Chapter 17 Clinical development
present and future
Introduction
Clinical development is the art of turning science into medicine. It is the point at which all the data from basic science, preclinical pharmacology and safety are put into medical practice to see whether scientific theory can translate into a valuable new medicine for patients. The fundamental purpose of the clinical development programme is to provide the clinical information to support the product labelling, which ultimately tells the healthcare professional and patient how to use the drug effectively and safely.
The segments of product label coming from clinical trials are the pharmacokinetics, the dosing regimen in the main population and in special populations, e.g. the elderly or those with hepatic and renal impairment, the clinical pharmacology/mechanism of action in man, drug interactions, contraindications, warnings, precautions, efficacy in the indication, safety and side effects. All this information has to be generated from a development programme designed to investigate these specific properties.
Clinical development has to satisfy the demands of regulators who will grant product approval, government organizations responsible for reimbursement in countries where healthcare is state subsidized, managed care organizations in the USA and also the marketing team who will sell the product. The needs are sometimes conflicting and a challenge of clinical development is to design a trials programme that not only demonstrates that the new drug is effective and safe, but also balances the various desires and requirements of the different parties, for the product profile.
Bringing new drugs to the market is not only complex but also costly, time and resource consuming. Taking into account failure of drugs in development to make it to market, Di Masi (2003) estimated the costs to be around US$802 million and Adams and Brantner (2006) made an estimate of US$868 million but within a range of $500 million to $2 billion, depending on the indication pursued and the company performing the development. Of this total amount the clinical development accounts for just over half, i.e. about US$480 million.
In spite of heavy investment in research and development, new product approvals have been decreasing in the last 10 years (Woodcock and Woosley, 2008). Pipelines of large pharmaceutical companies have been declining and the productivity of large pharmaceutical companies in new drug development has been diminishing in an environment of increasing costs of clinical development and increasing risk aversion of companies, regulators and the general public. New strategies are needed to overcome this problem, both in the way companies source and develop new drugs and also in the way regulators approach the evaluation of efficacy and safety of new medicines.
This new environment is leading to an evolution in clinical development strategy, with the type of indications being pursued and in the sourcing and development of new compounds. There is a move away from blockbuster medicines, i.e. ‘one size fits all’ in major indications, towards more specialized unmet medical needs and patient-tailored therapies, i.e. personalized medicine. The success of the human genome project was supposed to have heralded a new era for patient-specific drug development. However, for now, the art of medicine appears to continue to outwit the theory of basic science and drug development based purely upon genomic approaches has yet to live up to its promises.
There is an increasing trend for large pharmaceutical companies to collaborate with small pharmaceutical companies and biotech companies in order to enhance discovery pipelines and drug development productivity. Large companies have a great deal of resources to put behind development programmes but internal competition for resources, risk aversion and political pressures within these organizations can mean they are less flexible and creative in clinical development. Small pharmaceutical companies can provide the flexibility, innovation and creativity to complement the large pharmaceutical company development activities and there is an increasing trend for new drug development to be performed in partnership.
In the United Sates the Food and Drug Administration (FDA) published a white paper in 2004 (FDA, 2004a) that identified that the current methods of drug development were partly behind the decline of new drug applications and has set up the Critical Path Initiative (FDA, 2004b) in order to address some of the problems of low productivity and high late-stage attrition rates, the idea being to encourage novel approaches to clinical development in trial design and measuring outcomes. The Innovative Medicines Initiative (EFPIA, 2008), underway in Europe, is also looking to encourage public and private collaboration with small and large enterprises and academia, to share knowledge, enhance the drug discovery and development process, reduce late-stage attrition and, ultimately, bring good medicines to patients in a cost- and time-effective manner.
These factors are changing the way clinical development is being performed and will be performed in the future. In this chapter the conventional path of clinical development will be explained along with the new strategies for merging phases and using adaptive trial design to enhance the development of new medicines.
Clinical development phases
The conventional path of clinical development involves four phases:
• Phase I, pharmacokinetics, safety and tolerability and clinical pharmacology
• Phase IIa, exploratory efficacy
• Phase IIb, efficacy and dose range finding
• Phase III, pivotal efficacy and safety and larger population
A summary of these phases is given in Table 17.1.
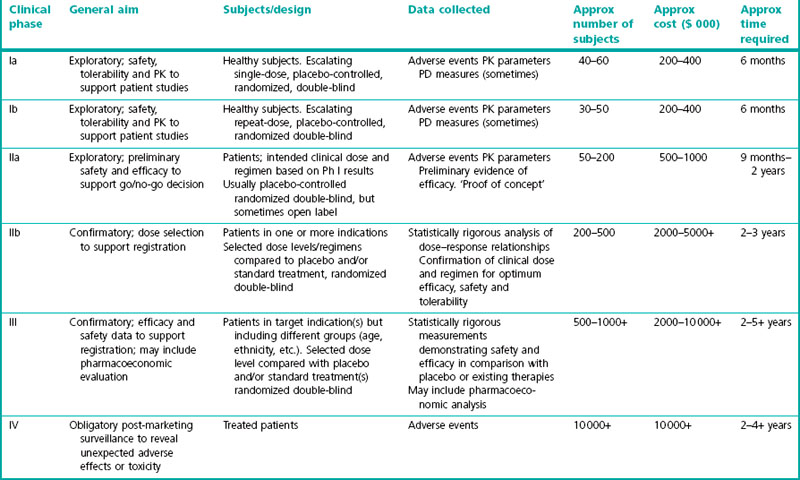
Traditionally these phases have been conducted in a step-wise manner with decisions to proceed to the next stage being made once the preceding stage was completed. However, more recently, the margins between phases are becoming less distinct as more seamless drug development programmes are being performed and adaptive trial designs adopted. Therefore, it may be more appropriate to describe the phases as: clinical pharmacology, including first in man – Phase I; exploratory, proof of concept – Phase IIa; confirmatory efficacy and dose range finding – Phase IIb; confirmatory, large scale efficacy and safety – Phase III; marketing authorization application, license extension and post-marketing surveillance – Phase IIIb/IV.
Phase I – Clinical pharmacology
Phase I is somewhat of a misnomer for, although the first studies in man are performed as part of Phase I, many of the other components of Phase I, for example drug–drug interactions, special populations and human radiolabelled studies, are conducted in parallel with later phase studies. Hence it is probably more appropriate to call these ‘clinical pharmacology studies’.
A typical Phase I programme may contain around 20 clinical pharmacology studies. The main objectives of the programme are to define the pharmacokinetics, metabolism and safety of the intended formulation given alone and with other drugs that have potential to interact either kinetically or dynamically with the new drug. How the drug is handled by certain populations, such as the elderly, ethnic groups or those with hepatic or renal impairment, is also studied. All of this information goes into the prescribing information to guide the safe and effective use and dosing of the drug. Some efficacy information can also be gathered in human pharmacological models in order to assist dose selection for trials in patients. The principal components are as follows:
• First in man single ascending dose pharmacokinetics and safety
• Multiple ascending repeat dose pharmacokinetics and safety
• Bioavailability absolute and bioequivalence of new formulations
• Absorption distribution metabolism excretion in man (radiolabelled studies)
• Drug–drug interaction studies
• Safety pharmacology, e.g. thorough QT studies and abuse liability studies
• Elderly pharmacokinetics and safety
• Ethnic groups pharmacokinetics and safety
• Hepatic and renal impairment, pharmacokinetics and safety.
Clinical pharmacology studies and in particular ‘first in man’ studies are conducted by specialist medical staff, trained in clinical pharmacology, in specialized units either within or close to a major hospital. The units are specifically equipped for the preparation and correct administration of the test drugs (and in some cases manufacture of the drug product), collection and storage of biological samples and management of subject safety, including full resuscitation facilities. The subjects selected for studies are usually enrolled from the unit’s subject volunteer panel. Typically a clinical pharmacology unit will advertise for subjects to join their database. People who are interested in taking part in trials are then carefully screened for their physical health, their ability to comprehend the requirements of taking part in the studies and their motivation to comply with the constraints of the studies, to check whether they are suitable to join the volunteer panel. In some countries (e.g. France), the subjects have to be registered on a national database to make sure they comply with laws governing participation in clinical trials. For most large professional units, the database will comprise a variety of healthy subjects including young men and women (18–45 years), older healthy subjects (over 55 years), subjects of non-Caucasian origin (e.g. Japanese) and subjects with genetic polymorphisms for CYP metabolism. As the subjects gain no medical benefit from taking part in the studies and have to undergo procedures which can be mildly unpleasant or inconvenient, they are paid for taking part. However, the amount they can be paid is restricted to be commensurate with the inconvenience and discomfort of the study and is not so high as to be an inducement to take part. The subjects’ reimbursement is reviewed and has to be approved by the ethics committee, before the study can go ahead. The number of studies that subjects can take part in annually is also restricted. Most protocols demand that a subject cannot be exposed to another investigational drug within 90 days of taking part in a study and so that limits how many studies in which a given subject can participate, in any one year.
First in man, single ascending dose, pharmacokinetics and safety
First dose in man (FDIM), also known as single ascending dose (SAD), is a red-letter day for the drug development team, when single doses of the drug are given to small cohorts of subjects in a sequential manner until the maximum tolerated dose is achieved.
Objectives
The objectives of the SAD study are to evaluate the safety (physiological effects on body systems), tolerability (occurrence of side effects) and pharmacokinetics of different doses of the test drug, and to identify the maximum tolerated dose (MTD). That is the dose at which either the occurrence of intolerable side effects and/or unacceptable safety findings are encountered. The MTD is important to identify for estimation of the therapeutic window, which is the dose range within which the drug is effective but safe and well tolerated. In some cases it is not possible to achieve the MTD, perhaps if the drug is very well tolerated, or has poor bioavailability, in which case the maximum feasible dose can be used to estimate the therapeutic window.
Subjects
In most cases the subjects in first in man studies will be conducted in healthy young men usually aged 18–45 years. The idea behind this is to have a fairly homogeneous population in which to study the effects of the new drug and so limit variability, and also to have a population who will be more able to withstand any unexpected toxicity caused by the test drug. Healthy subjects are those who have no underlying diseases that could interfere with the conduct of the study or confound the interpretation of the safety or pharmacokinetic data. Criteria for inclusion into the study based upon medical history, physical examination, use of concomitant medications, alcohol, cigarettes and recreational drugs, as well as the results of blood testing 12-lead ECG, blood pressure heart rate are laid out in the study protocol. Male subjects are generally preferred, because at this early stage of development, reproductive toxicology testing in animals will not have been completed and the risk to the fetus of female subjects who might be pregnant or become pregnant shortly before or after the study, has not been characterized. Once the segment 2 reproductive toxicology has been completed (see Chapter 15), female subjects of non-childbearing potential, i.e. post-menopausal, surgically sterilized, sexually abstinent or using effective methods of contraception, may be included in European studies. In the USA, female subjects may be included prior to reproductive toxicology data being available if they are of non-childbearing potential. In some cases it is not appropriate to use healthy young men, for example, studies of female hormone products or products for oncology, and in these cases the subjects will be selected from the appropriate population.
Design
The classical design of the SAD study is a double-blind, placebo-controlled sequential cohort design; where the first cohort takes the lowest dose and then the dose is escalated through subsequent cohorts, provided the tolerability and safety in the preceding cohort are acceptable. The size of each group is usually six to eight subjects, with two of the subjects being randomized to placebo. The use of placebo and the double-blinding, where neither the investigator nor the subject knows the treatment being taken, allows for a more objective evaluation of the safety and tolerability of the test drug.
The choice of doses to be administered in the SAD trials should be based on the highest dose at which no adverse effects were seen in the most sensitive species tested in toxicology studies (the no observed adverse effect level, or NOAEL; see Chapter 15) and on the nature of the toxicity observed. The starting dose should be at least 10-fold lower than the NOAEL, but specific guidelines exist for the accurate determination of the starting dose on a case-by-case basis (FDA; http:www.fda.gov/cber/gdlns/dose.htm).
The dose escalation plan will depend on the characteristics of the drug and its metabolites, and especially on the nature of any toxicity seen in preclinical toxicology testing at doses above the NOAEL. It will also be influenced by the relationship between dose, systemic exposure as determined by its pharmacokinetic (PK) profile in animals, and the pharmacodynamic (PD) effects observed in preclinical pharmacology studies. Each dose escalation step will be dependent on satisfactory safety data from the previous dose level, according to the clinical judgment of the investigator.
Usually the drug is administered only once to each subject so that each dose group comprises separate subjects. This has the advantage of maximizing the population exposed to the test drug. Sometimes, however, it may be appropriate for each subject to receive two or three of the planned doses at successive visits, with the proviso that each dose is administered only after the response to the preceding dose in the series has been evaluated. In this way the required number of subjects is reduced, but each has to attend more than once.
Typical dose escalation schedules from a starting dose of X are:
The dose escalation schedule is guided primarily by safety considerations. Dose escalation schedule 1, where the dose is increased exponentially, would be appropriate for a drug that has shown low toxicity in animal testing. Schedule 2, where the dose increments are constant, is more conservative and might be more appropriate for a drug which has a toxicology profile that calls for a more cautious approach to dose escalation in man. There are many other possible dose escalation patterns, and each is considered on its own merits. The ideal is to exceed the dose predicted to effective from preclinical pharmacology studies, by a good margin. A drug for which the MTD is close to the dose predicted for therapeutic effect is less likely to be successful than one that has a wide therapeutic margin.
As it is common for the PK profiles of orally administered drugs to be affected by the presence or absence of food in the stomach at the time of dosing, the food effect is usually investigated at the end of the SAD study. A dose level that is safe and well tolerated (e.g. one-quarter of the MTD) will be given to healthy subjects on two occasions once in the fed state (following a standard high-fat breakfast) and once fasted (overnight fast). The order of fed and fasted administration is randomized among the subjects and the two dose periods are separated by an appropriate washout period, so that the results of the second period are not affected by the drug administration on the first period. The results of the fed/fasted comparison will form the basis of the dosing instructions for all future studies with the drug.
Outcome measures
In the SAD study the principal outcome measures are safety, tolerability and pharmacokinetics.
Throughout the study, for an appropriate period after each dose, the subjects are intensively monitored for signs and/or symptoms of toxicity (adverse events). The safety parameters measured include, blood pressure, heart rate and rhythm, 12-lead ECG (intervals and morphology), body temperature, haematology, liver function and renal function, as well as observation for any other unwanted effects. Tolerability is evaluated by the documentation of adverse events which are collected throughout the study and then categorized by severity, duration, outcome and causality. If an adverse event meets certain specific criteria (for instance if it is life-threatening or necessitates hospitalization) it is classified as a serious adverse event (SAE) and must be reported without delay to the ethics committee, and usually also to the regulatory authority.
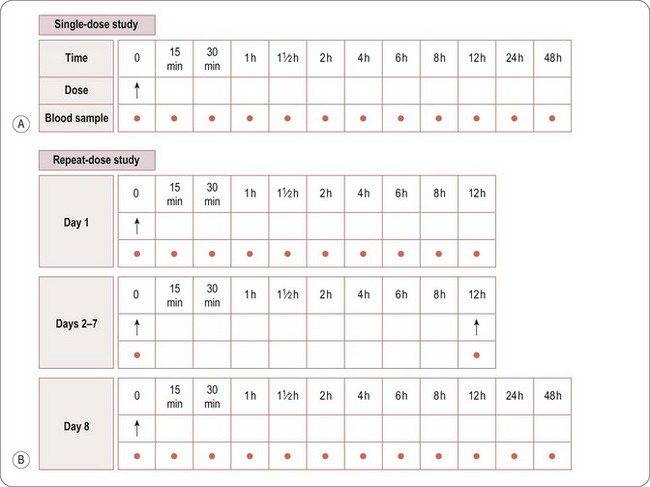
Whereas assessment of safety and tolerability is the primary objective, pharmacokinetic evaluation is the secondary objective of a SAD study. Blood samples will normally be taken before dosing and at specified intervals after dosing to measure the amount of drug in the blood or plasma at various time points after each dose. A typical sampling schedule is shown in Figure 17.1A. In addition, urine and/or faeces may be collected to measure the excretion of the drug via the kidneys and/or liver (in bile). The results enable the rate of absorption, metabolism and excretion of the drug to be explored, and the PK parameters to be estimated (see also Chapter 10).
The plasma or serum PK parameters usually derived are:
• Cmax: peak drug and/or metabolite(s) concentration
• Tmax: time to peak drug and/or metabolite(s) concentration
• AUC0–∞: area under the concentration–time curve of the drug and/or metabolite(s), extrapolated to infinity
• AUC0-T: area under the concentration–time curve of the drug and/or metabolite(s), calculated to a specific time point T
• t1/2: time taken for levels of drug and/or metabolite(s) to decrease by half (a measure of the rate of elimination of the drug from plasma).
Other PK parameters may also be determined, including:
• VD: volume of distribution of drug and/or metabolite(s)
• Cl: clearance of drug and/or metabolite, i.e. the volume of plasma/serum cleared of drug and/or metabolite(s) per unit time, e.g. mL/min, L/h
• MRT: mean residence time, i.e. the average time a drug molecule remains in the body after rapid i.v. injection.
Specialist pharmacokineticists perform the calculation of these parameters.
A comparison of the PK parameters at each dose level, e.g. AUC∞′, Cmax, will indicate whether they increase proportionately (linear kinetics) or disproportionately (non-linear kinetics) with increasing dose. This information will influence the selection of dose levels, regimen and duration of dosing for the multiple, ascending repeat-dose study (MAD). The single-dose PK data can also be used to predict the drug/metabolite(s) concentrations expected on repeated dosing, based on the assumption that the kinetics do not change with time.
Multiple ascending repeat-dose studies
After review of the safety data and the single-dose PK profile, two or three safe and well-tolerated dose levels are chosen and the multiple ascending (repeated-dose) study (MAD) is designed. Its purpose is to test safety, tolerability and PK when the drug is given repeatedly.
Design
A typical MAD study design will be double-blind, placebo-controlled with two or three cohorts of eight (six active two placebo) to 12 (eight active four placebo) subjects taking successively higher doses for several days. As for the SAD, the decision to escalate to the next dose level is based upon the safety and tolerability of the preceding dose level. The dose regimen in the MAD study is based upon the PK characteristics seen in the SAD and designed to give the PK profile necessary to allow the drug to exert its therapeutic effect and to achieve ‘steady state’ in which the drug’s input rate is balanced by its rate of elimination. The duration of the dosing is based upon the desire to achieve steady state but also to collect sufficient safety information to support use in Phase II studies. For indications where chronic dosing is required a duration of around 7–10 days is usual in MAD studies.
Outcome measures
Safety assessment is necessary under these conditions, as steady-state blood levels are usually higher than blood levels following a single administration. It is also important to know whether the PK of the drug and/or metabolite(s) changes on repeated dosing. For instance, saturation of elimination pathways (e.g. metabolizing enzyme systems) could cause the drug to accumulate to toxic levels in the body, or alternatively stimulation (induction) of drug-metabolizing enzyme systems could cause the levels of drug and/or metabolite(s) to decrease to subtherapeutic levels. A comparison of the predicted and observed plasma–serum concentration time curves will provide evidence of any such non-linear or time-dependent kinetics for the drug and/or metabolite(s).
The general requirements and procedures for MAD studies are the same as those for Phase I SAD. A typical Phase MAD blood sampling schedule is shown in Figure 17.1B. Blood samples for PK profiling will generally be taken on the first and last days of dosing, with additional single samples taken immediately before the first morning dose each day, to measure the levels of drug remaining in the blood immediately before the next dose is administered, i.e. the trough level.
The results of the Phase I SAD and MAD studies together support the decision as to whether to administer the drug to patients and, if so, at what dose and regimen and for how long.
Pharmacodynamic studies
Pharmacodynamic (PD) assessments may be included in the MAD studies, or specific PD studies can be performed separately. Usually prior to Phase IIa, the objective of these studies is to establish whether the drug has some pharmacological effect in man that may be relevant to its therapeutic effect, and to determine at what doses and plasma concentrations the effects are seen, with a view to optimizing dose selection for Phase IIa. Such studies are termed PK/PD studies.
This approach must still be treated with some caution, as the physiology in patients may differ from that in healthy subjects, and clinical efficacy may therefore not be reliably predicted from Phase I results. It is not uncommon for drugs that are highly effective in patients suffering from a certain condition to have little or no effect on the same body system in healthy subjects. This is particularly true of drugs acting on psychiatric diseases as these conditions are very difficult to emulate in healthy subjects. The ideal PD assessments in Phase I are those that have biological or surrogate markers that are measurable in healthy subjects and are relevant to drug’s mechanism of action and/or therapeutic effect. For example, the ability of a β-adrenoceptor antagonist to inhibit exercise-induced tachycardia, or the effect of a proton pump inhibitor on acid gastric secretion are relevant effects that can easily be measured objectively in volunteers. However, such markers are not always available (e.g. in the case of many psychiatric diseases) or may be misleading (pain is a good example because the endpoints are subjective rather than objective), and the interpretation of such data is usually approached with care. Nonetheless, Phase I PK/PD studies can be very useful to confirm that a new drug is actually having the pharmacological effect in man that was predicted from animal studies. As well as studying pharmacodynamics in healthy subjects, patients with a mild form of disease can be studied. An example is asthma, where a new drug could be studied for a short period of time in mild asthmatics to observe a pharmacological response, without necessarily intending to provide a long-term therapeutic benefit. Such subjects are known as patient volunteers and, like healthy subjects, as they are not entering the study to seek a cure for their disease, they can receive some financial compensation for their time and inconvenience.
Drug–drug interaction studies
The objective of drug–drug interaction studies (DDI) is to determine whether the test drug’s efficacy, safety or pharmacokinetics will be altered if it is given with other drugs that the target patient population may also be taking. The timing of drug interaction studies depends partly on the importance of understanding interactions prior to treating patients. Some DDIs may need to be performed prior to Phase IIa if the patient population in the study cannot be excluded from taking concomitant medications that might interact with the test drug. Otherwise DDIs can be conducted later in the development programme when more is known about the target treatment population and the efficacy and safety of the new drug. The choice of which DDI studies to perform is based on the following: the metabolism of the new drug (for example if it inhibits, induces or is metabolized by certain cytochrome p450 enzymes – see Chapter 10); the pharmacodynamic action of the drug; and which drugs the target population may be taking concomitantly. Drug interactions can occur on a metabolic level, so that the exposure to a certain dose may be changed by a drug interaction or on a pharmacodynamic level so that pharmacological effects may be increased or diminished by the interacting drug. It is important to known whether co-administration of the relevant drugs leads to a change in exposure or a change in pharmacodynamic effect of either the test drug or the interacting drug.
In a typical DDI study, subjects are dosed with the interacting drug until steady state is achieved and then a single dose of the test drug is given and the PK, safety, and sometimes PD effects are evaluated. The information from the DDI studies gives guidance as to whether any dose alterations are necessary when the new drug is co-administered with a drug with which it interacts and the information is included on the product label.
Absolute bioavailability and bioequivalence of new formulations
Absolute bioavailability studies are performed to compare the exposure of an intravenous preparation of the study drug which has 100% bioavailability, with the formulation intended for clinical use which is to be given by another route, e.g. oral or subcutaneous. The absolute bioavailability information is needed for the product label, but also assists further formulation development as modifications to the formulation can be made in order to optimize exposure to the drug. The studies are conducted in healthy subjects in a crossover fashion where each subject receives an intravenous dose and then one or more doses of the study drug given by its intended clinical route of administration. Standard pharmacokinetic parameters are measured and the absolute bioavailability calculated by comparing the pharmacokinetics of the intravenous and non-intravenous doses.
In the initial clinical pharmacology trials and in the exploratory efficacy studies it is not usual to use a formulation that would be the final commercial formulation. Development of a commercial formulation is performed in parallel with the early phase studies and the data from those studies guides the formulation development activity. Once a commercial type formulation or formulations has been identified, the safety and pharmacokinetics are compared with the prototype formulation in comparative bioavailability studies. As for absolute bioavailability, the studies are conducted in healthy subjects in a crossover fashion. If there is no more than 5% difference in exposure (AUC) between two formulations, they are considered to be bioequivalent. Dosing information obtained from exploratory efficacy studies using a prototype formulation which is bioequivalent to the commercial type formulation, can, therefore, be applied directly to confirmatory efficacy dose range finding studies. If the formulations are not bioequivalent, further multiple dose pharmacokinetics and pharmacodynamic studies may be needed to determined dosing regimens for the dose range finding studies. Depending on the intricacy of the formulation development, comparative bioavailability studies may need to be performed on more than one occasion.
Absorption distribution metabolism excretion (ADME) in man (radiolabelled studies)
The objective of the human ADME is to identify precisely the handling of the drug by the body and to look for and quantify metabolites that might also have effects on its efficacy and safety. The study involves giving a small number (usually four to six) of male subjects a dose of radiolabelled compound and then sampling blood urine and faeces over a period commensurate with its elimination half-life. Unless information about metabolite presence and activity is needed more urgently, the study is usually performed in late Phase II or early in Phase III.
Other safety pharmacology studies
The clinical pharmacology programme also contains studies to examine safety aspects, notably the ability of the new compound to cause QT prolongation, which may carry a risk of a potentially fatal ventricular arrhythmia, called torsade de pointes, or for some central nervous system drugs, abuse liability studies are needed. There are specific study designs to look for whether a drug has potential for abuse. Other studies such as effect on reaction time or driving ability may also be needed, particularly for central nervous system compounds.
The need for thorough QT studies has become a general requirement for all NCEs in the past few years, whether or not the compound demonstrates any potential to cause cardiac conduction abnormalities in vitro or in non-clinical studies. The studies are usually conducted in mid-stage of development when efficacy has been shown, the likely therapeutic dose is known and prior to large scale confirmatory studies. They are conducted in healthy male and female subjects with no evidence of heart disease or concomitant medication that could affect cardiac conduction. The study typically comprises four arms in a crossover, that is the test drug at the intended clinical dose, the test drug dosed at or near the MTD, an active comparator known to cause QTc prolongation, e.g. moxifloxacin and placebo. Drug dosing on each treatment day is followed by the collection of multiple ECGs at multiple time points, especially around Cmax, that are read by hand by a blinded central observer. As the studies require approximately 50 subjects and hundreds of ECGs to be read they are laborious and expensive. The Guidance ICH E14 from 2005 sets out the conduct and interpretation of thorough QT studies. However, the true predictiveness of these studies for torsade de pointes remains to be proven and it is likely that a number of useful drugs may be halted in development due to an observation of prolongation of QTc that may not represent a risk of dangerous arrhythmia to patients.
Special populations
In order to provide accurate dosing information in the product label, to cover administration to different patient types in the target population, the pharmacokinetics and safety are studied in patient subgroups. These include elderly subjects, specific ethnic groups, subjects belonging to a defined genetic subgroup for metabolism (fast or slow metabolizers) and also subjects with hepatic impairment and subjects with renal impairment. These studies are usually conducted once the clinically effective dose is fairly certain, as they are performed with the intended clinical dose. As many news drugs will be given to patients over 65 years old, elderly subjects are required in order to assess safety and kinetics in a group of healthy subjects more representative of the target patient population. Specific ethnic groups, on the other hand, will be required when a drug being developed in Caucasian populations is also being submitted for approval in a different population (e.g. Japanese). In this case, it is to determine whether significant differences exist in the pharmacokinetics and pharmacodynamics of the two ethnic groups, and whether different dosage regimens will be necessary. Specific genetic metabolic subtypes might be selected in order to ensure that where slow metabolizer subtypes exist this does not cause accumulation of the drug and related toxicity in those individuals. Finally, as most drugs are eliminated via the kidney and/or the liver, alteration of the function of these organs could change the kinetics and safety when the drug is given to patients with hepatic or renal impairment; hence these studies are performed to make dosing recommendations for those patients.
Phase IIa – Exploratory efficacy
Objectives
The exploratory efficacy studies have different objectives depending on whether the drug in development has a novel mechanism of action, i.e. is a first in class drug, or if it has a known mechanism and others in the class are already available for patient use in the indication, i.e. a ‘follow-on drug’. In the case of the former, The ‘proof of concept’ (PoC) studies in Phase IIa will be the first time the drug is tested in patients and this is when scientific theory is tested in clinical reality. It is interesting for the non-clinical and the clinical teams to see how translational the animal models turn out to be in patients. As it is the first time the drug is given to patients, safety evaluation is the key objective. The design of PoC for novel mechanisms is crucial to ensure that meaningful clinical effect can be identified, if it exists and likewise if there is no clinical effect this also needs to be identified, so that a decision about the future of the drug in the indication can be determined.
In the case of a follow-on drug with a known mechanism of action, the PoC studies will be more orientated towards establishing points of differentiation with drugs on the market that have the same mechanism of action. The existing product may have weaknesses of efficacy, side effects or pharmacokinetics which the follower drug can improve upon. An example of this is the class of oral triptans for acute treatment of migraine, where speed of onset of action and duration of action, manifest by a lower headache recurrence rate, were differentiators of interest compared to the market leader sumatriptan. Follow-on compounds in the class concentrated on having either a more rapid onset of action or a long half-life leading to lower headache recurrence. The design of studies for follow on compounds will be orientated towards drawing out these differentiating features rather than answering the question ‘does it work or not?’
Design consideration for first in class compounds – large pharma vs small pharma
Phase IIa PoC studies are small in size and designed carefully to answer the questions about whether the drug has any meaningful therapeutic activity in the case of first in class compounds, or whether it has any useful differentiating features in the case of follow-on compounds. As the studies are not fully statistically powered to demonstrate treatment differences it is important to determine a priori what are the criteria for success or failure in the study. This requires objectivity and an experienced development team.
The objectives of a large pharmaceutical company in conducting PoC with a novel mechanism of action may be different to those of a small pharmaceutical company. For the large company, the most important aspect will be to show as soon as possible that they have a novel mechanism of action that is safe and well tolerated and works in man, to feedback to the discovery teams working on the back-up programme. To that end they may choose a drug which is good enough for exploratory clinical trials, but may have features that are not optimal for a final commercial medicinal product, for example a short half-life or poor solubility. However, once PoC has been demonstrated with the novel mechanism of action they can accelerate development of back-up molecules, which have better properties to become medicinal products. On the other hand if the PoC fails, then this line of research can be abandoned.
For a small company with more limited resources and fewer pipeline programmes, the Phase IIa PoC studies may be make or break for the entire company. Start-up companies funded by venture capital may only have one lead product going into Phase IIa, no products on the market and other compounds in their pipeline may well be a long way from the clinic. In that case a great deal of the company’s fortune rides on the drug being tested in Phase IIa becoming a medicinal product. The small company may well have done their best to select a molecule with good drug-like properties which they believe could make it to the market, prior to entering into man. The objectives of Phase IIa for this type of company are to demonstrate convincing therapeutic effect with acceptable safety so that the compound can be advanced further into development. It is not generally the intention to go back to other molecules that might look a bit better, as in the case of large companies, unless there is a major problem with the lead compound. Whatever the size of the company, the basic tenet of Phase IIa is to gain robust data upon which to base decisions about the future of the drug. Given that the PoC studies are fundamental to the go/no-go decision for an entire development project in both large and small companies, the design of the studies is crucial.
Design
In most cases, a double-blind placebo-controlled study is an ideal approach as this allows for a more objective measure of efficacy and safety. However, there are notable exceptions, for example in oncology, where a comparison with, or add-on to a standard therapy is usually needed as it would be unethical to withhold known effective treatments from patients with cancer. It is possible to perform open-label small ‘look see’ studies in Phase IIa using historical comparison with efficacy of other drugs used in the indication, but this is not an ideal strategy as there is a substantial risk of bias if there is no control arm. The dosing regimen will have been determined from the Phase I studies and also from supporting non-clinical pharmacology data. There are numerous possibilities for choosing the dose, but the principal objective is to be as sure as possible that the dose or doses chosen for the study have a high likelihood of showing an effect if there is one and that the doses have acceptable safety and tolerability. A maximum tolerated dose approach is often used, especially for drugs with good safety and tolerability. Doses can be selected to achieve plasma concentrations that have been shown to be effective in animal studies or which are likely to give a particular receptor occupancy if PET studies have been performed previously. The number of dose groups in Phase IIa studies is usually limited to one or two active groups and a placebo group. The treatment groups may be parallel or the study can be done as a crossover, which is when the patient is randomized to receive each of the treatments in a random order, separated by a suitable washout period. The advantage of a crossover is that the patient acts as his/her own control and the number of patients can be reduced. The disadvantage is that there can be an order effect where the previous treatment affects the outcome of the subsequent treatment. Also crossovers are generally more suitable for indications where the outcome variables are rapidly measurable, e.g. pain, or have objective endpoints, e.g. blood pressure. Crossovers are less suitable for indications where the efficacy measurement is highly dependent on patient reported outcomes, e.g. anxiety, because they are more prone to bias from order effects. As the patients have to have more than one treatment the crossover study may also take longer to complete than a parallel group. However, if patient recruitment for a particular indication is difficult (for example rarer diseases or highly competitive therapeutic trial areas) a study with a larger parallel group population may take longer to complete. As they are exploratory, Phase IIa studies lend themselves to adaptive trial designs where the dosing may be adjusted during the study according to the response of the study population. Adaptive trial designs are discussed in more detail below.
In Phase IIa, as clinical data are required rapidly, the preclinical programme may well be lean, and focused on getting only the data needed to support initial study in humans, therefore the formulation development is likely to be incomplete. If the compound is highly soluble, an intravenous or simple oral formulation (solution or simple tablet) can be used in early Phase I and Phase IIa. Information from these studies, in particular the PK-PD data, will help with the development of a commercial type formulation, to be used in confirmatory efficacy studies. If the compound is poorly soluble more formulation development will have taken place prior to entry into man. Nevertheless, in Phase IIa it is not usual to use a final commercial type formulation as the results of the initial clinical studies do influence the final formulation development.
The duration of Phase IIa studies for novel drugs is influenced by the indication, but is usually shorter than for larger scale later phase studies, for two reasons. Firstly it is important to gain information on clinical effect as soon as possible so that decisions on compound development can be made. Secondly, the toxicology programme to support the shorter studies, e.g. up to 1 month in duration, will also be shorter. Clinical trials of 3 and 6 months dosing duration require toxicology studies in two species of corresponding duration to support the human dosing. To make this type of investment in a toxicology programme for a novel mechanism of action drug before its clinical effects are known, is not always desirable, particularly for smaller companies. For follow-on compounds, however, studies of more than 1 month in duration might be needed in Phase IIa for indications in which clinical differentiation will only become apparent after longer-term administration, for example in Parkinson’s disease or psychiatric indications.
Patients to be studied
The PoC is usually the first time that the drug will be used in patients with the relevant disease; therefore, careful patient selection is vital to the success of a PoC study, especially those of novel compounds. It important to be clear about what question is being asked in the study and what patient group should be targeted so that question can be answered. Typically in the exploratory phase, as the population is small, a more homogeneous patient group is selected to reduce variability that might dilute the possibility of seeing a result. The inclusion and exclusion criteria at this stage may be more restrictive than at later stages of development. In the exploratory efficacy phase less is known about the safety of the drug in diverse clinical situations and the drug–drug interactions will not have been fully explored. A subgroup of patients within a disease entity who are considered most likely to show a benefit can be studied at this early stage. For example, to evaluate the benefit of reflux inhibition with a novel drug in patients with gastroesophageal reflux disease (GORD), patients with non-erosive reflux disease and classical symptoms of heartburn and regurgitation would be selected, in order to increase the likelihood of seeing a response to the drug. Once convincing efficacy and satisfactory safety/tolerability have been demonstrated, subsequent studies can include patients with more severe disease, or more diverse symptoms. In the case of GORD this could extend to patients with erosive oesophagitis and symptoms other than heartburn and regurgitation that are thought to be due to GORD.
Outcome measures
Thorough safety and adverse event monitoring is performed in these initial patient studies, with close attention paid to looking for untoward effects in patients that might not have been seen in healthy subjects. With regards to efficacy, as the PoC studies have a relatively small population and short duration, it is necessary to have robust outcome measures. In the exploratory phase it can be tempting to try and answer a lot of questions in one clinical trial. This is not a good idea because putting too much into the trial increases the complexity of its execution and can confound the interpretation of the data. It is by far better to have one clear principal objective and, at most, two smaller less important objectives. The ease of having robust measurable endpoints depends a lot on the indication. Those indications with objective measures, e.g. hypertension and diabetes, are straightforward to demonstrate in terms of proof of concept. However, for licensing in such indications it is not sufficient to show that the drug lowers blood pressure or glucose alone, it has to be shown that the drug improves patient outcome in reducing mortality or cardiovascular morbidity, which presents a substantial challenge in late phase development. In the middle are pain-type indications, as pain is rapidly treatable. However, as one is relying on the patient to report the severity of pain, the outcome can be subject to bias and a high placebo response. For other indications where long-term treatment is needed to evaluate the full benefit, a good surrogate marker can be used in the PoC. One example would be in the case of the treatment of rheumatoid arthritis where measuring inflammatory mediators in the blood can indicate evidence of meaningful pharmacological effect. Another example comes from a study of the prevention of vasospasm post subarachnoid haemorrhage, with an endothelin antagonist. Ultimately it needs to be known if treatment with the drug improves patient outcome, i.e. mortality and morbidity, but in the PoC trial the occurrence of vasospasm was measured with angiography, transcranial Doppler and the presence of infarcts on CT scanning, to see if the drug was able to prevent physiological vasospasm and its anatomical sequelae.
Pharmacokinetic samples may also be taken in the Phase IIa studies to examine the PK-PD response and also to see if the pharmacokinetics in patients are different from healthy subjects; migraine patients, for example, have gastric stasis during an attack and this can alter the absorption of orally administered drugs. Phase IIa may be the first time that the PK–PD relationship is studied and the information can be used to guide the dose selection for Phase IIb.
In summary, the characteristics of Phase IIa PoC studies are short, focused, using carefully selected patient populations and robust endpoints to enable go/no-go decisions for new drugs in development.
Phase IIb to III dose range finding and confirmatory efficacy studies
Objectives
The purpose of this phase of development is to confirm the initial efficacy seen in the Phase IIa PoC studies and to provide the pivotal data which will support the application for market approval, as such extensive, comprehensive data from well-designed and adequately powered studies are required. The data from the confirmatory studies will be used to make the claims in the product label upon which the drug can be promoted. Therefore, during this part of the development, the clinical team needs to liaise closely with Regulatory and Marketing so that the trials can be designed to fulfil the desired product label. Usually the first step is a Phase IIb dose range finding study to confirm the preliminary safety and efficacy data and to explore the dose–response relationship in detail, to select the dose that has the optimal efficacy and safety profile to be used for large-scale Phase III efficacy and long-term safety studies. The eventual marketing strategy is a major guiding factor for the design of the Phase III programme, especially for follow-on drugs where product differentiation to existing treatments, whether it be on efficacy, safety or cost effectiveness, is crucial to its success.
Design
The standard design of Phase IIb dose-finding studies is parallel-group randomized double-blind and, placebo-controlled and/or active comparator controlled. The placebo group in theory allows for a comparison of active intervention with ‘no treatment’ and, therefore, should provide a clear view of the efficacy benefits and safety of the test drug. However, it is well documented that patients in clinical trials do have a response to placebo (Beecher, 1955). Strictly speaking, taking placebo does not mean that the patient has no treatment. The very act of taking part in a clinical trial and receiving extra medical attention can contribute to an improvement to a patient’s underlying condition. This is particularly true for psychiatric conditions or those exacerbated by stress or anxiety. Trials in psychiatry and those which rely heavily on patient reported outcomes have notoriously high placebo response rates. Occasionally the enthusiasm of the investigating physician for the new treatment can colour their view of the efficacy, which can also contribute to high placebo response rates. In some medical conditions, for example oncology or other serious conditions for which effective treatment exists, it is not appropriate for patients to receive placebo. In this case the new drug can be tested either against a gold standard, single active comparator or against a standard-care treatment regimen. In the latter case the test drug might also be added to standard care in one group while the other group just receives standard care alone. Another way to minimize placebo response in the active treatment phase, is to have a single blind, placebo run-in period, where all patients take placebo but only the investigator knows that they are on placebo. At the end of the period, those who continue to fulfil the predefined eligibility criteria can be randomized to the double-blind phase. For pivotal efficacy Phase IIb or III studies in Europe, it is a requirement to have at least one study with an active comparator arm. The choice of comparator will depend on marketing considerations and what the potential advantages the new drug can offer, whether it be in terms of safety, tolerability, efficacy or cost effectiveness. Active comparator studies may be designed to show superiority, equivalence or non-inferiority compared to the gold standard treatment. The choice will depend upon the efficacy and the safety of the drug. For example, if the drug is thought to have similar efficacy but a superior safety profile to the existing treatment an equivalence or non-inferiority design may be chosen for the primary efficacy, because the main objective is to demonstrate the better safety and tolerability. The treatment selected for comparison has to be justified to the regulatory authorities when submitting the trial application and can be discussed with them in advance, for example at an end of Phase II meeting. The only exception to this is for indications where no treatment is currently licensed. An example of this is levodopa-induced dyskinesia in Parkinson’s disease. Even so, the non-use of an active comparator needs to be justified in the clinical trial application.
These trials form the basis of the marketing authorization application and so they have to have a sufficient sample size to demonstrate efficacy with confidence. Also sufficient safety data to support exposure to the target patient population needs to be generated. Statistical calculations are made to ensure enough patients are enrolled to have robust efficacy results that will support the desired claims on the product label. In the case of a parallel group, multiple, dose-range finding Phase IIb study, although the power of the study might be lower than required for a Phase III pivotal efficacy study (e.g. 85% rather than 90%), sample size calculations have to take into account adjustment for comparisons of the multiple dose arms. Typically the studies will have several hundred patients or in the case of some cardiovascular intervention (e.g. hypertension) studies, a few thousand patients may be required. The dosing duration must be sufficient not only to demonstrate efficacy but also to establish safety. For indications where long-term chronic use is intended, even if it is intermittent dosing, the safety database should contain information on dosing for at least 12 months in an adequate number of patients. These data are often generated by enrolling patients from the Phase IIb and III studies into a long-term, open-label, extension study at the intended clinical dose.
The dose range for Phase IIb will be selected based upon the efficacy and safety data from the Phase I and Phase IIa studies. If the Phase IIb study is successful it will have identified the dose level and dosing regimen which gives the optimal balance between efficacy, safety and tolerability. This dosing regimen is subject to confirmation in Phase III.
Patients and study setting
For the later phases of development it is important to study the drug in a more ‘real-life’ setting, because the safety and efficacy data from these studies has to support the use in the patient population in the target market. Therefore, the trial eligibility criteria are expanded in Phases IIb and III to enroll patients that are more representative of the final target population. By this stage more safety and drug interaction data are available, so reducing the restrictions on patient recruitment can be done with more confidence. The countries, study sites and doctors selected for the late phase studies will generally be more diverse than in Phase IIa and influenced by marketing as well as regulatory needs. It is perfectly possible to apply for a product licence in a country or region without conducting pivotal efficacy trials there, because the trials are conducted according to ICH GCP so the trials should be acceptable as long as the data generated cover any intrinsic (genetic) or extrinsic (e.g. diet, medical practice) differences that exist in that region. However, for the major territories of USA, Europe and Japan it is usual to conduct at least one study in that region and, for Japan, bridging studies may be needed to demonstrate that the properties of the drug demonstrated in a ‘European type’ population are applicable to Japanese patients.
Outcome measures
In the large-scale confirmatory studies the surrogate endpoints of Phase IIa give way to demonstrating clinically meaningful benefit. This means that simply demonstrating a reduction in blood pressure, or preventing cerebral vasospasm or reflux events from occurring, to name but a few examples, has to be shown to provide a benefit on disease-free survival or a benefit to the patient’s ability to function in their daily life. It also has to be shown in many cases that the new medicine will be cost effective. This means that the outcome measures in Phase IIb and III are more orientated towards patient and physician reported outcomes. For example, in oncology tumour markers used in Phase IIa will give way to demonstrating survival over a predefined period of time. In hypertension, the measurement of blood pressure may need to be accompanied by data on the incidence of cardiovascular morbidity and mortality, and in neurology and psychiatry there are a host of validated questionnaires to demonstrate meaningful clinical benefit. In addition, in many cases pharmacoeconomic data needs to be collected, if not to justify marketing authorization, then to justify the pricing of the drug to reimbursement committees and managed care plans in the various countries where the drug is to be sold. As these reported outcomes have various factors that can influence them the sample size needed to show a positive effect is generally much larger than when objectively measured surrogate markers are used. For many indications where treatments exist there are outcome measures that are recognized by the regulatory authorities as being the gold standard efficacy measure for the particular indication. These measures are not always appropriate for drugs with novel mechanism of action or for a subset of patients within an indication. It is possible to stray from the path of the standard efficacy measure and use outcomes more adapted to the clinical scenario, but it requires good justification and careful negotiation with the relevant competent authorities.
Phase IIIb and IV studies
The data included in the submission package from the Phase I to III studies is very comprehensive; however, it is not possible to guarantee that all adverse effects that could be observed when the drug is on the market, have been identified in the pre-marketing studies; especially for those events that occur with an incidence of less than 1 in 10 000. Indeed there are many cases of drugs being withdrawn from the market for safety reasons, or having significant labelling amendments applied to them, once more information has become available. Regulatory authorities recognize that to ask companies to collect tens of thousands of patients’ safety data in the initial submission package would be costly, time consuming, and could cause unnecessary delay, or even prevent the marketing of useful new medicines. Therefore, in order to protect patient safety once the drug is on the market, the authorities request that the companies perform post-marketing safety surveillance, through specific studies designed to evaluate safety of the marketed drug and by means of filing Periodic Safety Update Reports (PSURS). The data for these reports are collected by the company’s pharmacovigilance group, whose members are specially trained in drug safety surveillance. The reports are required to be filed at regular intervals and include data from ongoing trials and spontaneous adverse event reports, which can arise from a variety of sources, e.g. doctors, patients, clinical trials, journal articles, etc.
Clinical trials collecting additional data can be started while the initial marketing authorization submission is being reviewed. Authorities will sometimes accept dossiers for review with limited duration safety data if the company continues the collection of data during the review period and has sufficient patient exposure by the time the authority comes to make their decision. Studies conducted in this peri-approval period are known as Phase IIIb studies. If a company wishes to expand the indication, they may also conduct additional pivotal efficacy studies in the peri-approval period or shortly after product launch and these studies also fall into the category of Phase IIIb.
The Phase IV studies, which take place once the product is on the market, may take the form of collecting safety data as required by authorities, but they may also be used to collect additional simple efficacy data which are used to support the marketing effort. Phase IV studies are generally large in scale, conducted at widespread sites but usually of simple design, and are orientated towards looking at how the drug is used in everyday practice within the confines of the existing product licence. As for any clinical trial, Phase IIIb and IV studies are subject to the conditions of ICH GCP.
Bridging studies
Data generated in the USA and/or Europe and used to support marketing approval of the drug in Japan present more problems, owing to the more significant intrinsic (e.g. genetic) differences between Caucasian and Japanese populations. An example is the greater frequency of polymorphisms in the cytochrome P450 enzyme CYP2A6 (Oscarson, 2001) seen in Chinese and Japanese populations, and the related differences in nicotine metabolism. It is therefore usually necessary for a study to be carried out to ‘bridge’ Caucasian data into Japan, i.e. to test the validity of Caucasian data in a Japanese population. This is done by studying the drug’s pharmacokinetics, and usually its pharmacodynamic properties, in both populations and analysing the results to determine whether they are comparable.
In considering the issue of extrapolating drug safety and efficacy data from one population to the other, the objective is to do this safely while not repeating clinical trials unnecessarily. Specific ICH guidelines exist for the extrapolation of data from one ethnic group to another (ICH, E5 latest update 2006). There are several strategies and approaches for exploring ethnic differences in drug response, but none is appropriate for all circumstances and each situation must be carefully evaluated, with a detailed understanding of the requirements of the target regulatory authority and its acceptance of foreign data.
Patient recruitment in efficacy studies
In exploratory efficacy studies, as the efficacy and safety in the patient population are yet to be determined, it is desirable to have a relatively homogeneous population in order to minimize variability which might confound the results. The entry criteria for patients in exploratory studies will therefore be more stringent than in the later phases where it is then desirable to study patients who are more representative of the population, which will use the drug once it is on the market. Patients can be recruited from the patient pool known to the clinic if they are regular outpatients or if they have already participated in a clinical trial in the indication. In some studies the patients may already be in the hospital (e.g. in studies of interventions in acute medical emergencies, such as myocardial infarction, stroke, or inflammatory bowel disease). Patients may be referred to the investigator from other clinics, or it is possible to source patients externally by means of advertising. There are various possibilities for advertising. Media such as radio and television are popular in the USA but less so in Europe, partly because the cost is not always commensurate with the return. Publicity in local papers and posters and fliers in hospital or general practice clinics are often effective, as are advertisements on a hospital website. The appropriateness of external advertising will depend on the indication. For common, straightforward ailments, such as migraine or allergies, external advertising can be a good way to recruit patients, but for more complex indications, for example in neurology or oncology, it may not be appropriate. The disadvantage of advertising is that the patient coming in ‘off the street’, is not already known to the investigator and so a thorough check of the patient’s medical history and trial eligibility, including likelihood of good compliance, needs to be made. All advertising materials, including the scripts for radio and television advertisements, have to be reviewed and approved by ethics committees before they can be used. Patients who take part in the efficacy trials may anticipate deriving some clinical benefit; therefore, they are not volunteers as such, and cannot be paid for their participation, as is the case for subjects in Phase I studies. However, the patients can receive a modest sum to cover out-of-pocket costs for transport and subsistence associated with the study clinic visits, the amount of which has to be approved by the ethics committee.
Clinical trials in children
As part of the overall clinical development plan it is now a requirement in the EU and USA to include a paediatric drug development plan. The objective is to have thorough testing of the safety, pharmacokinetics and efficacy of drugs that may also be used in children, so that correct dosing information, using an appropriate formulation, can be given for this population. In the past, companies tended to shy away from testing their drugs in children due to ethical concerns and also because the paediatric market was not seen to be particularly commercially attractive. Adult drugs were used off-label in children, often by extrapolating the adult dose to the weight of the child with no proper guidance in the product label for paediatric use. However, there are many medical conditions that occur in children as well as in adults. Examples of drugs commonly used by children as well as adults include anti-epileptics, asthma drugs, anti-inflammatory and anti-infectives. The therapeutic margin of some of these compounds can be quite narrow and so precise information about safety and dosing in children is very important. Differences in metabolism exist between adults and children and at different times in childhood, e.g. during adolescence, that may alter the kinetics or safety of the adult medicine. Also formulations used by adults many not be suitable for use in children. The purpose of the plan, therefore, is to ensure that companies will develop medicines that can also safely and effectively treat important conditions in children. The draft plan is usually requested by the end of Phase II. The timing of the start of the paediatric trials is usually within a year after the product licence for the adult use has been granted. However, this can be varied according to the indication or need. Exemptions to paediatric development can be obtained if the disease does not exist in children, for example Parkinson’s disease or Alzheimer’s disease, or in certain age categories, e.g. migraine, which does not really occur in children less than 6 years old.
Regulatory and ethical environment
In most parts of the world, clinical trials are a legal requirement before a new drug can be sold or any claims made for its therapeutic benefit or safety. All clinical trials, including Phase I studies, are subject to international, national and, sometimes, also local regulation. International regulatory requirements for human administration of a new active substance (NAS) are set out in a series of guidelines published by the International Committee on Harmonization (ICH; see Chapter 20). This committee was formed to harmonize the regulation of clinical trials in the three major pharmaceutical development regions (European Union, USA and Japan), with the aim of avoiding duplication of clinical research programmes when applying for approval in all three ICH regions. National and local regulations still exist and may vary from country to country within these regions, but ICH guidelines still apply and usually have the force of law. Clinical development of new drugs for registration in any of these regions must comply, and be seen to comply, with ICH guidelines if the data are to be accepted for registration purposes in the ICH regions, irrespective of where in the world they were generated. This means it is not possible to sidestep the requirements of the ICH guidelines by developing drugs outside the ICH regions in a manner that would not comply with them. As the EU, USA and Japan represent the three biggest world markets for new drugs, there is little incentive for non-compliance.
All human studies are performed according to strict ethical requirements. The Declaration of Helsinki (World Medical Association, 2008) and ICH Guidance E6 (CPMP/ICH/135/95) 2002 set the rules for all clinical development. In one sentence, the message comes through: ‘It is the duty of the physician in medical research to protect the life, health, privacy, and dignity of the human subject’. The major points of the Declaration of Helsinki and ICH E6 Good Clinical Practice are summarized below:
• The risks and potential benefits must be assessed before trials are initiated, and the benefits must outweigh the risks.
• The interests of the individual study subjects must take precedence over those of science or society.
• All trial subjects must freely give their informed consent prior to participation.
• Trials must be scientifically sound and clearly described in a trial protocol.
• The trial must be carried out according to the protocol, which must be reviewed and approved by a properly constituted ethics committee.
• Only properly qualified physicians may provide medical care to trial subjects, and all other staff involved in clinical trials must be appropriately educated, qualified and experienced for the tasks they carry out.
• Human administration must be supported by the results of adequate preclinical testing in compliance with ICH guidelines for the administration of drugs to man.
• Data from clinical trials must be recorded, handled and stored in a way that allows accurate reporting, interpretation and verification.
• Trial subjects’ privacy and confidentiality must be respected and assured.
• The material to be administered must be of acceptable quality and purity, as defined by the relevant ICH guidelines. This means both the drug substance and the formulated drug product must be manufactured in compliance with ICH Guidelines (2000) for good manufacturing practice (GMP) and used in accordance with the trial protocol.
• The trial must be registered in a publicly available database prior to the first subject being recruited.
Ethical procedures
The safety of subjects is always the paramount consideration in clinical trials. There are two main bodies responsible for evaluating proposals for trials: the national drug regulatory authority in the country where the trial will take place, and the local ethics committee (EC) or, in the USA, the institutional review board (IRB) of the clinic in which the study will be performed. All clinical trials must be approved by a properly convened and correctly functioning EC or IRB before they can be initiated. Ethics committees are composed of independent experts and lay members, who review the proposed study (protocol, investigator’s brochure, insurance arrangements, patient information sheet, informed consent documentation and any other patient facing materials, e.g. electronic or paper diaries, questionnaires, etc.) and decide whether it is justified on ethical grounds. They evaluate the study in terms of the risk to the subjects, the appropriateness of any remuneration offered to both subjects and the clinical investigator, the design of the study and its ability to fulfil its primary objective(s), the qualifications, experience and clinical trial performance of the clinical investigator, the text of any advertising used to recruit subjects, etc., and approve or reject the proposed trial on the basis of these ethical considerations. If it is approved, the EC/IRB remains involved with the trial until it is completed: for example, it must be informed of any serious adverse events (see below) that occur, and of any other major issues that cause concern during the study. Changes to an approved protocol may not normally be implemented without the EC/IRB approval.
In the case of regulatory authorities the degree of involvement varies from country to country (see Chapter 20), some reviewing all of the available data on the drug and some only requiring notification that EC/IRB approval has been given and that the trial will take place.
Clinical trial operations and quality assurance
As described above, all clinical trials have to be conducted to ICH Good Clinical Practice (GCP) by investigators and staff properly trained in the conduct of clinical trials. The sponsor has a duty to ensure that the trials are being conducted according to GCP both at the investigational site and within the sponsor’s organization. The constraints of ICH and their local rules place a large organizational and administrative burden on the trial centres, which need to be selected with care, based on inspection of their facilities and an assessment of the investigator’s and site staff’s qualifications, experience and availability to carry out the study to the standard required.
The sponsor has specific responsibilities to train those involved in the trial in the requirements of the study protocol and ICH GCP standards, and then to monitor the project on site to ensure compliance monitor the performance of the sites during the study. Study staff training and monitoring may be performed by the sponsor company or delegated to a subcontractor, i.e. a clinical research organization (CRO), in which case the Sponsor has to oversee the activities of the CRO to ensure and be seen to ensure, that they are compliant with GCP. The study monitoring is performed by specially trained clinical monitors who visit the sites regularly throughout the trial and, using specific written procedures, verify the study data, check that the study is being conducted in accordance with the protocol and that there are no breaches of GCP. To further ensure compliance, additional quality assurance (QA) is carried out in the form of an independent audit. Study sites will be selected for auditing. The clinical monitor is instrumental in identifying the sites for auditing. Typically those sites who are high recruiters or who have had particular problems with the study are selected. In addition, if the monitor suspects any irregularity in the data, or even fraud, a site may undergo a ‘for cause’ audit. Auditors are responsible for thoroughly inspecting the data and documentation files to validate the site and the data it produces in terms of ICH compliance.
Finally, the regulatory authority itself may choose to inspect one or more clinical sites and/or the files of the sponsoring company or its CRO, as a final check on data quality. Issues arising at this late stage cast suspicion on the whole dossier and can delay or prevent the granting of marketing approval. A detailed overview of the regulatory requirements for product development and licensing are given in Chapter 20.
Issues of confidentiality and disclosure
Since 2008 it has become a requirement for all clinical trials to be registered on a publicly accessible database before the first patient is enrolled. In the USA and Europe clinical trials can be registered on http//www.clinicaltrials.gov or http//pharmacos.eudra.org which provide details of the purpose and plan of the trial, the clinical centres and investigators involved, and the stage the trial has reached. For every trial a registration number (International Standardized Randomized Controlled Trial Number, ISRCTN) is assigned, allowing public access to information on all prospective studies involving experimental and registered compounds. Its main aim is to avoid unnecessary repetition of clinical trials. These databases do not, however, include the results of completed trials, which frequently remain unpublished.
Regulatory authorities (see Chapter 20) require detailed results of all clinical trials approved by them – including trials of marketed compounds – to be reported to them, but this information is not, in general, publicly accessible, except to the extent that the Summary Basis of Approval (USA) or Centralized Evaluation Report (EU) that is published when a drug is approved, includes a summary of the clinical trials results on which the approval was based. Clinical trial sponsors are under an obligation to report to the regulatory authority any safety issues that come to light, and in the case of marketed drugs the regulatory authority may respond by altering the terms of the marketing approval (for example by including a warning in the package insert, or, in an extreme case, by withdrawing the approval altogether). Publication of trial results in the open literature is not obligatory. Trial sponsors often choose to publish their data in refereed journals, although they are not obliged to do so, and both they and journal editors tend to give preference to positive rather than negative findings (Hopewell et al., 2009). Publication bias inevitably means that good news receives more publicity than does bad news. The most recent Declaration of Helsinki, from 2008, states that authors should publish trial results or make them publicly available whether they are positive, negative or inconclusive. GlaxoSmithKline, has a publicly available database of trial results summaries for its marketed drugs and www.clinicalstudyresults.org also contains study results for marketed compounds. For drugs that fail in development there is no obligation to put the results into a public database, nevertheless, there is increasing pressure for companies to publish data about their drugs in development whether positive or negative. Such transparency could help drug development in general as much can be learned from the development programmes of drugs that do not make it to market.
Seamless drug development with adaptive clinical trial design: the future of clinical development?
The disadvantage of the conventional step-wise approach to clinical development where drugs move to the next Phase once the preceding Phase is finished is that it is time-consuming, costly and drugs may fail in late Phase development. It is estimated currently that approximately 40% of drugs entering Phase III do not make it through registration (Di Masi et al., 2010). The success rate is somewhat dependent on the indication, with GI, CNS and cardiovascular drugs having the lowest overall success rates (Di Masi et al., 2010). This is a great waste of resources for the pharmaceutical industry and a missed opportunity for medical practice. Late-stage attrition rates may be reduced by adopting a more seamless approach to the clinical development programme using adaptive clinical trial designs so that ineffective drugs can be identified earlier and their development terminated and those drugs with promising efficacy can be accelerated through development, with a higher likelihood of successful registration.
There are a number of ways in which the development stages can be merged or overlapped. Broadly speaking the development path is looking to have initial safety testing with exploratory efficacy leading to a go/no-go decision and then confirmatory efficacy and safety, leading up to filing the registration dossier. It is becoming more common for initial Phase I protocols to contain more than one stage. For example, within the same protocol it is possible to do a single ascending dose study, a food effect crossover at a dose determined from the SAD, and then, based upon these results, go into the multiple ascending dose study, which itself might contain pharmacodynamic evaluations relevant to the desired indication. Using this combined approach saves time in making repeated applications to regulators and ethics committees. Provided the decision points to proceed to the next stage within the study are clear and subjects’ safety is maintained throughout, combination Phase I protocols can be approved at a single regulatory and ethical review. If efficacy in an indication can be shown with a single dose, an initial proof of concept study in patients could commence after the SAD and be conducted in parallel with the MAD, thereby overlapping Phase IIa with Phase I. This always assumes that safety and tolerability in the SAD were acceptable. A good example of this is acute treatment of migraine, where efficacy can be shown with a single dose of study medication to treat a single attack of migraine. For indications where repeat dosing and/or steady state plasma levels are considered necessary to see a treatment effect one can consider performing the MAD study in patients, again assuming that the level of tolerability is acceptable for proceding straight to patients after the SAD. Most likely a relevant surrogate marker of efficacy will be required as the full effect on symptom control or control of the disease may not be evident until after several weeks’ treatment. An example of this could be treatment of gastro-esophageal reflux with a reflux inhibitor. The effect of a study drug on the occurrence of reflux events and transient lower oesophageal sphincter relaxations, following a challenge meal, can be monitored with oesophageal impedance-pH monitoring and manometry respectively, on a single day at steady state. Clinical symptoms could be monitored as a secondary objective because a significant effect on clinical symptom control would not necessarily be anticipated with such a short duration study. In the case of oncology studies it is routine to look for clinical effects in patients using surrogate markers in clinical pharmacology studies, as it is not possible to test most anticancer drugs in healthy subjects.
Once a go/no-go decision has been achieved in the exploratory phase, dose range finding Phase IIb studies can be combined with Phase III to achieve the objective of finding the optimal dose and having adequate, well-controlled, pivotal efficacy and safety data. An example of this approach is to have a parallel group, dose range finding study that has sufficient sample size and power so that it can be considered pivotal. Patients on the optimal dose can then continue either into an open-label safety phase or generate further efficacy and safety data in a double-blind controlled study extension Adaptive trial designs are useful in this regard, because the optimal dose could be selected from several dose arms on an interim measure, and then the study continued with the optimal dose to achieve full power (Orloff et al., 2009). Using the seamless Phase II to Phase III design, the second pivotal efficacy study can be started with the optimal dose as soon as it has been identified and the start of this study would overlap with the end of, or continuation of the dose range finding study. As the sample size for this type of study is likely to be larger than a conventional stand-alone Phase IIb dose range finding study, there needs to be sufficient confidence that the results of exploratory studies will be replicated in the larger scale trials at the doses chosen.
Adaptive design trials are those where an interim adaptation of treatment or endpoint during the study is decided before the study is started. The a priori protocol design incorporates the changes within it. The FDA’s draft guidance from February 2010 (FDA, 2010) defines adaptive trial design studies as ‘a prospectively planned opportunity for modification of one or more specific aspects of the study design and hypotheses based on analysis of the data (usually interim data) from subjects in the study’.
The purpose is to make studies more efficient, either by having a shorter duration, fewer patients, more appropriate patients, increasing the likelihood of identifying drug activity if it exists, or making the study more informative, for example in understanding better the dose response, the patients, or part of the indication most likely to benefit.
The adaptations may include the following:
• Change of sample size to either increase or decrease it according to results of an interim analysis
• Reallocate treatment distribution; dose groups may be added or dropped according to predefined criteria for doing so
• Changing the treatment duration
• Refining the study entry criteria: certain patient types may be dropped if they appear to have no benefit, alternatively the population can be enriched with patient types who appear to be deriving benefit.
The types of adaptations used may differ according to whether the trial is in the exploratory or confirmatory phase (Orloff et al., 2009). For example, surrogate endpoints such as biomarkers may be used to evaluate patient progress in an exploratory study, whereas clinical outcome measures would be needed in a confirmatory study. In an exploratory study it may be possible to use a single (patient) blind approach to reallocate treatments. An example of this was a study of migraine. The investigator knew the treatment allocation but the patients did not. The first patient at each site was allocated to start on a mid-range dose of the treatment. If the patient had a headache response, the next patient was allocated the next dose down, if not, the next patient was allocated the next dose up. The doses were changed up or down by the investigator according to each patient’s response. The results of this study turned out to closely predict the dose that was chosen as the optimal effective dose by a subsequent, conventional, parallel, dose-range finding study.
It is crucial that making the adaptations while the study is ongoing does not compromise the integrity or outcome of the study. The FDA draft Guidance 2010 (FDA, 2010) lays out the major design and performance considerations to be taken into account for adaptive clinical trials. The study needs to be carefully designed and the protocol clearly written to incorporate any interim analyses, the conduct of which must be performed in such a way as to not compromise the study blinding, or influence interpretation of the data. Close liaison with statisticians, who will have to model the effects of proposed adaptations on statistical outcomes, will be required when designing the study. Likewise the regulatory affairs group needs to be involved to help craft a protocol that will be acceptable and justify the use of the adaptive design to competent authorities. This means that the study design and protocol writing stage may take longer than a conventional study, but the reward should come in the form of a study that may be shorter to perform and have a higher chance of a successful outcome.
Adaptive clinical trials lend themselves better to some indications than others. The ideal scenario is to have an indication where the effect of treatment is rapidly evident and objectively measurable. For example, as blood glucose and blood pressure are objective and respond rapidly to effective intervention, Phase IIa studies in diabetes or hypertension could use adaptive design to good advantage, to rapidly identify doses and the target patient population. Acute treatment of migraine is a good example of an indication where adaptive design could be used throughout the development. Migraine attacks occur frequently in the study population and the gold standard efficacy parameter ‘pain free at 2 hours post dose’ is easily identifiable, even though it relies on the patient to report it. Indications where drug benefit takes months or years to become apparent and rely heavily on patient reported outcome are more challenging. This includes studies in oncology, neurology and psychiatry. Nevertheless, in the exploratory phase it may well be possible to perform an adaptive design using surrogate markers, to improve the dose and patient selection for confirmatory efficacy in these indications.
To summarize, the schematic in Figure 17.2A shows the stages and timelines of a conventional clinical development path and Figure 17.2B shows how a new form of clinical development might look, using seamless development with adaptive clinical trial design.
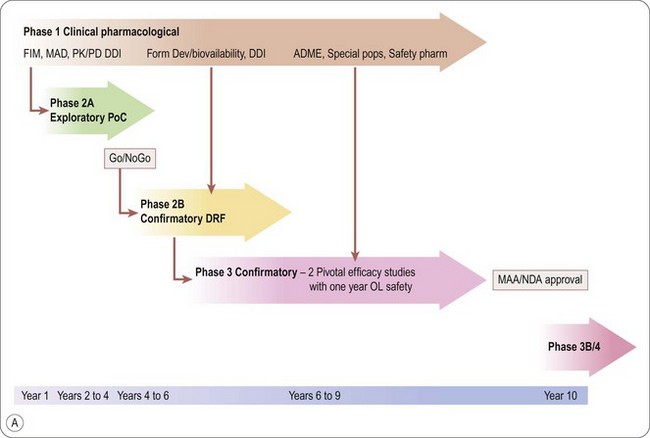
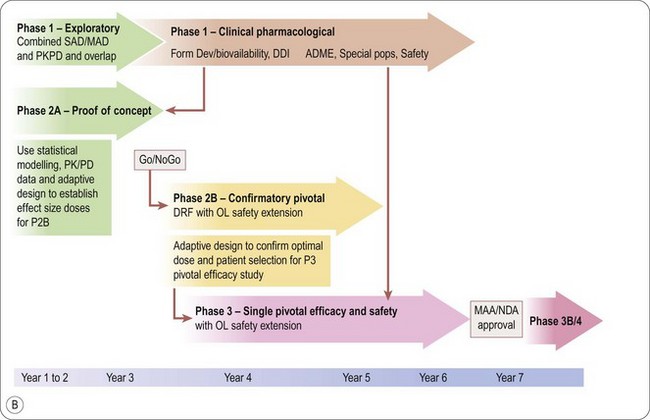
Conclusions
Clinical development is the cornerstone of bringing new medicines to the market. It is a complex, highly regulated, time-consuming and very costly part of the overall drug development process, with a substantial risk of failure. The traditional path of clinical development hitherto has served the pharmaceutical industry reasonably well and has enabled high-quality innovative medicines to be delivered to patients. However, the current process has a high attrition rate which is wasteful of financial, medical and patient resources. New initiatives are being launched at the input end to improve the sourcing of the drug development pipeline through public and private partnership, partnership with academia and partnership between small and large pharmaceutical companies and at the output end, by modifying the traditional clinical development pathway. It is hoped that adopting these new initiatives will enable companies to bring new medicines to the market and to patients more effectively and rapidly.
Adams CP, Brantner VV. Estimating the cost of new drug development: is it really 802 million dollars? Health Affairs. 2006;25:420–428.
Beecher HK. The powerful placebo. Journal of the American Medical Association. 1955;159:1602–1606.
Di Masi JA, Hansen RW, Grabowski HG. The price of innovation: new estimates of drug development costs. Journal of Health Economics. 2003;22:151–185.
Di Masi JA, Feldman L, Seckler A, et al. Trends in risks associated with new drug development: success rates for investigational drugs. Clinical Pharmacology and Therapeutics. 2010;87:272–277.
EFPIA. The Innovative Medicines Initiative. www.imi-europe.org, 2008.
FDA. US Food and Drug Administration 2004. Innovation or stagnation: challenge and opportunity on the critical path to new medicinal products www.fda.gov/oc/initiatives/criticalpath/whitepaper.html, 2004.
FDA. US Food and Drug Administration 2004. Innovation or stagnation: critical path opportunities report and list www.fda.gov/oc/initiatives/criticalpath/reports/opp_report.pdf, 2004.
FDA (2010) FDA Guidance for industry: adaptive design clinical trials for drugs and biologics. Feb 2010.
Hopewell S, Loudon K, Clarke MJ, et al. Publication bias in clinical trials due to statistical significance or direction of trial results. Cochrane Database Systematic Review. 1, 2009. MR000006
Orloff J, Douglas F, Pinheiro J, et al. The future of drug development: advancing clinical trial design. Nature Reviews Drug Discovery. 2009;8:949–957.
Oscarson M. Nicotine metabolism by the polymorphic cytochrome P450 2a6 (cyp2a6) enzyme: implications for interindividual differences in nicotine metabolism. Drug Metabolism and Disposition. 2001;29:91–95.
Woodcock J, Woosley R. The FDA critical path initiative and its influence on new drug development. Annual Review of Medicine. 2008;59:1–12.
World Medical Association. Declaration of Helsinki. Ethical principles for medical research involving human subjects. 59th WMA General Assembly, Seoul, October 2008 www.wma.net/en/30publications/10policies/b3/index.html, 2008.