Chapter 17 Eyes and Ears
Development of Eyes and Related Structures
Early eye development results from a series of inductive signals and is first evident at the beginning of the fourth week, when optic grooves appear in the cranial neural folds (Fig. 17-1A and B). As the neural folds fuse, the optic grooves evaginate to form hollow diverticula—optic vesicles—which project from the wall of the forebrain into the adjacent mesenchyme (Fig. 17-1C). Formation of the optic vesicles is induced by the mesenchyme adjacent to the developing brain. As the optic vesicles enlarge, their connections with the forebrain constrict to form hollow optic stalks (Fig. 17-1D).
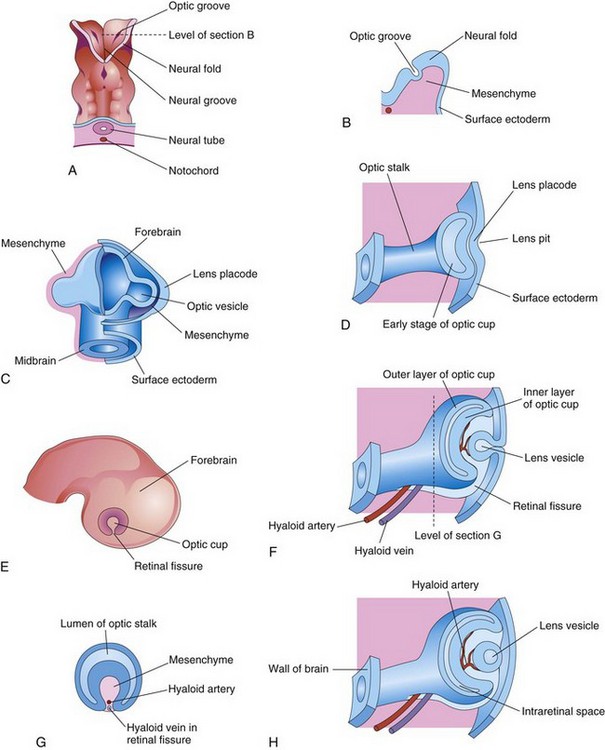
Figure 17–1 A, Dorsal view of the cranial end of an embryo at approximately 22 days, showing the optic grooves, the first indication of eye development. B, Transverse section of a neural fold, showing the optic groove. C, Forebrain of an embryo (approximately 28 days). D, F, and H, The developing eye, showing successive stages in the development of the optic cup and the lens vesicle. E, Lateral view of an embryo (approximately 32 days), showing the external appearance of the optic cup. G, Transverse section of the optic stalk, showing the retinal fissure and its contents.
An inductive signal passes from the optic vesicles and stimulates the surface ectoderm to thicken and form lens placodes, the primordia of the lenses (Fig. 17-1C). These placodes invaginate and sink deep to the surface ectoderm, forming lens pits (Figs. 17-1D and 17-2). The edges of each lens pit approach and fuse to form spherical lens vesicles (Fig. 17-1F and H), which soon lose their connection with the surface ectoderm. As the lens vesicles develop, the optic vesicles invaginate to form double-walled optic cups (Figs. 17-1H and 17-2), with the lens becoming infolded by the rim of the optic cup (Fig. 17-3A). By this stage, the lens vesicles have entered the cavities of the optic cups (Fig. 17-4). Linear grooves—retinal (optic) fissures—develop on the ventral surface of the optic cups and along the optic stalks (Figs. 17-1E to H and 17-3A to D). The retinal fissures contain vascular mesenchyme from which the hyaloid blood vessels develop. The hyaloid artery supplies the inner layer of the optic cup, the lens vesicle, and the mesenchyme in the optic cup (Figs. 17-1H and 17-3). As the edges of the retinal fissure fuse, the hyaloid vessels are enclosed within the primordial optic nerve (Fig. 17-3C to F). Distal parts of the hyaloid vessels eventually degenerate, but proximal parts persist as the central artery and vein of the retina (Fig. 17-5D).
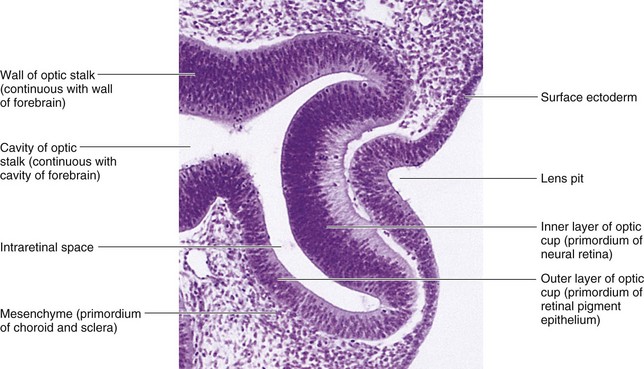
Figure 17–2 Photomicrograph of a sagittal section of the eye of an embryo at approximately 32 days. Observe the primordium of the lens (invaginated lens placode), the walls of the optic cup (primordium of the retina), and the optic stalk (primordium of the optic nerve).
(From Moore KL, Persaud TVN, Shiota K: Color Atlas of Clinical Embryology, 2nd ed. Philadelphia, WB Saunders, 2000.)
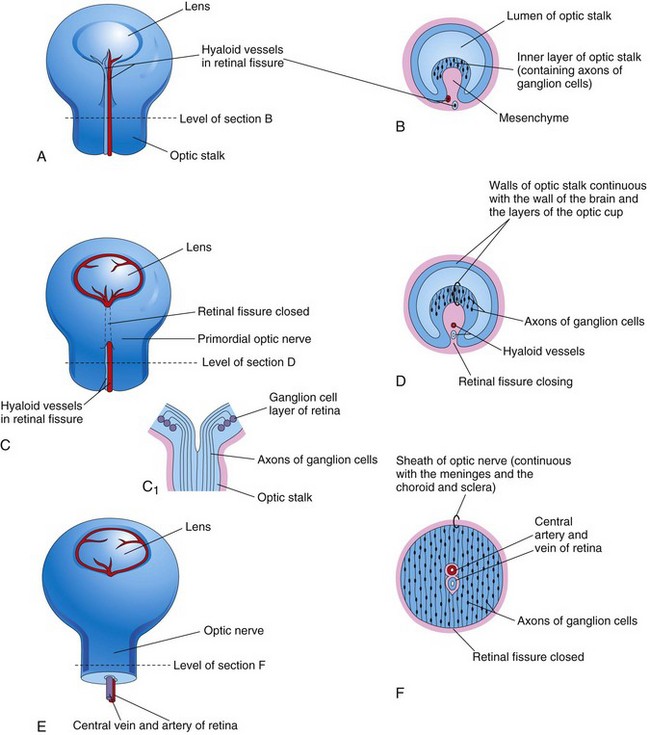
Figure 17–3 Closure of the retinal fissure and formation of the optic nerve. A, C, and E, Views of the inferior surface of the optic cup and optic stalk, showing progressive stages in the closure of the retinal fissure. C1, Longitudinal section of a part of the optic cup and optic stalk, showing the axons of the ganglion cells of the retina growing through the optic stalk to the brain. B, D, and F, Transverse sections of the optic stalk, showing successive stages in the closure of the retinal fissure and formation of the optic nerve.
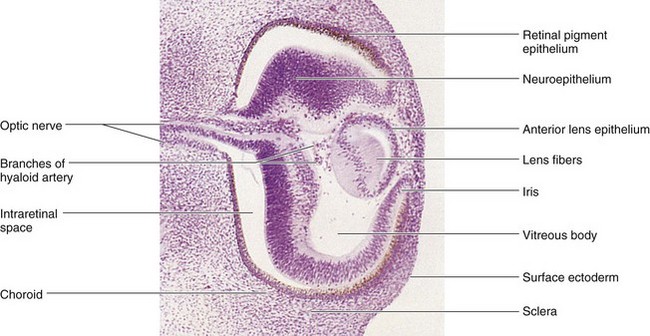
Figure 17–4 Photomicrograph of a sagittal section of the eye of an embryo at approximately 44 days. Observe that the posterior wall of the lens vesicle forms the lens fibers. The anterior wall does not change appreciably as it becomes the anterior lens epithelium.
(From Nishimura H [ed]: Atlas of Human Prenatal Histology. Tokyo, Igaku-Shoin, 1983.)
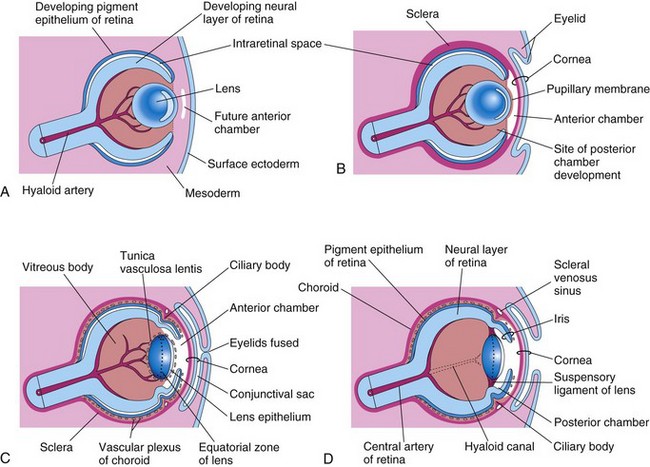
Figure 17–5 Sagittal sections of the eye, showing successive developmental stages of the lens, retina, iris, and cornea. A, At 5 weeks. B, At 6 weeks. C, At 20 weeks. D, Newborn infant.
Development of Retina
The retina develops from the walls of the optic cup (Figs. 17-1 and 17-2). In the invaginated optic cup, the deeper, thinner layer becomes the retinal pigment epithelium, whereas the more superficial, thicker layer differentiates into the neural retina. The two retinal layers are separated by an intraretinal space (Figs. 17-1H and 17-4), which is the original cavity of the optic cup. Before birth, this space gradually disappears as the two layers of the retina fuse (Fig. 17-5D). Because the optic cup is an outgrowth of the forebrain, the layers of the optic cup are continuous with the wall of the brain (Fig. 17-1H). Under the influence of the developing lens, the inner layer of the optic cup proliferates to form a thick neuroepithelium (Fig. 17-4). Subsequently, the cells of the inner layer closest to the retinal pigment epithelium differentiate into the neural retina, the light-sensitive region of the retina (Fig. 17-4). This region contains photoreceptors (rods and cones) and the cell bodies of neurons (e.g., ganglion cells). Because the optic vesicle invaginates as it forms the optic cup, the neural retina is “inverted”; that is, light-sensitive parts of the photoreceptor cells are adjacent to the retinal pigment epithelium. As a result, light must pass through the thickest part of the retina before reaching the receptors; however, because the retina is transparent, it does not form a barrier to light.
The axons of the ganglion cells in the superficial layer of the neural retina grow proximally in the wall of the optic stalk to the brain (Fig. 17-3A). The cavity of the optic stalk is gradually obliterated as the axons of the ganglion cells form the optic nerve (Fig. 17-3F). Myelination of the optic nerve fibers begins late in the fetal period and is completed by the 10th week after birth. Molecular studies have shown that the homeobox genes PAX6 and OTX2 regulate retinal differentiation and pigment formation, respectively.
Retinal Detachment
Congenital detachment of the retina occurs when the inner and outer layers of the optic cup do not fuse during the fetal period to form the retina and obliterate the intraretinal space (Figs. 17-3 and 17-5). The separation of the neural and pigmented layers may be partial or complete. Retinal detachment may result from unequal rates of growth of the two retinal layers; as a result, the layers of the optic cup are not in perfect apposition. Although separated from the retinal pigment epithelium, the neural retina retains its blood supply (central artery of retina). Normally, the retinal pigment epithelium becomes firmly fixed to the choroid, but its attachment to the neural retina is not firm; hence, retinal detachment is not uncommon.
Coloboma of Retina
Retinal coloboma is a defect that is characterized by a localized gap in the retina, usually inferior to the optic disc. The defect is bilateral in most cases. A typical coloboma results from defective closure of the retinal fissure.
Coloboma of Iris
In infants with coloboma of the iris, a defect in the inferior sector of the iris or the pupillary margin gives the pupil a keyhole appearance (Fig. 17-6). The coloboma may be limited to the iris, or it may extend deeper and involve the ciliary body and retina. A typical coloboma results from failure of closure of the retinal fissure during the sixth week. The defect may be genetically determined, or it may be caused by environmental factors. A simple coloboma of the iris is frequently hereditary and is transmitted as an autosomal dominant characteristic.

Figure 17–6 Coloboma of the left iris. Observe the defect in the inferior part of the iris (at the 6 o’clock position). The defect represents failure of fusion of the retinal fissure.
(Reprinted from Otolaryngologic Clinics of North America 40(1), Guercio J, Martyn L, Congenital malformations of the eye and orbit, 113-140, Copyright 2007, with permission from Elsevier.)
Development of Choroid and Sclera
The mesenchyme surrounding the optic cup differentiates into an inner, vascular layer—the choroid—and an outer, fibrous layer—the sclera (see Figs. 17-5C and 17-7). At the rim of the optic cup, the choroid forms the cores of the ciliary processes, consisting chiefly of capillaries supported by delicate connective tissue.
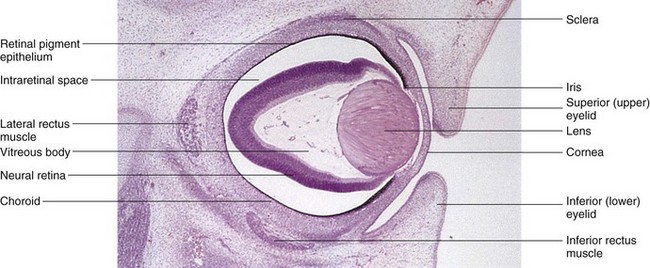
Figure 17–7 Photomicrograph of a sagittal section of the eye of an embryo (x50) at approximately 56 days. Observe the developing neural retina and the retinal pigment epithelium. Note the large intraretinal space between the two layers of the retina.
(From Moore KL, Persaud TVN, Shiota K: Color Atlas of Clinical Embryology, 2nd ed. Philadelphia, WB Saunders, 2000.)
Development of Ciliary Body
The ciliary body is a wedge-shaped extension of the choroid (Fig. 17-5C and D). Its medial surface projects toward the lens, forming the ciliary processes. The pigmented part of the ciliary epithelium is derived from the outer layer of the optic cup and is continuous with the retinal pigment epithelium. The nonpigmented part of the ciliary epithelium represents the anterior prolongation of the neural retina, in which no neural elements develop. The smooth ciliary muscle, responsible for focusing the lens and the connective tissue in the ciliary body, develop from mesenchyme at the edge of the optic cup between the anterior scleral condensation and the ciliary pigment epithelium.
Development of Iris
The iris develops from the rim of the optic cup, which grows inward and partially covers the lens (Fig. 17-5D). The epithelium of the iris represents both layers of the optic cup; it is continuous with the double-layered epithelium of the ciliary body and with the retinal pigment epithelium and neural retina. The connective tissue framework of the iris is derived from neural crest cells that migrate into the iris. The dilator pupillae and sphincter pupillae muscles of the iris are derived from the neuroectoderm of the optic cup. These smooth muscles result from a transformation of epithelial cells into smooth muscle cells.
Development of Lens
The lens develops from the lens vesicle, a derivative of the surface ectoderm (Fig. 17-1). The anterior wall of the lens vesicle becomes the subcapsular lens epithelium (Fig. 17-5C). The nuclei of the tall columnar cells that form the posterior wall of the lens vesicle undergo dissolution. These cells lengthen considerably to form highly transparent epithelial cells, the primary lens fibers. As these fibers grow, they gradually obliterate the cavity of the lens vesicle (Figs. 17-5A to C). The rim of the lens—the equatorial zone—is located midway between the anterior and the posterior poles of the lens. The cells in the equatorial zone are cuboidal; as they elongate, they lose their nuclei and become secondary lens fibers (Fig. 17-8). These fibers are added to the external sides of the primary lens fibers. Lens formation involves the expression of L-Maf (lens-specific Maf) and other transcription factors in the lens placode and vesicle. The transcription factors Pitx3 and GAT-3 are also essential for the formation of the lens.
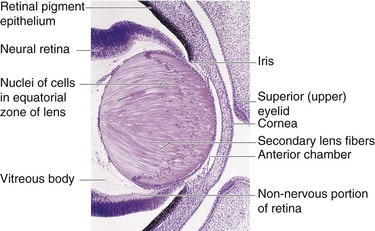
Figure 17–8 Photomicrograph of a sagittal section of a portion of the developing eye of an embryo at approximately 56 days. Observe that the lens fibers have elongated and obliterated the cavity of the lens vesicle.
(From Moore KL, Persaud TVN, Shiota K: Color Atlas of Clinical Embryology, 2nd ed. Philadelphia, WB Saunders, 2000.)
Although secondary lens fibers continue to form during adulthood and the lens increases in diameter as a result, the primary lens fibers must last a lifetime. The developing lens is supplied with blood by the distal part of the hyaloid artery (Figs. 17-4 and 17-5); however, it becomes avascular in the fetal period when this part of the artery degenerates (Fig. 17-5D). After this, the lens depends on diffusion from the aqueous humor in the anterior chamber of the eye, which bathes its anterior surface, and from the vitreous humor in other parts. The lens capsule is produced by the anterior lens epithelium. The lens capsule represents a greatly thickened basement membrane and has a lamellar structure. The former site of the hyaloid artery is indicated by the hyaloid canal in the vitreous body (Fig. 17-5D); this canal is usually inconspicuous in the living eye.
Persistence of Hyaloid Artery
The distal part of the hyaloid artery normally degenerates as its proximal part becomes the central artery of the retina. If part of the hyaloid artery persists distally, it may appear as a freely moving, nonfunctional vessel, a wormlike structure projecting from the optic disc, or as a fine strand traversing the vitreous body. In other cases, the hyaloids artery remnant may form a cyst.
The vitreous body forms within the optic cup (Fig. 17-5C). It is composed of vitreous humor, an avascular mass of transparent, gel-like, intercellular substance.
Development of Aqueous Chambers
The anterior chamber of the eye develops from a cleft-like space that forms in the mesenchyme located between the developing lens and the cornea (Fig. 17-5A and B). The posterior chamber of the eye develops from a space that forms in the mesenchyme posterior to the developing iris and anterior to the developing lens (Fig. 17-5D). After the lens is established, it induces the surface ectoderm to develop into the epithelium of the cornea and conjunctiva. When the pupillary membrane disappears and the pupil forms, the anterior and posterior chambers of the eye communicate with each other through the scleral venous sinus. This sinus encircles the anterior chamber and allows aqueous humor to flow from the anterior chamber of the eye to the venous system.
Congenital Glaucoma
Intraocular tension (abnormal elevation of intraocular pressure) occurs because of an imbalance between the production of aqueous humor and its outflow. This imbalance may result from abnormal development of the scleral venous sinus (Fig. 17-5D). Congenital glaucoma is usually genetically heterogeneous, but the condition may result from rubella infection during early pregnancy (see Fig. 19-16B). It has been shown that the gene CYP1B1 is responsible for the majority of primary congenital glaucomas.
Congenital Cataracts
In congenital cataracts, the lens is opaque and frequently appears grayish white. Blindness results. Many lens opacities are inherited, with dominant transmission being more common than recessive, or sex-linked, transmission. Some congenital cataracts are caused by teratogenic agents—particularly the rubella virus (see Fig. 19-16A)—that affect the early development of the lenses. The lenses are vulnerable to the rubella virus between the fourth and seventh weeks, when primary lens fibers are forming. Physical agents, such as radiation, can also damage the lens and produce cataracts (see Chapter 19).
Development of Eyelids
The eyelids develop during the sixth week from the neural crest–derived mesenchyme and from two folds of skin that grow over the cornea (Fig. 17-5B). The eyelids adhere to one another at approximately the 10th week and remain fused until the 26th to 28th weeks (Fig. 17-5C). The palpebral conjunctiva lines the inner surface of the eyelids. The eyelashes and glands in the eyelids are derived from the surface ectoderm (Chapter 18). The connective tissue and tarsal plates develop from mesenchyme in the developing eyelids. The orbicularis oculi muscle is derived from the mesenchyme in the second pharyngeal arch (see Chapter 10).
Congenital Ptosis of Eyelid
Drooping of the superior (upper) eyelids at birth is relatively common. Congenital ptosis may result from dystrophy of the levator palpebrae superioris muscle. Ptosis occurs more rarely as a result of prenatal injury or dystrophy of the superior division of the oculomotor nerve (CN III) that supplies this muscle. Congenital ptosis may be transmitted as an autosomal dominant trait.
Development of Lacrimal Glands
The lacrimal glands are derived from a number of solid buds from the surface ectoderm. The buds branch and canalize to form lacrimal excretory ducts and the alveoli of the glands. The lacrimal glands are small at birth and do not function fully until approximately 6 weeks; hence, the newborn infant does not produce tears when crying.
Development of EarS
The ears are composed of external, middle, and internal anatomical parts. The external and middle parts of the ears regulate the transference of sound waves from the exterior to the internal ears; this process converts the sound waves into nerve impulses. The internal ears are concerned with both hearing and balance.
Development of Internal Ears
The internal ears are the first of the three parts of the ear to develop. Early in the fourth week, a thickening of the surface ectoderm—the otic placodes—appears on each side of the embryo at the level of the caudal part of the hindbrain (Fig. 17-9A and B). Inductive influences from the notochord and paraxial mesoderm stimulate the surface ectoderm to form the otic placodes. Fibroblast growth factor 3 (FGF-3) may play a role in this process. Each otic placode soon invaginates and sinks deep to the surface ectoderm into the underlying mesenchyme, forming an otic pit (Fig. 17-9C and D). The edges of the otic pit come together and fuse to form an otic vesicle (Fig. 17-9E to G). The otic vesicle then loses its connection with the surface ectoderm, and a diverticulum grows from the otic vesicle and elongates to form the endolymphatic duct and sac (Fig. 17-10A to E). As the otic vesicle grows, two regions become visible:
• A dorsal utricular part from which the endolymphatic duct, utricle, and semicircular ducts arise
• A ventral saccular part, which gives rise to the saccule and cochlear duct, in which the spiral organ (of Corti) is located
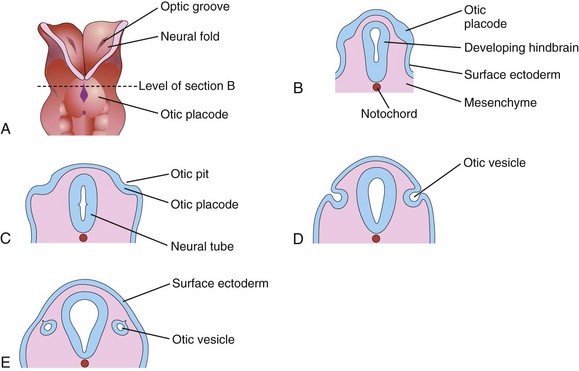
Figure 17–9 Early development of the internal ear. A, Dorsal view of a 4-week embryo (at approximately 22 days). B, C, D, and E, Schematic coronal sections, showing successive stages in the development of the otic vesicles.
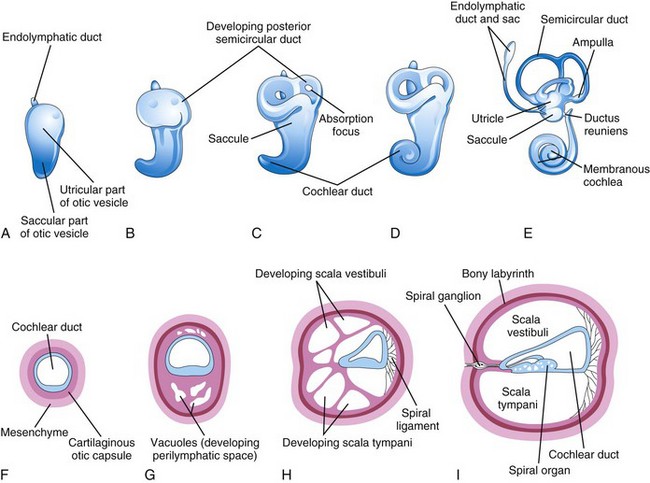
Figure 17–10 The otic vesicle, showing the development of the membranous and bony labyrinths of the internal ear. A to E, Lateral views, showing successive stages in the development of the otic vesicle into the membranous labyrinth from the fifth to eighth weeks. F to I, Sections through the cochlear duct, showing successive stages in the development of the spiral organ and the perilymphatic space from the eighth to 20th weeks.
Three disc-like diverticula grow out from the utricular part of the primordial membranous labyrinth. The central parts of these diverticula fuse and disappear (Fig. 17-10B to E). The peripheral unfused parts of the diverticula become the semicircular ducts, which are attached to the utricle and are later enclosed in the semicircular canals of the bony labyrinth. Localized dilatations, the ampullae, develop at one end of each semicircular duct (Fig. 17-10E). Specialized receptor areas—cristae ampullares—differentiate in these ampullae as well as in the utricle and saccule.
From the ventral saccular part of the otic vesicle, a tubular diverticulum—the cochlear duct—grows and coils to form the membranous cochlea (Fig. 17-10C to E). A connection of the cochlea with the saccule, the ductus reuniens, soon forms. The spiral organ differentiates from cells in the wall of the cochlear duct (Fig. 17-10F to I). Ganglion cells of the vestibulocochlear nerve (CN VIII) migrate along the coils of the membranous cochlea and form the spiral ganglion. Nerve processes extend from this ganglion to the spiral organ, where they terminate on hair cells. The cells in the spiral ganglion retain their embryonic bipolar condition.
Inductive influences from the otic vesicle stimulate the mesenchyme around the otic vesicle to differentiate into a cartilaginous otic capsule (Fig. 17-10F). The cartilaginous otic capsule later ossifies to form the bony labyrinth of the internal ear. Transforming growth factor β1 may play a role in modulating epithelial–mesenchymal interaction in the internal ear and directing the formation of the otic capsule. As the membranous labyrinth enlarges, vacuoles appear in the cartilaginous otic capsule that soon coalesce to form the perilymphatic space. As a result, the membranous labyrinth becomes suspended in perilymph (fluid in the perilymphatic space). The perilymphatic space related to the cochlear duct develops two divisions, the scala tympani and scala vestibuli (Fig. 17-10H and I). The internal ear reaches its adult size and shape by the middle of the fetal period (20-22 weeks).
Development of Middle Ears
Development of the tubotympanic recess (Fig. 17-11B) from the first pharyngeal pouch is described in Chapter 10. The proximal part of the tubotympanic recess forms the pharyngotympanic tube (auditory tube). The distal part of the tubotympanic recess expands and becomes the tympanic cavity (Fig. 17-11C), which gradually envelops the auditory ossicles (malleus, incus, and stapes), their tendons and ligaments, and the chorda tympani nerve. All these structures receive a virtually complete epithelial investment. An epithelial-type organizer, located at the tip of the tubotympanic recess, probably plays a role in the early development of the middle ear cavity by inducing programmed cell death—apoptosis. The malleus and the incus develop from the cartilage of the first pharyngeal arch, whereas the stapes develops from the cartilage of the second arch. The tensor tympani, the muscle attached to the malleus, is derived from the mesenchyme in the first pharyngeal arch, and the stapedius muscle is derived from the second pharyngeal arch. During the late fetal period, expansion of the tympanic cavity gives rise to the mastoid antrum, located in the temporal bone. The mastoid antrum is almost adult size at birth; however, no mastoid cells are present in newborn infants. By 2 years, the mastoid cells are well developed and produce conic projections of the temporal bones, the mastoid processes. The middle ear continues to grow through puberty.
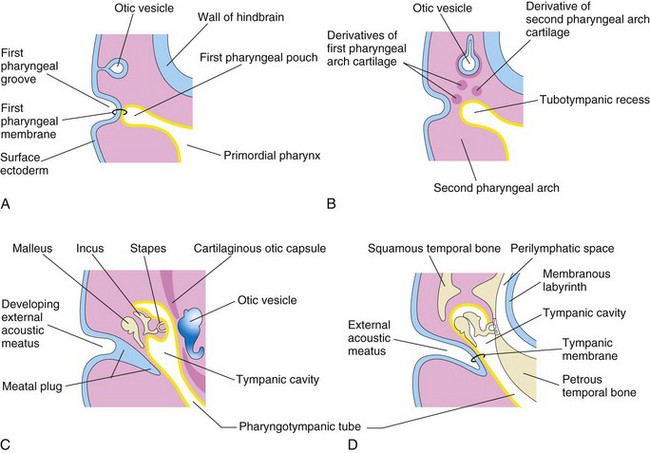
Figure 17–11 Development of the external and middle parts of the ear. A, At 4 weeks, showing the relation of the otic vesicle to the pharyngeal apparatus. B, At 5 weeks, showing the tubotympanic recess and pharyngeal arch cartilages. C, At a later stage, showing the tubotympanic recess (primordium of the tympanic cavity and mastoid antrum) beginning to envelop the ossicles. D, Final stage of ear development, showing the relation of the middle ear to the perilymphatic space and the external acoustic meatus.
Development of External Ears
The external acoustic meatus develops from the dorsal part of the first pharyngeal groove. The ectodermal cells at the bottom of this tube proliferate to form a solid epithelial plate, the meatal plug (Fig. 17-11C). Late in the fetal period, the central cells of this plug degenerate, forming a cavity that becomes the internal part of the external acoustic meatus (Fig. 17-11D). The primordium of the tympanic membrane is the first pharyngeal membrane, which separates the first pharyngeal groove from the first pharyngeal pouch (Fig. 17-11A). The external covering of the tympanic membrane is derived from the surface ectoderm, whereas its internal lining is derived from the endoderm of the tubotympanic recess.
The auricle develops from the mesenchymal proliferations in the first and second pharyngeal arches. Prominences—auricular hillocks—surround the first pharyngeal groove (Fig. 17-12A). As the auricle grows, the contribution of the first arch is reduced (Fig. 17-12B to D). The lobule (earlobe) is the last part to develop. The auricle is initially located at the base of the neck (Fig. 17-12A and B). As the mandible develops, the auricle assumes its normal position at the side of the head (Fig. 17-12C and D).
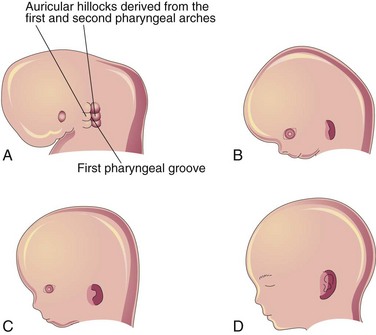
Figure 17–12 Development of the auricle of the external ear. A, At 6 weeks. Note that three auricular hillocks are located on the first pharyngeal arch and three are on the second arch. B, At 8 weeks. C, At 10 weeks. D, At 32 weeks.
Congenital Deafness
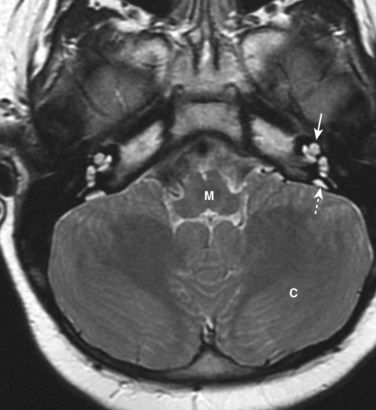
Approximately 1 in 1000 newborn infants have significant hearing loss. Congenital deafness may be the result of maldevelopment of the sound-conducting apparatus of the middle and external ears, or of the neurosensory structures in the internal ear. Enlargement of the vestibular aqueduct and endolymphatic duct is the most common congenital ear defect in children with hearing loss (Fig. 17-13). This defect is typically bilateral and is an autosomal recessive condition. Rubella infection during the critical period of development of the internal ear can cause maldevelopment of the spiral organ and deafness. Congenital fixation of the stapes results in conductive deafness in an otherwise normal ear. Failure of differentiation of the anular ligament, which attaches the base of the stapes to the oval window, results in fixation of the stapes to the bony labyrinth and loss of sound conduction.
Auricular Anomalies
Minor auricular deformities are common and may serve as indicators of a specific pattern of congenital anomalies. For example, the auricles are often low-set and abnormal in shape in infants with chromosomal syndromes, such as trisomy 18 (see Chapter 19), and in infants affected by maternal ingestion of certain drugs (e.g., trimethadione).
Auricular Appendages
Auricular appendages (skin tags) are common and result from the development of accessory auricular hillocks (Fig. 17-14). The appendages usually appear anterior to the auricle, more often unilaterally than bilaterally. The appendages, often with narrow pedicles, consist of skin but they may also contain some cartilage.
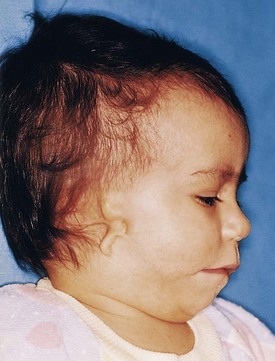
Figure 17–14 Child with a rudimentary auricle (microtia) and a preauricular tag. The external acoustic meatus is also absent.
(Courtesy of A.E. Chudley, M.D., Section of Genetics and Metabolism, Department of Pediatrics and Child Health, Children’s Hospital, University of Manitoba, Winnipeg, Manitoba, Canada.)
Microtia
Microtia (small auricle) results from suppressed development of the auricular hillocks (Fig. 17-14). This anomaly often serves as an indicator of associated anomalies, such as atresia of the external acoustic meatus and abnormalities of the middle ear.
Preauricular Sinuses
Shallow, pitlike cutaneous sinuses are occasionally located anterior to the auricle. These sinuses usually have pinpoint external openings. Some sinuses contain a vestigial cartilaginous mass. These defects are probably related to abnormal development of the auricular hillocks and defective closure of the dorsal part of the first pharyngeal groove. Preauricular sinuses are familial and are frequently bilateral.
Atresia of External Acoustic Meatus
Atresia (blockage) of the external acoustic meatus results from failure of the meatal plug to canalize (Fig. 17-11C). Usually the deep part of the canal is open but the superficial part is blocked by bone or fibrous tissue. Most cases are associated with the first arch syndrome (see Chapter 10). Often the auricle is also severely affected, and anomalies of the middle ear, internal ear, or both may be present. Atresia of the external acoustic meatus can occur bilaterally or unilaterally and usually results from autosomal dominant inheritance.
Absence of External Acoustic Meatus
Absence of the external acoustic meatus is rare (Fig. 17-14). This anomaly results from failure of inward expansion of the first pharyngeal groove and failure of the meatal plug to disappear.
Clinically Oriented Questions
1. If a woman has rubella (German measles) during the first trimester of pregnancy, what are the chances that the eyes and ears of the fetus will be affected? What is the most common manifestation of late fetal rubella infection? If a pregnant woman is exposed to rubella, can it be determined if she is immune to the infection?
2. Some believe that a good way of preventing the congenital anomalies caused by rubella is by the purposeful exposure of young girls to rubella. Is this the best way for a woman to avoid rubella infection during pregnancy? If not, what can be done to provide immunization against rubella infection?
3. It has been reported that deafness and tooth defects occurring during childhood may result from a condition called fetal syphilis. Is this true? If so, how could this happen? Can these congenital defects be prevented?
4. There are reports that blindness and deafness can result from herpes virus infections. Is this true? If so, which herpes virus is involved? What are the affected infant’s chances of normal development?
5. An article in the newspaper reported that methyl mercury exposure in utero can cause mental deficiency, deafness, and blindness. The article cited the eating of contaminated fish as the cause of the abnormalities. How might these anomalies be caused by methyl mercury?