years. Although our understanding of this phenomenon remains incomplete, it clearly met the criteria for inclusion as an “enabling hallmark” into the updated Hanahan and Weinberg model. The importance of inflammation was inferred from the earliest microscopic studies of cancer, but it was a seminal paper by Dvorak in 1986, in which he described tumors as “wounds that never heal,”87 that provided synthesis for the recurrent observation that tumors were often infiltrated by inflammatory cells of the innate (granulocytes, histiocytes, and macrophages) and the adaptive (lymphocytes) immune systems. Mechanistic distinctions remain to be defined between inflammation that favors tumor growth and inflammation that retards growth or eliminates the tumor,88-90 but we can say confidently that inflammation contributes to tumor growth and survival by supplying factors that sustain proliferation; factors that limit cell death; proangiogenic factors; extracellular matrix-modifying enzymes that facilitate angiogenesis, invasion, and metastasis; and other signals that lead to activation of EMT and other hallmark-facilitating programs.11 As noted earlier, inflammatory cells also release notably reactive oxygen species that are actively mutagenic for nearby cancer cells, accelerating their genetic evolution toward states of heightened malignancy.91
Reprogramming Energy Metabolism
In the early years of the twentieth century, Otto Warburg observed that cancer cells preferentially utilized glycolytic (anaerobic) rather than oxidative (aerobic) pathways to generate energy even under conditions of normal or high oxygen. This metabolic peculiarity of cancer cells, called the Warburg effect, seems to be driven by activated oncogenes and/or by loss of tumor suppressor genes that provide cancer cells with selective growth and survival advantages by conferring the hallmark capabilities of cell proliferation, avoidance of cytostatic controls, and attenuation of apoptosis. The reliance of cancer cells on glycolysis can be further accentuated under the hypoxic conditions. In fact, Warburg-like metabolism seems to be present in rapidly dividing embryonic tissues, suggesting a role in supporting large-scale biosynthetic programs that are required for active cell proliferation.
Cancer cells do not seem to enable the Warburg effect universally. Rather, much like other cells with high energetic demands, they seem to sort out into lactate-secreting (Warburg) and lactate-consuming cells, providing an efficient, albeit homeostatically disturbed, energy environment. Furthermore, it seems that oxygenation is not static in tumors but instead fluctuates temporally and regionally due to the instability and chaotic organization of tumor-associated neovasculature. Altered energy metabolism is proving to be as widespread in cancer cells as many of the other cancer-associated traits that have been accepted as hallmarks of cancer. This realization raises the question of whether deregulating cellular energy metabolism is therefore a core hallmark capability of cancer cells that is as fundamental as the six well-established core hallmarks. In fact, the redirection of energy metabolism is largely orchestrated by proteins that are involved in one way or another in programming the core hallmarks of cancer. When viewed in this way, aerobic glycolysis is simply another phenotype that is programmed by proliferation-inducing oncogenes and the designation of reprogrammed energy metabolism as an emerging hallmark seems most appropriate.
It is worth noting that this characteristic of tumor cells provides at least one important diagnostic advantage. Upregulation of the major glucose transporter, GLUT-1, is seen in virtually all tumors, making the cells efficient glucose scavengers. This can be exploited to image tumor cells noninvasively with precision by visualizing glucose uptake using positron-emission tomography (PET) with a radiolabeled analog of glucose (18F-fluorodeoxyglucose [18F-FDG]) as a reporter. The combination of PET with computed tomography (PET-CT) is now one of the most robust means to evaluate composition of tumors, minimal residual disease, and tumor-specific objective responses in patients receiving conventional and experimental therapies.
Evading Immune Destruction
Burnet and Thomas proposed the concept that the immune system can recognize and destroy incipient tumors (cancer immunosurveillance) in the 1950s.78 However, the hypothesis was far ahead of its time, and technologic obstacles impeded proof, so the theory fell into disfavor. In recent years, the immunosurveillance theory has gained traction anew because data strongly suggest that the immune system helps to maintain tumors at bay, and thus tumors must evade the immune response to survive. In its recent incarnation, the theory has been refined to incorporate the concept of immunoediting, in which the immune system destroys strongly antigenic tumor cells, providing weakly antigenic cells a survival advantage.78 Experimental evidence for this concept includes differences between tumors grown in immunocompetent (only weakly antigenic tumors survive) and immunocompromised mice (no selection against strongly antigenic tumors is observed), but it is unknown if immunoediting is operative in spontaneous cancers.
That the tumor microenvironment forms and maintains an immunosuppressive barrier provides more compelling evidence for the role of the immune system to limit tumor growth and metastasis. This immunosuppressive barrier includes cellular factors such as regulatory T cells (Tregs), myeloid-derived suppressor cells (MDSCs), and MSCs. Tregs, MDSCs, and MSCs can attack distinct and complementary antigen-specific (Tregs) and nonspecific (MDSCs and MSCs) facets of immune effector cell activation and function. Soluble factors, including transforming growth factor-β (TGF-β) and immunoglobulins, also contribute to the immunosuppressive barrier directly by disabling immune effector cells and indirectly by “educating” tumor-associated stromal cells, which in turn promotes secretion of stromal-derived factors that recruit additional inflammatory cells (tumor macrophages) and endothelial cells that further accelerate tumor growth.92 This is an active area of basic and clinical research in which companion animal oncology has been at the forefront, for example, through the generation and approval of the first active gene-based therapeutic cancer vaccine for canine melanoma.93
Epigenetic Events
Another observation is that events leading to cancer need not necessarily be caused by mutational events but instead can be caused by epigenetic changes. Epigenetic events are those that can alter phenotype without changing the genotype. Two well-characterized epigenetic mechanisms regulate gene expression. Gene silencing can occur by methylation of CpG residues in promoter regions, as well as by histone deacetylation. Both of these events interfere with the transcriptional machinery and repress gene expression. The effects of global changes in methylation or deacetylation (e.g., by inactivation of DNA methylases or histone deacetylases) remain incompletely understood, but silencing of specific genes by methylation is implicated in numerous cancers of humans and animals.2,94-96 One important observation is that most (or all) genes that are subject to silencing by methylation in specific cancers (e.g., CDKN2A in T-cell leukemia) are commonly inactivated by mutation or deletion in other cancers (e.g., CDKN2A in melanoma).
As is true for mutations, gene regulation by epigenetic methylation can occur sporadically or it can be heritable. Silencing of some tumor suppressor genes in sporadic cancers occurs more frequently by epigenetic methylation than by mutation or deletion. These different mechanisms of gene silencing are not equivalent, as they each result in specific tumor phenotypes. For example, data from our laboratories indicate that loss of canine chromosome 11, with resultant deletion of the INK4 tumor suppressor locus containing the CDKN2A, CDKN2B, and ARF genes, and methylation of CDKN2A are each associated with morphologically distinct types of T-cell lymphoma that have a different clinical presentation and prognosis.37,51
Genomic imprinting presents a unique example in which heritable epigenetic changes influence cancer predisposition. Genomic imprinting refers to a pattern of gene expression that is determined by the parental origin of the gene; in other words, unlike most genes in which both parental alleles are expressed, only one allele (specifically derived from the mother or from the father, depending on the gene) of an imprinted gene is expressed and the other one is permanently repressed. Epigenetic changes in Wilms’ tumor and in heritable colon cancer (among others) alter the expression of the imprinted allele, leading to loss of imprinting that causes overexpression of the insulin growth factor-2 (IGF2) gene.2,97
Cancer Stem Cells
The paradoxic nature of some cancers gave rise to the notion of a “cancer progenitor” or a CSC as far back as the 1960s. The best illustration for this concept was CML, where the bulk of the tumor consists of terminally differentiated neutrophils that are incapable of recreating the malignancy. However, it was apparent that there were multipotent stem cells in this tumor population. In 1994, Dick’s group proved conclusively that another type of leukemia, acute myelogenous leukemia (AML), was a hierarchically organized disease in which a small number of cells that were undetectable by conventional methods could be isolated from patients and made to recapitulate the full spectrum of the disease in an animal model.98 This gave rise to the CSC or “tumor-initiating cell” hypothesis, which is based on the concept that tumors are hierarchically organized into a subpopulation of cells that retain or acquire the capacity for self renewal and are capable and responsible for initiating and maintaining the tumor (Figure 1-8).99 Another subpopulation of cells that consists of the CSC progeny undergo partial to complete differentiation and lose the capability to support the tumor, albeit they still contribute to the morbidity of cancer. This hypothesis fundamentally altered the way we understand cancer but also gave rise to a debate regarding how widely this model applies. The competing hypothesis is based on a model where all the cells in a tumor possess an equal capacity for self-renewal and is commonly referred to as the stochastic model. According to this model, the process of cancer is driven entirely (or almost entirely) by environmental selection of favorable mutations; this model would necessarily predict that cancer is an inevitable outcome for multicellular organisms, and few, if any, long-lived animals would reach reproductive age.100 Thus this model must, by necessity, invoke the existence of protective mechanisms that are independent of cancer risk (e.g., efficient DNA repair mechanisms and immune surveillance).

• Figure 1-8 Multiple facets to cancer stem cell (CSC) self-renewal. Increasing evidence is emerging to support the notion that CSC self-renewal decisions can be guided by the activation of several pathways, including Wnt, Notch, Hedgehog, and others. A CSC may autonomously trigger the appropriate signaling cascade to maintain self-renewal with minimal niche support. It is likely that some CSCs need the appropriate microenvironment to provide the stimuli for uncontrolled self-renewal. Finally, some cancer cells have lost the capacity to self-renew regardless of stimulating molecules and hence cannot initiate a tumor. Rights were not granted to include this figure in electronic media. Please refer to the printed book. (Reprinted from O’Brien CA, Kreso A, Jamieson CHM: Cancer stem cells and self-renewal, Clin Cancer Res 16(12):3113–3120, 2010, with permission.) Clin Cancer Res
It is possible that the two models represent a continuum dependent on the extent to which CSCs undergo asymmetric versus symmetric divisions. Under conditions in which CSC divisions are primarily asymmetric, few CSCs would be apparent and the population would achieve a hierarchical organization, whereas under conditions in which CSCs underwent symmetric divisions, virtually every cell in the tumor would have CSC-like properties and the organization would be more consistent with a stochastic model. The prevailing opinion is that CSCs exist and are characterized both by peculiar phenotypes and defined sets of mutations of a small number of genes.101-103 Other mutations then endow their progeny with limited or extensive capacity to undergo programmed differentiation, thus resulting in the distinct clinical phenotypes that characterize acute and chronic leukemias or high-grade and low-grade solid tumors. The origin of CSCs is among the most important contemporary topics of investigation. CSCs may arise from mutations that occur in bona fide stem cells, they may arise by “de-differentiation” of somatic cells that acquire mutations that endow them with stem cell–like properties, or they may develop by fusion of a transformed cell and a bone marrow–derived stem cell.104 In companion animals, putative CSCs have been identified in hemangiosarcoma, osteosarcoma, brain tumors, and possibly lymphoma.105-108
As is true for the rest of cancer genetics, information in this field is rapidly evolving. Large-scale bioinformatics and conceptual advances are integrating the CSC theory into the mainstream of cancer research and biology, as well as into the design for new diagnostic and therapeutic strategies. For example, it appears that much like hematopoietic stem cells, the CSC niche favors oligoclonality and some genetic diversity. Thus clonal competition can ensue, giving rise to heterogeneous tumors and maintaining a reservoir of cells that can reestablish the tumor when a therapy effectively kills the predominant CSC clone and its progeny. Similarly, clonal competition can facilitate distant spread by selection of cells with different capabilities. An extreme example may be the potential for a single tumor cell (or a small population of oligoclonal cells in a tumor) to give rise to histologically distinct tumors—an event we have observed in xenotransplanted sarcomas.
Summary
The genetic basis of cancer is now beyond question. It is estimated that at least five to seven mutational events are required for overt malignant transformation, and genomic instability seems to be necessary to establish a self-renewing population of cells (possibly CSCs) whose progeny expand to cause clinical disease. Ultimately, a subpopulation endowed with metastatic properties that is drug resistant leads to death of the cancer patient. The rate and the flow of information is such that we predict the coming decade will see transformational changes in our perception of how genes interact with the macroenvironment at the organismal level and with the microenvironment in tumors. Although it is possible that cancer in higher vertebrates is an inevitable consequence of evolution,109 improvements in our understanding of fundamental mechanisms that account for malignant transformation and tumor progression will allow us to design strategies to improve quality of life and outcomes for cancer patients.
References
1. Vogelstein, B, Kinzler, KW. Cancer genes and the pathways they control. Nat Med. 2004;10:789–799.
2. Ponder, BA. Cancer genetics. Nature. 2001;411:336–341.
3. Ley, TJ, et al. DNA sequencing of a cytogenetically normal acute myeloid leukaemia genome. Nature. 2008;456:66–72.
4. Sjoblom, T, et al. The consensus coding sequences of human breast and colorectal cancers. Science. 2006;314:268–274.
5. Morin, RD, et al. Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma. Nature. 2011;476:298–303.
6. Pasqualucci, L, et al. Analysis of the coding genome of diffuse large B-cell lymphoma. Nat Genet. 2011;43:830–837.
7. Fearon, ER. Human cancer syndromes: clues to the origin and nature of cancer. Science. 1997;278:1043–1050.
8. Nagy, R, Sweet, K, Eng, C. Highly penetrant hereditary cancer syndromes. Oncogene. 2004;23:6445–6470.
9. Knudson, AG. Mutation and cancer: Statistical study of retinoblastoma. Proc Natl Acad Sci U S A. 1971;68:820–823.
10. Hahn, WC, Weinberg, RA. Modelling the molecular circuitry of cancer. Nat Rev Cancer. 2002;2:331–341.
11. Hanahan, D, Weinberg, RA. Hallmarks of cancer: The next generation. Cell. 2011;144:646–674.
12. Pihan, G, Doxsey, SJ. Mutations and aneuploidy: Co-conspirators in cancer? Cancer Cell. 2003;4:89–94.
13. Lingaas, F, et al. A mutation in the canine BHD gene is associated with hereditary multifocal renal cystadenocarcinoma and nodular dermatofibrosis in the German Shepherd dog. Hum Mol Genet. 2003;12:3043–3053.
14. Nickerson, ML, et al. Mutations in a novel gene lead to kidney tumors, lung wall defects, and benign tumors of the hair follicle in patients with the Birt-Hogg-Dube syndrome. Cancer Cell. 2002;2:157–164.
15. Trosko, JE. Commentary: Is the concept of “tumor promotion” a useful paradigm? Mol Carcinog. 2001;30:131–137.
16. Kopelovich, L. Hereditary adenomatosis of the colon and rectum: relevance to cancer promotion and cancer control in humans. Cancer Genet Cytogenet. 1982;5:333–352.
17. Heng, HH. Cancer genome sequencing: the challenges ahead. Bioessays. 2007;29:783–794.
18. Balmain, A. Cancer as a complex genetic trait: tumor susceptibility in humans and mouse models. Cell. 2002;108:145–152.
19. Amos, CI, et al. Genome-wide association scan of tag SNPs identifies a susceptibility locus for lung cancer at 15q25.1. Nat Genet. 2008;40:616–622.
20. Hung, RJ, et al. A susceptibility locus for lung cancer maps to nicotinic acetylcholine receptor subunit genes on 15q25. Nature. 2008;452:633–637.
21. Thorgeirsson, TE, et al. A variant associated with nicotine dependence, lung cancer and peripheral arterial disease. Nature. 2008;452:638–642.
22. Liu, JZ, et al. Meta-analysis and imputation refines the association of 15q25 with smoking quantity. Nat Genet. 2010;42:436–440.
23. Shah, PP, et al. Association of functionally important polymorphisms in cytochrome P4501B1 with lung cancer. Mutat Res. 2008;643:4–10.
24. Hecht, SS, Kassie, F, Hatsukami, DK. Chemoprevention of lung carcinogenesis in addicted smokers and ex-smokers. Nat Rev Cancer. 2009;9:476–488.
25. Jalas, JR, Hecht, SS, Murphy, SE. Cytochrome P450 enzymes as catalysts of metabolism of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone, a tobacco specific carcinogen. Chem Res Toxicol. 2005;18:95–110.
26. Loeb, LA. Mutator phenotype may be required for multistage carcinogenesis. Cancer Res. 1991;51:3075–3079.
27. Lindblad-Toh, K, et al. Breen, M, et al, eds Genes, dogs and cancer: Fifth Canine Cancer Conference. Orlando: IVIS; 2009;vol 5.
28. Ostrander, EA, Breen, M, et al, eds. Genes, dogs and cancer: Fifth Canine Cancer Conference, vol 5. Orlando: IVIS, 2009.
29. Pontius, JU, et al. Initial sequence and comparative analysis of the cat genome. Genome Res. 2007;17:1675–1689.
30. Thomas, R, et al. Microarray-based cytogenetic profiling reveals recurrent and subtype-associated genomic copy number aberrations in feline sarcomas. Chromosome Res. 2009;17:987–1000.
31. Hedan, B, et al. Molecular cytogenetic characterization of canine histiocytic sarcoma: A spontaneous model for human histiocytic cancer identifies deletion of tumor suppressor genes and highlights influence of genetic background on tumor behavior. BMC Cancer. 2011;11:201.
32. Thomas, R, et al. Influence of genetic background on tumor karyotypes: Evidence for breed-associated cytogenetic aberrations in canine appendicular osteosarcoma. Chromosome Res. 2009;17:365–377.
33. Modiano, JF, et al. Distinct B-cell and T-cell lymphoproliferative disease prevalence among dog breeds indicates heritable risk. Cancer Res. 2005;65:5654–5661.
34. Tamburini, BA, et al. Gene expression profiles of sporadic canine hemangiosarcoma are uniquely associated with breed. PLoS ONE. 2009;4:e5549.
35. Scott, MC, et al. Molecular subtypes of osteosarcoma identified by reducing tumor heterogeneity through an interspecies comparative approach. Bone. 2011;49:356–367.
36. Breen, M, Modiano, JF. Evolutionarily conserved cytogenetic changes in hematological malignancies of dogs and humans—man and his best friend share more than companionship. Chromosome Res. 2008;16:145–154.
37. Fosmire, SP, et al. Inactivation of the p16 cyclin-dependent kinase inhibitor in high-grade canine non-Hodgkin’s T-cell lymphoma. Vet Pathol. 2007;44:467–478.
38. Angstadt, AY, et al. Characterization of canine osteosarcoma by array comparative genomic hybridization and RT-qPCR: Signatures of genomic imbalance in canine osteosarcoma parallel the human counterpart. Genes Chromosomes Cancer. 2011;50(11):859–874.
39. Duesberg, P, Li, R. Multistep carcinogenesis: A chain reaction of aneuploidizations. Cell Cycle. 2003;2:202–210.
40. Duesberg, P, Rausch, C, Rasnick, D, et al. Genetic instability of cancer cells is proportional to their degree of aneuploidy. Proc Natl Acad Sci U S A. 1998;95:13692–13697.
41. Mani, RS, Chinnaiyan, AM. Triggers for genomic rearrangements: insights into genomic, cellular and environmental influences. Nat Rev Genet. 2010;11:819–829.
42. O’Donoghue, LE, et al. Expression profiling in canine osteosarcoma: identification of biomarkers and pathways associated with outcome. BMC Cancer. 2010;10:506.
43. Paoloni, M, et al. Canine tumor cross-species genomics uncovers targets linked to osteosarcoma progression. BMC Genomics. 2009;10:625.
44. Selvarajah, GT, et al. Gene expression profiling of canine osteosarcoma reveals genes associated with short and long survival times. Mol Cancer. 2009;8:72.
45. Hanahan, D, Weinberg, RA. The hallmarks of cancer. Cell. 2000;100:57–70.
46. Ruchatz, H, Coluccia, AM, Stano, P, et al. Constitutive activation of Jak2 contributes to proliferation and resistance to apoptosis in NPM/ALK-transformed cells. Exp Hematol. 2003;31:309–315.
47. Modiano, JF, Ritt, MG, Wojcieszyn, J, et al. Growth arrest of melanoma cells is differentially regulated by contact inhibition and serum deprivation. DNA Cell Biol. 1999;18:357–367.
48. Modiano, JF, Johnson, LD, Bellgrau, D. Negative regulators in homeostasis of naive peripheral T cells. Immunol Res. 2008;41:137–153.
49. Levine, RA, Fleischli, MA. Inactivation of p53 and retinoblastoma family pathways in canine osteosarcoma cell lines. Vet Pathol. 2000;37:54–61.
50. Levine, RA, Forest, T, Smith, C. Tumor suppressor PTEN is mutated in canine osteosarcoma cell lines and tumors. Vet Pathol. 2002;39:372–378.
51. Modiano, JF, Breen, M, Valli, VE, et al. Predictive value of p16 or Rb inactivation in a model of naturally occurring canine non-Hodgkin’s lymphoma. Leukemia. 2007;21:184–187.
52. Dickerson, EB, et al. Mutations of phosphatase and tensin homolog deleted from chromosome 10 in canine hemangiosarcoma. Vet Pathol. 2005;42:618–632.
53. Hammerman, PS, Fox, CJ, Thompson, CB. Beginnings of a signal-transduction pathway for bioenergetic control of cell survival. Trends Biochem Sci. 2004;29:586–592.
54. White, E, DiPaola, RS. The double-edged sword of autophagy modulation in cancer. Clin Cancer Res. 2009;15:5308–5316.
55. Ohyashiki, JH, Sashida, G, Tauchi, T, et al. Telomeres and telomerase in hematologic neoplasia. Oncogene. 2002;21:680–687.
56. Pang, LY, Argyle, DJ. Using naturally occurring tumours in dogs and cats to study telomerase and cancer stem cell biology. Biochim Biophys Acta. 2009;1792:380–391.
57. Ferrara, N, Kerbel, RS. Angiogenesis as a therapeutic target. Nature. 2005;438:967–974.
58. Folkman, J. The role of angiogenesis in tumor growth. Semin Cancer Biol. 1992;3:65–71.
59. Folkman, J. Tumor angiogenesis: therapeutic implications. N Engl J Med. 1971;285:1182–1186.
60. Chen, H, et al. In vitro and in vivo production of vascular endothelial growth factor by chronic lymphocytic leukemia cells. Blood. 2000;96:3181–3187.
61. Kini, AR, Kay, NE, Peterson, LC. Increased bone marrow angiogenesis in B cell chronic lymphocytic leukemia. Leukemia. 2000;14:1414–1418.
62. Molica, S, et al. Prognostic value of enhanced bone marrow angiogenesis in early B-cell chronic lymphocytic leukemia. Blood. 2002;100:3344–3351.
63. Juliusson, G, et al. Frequent good partial remissions from thalidomide including best response ever in patients with advanced refractory and relapsed myeloma. Br J Haematol. 2000;109:89–96.
64. Tosi, P, et al. Salvage therapy with thalidomide in multiple myeloma patients relapsing after autologous peripheral blood stem cell transplantation. Haematologica. 2001;86:409–413.
65. Jorgensen, JM. The role of angiogenesis in non-Hodgkin lymphoma. Dan Med Bull. 2005;52:254.
66. Ranieri, G, et al. Endothelial area and microvascular density in a canine non-Hodgkin’s lymphoma: an interspecies model of tumor angiogenesis. Leuk Lymphoma. 2005;46:1639–1643.
67. Preziosi, R, Sarli, G, Paltrinieri, M. Prognostic value of intratumoral vessel density in cutaneous mast cell tumors of the dog. J Comp Pathol. 2004;130:143–151.
68. Restucci, B, et al. Expression of vascular endothelial growth factor receptor Flk-1 in canine mammary tumours. J Comp Pathol. 2004;130:99–104.
69. Fidler, IJ. The pathogenesis of cancer metastasis: the ‘seed and soil’ hypothesis revisited. Nat Rev Cancer. 2003;3:453–458.
70. Kim, JW, et al. Rapid apoptosis in the pulmonary vasculature distinguishes non-metastatic from metastatic melanoma cells. Cancer Lett. 2004;213:203–212.
71. Wong, CW, et al. Intravascular location of breast cancer cells after spontaneous metastasis to the lung. Am J Pathol. 2002;161:749–753.
72. Wong, CW, et al. Apoptosis: an early event in metastatic inefficiency. Cancer Res. 2001;61:333–338.
73. Koshkina, NV, et al. Fas-negative osteosarcoma tumor cells are selected during metastasis to the lungs: the role of the Fas pathway in the metastatic process of osteosarcoma. Mol Cancer Res. 2007;5:991–999.
74. Krishnan, K, et al. Ezrin mediates growth and survival in Ewing’s sarcoma through the AKT/mTOR, but not the MAPK, signaling pathway. Clin Exp Metastasis. 2006;23:227–236.
75. Mueller, MM, Fusenig, NE. Friends or foes—bipolar effects of the tumour stroma in cancer. Nat Rev Cancer. 2004;4:839–849.
76. Nowell, PC. Mechanisms of tumor progression. Cancer Res. 1986;46:2203–2207.
77. Modiano, JF, et al. Fas ligand gene transfer for cancer therapy. Cancer Ther. 2004;2:561–570.
78. Dunn, GP, Bruce, AT, Ikeda, H, et al. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol. 2002;3:991–998.
79. Breivik, J. The evolutionary origin of genetic instability in cancer development. Semin Cancer Biol. 2005;15:51–60.
80. Nguyen, DH, et al. Radiation acts on the microenvironment to affect breast carcinogenesis by distinct mechanisms that decrease cancer latency and affect tumor type. Cancer Cell. 2011;19:640–651.
81. Boveri, T. Concerning the origin of malignant tumors. J Cell Sci. 2008;121:1–84.
82. Albertson, DG, Collins, C, McCormick, F, et al. Chromosome aberrations in solid tumors. Nat Genet. 2003;34:369–376.
83. Teixeira, MR, Heim, S. Multiple numerical chromosome aberrations in cancer: what are their causes and what are their consequences? Semin Cancer Biol. 2005;15:3–12.
84. Gollin, SM. Mechanisms leading to chromosomal instability. Semin Cancer Biol. 2005;15:33–42.
85. Rajagopalan, H, Lengauer, C. Aneuploidy and cancer. Nature. 2004;432:338–341.
86. Maser, RS, DePinho, RA. Connecting chromosomes, crisis, and cancer. Science. 2002;297:565–569.
87. Dvorak, HF. Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N Engl J Med. 1986;315:1650–1659.
88. Lin, WW, Karin, M. A cytokine-mediated link between innate immunity, inflammation, and cancer. J Clin Invest. 2007;117:1175–1183.
89. Mantovani, A, Allavena, P, Sica, A, et al. Cancer-related inflammation. Nature. 2008;454:436–444.
90. Bhatia, R, McGlave, PB, Dewald, GW, et al. Abnormal function of the bone marrow microenvironment in chronic myelogenous leukemia: role of malignant stromal macrophages. Blood. 1995;85:3636–3645.
91. Grivennikov, SI, Greten, FR, Karin, M. Immunity, inflammation, and cancer. Cell. 2010;140:883–899.
92. Erez, N, Truitt, M, Olson, P, et al. Cancer-associated fibroblasts are activated in incipient neoplasia to orchestrate tumor-promoting inflammation in an NF-kappaB-dependent manner. Cancer Cell. 2010;17:135–147.
93. Bergman, PJ. Anticancer vaccines. Vet Clin North Am Small Anim Pract. 2007;37:1111–1119. [vi-ii].
94. Wolffe, AP, Matzke, MA. Epigenetics: regulation through repression. Science. 1999;286:481–486.
95. Costello, JF. Comparative epigenomics of leukemia. Nat Genet. 2005;37:211–212.
96. Yu, L, et al. Global assessment of promoter methylation in a mouse model of cancer identifies ID4 as a putative tumor-suppressor gene in human leukemia. Nat Genet. 2005;37:265–274.
97. Cui, H, et al. Loss of IGF2 imprinting: a potential marker of colorectal cancer risk. Science. 2003;299:1753–1755.
98. Lapidot, T, et al. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. 1994;367:645–648.
99. O’Brien, CA, Kreso, A, Jamieson, CHM. Cancer stem cells and self-renewal. Clin Cancer Res. 2010;16(12):3113–3120.
100. Clarke, MF, Fuller, M. Stem cells and cancer: two faces of eve. Cell. 2006;124:1111–1115.
101. Huntly, BJ, Gilliland, DG. Leukaemia stem cells and the evolution of cancer-stem-cell research. Nat Rev Cancer. 2005;5:311–321.
102. Singh, SK, et al. Identification of human brain tumour initiating cells. Nature. 2004;432:396–401.
103. Smith, GH. Mammary cancer and epithelial stem cells: a problem or a solution? Breast Cancer Res. 2002;4:47–50.
104. Bjerkvig, R, Tysnes, BB, Aboody, KS, et al. Opinion: the origin of the cancer stem cell: current controversies and new insights. Nat Rev Cancer. 2005;5:899–904.
105. Lamerato-Kozicki, AR, Helm, KM, Jubala, CM, et al. Canine hemangiosarcoma originates from hematopoietic precursors with potential for endothelial differentiation. Exp Hematol. 2006;34:870–878.
106. Wilson, H, et al. Isolation and characterisation of cancer stem cells from canine osteosarcoma. Vet J. 2007;175(1):69–75.
107. Stoica, G, et al. Identification of cancer stem cells in dog glioblastoma. Vet Pathol. 2009;46(3):391–406.
108. Ito, D, et al. A tumor-related lymphoid progenitor population supports hierarchical tumor organization in canine B-cell lymphoma. J Vet Intern Med. 2011;25:890–896.
109. Modiano, JF, Breen, M. Shared pathogenesis of human and canine tumors—an inextricable link between cancer and evolution. Cancer Ther. 2008;6:239–246.
Section B
Chemical, Physical, and Hormonal Factors
In 1978, the United States Congress ordered development of the first Report on Carcinogens (RoC), a document designed to educate the public and health professionals on potential cancer hazards. The document is now required by law to be released every 2 years by the Secretary of the Department of Health and Human Services. The twelfth edition of the RoC, released in 2011, lists 240 potential carcinogens, of which 54 are categorized as known to be human carcinogens and 186 are categorized as reasonably anticipated to be human carcinogens.1 Although no such report exists for companion animals, one could reasonably assume that there would be considerable overlap between such a list and the potential carcinogens found in the RoC. The 2005 RoC was the first to include neutrons, x- and gamma-radiation, and viruses (human papillomavirus, hepatitis B virus, and hepatitis C virus). Although the list of carcinogens reportedly associated with cancer in companion animals is less extensive, this section will address chemical, physical, and hormonal factors that have been linked to carcinogenesis in pet animals. Viral carcinogenesis will be addressed in a separate section (see Section C below).
Chemical Factors
Despite ample evidence that secondhand smoke increases the risk of lung cancer in people,2,3 the data for this effect in companion animals are less compelling. One case-control study involving dogs with lung cancer from two veterinary hospitals showed only a weak relationship between living with a smoker and development of lung cancer, and the risk did not increase with an increased smoke exposure index.4 However, evidence for a relationship between exposure to environmental tobacco smoke (ETS) and development of other malignancies in companion animals is mounting.
Based on human data suggesting that smoking may increase the risk of non-Hodgkin’s lymphoma,5,6 Bertone et al examined the relationship between ETS exposure and development of feline lymphoma.7 In a case-control study of 80 cats with malignant lymphoma and 114 control cats with renal disease that presented to Tufts University School of Veterinary Medicine (TUSVM) between 1993 and 2000, the relative risk of lymphoma for cats with any household ETS exposure was 2.4. As has been reported for male smokers,8 the risk of lymphoma increased with increases in either duration or quantity of exposure. More recently, an Italian study of environmental risk factors for development of cancer in domestic animals demonstrated that ETS exposure increased the risk of lymphoma in dogs.9
Hypothesizing that inhalation and ingestion of carcinogenic compounds in ETS during grooming might also predispose cats in smoking households to development of oral squamous cell carcinoma (SCC), Bertone et al examined ETS and other environmental and lifestyle risk factors in cats with SCC.10 The study examined a population of 36 cats with histologically confirmed oral SCC and a control population of 112 cats with renal disease, all presenting to TUSVM between 1994 and 2000. Exposure to ETS was associated with a twofold but statistically insignificant increased risk of oral SCC.10 Interestingly, in a separate report, the investigators showed that SCC tissue from cats exposed to any ETS were 4.5 more likely to overexpress p53 and those from cats with 5 years or more of ETS exposure were 7 times more likely to overexpress p53.11 Although the findings did not reach statistical significance, the collective work of this group provides an intriguing suggestion that both ETS and mutations in the p53 gene may play a role in the etiology of feline oral SCC.
Pesticides, Herbicides, and Insecticides
In 1991, investigators at the National Cancer Institute (NCI) completed a case-control study to examine the relationship between exposure of dogs to the herbicide, 2,4-dichlorophenoxyacetic acid (2,4-D), and development of lymphoma.12 Dogs with a histologically confirmed diagnosis of lymphoma during a 4-year period were identified through the computerized medical record information from three veterinary teaching hospitals. Each case animal was age-matched with two control animals. The first control group consisted of dogs diagnosed with tumors other than lymphoma during the same time period and the second control group was a nontumor group, selected from all other dogs presenting to the hospital for conditions deemed unrelated to chemical exposure. Owners were questioned about household use of and potential pet exposure to commercial lawn care and owner-applied herbicides. A positive association was found between exposure to owner-applied 2,4-D or the use of commercial lawn care services and the development of canine lymphoma. The risk of lymphoma development doubled when owners applied 2,4-D liquid or granules to the lawn four or more times a year. After these findings were reported, an independent review panel was convened to assess the validity of the NCI study.13 This panel voiced concerns about the original study design, data analysis, and interpretation, concluding that a relationship between 2,4-D exposure and the development of canine lymphoma could not be established based on the reported data. In response, the original investigators reanalyzed their data, addressing many of the concerns raised by the scientific review panel.14 In their second study, Hayes et al used a more stringent definition of exposure to 2,4-D, including only cases in which the owner applied 2,4-D as the sole herbicide and did not use other lawn chemicals or lawn care services. Their second report did not show a statistically significant association between exposure to 2,4-D and development of lymphoma.14 However, they concluded that their results did indicate a dose-response relationship between disease incidence and number of yearly 2,4-D applications by dog owners. In a subsequent study conducted by researchers at Michigan State University, the original 1991 data was again reanalyzed using the more stringent definition of exposure and completing a dose-response analysis. The study, which was funded by a chemical industry task force, showed no dose-response relationship between number of 2,4-D applications and the occurrence of canine lymphoma.15 Although increased urinary excretion of 2,4-D has been demonstrated in dogs exposed to herbicide-treated lawns, a direct link between such exposure and development of lymphoma has not been shown.16 A 2011 case-control study conducted in Italy was designed to assess the effect of residential exposure to environmental pollutants on the risk of developing lymphoma.17 The investigators were unable to demonstrate an association between exposure to pesticides (which by their definition included herbicides) and development of lymphoma. They did, however, find that living in industrial areas and owner use of chemicals such as paints and solvents were significantly and independently associated with lymphoma.
Canine transitional cell carcinoma (TCC) of the urinary bladder is another malignancy that has been linked to environmental carcinogens including insecticides and herbicides. In a case-control study of 59 dogs with TCC and 71 age-matched and breed size–matched control dogs with other neoplasms or chronic disease, investigators compared the two populations to assess the effect of obesity, exposure to sidestream cigarette smoke and chemicals, and use of topical insecticides on risk of TCC.18 They reported an increased risk of TCC in dogs treated with topical insecticides, with an enhancement of this risk in overweight or obese dogs. In the aforementioned study of risk factors for oral SCC in cats, Bertone et al reported a significantly increased risk of oral SCC in cats that wore flea collars.10 However, newer topical spot-on flea and tick products have been evaluated in Scottish terrier populations due to the breed’s predisposition for development of TCC of the urinary bladder and have not been shown to increase the risk of TCC.19 Other studies of Scottish terriers have suggested that exposure to lawn and garden care products containing phenoxy herbicides, including 2,4-D, 4-chloro-2-methylphenoxy acetic acid (MCPA), and 2-(4-chloro-2-methylphenoxyl) propionic acid (MCPP), is associated with an increased risk of TCC.20 Although it has been difficult to prove a link between phenoxy herbicides and development of lymphoma or TCC, attempts to limit exposure of pets to these products is advised.
Cyclophosphamide
The cytotoxic alkylating agent, cyclophosphamide, has been implicated in the development of urinary bladder cancer in people and dogs.21-23 A known potential side effect of cyclophosphamide therapy is sterile hemorrhagic cystitis, which may develop due to the irritating effects of the drug’s metabolite, acrolein, on the bladder mucosa. Although the specific etiology is unknown, it is speculated that chronic inflammation secondary to acrolein exposure is the underlying event that leads to bladder cancer in some patients that have undergone cyclophosphamide therapy. The author has had the experience of treating a dog for lymphoma that was found to have concurrent but clinically occult TCC of the bladder prior to initiation of cyclophosphamide chemotherapy. If an abdominal ultrasound had not been performed on this dog as part of the initial staging procedures prior to chemotherapy, the bladder TCC may have been diagnosed at a later date and incorrectly attributed to administration of cyclophosphamide. This exemplifies the danger of assuming a causal relationship for potential carcinogens, especially in animals that have a prior malignancy.
Rural versus Urban Environment
Although several reports have identified differences in cancer incidence between companion animals living in urban versus rural settings, the underlying cause for these differences is unclear. An increased incidence of some canine cancers, including lymphoma, tonsillar SCC, and nasal carcinoma,17,18,24,25 has been reported in urban/industrial settings as compared with rural settings. However, the coexistence of multiple environmental carcinogens in the same setting makes discerning the “smoking gun” a difficult task. Nonetheless, the study of animals as sentinels of environmental health hazards has been recommended and provides supportive evidence for carcinogenic risk assessment across species.26-29 Results of a hospital-based case-control study conducted in Naples, Italy and nearby cities with known high levels of illegal waste dumping suggest that living in these sites of waste emission increases the risk of cancer development in dogs but not cats. This may relate to reduced exposure of cats to environmental carcinogens, as they are often exclusively indoor pets.9
Physical Factors
The relationship between sunlight exposure or ultraviolet irradiation and subsequent development of skin cancer is one of the better known examples of physical carcinogenesis. Recognized for its role in human SCC induction, sunlight has also been implicated as a cause of SCC in domestic animals and livestock—an implication that is strengthened by a clear dose-response relationship shown in both epidemiologic and experimental studies.28-31 In particular, light skin pigmentation and chronic sun exposure are associated with the development of facial, aural, and nasal planum SCC in white or partially white cats and may also play a similar role in some cutaneous SCC lesions in dogs. The portion of the ultraviolet spectrum most likely to be responsible for nonmelanotic skin lesions in people and animals is ultraviolet B (UV-B), which is in the range of 280 to 320 nm.28 Cumulative long-term exposure to UV-B may induce skin tumors directly through genetic mutations, including mutations in p53, and indirectly by impairing the response of the immune system to tumor antigens.28,32,33 Pets are at greatest risk of exposure to UV-B during the midday hours and should be protected from this exposure, especially if they are a lightly pigmented breed.
Trauma/Chronic Inflammation
Chronic inflammation may lead to cellular mutations that in turn cause neoplastic transformation. In four dogs with chronic pigmentary keratitis, neoplastic lesions of the cornea, including three SCC and one squamous papilloma, were reported.34 Although the underlying etiology of the keratitis could not be confirmed, the neoplastic transformation was likely related to chronic inflammation. Earlier reports have linked feline eye tumors to ocular trauma that induces secondary uveitis and lens rupture (see Chapter 31).35 Unlike the corneal tumors reported in dogs with pigmentary keratitis, the ocular lesions in cats were intraocular sarcomas. Despite the varied histology, the underlying etiology in all cases was thought to be related to inflammatory changes. Another companion animal malignancy thought to be associated with inflammation is vaccine-associated feline sarcoma (VAFS). This tumor type and its etiology are discussed in detail in Chapter 21.
Magnetic Fields
More than a quarter century ago, a potential link between chronic low-dose exposure to magnetic fields and development of childhood cancer was proposed.36 Since then, multiple studies have been conducted in an attempt to discern links between magnetic fields and a variety of human cancers ranging from hematopoietic malignancies to breast cancer. The extremely low frequency (<60 Hz) magnetic fields in question are ubiquitous in today’s society and are generated by household appliances, industrial machinery, and electrical power lines. Since pets share our environment and have similar exposure to magnetic fields, a similar risk of cancer development has been presumed for companion animals. In a 1995 study, the risk for development of lymphoma was found to be highest in dogs from households with the highest measured exposure to magnetic fields.37 The risk was related to both duration and intensity of exposure and was highest for dogs that spent more than 25% of the day outdoors. In the following year at the request of Congress, a report was published by the National Research Council (NRC) that reviewed over 500 studies on the subject of cancer risk and exposure to electromagnetic fields.38 The report concluded that, although a weak association has been shown between development of childhood leukemia and exposure to electromagnetic fields, no clear evidence exists to suggest that exposure to electromagnetic fields is a true threat to human health. To the author’s knowledge, no reports on the possible link between magnetic fields and cancer in companion animals have been published since the 1995 report, although the magnetic field debate continues in the human literature. The NRC report suggested that other factors, including air quality and proximity to high traffic density, may be more likely environmental causes of cancer than low frequency magnetic fields.
Radiation
The first report of cancer development after therapeutic irradiation in a dog dates back over 25 years, when orthovoltage radiation was considered state-of-the-art.39 At that time, the term malignant transformation was used to describe the development of epithelial malignancies at the site of prior irradiation for acanthomatous epulides in four dogs. Following a review of more recent cases with megavoltage irradiation, the author of the original report has since suggested that the concept of malignant transformation should be discarded, in that the occurrence of second tumors was not likely due to a true transformation of epulides into carcinomas.40 Rather, the relatively high rate of carcinomas at previously irradiated sites for epulides is a result of less effective forms of irradiation or misclassification of the tumor type. Radiation carcinogenesis is considered the cause of second tumors arising in radiation fields. In human oncology, most tumors occurring in heavily irradiated treatment fields are mesenchymal, rather than epithelial, in origin.41-43 Several reports of sarcomas occurring in sites of prior radiation can be found in the veterinary literature, as well,39,44-46 with the most recent being a retrospective review of 57 dogs undergoing definitive megavoltage radiation therapy with 60Cobalt photons for acanthomatous epulis.40 In the latter report, McEntee et al describe the development of a second tumor (one sarcoma and one osteosarcoma) in 2 of the 57 irradiated dogs, occurring 5.2 and 8.7 years after the initial treatment, respectively. The overall incidence of second tumors was lower in their study than in previous reports (3.5% versus up to 18%).39,47 The fact that no epithelial tumors were reported may indicate a more efficient targeting of a subpopulation of malignant epithelial cells by megavoltage radiotherapy, as compared to orthovoltage. The report suggests that the risk of second tumors at sites of radiation therapy is primarily of clinical concern for young dogs expected to enjoy long-term survival. Second tumors have also been reported in at least six people who have undergone stereotactic radiosurgery.48 As this radiation technique becomes more commonplace in veterinary medicine, the possibility of second tumors may need to be considered in companion animals undergoing stereotactic radiosurgery.
Surgery and Implanted Devices
The development of sarcomas at the site of metallic implants has been reported in people, dogs, and laboratory animal models.49,50 However, it is often difficult to discern if sarcoma development is related to fracture fixation devices or to other factors, including wound healing complications and osteomyelitis. The largest veterinary study examining the relationship between metallic implants and tumor development in dogs was published in 1993 by Li et al.50 The authors reported on 222 dogs that developed tumors of any kind after fracture fixation, compared to 1635 dogs that underwent fracture fixation without subsequent tumor development. They concluded that use of metallic implants was not a risk factor for bone tumor development. Other types of implants and foreign materials related to surgery are sporadically implicated in carcinogenesis in human and veterinary case reports. Published examples include one dog that developed a myxoma at the site of a subcutaneous pacemaker and another that developed a jejunal osteosarcoma associated with a surgical sponge presumably not retrieved during an abdominal surgery 6 years prior.51,52
Asbestos
Asbestos exposure is a known risk factor for development of mesothelioma in people.53 In fact, an estimated 60% to 88% of all cases of human mesothelioma are attributable to asbestos exposure.53 A similar association has been found for dogs whose owners have an asbestos-related occupation or hobby.54 This association was further supported by a study in which significantly more asbestos bodies were found in dogs with mesothelioma than in control dogs.55 Pericardial mesothelioma was reported in five golden retrievers with histories of chronic idiopathic hemorrhagic pericardial effusion, suggesting that other factors, including breed predispositions and chronic inflammation unrelated to asbestos exposure, may be involved in the etiology of mesothelioma affecting the pericardium.56
Hormonal Factors
Canine Mammary Cancer
Canine mammary cancer is a well-established model of hormonal carcinogenesis in domestic animals (see Chapter 27). The most common neoplasm of female intact dogs, mammary tumors affect approximately 260/100,000 dogs in the United States each year.57,58 Dogs spayed before their first estrous cycle have a greatly reduced risk of developing breast cancer, with the risk rising to 26% for dogs that are spayed after their second estrus.59,60 Mammary tumors primarily affect late middle-aged (9 to 11 years) female intact dogs, with an increased incidence beginning at approximately 6 years of age.61 Sexual steroid hormones likely have their primary effect on target cells during the very early stages of mammary carcinogenesis in dogs; thus, the protective effect of spaying is lost with time.62-68 In addition to the influence of ovarian hormones on breast cancer development, the use of medroxyprogesterone acetate (progestin and estrogen combination) products to prevent estrus or to treat pseudopregnancy has been linked to an increased incidence of mammary tumor development in dogs.69-71
Progestin-induced growth hormone (GH) excess in dogs originates in the mammary gland. Within the mammary gland, the gene encoding GH may act in an autocrine/paracrine fashion, effecting cyclic epithelial changes and, perhaps, carcinogenesis. Research to determine the mechanism of progestin-induced mammary GH expression in dogs has led to the cloning and cellular localization of the canine progesterone receptor (PR).72 The investigators concluded that within the same mammary gland cell, the activated PR may transactivate GH expression and function as a prerequisite transcription factor. However, this regulation may be lost during malignant transformation. Mammary GH expression has been reported in people as well, suggesting that evaluation of links between this hormone and mammary carcinogenesis may have implications for both species.73,74
Feline Mammary Cancer
Both estrogen and progesterone are thought to play important roles in feline mammary carcinogenesis, although the underlying mechanisms are less clear than for dogs. Prior studies have shown that intact female cats and those cats that are exposed regularly to progestin are at an increased risk for mammary cancer development. The literature also suggests that, as is the case in dogs, ovariectomy may be protective against feline mammary gland tumor development in cats.57,75-77 In one study, cats ovariectomized at 6 months of age had an approximate sevenfold reduction in risk of mammary tumor development compared to intact cats.57 What has been lacking in the veterinary literature is an epidemiologic study of cats with age-matched controls for comparison to specifically investigate the effects that spaying and age of spay have on the risk of feline mammary carcinoma development. Overley et al attempted to address these issues in a retrospective study that compared a population of 308 cats with biopsy-proven mammary carcinoma diagnosed between 2000 and 2001 and a control population of 400 female cats not diagnosed with mammary tumors but from the same biopsy service population as the affected cats. Cats from the two groups were frequency-matched by age and year of diagnosis.77 The study reported a 91% reduction in risk for those spayed prior to 6 months of age and an 86% reduction in risk for those spayed prior to 1 year of age, compared to intact cats. Although the study was retrospective in nature and relied on questionnaire data from a survey with a 58% response rate, the manuscript is the first published report attempting to age-match controls and evaluate age at time of spay as a risk factor for mammary tumor development in cats. Although further epidemiologic evaluation and prospective assessment are needed to confirm these findings, the reported results provide some justification for recommending ovariohysterectomy prior to 1 year of age in cats.
Lymphoma
Surveillance, Epidemiology and End Results (SEER) data indicate that non-Hodgkin’s lymphoma is approximately 50% more common among men than women.78 Although a similar male predisposition is reported for canine lymphoma, the underlying role of gender in lymphoma etiology remains elusive. The author and others undertook a population-based study using the Veterinary Medical Database (VMDB) to determine the relationship between gender and development of canine lymphoma.79 Data from 1980 to 2000 were retrieved from the VMDB and sorted by gender and reproductive status. In the statistical analysis, spayed or neutered dogs diagnosed with lymphoma were compared to intact dogs seen each year in each gender category. The VMDB included nearly 15,000 lymphoma cases in a population of over 1.2 million dogs. These data suggest that intact females were significantly less likely to develop lymphoma than were other gender groups. Based on this initial data, we propose that further examination of the role of estrogen in the development or prevention of canine lymphoma is warranted.
Androgens/Testosterone
Perianal Adenoma
Perianal adenoma is androgen dependent and occurs primarily in intact male dogs, whereas perianal adenocarcinoma occurs in both intact and castrated males. Perianal adenomas may also develop in female dogs secondary to testosterone secretion from the adrenal gland.80 The majority of these tumors in male dogs resolve after castration, a fact that lends further support to the assertion that androgens are involved in the etiology of this tumor (see Chapter 22).81
Prostate Cancer
Although there is a well-established link between presence of testosterone and development of benign prostatic hyperplasia (BPH) in dogs and man, prostatic cancer risk is not higher in intact dogs compared to those that are castrated.82 To the contrary, neutered dogs have been shown to be at increased risk. Castration is likely not an initiating event, but is thought to favor tumor progression.83-86 A clear relationship between age at castration and risk of prostate cancer development is yet to be determined (see Chapter 28).
References
1. Report on Carcinogens: ed 12, 2011; Available at http://ntp.niehs.nih.gov
2. Leonard, CT, Sachs, DPL. Environmental tobacco smoke and lung cancer incidence. Curr Opin Pulm Med. 1999;5:189.
3. Hackshaw, AK, Law, MR, Wald, NJ. The accumulated evidence on lung cancer and environmental tobacco smoke. Br Med J. 1997;315:980.
4. Reif, JS, Dunn, K, Ogilvie, GK, et al. Passive smoking and canine lung cancer risk. Am J Epidemiol. 1992;135:234.
5. Herrinton, LJ, Friedman, GD. Cigarette smoking and risk of non-Hodgkin’s lymphoma subtypes. Cancer Epidemiol Biomarkers Prev. 1998;7:25.
6. Linet, MS, McLaughlin, JK, Hsing, AW, et al. Is cigarette smoking a risk factor for non-Hodgkin’s lymphoma or multiple myeloma? Results from the Lutheran Brotherhood Cohort Study. Leuk Res. 1992;16:621.
7. Bertone, ER, Snyder, LA, Moore, AS. Environmental tobacco smoke and risk of malignant lymphoma in pet cats. Am J Epidemiol. 2002;156:268.
8. Freedman, DS, Tolbert, PE, Coates, R. Relation of cigarette smoking to non-Hodgkin’s lymphoma among middle-aged men. Am J Epidemiol. 1998;148:833.
9. Marconato, L, Leo, C, Girelli, R, et al. Association between waste management and cancer in companion animals. J Vet Intern Med. 2009;23:564.
10. Bertone, ER, Snyder, LA, Moore, AS. Environmental and lifestyle risk factors for oral squamous cell carcinoma in domestic cats. J Vet Intern Med. 2003;17:557.
11. Snyder, LA, Bertone, ER, Jakowski, RM, et al. p53 expression and environmental tobacco smoke exposure in feline oral squamous cell carcinoma. Vet Pathol. 2004;41:209.
12. Hayes, HM, Tarone, RE, Cantor, KP, et al. Case-control study of canine malignant lymphoma: Positive association with dog owner’s use of 2,4-dichlorophenoxyacetic acid herbicides. J Natl Cancer Inst. 1991;83:1226.
13. Carlo, GL, Cole, P, Miller, AM, et al. Review of a study reporting an association between 2,4-dichlorophenoxyacetic acid and canine malignant lymphoma: Report of an expert panel. Reg Toxicol Pharmacol. 1992;10:245.
14. Hayes, HM, Tarone, RE, Cantor, KP. On the association between canine malignant lymphoma and opportunity for exposure to 2,4-dichlorophenoxyacetic acid. Environ Res. 1995;70:119.
15. Kaneene, JB, Miller, R. Re-analysis of 2,4-D use and the occurrence of canine malignant lymphoma. Vet Hum Toxicol. 1999;41:164.
16. Reynolds, PM, Reif, JS, Ramsdell, HS. Canine exposure to herbicide-treated lawns and urinary excretion of 2,4-dichlorophenoxyacetic acid. Cancer Epidemiol Biomarkers Prev. 1994;3:233.
17. Gavazza, A, Presciuttini, S, Barale, R, et al. Association between canine malignant lymphoma, living in industrial areas, and use of chemical by dog owners. J Vet Intern Med. 2001;15:190.
18. Glickman, LT, Schofer, FS, McKee, LJ, et al. Epidemiologic study of insecticide exposures, obesity, and risk of bladder cancer in household dogs. J Toxicol Environ Health. 1989;28:407.
19. Raghavan, M, Knapp, DW, Dawson, MH, et al. Topical flea and tick pesticides and the risk of transitional cell carcinoma of the urinary bladder in Scottish Terriers. J Am Vet Med Assoc. 2004;225:389.
20. Glickman, LT, Raghavan, M, Knapp, DW, et al. Herbicide exposure and the risk of transitional cell carcinoma of the urinary bladder in Scottish Terriers. J Am Vet Med Assoc. 2004;224:1290.
21. Baker, GL, Kahl, LE, Zee, BC, et al. Malignancy following treatment of rheumatoid arthritis with cyclophosphamide. Long-term case-control follow-up study. Am J Med. 1987;83:1.
22. Weller, RE, Wolf, AM, Oyejide, A. Transitional cell carcinoma of the bladder associated with Cyclophosphamide administration. J Am Anim Hosp Assoc. 1979;15:733.
23. Macy, DW, Withrow, SJ, Hoopes, J. Transitional cell carcinoma of the bladder associated with Cyclophosphamide administration. J Am Anim Hosp Assoc. 1983;19:965.
24. Reif, JS, Bruns, C, Lower, KS. Cancer of the nasal cavity and paranasal sinuses and exposure to environmental tobacco smoke in pet dogs. Am J Epidemiol. 1998;147:488.
25. Reif, JS, Cohen, D. The environmental distribution of canine respiratory tract neoplasms. Arch Environ Health. 1971;22:136.
26. Hayes, HM, Hoover, R, Tarone, RE. Bladder cancer in pet dogs: A sentinel for environmental cancer? Am J Epidemiol. 1981;114:229.
27. van der Schalie, WH, Gardner, HS, Jr., Bantle, JA, et al. Animals as sentinels of human health hazards of environmental chemicals. Environ Health Perspect. 1999;107:309.
28. Bukowski, JA, Wartenberg, D, Goldschmidt, M. Environmental causes for sinonasal cancers in pet dogs, and their usefulness as sentinels of indoor cancer risk. J Toxicol Environ Health A. 1998;54(7):579.
29. Buckowski, JA, Wartenberg, D. An alternative approach for investigating the carcinogenicity of indoor air pollution: Pets as sentinels of environmental cancer risk. Environ Health Perspect. 1997;105:1312.
30. Fu, W, Cockerell, CJ. The actinic (solar) keratosis: A 21st-century perspective. Arch Dermatol. 2003;139:66.
31. Kahn, SG, Bicker, DR, Mukhtar, H, et al. Ras p21 farnesylation in ultraviolet B radiation-induced tumors in the skin of SKH-1 hairless mice. J Invest Dermatol. 1994;102:754.
32. Lowe, NJ, Weingarten, D, Wortzman, M. Sunscreens and phototesting. Clin Derm. 1988;6:40.
33. Hargis, AM. A review of solar-induced lesions in domestic animals. Comp Cont Edu. 1981;3:287.
34. Bernays, ME, Flemming, D, Peiffer, RL, Jr. Primary corneal papilloma and squamous cell carcinoma associated with pigmentary keratitis in four dogs. J Am Vet Med Assoc. 1999;214:215.
35. Hakanson, N, Shively, JN, Reed, RE. Intraocular spindle cell sarcoma following ocular trauma in a cat: Case report and literature review. J Am Anim Hosp Assoc. 1990;26:63.
36. Wertheimer, N, Leeper, E. Electrical wiring configurations and childhood cancer. Am J Epidemiol. 1979;109:273.
37. Reif, JS, Lower, KS, Ogilvie, GK. Residential exposure to magnetic fields and risk of canine lymphoma. Am J Epidemiol. 1995;141:352.
38. National Research Council Report. Possible health effects of exposure to residential electric and magnetic fields. Washington, DC: National Academy Press; 1996.
39. Thrall, DE. Orthovoltage radiotherapy of acanthomatous epulides in 39 dogs. J Am Vet Med Assoc. 1984;184:826.
40. McEntee, MC, Page, RL, Theon, A, et al. Malignant tumor formation in dogs previously irradiated for acanthomatous epulis. Vet Radiol Ultrasound. 2004;45:357.
41. Kuttesch, JF, Wexler, LH, Marcus, RB, et al. Second malignancies after Ewing’s sarcoma: radiation dose-dependency of secondary sarcomas. J Clin Oncol. 1996;14:2818.
42. Hall, EJ, Wuu, CS. Radiation-induced second cancers: the impact of 3D-CRT and IMRT. Int J Radiat Oncol Biol Phys. 2003;56:83.
43. Suit, H, Goldberg, S, Niemierko, A, et al. Secondary carcinogenesis in patients treated with radiation: A review of data on radiation-induced cancers in human, non-human primate, canine, and rodent subjects. Radiation Res. 2007;167(1):12.
44. White, RAS, Jefferies, AR, Gorman, NT, et al. Sarcoma development following irradiation of acanthomatous epulis in two dogs. Vet Rec. 1986;118:668.
45. McChesney, SL, Withrow, SJ, Gillette, EL, et al. Radiotherapy in soft tissue sarcomas in dogs. J Am Vet Med Assoc. 1989;194:60.
46. Gillette, SM, Gillette, EL, Powers, BE, et al. Radiation-induced osteosarcoma in dogs after external beam or intraoperative radiation therapy. Cancer Res. 1990;50:54.
47. Theon, AP, Rodriquez, C, Griffey, S, et al. Analysis of prognostic factors and patterns of failure in dogs with periodontal tumors treated with megavoltage irradiation. J Am Vet Med Assoc. 1997;210:785.
48. Loeffler, JS, Niemierko, A, Chapman, P. Second tumors after radiosurgery: Tip of the iceberg or a bump in the road? Neurosurgery. 2003;52:1436.
49. Lewis, CG, Sunderman, FW, Jr. Metal carcinogenesis in total joint arthroplasty: animal models. Clin Orthop. 1996;329S:S264.
50. Li, XQ, Hom, DL, Black, J, et al. Relationship between metallic implants and cancer: A case control study in a canine population. Vet Comp Orthop Traumatol. 1993;6:70.
51. Rowland, PH, Moise, NS, Severson, D. Myxoma at the site of a subcutaneous pacemaker in a dog. J Am Anim Hosp Assoc. 1991;27:649.
52. Pardo, AD, Adams, WH, McCracken, D, et al. Primary jejunal osteosarcoma associated with a surgical sponge. J Am Vet Med Assoc. 1990;196:935.
53. Orenstein, MR, Schenker, MB. Environmental asbestos exposure and mesothelioma. Curr Opin Pulm Med. 2000;6:371.
54. Glickman, LT, Domanski, LM, Maguire, TG, et al. Mesothelioma in pet dogs associated with exposure of their owners to asbestos. Environ Res. 1983;32:305.
55. Harbison, ML, Godleski, JJ. Malignant mesothelioma in urban dogs. Vet Pathol. 1983;20:531.
56. Machida, N, Tanaka, R, Takemura, N, et al. Development of pericardial mesothelioma in golden retrievers with a long-term history of idiopathic haemorrhagic pericardial effusion. J Comp Pathol. 2004;131:166.
57. Dorn, CA, Taylor, DON, Schneider, R. Survey of animal neoplasms in Alameda and Contra Costa Counties, California II. Cancer morbidity in dogs and cats from Alameda County. J Natl Cancer Inst. 1968;40:307.
58. Moulton, JE. Tumors of the mammary gland. In Moulton JE, ed.: Tumours in domestic animals, ed 3, Berkeley: University of California Press, 1990.
59. Schneider, R, Dorn, CR, Taylor, DON. Factors influencing canine mammary tumor development and postsurgical survival. J Natl Cancer Inst. 1969;43:1249.
60. Brodey, RS, Goldschmidt, MH, Roszel, JR. Canine mammary neoplasms. J Am Anim Hosp Assoc. 1983;19:61.
61. Perez Alenza, MD, Pena, L, del Castillo, N, et al. Factors influencing the incidence and prognosis of canine mammary tumours. J Small Anim Pract. 2000;41:287.
62. MacEwen, EG, Patnaik, AK, Harvey, HJ, et al. Estrogen receptors in canine mammary tumors. Cancer Res. 1982;42:2255.
63. Mialot, JP, Andre, F, Martin, PM, et al. Etude de receptors des hormones steroids dans les tumeurs mammaries de la chienne II: correlations avec quelques caracteristiques cliniques. Recueil Medicine Veterinaire. 1982;158:513.
64. Monson, KR, Malbica, JO, Hubben, K. Determination of estrogen receptors in canine mammary tumors. Am J Vet Res. 1987;38:1937.
65. Donnay, I, Rauis, J, Devleeschower, N, et al. Comparison of estrogen and progesterone receptor expression in normal and mammary tissues from dogs. Am J Vet Res. 1995;56:1188.
66. Elling, H, Ungemach, FR. Simultaneous occurrence for receptors of estradiol, progesterone and dihydrotestosterone in canine mammary tumors. J Cancer Res Clin Oncol. 1983;105:231.
67. Sartan, EA, Barnes, S, Kwapien, R, et al. Estrogen and progesterone receptor status of mammary carcinomas and correlations with clinical outcome in the dog. Am J Vet Res. 1992;53:2196.
68. Rutteman, GR, Misdorp, W, Blankenstein, NMA, et al. Oestrogen and progestin receptors in mammary tissue of the female dog: different receptor profile in nonmalignant and malignant states. Breast Cancer. 1988;58:594.
69. Rutteman, GR. Hormones and mammary tumour disease in the female dog: An update. In Vivo. 1990;4:33.
70. Stovring, M, Moe, L, Glattre, E. A population-based case-control study of canine mammary tumors and clinical use of medroxyprogesterone acetate. Acta Pathologica Microbiologica Immunologica Scandinavica. 1997;105:590.
71. Zanninovic, P, Simcic, V. Epidemiology of mammary tumors in dogs. Eur J Comp Anim Pract. 1994;IV:67.
72. Lantinga-van Leeuwen, IS, van Garderen, E, Rutteman, GR, et al. Cloning and cellular localization of the canine progesterone receptor: Co-localization with growth hormone in the mammary gland. J Steroid Biochem Molec Biol. 2000;75:219.
73. Mol, JA, Lantinga-van Leeuwen, I, van Garderen, E, et al. Progestin-induced mammary growth hormone (GH) production. Adv Exp Med Biol. 2000;480:71.
74. Rijnberk, A, Kooistra, HS, Mol, JA. Endocrine diseases in dogs and cats: Similarities and differences with endocrine diseases in humans. Growth Horm IGF Res. 2003;13(suppl A):S158.
75. Hayes, HM, Milne, KL, Mandell, CP. Epidemiological features of feline mammary carcinoma. Vet Rec. 1981;108:476.
76. Misdorp, W, Romijin, A, Hart, AAM. Feline mammary tumors: A case-control study of hormonal factors. Anticancer Res. 1991;11:1793.
77. Overley, B, Shofer, FS, Goldschmidt, MH, et al. Association between ovarihysterectomy and feline mammary carcinoma. J Vet Intern Med. 2005;19:560.
78. National Cancer Institute. Surveillance, epidemiology and end results program public use data (1973-2000). Bethesda: The Institute; 2003.
79. Villamil, JA, Henry, CJ, Hahn, AW, et al. Hormonal and sex impact on the epidemiology of canine lymphoma. J Cancer Epidemiol. 2009:591753. [Epub 2010 Mar 14].
80. Dow, SW, Olson, PN, Rosychuk, RAW, et al. Perianal adenomas and hypertestosteronemia in a spayed bitch with pituitary-dependent hyperadrenocorticism. J Am Vet Med Assoc. 1988;192:1439.
81. Wilson, GP, Hayes, HM. Castration for treatment of perianal gland neoplasms in the dog. J Am Vet Med Assoc. 1979;174:1301.
82. Waters, DJ, Sakr, WA, Hayden, DW, et al. Workgroup 4: Spontaneous prostate carcinoma in dogs and nonhuman primates. Prostate. 1998;36:64.
83. Madewell, BR, Gandour-Edwards, R, White, RWD, et al. Canine prostatic intraepithelial neoplasia: Is the comparative model relevant? Prostate. 2004;58:314.
84. Teske, E, Naan, EC, von Dijk, EM, et al. Canine prostate carcinoma: epidemiological evidence of an increased risk in castrated dogs. Mol Cell Endocrinol. 2002;197:251.
85. Sorenmo, KU, Goldschmidt, M, Shofer, F, et al. Immunohistochemical characteristics of canine prostatic carcinoma and correlation with castration status and castration time. Vet Comp Oncol. 2003;1:48.
86. Bryan, JN, Keeler, MR, Henry, CJ, et al. A population study of neutering status as a risk factor for canine prostate cancer. Prostate. 2007;67:1174.
Section C
Cancer-Causing Viruses
Dennis W. Macy and Carolyn J. Henry
Both DNA- and RNA-containing viruses are known to cause cancer. An initial step in malignant transformation of normal cells by most tumor viruses is integration of all or part of the viral DNA (or DNA copy of retroviral RNA) into the host cell genome. For some viruses, specific viral genes (oncogenes) have been identified that lead to malignant transformation when expressed in normal cells. Other viruses, through the process of integration, activate the expression of normal cellular genes, leading to overexpression or inactivation of genes, resulting in cellular transformation or uncontrolled growth.1
Tumor-Causing Viruses of Dogs
Papillomaviruses are oncogenic, contagious, and infectious and have been described in several animal species.2 Papillomaviruses are considered species specific, and isolates of humans, cattle, and dogs lack serologic cross-reactivity.2 However, cross-infection with other species can occur. For example, the coyote can be infected with dog isolates, and bovine papillomaviruses type 1 and type 2 have been reported to infect horses.3 In addition, bovine papillomaviruses have been isolated from tumors in cats, indicating a unique cross-species infection in a dead-end host.4
Four or possibly more papillomaviruses infect dogs and are responsible for a wide spectrum of clinical syndromes. Papillomaviruses of the family Papovaviridae produce benign, mucocutaneous, and cutaneous canine papillomas and in rare cases transform into SCCs.5,6
The canine papillomaviruses are naked DNA viruses; they are larger than the canine parvoviruses but similar in structure. Electron microscopy has been used to detect the virus in infected tissues. Like other papillomaviruses, canine papillomaviruses are resistant, acid stable, and relatively thermostable.7 Only a limited sequence homology exists between the DNA sequences of papillomaviruses of different species, but substantial sequence homology exists between isolates from any given species.2
Pathogenesis
Papillomas develop subsequent to introduction of papillomavirus through breaks in the epithelium. Different viruses derived from the same species are believed to correlate with the type of clinical disease produced by the virus (i.e., oral versus cutaneous isolates). However, this feature of papillomaviruses has yet to be proven for the dog, and experimentally, ocular isolates have produced oral papillomas.8-12 The presence and location of mature, complete viruses on the surface of papillomas are believed to aid its transmission to adjacent epithelial tissues.2 In contrast to other oncogenic or transforming DNA viruses, papillomaviruses rarely integrate into the cellular genome and remain episomal.2
Infection of epithelial cells results in a marked increase in cellular mitosis and hyperplasia of cells with a strand of spongiosum, with subsequent degeneration and hyperkeratinization.13 Clinical evidence of hyperplasia and hyperkeratinization usually manifests 4 to 6 weeks after infection.13 Canine papillomas generally persist for 4 to 6 months in the mouth and 6 months to 1 year on the skin before undergoing spontaneous regression, and multiple warts generally regress simultaneously.13 Although antibodies are produced against the papillomavirus, antibody levels do not appear to correlate with either growth or regression of the papilloma; the mechanism of induction or regression remains unknown, although it is thought in most cases to be associated with cellular immunity.14 The development of multiple papillomavirus-associated epidermal hematomas and SCC in situ in a dog after treatment with prednisone and cyclosporine has been reported.15
Clinical Features
Papillomas may be referred to as warts, verruca vulgaris, squamous cell papillomas, or cutaneous papillomatosis. Papillomas caused by the papillomavirus usually are multiple and frequently infect young dogs. In the dog, multiple papillomatosis most frequently is seen in the oral cavity, involving the labial margins, tongue, pharyngeal mucosa, hard palate, and epiglottis (Figure 1-9).14 Four to 8 weeks after infection, small, pale, smooth, elevated lesions appear; these quickly develop a cauliflower-like appearance, with fine, white frons extending from the surface of the lesions. Multiple sites of susceptible tissue in the oral cavity appear to be affected early in the course of the disease; as many as 50 to 100 tumors may be present at the time of diagnosis.14 The primary complaints of owners of infected dogs are halitosis, ptyalism, hemorrhage, and difficulty eating. Most oral cavity papillomas start regressing after 4 to 8 weeks. However, some oral lesions may show incomplete regression, and some have been known to persist up to 24 months.14
Ocular papillomas, which are less numerous than the oral type, appear on the conjunctiva, cornea, and eyelid margins (Figure 1-10). Ocular papillomas also occur less often than oral lesions.14 Experimentally, viruses isolated from ocular lesions can produce oral papillomatosis, although whether this occurs in nature is unknown.8 Ocular papillomatosis most frequently occurs in dogs 6 months to 4 years of age, but it occasionally is reported in older dogs.
Multiple cutaneous papillomatosis is thought to be of viral origin (Figure 1-11); however, evidence suggests that it is not the same strain of papillomavirus that produces oral papillomatosis in the dog.8 Multiple skin papillomatosis affects a much broader age range of canine patients, and regression of the lesion is prolonged, sometimes taking years.14 A rare form of cutaneous papillomatosis, in which the lesion appears in the interdigital areas of the pad, has been described in greyhounds, particularly young ones (12 to 18 months of age).12 Canine pigmented plaques have been associated with papillomaviruses in miniature schnauzers and pugs.16 The lesions may or may not regress and are considered premalignant.
Although papillomatosis should be considered a benign disease, in rare cases, oral and corneal papillomas have transformed into SCCs.5,6
Treatment
Most clinicians elect not to treat papillomatosis because of the lack of proven efficacy of recommended treatments and the expected spontaneous regression of these tumors. However, if the number of papillomas increases or if the animal has significant difficulty eating, owners often request treatment. Surgical excision, cryosurgery, laser surgery, or electrosurgery for just a few lesions has resulted in regression of the remaining papillomas, presumably through immunologic mechanisms.14,17,18 The exact mechanism by which regression of papillomas occurs is unknown. Serum from dogs in which papillomas have undergone spontaneous regression not only fails to produce tumor regression when administered to infected animals, it actually enhances existing tumor growth. However, administration of immune lymphocytes from dogs in which tumors have regressed has been shown to enhance regression.7 This effect may be a result of induction of blocking antigen-antibody factors, which may impede cytotoxic lymphocyte action on target cells. CD4 lymphocytes activate macrophages and have been shown to inhibit the virus in dogs.7 Interferon also has been tried, with some success (1 to 3 million IU/m2 given subcutaneously three times a week [Monday-Wednesday-Friday]), and chemotherapy of resistant lesions has produced variable results.19 Corticosteroids should be avoided because they are thought to contribute to the dissemination of papillomas.20
Most systemic chemotherapeutic agents (e.g., bleomycin, vincristine, cyclophosphamide, and doxorubicin) have failed to cause regression of papillomas. However, etretinate (1 mg/kg given orally daily) has been effective in some dogs with persistent papillomas.21,22 Topical or intralesional compounds containing 5-fluorouracil (5-FU) have been used in both humans and dogs to treat papillomas. In the past, autogenous wart vaccines have been recommended but have proved of little value in the treatment of resistant papillomatosis of the dog.14 In at least one study, papillomavirus vaccines have been shown to prevent the development of oral papillomas in the dog; however, cutaneous neoplasms at the injection sites have been attributed to administration of the vaccines.11,23,24
Tumor-Causing Viruses of the Cat
Feline viral papillomatosis is a rarely recognized condition caused by a papillomavirus specific to the cat. The feline isolates Felis domesticus papillomavirus type 1 (FdPV-1), FdPV-JM, and FdPV-MY are genomically very similar to canine isolates but are considered species specific.25 Papillomavirus-associated lesions have been reported in six species of felids besides the domestic cat: the mountain lion, Florida panther, bobcat, Asian lion, snow leopard, and clouded leopard.26,27 Unlike in the domestic cat, in which the lesions commonly affect areas of haired skin, papillomas in exotic species most often are found in the oral cavity, similar to those in the dog.26,27 Despite the clinical similarities, genetic and antigenic studies indicate that each species of felid is infected by a unique papillomavirus.
Pathogenesis
In cats, as in other species, papillomas are believed to develop after the virus is introduced through lesions or abrasions in the skin. Unlike in the dog, most feline case reports involve older cats (6 to 13 years of age), although papillomavirus lesions have been reported in kittens 6 to 7 months old.28,29 As in other species, impaired T-cell function likely plays a significant role in lesion formation. Papillomas in cats that are receiving immunosuppressive therapy or are infected with the feline immunodeficiency virus (FIV) support this hypothesis.
Although papillomas most frequently are benign lesions, recent studies have associated the papillomavirus with SCCs and other malignant neoplasms in cats30; specifically, papillomavirus has been isolated from 30 of 63 squamous cell carcinoma in situ skin lesions. Through the use of polymerase chain reaction (PCR) techniques, papillomaviruses have been found in 17 of 19 and 9 of 12 fibropapillomas in cats.31,32 Although a cause-and-effect relationship has yet to be proved for carcinoma in situ, Bowen’s disease, fibropapillomas, and papillomaviruses, the evidence is compelling.30-32 Over 100 papillomaviruses occur in humans and are believed responsible for cervical cancer, between 25% and 60% of SCCs of the oral cavity, and rarely even some that occur on the skin surface.33
Bovine papillomaviruses may also play a role in the pathogenesis of feline cutaneous fibropapillomas. In a study of 20 cats with fibropapillomas, more than half were known to have exposure to cattle, and all were within an area with dairy farms.31 In one isolate, the nucleotide sequence was similar to that of the bovine papillomavirus. Injection of that isolate back into bovine skin resulted in asymptomatic infection. Also, although papillomaviruses generally are species specific, an association between bovine papillomavirus types 1 and 2 has been suggested as causes of equine sarcoids.34
Clinical Features
Lesions in the cat differ from those in the dog because they are more like plaques than warts. The plaques are several millimeters in diameter, may be white or pigmented, and are scaly or greasy. Also, lesions in the cat usually affect haired skin rather than the mucous membrane locations common for oral and ocular papillomas of the dog and wild felids.26,27
Diagnosis
Definitive diagnosis depends on histopathologic, immunohistochemical, or electron microscope (EM) examination of excised lesions. Histologic features include proliferation of all cell layers with little or no inflammation. Typically the epidermal hyperplasia is accompanied by acanthosis, hypergranulosis, hyperkeratosis, and ballooning degeneration of cells of the stratum spinosum and stratum granulosum. Amphophilic cytoplasmic inclusion structures may be present in cells of the upper stratum granulosum. EM findings in the lesions include intranuclear particles within keratinized cells in the superficial epithelial strata of the plaques. Immunohistology can be performed on sections using band-reactive, genus-specific antiserum. Interestingly, the histologic features of the feline fibropapilloma are very similar to those of equine sarcoids, with characteristic fibroblastic proliferation, hyperplasia of epidermis, and rete ridges.31 PCR also has demonstrated papillomavirus DNA in the lesions.
Treatment
Surgical excision is generally used; however, parenteral alpha interferon has been suggested as an alternative. Medications containing 5-FU that are used in humans and dogs should NOT be used in cats. Imiquimod 5% cream (Aldara) is a novel immune-response modifier (IRM) that has been used in humans with Bowen’s disease and has recently been used in cats with the same disease. Although 41% of cats treated with imiquimod developed some level of toxicity, most adverse events were manageable.35
Retroviruses
Retroviral infections are considered the number one infectious cause of morbidity and mortality in the domestic cat. Before the vaccine was developed and routine testing and control measures became widespread, the feline leukemia virus (FeLV) was associated with one-third of deaths in cats.36,37 The cat is believed to be affected by the largest number of retroviruses of any companion animal, and these viruses produce a wide spectrum of diseases, including cancer.38-40
The cat has both endogenous and exogenous retroviruses. The endogenous retroviruses generally are considered nonpathogenic, are present in the host DNA, and are passed from generation to generation genetically, as are other chromosomal genes. The exogenous retroviruses include both pathogenic and nonpathogenic viruses and are passed horizontally and vertically between cats. Pathogenic exogenous retroviruses include FeLV and FIV.41 The exogenous RNA sequences of FeLV play the most important role in tumorigenesis in the cat.40 Another pathogenic retrovirus, the feline sarcoma virus (FeSV), arises from the combination of exogenous FeLV and proto-oncogenes in the cat’s genome.42 Feline syncytium-forming virus (FeSFV), also called the feline foamy virus, is a nonpathogenic exogenous retrovirus.36
FeLV is believed to have been contracted from the ancestral rat approximately 10 million years ago.43 The ancestral source of other retroviruses is unknown. The three pathogenic retroviruses of clinical importance are FeLV, FIV, and, in rare cases, FeSV.
Feline Leukemia Virus
The retrovirus FeLV belongs to the subfamily oncornavirus, or tumor-producing RNA viruses. Like other retroviruses, it has a single strand of RNA and an enzyme, reverse transcriptase (RT), that synthesizes DNA from the virus RNA template. Nondomestic felids, including the cheetah and bobcat, can be infected by FeLV; however, it is not considered enzootic in wild felids except for European wild cats in France and Scotland.44,45
The basic FeLV proteins include the envelope proteins and the core proteins, several of which are important clinically. Two envelope proteins, the P15E and the GP70 glycoproteins, have particular clinical significance.46-48 P15E is thought to be one of the mediators of immunosuppression in FeLV-infected cats.49 The glycoprotein of the envelope GP70 may contain three subgroup antigens, A, B, and C.50,51 An individual cat may have combinations of viruses with these subgroup viral antigens. Considerable antigenic variation exists within subgroups, which can affect the biologic properties of the individual isolates or strains of FeLV.40,50 These subgroup antigens bind the virion to receptors on the surface of cells. The specific characters of these proteins also predict the pathogenicity, host range, infectivity, and other biologic properties of the virus.40,50 The antibodies produced against envelope proteins can be neutralizing and thus can prevent infection. Envelope proteins are very important components of FeLV vaccines.
Core proteins (capsids) include P15C, P12, P10, and P27. P27 is quite soluble and can be found in large amounts in the cytoplasm of cells and bodily fluids, such as tears and serum.46-48 P27 is the antigen that is detected in immunofluorescent assay (IFA) tests and enzyme-linked immunosorbent assays (ELISAs), which are commonly used in the diagnosis of FeLV infection.51
Transmission
FeLV is an enveloped virus and is considered very fragile. Desiccation rapidly reduces the amount of viable virus in saliva, and inactivation occurs in 1 to 2 hours. In exudates or blood, the virus may be viable for only 48 hours (at 37° C) or 1 to 2 weeks (at 22° C).52 Like most retroviruses, FeLV is rapidly inactivated by heating and most disinfectants.47 Given these characteristics, environmental contamination (e.g., examination tables, cages, and waiting rooms) is unlikely to be a potential source of FeLV infection.40 Although saliva may contain up to 100,000 virus particles per milliliter, prolonged, intimate contact with infected cats usually is required for transmission. The factors most frequently incriminated in the transmission of FeLV are licking, biting, grooming, and sharing of litter pans, food bowls, and water dishes. Intimate contact is enhanced in catteries and multiple-cat households, where infection rates may be very high.49
Although cats may be infected with FeLV subgroups A, B, or C or other recombinants, only subgroup A has been found in cell-free fluids and is thought to be associated with natural transmission of FeLV. Subgroups B and C and other recombinants are more cell associated and are not thought to be transmitted in nature.53-56
Before vaccines and routine testing became available, the overall prevalence of FeLV infection in the United States was estimated at 1% to 3% of the population.38,39 The prevalence of FeLV infection was less than 1% in single-cat households and as high as 30% in multiple-cat households.57 The incidence of FeLV-positive test results in sick cats in the United States was approximately 11.5%.58 Several studies have reported a decline in the prevalence of FeLV by as much as 50% over the past 20 years.37,59,60
The FeLV subgroups are characterized by their cross-interference with homologous but not heterologous subgroups of FeLV and by their host range and other factors. All naturally infected FeLV cats have subgroup A, 50% of infected cats have a combination of subgroups A and B, and 1% of infected cats in nature have a mixture of subgroup C either as AC or ABC.38,58,61
The relevance of subgroups in strains is essential to an understanding of the biodiversity of the clinical disease caused by FeLV infection. Although subgroups A, B, and C maintain 85% genomic homology, cats infected with various combinations of these subgroups may manifest vastly different diseases.
Subgroup A has a variety of strains that range from nonpathogenic to very pathogenic.62 Although most strains of subgroup A have limited pathogenicity, their pathogenicity increases dramatically if they are present with other subgroups.
Subgroup B is created when subgroup A recombines with endogenous FeLV envelopes at sequences already in the feline genome.63-65 Each recombination is unique, resulting in many strains of FeLV-B. The combination of subgroups A and B is more contagious and pathogenic than subgroup A alone.58,61,62 Cats infected with subgroups A and B often develop thymic lymphoma and myeloproliferative disease.63
Subgroup C arises from the mutation of subgroup A.66 Cats may be infected with a combination of C and other subgroups, although these combinations are uncommon and are found in only about 1% of naturally infected cats. FeLV-C is antigenically similar to the associated membrane antigen (feline oncornavirus-associated cell membrane antigen [FOCMA]), and cats carrying FeLV-C have developed severe erythroid hypoplasia and anemia and usually die within 1 to 2 months.53 Further complicating the biodiversity of subgroups and strains is the fact that subgroups A and B can recombine with proto-oncogenes such as MYC or TCR, producing FeLV-MYC or FeLV-TCR.36 Both of these recombinants are considered more potent tumor producers than their nonrecombinant FeLV parent. Another subgroup, T, is highly cytolytic for T lymphocytes and causes severe immunosuppression.54-56
The Rickard strain of FeLV (FeLV-R), although similar to MYC-containing recombinant strains in its ability to produce mediastinal lymphoma rapidly, does not recombine with the MYC gene.36,67 Instead, it obtains some of the biologic effects by integrating adjacent to the C-MYC gene, causing its overexpression.36
Feline Oncornavirus-Associated Cell Membrane Antigen
FOCMA is a protein found on the surface of FeLV and FeLV-induced neoplasms but not on nonneoplastic feline cells.68,69 FOCMA is detected serologically when cells expressing it react to immunoglobulins produced in cats that have regressed FeSV-induced fibrosarcoma or FeLV infection. The presence of FOCMA antibody is determined by the ability of the serum to react with FL74 cells, a transformed infected feline lymphocyte line.70 Antibodies to FOCMA protect against neoplastic and myeloproliferative disease. Some FeLV vaccines contain FOCMA and elicit an anti-FOCMA response.71 The relative importance of this in preventing disease in vaccinates is unknown.
Neoplastic Diseases Caused by Feline Leukemia Virus
We have much to learn about the genetic basis for the vast diversity of tumor types produced by FeLV and its recombinants. We now know that FeLV, through one or another of its recombinants, may cause virtually any hematopoietic neoplasm in the cat. The only hematopoietic neoplasms not yet associated with FeLV in nature are mast cell leukemia, eosinophilic leukemia, plasma cell tumors, and polycythemia vera.36
Although FeLV infection is considered the most significant infectious cause of morbidity and mortality in cats, only 20% of cats persistently infected with FeLV develop lymphoid cancer.72,73 The cat has the highest incidence of hematopoietic neoplasms of domestic animals, and the prevalence of lymphoma ranges from 44 to 200 cases per 100,000 cats, six times the rate of this disease in humans.36 Twenty years ago, 70% of lymphomas in cats were believed to be caused by FeLV.
Some cancers are more commonly associated with FeLV infection than others. Large, granular lymphoma and globular leukocyte tumors usually test negative for FeLV,74,75 whereas 70% to 90% of cats with nonlymphoid hematopoietic neoplasia (myeloproliferative disease) test positive for FeLV.36 The percentage of lymphomas that test positive for FeLV also varies, depending on the anatomic location of the tumor.76-78 Cats with spinal, mediastinal, ocular, and renal lymphoma frequently tested positive for FeLV prior to routine vaccination (more than 70%).79 Extranodal sites such as lymphomas of the nasal cavity and the alimentary form of lymphoma frequently test negative for FeLV infection.36 Over the past 20 years, the multicentric FeLV-positive form has declined in young cats, and the FeLV-negative alimentary form in older cats has increased.80-82 Although the alimentary form most often is FeLV negative, as assessed by IFA and ELISA testing, some of these lesions have been shown by PCR to be FeLV positive, which suggests that the disease may be related to previous FeLV exposure.
Although not all lymphomas are caused by FeLV, the relative risk of developing lymphoma is 62 times higher in FeLV-positive cats, and cats that are FeLV negative but that have had previous exposure to FeLV have a fortyfold increase in the risk of developing lymphoma.83 Most spontaneous lymphomas of cats that test positive for FeLV arise from T cells, whereas FeLV-negative lymphoma frequently is of alimentary or B-cell origin.84,85 The time from infection to tumor development varies and may depend on the age at which the cat is infected or on other factors, such as strain, anatomic location, and viral subgroup.36 The range from the time of experimental infection to tumor production is 1 to 23 months (mean, 5.3 months).86,87 The younger the cat when infected with FeLV, the shorter the time to the development of neoplastic disease. Some cats infected with FeLV die of immunosuppressive disease before tumors have a chance to develop.
Treatment of Feline Leukemia Virus Infections
Although no effective treatment exists to eliminate established FeLV infection in cats, a variety of antiviral and biologic response modifiers have been used to manage retroviral infections in cats and humans. The mainstay of therapy for cats infected with FeLV or other retroviruses is supportive care.88-90 Maintaining hydration and nutritional status not only prolongs life but also enhances the patient’s quality of life. The cat should be kept in a humid environment to reduce the chance of water loss. Appetite stimulants and placement of gastrostomy tubes may facilitate nutritional therapy. The cat’s requirement for B vitamins is eight times that of the dog, and dietary concentrations must be maintained to maintain appetite. Semimoist cat foods often contain propylene glycol, which can shorten red blood cell survival. These foods, although often quite palatable, should not be used for the nutritional management of cats infected with FeLV.89 Many cats with FeLV are anemic, but administering erythropoietin is not helpful because endogenous erythropoietin levels usually are 20 times normal.91
Biologic-Response Modifiers
A variety of biologic-response modifiers (BRMs) has been used in cats infected with FeLV,92-100 but none has shown benefit in controlled trials. A few of the most popular are discussed here.
Interferons have been studied extensively for the management of FeLV infection, but the results have been mixed. In one study, oral and parenteral doses of either human recombinant interferon-α or bovine interferon failed to alter the viremia or result in clinical improvement. However, some uncontrolled studies have shown improvement in the clinical status of cats treated with oral interferon-α.99 Controlled trials are needed to establish the true efficacy of these products. Orally administered interferon probably is inactivated by gastric acid in the stomach. Parenterally administered interferons from other species (i.e., bovine and human) are likely to have temporary activity because of the production of neutralizing antibodies.
Carrisyn (Acemannan) is a BRM designed to enhance macrophage phagocytosis and cell killing. Viremic cats treated with carrisyn have been reported to improve clinically; however, the studies reporting these results have not been well controlled, and the observed benefit may be due to the natural waxing and waning clinical course commonly observed in FeLV-positive cats.100
Lymphocyte T-cell immunomodulator (LTCI) has recently become commercially available for the treatment of cats infected with FeLV and FIV. The true efficacy of this product, if any, awaits the results of controlled trials in cats.101
The apparent positive effect of many of the BRMs, which, in fact, may be due to the anabolic effect observed with some of these cytokines, is thought to be based on endorphin release rather than a direct effect on the viral infection.
Reverse Transcriptase Inhibitors
Drugs that inhibit RT and retrovirus integration into the host cell have been evaluated for their potential use in the treatment of FeLV-positive cats. The drugs evaluated include suramin (a polyionic dye used to treat filariasis in humans), nucleoside analogs (AZT, DDC, DDA, and PNEA), glucose homopolymers, dextran sulfate, phosphonate, and others.102-105 More detailed descriptions of these therapies are provided elsewhere.102,105 In general, most of these agents have shown efficacy in vitro against FeLV, the human immunodeficiency virus (HIV), and in some cases, FIV. Most of these drugs result in some reduction in viremia in vivo, but none are capable of reversing established viremia, although some may prevent viremia if administered prophylactically. Most of these drugs cause significant toxicities at the dosages needed to produce antiviral effects and therefore have not gained popularity in clinical practice. AZT (zidovudine) is the most widely studied RT inhibitor.106 AZT inhibits FeLV reverse transcriptase when administered at a dosage of 10 to 20 mg/kg in daily divided doses. AZT prevents viremia if given within 72 hours of exposure to FeLV. The antiviral effects of AZT appear to be synergistic with interferon.107,108 Reversal of established experimental FeLV viremia through adoption transfer of lectin/IL-2–activated lymphocytes, interferon-α, and AZT has been reported.
Prevention and Control
The most effective means of preventing FeLV infection is to eliminate contact with viremic cats. The test and removal program is the most effective means of controlling FeLV in multiple-cat households.109 The program consists of closing the household or cattery to new cats, testing the remaining cats every 3 months, and removing all animals that test positive. When all cats test negative for two consecutive sessions, the facility is determined to be FeLV free. New cats may enter the household or facility only after a 3-month quarantine and two negative FeLV tests. The test and removal system has been shown to reduce the incidence of FeLV in a variety of settings and geographic locations.109
Prevention by Vaccination
Vaccinations help control or eliminate many infectious diseases in veterinary medicine. The first commercial FeLV vaccine was introduced in 1985. Since then, seven FeLV vaccine products from six companies have been licensed for sale to veterinarians in the United States. Despite the fact that FeLV vaccines have been available for a decade, a survey of US veterinary teaching hospitals in 1991 found that only two of the 22 teaching hospitals considered FeLV vaccination to be part of their routine feline preventive medicine program.110 The principal concern has been the perceived lack of efficacy of the FeLV vaccines. Some studies of available vaccines have reported efficacies ranging from 0 to 100%.111 In addition to efficacy questions, it has been established that soft tissue sarcomas may develop after FeLV and rabies vaccination.112
FeLV vaccination issues are discussed elsewhere.71 However, several comments regarding FeLV vaccines should help practitioners decide whether to use FeLV vaccines in their practice. FeLV vaccines may contain two or three subgroup antigens. Because only subgroup A is transmitted contagiously between cats, vaccines need only to contain subgroup A. Their primary means of protecting against tumor development is preventing persistent viremia. If a vaccine protects against persistent FeLV infection, it need not contain FOCMA. The value of FOCMA in FeLV vaccines has yet to be proved. Vaccines should protect against a variety of strains of subgroup A, and none of the available vaccines contain more than one strain of subgroup A. Differences in published comparative studies of vaccine efficacy probably are related to differences in vaccine strains and to the challenge strains used. Vaccines that contain adjuvants enhance immunity but at the expense of producing local inflammatory reactions at the injection site.113 These local reactions may lead to the development of soft tissue sarcomas. However, the development of soft tissue sarcomas after vaccination, either with rabies or FeLV vaccines, is thought to occur in only 1 in 1000 to 1 in 10,000 vaccinates.112 Nonadjuvanted FeLV vaccines have shown little or no inflammatory reaction 21 days after administration.113 A canarypox-vectored FeLV nonadjuvanted vaccine is available that stimulates both cellular and humoral immunity without significant injection site inflammation. Clinical discretion should be used when recommending FeLV vaccines.
The American Association of Feline Practitioners (AAFP) does not consider FeLV vaccine a core vaccine, and only cats at significant risk should be vaccinated. Cats under 12 weeks of age have an 85% chance of becoming persistently infected, whereas cats over 6 months of age have only a 15% chance of becoming persistently infected after challenge. Age-acquired immunity of adult cats is associated with improved macrophage function and reduced viral receptors on target tissues. Annual vaccination of adult cats also appears to be a questionable practice, given age-acquired immunity and the risk of vaccine-associated sarcomas.
Feline Sarcoma Virus
FeSVs are true hybrids that result from the rare recombination of FeLV DNA provirus with cat proto-oncogenes. Cats have at least 30 proto-oncogenes.36,114,115 Proto-oncogenes have many biologic functions; when they are altered and activated inappropriately, they are called oncogenes, which can play a key role in the development of cancerous phenotypes. Proto-oncogenes can be activated by mutations that produce chromosomal translocations such as those that may be associated with inflammation and vaccine-associated sarcomas, or by incorporation into a retrovirus, such as FeLV.114-116 When FeLV-derived DNA inserts near a proto-oncogene and takes up the proto-oncogene into the FeLV provirus, formation of FeSV results. In the process, part of the FeLV GAG gene, most of the FeLV envelope gene, and all of the pole genes are lost.115 The loss of these vital components makes FeSV dependent on FeLV as a helper virus for replication. Cats that have FeSV always test FeLV positive. Because several different recombinations may recur with several different proto-oncogenes, each recombination is a unique event, and each isolate is distinct.116 Despite this phenotypic heterogeneity, the recombinations transform fibroblasts, and all produce fibrosarcomas.
Natural transmission of FeSV between cats has not been described, and as with other FeLV recombinants (e.g., FeLV-B), transmission of the recombinant product is not thought to occur in nature. Some cats are capable of rejecting transformed cells and producing FOCMA antibody.90,91 FOCMA is important in the experimental response of cats to FeSV because it has been associated with tumor regression and failure to develop tumors.117,118 Cats that fail to develop antibodies against FOCMA die quickly of fast-growing sarcomas.119
Clinical Features of Feline Sarcoma Virus and Induced Fibrosarcomas
Only 2% of fibrosarcomas of cats are virally induced.42 In contrast to the solitary, slow-growing, nonvirally induced sarcomas seen in older cats, FeSV-induced tumors are multicentric and are found most frequently in young cats.120 They are characterized by rapid growth, including doubling times as short as 12 to 72 hours.36 This rapid growth often is accompanied by superficial ulceration. Lesions frequently occur at sites of previous bite wounds.36 Metastasis to the lungs or other organs occurs with approximately 30% of virally induced fibrosarcomas in cats. Hypercalcemia was observed in association with multicentric fibrosarcomas in one cat with FeSV.36 Virally induced fibrosarcomas are always FeLV positive; this helps differentiate them from vaccine-associated sarcomas, which have growth characteristics similar to those of virally induced tumors. Cats with multicentric FeSV-induced tumors have a very poor prognosis. Doxorubicin, vincristine, vinblastine, lomustine (CCNU), and radiotherapy have been used to treat vaccine-associated sarcomas in the cat.121 Although radiotherapy often is used in combination with surgery, recurrence both within and outside the treatment fields is common.
Feline Immunodeficiency Virus
FIV, which is classified as a retrovirus in the subfamily Lentivirinae, is distinct from other retroviruses that infect cats. Like other retroviruses, FIV is an enveloped, single-stranded RNA virus in which the RNA is copied into the DNA in the infected host by RT in the virus.
The nucleotide sequence of several FIV isolates has been determined, and genetic homology falls between 36% and 97%. Despite this homology, significant differences in pathogenicity and infectivity exists between FIV strains.122,123 Although lentiviruses are known to infect wild felids, they are antigenically distinct from domestic cat isolates; they also are well adapted to their host and seldom cause clinical disease.
Transmission
FIV is present in all bodily fluids of infected cats, similar to FeLV, but at much lower concentrations. FIV is mainly cell associated and is present in relatively low concentrations in the blood, although high amounts can be found in the saliva.124,125 FIV is not thought to be very infectious and is mainly transmitted through biting during cat fights.126,127
Feline Immunodeficiency Virus–Associated Neoplasms
The prevalence of neoplasms in FIV-positive cats ranges from 1% to 62%.83,128,129 Lymphomas and myeloid tumors (myelogenous leukemia, myeloproliferative disease) and a few carcinomas and sarcomas are the neoplasms most commonly linked to FIV infection. One study found that cats infected with FIV and FeLV are 5.6 times more likely to develop lymphoma or leukemia than if they had been infected with either virus alone. Cats with combined infections had a 77.3% greater likelihood of developing lymphoma or leukemia than noninfected cats.83 Lymphoreticular neoplasms have been linked to HIV infection in humans and simian immunodeficiency virus (SIV) infection in nonhuman primates. In contrast to FeLV-associated lymphomas, FIV-associated lymphomas most often develop in extranodal sites and occur in older cats (mean age, 8.7 years).83 Myeloproliferative disease also has been observed in cats naturally and in cats experimentally infected with FIV.83,130,131
Although lentiviruses such as FIV have not been thought to be oncogenic in themselves, they are markedly immunosuppressive and affect normal immunosurveillance of cancerous cells. FIV-positive cats with lymphoma have extremely low CD4 lymphocyte counts.121 SCCs of the skin have been linked to FIV infection in two geographic areas, California and Colorado, but this association is believed to be due to a co-risk behavior (outdoor cats) rather than to any direct viral contribution to tumor development.132,133 Other reports have linked FIV infection to oral SCC, mammary carcinoma, fibrosarcoma, myeloproliferative disease, and histiocytic mast cell disease.128,134,135 The nature of these associations awaits further investigation.
Treatment
The same treatment considerations in the management of cats with FeLV can be applied to the treatment of FIV-positive cats. The most widely applied treatments have been the RT inhibitors and interferon (see the earlier discussion on the treatment of FeLV). As in the treatment of FeLV, FIV-positive cats remain positive despite these therapies. A single inactivated FIV vaccine has been licensed for use in domestic cats. However, this is an adjuvanted vaccine, and it may be associated with an increased risk of vaccine-associated sarcoma. The primary concern with the vaccine is that it generates antibodies that cross-react with the currently recommended antibody-based diagnostic test for FIV infection. PCR-based tests are not currently considered reliable for diagnosis of FIV, and antibody-based testing remains the gold standard. It is important to note that the AAFP does not recommend the use of an FIV vaccine in domestic cats.
Comparative Aspects
The association between human viruses and certain cancers has been established on the basis of epidemiologic, clinical, and molecular studies.1 Human T-cell leukemia virus (HTLV-1) has been linked to adult T-cell leukemia. The human papillomavirus (HPV) is associated with cervical cancer. The Epstein-Barr virus (EBV) is related to the development of Burkitt’s lymphoma and nasopharyngeal carcinoma, and human hepatitis B and C viruses are associated with hepatocellular carcinoma. The human herpes virus (HHV) is implicated in the development of Kaposi’s sarcoma.
References
1. Benchimol, S, Minden, MD. Viruses, oncogenes, and tumor suppressor genes. In Tannock IF, Hill RP, eds.: The basis science of oncology, ed 3, New York: McGraw-Hill, 1998.
2. Pfister, HH. Biology and biochemistry of papillomaviruses. Rev Physiol Biochem Pharmacol. 1984;99:111–181.
3. Sundberg, JP, Reszler, AA, Williams, ES, et al. An oral papillomavirus that infected one coyote and three dogs. Vet Pathol. 1991;28:87–88.
4. Munday, JS, Knight, CG. Amplification of feline sarcoid-associated papillomaviruses DNA sequences from bovine skin. Vet Dermatol. 2010;21:341.
5. Belkin, PV. Ocular lesions in canine oral papillomatosis. Vet Med Small Anim Clin. 1979;74:1520–1524.
6. Watrach, AM, Small, E, Case, MT. Canine papilloma: progression of oral papilloma to carcinoma. J Natl Cancer Inst. 1970;45:915–920.
7. Nicholls, PE, Starley, MA. The immunology of animal papillomaviruses. Vet Immunol Immunopathol. 2000;73:101–127.
8. Tokita, H, Konishi, S. Studies on canine oral papillomatosis. II. Oncogenicity of canine oral papilloma virus to various tissues of dog with special reference to eye tumor. Jpn J Vet Sci. 1975;37:109–120.
9. Hare, CL, Howard, EB. Canine conjunctiva-corneal papillomatosis. J Am Anim Hosp Assoc. 1977;13:688–690.
10. Bonney, CH, Koch, SA, Conter, AW, et al. Case report: A conjunctivocorneal papilloma with evidence of a viral etiology. J Small Anim Pract. 1980;21:183–188.
11. Bregman, CL, Hirth, RS, Sundberg, JP, et al. Cutaneous neoplasms in dogs associated with canine oral papillomavirus. Vet Pathol. 1987;24:477–487.
12. Davis, PE, Huxtable, CRR, Sabcine, M. Dermal papillomas in the racing greyhound. Aust J Dermatol. 1976;17:13–16.
13. Theilen, GH, Madewell, BR. Papillomatosis and fibromatosis. In: Theilen GH, Madewell BR, eds. Veterinary cancer medicine. Philadelphia: Lea & Febiger, 1987.
14. Calvert, CA. Canine viral papillomatosis. In: Greene GE, ed. Infectious diseases of the dog and cat. Philadelphia: WB Saunders, 1991.
15. Callan, MB, Preziosi, D, Mauldin, E. Multiple papillomavirus-associated epidermal hamartomas and squamous cell carcinomas in situ in a dog following treatment with prednisone and cyclosporine. Vet Dermatol. 2005;16:338.
16. Nagata, M. Canine papillomatosis. In: Bonagura JD, ed. Kirk’s current veterinary therapy XIII. Philadelphia: WB Saunders, 2000.
17. Bonney, CH, Koch, SA, Dice, PF, et al. Papillomatosis of conjunctiva and adnexa in dogs. J Am Vet Med Assoc. 1980;176:48.
18. Kuntsi-Vaattovaara, H, Verstraete, FJM, Newsome, JT, et al. Resolution of persistent oral papillomatosis in a dog after treatment with a recombinant canine oral papillomavirus vaccine. Vet Comp Oncol. 2003;1:57.
19. Bomholt, A. Interferon therapy for laryngeal papillomatosis in adults. Arch Otolaryngol. 1983;109:550–552.
20. Sundberg, JP, Smith, EK, Herron, AJ, et al. Cutaneous verrucosis in a Chinese Shar pei dog. Vet Pathol. 1994;31:183.
21. Nagata, M. Canine papillomatosis. In: Bonagura JD, ed. Kirk’s current veterinary therapy XIII. Philadelphia: WB Saunders, 2000.
22. Nagata, M, Nanko, H, Moriyana, A, et al. Pigmented plaques associated with papillomavirus infection in dogs: is this epidermodysplasia verruciformis? Vet Dermatol. 1995;6:179.
23. Bregman, CL, Hinth, RS, Sundberg, JP, et al. Cutaneous neoplasms in dogs associated with canine oral papillomavirus vaccine. Vet Pathol. 1987;24:477.
24. Meunier, LD. Squamous cell carcinoma in beagles subsequent to canine oral papillomavirus vaccine. Lab Anim Sci. 1990;40:568.
25. Munday, JS, Willis, KA, Kiupel, M, et al. Amplification of three different papillomaviral DNA sequences from a cat with viral plaques. Vet Dermatol. 2008;19:400.
26. Schulman, FY, Krafft, AE, Janczewski, T, et al. Cutaneous fibropapilloma in a mountain lion (Felis concolor). J Zoo Wildl Med. 2003;34:179.
27. Sundberg, JP, Van Ranst, M, Montali, R, et al. Feline papillomas and papillomaviruses. Vet Pathol. 2000;37:1.
28. Egberink, HF, Horzinek, MC. Feline viral papillomatosis. In Greene CE, ed.: Infectious diseases of the dog and cat, ed 2, Philadelphia: WB Saunders, 1998.
29. Lozano-Alarcon, F, Lewis, TP, Clark, EG, et al. Persistent papillomavirus infection in a cat. J Am Anim Hosp Assoc. 1996;32:392.
30. LeClerc, SM, Clark, EG, Haines, DM. Papillomavirus infection in association with feline cutaneous squamous cell carcinoma in situ. Proc Am Assoc Vet Derm/Am Coll Vet Derm. 1997;13:125.
31. Schulman, FY, Krafft, AE, Janczewski, T. Feline cutaneous fibropapillomas: clinicopathologic findings and association with papillomavirus infection. Vet Pathol. 2001;38:291.
32. Teifke, JP, Kidney, BA, Lohr, CV, et al. Detection of papillomavirus DNA in mesenchymal tumour cells and not in the hyperplastic epithelium of feline sarcoids. Vet Dermatol. 2003;14:1. [47].
33. Munday, JS, Kiuppel, M. Papillomavirus-associated cutaneous neoplasia in mammals. Vet Pathol. 2010;47:254.
34. Angelos, JA, Marti, E, Lazary, S, et al. Characterization of BPV-like DNA in equine sarcoids. Arch Virol. 1991;119:95.
35. Gill, VL, Bergman, PJ, Baer, KE, et al. Use of Imiquimod cream (Aldara) in cats with multicentric squamous cell carcinoma in situ: 12 cases (2002-2005). Vet Compar Oncol. 2008;16:55.
36. Rojko, JL, Hardy, WD, Jr. Feline leukemia virus and other retroviruses. In Sherding RG, ed.: The cat: Diseases and clinical management, ed 2, New York: Churchill Livingstone, 1994.
37. Cotter, SM. Feline viral neoplasia. In Greene CE, ed.: Infectious diseases of the dog and cat, ed 2, Philadelphia: WB Saunders, 1998.
38. Hardy, WD, Jr., Hess, PW, MacEwen, EG, et al. Biology of feline leukemia virus in the natural environment. Cancer Res. 1976;36:582.
39. Essex, M. Feline leukemia and sarcoma viruses. In: Klein G, ed. Viral oncology. New York: Raven Press, 1980.
40. Hardy, WD, Jr. The feline leukemia virus. J Am Anim Hosp Assoc. 1981;17:951.
41. Pedersen, NC. Feline immunodeficiency virus. In: Schellekens LT, Horzinek MC, eds. Animal models in AIDS. Amsterdam: Elsevier, 1990.
42. Hardy, WD, Jr. The feline sarcoma viruses. J Am Anim Hosp Assoc. 1981;17:981.
43. Benveniste, RE, Sherr, CJ, Todaro, GJ. Evolution of type C viral genes: origin of feline leukemia virus. Science. 1975;190:886.
44. Daniels, MJ, Golder, MC, Jarrett, O, et al. Feline viruses in wildcats from Scotland. J Wildl Dis. 1999;35:121.
45. Marker, L, Munson, L, Basson, PA, et al. Multicentric T-cell lymphoma associated with feline leukemia virus infection in a captive Namibian cheetah (Acinonyx jubatus). J Wildl Dis. 2003;39:690.
46. Schafer, W, Bolognesi, DP. Mammalian type C oncornaviruses: relationships between viral structure and cell surface antigens and their possible significance in immunological defense mechanisms. Contemp Top Immunobiol. 1977;6:127.
47. Bolognesi, DP, Montelaro, RC, Frank, H, et al. Assembly of type C oncornaviruses: A model. Science. 1978;199:183.
48. Hardy, WD, Jr. Immunology of oncornaviruses. Vet Clin North Am Small Anim Pract. 1974;4:133.
49. Sarma, PS, Log, T. Subgroup classification of feline leukemia and sarcoma viruses by viral interference and neutralization tests. Virology. 1973;54:160.
50. Sarma, PS, Log, T, Jain, D, et al. Differential host range of viruses of feline leukemia-sarcoma complex. Virology. 1975;64:438.
51. Hardy, WD, Jr., Hirshaut, Y, Hess, P. Detection of the feline leukemia virus and other mammalian oncornaviruses by immunofluorescence. In: Dutcher RM, Chieco-Bianchi L, eds. Unifying concepts of leukemia. Basel: S Karger, 1973.
52. Francis, DP, Essex, M, Gayzagian, D. Feline leukemia virus: Survival under home and laboratory conditions. J Clin Microbiol. 1979;9:154.
53. Dornsife, RE, Gasper, PW, Mullins, JI. Induction of aplastic anemia by intrabone marrow inoculation of a molecularly cloned feline retrovirus. Leuk Res. 1989;13:745.
54. Hartmann, K, Werner, RM, Egberink, H, et al. Comparison of six in-house tests for the rapid diagnosis of feline immunodeficiency and feline leukaemia virus infections. Vet Rec. 2001;149:317.
55. Lauring, AS, Anderson, MM, Overbaugh, J. Specificity in receptor usage by T-cell–tropic feline leukemia viruses: implications for the in vivo tropism of immunodeficiency-inducing variants. J Virol. 2001;75:8888.
56. Lauring, AS, Cheng, HH, Eiden, MV, et al. Genetic and biochemical analyses of receptor and cofactor determinants for T-cell–tropic feline leukemia virus infection. J Virol. 2002;76:8069.
57. Essex, M, Cotter, SM, Sliski, AH, et al. Horizontal transmission of feline leukaemia under natural conditions in a feline leukaemia cluster household. Int J Cancer. 1977;19:90.
58. Vail, DM, Moore, AS, Ogilvie, GK, et al. Feline lymphoma (145 cases): proliferation indices, CD3 immunoreactivity and their association with prognosis in 90 cats receiving therapy. J Vet Intern Med. 1998;12:349.
59. Louwerens, M, London, CA, Pedersen, NC, et al. Feline lymphoma in the post-feline leukemia virus era. J Vet Intern Med. 2005;19:329.
60. Jarrett, O, Hardy, WD, Jr., Golder, MC, et al. The frequency of occurrence of feline leukaemia virus subgroups in cats. Int J Cancer. 1978;21:334.
61. Jarrett, O, Russell, PH. Differential growth and transmission in cats of feline leukaemia viruses of subgroups A and B. Int J Cancer. 1978;21:466.
62. Rosenburg, Z, Pederson, FF, Haseltine, WA. Comparative analysis of the genome of feline leukemia virus. J Virol. 1980;35:542.
63. Tzavaras, T, Stewart, M, McDougall, A, et al. Molecular cloning and characterization of a defective recombinant feline leukaemia virus associated with myeloid leukaemia. J Gen Virol. 1990;71:343.
64. Stewart, MA, Warnock, M, Wheeler, A, et al. Nucleotide sequences of a feline leukemia virus subgroup: A envelope gene and long terminal repeat and evidence for the recombinational origin of subgroup B viruses. J Virol. 1986;58:825.
65. Elder, JM, Mullins, JI. Nucleotide sequence of the envelope gene of Gardner-Arnstein feline leukemia virus B reveals unique sequence homologies with a murine mink cell focus-forming virus. J Virol. 1983;46:871.
66. Rigby, MA, Rojko, JL, Stewart, MA, et al. Partial dissociation of subgroup C phenotype and in vivo behaviour in feline leukaemia viruses with chimeric envelope genes. J Gen Virol. 1992;73:2839.
67. Heding, LD, Schaller, JP, Blakeslee, JR, et al. Inactivation of tumor cell–associated feline oncornavirus for preparation of an infectious virus–free tumor cell immunogen. Cancer Res. 1976;36:1647.
68. Essex, M, Klein, G, Snyder, SP, et al. Feline sarcoma virus (FSV)–induced tumors: correlation between humoral antibody and tumor regression. Nature. 1971;233:195.
69. Vedbrat, S, Rasheed, S, Lutz, H, et al. Feline oncornavirus–associated cell membrane antigen: a viral and not a cellularly coded transformation-specific antigen of cat lymphomas. Virology. 1983;124:445.
70. Snyder, HW, Singhal, MC, Zuckerman, EE, et al. The feline oncornavirus associated cell membrane antigen (FOCMA) is related to, but distinguishable from, FeLV-C gp70. Virology. 1983;131:315.
71. Macy, DW. Vaccination against feline retroviruses. In: August J, ed. Consultations in feline internal medicine. Philadelphia: WB Saunders, 1994.
72. Dorn, CR, Taylor, DON, Schneider, R, et al. Survey of animal neoplasms in Alameda and Contra Costa counties, California. II. Cancer morbidity in dogs and cats from Alameda county. J Natl Cancer Inst. 1968;40:307.
73. Schneider, R. Comparison and age- and sex-specific incidence rate patterns of the leukemia complex in the cat and the dog. J Natl Cancer Inst. 1983;70:971.
74. Goitsuka, R, Tsuji, M, Matsumoto, Y, et al. A case of feline large granular lymphoma. Jpn J Vet Sci. 1988;50:593.
75. Finn, JP, Schwartz, LW. A neoplasm of globule leukocytes in the intestine of a cat. J Comp Pathol. 1972;82:323.
76. Hardy, WD, Jr., McClelland, AJ, Zuckerman, EE, et al. Development of virus non-producer lymphosarcomas in pet cats exposed to FeLV. Nature. 1980;288:90.
77. Hardy, WD, Jr., Zuckerman, EE, McClelland, AJ, et al. The immunology and epidemiology of FeLV-nonproducer feline lymphosarcomas. Cold Spring Harbor Conf Cell Prolif. 1980;7:677.
78. Hardy, WD, Jr. Hematopoietic tumors of cats. J Am Anim Hosp Assoc. 1981;17:921.
79. Spodnick, GJ, Berg, J, Moore, FM, et al. Spinal lymphoma in cats: 21 cases (1976-1989). J Am Vet Med Assoc. 1992;200:373.
80. Hartmann K, Gerle K, Leutenegger C, et al: Feline leukemia virus: Most important oncogene in cats? Abstracts from the Fourth International Feline Retrovirus Research Symposium, Glasgow, 1998.
81. Cotter, SM. Feline viral neoplasia. In: Greene CE, ed. Infectious diseases of the dog and cat. Philadelphia: WB Saunders, 1990.
82. Teske, E, van Straten, G, van Noort, R, et al. Chemotherapy with cyclophosphamide, vincristine, and prednisolone (COP) in cats with malignant lymphoma: New results with an old protocol. J Vet Intern Med. 2002;16:179.
83. Shelton, GH, Grant, CK, Cotter, SM, et al. Feline immunodeficiency virus and feline leukemia virus infections and their relationships to lymphoid malignancies in cats: A retrospective study (1968-1988). J Acquir Immune Defic Syndr. 1990;3:623.
84. Hardy, WD, Jr., Zuckerman, BE, MacEwen, EG, et al. A feline leukemia and sarcoma virus–induced tumor-specific antigen. Nature. 1977;270:249.
85. Hardy, WD, Jr., Zuckerman, EE, Essex, M, et al. Feline oncornavirus–associated cell membrane antigen: an FeLV- and FeSV-induced tumor-specific antigen. In: Clarkson B, Marks PA, Till JE, eds. Differentiation of normal and neoplastic hematopoietic cells. Cold Spring Harbor, New York: Cold Spring Harbor Laboratory Press, 1978.
86. Francis, DP, Cotter, SM, Hardy, WD, Jr., et al. Comparison of virus-positive and virus-negative cases of feline leukemia and lymphoma. Cancer Res. 1979;39:3866.
87. McClelland, AJ, Hardy, WD, Jr., Zuckerman, EE. Prognosis of healthy feline leukemia virus infected cats. Dev Cancer Res. 1980;4:121.
88. Cotter, SM. Feline leukemia virus: pathophysiology, prevention and treatment. Cancer Invest. 1992;10:173.
89. Cotter, SM. Management of healthy feline leukemia virus–positive cats. J Am Vet Med Assoc. 1991;199:1470.
90. August, JR. Husbandry practices for cats infected with feline leukemia virus or feline immunodeficiency virus. J Am Vet Med Assoc. 1991;199:1474.
91. Kociba, GJ, Lange, RD, Dunn, CD, et al. Serum erythropoietin changes in cats with feline leukemia virus–induced erythroid aplasia. Vet Pathol. 1983;20:548.
92. Snyder, HW, Jr., Singhal, MC, Hardy, WD, Jr., et al. Clearance of feline leukemia virus from persistently infected pet cats treated by extracorporeal immunoadsorption is correlated with an enhanced antibody response to FeLV gp70. J Immunol. 1984;132:1538.
93. MacEwen EG: Current immunotherapeutic approaches in small animals, Proceedings of the Kal Kan Symposium on Oncology, Columbus, 1986.
94. Kitchen, LW, Mather, FJ. Hematologic effects of short-term oral diethylcarbamazine treatment given to chronically feline leukemia virus infected cats. Cancer Lett. 1989;45:183.
95. Kitchen, LW, Cotter, SM. Effect of diethylcarbamazine on serum antibody to feline oncornavirus–associated cell membrane antigen in feline leukemia virus–infected cats. J Clin Lab Immunol. 1988;25:101.
96. Barta, O. Immunoadjuvant therapy. In: Kirk RW, Bonagura JD, eds. Kirk’s current veterinary therapy XI: Small animal practice. Philadelphia: WB Saunders, 1992.
97. Elmslie, RE, Ogilvie, GK, Dow, SW, et al. Evaluation of a biologic response modifier derived from Serratia marcescens: effects on feline macrophages and usefulness for the prevention and treatment of viremia in feline leukemia virus–infected cats. Mol Biother. 1991;3:231.
98. Gasper, PW, Fulton, R, Thrall, MA. Bone marrow transplantation: Update and current considerations. In: Kirk RW, Bonagura JD, eds. Kirk’s current veterinary therapy XI: Small animal practice. Philadelphia: WB Saunders, 1992.
99. Tompkins, MB, Cummins, JM. Response of feline leukemia virus–induced nonregenerative anemia to oral administration of an interferon-containing preparation. Feline Pract. 1982;12:6.
100. Tizard, I. Use of immunomodulators as an aid to clinical management of feline leukemia virus–infected cats. J Am Vet Med Assoc. 1991;199:1482.
101. Gingerich, DA. Lymphocyte T-cell immunomodulator (LTCI): Review of the immunopharmacology of a new veterinary biologic. Intern J Applied Res Vet Med. 2008;6:61.
102. Polas, PV, Swenson, CL, Sams, R, et al. In vitro and in vivo evidence that the antiviral activity of 2-3-dideoxycytidine is target cell–dependent in a feline retrovirus animal model. Antimicrob Agents Chemother. 1990;34:1414.
103. Zeidner, NS, Strobel, JD, Perigo, NA, et al. Treatment of FeLV-induced immunodeficiency syndrome (FeLV-FAIDS) with controlled release capsular implantation of 2′,3′-dideoxycytidine. Antiviral Res. 1989;11:147.
104. DeClerq, E, Sakuma, T, Baba, M, et al. Antiviral activity of phosphonymethoxyalkyl derivatives of purines and pyrimidines. Antiviral Res. 1987;8:261.
105. Hoover, EA, Ebner, JP, Zeidner, NS, et al. Early therapy of feline leukemia virus infection (FeLV-FAIDS) with 9-(2-phosphonylmethoxyethyl) adenine (PMEA). Antiviral Res. 1991;16:77.
106. Hoover, EA, Zeidner, NS, Mullins, JI. Therapy of presymptomatic FeLV-induced immunodeficiency syndrome with AZT in combination with α-interferon. Ann NY Acad Sci. 1990;616:258.
107. Zeidner, NS, Myles, MH, Mathiason-DuBard, CK, et al. α-Interferon (2b) in combination with zidovudine for the treatment of presymptomatic feline leukemia virus–induced immunodeficiency syndrome. Antimicrob Agents Chemother. 1990;34:1749.
108. Zeidner, NS, Mathiason-DuBard, CK, Hoover, EA. Reversal of feline leukemia virus infection by adoptive transfer of lectin/interleukin-2–activated lymphocytes, interferon-α and zidovudine. J Immunother. 1993;14:22.
109. Hardy, WD, Jr., McClelland, AJ, Zuckerman, EE, et al. Prevention of the contagious spread of feline leukaemia virus and the development of leukaemia in pet cats. Nature. 1976;263:326.
110. Macy DW: Unpublished data. 1992.
111. Legendre, AM, Hawks, DM, Sebring, R. Comparison of the efficacy of three commercial FeLV vaccines in a natural challenge. J Am Vet Med Assoc. 1991;199:1446.
112. Hendrick, WH, Kass, PH, McGill, LD, et al. Postvaccinal sarcomas in cats. J Natl Cancer Inst. 1994;86:5. [341].
113. Macy DW: Unpublished data, 1994.
114. Varmus, HE. Form and function of retroviral proviruses. Science. 1982;216:812.
115. Coffin, JM. Structure, replication and recombination of retrovirus genomes: some unifying hypotheses. J Gen Virol. 1979;42:1.
116. Besmer, P. Acute transforming feline retroviruses. Contemp Top Microbiol Immunol. 1993;107:1.
117. Essex, M, Klein, G, Snyder, SP, et al. Feline sarcoma virus (FSV)–induced tumors: correlation between humoral antibody and tumor regression. Nature. 1971;233:195.
118. Essex, M, Klein, G, Synder, SP, et al. Antibody to feline oncornavirus–associated cell membrane antigen in neonatal cats. Int J Cancer. 1971;8:384.
119. Essex, M, Cotter, SM, Hardy, WD, Jr., et al. Feline oncornavirus–associated cell membrane antigen. IV. Antibody titers in cats with naturally occurring leukemia, lymphoma, and other diseases. J Natl Cancer Inst. 1975;55:463.
120. Patnaik, AK, Liu, SK, Hurvitz, AI, et al. Nonhematopoietic neoplasms in cats. J Natl Cancer Inst. 1975;54:855.
121. Macy DW: Unpublished data, 1992.
122. Sieblink HJ, Chu I, Rimmelzwaan GF, et al: Isolation and partial characterization of infectious molecular clones of feline immunodeficiency virus directly obtained from bone marrow DNA of a naturally infected cat. Paper presented at the First International Conference of Feline Immunodeficiency Virus Researchers, 1990, Davis, CA.
123. Hoise, MJ, Jarrett, O. Serological responses of cats to feline immunodeficiency virus. AIDS. 1990;4:215.
124. Yamamoto, JK, Sparger, E, Ho, EW, et al. Pathogenesis of experimentally-induced feline immunodeficiency virus infection in cats. Am J Vet Res. 1988;49:1246.
125. Yamamoto, JK, Hansen, H, Ho, EW, et al. Epidemiologic and clinical aspects of feline immunodeficiency virus infection in cats from the continental United States and Canada and possible mode of transmission. J Am Vet Med Assoc. 1989;194:213.
126. North, TW, North, GLT, Pedersen, NC. Feline immunodeficiency virus: A model for reverse transcriptase–targeted chemotherapy for acquired immune deficiency syndrome. Antimicrob Agents Chemother. 1989;33:915.
127. Fleming, EJ, McCaw, DL, Smith, JA, et al. Clinical hematologic and survival data from cats infected with feline immunodeficiency virus: 42 cases (1983-1988). J Am Vet Med Assoc. 1991;199:913.
128. Ishida, T, Wahiza, T, Toriyabe, K, et al. Feline immunodeficiency virus infection in cats in Japan. J Am Vet Med Assoc. 1989;194:221.
129. Sabine, M, Michelsen, J, Thomas, F, et al. Feline AIDS. Aust Vet Pract. 1988;18:105.
130. Pedersen, NC, Ho, EW, Brown, ML, et al. Isolation of a T-lymphotropic virus from domestic cats with an immunodeficiency-like syndrome. Science. 1987;235:790.
131. Swinney, GR, Pauli, JV, Hones, BE, et al. Feline T-lymphotrophic virus (FTLV) in cats in New Zealand. N Z Vet J. 1989;37:41.
132. Hutson, CA, Rideout, BA, Pedersen, NC. Neoplasia associated with feline immunodeficiency virus infection in cats of Southern California. J Am Vet Med Assoc. 1991;199:1357.
133. Macy DW, Podolsiki CL, Collins J: Prevalence of FeLV and FIV high risk cats in northeastern Colorado. Paper presented at the Tenth Annual Conference of the Veterinary Cancer Society, 1990, Auburn, AL.
134. Pedersen, WC, Barlough, JE. Clinical overview of feline immunodeficiency virus. J Am Vet Med Assoc. 1991;199:1298.
135. Neu, H. FIV (FTLV)–Infektion der Katze: II Falle Beitrag zur epidemiologic, klinischen Symptomatologie undzum Krankheitsverlauf. Kleintierpraxis. 1989;34:373.