Drug Action and Handling
1 Differentiate dose, potency, and efficacy in the context of the actions of drugs.
2 Explain the pharmacologic effect of a drug.
3 Discuss the major steps of pharmacokinetics: absorption, distribution, metabolism, and excretion.
4 Summarize the various routes of drug administration.
5 Provide example of factors that may alter the effect of a drug.
To discuss the drugs used in dentistry or those that patients may be taking when they come to the dental office, the dental health care worker must be familiar with some basic principles of pharmacology. This chapter discusses the action of drugs in the body and methods of drug administration. Chapter 3 considers the problems or adverse reactions these drugs can cause. By understanding how drugs work, what effects they can have, and what problems they can cause, the dental health care worker can better communicate with the patient and other health care providers about medications the patient may be taking or may need to have prescribed for dental treatment.
Drugs are broadly defined as chemical substances used for the diagnosis, prevention, or treatment of disease or for the prevention of pregnancy. Most drugs are differentiated from inert chemicals and chemicals necessary for the maintenance of life processes (e.g., vitamins) by their ability to act selectively in biologic systems to accomplish a desired effect. Historically, drugs were discovered by randomly searching for active components among plants, animals, minerals, and the soil. Today, organic synthetic chemistry researchers are primarily responsible for developing new drugs. Parent compounds that exhibit known pharmacologic activity are chemically modified to produce congeners or analogs: agents of a similar chemical structure with a similar pharmacologic effect. This technique of modifying a chemical molecule to provide more useful therapeutic agents has evolved from studies of the relationship between the chemical structure and the biologic activity called structure-activity relationship (SAR).
CHARACTERIZATION OF DRUG ACTION
Dose-response curve, potency, and efficacy are terms used to measure drug response or action.
Log Dose Effect Curve
When a drug exerts an effect on biologic systems, the effect can be related quantitatively to the dose of the drug given. If the dose of the drug is plotted against the intensity of the effect, a curve will result (Figure 2-1). If this curve is replotted using the log of the dose (log dose) versus the response, another curve is produced from which the potency and efficacy of a drug’s action may be determined (Figure 2-2).
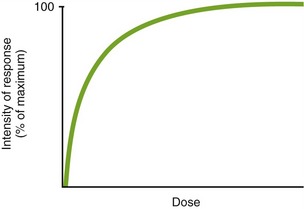
FIGURE 2-1 Dose effect curve. The x-axis (horizontal) is an increasing dose of the drug, and the y-axis (vertical) is an increasing effect of the drug.
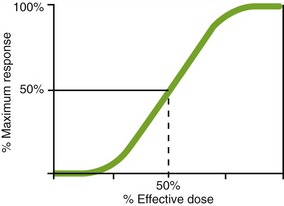
FIGURE 2-2 Log dose effect curve. As the dose is increased (going to the right on the x-axis), the effect (the y-axis) is zero at first, then there is a small effect, and finally the effect quickly increases. Around the dose where the line is increasing sharply is the therapeutic range of the compound. Finally, the curve plateaus (flattens out). This is the maximum response a drug can exhibit.
Potency
The potency of a drug is a function of the amount of drug required to produce an effect. The potency of a drug is shown by the location of that drug’s curve along the log-dose axis (x-axis). The curves in Figure 2-3 illustrate two drugs with different potencies. The potency of drug A is greater because the dose required to produce its effect is smaller. The potency of B is less than A because B requires a larger dose to produce its effect.
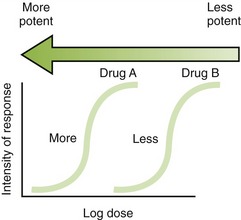
FIGURE 2-3 Potency of agent. The arrow is shaded proportional to increasing potency. (Dark shading, very potent; light shading, low potency.)
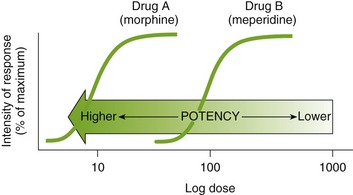
As an example of different potencies, three alcoholic beverages are compared: bourbon, beer, and wine cooler (or spritzer). One ounce of bourbon contains the same amount of alcohol as one beer (12 oz) or as one wine cooler or spritzer (16 oz [depends on dilution]). All of these drinks could equally inebriate an individual (produce adverse reactions). To produce a similarly drunk individual, the same amount of alcohol would have to be ingested. However, this amount would be contained in a different volume of fluid, depending on its concentration (or potency).* Therefore, when someone says “I’m not drunk because I just drank beer,” the statement is false. The absolute potency of a drug is immaterial as long as an appropriate dose is administered. Both meperidine and morphine have the ability to treat severe pain, but approximately 100 mg of meperidine would be required to produce the same action as 10 mg of morphine. Thus the absolute potency of oral morphine is 10 times that of oral meperidine, or meperidine is one-tenth as potent as morphine, even though both agents can relieve intense pain (equal efficacy, as explained next). In Figure 2-4, the curve for drug B (meperidine) is to the right of the curve for drug A (morphine) because the dose of meperidine needed to produce pain relief is larger (10 times larger) than that for morphine. The potency of different drugs that elicit similar effects can be compared by observing the dose that produces 50% (drop a vertical line down from the center of the curve) of the total, or maximum, effect.
Efficacy
Efficacy is the maximum intensity of effect or response that can be produced by a drug. Administering more drug will not increase the efficacy of the drug but can often increase the probability of an adverse reaction. The efficacy of a drug increases as the height of the curve increases (Figure 2-5). The efficacy of the drugs whose curves are illustrated in Figure 2-5 are shown by the height of the curve when it plateaus (levels out horizontally). It is shaded from least (light) to most (dark) potent. The efficacy of any drug is a major descriptive characteristic indicating its action. For example, the efficacy of drug B (meperidine) and drug A (morphine) is about the same because both drugs relieve severe pain.
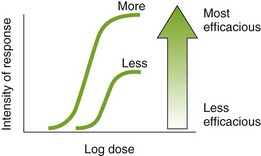
If one “drink” of both bourbon (1 oz) and beer (12 oz) were ingested, they could produce equal “silliness” in an individual. If very large doses of either agent were ingested, unconsciousness could be produced. Both are equally efficacious, but they differ in their potency. The efficacy and the potency of a drug are unrelated.
Because death is the endpoint when measuring the lethal dose, the median lethal dose (LD50) is the dose when one-half of the subjects die. For obvious reasons, the LD50 is only determined in animals.
Chemical Signaling Among Cells
For the autonomic nervous system to function, messages from the brain must be transmitted to many parts of the body commanding the parts to “do something” (e.g., enlarge pupil or sweat). Complex mechanisms for transmitting these messages allow for amplification or damping of the effect, depending on a multitude of factors. The complexity allows for very fine tuning of the body’s functions. Neurotransmitters are chemicals responsible for transporting a wide variety of messages across the synapse (space between nerve and receptor). Chemical signaling involves release of neurotransmitters, local substances, and hormone secretion.
 NEUROTRANSMITTERS
NEUROTRANSMITTERS
The messengers that move the electrical impulses from a nerve are transmitted across the synapse via neurotransmitters. The neurotransmitters are released and quickly travel across the synapse to the receptor (Figure 2-6). There are at least 50 different agents that transmit messages. Examples of neurotransmitters include acetylcholine, norepinephrine/epinephrine, dopamine, serotonin, γ-aminobutyric acid (GABA), and histamine.
LOCAL
Some organs secrete chemicals that work near them. These chemicals are not released into the systemic circulation. Prostaglandins and histamine are examples. For example, a person wears a nickel-containing watch and a red spot appears on the skin beneath the watch. This localized allergic reaction is caused by release of inflammation-producing substances, such as histamine, at that point on the skin. Because the reaction is localized, the patient’s nose does not begin to run. Prostaglandins contract the uterine muscles and become important as a baby is born. When prostaglandins are released in the uterus, they produce menstrual “cramps,” and when released in the stomach, they protect its lining.
MECHANISM OF ACTION OF DRUGS
After drugs have been distributed to their site of action, they elicit a pharmacologic effect. The pharmacologic effect occurs because of a modulation in the function of an organism. Drugs do not impart a new function to the organism; they merely produce either the same action as an endogenous agent or block the action of an endogenous agent. This signaling mechanism has two functions: amplification of the signal and flexible regulation. The presence of very fine controls to modulate the body’s function allows the regulation of certain reactions, slowing or speeding them.
Nerve Transmission
Within a nerve, the transmission of impulses travels along the nerve, producing a nerve action potential. The action potential is triggered by the neurotransmitter released at the previous synapse. Drugs that interfere with this process, such as local anesthetics (see Chapter 10), block messages from being sent. The processes involved in the drug’s effect begin the drug-receptor interaction. The receptors, macromolecular chemical structures, interact with both endogenous substances and drugs. This drug-receptor interaction results in a conformational (shape) change, which may allow the drug inside the cell to produce its effect or may cause the release of a second messenger, which then produces the effect. Many of the effects involve altering enzyme-regulated reactions or regulatory processes for protein synthesis after a series of reactions, similar to a chain reaction. These steps in the process of communicating are briefly discussed.
Receptors
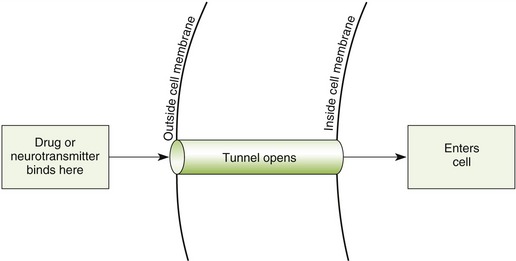
Once a drug passes through the biologic membrane, it is carried to many different areas of the body, or site of action, to exert its therapeutic effect or adverse effect. For the drug to exert its effects, it must bind with the receptor site on the cell membrane. Drug receptors appear to consist of many large molecules that exist either on the cell membrane or within the cell itself (Figure 2-7). More than one receptor type or identical receptors can be found at the site of action. Usually, a specific drug will bind with a specific receptor in a lock-and-key fashion. Many drug-receptor interactions consist of weak chemical bonds, and the energy formed during this interaction is very low. As a result, the bonds can be formed and broken easily. Once a bond is broken, another drug molecule immediately binds to the receptor.
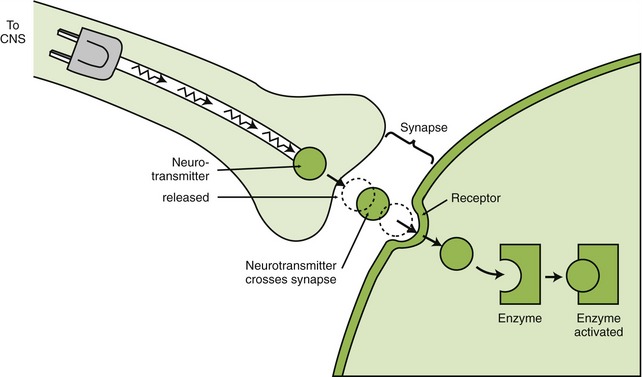
FIGURE 2-7 The neurotransmitter is transmitting the message (like electricity) across the synapse (space where nerve is absent). The neurotransmitter then interacts with the receptor (shaped to fit together), which then may signal an enzyme to be synthesized or activated.
Different drugs often compete for the same receptor sites. The drug with the stronger affinity for the receptor will bind to more receptors than the drug with the weaker affinity (Figure 2-8). More of the drug with the weaker affinity will be required to produce a pharmacologic response. Drugs with a stronger affinity for receptor sites are more potent than drugs with weaker affinities for the same site.
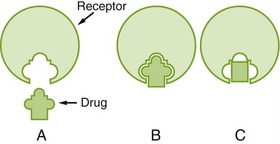
FIGURE 2-8 A, Drugs act by forming a chemical bond with specific receptor sites, similar to a lock and key. B, The better the “fit,” the better the response. Drugs with complete attachment and response are called agonists. C, Drugs that attach but do not elicit a response are called antagonists. (From Clayton BD, Stock YN, Harroun RD: Basic pharmacology for nurses, ed 14, St Louis, 2007, Mosby.)
AGONISTS AND ANTAGONISTS
When a drug combines with a receptor, it alters the function of the organism. It may produce enhancement or inhibition of the function. Drugs that combine with the receptor may be classified as either agonists or antagonists (Figure 2-9).
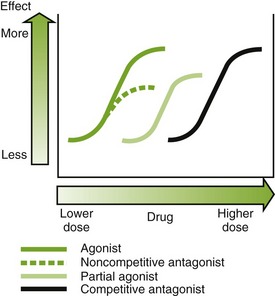
Agonist: An agonist is a drug that (1) has affinity for a receptor, (2) combines with the receptor, and (3) produces an effect. Naturally occurring neurotransmitters are agonists.
Antagonist: An antagonist counteracts the action of the agonist. The following are three different types of antagonists:
• A competitive antagonist is a drug that (1) has affinity for a receptor, (2) combines with the receptor, and (3) produces no effect. This causes a shift to the right in the dose-response curve (see Figure 2-9). The antagonist competes with the agonist for the receptor, and the outcome depends on the relative affinity and concentrations of each agent. If the concentration of the agonist is increased, the competitive antagonism can be overcome and vice versa.
• Noncompetitive antagonists bind to a receptor site that is different from the binding site for the agonist. This reduces the maximal response of the agonist (see Figure 2-9).
• A physiologic antagonist has affinity for a different receptor site than the agonist. This decreases the maximal response of the agonist by producing an opposite effect via different receptors.
Transport carriers are systems that are available for moving neurotransmitters or drugs into the cell. In the process of making a neurotransmitter, the precursors (chemical to make a neurotransmitter) must be taken into the cell by an active transport pump (requires adenosine triphosphatase [ATPase]). For example, the precursor for norepinephrine is tyramine, so it must be pumped into the cell. After the neurotransmitter is synthesized, it is placed in little “suitcases” called granules. These go to the membranes and await the signal to “dump” their contents into the synapse. After the neurotransmitter is released, there are three paths that it can take. It can be broken down by enzymes designed to terminate the neurotransmitter’s effect, it can migrate to the receptor and interact to produce an effect, or it can be taken up by the presynaptic nerve ending (reuptake). Reuptake is an easy way (requires little energy) to recover the neurotransmitter for future use because it is as easy as vacuuming up dirt.
PHARMACOKINETICS
Pharmacokinetics is the study of how a drug enters the body, circulates within the body, is changed by the body, and leaves the body. Factors that influence the movement of a drug are divided into four major steps: absorption, distribution, metabolism, and excretion (ADME).
Passage Across Body Membranes
The amount of drug passing through a cell membrane and the rate at which a drug moves are important in describing the time course of action and the variation in individual response to a drug. Before a drug is absorbed, transported, distributed to body tissues, metabolized, and subsequently eliminated from the body, it must pass through various membranes such as cellular membranes, blood capillary membranes, and intracellular membranes. Although these membranes have variable functions, they share certain physicochemical characteristics that influence the passage of drugs across their borders.
These membranes are composed of lipids (fats), proteins, and carbohydrates. The membrane lipids make the membrane relatively impermeable to ions and polar molecules. Membrane proteins make up the structural components of the membrane and help move the molecules across the membrane during the transport process. Membrane carbohydrates are combined with either proteins or lipids. The lipid molecules orient themselves so that they form a fluid bimolecular leaflet structure with the hydrophobic (lipophilic) ends of the molecules shielded from the surrounding aqueous environment and the hydrophilic ends in contact with the water. The various proteins are embedded in and layered onto this fluid lipid bilayer, forming a mosaic. Studies of the ability of substances to penetrate this membrane have indicated the presence of a system of pores or holes through which lower-molecular-weight and smaller size chemicals can pass.
The physicochemical properties of drugs that influence the passage of drugs across biologic membranes are lipid solubility, degree of ionization, and molecular size and shape. The mechanisms of drug transfer across biologic membranes are passive transfer and specialized transport.
PASSIVE TRANSFER
Lipid-soluble substances move across the lipoprotein membrane by a passive transfer process called simple diffusion. This type of transfer is directly proportional to the concentration gradient (difference) of the drug across the membrane and the degree of lipid solubility. For example, a highly lipid-soluble compound will attain a higher concentration at the membrane site and will readily diffuse across the membrane into an area of lower concentration (Figure 2-10). A water-soluble agent will have difficulty passing through a membrane.
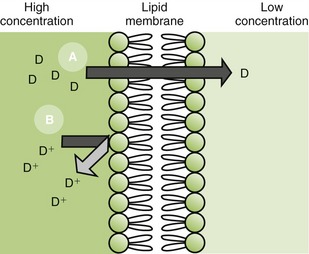
FIGURE 2-10 Passage of drug and metabolite through membranes. A, Lipid soluble, nonionized: drug easily passes through the cell membrane from area of high to low drug concentration. B, Water soluble, ionized: drug cannot pass through the cell membrane. D, Drug.
Water-soluble molecules small enough to pass through the membrane pores may be carried through the pores by the bulk flow of water. This process of filtration through single-cell membranes may occur with drugs having molecular weights of 200 or less. However, drugs with molecular weights of 60,000 can “filter” through capillary membranes.
SPECIALIZED TRANSPORT
Certain substances are transported across cell membranes by processes that are more complex than simple diffusion or filtration. These processes include the following:
• Active transport is a process by which a substance is transported against a concentration gradient or electrochemical gradient. This action is blocked by metabolic inhibitors. Active transport is believed to be mediated by transport “carriers” that furnish energy for the transportation of the drug.
• Facilitated diffusion does not move against a concentration gradient. This phenomenon involves the transport of some substances, such as glucose, into cells. It has been suggested that the process of pinocytosis may explain the passage of macromolecular substances into the cells.
Absorption
Absorption is the process by which drug molecules are transferred from the site of administration to the circulating blood. This process requires the drug to pass through biologic membranes.
The following factors influence the rate of absorption of a drug:
• The physicochemical factors discussed previously.
• The site of absorption, which is determined by the route of administration. For example, one advantage of the oral route is the large absorbing area presented by the intestinal mucosa.
• The drug’s solubility. Drugs in solution are more rapidly absorbed than insoluble drugs.
EFFECT OF IONIZATION
Drugs that are weak electrolytes dissociate in solution and equilibrate into a nonionized form and an ionized form. The nonionized, or uncharged, portion acts like a nonpolar, lipid-soluble compound that readily crosses body membranes (see Figure 2-10). The ionized portion will traverse these membranes with greater difficulty because it is less lipid soluble.
The pH of the tissues at the site of administration and the dissociation characteristics (pKa) of the drug will determine the amount of drug present in the ionized and nonionized state. The proportion in each state will determine the ease with which the drug will penetrate the tissues.
Weak Acids: When the pH at the site of absorption increases, the hydrogen ion concentration simply falls. This results in an increase in the ionized form (A−), which cannot easily penetrate tissues.
Conversely, if the pH of the site falls, the hydrogen ion concentration will rise. This results in an increase in the un-ionized form (HA), which can more easily penetrate tissues.
Weak Bases: If the pH of the site rises, the hydrogen ion concentration will fall. This results in an increase in the un-ionized form (B), which can more easily penetrate tissues. Conversely, if the pH of the site falls, the hydrogen ion concentration will rise. This results in an increase in the ionized form (BH+), which cannot easily penetrate tissues. In summary, weak acids are better absorbed when the pH is less than the pKa, whereas weak bases are better absorbed when the pH is greater than the pKa.
This dissociation also explains the fact that in the presence of infection the acidity of the tissue increases (and the pH decreases) and the effect of local anesthetics decreases. In the presence of infection, the [H+] increases because of accumulating waste products in the infected area. The increase in [H+] (decrease in pH) leads to an increase in ionization and a decrease in penetration of the membrane. This reduced penetration reduces the clinical effect of the local anesthetic.
ORAL ABSORPTION
The dose form of a drug is an important factor influencing absorption of drugs administered via the oral route. Unless the drug is administered as a solution, the absorption of the drug in the gastrointestinal tract involves a release from a dose form such as a tablet or capsule. This release requires the following steps before absorption can take place:
• Disruption: The initial disruption of a tablet coating or capsule shell is necessary.
• Disintegration: The tablet or capsule contents must disintegrate (break apart).
• Dispersion: The concentrated drug particles must be dispersed (spread) throughout the stomach or intestines.
• Dissolution: The drug must be dissolved (in solution) in the gastrointestinal fluid.
A drug in solution skips these four steps, so it usually has a quicker onset of action.
ABSORPTION FROM INJECTION SITE
Absorption of a drug from the site of injection depends on the solubility of the drug and the blood flow at that site. For example, drugs with low water solubility, such as some penicillin salts, are absorbed very slowly after intramuscular injection. Absorption at injection sites is also affected by the dose form. Drugs in suspension are absorbed much more slowly than those in solution. Certain insulin preparations are formulated in suspension form to decrease their absorption rate and prolong their action. Drugs that are least soluble will have the longest duration of action.
Distribution
All drugs occur in two forms in the blood: bound to plasma proteins and the free drug. The free drug is the form that exerts the pharmacologic effect. The bound drug is a reservoir (place to store) for the drug. The proportion of drug in each form depends on the properties of that specific drug (percent protein bound). Within each compartment (e.g., blood, brain), the drug is split between the bound drug and the free drug. Only the free drug can pass across cell membranes.
For a drug to exert its activity, it must be made available at its site of action in the body. The mechanism by which this is accomplished is distribution, which is the passage of drugs into various body fluid compartments such as plasma, interstitial fluids, and intracellular fluids. The manner in which a drug is distributed in the body will determine how rapidly it produces the desired response; the duration of that response; and in some cases, whether a response will be elicited at all.
Drug distribution occurs when a drug moves to various sites in the body, including its site of action in specific tissues. However, drugs are also distributed to areas where no action is desired (nonspecific tissues). Some drugs, because of their characteristics, are poorly distributed to certain regions of the body. Other drugs are distributed to their site of action and then redistributed to another tissue site. The distribution of a drug is determined by several factors such as the size of the organ, the blood flow to the organ, the solubility of the drug, the plasma protein-binding capacity, and the presence of certain barriers (blood-brain barrier, placenta).
DISTRIBUTION BY PLASMA
After a drug is absorbed from its site of administration, it is distributed to its site of action by the blood plasma. Therefore the biologic activity of a drug is related to the concentration of the free, or unbound, drug in the plasma. Drugs are bound reversibly to plasma proteins such as albumin and globulin. The drug that is bound to the protein does not contribute to the intensity of the drug action because only the unbound form is biologically active. The bound drug is considered a storage site. If one drug is highly bound, another administered drug that is highly bound may displace the first drug from its plasma protein-binding sites, increasing the effect of the first drug. This is one mechanism of drug interaction.
Half-Life
The half-life (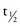 ) of a drug is the amount of time that passes for the concentration of a drug to fall to one-half of its blood level (Figure 2-11). When the half-life of a drug is short, it is quickly removed from the body and its duration of action is short. When the half-life of a drug is long, it is slowly removed from the body and its duration of action is long.
) of a drug is the amount of time that passes for the concentration of a drug to fall to one-half of its blood level (Figure 2-11). When the half-life of a drug is short, it is quickly removed from the body and its duration of action is short. When the half-life of a drug is long, it is slowly removed from the body and its duration of action is long.
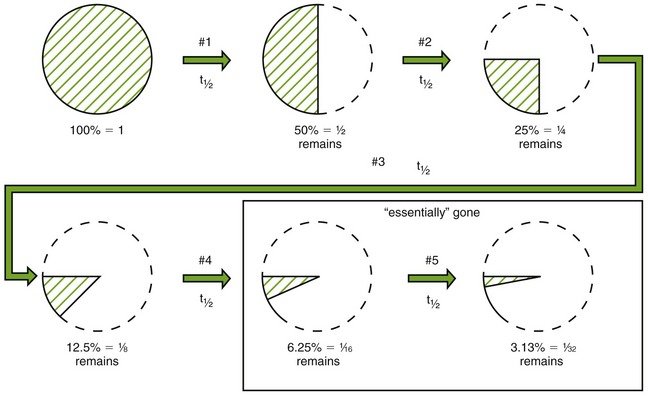
FIGURE 2-11 First-order kinetics. Half-life constant throughout usual doses. Half of the dose of the drug in the body is removed with each half-life. #1, #2 … #5, Number of half-lives that have passed.
Figure 2-11 shows the percent of a drug remaining after each of four and five half-lives. Because only 3% to 6% remains after four or five half-lives, respectively, we can say that the drug is essentially gone. Conversely, it takes about four or five half-lives of repeated dosing for a drug’s level to build up to a steady state (level amount) in the body. If the half-life of a drug is 1 hour, then in 4 or 5 hours the drug would be mostly gone from the body. In 4 hours, 94% of the drug would be gone. However, if the half-life of a drug is 60 hours, then it would take 240 (10 days) to 300 hours (12 days) for that drug to be eliminated from the body. Even after discontinuing a drug with a long half-life, its effect can take several days to dissipate, depending on its half-life.
Blood-Brain Barrier
The tissue sites of distribution should be considered before administration. For example, for drugs to penetrate the central nervous system (CNS), they must cross the blood-brain barrier. The passage of a drug across this barrier is related to the drug’s lipid solubility and degree of ionization. The endothelium of this barrier contains a cell layer and a basement membrane. The welding of the endothelial cells together prevents the formation of clefts, gaps, or pores that might allow the penetration of certain drugs. To diffuse transcellularly, the drug must penetrate the epithelial and basement membrane cells. Thiopental, a highly lipid-soluble, nonionized drug, easily penetrates the blood-brain barrier to gain access to the cerebrospinal fluid and induce sleep within seconds after intravenous administration. In contrast, a highly ionized compound such as hexamethonium would not be likely to cross this barrier and therefore would produce few if any effects on the brain.
PLACENTA
The passage of drugs across the placenta involves simple diffusion in accordance with their degree of lipid solubility. Although the placenta may act as a selective barrier against a few drugs, most drugs pass easily across the placental barrier. Lipid-soluble drugs penetrate this membrane most easily. Therefore when agents are administered to the mother, they are concomitantly administered to the fetus. The term barrier is a misnomer.
ENTEROHEPATIC CIRCULATION
Drugs are typically absorbed via the intestines, are distributed through the serum, pass to specific and nonspecific sites of action, come to the liver, and are metabolized before being excreted via the kidneys. When a drug undergoes enterohepatic circulation, the process varies. The steps are the same until the drug is metabolized. At that point, the metabolite is secreted via the bile into the intestine. The metabolite is broken down by enzymes and releases the drug. The drug is then absorbed again, and the process continues. After being taken up by the liver the second time, these drugs are again secreted into the bile. This circular pattern continues with some drug escaping with each passing. This process prolongs the effect of a drug. If enterohepatic circulation is blocked, the level of the drug in the serum will fall.
Redistribution
Redistribution of a drug is the movement of a drug from the site of action to nonspecific sites of action. A drug’s duration of action can be affected by redistribution of the drug from one organ to another. If redistribution occurs between specific sites and nonspecific sites, a drug’s action will be terminated. For example, thiopental produces sleep within seconds, but the effect is terminated within a few minutes. This is because the drug is first distributed to the CNS (sleep), subsequently redistributed through the plasma to the muscle (action terminated), and finally reaches the fat depots of the body (no action still).
Metabolism (Biotransformation)
Metabolism, which is also known as biotransformation, is the body’s way of changing a drug so that it can be more easily excreted by the kidneys. Many drugs undergo metabolic transformation or change, most commonly in the liver. The metabolite (metabolic product) formed is usually more polar (ionized) and less lipid soluble than its parent compound. This means that renal tubular reabsorption of the metabolite will be reduced because reabsorption favors lipid-soluble compounds. Metabolites are also less likely to bind to plasma or tissue proteins and less likely to be stored in fat tissue. Decreased renal tubular reabsorption, decreased binding to the plasma or tissue proteins, and decreased fat storage cause the metabolite to be excreted more easily. Drug metabolism is an enzyme-dependent process that has developed through evolution.
Drugs can be metabolized in one of three of the following different means (Figure 2-12):
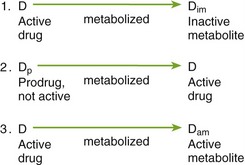
• Active to inactive: By metabolism, an inactive compound may be formed from an active parent drug. This is the most common type of reaction in drug biotransformation. Agents that interfere with the metabolism of certain drugs will increase the blood level of the drugs whose metabolism is inhibited. An example of this is doxycycline. Doxycycline itself is the active compound and is metabolized by the liver into a metabolite without activity.
• Inactive to active: An inactive parent drug may be transformed into an active compound. The inactive compound is then termed a prodrug. Interference with the metabolism of this drug will delay its onset of action because it will be harder for the active compound to be formed. For example, acyclovir is an antiviral agent. To be effective, it must be taken into the cell and converted to its active metabolite.
• Active to active: An active parent drug may be converted to a second active compound, which is then converted to an inactive product. The total effect of such a drug would be the addition of the effect of the parent drug plus the effect of the active drug metabolite. When an active metabolite is formed, the action of the drug is prolonged. For example, diazepam (Valium), an active antianxiety agent, is metabolized into its active metabolite, desmethyldiazepam. Diazepam’s action is prolonged because of its own effect combined with that of its active metabolite.
Although the rates and pathways of drug metabolism vary among species, most studies indicate that drug biotransformation in laboratory animals is similar to that in humans. Many synthetic mechanisms of drug metabolism occur in the body to form metabolites.
FIRST-PASS EFFECT
Metabolism of drugs may be divided into two general types: phase I and phase II. If a drug has no functional groups with which to combine, then the drug must undergo a phase I reaction.
Phase I: In phase I reactions, lipid molecules are metabolized by the three processes of oxidation, reduction, and hydrolysis.
Oxidation: When a drug is administered that does not possess an appropriate functional group suitable for combining with body acids (conjugation), the body has more difficulty detoxifying that drug. An enzyme system responsible for the oxidative metabolism of many drugs is located in the liver. The enzymes are located in the endoplasmic reticulum and are termed microsomal enzymes because they are found in the microsomal fraction as prepared from liver homogenates. A variety of oxidative reactions, such as hydroxylation or the incorporation of oxygen into the substrate molecule, occur in these hepatic microsomal enzymes.
Oxidative processes involving enzymes other than those of the hepatic microsomal enzymes can take place. Some compounds are oxidatively deaminated by enzymes located in the liver, kidney, and nervous tissue. Other agents are detoxified by specific oxidative enzymes.
Hydrolysis: Some ester compounds are metabolized by hydrolysis. Hydrolytic enzymes, which are found in plasma and a variety of tissues, break up esters and add water. The ester local anesthetics are inactivated by plasma cholinesterases.
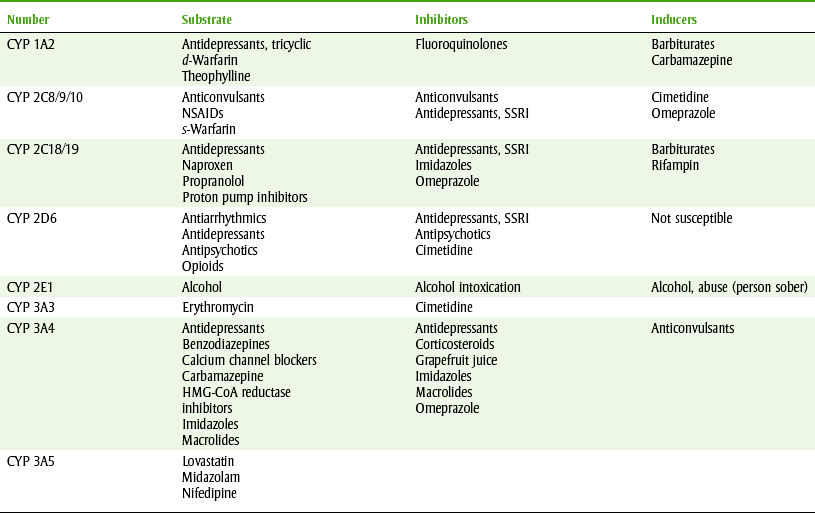
Microsomal Enzymes: Phase I reactions are carried out by the microsomal or cytochrome P-450 enzymes, which are also known as the mixed function oxidases in the liver. The concentration of these enzymes can be affected by drugs and environmental substances. Phase I metabolism may be affected by other drugs that alter microsomal enzyme inhibition or induction.
Induction: The P-450 hepatic microsomal enzymes can be induced (the amount of enzyme increased) by some drugs and by smoking tobacco. Because drugs that cause enzyme induction cause other drugs to be more quickly metabolized, the metabolized drugs will have reduced pharmacologic effects. More recent evidence has discovered that the hepatic enzymes can be divided into many categories called isoenzymes. Examples of isoenzymes include cytochrome P-450 2D6 and 3A4. Table 2-1 lists samples of drugs that are substrates of these enzymes and drugs that either induce or inhibit these isoenzymes. For example, phenobarbital stimulates the production of microsomal enzymes that normally metabolize the anticoagulant warfarin. Thus administering phenobarbital to a patient taking warfarin can decrease warfarin’s anticoagulant response because the metabolism of the anticoagulant is stimulated by phenobarbital (Figure 2-13).
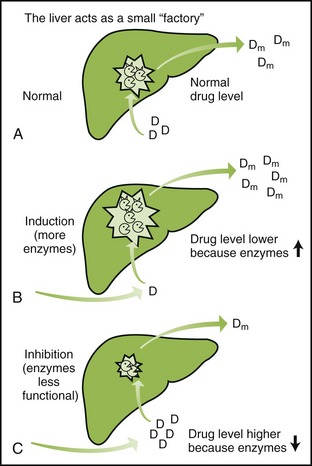
FIGURE 2-13 Alteration of drug metabolism induction and inhibition. Enzyme induction and inhibition alter the blood levels of drugs metabolized by the hepatic enzymes. A, Normal. The liver is metabolizing drugs at the normal rate producing the normal effects. B, Induction. With enzyme induction (stimulation), increase in the enzymes causes the drug to be more quickly metabolized, and blood level of the drug and its effect are decreased (assume that metabolite is inactive). Induction = effect. C, Inhibition. With enzyme inhibition, the metabolism of drugs is slower (weaker enzymes) and the blood level of the drug that is metabolized is increased. Inhibition = effect.
Some drugs, such as valproate and carbamazepine (anticonvulsants), can also stimulate their own metabolism. The tolerance that patients develop to certain drugs can be explained, at least in part, by an increased ability to metabolize the drug because of stimulation of microsomal enzymes.
Inhibition: Inhibition of the metabolism of certain drugs may occur through several mechanisms. With inhibition, the blood levels and action of the drugs metabolized by these enzymes will be increased. Examples of drugs that inhibit the metabolism of other drugs are erythromycin and cimetidine. Inhibiting the microsomal enzymes would result in an increase in the effect of the drugs metabolized by the liver enzymes (see Table 2-1).
Phase II: Phase II reactions involve conjugation with the following agents: glucuronic acid, sulfuric acid, acetic acid, or an amino acid. The most common conjugation occurs with glucuronic acid. This conjugation is termed glucuronidation. Glucuronic acid, which is a substance normally occurring in the body, may be transferred to a drug molecule that has an appropriate functional group to accept it. Functional groups that may be involved include ethers, alcohols, aromatic amines, and carboxylic acid. This mechanism, either alone or in combination with a phase I reaction, allows the body to convert a lipid-soluble drug to a more polar compound. The enzymes that mediate the conjugation are termed transferases.
EXCRETION
Although drugs may be excreted by any of several routes that have direct access to the external environment, renal (kidney) excretion is the most important. Extrarenal routes include the lungs, bile, gastrointestinal tract, sweat, saliva, and milk. Drugs may be excreted unchanged or as metabolites.
Kinetics: Kinetics is the mathematical representation of the way in which drugs are removed from the body. The most common mechanism is first-order kinetics (see Figure 2-11).
A few drugs, such as aspirin and alcohol, exhibit zero-order kinetics. With zero-order kinetics, the rate of metabolism remains constant over time, and the same amount of drug is metabolized per unit of time regardless of dose. Zero-order kinetics occurs because the enzymes that metabolize these drugs can become saturated at usual therapeutic doses. If the dose of the drug is increased, the metabolism cannot increase above its maximum rate. With small doses, drugs with zero-order kinetics can metabolize the drug without buildup. With high doses, the metabolism of the drug cannot increase and the duration of action of the drug can be greatly prolonged (Figure 2-14). Small changes in the dose of these drugs may produce a large change in the serum concentration, leading to unexpected toxicity.
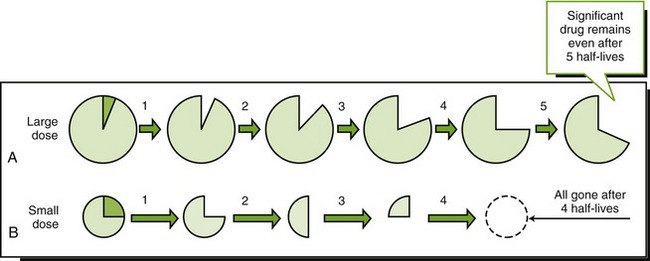
FIGURE 2-14 Zero-order kinetics. Large dose (A). Small dose (B). Disappearance of a drug whose metabolism is saturable: With small dose (B), the drug is metabolized more quickly than when a large dose (A) is given. With large dose the metabolism cannot increase, so it takes a long time for the body to clear the drug. The half-life varies with the dose of the drug.
Renal Route: Elimination of substances in the kidney can occur by the following three routes:
1. Glomerular filtration: Either the unchanged drug or its metabolites are filtered through the glomeruli and concentrated in the renal tubular fluid. This filtration process depends on the amount of plasma protein binding and the glomerular filtration rate. Bound drugs cannot be filtered and remain in the systemic circulation. Most drugs are managed by this mechanism.
2. Active tubular secretion: Active secretion transports the drug from the bloodstream across the renal tubular epithelial cells and into the renal tubular fluid. Glomerular filtration and active tubular secretion are relatively nonselective, and several compounds, both exogenous and endogenous (naturally occurring), can compete for transport.
3. Passive tubular diffusion: With most drugs, passive tubular diffusion (also termed passive reabsorption) plays a part in regulating the amount of drug in the tubular fluid. This process favors the reabsorption of nonionized, lipid-soluble compounds. The more ionized, less lipid-soluble metabolites have more difficulty penetrating the cell membranes of the renal tubules and are likely to be retained in the tubular fluid and eliminated in the urine. This process is also influenced by the urinary pH, which affects the amount of ionized and nonionized drug in the tubular fluid. By altering the pH of the urine, drug excretion can be favored in cases of poisoning or can be inhibited when a prolongation of the drug effect is desired. Weakly ionized acids or bases are excreted in the following fashion:
Extrarenal Routes: Certain drugs may be partially or completely eliminated via routes other than the kidney by the lungs. Gases used in general anesthesia are excreted across the lung tissue by a process of simple diffusion. Alcohol is also partially excreted from the lungs. (One can smell alcohol on someone’s breath if they have been drinking alcohol.) This fact is used when testing a driver’s breath for the presence of alcohol (Breathalyzer).
Biliary Excretion: Biliary excretion is the major route by which systemically absorbed drugs enter the gastrointestinal tract and are eliminated in the feces. Drugs excreted in the bile may also be reabsorbed from the intestines. Thus enterohepatic circulation, discussed earlier, prolongs a drug’s action.
Other: Two minor routes of elimination are in the milk and the sweat. The distribution of drugs in milk may be a potential source of undesirable effects for the nursing infant. Chapter 25 discusses dental drugs that can be given to nursing mothers.
Saliva: Drugs can also be excreted into the saliva. After drugs are excreted in the saliva, they are usually swallowed and their fate is the same as drugs ingested orally. The following drugs have been detected at significant levels in saliva after oral ingestion: aspirin, phenytoin, ampicillin, diazepam, penicillin VK, and phenobarbital. Present evidence suggests that most drugs that are secreted in the salivary glands enter saliva by simple diffusion, and their passage depends mainly on the lipid solubility of the drug. Thus a drug with high lipid solubility at plasma and salivary pH will readily enter saliva from plasma.
Drug levels in saliva have been studied to see if they can be used to monitor therapy with certain agents. For example, antiepileptic drug monitoring is essential for the rational treatment of epilepsy, and the measurement of these drugs in plasma is now routine. Assay of salivary concentrations of these drugs may become a reliable method that is not invasive for predicting plasma levels. More study is needed before salivary levels can replace measuring the blood levels of the drug.
Gingival Crevicular Fluid: Drugs may also be excreted in the gingival crevicular fluid (GCF). Drugs excreted in the GCF produce a higher level of drug in the gingival crevices, which can increase their usefulness in the treatment of periodontal disease. Some drugs, such as the tetracyclines, are concentrated in the GCF. This means that the drug level of tetracycline in the GCF will be several times (four or more times) higher than the blood level. This property makes the systemic use of a drug more effective within the gingival sulcus than one that is not concentrated.
ROUTES OF ADMINISTRATION AND DOSE FORMS
The route of administration of a drug affects both the onset and duration of response. Onset refers to the time it takes for the drug to begin to have its effect. Duration is the length of a drug’s effect. The routes of administration can be classified as enteral or parenteral. Drugs given by the enteral route are placed directly into the gastrointestinal tract by oral or rectal administration. Parenteral administration bypasses the gastrointestinal tract and includes various injection routes, inhalation, and topical administration. In practice, the term parenteral usually refers to an injection.
Although oral administration is considered the safest, least expensive, and most convenient route, the parenteral route has certain advantages. The injection results in fast absorption, which produces a rapid onset and a more predictable response than oral administration. The parenteral route is useful for emergencies, unconsciousness, lack of cooperation, or nausea. Some drugs must be administered by injection to remain active. The disadvantages of the parenteral route include the facts that asepsis must be maintained to prevent infection, an intravascular injection can occur by accident, administration by injection is more painful, it is difficult to remove the drug, adverse effects may be more pronounced, and self-medication is difficult. Parenteral therapy is also more dangerous and more expensive than oral medication. Figure 2-15 illustrates several common forms of drug administration.
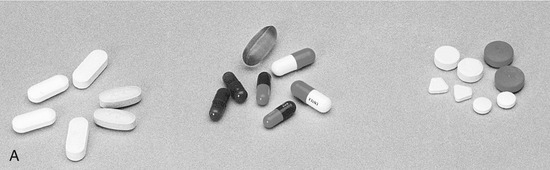
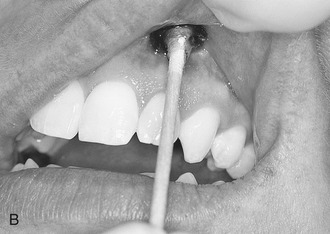


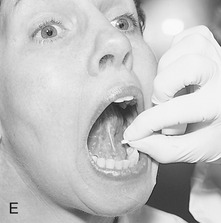
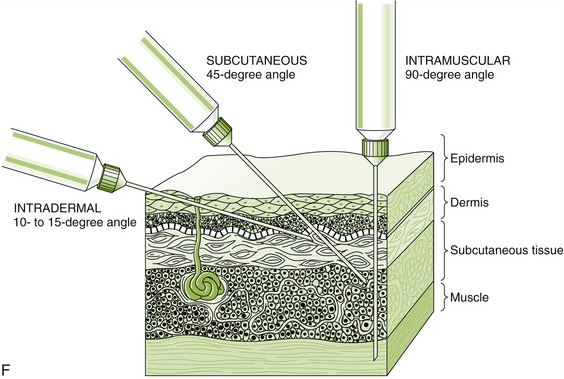
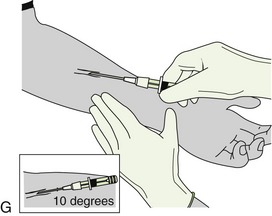
FIGURE 2-15 Routes of drug administration. A, Oral route in the form of pills, tablets, capsules, or liquids. B, Topical route by applying on the surface of the mucosa or skin. C, Transdermal route through a patch that continuously releases a controlled quantity of a medication through the skin. D, Inhalation route by breathing in a gaseous substance. E, Sublingual route by placing medication under the tongue (absorption takes place through the oral mucosa). F, Injection route. The type of drug determines how the injection is given: subcutaneous, directly under the skin; intramuscular, into a muscle; intradermal, into the skin. G, Example of an intravenous push medication administration. (A, From Young AP, Proctor DB: Kinn’s the medical assistant: an applied learning approach, ed 10, St. Louis, 2007, Saunders; B, from Daniel SJ, Harfst SA, Wilder RS: Mosby’s dental hygiene: concepts, cases, and competencies, ed 2, St. Louis, 2008, Mosby; C-F, from Chester GA: Modern medical assisting, Philadelphia, 1998, Saunders; G, from Clayton BD, Stock YN, Harroun RD: Basic pharmacology for nurses, ed 14, St Louis, 2007, Mosby.)
ORAL ROUTE
The oral route of administration is the simplest way to introduce a drug into the body. It allows the use of many different dose forms to obtain the desired results; tablets, capsules, and liquids are conveniently given. An advantage of this route is the large absorbing area present in the small intestine. Oral administration produces a slower onset of action than parenterally administered agents. One disadvantage of this route is that stomach and intestinal irritation may result in nausea and vomiting. Another disadvantage is that certain drugs, such as insulin, are inactivated by gastrointestinal tract acidity or enzymes.
When drugs are given orally, they are absorbed through the intestinal wall and then pass through the hepatic (liver) portal circulation, which can inactivate some drugs. This is termed the first-pass effect because the drug passes through the liver first before it circulates in the systemic circulation. During the drug’s first pass through the liver, it is metabolized (amount metabolized varies) and the amount of drug available to produce a systemic effect is reduced. Drugs with a high first-pass effect have a larger oral-to-parenteral dose ratio. This means that the dose required for an equivalent effect orally is much greater than the dose needed when used parenterally. Because morphine has a high first-pass effect, the oral dose needed to produce an equivalent effect is much larger than its parenteral dose.
Blood levels obtained after oral administration are less predictable than those obtained parenterally. The presence of food in the stomach, the pathologic condition of the gastrointestinal tract, the effects of gastric acidity, and passage through the hepatic portal circulation can alter blood levels. Drug interactions can occur when two drugs are present in the stomach. The oral route necessitates greater patient cooperation.
RECTAL ROUTE
Drugs may be given rectally as suppositories, creams, or enemas. Rectal administration can be used if a patient is vomiting or unconscious. This route may be used for either a local (e.g., hemorrhoids) or a systemic (e.g., antiemetic) effect. Because most drugs are poorly and irregularly absorbed rectally, this route is not often used to achieve a systemic drug effect. In addition, patient acceptance of this route is poor.
INTRAVENOUS ROUTE
Intravenous administration produces the most rapid drug response, with an almost immediate onset of action. Because the injection is made directly into the blood, the absorption phase is bypassed. Another advantage of the intravenous route is that it produces a more predictable response than oral administration because factors that affect drug absorption have been eliminated. It is also the route of choice for an emergency situation. The disadvantages of administration include phlebitis caused by local irritation, drug irretrievability (cannot get it back), allergy, and side effects related to high plasma concentrations of the drug.
INTRAMUSCULAR ROUTE
Absorption of drugs injected into the muscle occurs because of the high blood flow through skeletal muscles. Somewhat irritating drugs may be tolerated if given by the intramuscular route. This route may also be used for injection of suspensions to provide a sustained effect. Injections are usually made in the deltoid region or gluteal mass.
SUBCUTANEOUS ROUTE
The subcutaneous route involves the injection of solutions or suspensions of drugs into the subcutaneous areolar tissue to gain access to the systemic circulation. If irritating solutions are injected, sterile abscesses may result. Insulin is commonly administered by this route.
INTRADERMAL ROUTE
Small amounts of drugs, such as local anesthetics, can be injected into the epidermis of the skin to provide local anesthesia. With this type of injection, a small bump (bleb) rises as the liquid is injected just under the skin. The tuberculosis skin test is performed using the intradermal route.
INTRATHECAL ROUTE
Intrathecal administration involves the injection of solutions into the spinal subarachnoid space. This may be used for spinal anesthesia or for the treatment of certain forms of meningitis.
INTRAPERITONEAL ROUTE
The intraperitoneal route involves placing fluid into the peritoneal cavity, where exchange of substances can occur. A drug may be absorbed through the mesenteric veins. This route of administration is also used for peritoneal dialysis. In this case, the substances are passing from the body to the fluid. Large volumes of fluids are slowly run into the peritoneal cavity. A waiting period of several hours allows the waste products from the body to be exchanged with the fluid in the peritoneal cavity. The fluid is removed, and the body’s waste products are carried out in the fluid. This process is used as a substitute for the failing kidney to manage patients with renal failure.
INHALATION ROUTE
The inhalation route may be used in the administration of the gaseous, microcrystalline, liquid, or powdered form of drugs. This route of administration may be used for either local or systemic effects. An example of inhalers being used for their local effects are those used to treat asthma. After inhalation, the drug is deposited on the bronchiolar endothelium and exerts its action by producing bronchodilation or reducing inflammation. Inhalation of aerosolized liquid in fine droplets also produces a local effect. Today’s oral metered dose inhalers contain hydrofluorocarbons that do not harm the ozone. These inhalers contain finely powdered drugs that are also inhaled into the lungs. One advantage of the use of the powdered form is that inhalation must continue until the visible powder is gone. General anesthetics in the form of volatile liquids, such as isoflurane, or gases, such as nitrous oxide (N2O) and oxygen, are examples of the use of the inhalation route for systemic effects. This route of administration is popular for the abuse of many drugs (smoked or even inhaled) because of the quick onset of action and the lack of need for needles.
TOPICAL ROUTE
Topical routes consist of application to body surfaces. Topical applications are administered to the skin, the oral mucosa, and even sublingually (under the tongue). The inhalation route could even be called topical. Drugs used topically may be intended to produce either local or systemic effects.
Because most drugs do not penetrate intact skin, application to the skin is generally used for local effects. Corticosteroids are applied to inflamed or irritated skin. Intravaginal creams or suppositories or solutions or suspensions instilled into the eye or ear are other ways to administer topical agents to produce local effects.
Rarely, systemic side effects (unintended) can occur from the topical administration of drugs for their local effect. One example is the administration of topical corticosteroids over a large proportion of the body, resulting in symptoms of systemic toxicity (Cushing’s syndrome). If an occlusive dressing (commercial plastic wrap or a plastic suit) is used or if the surface is abraded, inflamed, or sloughing, the chance of side effects increases dramatically. In the oral cavity, interruptions in the mucous membranes or mucosal inflammation increase the likelihood of a systemic effect. Local anesthetics sprayed into the mouth may be adsorbed and produce a blood level equivalent to that produced by intravenous administration.
Examples of drugs applied topically for a systemic effect include transdermal patches and sublingual spray or tablet administration. Drugs that often produce allergic reactions should not be administered topically because sensitization occurs more readily than when used orally.
Subgingival Strips and Gels: A dental-specific topical application involves the placement of drug-impregnated strips or gels subgingivally. Systemic effects are minimized because small doses can be used when drugs are administered via this route. Doxycycline gel (Atridox) and a chlorhexidine-containing chip (PerioChip) are examples of agents administered into the gingival crevice.
Transdermal Patch: Transdermal drug delivery systems (drug patches) are designed to provide continuous controlled release of medication through a semipermeable membrane over a given period after application of drug to the intact skin. This eliminates the need for repeated oral dosing. Examples of patches on the market include scopolamine (Transderm-Scop), nitroglycerin (Transderm-Nitro, Nitrodisc, Nitro-Dur), clonidine (Catapres-TTS), estrogens (Estraderm, Climara), fentanyl (Duragesic), and nicotine transdermal patches (NicoDerm, Nicobid).
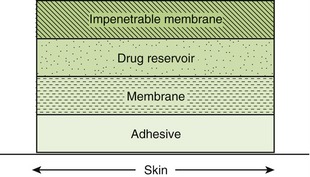
Most patches consist of several layers. Beginning with the skin, the layers are as follows: adhesive (to stick to the skin), membrane (to control the rate of drug release), drug reservoir (where the drug is stored), and a backing that is impenetrable to the drug (to keep the drug from evaporating into the air) (Figure 2-16). Before use, the protective backing must be removed. The most common problems with transdermal patches are local irritation, erythema, and edema. These problems can be minimized by rotating the location of the patch. Patches are designed to be changed daily, every few days, or weekly, depending on the drug.
Topical Anesthesia: Topical anesthetics are applied directly to the mucous membranes and rapidly absorbed into the systemic circulation, providing the patient with injection-free local anesthesia. An example of this type of anesthesia is the combination of lidocaine and prilocaine (Oraqix).
Two ways in which drugs can be applied topically are sublingual (SL) and buccal routes. The mucous membranes of the oral cavity provide a convenient absorbing surface for the systemic administration of drugs, which can be placed under the tongue (sublingual) or on other areas of the oral mucosa (buccal pouch). Absorption of many drugs into the systemic circulation occurs rapidly. An example of this effect is the fast onset of action of nitroglycerin sublingual tablets to treat acute anginal pain. Drugs that are susceptible to degradation by the gastrointestinal tract and even by the liver, such as testosterone, are safely administered as sublingual tablets because they avoid both the first-pass effect and gastrointestinal acid and enzymes.
OTHER ROUTES
Drugs, such as progestins (Norplant), can be implanted under the skin to release a drug over a prolonged duration (5 years). Pumps that deliver drugs intravenously can be implanted in the body. When insulin pumps are used, they can be programmed externally using a calculator-like keyboard.
Dose Forms
Table 2-2 lists the usual dose forms. The most commonly used dose forms in dentistry are the tablet and capsule given orally. Liquid solutions or suspensions are often prescribed for children. Sometimes drugs are given in solution or suspension when a liquid form is desired. Liquid forms of a drug are often used for children. For injection, the drug may be in solution, such as a local anesthetic, or it may be in a suspension, such as procaine penicillin G, when a longer duration of action is desired. Mouthwashes containing alcohol are also recommended by dental health care workers. Elixirs (contain alcohol) and syrups (contain sugar) are children’s dose forms.
TABLE 2-2
| Form | Definition | Example |
| Tablet | Molded or compressed medicinal substance with inert binder included to make a hard mass | Acetaminophen tablet |
| Capsule | Gelatin shell that disintegrates in water to administer solids or liquids | Tetracycline capsule |
| Pill | Globular or ovoid dose form made by incorporating medicinal agents with other binders to make a plastic mass; obsolete | Ferrous carbonate pill |
| Lozenge, troche | Flavored dose form designed to be held in the mouth to dissolve or disintegrate slowly | Cough drop |
| Suppository | Single-dose medication in waxy or fatty conical or ovoid shape that liberates active ingredient after insertion into the rectum or vagina for local or systemic effects | Glycerin suppository |
| Solution | One-phase system of two or more chemical components | Saline water |
| Elixir | Sweetened hydroalcoholic solution containing flavoring materials | Acetaminophen and codeine elixir; diphenhydramine |
| Syrup | Nearly saturated aqueous solution of sugar | Dextromethorphan syrup |
| Tincture | Alcoholic or hydroalcoholic solution of drugs | Iodine tincture |
| Spirit | Solution of volatile substance in alcohol | Aromatic ammonia spirit |
| Lotion | Liquid suspension that can be protective | Hand lotion |
| Emulsion | Preparation of two immiscible liquids, usually water and oil, one dispersed as small globules in the other | Liquid petrolatum emulsion |
| Suspension | Dispersion containing finely divided insoluble material suspended in a liquid medium | Amoxicillin suspension |
| Cream | Emulsions that contain an oily and aqueous phase; external phase aqueous | Hydrocortisone cream |
| Ointment | Semisolid preparation for external use that is a consistency that can be applied by rubbing; external phase oily | Hydrocortisone ointment, A+D ointment |
| Transdermal patch | A permeable polymer membrane backed with a drug reservoir designed to provide controlled release of medication over a given period after application to intact skin | Nitroglycerin, scopolamine, fentanyl, nicotine |
| Aerosol spray | Solution of volatile liquids with a propellant that delivers drugs to area | Albuterol inhaler, foot spray |
| Intradermal implant | Small pellets implanted under skin that allow drugs to be released slowly | Norplant |
| Micropump | An implanted pump that delivers drug via a needle | Insulin pump |
FACTORS THAT ALTER DRUG EFFECTS
When a drug is administered, the following factors may influence or modify the drug’s effect:
• Patient compliance: Through either lack of understanding or lack of motivation, patients often take medication incorrectly or not at all. Sometimes this may result from faulty communication, inadequate patient education, or the patient’s health belief system. Thus poor patient compliance can be an important factor in a therapeutic failure.
• Psychologic factors: The attitude of the prescriber and the dental staff can affect the efficacy of the drug prescribed. A placebo is a dose form that looks like the active agent but contains no active ingredients (the “sugar pill”). The magnitude of the placebo effect depends on the patient’s perception, and there is large individual variation. Health care providers can maximize the drug’s effect to achieve an improved therapeutic result by talking up the drug. This may account for the popularity of herbs and plants.
• Tolerance: A patient may exhibit tolerance to many drugs, including the sedative-hypnotics and the opioids. Drug tolerance is defined as the need for an increasingly larger dose of the drug to obtain the same effects as the original dose or the decreased effect produced after repeated administration of a given dose of the drug. When a patient becomes tolerant to one drug, tolerance to other drugs with similar pharmacologic actions occurs. This is termed cross-tolerance. If tolerance develops, a normal sensitivity to the drug’s effect may be restored by ceasing administration of the drug. Tachyphylaxis is the very rapid development of tolerance, often within hours.
• Pathologic state: Diseased patients may respond to the administration of medication differently than other patients. For example, patients with hyperthyroidism are extremely sensitive to the toxic effects of epinephrine. Hepatic or renal disease influences the metabolism and excretion of drugs, potentially leading to an increased duration of drug action. With repeated doses in diseased patients, the serum level of a drug may become toxic.
• Time of administration: The time a drug is administered, especially in relation to meals, alters the response to that drug. Certain drugs with a sedative action are best administered at bedtime to minimize the sedation experienced by the patient.
• Route of administration: The effect of the route of administration on the onset and duration of action of a drug was discussed previously.
• Sex: The sex of the patient can alter a drug’s effect. Women may be more sensitive than men to certain drugs, perhaps because of their smaller size or their hormones. Pregnancy alters the effect of certain drugs. Women of child-bearing age should avoid teratogenic drugs, and the oral health care provider should determine whether the patient is pregnant before administering any agent.
• Genetic variation: Many differences in patient response to drugs have been associated with variations in ability to metabolize certain drugs. This difference may account for the fact that certain populations have a higher incidence of adverse effects to some drugs—a genetic predisposition.
• Drug interactions: A drug’s effect may be modified by previous or concomitant administration of another drug. There are many mechanisms by which drug interactions may modify a patient’s treatment.
• Age and weight: The dose of a drug administered to children should be reduced from the adult dose. Age or weight has been suggested as a method of calculating a child’s dose. Because of the great variability of weight in relation to age, the child’s weight should be used to determine the child’s dose. Because a child is not just a small adult, the manufacturer’s recommendations for children’s dosing would be best. Older adults may respond differently to drugs than younger patients. Whether this is solely because of changes in renal or liver function or whether being elderly patients predisposes this sensitivity is controversial.
• Environment: The environment contains many substances that may affect the action of drugs. Smoking induces enzymes, so higher doses of benzodiazepines are needed to produce the same effect as compared to nonsmokers. Some chemical contaminants, such as pesticides or solvents, can have an effect on a drug’s action.
• Other: The action of drugs can be altered by the patient-provider interaction. If the patient “believes” in the substance or process (drug/herb//incantation) being used, the patient’s opinion will enhance the drug’s effect. The attitude of both the patient and the provider can alter the physiology of the body. These actions may account for the positive effect of many mental exercises (e.g., meditation).
CLINICAL SKILLS ASSESSMENT
1. Define and differentiate between the potency and efficacy of a drug.
2. Describe the dose-response curve using the terms ED50 (effective dose) and LD50 (lethal dose).
3. Define the term pharmacokinetics. Name the four categories involved.
4. Define the major routes of drug administration, including the following:
5. State the dose forms most often used in dentistry.
6. Explain the influence of pH on the dissociation characteristics of weak acids and weak bases.
7. Explain each of the steps involved in oral absorption, including the following:
8. Define the 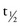 , or half-life, of a drug and state its significance.
, or half-life, of a drug and state its significance.
9. Though an elderly patient appears healthy and weighs 110 pounds, what are your concerns regarding drug distribution?
10. Define the following terms:
11. What are the different ways that drugs can be metabolized?
12. State the major route of drug excretion.
13. Explain how metabolism can be altered by an effect on liver microsomal enzymes.
 Please visit http://evolve.elsevier.com/Haveles/pharmacology for review questions and additional practice and reference materials.
Please visit http://evolve.elsevier.com/Haveles/pharmacology for review questions and additional practice and reference materials.
*Ignoring the effect on the stomach of different nonalcoholic fluids or food ingested.