14 Mechanisms of Bacterial Pathogenesis
To a bacterium, the human body is a collection of environmental niches that provide the warmth, moisture, and food necessary for growth. Bacteria have traits that enable them to enter (invade) the environment, remain in a niche (adhere or colonize), gain access to food sources (degradative enzymes), and escape clearance by host immune and nonimmune protective responses (e.g., capsule). When sufficient numbers of bacteria are present (quorum), they turn on functions to support the colony, including production of a biofilm. Unfortunately, many of the mechanisms that bacteria use to maintain their niche and the byproducts of bacterial growth (e.g., acids, gas) cause damage and problems for the human host. Many of these traits are virulence factors, which enhance the ability of bacteria to cause disease. Although many bacteria cause disease by directly destroying tissue, some release toxins, which are then disseminated by the blood to cause system-wide pathogenesis (Box 14-1). The surface structures of bacteria are powerful stimulators of host responses (acute phase: interleukin-1 [IL-1], IL-6, tumor necrosis factor-α [TNF-α]), which can be protective but are often major contributors to the disease symptoms (e.g., sepsis). Production of disease results from the combination of damage caused by the bacteria and the consequences of the innate and immune responses to the infection (Box 14-2).
Box 14-2
Bacterial Disease Production
1. Disease is caused by damage produced by the bacteria plus the consequences of innate and immune responses to the infection.
2. The signs and symptoms of a disease are determined by the function and importance of the affected tissue.
3. The length of the incubation period is the time required for the bacteria and/or the host response to cause sufficient damage to initiate discomfort or interfere with essential functions.
Not all bacteria or bacterial infections cause disease; however, some always cause disease. The human body is colonized with numerous microbes (normal flora), many of which serve important functions for their hosts. Normal flora bacteria aid in the digestion of food, produce vitamins (e.g., vitamin K), protect the host from colonization with pathogenic microbes and activate host innate and immune responses. These endogenous bacteria normally reside in locations such as the gastrointestinal (GI) tract, mouth, skin, and upper respiratory tract, which can be considered to be outside the body (Figure 14-1). The composition of the normal flora can be disrupted by antibiotic treatment, diet, stress, and changes in the host response to the flora. The loss of controlling bacteria with broad spectrum antibiotic treatment often allows the outgrowth of Clostridium difficile, which causes pseudomembranous colitis. An altered normal flora can lead to inappropriate immune responses, causing inflammatory bowel diseases.
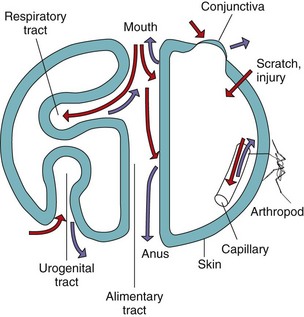
Figure 14-1 Body surfaces as sites of microbial infection and shedding. Red arrows indicate infection; purple arrows indicate shedding.
(Modified from Mims C, et al: Medical microbiology, London, 1993, Mosby-Wolfe.)
Normal flora bacteria cause disease if they enter normally sterile sites of the body. Virulent bacteria have mechanisms that promote their growth in the host at the expense of the host’s tissue or organ function. Opportunistic bacteria take advantage of preexisting conditions, such as immunosuppression, to grow and cause serious disease. For example, burn victims and the lungs of patients with cystic fibrosis are at higher risk to Pseudomonas aeruginosa infection, and patients with the acquired immunodeficiency syndrome (AIDS) are very susceptible to infection by intracellularly growing bacteria, such as the mycobacteria.
Disease results from the damage or loss of tissue or organ function due to the infection or the host inflammatory responses. The signs and symptoms of a disease are determined by the change to the affected tissue. Systemic responses are produced by toxins and the cytokines produced in response to the infection. The seriousness of the disease depends on the importance of the affected organ and the extent of the damage caused by the infection. Infections of the central nervous system are always serious. The bacterial strain and inoculum size are also major factors in determining whether disease occurs; however, the threshold for disease production is different for different bacteria (e.g., less than 200 Shigella are required for shigellosis but 108 Vibrio cholerae or Campylobacter organisms are required for disease of the GI tract). Host factors can also play a role. For example, although a million or more Salmonella organisms are necessary for gastroenteritis to become established in a healthy person, only a few thousand organisms are necessary in a person whose gastric pH has been neutralized with antacids or other means. Congenital defects, immunodeficiency states (see Chapter 10), and other disease-related conditions might also increase a person’s susceptibility to infection. The longer a bacterium remains in the body, the greater its numbers, its ability to spread, its potential to cause tissue damage and disease, and the larger the host response.
Many of the virulence factors consist of complex structures or activities that are only expressed under special conditions (see Figure 13-9). The components for these structures are often encoded together in a pathogenicity island. Pathogenicity islands are large genetic regions in the chromosome or on plasmids that contain sets of genes encoding numerous virulence factors that may require coordinated expression. These genes may be turned on by a single stimulus (e.g., the temperature of the gut, pH of a lysosome). A pathogenicity island is usually within a transposon and can be transferred as a unit to different sites within a chromosome or to other bacteria. For example, the SPI-2 pathogenicity island of Salmonella is activated by the acidic pH of a phagocytic vesicle within a macrophage. This promotes the expression of approximately 25 proteins that assemble into a syringe-like molecular device (type III secretion device) that injects proteins into the host cell to facilitate the bacteria’s intracellular survival and growth. Similarly, the biofilm produced by Pseudomonas is triggered when there are sufficient bacteria (a quorum) producing sufficient amounts of N-acyl homoserine lactone (AHL) to trigger expression of the genes for polysaccharide production.
Entry into the Human Body
For infection to become established, bacteria must first gain entry into the body (Table 14-1; see Figure 14-1). Natural defense mechanisms and barriers, such as skin, mucus, ciliated epithelium, and secretions containing antibacterial substances (e.g., lysozyme, defensins) make it difficult for bacteria to gain entry into the body. However, these barriers are sometimes broken (e.g., a tear in the skin, a tumor or ulcer in the bowel), providing a portal of entry for the bacteria, or the bacteria may have the means to compromise the barrier and invade the body. On invasion, the bacteria can travel in the bloodstream to other sites in the body.
Table 14-1 Bacterial Port of Entry
| Route | Examples |
|---|---|
| Ingestion | Salmonella spp., Shigella spp., Yersinia enterocolitica, enterotoxigenic Escherichia coli, Vibrio spp., Campylobacter spp., Clostridium botulinum, Bacillus cereus, Listeria spp., Brucella spp. |
| Inhalation | Mycobacterium spp., Nocardia spp., Mycoplasma pneumoniae, Legionella spp., Bordetella, Chlamydophila psittaci, Chlamydophila pneumoniae, Streptococcus spp. |
| Trauma | Clostridium tetani |
| Needlestick | Staphylococcus aureus, Pseudomonas spp. |
| Arthropod bite | Rickettsia, Ehrlichia, Coxiella, Francisella, Borrelia spp., Yersinia pestis |
| Sexual transmission | Neisseria gonorrhoeae, Chlamydia trachomatis, Treponema pallidum |
The skin has a thick, horny layer of dead cells that protects the body from infection. However, cuts in the skin, produced accidentally or surgically or kept open with catheters or other surgical appliances, provide a means for the bacteria to gain access to the susceptible tissue underneath. For example, Staphylococcus aureus and Staphylococcus epidermidis, which are a part of the normal flora on skin, can enter the body through breaks in the skin and pose a major problem for people with indwelling catheters and intravenous lines.
The mouth, nose, respiratory tract, ears, eyes, urogenital tract, and anus are sites through which bacteria can enter the body. These natural openings in the skin, and their associated body cavities are protected by natural defenses such as the mucus and ciliated epithelium that line the upper respiratory tract, the lysozyme and other antibacterial secretions in tears and mucus, and the acid and bile in the GI tract. However, many bacteria are unaffected or have the means to evade these defenses. For example, the outer membrane of the gram-negative bacteria makes these bacteria more resistant to lysozyme, acid, and bile. The enterobacteria are thus enabled to colonize the GI tract. A break in the normal barrier can allow entry of these endogenous bacteria to normally sterile sites of the body, such as the peritoneum and the bloodstream, to cause disease. An example of this is the patient whose colon tumor was diagnosed after detection of a septicemia (blood-borne infection) caused by enteric bacteria.
Colonization, Adhesion, and Invasion
Different bacteria colonize different parts of the body. This may be closest to the point of entry or due to the presence of optimal growth conditions at the site. For example, Legionella is inhaled and grows in the lungs but does not readily spread because it cannot tolerate high temperatures (e.g., 35° C). Colonization of sites that are normally sterile implies the existence of a defect in a natural defense mechanism or a new portal of entry. Patients with cystic fibrosis have such defects because of the reduction in their ciliary mucoepithelial function and altered mucosal secretions; as a result, their lungs are colonized by S. aureus and P. aeruginosa. In some cases, colonization requires special structures and functions to remain at the site, survive, and obtain food.
Bacteria may use specific mechanisms to adhere to and colonize different body surfaces (Table 14-2). If the bacteria can adhere to epithelial or endothelial cell linings of the bladder, intestine, and blood vessels, they cannot be washed away, and this adherence allows them to colonize the tissue. For example, natural bladder function eliminates any bacteria not affixed to the bladder wall. Escherichia coli and other bacteria have adhesins that bind to specific receptors on the tissue surface and keep the organisms from being washed away. Many of these adhesin proteins are present at the tips of fimbriae (pili) and bind tightly to specific sugars on the target tissue; this sugar-binding activity defines these proteins as lectins. For example, most E. coli strains that cause pyelonephritis produce a fimbrial adhesin termed the P fimbriae. This adhesin can bind to α-D-galactosyl-β-D-galactoside (Gal-Gal), which is part of the P blood group antigen structure on human erythrocytes and uroepithelial cells. Neisseria gonorrhoeae pili are also important virulence factors; they bind to oligosaccharide receptors on epithelial cells. Yersinia organisms, Bordetella pertussis, and Mycoplasma pneumoniae express adhesin proteins that are not on fimbriae. Streptococcus pyogenes uses lipoteichoic acid and the F protein (binds to fibronectin) to bind to epithelial cells.
Table 14-2 Examples of Bacterial Adherence Mechanisms
| Microbe | Adhesin | Receptor |
|---|---|---|
| Staphylococcus aureus | LTA | Unknown |
| Staphylococcus spp. | Slime | Unknown |
| Streptococcus, group A | LTA–M protein complex | Fibronectin |
| Streptococcus pneumoniae | Protein | N-Acetylhexosamine-galactose |
| Escherichia coli | Type 1 fimbriae | D-Mannose |
| Colonization factor antigen fimbriae | GM ganglioside 1 | |
| P fimbriae | P blood group glycolipid | |
| Neisseria gonorrhoeae | Fimbriae | GD1 ganglioside |
| Treponema pallidum | P1, P2, P3 | Fibronectin |
| Chlamydia trachomatis | Cell surface lectin | N-Acetylglucosamine |
| Mycoplasma pneumoniae | Protein P1 | Sialic acid |
| Vibrio cholerae | Type 4 pili | Fucose and mannose |
LTA, Lipoteichoic acid.
A special bacterial adaptation that facilitates colonization, especially of surgical appliances such as artificial valves or indwelling catheters, is a biofilm. Bacteria in biofilms are bound within a sticky web of polysaccharide that binds the cells together and to the surface. Production of a biofilm requires sufficient numbers of bacteria (quorum). When P. aeruginosa determine that the colony size is large enough (quorum sensing) they produce a biofilm. Dental plaque is another example of a biofilm. The biofilm matrix can also protect the bacteria from host defenses and antibiotics.
Although bacteria do not have mechanisms that enable them to cross intact skin, several bacteria can cross mucosal membranes and other tissue barriers to enter normally sterile sites and more susceptible tissue. These invasive bacteria either destroy the barrier or penetrate into the cells of the barrier. Shigella, Salmonella, and Yersinia organisms are enteric bacteria that use fimbriae to bind to M (microfold) cells of the colon and then inject proteins into the M cell that stimulate the cell membrane to surround and take in the bacteria. These bacteria produce a type III secretion device that resembles a molecular syringe that injects pore-forming factors and effector molecules into the host cells. The effector proteins can facilitate uptake and invasion, promote the intracellular survival and replication of the bacteria, or the apoptotic death of the host cell. Enteropathogenic E. coli secretes proteins into the host cell that create a portable docking system for itself and Salmonella uses the device to promote its uptake into a vesicle and live intracellularly within the macrophage (see animations developed by the Howard Hughes Medical Institute; website listed in references). Shigella uses a type III secretion device to enter cells; once inside cells, the organism causes cellular actin to polymerize and push the Shigella into an adjacent cell. Listeria monocytogenes causes the polymerization of actin at its rear to propel the bacteria around the cell and into an adjacent cell, as if on the top of a battering ram.
Pathogenic Actions of Bacteria
Tissue Destruction
Byproducts of bacterial growth, especially fermentation, include acids, gas, and other substances that are toxic to tissue. In addition, many bacteria release degradative enzymes to break down tissue, thereby providing food for the growth of the organisms and promoting the spread of the bacteria. For example, Clostridium perfringens organisms are part of the normal flora of the GI tract but are also opportunistic pathogens that can establish infection in oxygen-depleted tissues and cause gas gangrene. These anaerobic bacteria produce enzymes (e.g., phospholipase C, collagenase, protease, and hyaluronidase), several toxins, and acid and gas from bacterial metabolism, which destroy the tissue. Staphylococci produce many different enzymes that modify the tissue environment. These enzymes include hyaluronidase, fibrinolysin, and lipases. Streptococci also produce enzymes, including streptolysins S and O, hyaluronidase, DNAases, and streptokinases.
Toxins
Toxins are bacterial products that directly harm tissue or trigger destructive biologic activities. Toxins and toxin-like activities are degradative enzymes that cause lysis of cells or specific receptor-binding proteins that initiate toxic reactions in a specific target tissue. In addition, endotoxin (lipid A portion of lipopolysaccharide) and superantigen proteins promote excessive or inappropriate stimulation of innate or immune responses. In many cases, the toxin is completely responsible for causing the characteristic symptoms of the disease. For example, the preformed toxin present in food mediates the food poisoning caused by S. aureus and Bacillus cereus and the botulism caused by Clostridium botulinum. The symptoms caused by preformed toxin occur much sooner than for other forms of gastroenteritis because the effect is like eating a poison, and the bacteria do not need to grow for the symptoms to occur. Because a toxin can be spread systemically through the bloodstream, symptoms may arise at a site distant from the site of infection, such as occurs in tetanus, which is caused by Clostridium tetani.
Exotoxins
Exotoxins are proteins that can be produced by gram-positive or gram-negative bacteria and include cytolytic enzymes and receptor-binding proteins that alter a function or kill the cell. In many cases, the toxin gene is encoded on a plasmid (tetanus toxin of C. tetani, heat-labile [LT] and heat-stabile [ST] toxins of enterotoxigenic E. coli), or a lysogenic phage (Corynebacterium diphtheriae and C. botulinum).
Cytolytic toxins include membrane-disrupting enzymes, such as the α-toxin (phospholipase C) produced by C. perfringens, which breaks down sphingomyelin and other membrane phospholipids. Hemolysins insert into and disrupt erythrocyte and other cell membranes. Pore-forming toxins, including streptolysin O, can promote leakage of ions and water from the cell and disrupt cellular functions or cell lysis.
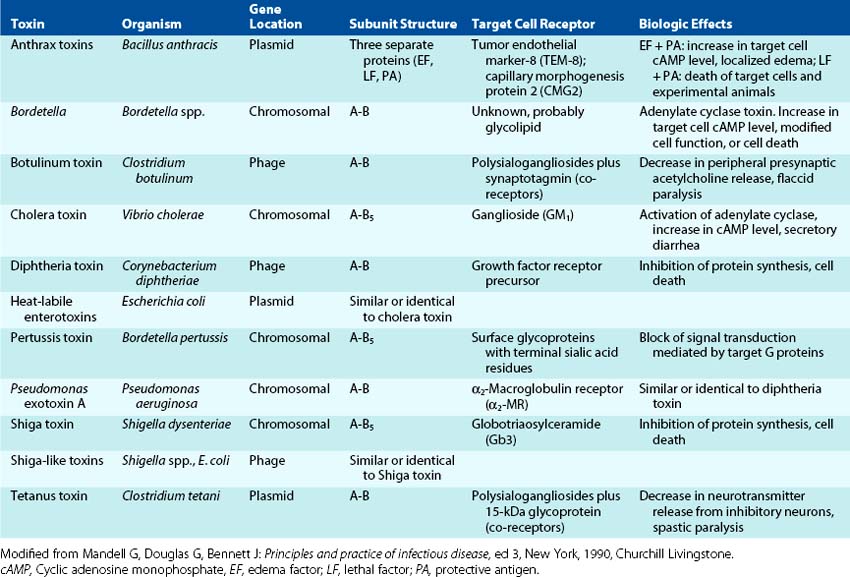
Many toxins are dimeric with A and B subunits (A-B toxins). The B portion of the A-B toxins binds to a specific cell surface receptor, and then the A subunit is transferred into the interior of the cell, where it acts to promote cell injury (B for binding, A for action). The tissues targeted by these toxins are very defined and limited (Figure 14-2; Table 14-3). The biochemical targets of A-B toxins include ribosomes, transport mechanisms, and intracellular signaling (cyclic adenosine monophosphate [cAMP] production, G protein function), with effects ranging from diarrhea to loss of neuronal function to death. The functional properties of cytolytic and other exotoxins are discussed in greater detail in the chapters dealing with the specific diseases involved.
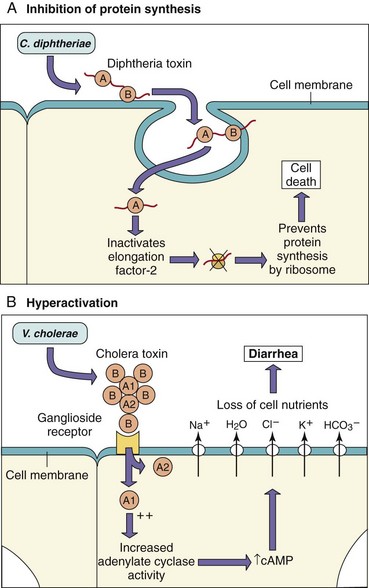
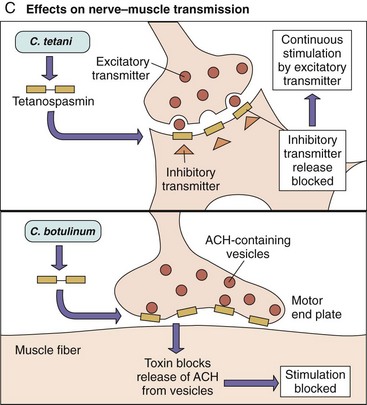
Figure 14-2 A-C, The mode of action of dimeric A-B exotoxins. The bacterial A-B toxins often consist of a two-chain molecule. The B chain binds and promotes entry of the A chain into cells, and the A chain has inhibitory activity against some vital function. ACH, Acetylcholine; cAMP, cyclic adenosine monophosphate.
(Modified from Mims C, et al: Medical microbiology, London, 1993, Mosby-Wolfe.)
Superantigens are a special group of toxins (Figure 14-3). These molecules activate T cells by binding simultaneously to a T-cell receptor and a major histocompatibility complex class II (MHC II) molecule on an antigen-presenting cell without requiring antigen. Superantigens activate large numbers of T cells to release large amounts of interleukins (cytokine storm), including IL-1, TNF, and IL-2, causing life-threatening autoimmune-like responses. This superantigen stimulation of T cells can also lead to death of the activated T cells, resulting in the loss of specific T-cell clones and the loss of their immune responses. Superantigens include the toxic shock syndrome toxin of S. aureus, staphylococcal enterotoxins, and the erythrogenic toxin A or C of S. pyogenes.
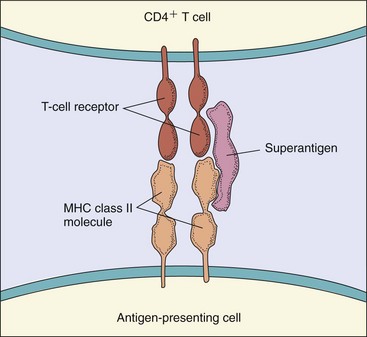
Endotoxin and Other Cell Wall Components
The presence of bacterial cell wall components acts as a signal of infection that provides a powerful multialarm warning to the body to activate the host’s protective systems. The molecular patterns in these structures (pathogen-associated molecular patterns [PAMPs]) bind to Toll-like receptors (TLRs) and other molecules and stimulate the production of cytokines (see Chapters 8 and 10). In some cases, the host response is excessive and may even be life threatening. The lipid A portion of lipopolysaccharide (LPS) produced by gram-negative bacteria is a powerful activator of acute-phase and inflammatory reactions and is termed endotoxin. It is important to appreciate that endotoxin is not the same as exotoxin and that only gram-negative bacteria make endotoxin. Weaker, endotoxin-like responses may occur to gram-positive bacterial structures, including teichoic and lipoteichoic acids.
Gram-negative bacteria release endotoxin during infection. Endotoxin binds to specific receptors (CD14 and TLR4) on macrophages, B cells, and other cells and stimulates the production and release of acute-phase cytokines, such as IL-1, TNF-α, IL-6, and prostaglandins (Figure 14-4). Endotoxin also stimulates the growth (mitogenic) of B cells.
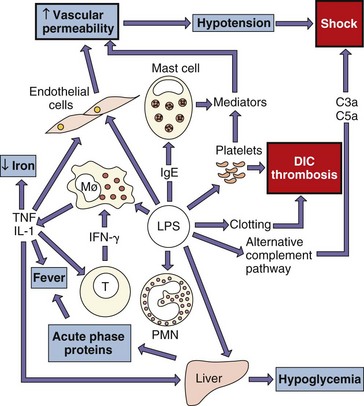
Figure 14-4 The many activities of lipopolysaccharide (LPS). This bacterial endotoxin activates almost every immune mechanism, as well as the clotting pathway, which together make LPS one of the most powerful immune stimuli known. DIC, Disseminated intravascular coagulation; IFN-γ, interferon-γ; IgE, immunoglobulin E; IL-1, interleukin-1; PMN, polymorphonuclear (neutrophil) leukocytes; TNF, tumor necrosis factor.
(Modified from Mims C, et al: Medical microbiology, London, 1993, Mosby-Wolfe.)
At low concentrations, endotoxin stimulates the development of protective responses, such as fever, vasodilation, and the activation of immune and inflammatory responses (Box 14-3). However, the endotoxin levels in the blood of patients with gram-negative bacterial sepsis (bacteria in the blood) can be very high, and the systemic response to these can be overpowering, resulting in shock and possibly death. High concentrations of endotoxin can also activate the alternative pathway of complement and production of anaphylotoxins (C3a, C5a), contributing to vasodilation and capillary leakage. In combination with TNF-α and IL-1, this can lead to hypotension and shock. Disseminated intravascular coagulation (DIC) can also result from the activation of blood coagulation pathways. The high fever, petechiae (skin lesions resulting from capillary leakage), and potential symptoms of shock (resulting from increased vascular permeability) associated with Neisseria meningitidis infection can be related to the large amounts of endotoxin released during infection.
Immunopathogenesis
In many cases, the symptoms of a bacterial infection are produced by excessive innate, immune, and inflammatory responses triggered by the infection. When limited and controlled, the acute-phase response to cell wall components is a protective antibacterial response. However, these responses also cause fever and malaise, and when systemic and out of control, the acute-phase response and inflammation can cause life-threatening symptoms associated with sepsis and meningitis (see Figure 14-4). Activated neutrophils, macrophage, and complement can cause damage at the site of the infection. Activation of complement can also cause release of anaphylotoxins that initiate vascular permeability and capillary breakage. Cytokine storms generated by superantigens and endotoxin can cause shock and disruption of body function. Granuloma formation induced by CD4 T cells and macrophages in response to Mycobacterium tuberculosis can also lead to tissue destruction. Autoimmune responses can be triggered by bacterial proteins, such as the M protein of S. pyogenes, which antigenically mimics heart tissue. The anti-M protein antibodies cross-react with and can initiate damage to the heart to cause rheumatic fever. Immune complexes deposited in the glomeruli of the kidney cause poststreptococcal glomerulonephritis. For Chlamydia, Treponema (syphilis), Borrelia (Lyme disease), and other bacteria, the host immune response is the principal cause of disease symptoms in patients.
Mechanisms for Escaping Host Defenses
Bacteria are parasites, and evasion of host protective responses is a selective advantage. Logically, the longer a bacterial infection remains in a host, the more time the bacteria have to grow and also cause damage. Therefore bacteria that can evade or incapacitate the host defenses have a greater potential for causing disease. Bacteria evade recognition and killing by phagocytic cells, inactivate or evade the complement system and antibody, and even grow inside cells to hide from host responses (Box 14-4).
The capsule is one of the most important virulence factors (Box 14-5). These slime layers function by shielding the bacteria from immune and phagocytic responses. Capsules are typically made of polysaccharides, which are poor immunogens. The S. pyogenes capsule, for example, is made of hyaluronic acid, which mimics human connective tissue, thereby masking the bacteria and keeping them from being recognized by the immune system. The capsule also acts like a slimy football jersey, in that it is hard to grasp and tears away when grabbed by a phagocyte. The capsule also protects a bacterium from destruction within the phagolysosome of a macrophage or leukocyte. All of these properties can extend the time bacteria spend in blood (bacteremia) before being eliminated by host responses. Mutants of normally encapsulated bacteria that lose the ability to make a capsule also lose their virulence; examples of such bacteria are Streptococcus pneumoniae and N. meningitidis. A biofilm, which is made from capsular material, can prevent antibody and complement from getting to the bacteria.
Box 14-5
Examples of Encapsulated Microorganisms
Bacteria can evade antibody responses by antigenic variation, by inactivation of antibody or by intracellular growth. N. gonorrhoeae can vary the structure of surface antigens to evade antibody responses and also produces a protease that degrades immunoglobulin A (IgA). S. aureus makes an IgG-binding protein, protein A, which prevents antibody from activating complement or being an opsonin and masks the bacteria from detection. Bacteria that grow intracellularly include mycobacteria, francisellae, brucellae, chlamydiae, and rickettsiae (Box 14-6). Unlike most bacteria, control of these infections requires T-helper cell immune responses to activate macrophages to kill or create a wall (granuloma) around the infected cells (as for M. tuberculosis).
Bacteria evade complement action by preventing access of the components to the membrane, masking themselves, and by inhibiting activation of the cascade. The thick peptidoglycan of gram-positive bacteria and the long O antigen of LPS of most gram-negative bacteria (not Neisseria species) prevent the complement from gaining access and protects the bacterial membrane from being damaged. By degrading the C5a component of complement, S. pyogenes can limit the chemotaxis of leukocytes to the site of infection. To compensate for the lack of O antigen, N. gonorrhoeae attaches sialic acid to its lipooligosaccharide (LOS) to inhibit complement activation.
Phagocytes (neutrophil, macrophage) are the most important antibacterial defense, but many bacteria can circumvent phagocytic killing in various ways. They can produce enzymes capable of lysing phagocytic cells (e.g., the streptolysin produced by S. pyogenes or the α-toxin produced by C. perfringens). They can inhibit phagocytosis (e.g., the effects of the capsule and the M protein produced by S. pyogenes) or block intracellular killing. Bacterial mechanisms for protection from intracellular killing include blocking phagolysosome fusion to prevent contact with its bactericidal contents (Mycobacterium species), capsule-mediated or enzymatic resistance to the bactericidal lysosomal enzymes or substances, and the ability to exit the phagosome into the host cytoplasm before being exposed to lysosomal enzymes (Table 14-4 and Figure 14-5). Production of catalase by staphylococci can break down the hydrogen peroxide produced by the myeloperoxidase system. Many of the bacteria that are internalized but survive phagocytosis can use the cell as a place to grow and hide from immune responses and as a means of being disseminated throughout the body.
Table 14-4 Methods That Circumvent Phagocytic Killing
| Method | Example |
|---|---|
| Inhibition of phagolysosome fusion | Legionella spp., Mycobacterium tuberculosis, Chlamydia spp. |
| Resistance to lysosomal enzymes | Salmonella typhimurium, Coxiella spp., Ehrlichia spp., Mycobacterium leprae, Leishmania spp. |
| Adaptation to cytoplasmic replication | Listeria, Francisella, and Rickettsia spp. |
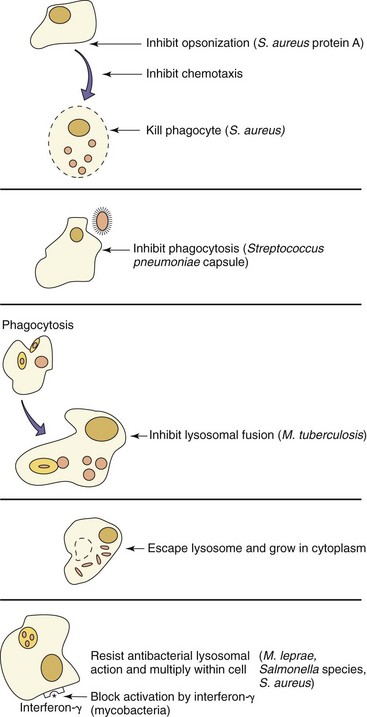
Figure 14-5 Bacterial mechanisms for escaping phagocytic clearance. Selected examples of bacteria that use the indicated antiphagocytic mechanisms are given.
S. aureus can also escape host defenses by walling off the site of infection. S. aureus can produce coagulase, an enzyme that promotes the conversion of fibrin to fibrinogen to produce a clotlike barrier; this feature distinguishes S. aureus from S. epidermidis. S. aureus and S.pyogenes and other bacteria are pyogenic (pus formers), and pus formation upon the death of neutrophils limits antibody or antibiotic access to the bacteria. M. tuberculosis is able to survive in a host by promoting the development of a granuloma, within which viable bacteria may reside for the life of the infected person. The bacteria may resume growth if there is a decline in the immune status of the person.
Summary
The primary virulence factors of bacteria are the capsule, adhesins, invasins, degradative enzymes, toxins, and mechanisms for escaping elimination by host defenses. Bacteria may only have one virulence mechanism. For example, C. diphtheriae has only one virulence mechanism, which is diphtheria toxin. Other bacteria express many virulence factors. S. aureus is an example of such a bacterium; it expresses adhesins, degradative enzymes, toxins, catalase, and coagulase, which are responsible for producing a spectrum of diseases. In addition, different strains within a bacterial species may express different virulence mechanisms. For example, the symptoms and sequelae of gastroenteritis (diarrhea) caused by E. coli may include invasion and bloody stools, cholera-like watery stools, and even severe hemorrhagic disease, depending on the specific infecting strain.
1. Name three routes by which exogenous pathogens can infect a person. List five examples of organisms that use each route.
2. How are microbes able to resist immunologic clearance? Give at least one specific example of each mechanism.
3. What are the two general types of exotoxins? List examples of each type.
1. (1) Ingestion. Examples: Salmonella, Shigella, Bacillus cereus, E. coli, Vibrio species
(2) Inhalation. Examples: Mycobacterium species, Mycoplasma pneumoniae, Legionella species, Bordetella, Streptococcus, Chlamydia pneumoniae
(3) Arthropod bite. Examples: Rickettsia, Ehrlichia, Coxiella, Francisella, Borrelia burgdorferi
2. Encapsulation. Example: antiphagocytic: Streptococcus pneumoniae
Intracellular growth. Example: Francisella tularensis
Antiimmunoglobulin proteases. Example: Neisseria gonorrhoeae
IgG binding proteins. Example: Staphylococcus protein A
Inhibition of phagolysosome fusion. Example: Legionella, Mycobacterium tuberculosis
Resistence to lysosomal enzymes. Example: Salmonella typhimurium
3. (1) Degradative enzymes. Example: α-toxin (phospholipase C from C. perfringens)
(2) A-B toxins. Example: tetanus toxoid
(3) Superantigens: toxic shock syndrome toxin from S. aureus