Chapter 10 Immunity and infection
Bacterial, viral, parasitic and fungal infections are major causes of morbidity and mortality worldwide, especially in poorer societies with less access to medicines and vaccines, greater exposure to infectious agents and poorer nutrition. Infectious and parasitic diseases were responsible for 29.6% of the world’s disease burden in 1999, according to the World Health Organization (Table 10.1).
Table 10.1 Leading causes of infectious diseases worldwide
| Infectious disease | Cause | Annual deaths |
|---|---|---|
| Acute respiratory infections (mostly pneumonia) | Bacterial or viral | 4 300 000 |
| Diarrhoeal diseases | Bacterial or viral | 3 200 000 |
| Tuberculosis | Bacterial | 3 000 000 |
| Hepatitis B | Viral | 1 000 000–2 000 000 |
| Malaria | Protozoan | 1 000 000 |
| AIDS | Viral | 1 000 000 |
| Measles | Viral | 900 000 |
| Neonatal tetanus | Bacterial | 600 000 |
| Pertussis (whooping cough) | Bacterial | 360 000 |
AIDS, acquired immune deficiency syndrome.
From The World Health Report (1999). WHO, Geneva.
All of the immunological mechanisms described in the previous two chapters are called upon to limit and eliminate infectious agents. However, pathogens have developed a remarkable variety of strategies to evade the host’s immune defences, and the immune response itself may damage host tissues.
Immunity to bacteria
Summary of defence mechanisms
Bacterial evasion strategies
Many bacteria have developed ways of interfering with phagocytosis. Encapsulated bacteria do not display sugar molecules for recognition by receptors on phagocytes. They are only phagocytosed when coated with antibodies, so can proliferate in non-immune individuals in the first few days after infection. Even when taken up by phagocytes, many encapsulated bacteria resist digestion (e.g. Haemophilus influenzae, Streptococcus pneumoniae, Klebsiella pneumoniae, Pseudomonas aeruginosa) or can even kill phagocytes (e.g. streptococci, staphylococci, Bacillus anthracis). Mycobacteria, listeria and Brucella spp. are able to survive within the cytoplasm of non-activated macrophages and can only be killed by a cell-mediated immune response driven by TH1 macrophage-activating lymphokines.
Damage caused by immune responses to bacteria
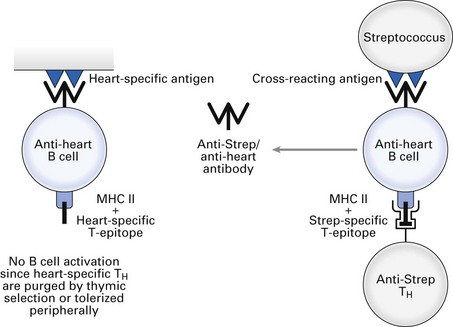
Group A β-haemolytic streptococci cause sore throat and scarlet fever, which resolve on induction of specific antibody. Certain components of some strains of streptococci contain epitopes that are cross-reactive with epitopes present on heart tissue. Antibodies that eliminate the infecting bacteria can bind to heart tissue and cause complement-mediated lysis and antibody-dependent cellular cytotoxicity (rheumatic heart disease). Furthermore, circulating immune complexes can deposit in synovia and glomeruli, causing complement-mediated joint pain and glomerulonephritis, respectively. Induction of cross-reacting anti-heart antibody by group A streptococci is illustrated in Fig. 10.1 (see also Fig. 23.2).
Persistent infection of macrophages, e.g. with Mycobacterium tuberculosis or Mycobacterium leprae, provokes a chronic, local, cell-mediated immune reaction due to continuous release of antigen. Lymphokine production causes large numbers of macrophages to accumulate, many of which give rise to epithelioid cells or fuse to form giant cells (syncytia). These giant cells release high concentrations of lytic enzymes, which destroy the surrounding tissue. Incorporation of fibroblasts also occurs, and the persisting pathogen becomes walled off inside a fibrotic, necrotic granuloma. Because the macrophages in a granuloma are activated, this mechanism also enhances the activation of T-helper cells. Granulomas may replace extensive areas of normal tissue, e.g. in the lungs of tuberculosis patients.
Immunity to viruses
Viruses cannot proliferate outside a host cell. The infectious virion must attach to a suitable cell via a specific membrane receptor and enter the cell cytoplasm. Viral replication may or may not destroy the host cell. Viral genes may become incorporated within the host cell genome and remain in a state of latency for long periods. In some cases, integrated viral genes activate cellular oncogenes and induce malignant transformation.
Summary of defence mechanisms
Viral evasion strategies
Certain viruses can modify the structure of components that are targets for the immune response (antigenic variation). Point mutations in the genes encoding viral antigens cause minor structural changes (antigenic drift), while exchange of large segments of genetic material with other viruses changes the whole structure of the antigen (antigenic shift). Antigenic drift of influenza A virus haemagglutinin occurs before each winter’s minor influenza epidemic, while major epidemics, such as those of 1918, 1957, 1968 and 1977, were the result of antigenic shift of haemagglutinin and/or neuraminidase.
Viruses that can integrate their genes within the host cell genome, such as human herpesviruses, provoke only low-level immunity, which fails to clear the latently infected cells. Viruses that infect cells of the immune system may inhibit their function, e.g. Epstein–Barr virus (B cells); measles, human T lymphotropic virus type I, human immunodeficiency virus (HIV) (T cells); dengue, lassa, Marburg–Ebola, HIV (macrophages).
Some herpesviruses and poxviruses can secrete proteins that mimic and interfere with key immune regulators such as cytokines and cytokine receptors.
Damage caused by immune responses to viruses
Epstein–Barr virus is a potent T cell-independent polyclonal activator of B cells. It induces B cells, including those with anti-self BCRs which are normally inactive due to purging of the corresponding anti-self T-helper cells, to secrete antibodies. Several viruses, notably hepatitis B virus, can cause chronic autoimmune disease due to release of previously sequestered (i.e. non-tolerogenic) self antigens following tissue damage. Complexes of antivirus antibodies with antigen can activate complement in the blood vessels, joints and glomeruli, causing vasculitis, arthritis and glomerulonephritis. Cytotoxic T cells may destroy essential host cells displaying viral antigens, e.g. coxsackievirus (myocarditis), mumps virus (meningoencephalitis) and viruses causing damage to the myelin nerve sheath (postviral polyneuritis).
HIV and AIDS
At the end of the year 2008, approximately 40 million people worldwide had become infected with HIV and approximately 25 million had died of the acquired immune deficiency syndrome (AIDS) (see also Chapter 30). The virus causes depletion of CD4+ T-helper lymphocytes over many years. Patients eventually succumb to opportunistic infections (Pneumocystis carinii, M. tuberculosis, atypical mycobacteria, Histoplasma, Coccidioides, Cryptococcus, Cryptosporidium and Toxoplasma spp., herpes simplex, cytomegalovirus) and may develop Kaposi’s sarcoma, B cell lymphomas and other malignancies. Infection of the brain by HIV can cause dementia and encephalitis.
The major route of transmission of HIV is by sexual intercourse: male to female, female to male and male to male. It can also be transmitted from mother to foetus across the placenta, during delivery or by breast-feeding. Direct injection into the blood stream, e.g. by multiple use of needles and syringes for injection of drugs, also transmits HIV.
The life cycle of HIV is shown in Figure 10.2. The virus gains entry into target cells by binding its surface gp120 molecule (glycoprotein of 120 kDa) to CD4 on T-helper cells and a subset of macrophages. The latter can also take up opsonized HIV via Fc or complement receptors. A coreceptor is also required for infection of target cells: CXCR4, also known as fusin or LESTR, is the receptor for the chemokine SDF-1 and is the coreceptor for infection of T cells by HIV; CCR5, the receptor for chemokines RANTES, MIP-1α and MIP-1p, is the coreceptor for infection of macrophages. Viral gp41 causes fusion with the cell membrane and injection into the target cell of two strands of viral genetic information, which is RNA. One strand is destroyed by viral ribonuclease H and viral reverse transcriptase converts the surviving strand into a DNA copy. This forms the template for synthesis of the complementary second strand by cellular DNA polymerase. The double-stranded DNA is then integrated into host cell DNA by viral integrase.
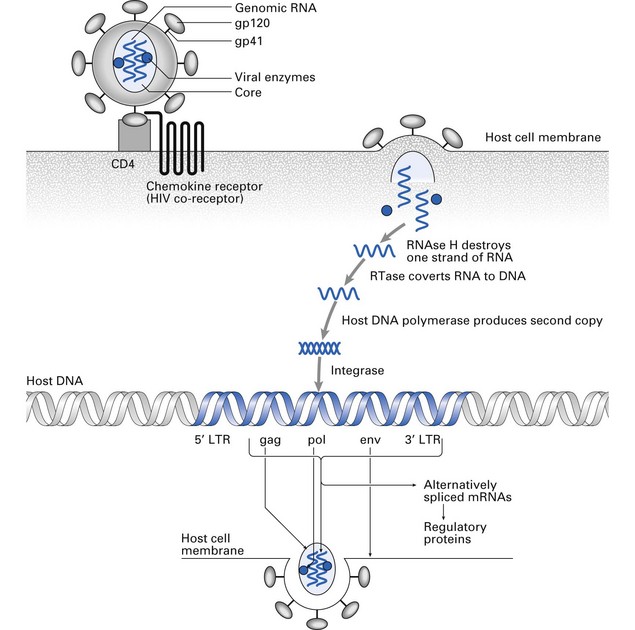
Fig. 10.2 Life cycle of human immunodeficiency virus (HIV). LTR, long terminal repeat; mRNA, messenger RNA; RNAse, ribonuclease; RTase, reverse transcriptase.
Research into the pathogenic process that underlies the progression of HIV to AIDS has revealed a dynamic interplay between the virus and the immune system. The virus is usually transmitted from person to person within macrophages (infected macrophages are more numerous than infected T-helper cells in genital secretions) or as cell-free virus. Infected macrophages contain HIV virions within cytoplasmic vacuoles; probably IL-6 and TNF-α produced in response to phagocytic uptake induce constant slow production of virions from integrated proviral DNA. When infected macrophages enter the new host, they are destroyed, releasing HIV. Dendritic cells transport HIV to draining lymph nodes where they infect CD4+ cells.
Proliferation of HIV within lymph nodes occurs throughout the long period of clinical latency, even though the patient remains well and is not deficient in T-helper cells. Eventually, the lymph node architecture becomes damaged, and generalized release of HIV causes rapid destruction of T-helper cells.
Budding of HIV from an infected T-helper cell destroys the integrity of the cell membrane. In addition to this direct form of killing of infected cells, HIV can apparently destroy or inactivate uninfected T-helper cells by various indirect mechanisms, most of which remain theoretically possible rather than of proven clinical relevance. These possible pathogenic mechanisms are shown in Figures 10.3-10.7.
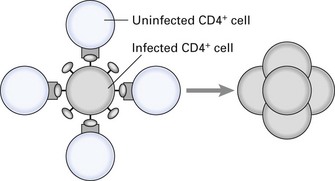
Fig. 10.3 Giant cell formation induced by human immunodeficiency virus (HIV). Glycoprotein gp120 present on the surface of HIV-infected cells binds to CD4 on uninfected cells; gp41 induces fusion of adjacent cells and production of non-functional, infected giant cells (syncytia). Thus, HIV can pass from cell to cell without being exposed to the host’s immune response.
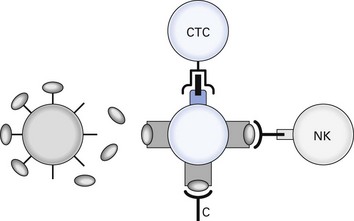
Fig. 10.4 Release of soluble gp120. Glycoprotein gp120 released from human immunodeficiency virus (HIV)-infected cells can bind to CD4 of uninfected cells, which can then be destroyed by antibody-dependent complement or natural killer (NK) cell cytotoxicity after binding of anti-gp120 antibody. Processing of gp120 on to major histocompatibility complex I (MHC I) forms a target structure for cytotoxic T cells (CTC).
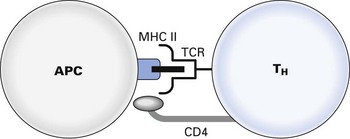
Fig. 10.5 Inhibition of T-helper cell (TH) activation: gp120 inhibits CD4–MHC II and TCR–MHC II–peptide interaction. APC, antigen-presenting cell; MHC II, major histocompatibility complex II; TCR, T cell receptor.
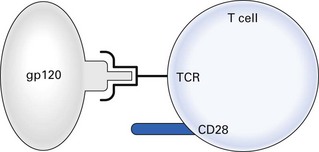
Fig. 10.6 Inactivation of T cell: gp120 has sequence homology with major histocompatibility complex II (MHC II) and can bind to T cell receptors (TCRs). Transmission of signal 1 without signal 2 (B7-CD28) inactivates the cell or induces apoptosis.
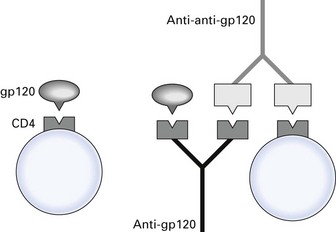
Fig. 10.7 Induction of anti-CD4 antibody. Anti-idiotypic antibody against anti-gp120 cross-reacts with CD4. Antibody-coated CD4-positive cells are destroyed by antibody-dependent complement or cellular cytotoxicity.
Throughout the period of clinical latency, there is massive infection, destruction and replacement of CD4+ T cells, with billions of new cells being infected and killed every day. Ultimately, the processes leading to replacement of T-helper cells become exhausted, the cell number drops and immune function deteriorates. The virus may reduce replenishment of T cells from haematopoietic stem cells following infection of the bone marrow. Furthermore, destroyed CD4+ cells can be replaced by CD4+ or CD8+ T cells with equal likelihood, so the representation of the latter gradually increases compared with the former and immune suppressor activity will come to dominate over helper activity. Virus-infected macrophages are deficient in IL-12 production and therefore cannot induce TH1 responses. Instead, the dominant TH2 response leads to hypergammaglobulinaemia and production of autoantibodies and B cell lymphomas.
Newly available, highly active drug combinations interfere with virus proliferation and T cell destruction and delay disease progression. The prototype of the drugs that interfere with reverse transcription is zidovudine, or AZT. Current treatment for AIDS is a combination therapy, using regimens designated highly active antiretroviral therapy (HAART) (see Chapter 30).
Immunity to parasites
Summary of defence mechanisms
Parasite evasion strategies
T. brucei possesses variant surface glycoproteins (VSGs). There are several genes for different VSGs, only one of which is expressed at any given time. After antibodies have been produced against one VSG, it is shed from the surface and a new VSG gene is expressed (antigenic variation). Leishmania cap off their surface antigens when exposed to antibody (antigenic modulation). Schistosomes synthesize host-like antigens such as α2-macroglobulin to mask their own foreignness and also adsorb host molecules, such as red blood cell antigens, MHC antigens, complement factors and immunoglobulins, on to their surface (antigenic disguise).
Parasites have various immune suppressor capabilities: T. brucei induces T-suppressor cell activation. Plasmodium and Leishmania release soluble antigens that intercept antiparasite antibodies and saturate phagocytes. T. cruzi produces molecules that inhibit or accelerate the decay of C3 convertases. Leishmania downregulates MHC II expression on parasitized macrophages, reducing their ability to present antigenic peptides to CD4+ T cells. Toxoplasma prevents fusion of phagocytic vacuoles with lysosomes. Leishmania inhibits the respiratory burst of macrophages. Schistosomes release peptidases that cleave bound immunoglobulin, and other factors that inhibit T cell proliferation, release of IFN-γ and eosinophil activation.
Damage caused by immune responses to parasites
Helminths induce not only parasite-specific IgE but also polyclonal IgE, which can give rise to manifestations of allergy such as urticaria and angioedema. Sudden release of large amounts of parasite antigen can trigger fatal anaphylactic shock.
Parasite antigens that cross-react with host antigens (e.g. T. cruzi antigens cross-reactive with cardiac antigens) and parasites coated with host antigens can induce autoimmune attack against host tissues (cf. Fig. 10.1).
Circulating immune complexes containing parasite antigens cause some of the tissue damage seen in malaria, trypanosomiasis and schistosomiasis. Portal fibrosis and pulmonary hypertension in schistosomiasis are due to T cell-mediated granulomatous responses to schistosome eggs.
Immunity to fungi
Fungal infections may be superficial (e.g. ringworm caused by Trichophyton rubrum, oral thrush and vulvovaginitis caused by Candida albicans), subcutaneous (e.g. abscesses and ulceration caused by Sporothrix schenckii) or systemic (e.g. histoplasmosis, coccidioidomycosis, systemic candidiasis, cryptococcosis, aspergillosis).
In healthy individuals, and even in immunodeficient patients with defects in antibody production, fungal infections generally remain localized and resolve rapidly. In contrast, patients with T cell or neutrophil defects may suffer chronic infections, indicating that these are the important effector cells in immunity to fungi.
Production of antifungal antibodies may result in IgE-mediated allergic disease, e.g. allergic bronchopulmonary aspergillosis, or IgG-mediated immune complex disease, e.g. when aspergillus grows to form an aspergilloma in pre-existing lung cavities. Histoplasma, Coccidioides and Cryptococcus can induce granuloma formation in the lungs.
Vaccination
Natural infection often produces lifelong protection, with development of memory T and B cells able to respond rapidly on subsequent challenges with the same agent. Many infections cause severe clinical symptoms and even death, which could be prevented by inducing memory cells before exposure to pathogens occurs (Fig. 10.8).
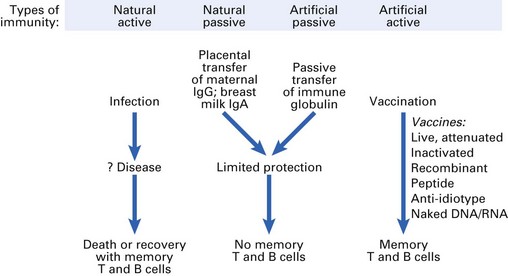
Passive immunization
Passive transfer of maternal antibodies during pregnancy and breast-feeding provides limited protection for the newborn baby, but following catabolism of these antibodies protection is lost. Vaccination to induce memory B cells is not successful during the neonatal period because maternal antibodies neutralize vaccine antigens, although induction of memory T cells can be achieved at this time.
Short-term protection can also be achieved later in life by passive transfer of immune globulin. Short-term passive immunization using hyperimmune globulin or monoclonal antibodies can be useful as post-exposure prophylaxis, e.g. following exposure to rabies virus.
Active immunization
Memory T and B cells can be induced most successfully using live vaccines containing microorganisms that have been attenuated to reduce their virulence. A single dose is usually sufficient to induce both systemic and mucosal immunity. Immunocompromised patients must not be given live vaccines because of the danger of disseminated infection.
Inactivated vaccines consist of killed whole organisms, products of organisms or subunits of organisms. Since there is no replication of the organism to provide immune stimulation over several days, inactivated vaccines must be given in multiple doses in the presence of an adjuvant. The most widely used adjuvant for human vaccines is alum, which forms a precipitate with protein antigens from which the antigens are slowly released to the immune system.
Toxoids consist of bacterial exotoxins rendered harmless by treatment with formaldehyde. Antigenicity can be increased by combination with suspensions of other bacteria containing endotoxins, for example, diphtheria–tetanus–pertussis triple vaccine (see also Chapter 36). Vaccination with the toxoid induces antitoxoid antibodies that are capable of binding to the toxin and neutralizing its effect.
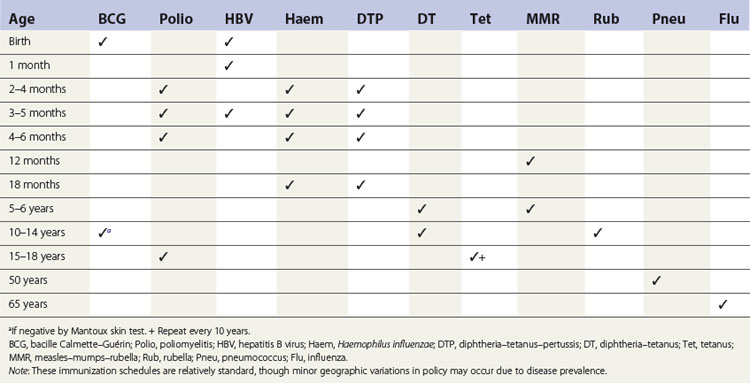
Vaccines currently available for active immunization are shown in Table 10.2, and recommended vaccination schedules are shown in Table 10.3.
Table 10.2 Currently available active immunizing agents
| Vaccine | Formulation |
|---|---|
| Anthrax | Inactivated Bacillus anthracis |
| BCG (tuberculosis)a | Live attenuated Mycobacterium bovis |
| Cholera | Inactivated Vibrio cholerae |
| Diphtheriaa | Toxoid |
| Haemophilus influenzae | Capsular polysaccharide conjugated to protein |
| Hepatitis B virusa | Recombinant viral protein |
| Influenzaa | Inactivated virus |
| Measlesa | Live attenuated virus |
| Meningococcus | Capsular polysaccharide |
| Mumpsa | Live attenuated virus |
| Pertussis | Killed whole Bordetella pertussis |
| Plague | Inactivated Yersinia pestis |
| Pneumococcus | Capsular polysaccharide of Streptococcus pneumoniae |
| Polioa | Inactivated or live attenuated virus |
| Rabies | Inactivated virus |
| Rubellaa | Live attenuated virus |
| Tetanusa | Toxoid |
| Typhoid | Inactivated or live attenuated Salmonella typhi |
| Yellow fever | Live attenuated virus |
BCG, bacille Calmette–Guérin.
New approaches to vaccine development
Proven, effective vaccines are still not available against many of today’s leading killers, notably malaria, parasitic diseases and HIV. Even the vaccines in regular use cannot be considered 100% effective. Most of them induce antibody effectively but are less able to stimulate cell-mediated immunity. It has even been suggested that current vaccines given early in life polarize cytokine production towards type 2 rather than type 1 responses and that the increasing prevalence of asthma and allergies could be partly a consequence of immunization with IL-4-inducing vaccines. However, new approaches to vaccine development promise greater control of infectious diseases in the not-too-distant future.
One way of improving the efficacy of inactivated vaccines would be to develop more effective adjuvants. Freund’s complete adjuvant, which contains oil, detergent and mycobacteria, stimulates powerful B cell and T cell responses in experimental animals, but is too toxic for human use. The active principal of mycobacteria, muramyl dipeptide, strongly enhances macrophage activity, is non-toxic and may become useful in human vaccines. Immunostimulating complexes (ISCOMs), prepared from saponin, cholesterol and phosphatidylcholine, provide a vehicle for presenting proteins to the immune system and induce T and B cell memory.
Inactivated vaccines made from whole microorganisms may contain proteins that stimulate both protective and non-protective – or even suppressive – immune responses. Subunit vaccines containing only protection-inducing proteins should be much more effective than cruder preparations.
Modern subunit vaccines are now being produced by recombinant DNA technology (Fig. 10.9). Candidate protein antigens must first be identified and purified so that a partial amino acid sequence can be determined. An oligonucleotide probe consisting of the corresponding nucleotide sequence is then constructed and labelled with a radioisotope.
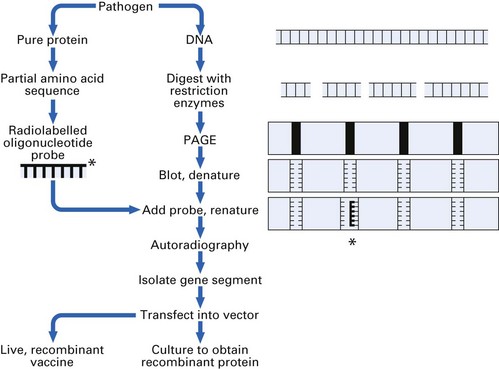
Next, DNA is extracted from the pathogen and digested with restriction enzymes, and the DNA fragments are separated by polyacrylamide gel electrophoresis (PAGE). After blotting on to nitrocellulose, the DNA is denatured by heating. The probe is added and binds to its complementary sequence when the temperature is lowered, thereby identifying the relevant gene segment. Autoradiography reveals its position on the blot and the original polyacrylamide gel can be sliced to obtain the gene. This is then transfected into the DNA of suitable host cells (bacterial, yeast, insect or human). When the host cells are cultured, recombinant as well as host proteins are synthesized.
This technology is particularly useful for producing antigenic proteins from viruses that are difficult to culture, and a highly effective recombinant hepatitis B vaccine is already in routine use. DNA vaccines offer advantages over many of the existing vaccines because the encoded protein is expressed in its natural form in the host. DNA vaccines cause prolonged expression of the antigen that generates significant immunological memory.
Synthetic peptide vaccines containing only relevant epitopes of the antigenic protein have also been produced and shown to be effective in animal models. In theory, it should be possible to construct vaccines containing both B cell and T cell epitopes on a carrier molecule such as poly-l-lysine. For pathogens that undergo antigenic variation, notably HIV, it might be possible to construct peptide vaccines containing sufficiently large arrays of peptides to protect against most variants of the pathogen. Since peptides are usually not as immunogenic as proteins, adjuvants and conjugates have been used to assist in raising protective immunity to synthetic peptides.
Live recombinant vaccines also hold considerable promise. Gene segments coding for pathogen proteins can be inserted into attenuated vectors such as vaccinia, bacille Calmette–Guérin (BCG) or adenovirus. Immunizing pathogen proteins are released during the time the vector replicates in the host. Live, replication-incompetent microorganisms can be engineered for use as vaccines by removing some of the genes involved in replication, though later reversion to full pathogenicity would be difficult to rule out.
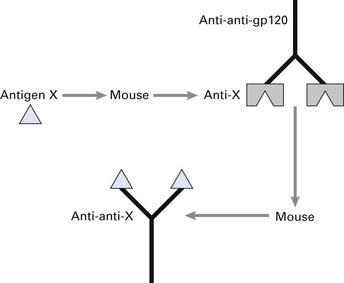
Anti-idiotypic antibodies can be used as vaccines instead of pathogen proteins. The protein antigen (X) is used to raise monoclonal antibodies in mice; V regions of anti-X are then used to immunize a second mouse. The resultant monoclonal anti-anti-X has similar antigenic properties to X itself (Fig. 10.10). By isolating the V genes from the hybridoma producing anti-anti-X, anti-idiotypic vaccines can be produced using recombinant DNA technology.
Recent progress in production of genetic vaccines using RNA or DNA coding for specific pathogen proteins has been encouraging. When injected intramuscularly, the genetic information remains unintegrated in muscle cells but gives long-term expression of properly folded and glycosylated immunogenic protein and strong cell-mediated, as well as humoral, immunity. In mice, DNA vaccines have been shown to induce protective cell-mediated immunity against leishmaniasis, tuberculosis and malaria. A trial of a DNA vaccine against malaria in humans showed induction of malaria-specific cytotoxic T cells. Recombinant DNA technology, coupled with the identification of viral and bacterial epitopes for T and B cell responses, will lead to vaccines of the future that are safe, easy to administer and affordable to a vast majority of the world population, especially in developing nations.
Key facts
Janeway C.A.Jr., Travers P., Walport M., Shlomchik M.J. Immunobiology, 5th ed. New York: Garland Publishing; 2001.
Mims C., Playfair J., Roitt I., Wakelin D., Williams R. Vaccination, Ch. 15. Medical microbiology, 2nd ed. St Louis: Mosby Year Book. 1998.
Powell M.F., Newman H.J., editors. Vaccine design. New York: Plenum, 1995.
Roitt I.M. Roitt’s essential immunology, 11th ed. Oxford: Blackwell; 2006.
Roitt I., Brostoff J., Male D. Vaccination, Ch. 19. Immunology, 7th ed. London: Mosby. 2006.
Review questions (answers on p. 352)
Please indicate which answers are true, and which are false.