10 Immune Responses to Infectious Agents
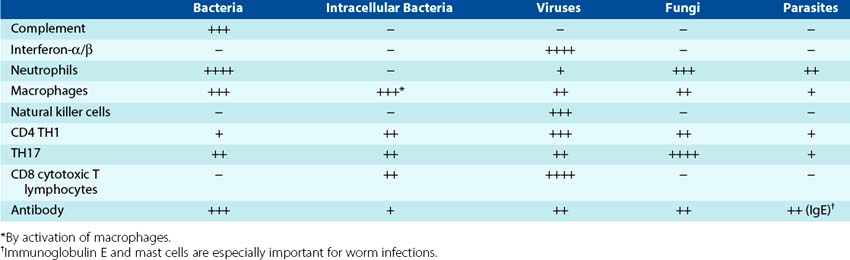
The previous chapters in this section introduced the different immunologic actors and their characteristics. This chapter describes the different roles they play in host protection from infection, their interactions, and the immunopathogenic consequences that may arise as a result of the response (Box 10-1). Most infections are controlled by innate responses before immune responses can be initiated, but immune responses are necessary to resolve the more troublesome infections. The importance of each of the components of the host response differs for different infectious agents (Table 10-1), and their importance becomes obvious when it is genetically deficient or is inhibited by chemotherapy, disease, or infection (e.g., acquired immunodeficiency syndrome [AIDS]).
Box 10-1
Summary of the Immune Response
The drama of the host response to infection unfolds in several acts after an infectious challenge, with certain differences depending upon the microbial villain. The actors include cells of the innate response, including neutrophils; monocyte-macrophage lineage cells, immature dendritic (iDCs), and dendritic cells (DCs); natural killer (NK) cells; the T and B lymphocytes of the antigen-specific response; and other cells. These cells are distinguished by their outer structures, their costumes, which also define their roles in the immune response. Act 1 starts at the site of infection and involves innate responses. Activation of complement releases the “a” fragments, C3a, C4a, and C5a, which attract the actors to the site of infection. Neutrophils and, later, activated macrophages act directly on bacteria and infection. Type 1 interferons limit virus replication, activate NK cells, and also facilitate the development of subsequent T-cell responses. The NK cells provide early responses to infection and kill virally infected and tumor cells. The NK cells return in Act 2 to kill cells decorated with antibody (antibody-dependent cellular cytotoxicity [ADCC]). DCs bridge the gap between the innate and the antigen-specific protective responses by first producing cytokines to enhance the action and then by taking their phagocytosed and pinocytosed cargo to the lymph node as the only antigen-presenting cell (APC) that can initiate an immune response. Act 2 commences in the lymph node, where the mature DCs present antigen to the T lymphocytes. The plot of this story may proceed to reinforce local-site inflammatory responses (TH17, TH1) or initiate systemic, humoral responses (TH2), depending on the cytokine dialogue of the DC and the T cell. The T cells play a central role in activating and controlling (helping) immune and inflammatory responses through the release of cytokines. In Act 3, the cast of T cells and B cells increase in number and terminally differentiate into effector and plasma cells to deliver antigen-specific cellular and antibody immune responses. Macrophages and B cells refine and strengthen the direction of the response as APCs. Certain members of the B- and T-cell cast maintain a low profile and become memory cells to be able to replay the drama more quickly and efficiently in the future. Specific cellular actors, the receptor-ligand interactions between the actors, and the cytokine dialogue determine the drama that unfolds during the immune response.
Human beings have three basic lines of protection against infection by microbes to block entry, spread in the body, and inappropriate colonization.
1. Natural barriers, such as skin, mucus, ciliated epithelium, gastric acid, and bile, restrict entry of the agent.
2. Innate, antigen-nonspecific immune defenses such as fever, interferon, complement, neutrophils, macrophages, dendritic cells (DCs), and natural killer (NK) cells provide rapid local responses to act at the infection site in order to restrict the growth and spread of the agent.
3. Adaptive, antigen-specific immune responses, such as antibody and T cells, reinforce the innate protections and specifically target, attack, and eliminate the invaders that succeed in passing the first two defenses.
Usually, barrier functions and innate responses are sufficient to control most infections before symptoms or disease occurs. Initiation of a new antigen-specific immune response takes time, and infections can grow and spread during this time period. Prior immunity and immune memory elicited by infection or vaccination can activate quickly enough to control most infections.
Antibacterial Responses
Figure 10-1 illustrates the progression of protective responses to a bacterial challenge. Protection is initiated by activation of innate and inflammatory responses on a local basis and progresses to acute-phase and antigen-specific responses on a systemic scale. The response progresses from soluble antibacterial factors (peptides and complement) to cellular responses and then soluble antibody responses. The most important antibacterial host response is phagocytic killing by neutrophils and macrophages. Complement and antibody facilitate the uptake of microbes by phagocytes and TH17, and TH1 CD4 T-cell responses enhance and regulate their function. A summary of antibacterial responses is presented in Box 10-2.
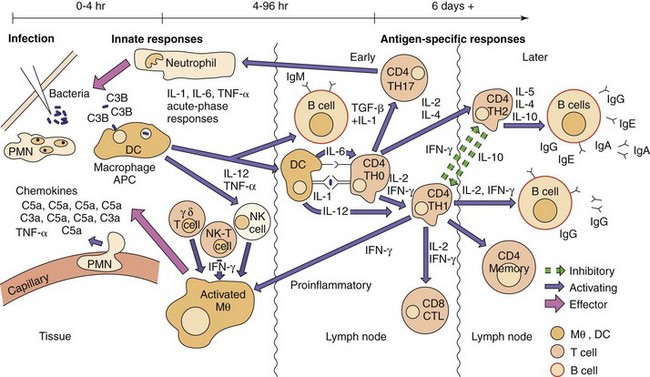
Figure 10-1 Antibacterial responses. First, innate antigen-nonspecific responses attract and promote polymorphonuclear neutrophil (PMN) and macrophage (Mθ) responses. Dendritic cells (DCs) and antigen reach the lymph node to activate early immune responses (TH17, TH1, and IgM). Later, TH2 systemic antibody responses and memory cells are developed. The time course of events is indicated at the top of the figure. APC, Antigen-presenting cell; CTL, cytotoxic T lymphocyte; IFN-γ, interferon-γ; IL, interleukin; TGF-β, transforming growth factor-β; TH, T helper (cell); TNF-α, tumor necrosis factor-α.
Box 10-2
Summary of Antibacterial Responses
Initiation of the Response
Once past the barriers, bacterial cell surfaces activate the alternative or lectin pathways of complement that are present in interstitial fluids and serum. The complement system (see Chapter 8) is a very early and important antibacterial defense. The alternative complement pathway (properdin) can be activated by teichoic acid, peptidoglycan, and lipopolysaccharide (LPS) in the absence of antibody and, with mannose-binding protein, can activate the lectin complement pathway. Later, when immunoglobulin (Ig) M or IgG is present, the classical complement pathway is activated. All three pathways converge to generate a C3 convertase to cleave C3 into C3a, C3b, and C3d and the C5 convertase to produce C5a. The “a” fragments activate, attract, and promote anaphylaxis by recruiting neutrophils and macrophages to the site of infection. C3b promotes its phagocytosis as an opsonin. The membrane attack complex (MAC) can directly kill gram-negative bacteria and, to a much lesser extent, gram-positive bacteria (the thick peptidoglycan of gram-positive bacteria shields them from the components). Neisseria are especially sensitive to complement lysis due to the truncated structure of lipooligosaccharide in the outer membrane. Complement facilitates elimination of all bacteria by producing
1. Chemotactic factors (C5a) to attract neutrophils and macrophages to the site of infection
2. Anaphylotoxins (C3a, C4a, and C5a) to stimulate mast cell release of histamine and thereby increase vascular permeability, allowing access to the infection site
3. Opsonins (C3b), which bind to bacteria and promote their phagocytosis
Bacterial cell wall molecules (teichoic acid and peptidoglycan fragments of gram-positive bacteria and lipid A of LPS of gram-negative bacteria) also activate pathogen-associated molecular pattern (PAMP) receptors, including the cell surface Toll-like receptors (TLRs) and the cytoplasmic peptidoglycan receptors—nucleotide-binding oligomerization domain protein (NOD)1, NOD2, and cryopyrin (Box 10-3). Lipid A (endotoxin) binds to TLR4 and other PAMP receptors and is a very strong activator of DCs, macrophages, B cells, and selected other cells (e.g., epithelial and endothelial cells). Binding of these PAMPs to receptors on epithelial cells, macrophages, Langerhans cells, and DCs activate kinase cascades that activate the inflammasome and also promote cytokine production (including the acute-phase cytokines, interleukin (IL)-1, IL-6, and tumor necrosis factor [TNF]), protective responses, and maturation of DCs. The inflammasome promotes the cleavage of IL-1β and IL-18 to reinforce local inflammation. NK cells, NKT cells, and γ/δ T cells residing in tissue also respond, produce cytokines, and reinforce cellular responses.
Box 10-3
Bacterial Components That Activate Protective Responses
IL-1 and TNF-α enhance the inflammatory response by locally stimulating changes in the tissue, promoting diapedesis of neutrophils and macrophages to the site, and activating these cells and activating systemic responses. IL-1 and TNF-α are endogenous pyrogens, inducing fever, and also induce the acute-phase response. The acute-phase response can also be triggered by inflammation, tissue injury, prostaglandin E2, and interferons associated with infection. The acute-phase response promotes changes that support host defenses and include fever, anorexia, sleepiness, metabolic changes, and production of proteins. Acute-phase proteins that are produced and released into the serum include C-reactive protein, complement components, coagulation proteins, LPS-binding proteins, transport proteins, protease inhibitors, and adherence proteins. C-reactive protein complexes with the polysaccharides of numerous bacteria and fungi and activates the complement pathway, facilitating removal of these organisms from the body through greater phagocytosis. The acute-phase proteins reinforce the innate defenses against infection.
Immature DCs (iDCs), macrophages, and other cells of the macrophage lineage will produce IL-23 and IL-12 in addition to the acute-phase cytokines. IL-12 activates NK cells at the site of infection, which can produce interferon-γ (IFN-γ) to further activate macrophages and DCs. IL-12 and IL-23 activate TH1 and TH17 immune responses, respectively, to reinforce macrophages and neutrophil function. Epithelial cells also respond to PAMPs and release cytokines to promote natural protections.
These actions initiate local, acute inflammation. Expansion of capillaries and increased blood flow brings more antimicrobial agents to the site. Increase in permeability and alteration of surface molecules of the microvasculature structure allows access for fluid, plasma proteins, and attract and facilitate leukocyte entry into the site of infection. Kinins and clotting factors induced by tissue damage (e.g., factor XII [Hageman factor], bradykinin, fibrinopeptides) are also involved in inflammation. These factors increase vascular permeability and are chemotactic for leukocytes. Products of arachidonic acid metabolism also affect inflammation. Cyclooxygenase-2 (COX-2) and 5-lipooxygenase convert arachidonic acid to prostaglandins and leukotrienes, respectively, which can mediate essentially every aspect of acute inflammation. The course of inflammation can be followed by rapid increases in serum levels of acute-phase proteins, especially C-reactive protein (which can increase a thousand fold within 24 to 48 hours) and serum amyloid A. Although these processes are beneficial, they also cause pain, redness, heat, and swelling and promote tissue damage. Tissue damage is caused to some extent by complement and macrophages but mostly by neutrophils. When triggered at a systemic level, these same functions can lead to septic shock, due in large part to the leakage of large amounts of fluid into tissue.
Phagocytic Responses
C3a, C5a, bacterial products (e.g., formyl-methionyl-leucyl-phenylalanine [f-met-leu-phe]), and chemokines produced by epithelial cells, Langerhans cells, and other cells in skin and mucous epithelium are powerful chemoattractants for neutrophils, macrophages, and later in the response, lymphocytes. The chemokines and tumor necrosis factor-α (TNF-α) cause the endothelial cells lining the capillaries (near the inflammation) and the leukocytes passing by to express complementary adhesion molecules (molecular “Velcro”) to promote diapedesis (see Figure 8-7). Polymorphonuclear neutrophils (PMNs), monocytes, and occasionally eosinophils are the first cells to arrive at the site in response to infection; they are followed later by macrophages. Recruitment of immature band forms of neutrophils from the bone marrow during infection is indicated by a “left shift” in the complete blood count. Neutrophils are recruited and activated by the TH17 response and macrophages, and DCs are activated by IFN-γ produced by NK cells and CD4 TH1 T cells.
Bacteria are bound to the neutrophils and macrophages with receptors for bacterial carbohydrates (lectins [specific sugar-binding proteins]), fibronectin receptors (especially for Staphylococcus aureus), and receptors for opsonins, including complement (C3b), C-reactive protein, mannose-binding protein, and the Fc portion of antibody. The microbes are internalized in a phagocytic vacuole that fuses with primary lysosomes (macrophages) or granules (PMNs) to allow inactivation and digestion of the vacuole contents. Phagocytic killing may be oxygen dependent or oxygen independent, depending on the antimicrobial chemicals produced by the granules (see Figure 8-8 and Box 8-5).
In the neutrophil, microorganisms are killed by hydrogen peroxide and superoxideion produced by nicotinamide adenine dinucleotide phosphate reduced (NADPH) oxidase and hypochlorous ions generated by myeloperoxidase. Nitric oxide produced by neutrophils and activated macrophages has antimicrobial activity and is also a major second messenger molecule (like cyclic adenosine monophosphate [cAMP]) that enhances the inflammatory and other responses. Oxygen-independent killing in the neutrophils occurs upon fusion of the phagosome with azurophilic granules containing cationic proteins (e.g., cathepsin G) and specific granules containing lysozyme and lactoferrin. These proteins kill gram-negative bacteria by disrupting their cell membrane integrity, but they are far less effective against gram-positive bacteria, which are killed principally through the oxygen-dependent mechanism.
The neutrophils contribute to the inflammation in several ways. Prostaglandins and leukotrienes are released and increase vascular permeability, cause swelling (edema) and stimulate pain receptors. In addition, during phagocytosis, the granules may leak their contents to cause tissue damage. The neutrophils have short lives, and dead neutrophils produce pus.
In contrast to neutrophils, macrophages have long lives, but the cells must be activated (made angry) with IFN-γ (best) in order to kill phagocytized microbes. Granulocyte-macrophage colony-stimulating factor (GM-CSF), TNF-α, and lymphotoxin (TNF-β) maintain the antimicrobial action. Early in the infection IFN-γ is produced by NK and NKT cells and later by CD4 T cells. In addition to the tissue macrophages, splenic macrophages are important for clearing bacteria, especially encapsulated bacteria, from blood. Asplenic (congenitally or surgically) individuals are highly susceptible to pneumonia, meningitis, and other manifestations of Streptococcus pneumoniae, Neisseria meningitidis, and other encapsulated bacteria.
Antigen-Specific Response to Bacterial Challenge
On ingestion of bacteria and after stimulation of TLRs by bacterial components, Langerhans cells and iDCs become mature, cease to phagocytize, and move to the lymph nodes to process and deliver their internalized antigen for presentation to T cells (Figure 10-2). Antigenic peptides (having more than 11 amino acids) produced from phagocytosed proteins (exogenous route) are bound to class II major histocompatibility complex (MHC) molecules and presented by these antigen-presenting cells (APCs) to naïve CD4 TH0 T cells. The CD4 T cells are activated by a combination of (1) antigenic peptide in the cleft of the MHC II molecule with the T-cell antigen receptor (TCR) and with CD4, (2) co-stimulatory signals provided by the interaction of B7 molecules on the DC with CD28 molecules on the T cells, and (3) IL-6, and other cytokines produced by the DC. The TH0 cells produce IL-2, IFN-γ, and IL-4. Simultaneously, bacterial molecules with repetitive structures (e.g., capsular polysaccharide) interact with B cells expressing surface IgM and IgD specific for the antigen and activate the cell to grow and produce IgM. Microbial cell wall polysaccharides, especially LPS and also the C3d component of complement, activate B cells and promote the specific IgM antibody responses. Swollen lymph nodes are an indication of lymphocyte activation in response to antigenic challenge.
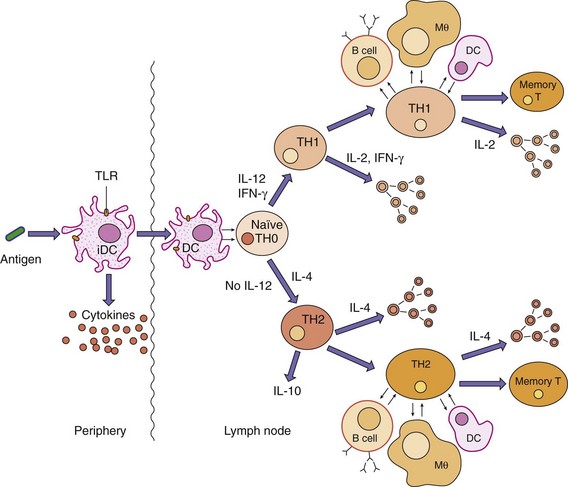
Figure 10-2 Initiation and expansion of specific immune responses. Immature dendritic cells (iDCs) at the site of infection acquire bacteria and debris, bacterial components activate the cell through Toll-like receptors (TLRs), and then dendritic cells (DCs) mature, move to the lymph node, and present antigen to naïve T cells to initiate the antigen-specific response. During a secondary or memory response, B cells, macrophages, and DCs can present antigen to initiate the response. IL, Interleukin; IFN-γ, interferon-γ; Mθ, macrophage; TH, T helper (cell).
Early responses are also provided by γ/δ T cells, NKT cells and innate lymphoid cells (including NK cells). γ/δ T cells in tissue and in the blood sense phosphorylated amine metabolites from some bacteria (Escherichia coli, mycobacteria) but not others (streptococci, staphylococci). DCs can present bacterial glycolipids to activate NKT cells. These T cells and innate lymphoid cells produce IFN-γ, which activate macrophages and DCs to enforce local cellular inflammatory reactions.
The conversion of TH0 cells to TH17 and TH1 cells initiates the expansion of the host response. Acute-phase cytokines IL-1 and TNF-α together with TGF-β promote the development of CD4 TH17 T cells. TH17 cells produce IL-17 and TNF-α to activate epithelial cells and neutrophils and also promote production of antimicrobial peptides. TH17 responses are important for early antibacterial responses and antimycobacterial responses. A balance of TH17 and Treg responses are also important to regulate the populations of intestinal flora.
DCs producing IL-12 promote TH1 responses. CD4 TH1 T cells (1) promote and reinforce inflammatory responses (e.g., IFN-γ activation of macrophage) and growth of T and B cells (IL-2) to expand the immune response, and (2) promote B cells to produce complement-binding antibodies (IgM, IgG upon class switching). These responses are important for the early phases of an antibacterial defense. TH1 responses are also essential for combating intracellular bacterial infections and mycobacteria, which are hidden from antibody. IFN-γ activates macrophage to kill the phagocytized microbe. Chronic stimulation of CD4 TH1 T cells by macrophages expressing microbial (mycobacterial or histoplasmic) antigen and production of IFN-γ may cause the transformation of other macrophages into epithelioid cells and giant cells, which can surround the infection and produce a granuloma. CD8 T cells are not very important for antibacterial immunity.
CD4 TH2 T-cell responses occur in the absence of IL-12 at more distant lymph nodes. These responses are also initiated by DCs and are sustained by the B-cell presentation of antigen. Binding of antigen to the cell surface antibody on B cells activates the B cells and also promotes uptake, processing of the antigen, and presentation of antigenic peptides on class II MHC molecules to the CD4 TH2 cell. The TH2 cell produces IL-4, IL-5, IL-6, IL-10, and IL-13, which enhance IgG production and, depending on other factors, the production of IgE or IgA. The TH2 response also promotes terminal differentiation of B cells to plasma-cell antibody factories.
CD4+CD25+ regulatory T cells (Treg) prevent spurious activation of naïve T cells, curtail both TH1 and TH2 responses, and promote the development of some of the antigen-specific cells into memory T cells. Only DCs can override the Treg block to naïve T cell activation.
Antibodies are the primary protection against extracellular bacteria and reinfection and promotes the clearance and prevents the spread of bacteria in the blood. Antibody promotes complement activation, opsonizes bacteria for phagocytosis, blocks bacterial adhesion, and neutralizes (inactivates) exotoxins (e.g., tetanospasmin, botulinum toxin) and other cytotoxic proteins produced by bacteria (e.g., degradative enzymes). Vaccine immunization with inactivated exotoxins (toxoids) is the primary means of protection against the potentially lethal effects of exotoxins.
IgM antibodies are produced early in the antibacterial response. IgM bound to bacteria activates the classical complement cascade, promoting both the direct killing of gram-negative bacteria and the inflammatory responses. IgM is usually the only antibody produced against capsular carbohydrates. The large size of IgM limits its ability to spread into the tissue. Later in the immune response, T-cell help promotes differentiation of the B cell and immunoglobulin class switching to produce IgG. IgG antibodies are the predominant antibody, especially on rechallenge. IgG antibodies fix complement and promote phagocytic uptake of the bacteria through Fc receptors on macrophages. The production of IgA requires TH2 cytokines and other factors. IgA is the primary secretory antibody and is important for protecting mucosal membranes. Secretory IgA acquires the secretory component that promotes interaction and passage of IgA through mucosal epithelial cells. IgA neutralizes the binding of bacteria and their toxins at epithelial cell surfaces.
A primary antigen-specific response to bacterial infection takes at least 5 to 7 days. Movement of the DC to the lymph node may take 1 to 3 days, followed by activation, expansion, and maturation of the response. On rechallenge to infection, long-lived plasma cells may still be producing antibody. Memory T cells can respond quickly to antigen presentation by DC, macrophage, or B cells, not just DC; memory B cells are present to respond quickly to antigen; and the secondary antibody response occurs within 2 to 3 days.
Intestinal Immune Responses
The intestinal flora is constantly interacting with and being regulated by the innate and immune systems of the gut-associated lymphoid tissue. Similarly, the immune response is shaped by its interaction with intestinal flora as regulatory cells limit the development of autoimmune responses and inflammation. DCs, innate lymphoid cells, Treg, TH17, TH1, and other T cells and B cells in Peyer patches and intestinal lymphoid follicles monitor the bacteria within the gut. These cells and epithelial and other cells lining the gut produce antimicrobial peptides and plasma cells secrete IgA into the gut to maintain a healthy mixture of bacteria. At the same time, regulatory cells prevent the development of detrimental or excessive immune responses to the contents of the gut. Alterations in the microbial flora or its interaction with the innate and immune cells can disrupt the system and result in inflammatory bowel diseases. For example, the absence or a mutation in the NOD2 receptor for peptidoglycan enhances chances for certain types of Crohn disease.
Bacterial Immunopathogenesis
Activation of the inflammatory and acute-phase responses can initiate significant tissue and systemic damage. Activation of macrophages and DCs in the liver, spleen, and blood by endotoxin can promote release of TNF-α into the blood, causing many of the symptoms of sepsis, including hemodynamic failure, shock, and death (see Cytokine Storm section and Chapter 14). Although IL-1, IL-6, and TNF-α promote protective responses to a local infection, these same responses can be life threatening when activated by systemic infection. Increased blood flow and fluid leakage can lead to shock when it occurs throughout the body. Antibodies produced against bacterial antigens that share determinants with human proteins can initiate autoimmune tissue destruction (e.g., antibodies produced in poststreptococcal glomerulonephritis and rheumatic fever). Nonspecific activation of CD4 T cells by superantigens (e.g., toxic shock syndrome toxin of S. aureus) promotes the production of large amounts of cytokines and, eventually, the death of large numbers of T cells. The sudden, massive release of cytokines (“cytokine storm”) can cause shock and severe tissue damage (e.g., toxic shock syndrome) (see Cytokine Storm section and Chapter 14).
Bacterial Evasion of Protective Responses
The mechanisms used by bacteria to evade host-protective responses are discussed in Chapter 14 as virulence factors. These mechanisms include (1) the inhibition of phagocytosis and intracellular killing in the phagocyte, (2) inactivation of complement function, (3) cleavage of IgA, (4) intracellular growth (avoidance of antibody), and (5) change in bacterial antigenic appearance. Some microorganisms, including but not limited to mycobacteria (also Listeria and Brucella species), survive and multiply within macrophages and use the macrophages as a protective reservoir or transport system to help spread the organisms throughout the body. However, cytokine-activated macrophages can kill the intracellular pathogens.
Antiviral Responses
Host Defenses against Viral Infection
The immune response is the best and, in most cases, the only means of controlling a viral infection (Figure 10-3; Box 10-4). Unfortunately, it is also the source of pathogenesis for many viral diseases. The humoral and cellular immune responses are important for antiviral immunity. The ultimate goal of the immune response in a viral infection is to eliminate both the virus and the host cells harboring or replicating the virus. Interferons, NK cells, CD4 TH1 responses, and CD8 cytotoxic killer T cells are more important for viral infections than for bacterial infections. Failure to resolve the infection may lead to persistent or chronic infection or death.
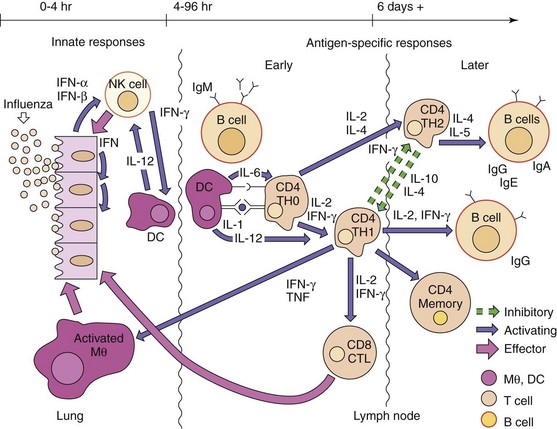
Figure 10-3 Antiviral responses. The response to a virus (e.g., influenza virus) initiates with interferon production and action and natural killer (NK) cells. Activation of antigen-specific immunity resembles the antibacterial response, except that CD8 cytotoxic T lymphocytes (CTLs) are important antiviral responses. The time course of events is indicated at the top of the figure. IFN, Interferon, IL, interleukin; Mθ, macrophage; TH, T helper (cell); TNF, tumor necrosis factor.
Box 10-4
Summary of Antiviral Responses
T Cells
T cells are essential for controlling enveloped and noncytolytic viral infections
T cells recognize viral peptides presented by MHC molecules on cell surfaces
Antigenic viral peptides (linear epitopes) can come from any viral protein (e.g., glycoproteins, nucleoproteins)
CD4 TH1 responses are more important than TH2 responses
CD8 cytotoxic T cells respond to viral peptide: class I MHC protein complexes on the infected cell surface
CD4 TH2 responses are important for the maturation of the antibody response
CD4 TH2 responses may be detrimental if they prematurely limit the TH1 inflammatory and cytolytic responses
Antibody
Antibody neutralizes extracellular virus:
Antibody opsonizes virus for phagocytosis
Antibody promotes killing of target cell by the complement cascade and antibody-dependent cellular cytotoxicity
Antibody resolves lytic viral infections
Antibody blocks viremic spread to target tissue
IgM is an indicator of recent or current infection
IgG is a more effective antiviral than IgM
Secretory IgA is important for protecting mucosal surfaces
Resolution requires elimination of free virus (antibody) and the virus-producing cell (viral or immune cell mediated lysis).
DC, Dendritic cell; IFN, interferon; Ig, immunoglobulin; MHC, major histocompatibility complex; NK, natural killer.
Innate Defenses
Body temperature, fever, interferons, other cytokines, the mononuclear phagocyte system, and NK cells provide a local rapid response to viral infection and also activate the specific immune defenses. Often the nonspecific defenses are sufficient to control a viral infection, thus preventing the occurrence of symptoms.
Viral infection can induce the release of cytokines (e.g., TNF, IL-1) and interferon from infected cells, iDCs, and macrophages. Viral RNA (especially dsRNA), DNA, and some viral glycoproteins are potent activators of TLRs, and viral nucleic acids can also trigger vesicular and cytoplasmic pathogen pattern receptors to initiate these interferon and cytokine responses. Interferons and other cytokines trigger early local and systemic responses. Induction of fever and stimulation of the immune system are two of these systemic effects.
Body temperature and fever can limit the replication of or destabilize some viruses. Many viruses are less stable (e.g., herpes simplex virus) or cannot replicate (rhinoviruses) at 37° C or higher.
Cells of the dendritic and mononuclear phagocyte system phagocytose the viral and cell debris from virally infected cells. Macrophages in the liver (Kupffer cells) and spleen rapidly filter many viruses from the blood. Antibody and complement bound to a virus facilitate its uptake and clearance by macrophages (opsonization). DCs and macrophages also present antigen to T cells and release IL-1, IL-12, and IFN-α to expand the innate and initiate the antigen-specific immune responses. Plasmacytoid DCs in the blood produce large amounts of IFN-α in response to a viremia. Activated macrophages can also distinguish and kill infected target cells.
NK cells are activated by IFNs-α and -β and IL-12 to kill virally infected cells. Viral infection may reduce the expression of MHC antigens to remove inhibitory signals or may alter the carbohydrates on cell surface proteins to provide cytolytic signals to the NK cell.
Interferon
Interferon was first described by Isaacs and Lindemann as a very potent factor that “interferes with” the replication of many different viruses. Interferon is the body’s first active defense against a viral infection, an “early warning system.” In addition to activating a target-cell antiviral defense to block viral replication, interferons activate the immune response and enhance T-cell recognition of the infected cell. Interferon is a very important defense against infection, but it is also a cause of the systemic symptoms associated with many viral infections, such as malaise, myalgia, chills, and fever (nonspecific flulike symptoms), especially during viremia. Type 1 interferon is also a factor in causing systemic lupus erythematosus.
Interferons comprise a family of proteins that can be subdivided according to several properties, including size, stability, cell of origin, and mode of action (Table 10-2). IFN-α and IFN-β are type I interferons that share many properties, including structural homology and mode of action. B cells, epithelial cells, monocytes, macrophages, and iDCs make IFN-α. Plasmacytoid DCs in blood produce large amounts in response to viremia. Fibroblasts and other cells make IFN-β in response to viral infection and other stimuli. IFN-λ (interferon lambda) is a type III interferon with activity similar to IFN-α and is important for antiinfluenza responses. IFN-γ is a type II interferon, a cytokine produced by activated T and NK cells that occurs later in the infection. Although IFN-γ inhibits viral replication, its structure and mode of action differ from those of the other interferons. IFN-γ is also known as macrophage activation factor and is the defining component of the TH1 response.
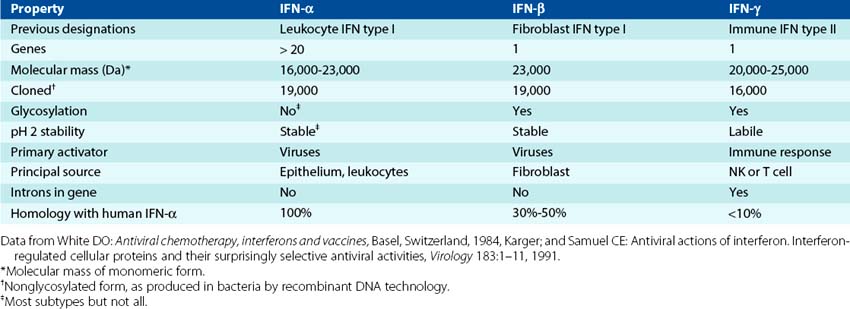
The best inducer of IFN-α and IFN-β production is dsRNA, produced as the replicative intermediates of RNA viruses or from the interaction of sense/antisense messenger RNAs (mRNAs) for some DNA viruses (Box 10-5). One dsRNA molecule per cell is sufficient to induce the production of interferon. Interaction of some enveloped viruses (e.g., herpes simplex virus and human immunodeficiency virus [HIV]) with iDCs can promote production of IFN-α. Alternatively, inhibition of protein synthesis in a virally infected cell can decrease the production of a repressor protein of the interferon gene, allowing production of interferon. Nonviral interferon inducers include the following:
1. Intracellular microorganisms (e.g., mycobacteria, fungi, protozoa)
2. Activators of certain TLRs or mitogens (e.g., endotoxins, phytohemagglutinin)
3. Double-stranded polynucleotides (e.g., poly I:C, poly dA:dT)
4. Synthetic polyanion polymers (e.g., polysulfates, polyphosphates, pyran)
5. Antibiotics (e.g., kanamycin, cycloheximide)
6. Low-molecular-weight synthetic compounds (e.g., tilorone, acridine dyes)
Box 10-5
Type I Interferons
Mechanism of Action
Initial infected cell or plasmacytoid dendritic cell releases interferon
Interferon binds to a specific cell surface receptor on another cell
Interferon induces the “antiviral state”:
Viral infection of the cell activates these enzymes
Protein synthesis inhibited to block viral replication
IFN-α, IFN-β, and IFN-λ can be induced and released within hours of infection (Figure 10-4). The interferon binds to specific receptors on the neighboring cells and induces the production of antiviral proteins–the antiviral state. However, these antiviral proteins are not activated until they bind dsRNA. The major antiviral effects of interferon are produced by two enzymes, 2′,5′-oligoadenylate synthetase (an unusual polymerase) and protein kinase R (PKR) (Figure 10-5), and for influenza, the mx protein is also important. Viral infection of the cell and production of dsRNA activate these enzymes and trigger a cascade of biochemical events that leads to (1) the inhibition of protein synthesis by PKR phosphorylation of an important ribosomal initiation factor (elongation initiation factor 2-α [eIF-2α]) and (2) the degradation of mRNA (preferentially, viral mRNA) by ribonuclease L, activated by 2′,5′-oligoadenosine. This process essentially puts the cellular protein synthesis factory “on strike” and prevents viral replication. It must be stressed that interferon does not directly block viral replication. The antiviral state lasts for 2 to 3 days, which may be sufficient for the cell to degrade and eliminate the virus without being killed.
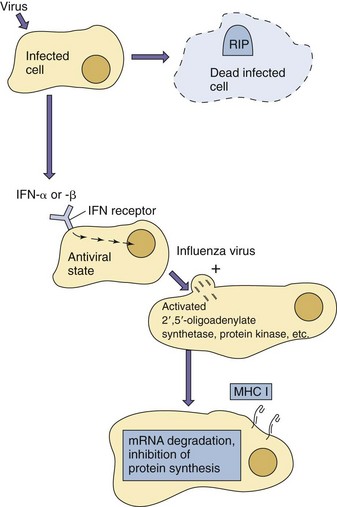
Figure 10-4 Induction of the antiviral state by interferon (IFN)-α or IFN-β. Interferon is produced in response to viral infection but does not affect the initially infected cell. The interferon binds to a cell surface receptor on other cells and induces production of antiviral enzymes (antiviral state). The infection and production of double-stranded RNA activates the antiviral activity. MHC I, Major histocompatibility antigen type 1.
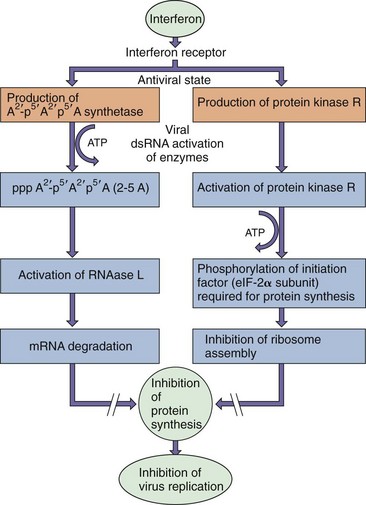
Figure 10-5 The two major routes for interferon inhibition of viral protein synthesis. One mechanism involves the induction of an unusual polymerase (2′,5′-oligoadenylate synthetase [2-5A]) that is activated by double-stranded RNA (dsRNA). The activated enzyme synthesizes an unusual adenine chain with a 2′,5′-phosphodiester linkage. The oligomer activates RNAase L that degrades messenger RNA (mRNA). The other mechanism involves the induction of protein kinase R (PKR), which prevents assembly of the ribosome by phosphorylation of the elongation initiation factor (eIF-2α) to prevent initiation of protein synthesis from capped mRNAs. ATP, Adenosine triphosphate.
Interferons stimulate cell-mediated immunity by activating effector cells and enhancing recognition of the virally infected target cell. Type I IFNs activate NK cells and assist in activation of CD8 T cells. IFN and activated NK cells provide an early, local, natural defense against virus infection. IFN-α and IFN-β increase the expression of class I MHC antigens, enhancing the cell’s ability to present antigen and making the cell a better target for cytotoxic T cells (CTLs). Activation of macrophages by IFN-γ promotes production of more IFN-α and IFN-β, secretion of other biologic response modifiers, phagocytosis, recruitment, and inflammatory responses. IFN-γ increases the expression of class II MHC antigens on the macrophage to help promote antigen presentation to T cells. Interferon also has widespread regulatory effects on cell growth, protein synthesis, and the immune response. All three interferon types block cell proliferation at appropriate doses.
Genetically engineered recombinant interferon is being used as an antiviral therapy for some viral infections (e.g., human papilloma and hepatitis C viruses). Effective treatment requires the use of the correct interferon subtype(s) and its prompt delivery at the appropriate concentration. IFN-β is used for treatment of multiple sclerosis. Interferons have also been used in clinical trials for the treatment of certain cancers. However, interferon treatment causes flulike side effects, such as chills, fever, and fatigue.
Antigen-Specific Immunity
Humoral immunity and cell-mediated immunity play different roles in resolving viral infections (i.e., eliminating the virus from the body). Humoral immunity (antibody) acts mainly on extracellular virions, whereas cell-mediated immunity (T cells) is directed at the virus-producing cell.
Humoral Immunity
Practically all viral proteins are foreign to the host and are immunogenic (i.e., capable of eliciting an antibody response). However, not all immunogens elicit protective immunity.
Antibody blocks the progression of disease through the neutralization and opsonization of cell-free virus. Protective antibody responses are generated toward the viral capsid proteins of naked viruses and the glycoproteins of enveloped viruses that interact with cell surface receptors (viral attachment proteins). These antibodies can neutralize the virus by preventing viral interaction with target cells or by destabilizing the virus, thus initiating its degradation. Binding of antibody to these proteins also opsonizes the virus, promoting its uptake and clearance by macrophages. Antibody recognition of infected cells can also promote antibody-dependent cellular cytotoxicity (ADCC) by NK cells. Antibodies to other viral antigens may be useful for serologic analysis of the viral infection.
The major antiviral role of antibody is to prevent the spread of extracellular virus to other cells. Antibody is especially important in limiting the spread of the virus by viremia, preventing the virus from reaching the target tissue for disease production. Antibody is most effective at resolving cytolytic infections. Resolution occurs because the virus kills the cell factory and the antibody eliminates the extracellular virus. Antibody is the primary defense initiated by most vaccines.
T-Cell Immunity
T cell–mediated immunity promotes antibody and inflammatory responses (CD4 helper T cells) and kills infected cells (cytotoxic T cells [primarily CD8 T cells]). The CD4 TH1 response is generally more important than TH2 responses for controlling a viral infection, especially noncytolytic and enveloped viruses. CD8 killer T cells promote apoptosis in infected cells after their T-cell receptor binds to a viral peptide presented by a class I MHC protein. The peptides expressed on class I MHC antigens are obtained from viral proteins synthesized within the infected cell (endogenous route). The viral protein from which these peptides are derived may not elicit protective antibody (e.g., intracellular or internal virion proteins, nuclear proteins, improperly folded or processed proteins [cell trash]). For example, the matrix and nucleoproteins of the influenza virus and the infected cell protein 4 (ICP4) (nuclear) of herpes simplex virus are targets for CTLs but do not elicit protective antibody. An immune synapse formed by interactions of the TCR and MHC I, the co-receptors, and adhesion molecules creates a space into which perforin, a complement-like membrane pore former, and granzymes (degradative enzymes) are released to induce apoptosis in the target cell. Interaction of the Fas ligand protein on CD4 or CD8 T cells with the Fas protein on the target cell can also promote apoptosis. CTLs kill infected cells and, as a result, eliminate the source of new virus.
The CD8 T-cell response probably evolved as a defense against virus infection. Cell-mediated immunity is especially important for resolving infections by syncytia-forming viruses (e.g., measles, herpes simplex virus, varicella-zoster virus, HIV), which can spread from cell to cell without exposure to antibody; and by noncytolytic viruses (e.g., hepatitis A and measles viruses). CD8 T cells also interact with neurons to control, without killing, the recurrence of latent viruses (herpes simplex virus, varicella-zoster virus, and JC papillomaviruses).
Immune Response to Viral Challenge
Primary Viral Challenge
The innate host responses are the earliest responses to viral challenge and are often sufficient to limit viral spread (see Figure 10-3). The type 1 interferons produced in response to most viral infections initiates the protection of adjacent cells, enhances antigen presentation by increasing the expression of MHC antigens, and initiates the clearance of infected cells by activating NK cells and antigen-specific responses. Virus and viral components released from the infected cells are phagocytosed by and activate iDCs to produce cytokines and then move to the lymph nodes. Macrophages in the liver and spleen are especially important for clearing virus from the bloodstream (filters). These phagocytic cells degrade and process the viral antigens. DCs present the appropriate peptide fragments bound to class II MHC antigens to CD4 T cells and can also cross-present these antigens on MHC I molecules to CD8 T cells to initiate the response. The APCs also release IL-1, IL-6, and TNF and, with IL-12, promote activation of helper T cells and specific cytokine production (TH1 response). The type 1 interferons and these cytokines induce the prodromal flulike symptoms of many viral infections. The activated T cells move to the site of infection and B-cell areas of the lymph node, and macrophages and B cells present antigen and become stimulated by the T cells.
Antiviral antigen-specific responses are similar to antibacterial antigen-specific responses, except that the CD8 T cell plays a more important role. IgM is produced approximately 3 days after infection. Its production indicates a primary infection. IgG and IgA are produced 2 to 3 days after IgM. Secretory IgA is made in response to a viral challenge of mucosal surfaces at the natural openings of the body (i.e., eyes, mouth, and respiratory and gastrointestinal systems). Activated CD4 and CD8 T cells are present at approximately the same time as serum IgG. During infection, the number of CD8 T cells specific for antigen may increase 50,000 to 100,000 fold. The antigen-specific CD8 T cells move to the site of infection and kill virally infected cells. Recognition and binding to class I MHC viral-peptide complexes promotes apoptotic killing of the target cells, either through the release of perforin and granzymes (to disrupt the cell membrane) or through the binding of the Fas ligand with Fas on the target cell. Resolution of the infection occurs later, when sufficient antibody is available to neutralize all virus progeny or when cellular immunity has been able to reach and eliminate the infected cells. For the resolution of most enveloped and noncytolytic viral infections, TH1-mediated responses are required to kill the viral factory and resolve infection.
Viral infections of the brain and the eye can cause serious damage because these tissues cannot repair tissue damage and are immunologically privileged sites of the body. TH1 responses are suppressed to prevent the serious tissue destruction that accompanies extended inflammation. These sites depend on innate, cytokine, TH17, and antibody control of infection.
Cell-mediated and IgG immune responses do not arise until 6 to 8 days after viral challenge. For many viral infections, this is after innate responses have controlled viral replication. However, for other viral infections, this period allows the virus to expand the infection, spread through the body and infect the target tissue, and cause disease (e.g., brain: encephalitis, liver: hepatitis). The response to the expanded infection may require a larger and more intense immune response, which often includes the immunopathogenesis and tissue damage that cause disease symptoms.
Secondary Viral Challenge
In any war, it is easier to eliminate an enemy if its identity and origin are known and if establishment of its foothold can be prevented. Similarly, in the human body, prior immunity, established by prior infection or vaccination, allows rapid, specific mobilization of defenses to prevent disease symptoms, promote rapid clearance of the virus, and block viremic spread from the primary site of infection to the target tissue to prevent disease. As a result, most secondary viral challenges are asymptomatic. Antibody and memory B and T cells are present in an immune host to generate a more rapid and extensive anamnestic (booster) response to the virus. Secretory IgA is produced quickly to provide an important defense to reinfection through the natural openings of the body, but it is produced only transiently.
Host, viral, and other factors determine the outcome of the immune response to a viral infection. Host factors include genetic background, immune status, age, and the general health of the individual. Viral factors include viral strain, infectious dose, and route of entry. The time required to initiate immune protection, the extent of the response, the level of control of the infection, and the potential for immunopathology (see Chapter 45) resulting from the infection differ after a primary infection and a rechallenge.
Viral Mechanisms for Escaping the Immune Response
A major factor in the virulence of a virus is its ability to escape immune resolution. Viruses may escape immune resolution by evading detection, preventing activation, or blocking the delivery of the immune response. Specific examples are presented in Table 10-3. Some viruses even encode special proteins that suppress the immune response.
Table 10-3 Examples of Viral Evasion of Immune Responses
| Mechanism | Viral Examples | Action |
|---|---|---|
| Humoral Response | ||
| Hidden from antibody | Herpesviruses, retroviruses | Latent infection |
| Herpes simplex virus, varicella-zoster virus, paramyxoviruses, human immunodeficiency virus | Cell-to-cell infection (syncytia formation) | |
| Antigenic variation | Lentiviruses (human immunodeficiency virus) | Genetic change after infection |
| Influenza virus | Annual genetic changes (drift) Pandemic changes (shift) |
|
| Secretion of blocking antigen | Hepatitis B virus | Hepatitis B surface antigen |
| Decay of complement | Herpes simplex virus | Glycoprotein C, which binds and promotes C3 decay |
| Interferon | ||
| Block production | Hepatitis B virus | Inhibition of IFN transcription |
| Epstein-Barr virus | IL-10 analogue (BCRF-1) blocks IFN-γ production | |
| Block action | Adenovirus | Inhibits up-regulation of MHC expression, VA1 blocks double-stranded RNA activation of interferon- induced protein kinase (PKR) |
| Herpes simplex virus | Inactivates PKR and activates phosphatase (PP1) to reverse inactivation of initiation factor for protein synthesis | |
| Immune Cell Function | ||
| Impairment of DC function | Measles, hepatitis C | Induction of IFN-β, which limits DC function |
| Impairment of lymphocyte function | Herpes simplex virus | Prevention of CD8 T-cell killing |
| Human immunodeficiency virus | Kills CD4 T cells and alters macrophages | |
| Measles virus | Suppression of NK, T, and B cells | |
| Immunosuppressive factors | Epstein-Barr virus | BCRF-1 (similar to IL-10) suppression of CD4 TH1 helper T-cell responses |
| Decreased Antigen Presentation | ||
| Reduced class I MHC expression | Adenovirus 12 | Inhibition of class I MHC transcription; 19-kDa protein (E3 gene) binds class I MHC heavy chain, blocking translocation to surface |
| Cytomegalovirus | H301 protein blocks surface expression of β2-microglobulin and class I MHC molecules | |
| Herpes simplex virus | ICP47 blocks TAP, preventing peptide entry into ER and binding to class I MHC molecules | |
| Inhibition of Inflammation | ||
| Poxvirus, adenovirus | Blocking of action of IL-1 or tumor necrosis factor | |
DC, Dendritic cell; ER, endoplasmic reticulum; ICP47, infected cell protein 47; IFN, interferon; IL, interleukin; MHC I, major histocompatibility complex, antigen type 1; NK, natural killer; PMN, polymorphonuclear neutrophil; TAP, transporter associated with antigen production.
Viral Immunopathogenesis
The symptoms of many viral diseases are the consequence of cytokine action or overzealous immune responses. The flulike symptoms of influenza and any virus that establishes a viremia (e.g., arboviruses) are a result of the interferon and other cytokine responses induced by the virus. Antibody interactions with large amounts of viral antigen in blood, such as occurs with hepatitis B virus infection, can lead to immune complex diseases. The measles rash, the extensive tissue damage to the brain associated with herpes simplex virus encephalitis (-itis means “inflammation”), and the tissue damage and symptoms of hepatitis are a result of cell-mediated immune responses. The more aggressive NK-cell and T-cell responses of adults exacerbate some diseases that are benign in children, such as varicella-zoster virus, Epstein-Barr virus infectious mononucleosis, and hepatitis B infection. Yet, the lack of such a response in children makes them prone to chronic hepatitis B infection because the response is insufficient to kill the infected cells and resolve the infection. Virus infections may also provide the initial activation trigger that allows the immune system to respond to self-antigens and cause autoimmune diseases.
Specific Immune Responses to Fungi
The primary protective responses to fungal infection are initiated by fungal cell wall carbohydrates binding to TLRs and the dectin-1 lectin and is delivered by neutrophils, macrophages, and antimicrobial peptides produced by the neutrophils, epithelial, and other cells. CD4 T-cell TH17 and TH1 responses stimulate the neutrophil and macrophage responses. Patients deficient in neutrophils or these CD4 T cell-mediated responses (e.g., patients with AIDS) are most susceptible to fungal (opportunistic) infections. Defensins and other cationic peptides may be important for some fungal infections (e.g., mucormycosis, aspergillus), and nitric oxide may be important against Cryptococcus and other fungi. Antibody, as an opsonin, may facilitate clearance of the fungi.
Specific Immune Responses to Parasites
It is difficult to generalize about the mechanisms of antiparasitic immunity because there are many different parasites that have different forms and reside in different tissue locations during their life cycles (Table 10-4). Stimulation of CD4 TH1, TH17, CD8 T-cell, and macrophage responses are important for intracellular infections, and TH2 antibody responses are important for extracellular parasites in blood and fluids. IgE, eosinophil, and mast cell action are especially important for eliminating worm (cestode and nematode) infections. The efficiency of control of the infection may depend on which response is initiated in the host. Dominance of a TH2 response to Leishmania infections results in the inhibition of TH1 activation of macrophages, inability to clear intracellular parasites, and a poor outcome. This observation provided the basis for the discovery that TH1 and TH2 responses are separate and antagonistic. Parasites have developed sophisticated mechanisms for avoiding immune clearance and often establish chronic infections.
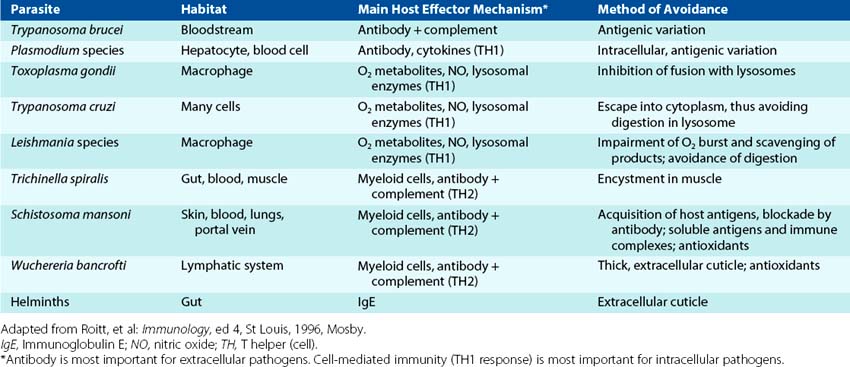
Extracellular parasites, such as Trypanosoma cruzi, Toxoplasma gondii, and Leishmania species, are phagocytosed by macrophage. Antibody may facilitate the uptake of (opsonize) the parasites. Killing of the parasites follows activation of the macrophage by IFN-γ (produced by NK, γ/δ T, or CD4 TH1 cells) or TNF-α (produced by other macrophages) and induction of oxygen-dependent killing mechanisms (peroxide, superoxide, nitric oxide). The parasites may replicate in the macrophage and hide from subsequent immune detection unless the macrophage is activated by TH1 responses.
TH1 production of IFN-γ and activation of macrophages are also essential for defense against intracellular protozoa and for the development of granulomas around Schistosoma mansoni eggs and worms in the liver. The granuloma, formed by layers of inflammatory cells, protects the liver from toxins produced by the eggs. However, the granuloma also causes fibrosis, which interrupts the venous blood supply to the liver, leading to hypertension and cirrhosis.
Neutrophils phagocytize and kill extracellular parasites through both oxygen-dependent and oxygen-independent mechanisms. Eosinophils localize near parasites, bind to IgG or IgE on the surface of larvae or worms (e.g., helminths, S. mansoni, and Trichinella spiralis), degranulate by fusing their intracellular granules with the plasma membrane, and release the major basic protein into the intercellular space. The major basic protein is toxic to the parasite.
For parasitic worm infections, cytokines produced by epithelial cells and CD4 TH2 T cells are very important for stimulating the production of IgE and the activation of mast cells (Figure 10-6). IgE bound to Fc receptors on mast cells targets the cells to antigens of the infecting parasite. In the lumen of the intestine, antigen binding and cross-linking of the IgE on the mast cell surface stimulate the release of histamine and substances toxic to the parasite and promote mucus secretion to coat and promote expulsion of the worm.
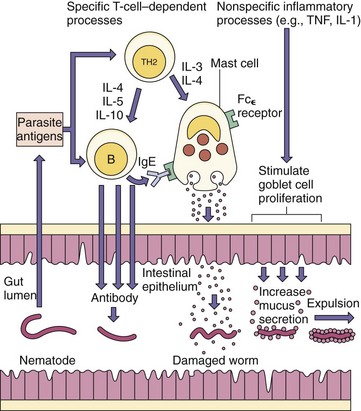
Figure 10-6 Elimination of nematodes from the gut. TH2 responses are important for stimulating the production of antibody. Antibody can damage the worm. Immunoglobulin E (IgE) is associated with mast cells, the release of histamine, and toxic substances. Increased mucus secretion also promotes expulsion. IL, Interleukin; TNF, tumor necrosis factor.
(From Roitt I, et al: Immunology, ed 4, St Louis, 1996, Mosby.)
IgG antibody also plays an important role in antiparasitic immunity, as an opsonin and by activating complement on the surface of the parasite.
Malaria poses an interesting challenge for the immune response. Protective antibodies are made toward attachment and other surface proteins, but these differ for each of the stages of the parasite’s development. TH1 responses and CTLs may be important during liver phases of infection. While in the erythrocyte, the parasite is hidden from antibody, unrecognizeable by CTLs but can stimulate NK- and NKT-cell responses. Cytokines, especially TNF-α, produced by these cells promote protection but also immunopathogenesis. Immune complexes containing malarial components and cell debris released upon erythrocyte lysis can clog small capillaries and activate type II hypersensititivity reactions (see later) and promote inflammatory tissue damage.
Evasion of Immune Mechanisms by Parasites
Animal parasites have developed remarkable mechanisms for establishing chronic infections in the vertebrate host (see Table 10-4). These mechanisms include intracellular growth, inactivation of phagocytic killing, release of blocking antigen (e.g., Trypanosoma brucei, Plasmodium falciparum), and development of cysts (e.g., protozoa: Entamoeba histolytica; helminths: T. spiralis) to limit access by the immune response. The African trypanosomes can reengineer the genes for their surface antigen (variable surface glycoprotein) and therefore change their antigenic appearance. Schistosomes can coat themselves with host antigens, including MHC molecules.
Other Immune Responses
Antitumor responses and rejection of tissue transplants are primarily mediated by T cells. CD8 cytolytic T cells recognize and kill tumors expressing peptides from embryologic proteins, mutated proteins, or other proteins on class I MHC molecules (endogenous route of peptide presentation). These proteins may be expressed inappropriately by the tumor cell, and the host immune response may not be tolerized to them. In addition, IL-2 treatment in vitro generates lymphokine-activated killer (LAK) cells and NK cells that target tumor cells, and IFN-γ-activated (“angry”) macrophages can also distinguish and kill tumor cells.
T-cell rejection of allografts used for tissue transplants is triggered by recognition of foreign peptides expressed on foreign class I MHC antigens. In addition to host rejection of the transplanted tissue, cells from the donor of a blood transfusion or a tissue transplant can react against the new host in a graft-versus-host (GVH) response. An in vitro test of T-cell activation and growth in a GVH-like response is the mixed lymphocyte reaction. Activation is usually measured as DNA synthesis.
Immunopathogenesis
Hypersensitivity Responses
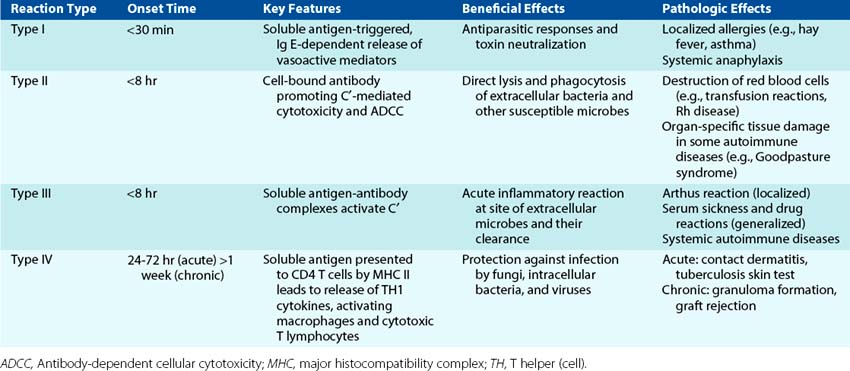
Once activated, the immune response is sometimes difficult to control and causes tissue damage. Hypersensitivity reactions are responsible for many of the symptoms associated with microbial infections, especially viral infections. Hypersensitivity reactions occur to people who have already established immunity to the antigen. The mediator and the time course primarily distinguish the four types of hypersensitivity responses (Table 10-5).
Type I hypersensitivity is caused by IgE and is associated with allergic, atopic, and anaphylactic reactions (Figure 10-7). IgE allergic reactions are rapid-onset reactions. IgE binds to Fc receptors on mast cells and becomes the cell surface receptor for antigens (allergens). Cross-linking of several cell surface IgE molecules by an allergen (e.g., pollen) triggers degranulation, releasing chemoattractants (cytokines, leukotrienes) to attract eosinophils, neutrophils, and mononuclear cells; activators (histamine, platelet-activating factor, tryptase, kininogenase) to promote vasodilation and edema; and spasmogens (histamine, prostaglandin D2, leukotrienes) to directly affect bronchial smooth muscle and promote mucus secretion. Desensitization (allergy shots) produces IgG to bind the allergen and prevent allergen binding to IgE. After 8 to 12 hours, a late-phase reaction develops because of the infiltration of eosinophils and CD4 T cells and cytokine reinforcement of inflammation.
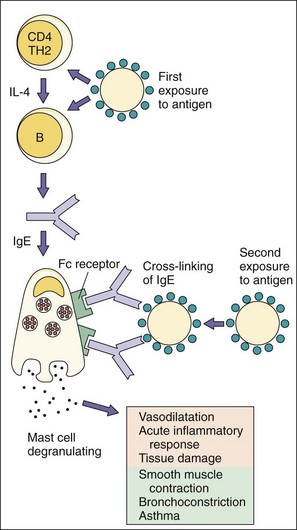
Figure 10-7 Type I hypersensitivity: immunoglobulin E (IgE)–mediated atopic and anaphylactic reactions. IgE produced in response to the initial challenge binds to Fc receptors on mast cells and basophils. Allergen binding to the cell surface IgE promotes the release of histamine and prostaglandins from granules to produce symptoms. Examples are hay fever, asthma, penicillin allergy, and reaction to bee stings. IL, Interleukin; TH, T helper (cell).
Type II hypersensitivity is caused by antibody binding to cell surface molecules and the subsequent activation of cytolytic responses by the classic complement cascade or by cellular mechanisms (Figure 10-8). These reactions occur as early as 8 hours following a tissue or blood transplant or as part of a chronic disease. Examples of these reactions are autoimmune hemolytic anemia, and Goodpasture syndrome (lung and kidney basement membrane damage). Another example is hemolytic disease of newborns (blue babies), which is caused by the reaction of maternal antibody generated during the first pregnancy to Rh factors on fetal erythrocytes of a second baby (Rh incompatibility). Antireceptor antibody activation or inhibition of effector functions are also considered type II responses. Myasthenia gravis is due to antibodies to acetylcholine receptors on neurons, Graves disease results from antibody stimulation of the thyroid-stimulating hormone (TSH) receptor, while some forms of diabetes can result from antibodies blocking the insulin receptor.
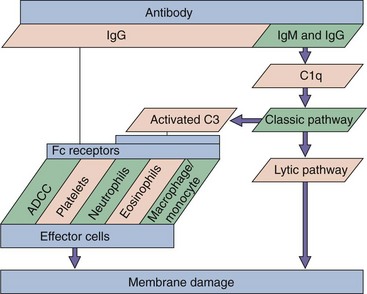
Figure 10-8 Type II hypersensitivity: mediated by antibody and complement. Complement activation promotes direct cell damage through the complement cascade and by the activation of effector cells. Examples are Goodpasture syndrome, the response to Rh factor in newborns, and autoimmune endocrinopathies. ADCC, Antibody-dependent cellular cytotoxicity; Ig, immunoglobulin.
Type III hypersensitivity responses result from activation of complement by immune complexes (Figure 10-9). In the presence of an abundance of soluble antigen in the bloodstream, large antigen-antibody complexes form, become trapped in capillaries (especially in the kidney), and then initiate the classical complement cascade. Activation of the complement cascade initiates inflammatory reactions. Immune complex disease may be caused by persistent infections (e.g., hepatitis B, malaria, staphylococcal infective endocarditis), autoimmunity (e.g., rheumatoid arthritis, systemic lupus erythematosus), or persistent inhalation of antigen (e.g., mold, plant, or animal antigens). For example, hepatitis B infection produces large amounts of hepatitis B surface antigen, which may promote formation of immune complexes that lead to glomerulonephritis. Type III hypersensitivity reactions can be induced in presensitized people by the intradermal injection of antigen to cause an Arthus reaction, a skin reaction characterized by redness and swelling. Serum sickness, extrinsic allergic alveolitis (a reaction to inhaled fungal antigen), and glomerulonephritis result from type III hypersensitivity reactions.
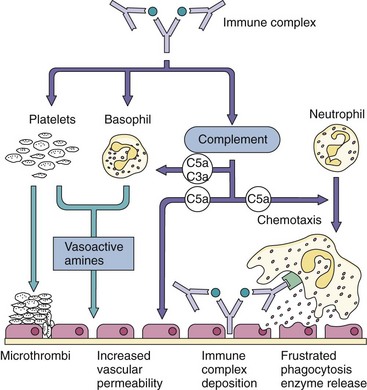
Figure 10-9 Type III hypersensitivity: immune complex deposition. Immune complexes can be trapped in the kidney and elsewhere in the body, can activate complement, and can cause other damaging responses. Examples are serum sickness, nephritis associated with chronic hepatitis B infection, and Arthus reaction.
Type IV hypersensitivity responses are TH1-mediated delayed-type hypersensitivity (DTH) inflammatory responses (Figure 10-10 and Table 10-6). It usually takes 24 to 48 hours for antigen to be presented to circulating CD4 T cells, for them to move to the site, and then activate macrophages to induce the response. Although essential for the control of fungal infections and intracellular bacteria (e.g., mycobacteria), DTH is also responsible for contact dermatitis (e.g., cosmetics, nickel) and the response to poison ivy. Intradermal injection of tuberculin antigen (purified protein derivative) elicits firm swelling that is maximal 48 to 72 hours after injection and indicative of prior exposure to Mycobacterium tuberculosis (Figure 10-11). Granulomas form in response to continued stimulation by the intracellular growth of M. tuberculosis. These structures consist of epithelioid cells created from chronically activated macrophages, fused epithelioid cells (multinucleated giant cells) surrounded by lymphocytes, and fibrosis caused by the deposition of collagen from fibroblasts. The granulomas restrict the spread of M. tuberculosis as long as CD4 T cells can provide IFN-γ. Granulomatous hypersensitivity occurs with tuberculosis, leprosy, schistosomiasis, sarcoidosis, and Crohn disease.
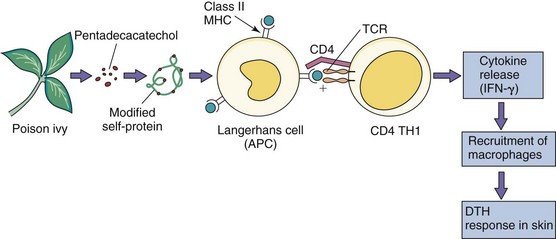
Figure 10-10 Type IV hypersensitivity: delayed-type hypersensitivity (DTH) mediated by CD4 T cells (TH1). In this case, chemically modified self-proteins are processed and presented to CD4 T cells, which release cytokines (including interferon-γ [IFN-γ]) that promote inflammation. Other examples of DTH are the tuberculin response (purified protein derivative test) and reaction to metals, such as nickel. APC, Antigen-presenting cell; TCR, T-cell receptor.
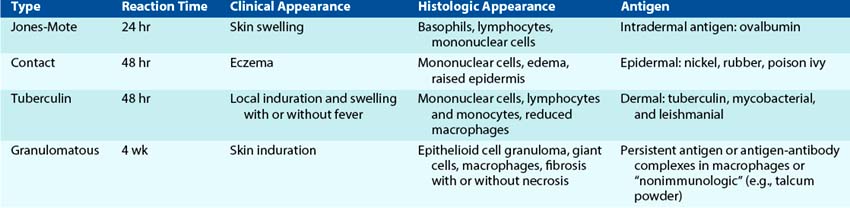
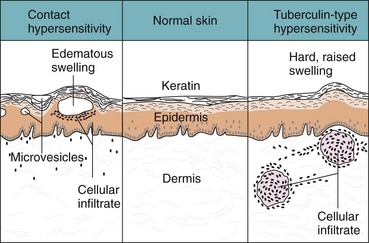
Figure 10-11 Contact and tuberculin hypersensitivity responses. These type IV responses are cell mediated but differ in the site of cell infiltration and in the symptoms. Contact hypersensitivity occurs in the epidermis and leads to the formation of blisters; tuberculin-type hypersensitivity occurs in the dermis and is characterized by swelling.
Cytokine Storm
Sepsis; toxin-mediated shock syndrome (e.g., induced by Staphylococcus toxic shock syndrome toxin); some virus infections, such as severe acute respiratory syndrome (SARS) and influenza; and graft-versus-host disease induce an overwhelming stimulation of innate and/or immune responses, producing excessive amounts of cytokines that disrupt the physiology of the body. The consequences are multisystem dysregulation, rash, fever, and shock. Superantigens clamp together TCRs with MHC II molecules on antigen-presenting cells to activate up to 20% of T cells. This triggers uncontrolled release of excess T cell– and macrophage-produced cytokines until the T cell dies of apoptosis. Bacteria, endotoxin, or viruses in blood can promote production of large amounts of acute-phase cytokines and type 1 interferons by plasmacytoid DCs, and certain viruses are very potent activators of interferon and cytokine production. Large amounts of TNF-α are produced during cytokine storms. TNF-α can promote inflammatory processes, such as enhanced vascular leakage and activation of neutrophils, that can be beneficial on a local level but, on a systemic level, will lead to fever, chills, aches, stimulation of coagulation pathways, elevated liver enzymes, loss of appetite, enhanced metabolism, weight loss, and potentially shock.
Autoimmune Responses
Normally a person is tolerized to self-antigens during the development of T cells and B cells and by Treg cells. However, deregulation of the immune response may be initiated by cross-reactivity with microbial antigens (e.g., group A streptococcal infection, rheumatic fever), polyclonal activation of lymphocytes induced by tumors or infection (e.g., malaria, Epstein-Barr virus infection), excessive cytokine production (e.g., type 1 interferons and systemic lupus erythematosus), or a genetic predisposition toward expression of self-antigenic peptides (MHC association) or lack of tolerization to specific antigens.
Autoimmune diseases result from the presence of autoantibodies, activated T cells, and hypersensitivity reactions. People with certain MHC antigens are at higher risk for autoimmune responses (e.g., HLA-B27: juvenile rheumatoid arthritis, ankylosing spondylitis). Once initiated, a cycle is established between antigen-presenting cells and T cells, which produce cytokines to promote inflammation and tissue damage and more self-antigen. TH17 responses are responsible for rheumatoid arthritis and other diseases.
Immunodeficiency
Immunodeficiency may result from genetic deficiencies, starvation, drug-induced immunosuppression (e.g., steroid treatment, cancer chemotherapy, chemotherapeutic suppression of tissue graft rejection), cancer (especially of immune cells), or disease (e.g., AIDS) and naturally occurs in neonates and pregnant women. Deficiencies in specific protective responses put a patient at high risk for serious disease caused by infectious agents that should be controlled by that response (Table 10-7). These “natural experiments” illustrate the importance of specific responses in controlling specific infections.
Table 10-7 Infections Associated with Defects in Immune Responses
| Defect | Pathogen |
|---|---|
| Induction by physical means (e.g., burns, trauma) | Pseudomonas aeruginosa |
| Staphylococcus aureus | |
| Staphylococcus epidermidis | |
| Streptococcus pyogenes | |
| Aspergillus species | |
| Candida species | |
| Splenectomy | Encapsulated bacteria and fungi |
| Granulocyte and monocyte defects in movement, phagocytosis, or killing or decreased number of cells (neutropenia) | S. aureus |
| S. pyogenes | |
| Haemophilus influenzae | |
| Gram-negative bacilli | |
| Escherichia coli | |
| Klebsiella species | |
| P. aeruginosa | |
| Nocardia species | |
| Aspergillus species | |
| Candida species | |
| Individual components of complement system | S. aureus |
| Streptococcus pneumoniae | |
| Pseudomonas species | |
| Proteus species | |
| Neisseria species | |
| T cells | Cytomegalovirus |
| Herpes simplex virus | |
| Herpes zoster virus | |
| Human herpesvirus 8 | |
| Listeria monocytogenes | |
| Mycobacterium species | |
| Nocardia species | |
| Aspergillus species | |
| Candida species | |
| Cryptococcus neoformans | |
| Histoplasma capsulatum | |
| Pneumocystis jirovecii | |
| Strongyloides stercoralis | |
| B cells | Enteroviruses |
| S. aureus | |
| Streptococcus species | |
| H. influenzae | |
| Neisseria meningitidis | |
| E. coli | |
| Giardia lamblia | |
| P. jiroveci | |
| Combined immunodeficiency | See pathogens listed for T cells and B cells |
Immunosuppression
Immunosuppressive therapy is important for reducing excessive inflammatory or immune responses or for preventing the rejection of tissue transplants by T cells. Therapy addresses the symptoms, the activator or the mediator of the response. Aspirin and nonsteroidal antiinflammatory drugs (NSAIDs) target the cyclooxygenases that generate inflammatory prostaglandins (e.g., PGD2) and pain. Other antiinflammatory treatments target the production and action of TNF-α, IL-12, and IL-1. Corticosteroids prevent their production by macrophages and may be toxic to T cells. Soluble forms of the TNF-α receptor and antibody to TNF-α can be used to block the binding of TNF-α and prevent its action. Antibodies to other cytokines, adhesion proteins on T cells or antigen-presenting cells, and antagonists of CD28 can block T-cell activation of inflammatory, antitissue graft, and other responses. Immunosuppressive therapy for transplantation generally inhibits the action or causes the lysis of T cells. Cyclosporin, tacrolimus (FK-506), and rapamycin prevent the activation of T cells (see Figure 9-5). Anti–CD40 ligand and anti–IL-2 prevent activation of T cells, whereas anti-CD3 promotes complement lysis of T cells to suppress T-cell responses. Anti-TNF-α therapies increase risk of M. tuberculosis disease and anti–α4 integrin cell adhesion molecule increases the risk of JC virus reactivation disease (progressive multifocal leukoencephalopathy).
Hereditary Complement Deficiencies and Microbial Infection
Inherited deficiencies of C1q, C1r, C1s, C4, and C2 components are associated with defects in activation of the classic complement pathway that lead to greater susceptibility to pyogenic (pus-producing) staphylococcal and streptococcal infections (Figure 10-12). These bacteria are not controlled by γ/δ T cells. A deficiency of C3 leads to a defect in activation of both the classical and the alternative pathways, which also results in a higher incidence of pyogenic infections. Defects of the properdin factors impair activation of the alternative pathway, which also results in an increased susceptibility to pyogenic infections. Finally, deficiencies of C5 through C9 are associated with defective cell killing, which raises the susceptibility to disseminated infections by Neisseria species.
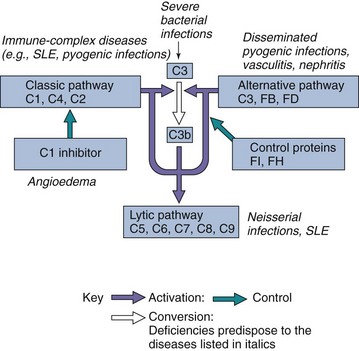
Figure 10-12 Consequences of deficiencies in the complement pathways. Factor B binds to C3b on cell surfaces, and the plasma serine protease D cleaves and activates B-C3b as part of the alternative pathway. Factors FI and FH limit the inappropriate activation of complement. FH binds to C3b and prevents activation and is a cofactor for FI. FI is a serine protease that cleaves C3b and C4b. SLE, Systemic lupus erythematosus.
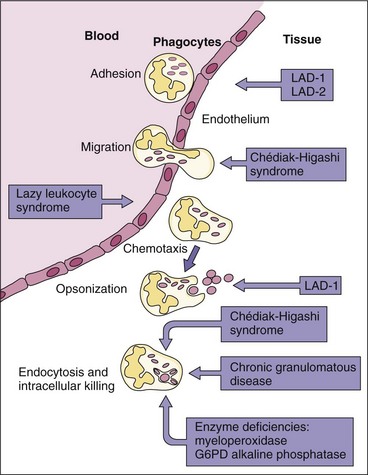
Defects in Phagocyte Action
People with defective phagocytes are more susceptible to bacterial infections but not to viral or protozoal infections (Figure 10-13). The clinical relevance of oxygen-dependent killing is illustrated by chronic granulomatous disease in children who lack the enzymes, such as NADPH oxidase, to produce superoxide anions. Although phagocytosis is normal, these children have an impaired ability to oxidize NADPH and destroy bacteria through the oxidative pathway. In patients with Chédiak-Higashi syndrome, the neutrophil granules fuse when the cells are immature in the bone marrow. Thus neutrophils from these patients can phagocytose bacteria but have greatly diminished ability to kill them. Granulomas are formed around the infected phagocyte to control the infection. Asplenic individuals are at risk for infection with encapsulated organisms because such people lack the filtration mechanism of spleen macrophages. Other deficiencies are shown in Figure 10-13.
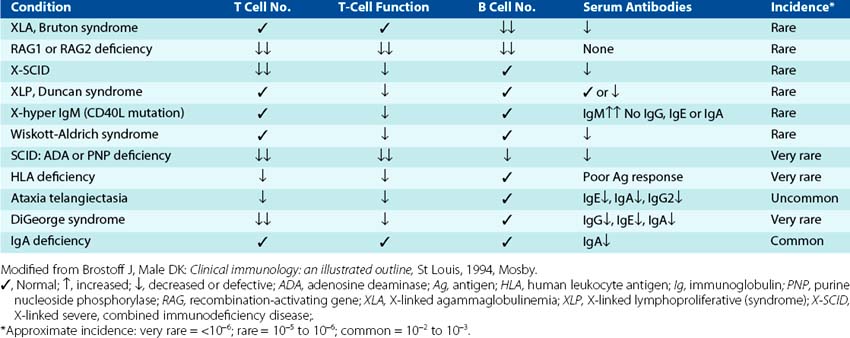
Deficiencies in Antigen-Specific Immune Responses
People deficient in T-cell function are susceptible to opportunistic infections by (1) viruses, especially enveloped and noncytolytic viruses and recurrences of viruses that establish latent infections; (2) intracellular bacteria; (3) fungi; and (4) some parasites. T-cell deficiencies can also prevent the maturation of B-cell antibody responses. T-cell deficiencies can arise from genetic disorders (e.g., X-linked immunodeficiency syndrome, Duncan disease, DiGeorge syndrome) (Table 10-8), infection (e.g., HIV and AIDS), cancer chemotherapy, or immunosuppressive therapy for tissue transplantation.
The T-cell response of neonates is deficient but is supplemented by maternal IgG. Insufficient TH1 responses and deficiency in IFN-γ puts them at high risk to infections by herpesviruses. Similarly, the less-pronounced cell-mediated immune and inflammatory responses of children decrease the severity (in comparison with adults) of herpes (e.g., infectious mononucleosis, chickenpox) and hepatitis B infections but also increase the potential for the establishment of a chronic hepatitis B virus infection because of incomplete resolution. Pregnancy also induces immunosuppressive measures to prevent rejection of the fetus (a foreign tissue).
B-cell deficiencies may result in a complete lack of antibody production (hypogammaglobulinemia), inability to undergo class switching, or inability to produce specific subclasses of antibody. People deficient in antibody production are very susceptible to bacterial infection. IgA deficiency, which occurs in 1 of 700 whites, results in a greater susceptibility to respiratory infections.
1. Describe the types of immune responses that would be generated to the following different types of vaccines. Consider the route of processing and presentation of the antigens and the cells and cytokines involved in generating each response.
2. Reproduce (i.e., write out on a separate piece of paper) the following table and fill in the appropriate columns:
| Immunodeficiency Disease | Immune Defect | Susceptibility to Specific Infections |
|---|---|---|
| Chédiak-Higashi syndrome | ||
| Chronic granulomatous disease | ||
| Complement C5 deficiency | ||
| Complement C3 deficiency | ||
| Complement C1 deficiency | ||
| Complement C6, C7, C8, or C9 deficiency | ||
| IgA deficiency | ||
| X-linked agammaglobulinemia | ||
| X-linked T-cell deficiency | ||
| AIDS | ||
| DiGeorge syndrome |
1. a. A TH2 response, which is predominantly an antibody response, will be generated to the bolus of tetanus toxoid protein presented in an “unnatural” manner. Lymph will bring the antigen to lymph nodes, where DCs will present the protein to CD4 T cells. CD4 T cells will make IL-4, IL-5, IL-6, and IL-10 and present antigen to B cells to promote class switching to TH2-related antibody production. Memory will not be efficient.
b. The inactivated polio vaccine will elicit a similar response as the tetanus toxoid.
c. Initially, a TH1 response will be generated to cells infected with the attenuated virus, which will naturally progress to TH2 and memory responses. The measles virus will activate IFN-α responses, followed by NK- and NKT-cell responses. The NK and NKT cells will make small amounts of IFN-γ. DCs will become activated, process the measles viral proteins, present antigen to CD4 and CD8 T cells while producing IL-12 to promote the generation of more IFN-γ by these T cells. Production of IL-2 by CD4 T cells will promote the growth of T and B cells, including CD8 T cells. IFN-γ will also promote a class switch for B cells from IgM to IgG production. Later, the response will include a TH2 response with the maturation of the IgG response. Long-term memory cells will also be elicited.
| Immunodeficiency Disease | Immune Defect | Susceptibility to Specific Infections |
|---|---|---|
| Chédiak-Higashi syndrome | Impaired release of lysosome contents into phagosome, delayed killing of phagocytized bacteria | Pyogenic infections (Staphylococcus and Streptococcus) |
| Chronic granulomatous disease | Inability to generate hydrogen peroxide for killing phagocytized bacteria | Recurrent infections with gram-negative and gram-positive bacteria, especially S. aureus and P. aeruginosa |
| Complement C5 deficiency | Decreased chemotaxis and bacterial killing | Bacterial infections |
| Complement C3 deficiency | Inhibition of complement cascade. C3 is the central character of both the classical and properidin pathways | Staphylococcus, Streptococcus, and other gram-positive infections |
| Complement C1 deficiency | Inhibition of classical pathway | Bacterial infections |
| Complement C6, C7, C8 or C9 deficiency | Inability to form membrane attack complex | Neisseria infections |
| IgA deficiency | Defective B cell; insufficient cytokine production; mutation in J or secretory chains | Respiratory and gastrointestinal infections |
| X-linked agammaglobulinemia | CD40 deficiency (T-cell help disorder); defective pre–B-cell maturation | Bacterial and other infections. Cannot undergo immunoglobulin class switch |
| X-linked T-cell deficiency | Defective receptor shared by IL-2, IL-7, IL-4, IL-9, IL-15 cytokines or signaling from the receptor. | Intracellular bacteria, viruses (especially herpes, JC), fungi. Cannot undergo immunoglobulin class switch |
| AIDS | CD4 T cell targeted killing by HIV | Intracellular bacteria, viruses (especially herpes, JC), fungi, and some parasites |
| DiGeorge syndrome | T-cell maturation | Intracellular bacteria, viruses (especially herpes, JC), fungi. Cannot undergo immunoglobulin class switch |