Chapter 20 Regulatory affairs
Introduction
This chapter introduces the reader to the role of the regulatory affairs (RA) department of a pharmaceutical company, outlining the process of getting a drug approved, and emphasizing the importance of interactions of regulatory affairs with other functions within the company, and with the external regulatory authorities.
To keep this chapter to a reasonable size the typical examples given refer to the first registration of a new chemical compound. The same way of reasoning also applies, however, to any subsequent change to the approval of products. Depending on the magnitude of the change, the new documentation that needs to be compiled, submitted and approved by health authorities is variable, ranging from a few pages of pharmaceutical data (e.g. for an update to product stability information) to a complete new application for a new clinical use in a new patient group in a new pharmaceutical form.
It needs also to be said that, as every drug substance and every project is unique, the views expressed represent the opinion of the authors and are not necessarily shared by others active in the field.
Brief history of pharmaceutical regulation
Control of pharmaceutical products has been the task of authorized institutions for thousands of years, and this was the case even in ancient Greece and Egypt.
From the Middle Ages, control of drug quality, composition purity and quantification was achieved by reference to authoritative lists of drugs, their preparation and their uses. These developed into official pharmacopoeias, of which the earliest was probably the New Compound Dispensatory of 1498 issued by the Florentine guild of physicians and pharmacists.
The pharmacopoeias were local rules, applicable in a particular city or district. During the 19th century national pharmacopoeias replaced local ones, and since the early 1960s regional pharmacopoeias have successively replaced national ones. Now work is ongoing to harmonize – or at least mutually recognize – interchangeable use of the US Pharmacopeia, the European Pharmacopoeia and the Japanese Pharmacopoeia.
As described in Chapter 1, the development of experimental pharmacology and chemistry began during the second half of the 19th century, revealing that the effect of the main botanical drugs was due to chemical substances in the plants used. The next step, synthetic chemistry, made it possible to produce active chemical compounds. Other important scientific developments, e.g. biochemistry, bacteriology and serology during the early 20th century, accelerated the development of the pharmaceutical industry into what it is today (see Drews, 1999).
Lack of adequate drug control systems or methods to investigate the safety of new chemical compounds became a great risk as prefabricated drug products were broadly and freely distributed. In the USA the fight against patent medicines led to the passing of the US Pure Food and Drugs Act against misbranding as long ago as 1906. The Act required improved declaration of contents, prohibited false or misleading statements, and required content and purity to comply with labelled information. A couple of decades later, the US Food and Drug Administration (FDA) was established to control US pharmaceutical products.
Safety regulations in the USA were, however, not enough to prevent the sale of a paediatric sulfanilamide elixir containing the toxic solvent diethylene glycol. In 1937, 107 people, both adults and children, died as a result of ingesting the elixir, and in 1938 the Food Drug and Cosmetics Act was passed, requiring for the first time approval by the FDA before marketing of a new drug product.
The thalidomide disaster further demonstrated the lack of adequate drug control. Thalidomide (Neurosedyn®, Contergan®) was launched during the last years of the 1950s as a non-toxic treatment for a variety of conditions, such as colds, anxiety, depression, infections, etc., both alone and in combination with a number of other compounds, such as analgesics and sedatives.
The reason why the compound was regarded as harmless was the lack of acute toxicity after high single doses. After repeated long-term administration, however, signs of neuropathy developed, with symptoms of numbness, paraesthesia and ataxia. But the overwhelming effects were the gross malformation in infants born to mothers who had taken thalidomide in pregnancy: their limbs were partially or totally missing, a previously extremely rare malformation called phocomelia (seal limb). Altogether around 12 000 infants were born with the defect in those few years. Thalidomide was withdrawn from the market in 1961/62.
This catastrophe became a strong driver to develop animal test methods to assess drug safety before testing compounds in humans. Also it forced national authorities to strengthen the requirements for control procedures before marketing of pharmaceutical products (Cartwright and Matthews, 1991).
Another blow hit Japan between 1959 and 1971. The SMON (subacute myelo-optical neuropathy) disaster was blamed on the frequent Japanese use of the intestinal antiseptic clioquinol (Entero-Vioform®, Enteroform® or Vioform®). The product had been sold without restrictions since early 1900, and it was assumed that it would not be absorbed, but after repeated use neurological symptoms appeared, characterized by paraesthesia, numbness and weakness of the extremities, and even blindness. SMON affected about 10 000 Japanese, compared to some 100 cases in the rest of the world (Meade, 1975).
These tragedies had a strong impact on governmental regulatory control of pharmaceutical products. In 1962 the FDA required evidence of efficacy as well as safety as a condition for registration, and formal approval was required for patients to be included in clinical trials of new drugs.
In Europe, the UK Medicines Act 1968 made safety assessment of new drug products compulsory. The Swedish Drug Ordinance of 1962 defined the medicinal product and required a clear benefit–risk ratio to be documented before approval for marketing. All European countries established similar controls during the 1960s.
In Japan, the Pharmaceutical Affairs Law enacted in 1943 was revised in 1961,1979 and 2005 to establish the current drug regulatory system, with the Ministry of Health and Welfare assessing drugs for quality, safety and efficacy.
The 1960s and 1970s saw a rapid increase in laws, regulations and guidelines for reporting and evaluating the risks versus the benefits of new medicinal products. At the time the industry was becoming more international and seeking new global markets, but the registration of medicines remained a national responsibility.
Although different regulatory systems were based on the same key principles, the detailed technical requirements diverged over time, often for traditional rather than scientific reasons, to such an extent that industry found it necessary to duplicate tests in different countries to obtain global regulatory approval for new products. This was a waste of time, money and animals’ lives, and it became clear that harmonization of regulatory requirements was needed.
European (EEC) efforts to harmonize requirements for drug approval began 1965, and a common European approach grew with the expansion of the European Union (EU) to 15 countries, and then 27. The EU harmonization principles have also been adopted by Norway and Iceland. This successful European harmonization process gave impetus to discussions about harmonization on a broader international scale (Cartwright and Matthews, 1994).
International harmonization
The harmonization process started in 1990, when representatives of the regulatory authorities and industry associations of Europe, Japan and the USA (representing the majority of the global pharmaceutical industry) met, ostensibly to plan an International Conference on Harmonization (ICH). The meeting actually went much further, suggesting terms of reference for ICH, and setting up an ICH Steering Committee representing the three regions.
The task of ICH was ‘… increased international harmonization, aimed at ensuring that good quality, safe and effective medicines are developed and registered in the most efficient and cost-effective manner. These activities are pursued in the interest of the consumer and public health, to prevent unnecessary duplication of clinical trials in humans and to minimize the use of animal testing without compromising the regulatory obligations of safety and effectiveness’ (Tokyo, October 1990).
ICH has remained a very active organization, with substantial representation at both authority and industry level from the EU, the USA and Japan. The input of other nations is provided through World Health Organization representatives, as well as representatives from Switzerland and Canada.
ICH conferences, held every 2 years, have become a forum for open discussion and follow-up of the topics decided. The important achievements so far are the scientific guidelines agreed and implemented in the national/regional drug legislation, not only in the ICH territories but also in other countries around the world. So far some 50 guidelines have reached ICH approval and regional implementation, i.e. steps 4 and 5 (Figure 20.1). For a complete list of ICH guidelines and their status, see the ICH website (website reference 1).
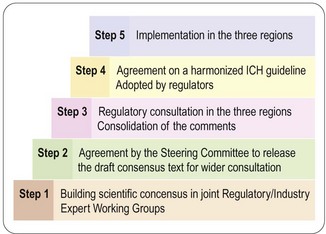
The process described in Figure 20.1 is very open, and the fact that health authorities and the pharmaceutical industry collaborate from the start increases the efficiency of work and ensures mutual understanding across regions and functions; this is a major factor in the success of ICH.
Roles and responsibilities of regulatory authority and company
The basic division of responsibilities for drug products is that the health authority is protecting public health and safety, and the pharmaceutical company is responsible for all aspects of the drug product. The regulatory approval of a pharmaceutical product permits marketing and is a contract between the regulatory authority and the pharmaceutical company. The conditions of the approval are set out in the dossier and condensed in the prescribing information. Any change that is planned must be forwarded to the regulatory authority for information and, in most cases, new approval before being implemented.
To protect the public health, regulatory authorities also develop regulations and guidelines for companies to follow in order to achieve a balance between the possible risks and therapeutic advantages to patients. The authorities’ work is partly financed by fees paid by pharmaceutical companies. Fees may be reduced, under certain conditions, to stimulate research. This may be driven, e.g., by company size or size of target patient groups.
 approves clinical trial applications
approves clinical trial applications
gives procedural and scientific advice to companies during drug development
approves for marketing drugs that have been scientifically evaluated to provide evidence of a satisfactory benefit/risk ratio
monitors the safety of the marketed product, based on (a) reports of adverse reactions from healthcare providers, and (b) from compiled and evaluated safety information from the company that owns the product
can withdraw the licence for marketing in serious cases of non-compliance (e.g. failure on inspections, failure of adequate additional warnings in prescribing information after clinical adverse reactions are reported, or failure of the company to consider serious findings in animal studies).
owns the documentation that forms the basis for assessment, is responsible for its accuracy and correctness, for keeping it up to date, and for ensuring that it complies with standards set by current scientific development and the regulatory authorities
collects, compiles and evaluates safety data, and submits reports to the regulatory authorities at regular intervals – and takes rapid action in serious cases. This might involve the withdrawal of the entire product or of a product batch (e.g. tablets containing the wrong drug or the wrong dose), or a request to the regulatory authority for a change in prescribing information
has a right to appeal and to correct cases of non-compliance.
The role of the regulatory affairs department
The regulatory affairs (RA) department of a pharmaceutical company is responsible for obtaining approval for new pharmaceutical products and ensuring that approval is maintained for as long as the company wants to keep the product on the market. It serves as the interface between the regulatory authority and the project team, and is the channel of communication with the regulatory authority as the project proceeds, aiming to ensure that the project plan correctly anticipates what the regulatory authority will require before approving the product. It is the responsibility of RA to keep abreast of current legislation, guidelines and other regulatory intelligence. Such rules and guidelines often allow some flexibility, and the regulatory authorities expect companies to take responsibility for deciding how they should be interpreted. The RA department plays an important role in giving advice to the project team on how best to interpret the rules. During the development process sound working relations with authorities are essential, e.g. to discuss such issues as divergence from guidelines, the clinical study programme, and formulation development.
Most companies assess and prioritize new projects based on an intended Target Product Profile (TPP). The RA professional plays a key role in advising on what will be realistic prescribing information (‘label’) for the intended product. As a member of the project team RA also contributes to designing of the development programme. The RA department reviews all documentation from a regulatory perspective, ensuring that it is clear, consistent and complete, and that its conclusions are explicit. The department also drafts the core prescribing information that is the basis for global approval, and will later provide the platform for marketing. The documentation includes clinical trials applications, as well as regulatory submissions for new products and for changes to approved products. The latter is a major task and accounts for about half of the work of the RA department.
An important proactive task of the RA is to provide input when legislative changes are being discussed and proposed. In the ICH environment there is a greater possibility to exert influence at an early stage.
The drug development process
An overview of the process of drug development is given in Chapters 14–18 and summarized in Figure 20.2. As already emphasized, this sequential approach, designed to minimize risk by allowing each study to start only when earlier studies have been successfully completed, is giving way to a partly parallel approach in order to save development time.
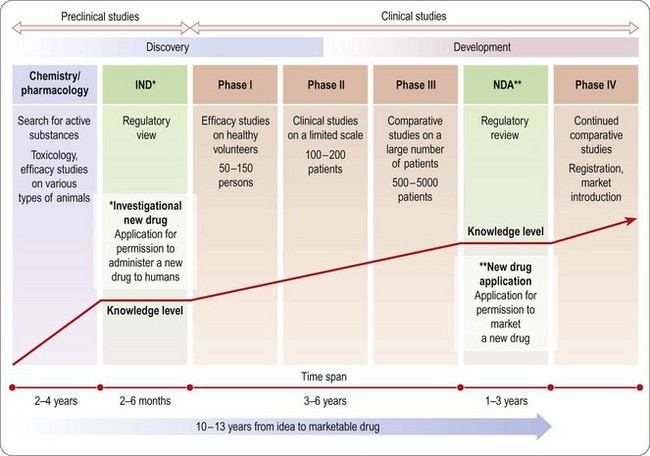
All studies in the non-clinical area – chemistry, pharmacology, pharmacokinetics, pharmaceutical development and toxicology – aim to establish indicators of safety and efficacy sufficient to allow studies and use in man. According to ICH nomenclature, documentation of chemical and pharmaceutical development relates to quality assessment, animal studies relate to safety assessment, and studies in humans relate to efficacy.
Quality assessment (chemistry and pharmaceutical development)
The quality module of a submission documents purity and assay for the drug substance, and purity data for all the inactive ingredients. The formulation must fulfil requirements for consistent quality and allow storage, and the container must be shown to be fit for its purpose. These aspects of a pharmaceutical product have to be kept under control throughout the development process, as toxicology and pharmacology results are reliable only for substances of comparable purity. Large-scale production, improved synthetic route, different raw material supply, etc., may produce a substance somewhat different from the first laboratory-scale batches. Any substantial change must be known and documented.
The formulation of a product is a challenge. For initial human studies, simple i.v. and oral solutions are needed for straightforward results, whereas for the clinical programme in patients, bioequivalent formulations are essential for comparison of results across studies, and so it is preferable to have access to the final formulation already during Phase II.
If the formulation intended for marketing cannot be completed until late in the clinical phase, bioequivalence studies showing comparable results with the preliminary and final market formulations will be necessary to support the use of results with the preliminary formulation. There may even be situations when clinical studies must be repeated.
The analytical methods used and their validation must be described. Manufacturing processes and their validation are also required to demonstrate interbatch uniformity. However, full-scale validation may be submitted when sales production has eventually started.
Studies on the stability of both substance and products under real-life conditions are required, covering the full time of intended storage. Preliminary stability data are sufficient for the start of clinical studies. The allowable storage time can be increased as data are gathered and submitted. Even marketing authorizations can be approved on less than real-time storage information, but there is a requirement to submit final data when available.
Inactive ingredients as well as active substances need to be documented, unless they are well known and already documented. Even then it may become necessary to perform new animal studies to support novel uses of commonly used additives.
Although the quality module of the documentation is the smallest, the details of requirements and the many changes needed during development and maintenance of a product make it the most resource intensive module from a regulatory perspective. Also, legislation differs most in this area, so it will often be necessary to adapt the documentation for the intended regional submission. RA professionals, however, try to convince regulatory authorities not to create local rules to avoid, as far as possible, different interpretations and duplicate work.
As previously said, all changes to the originally submitted dossier must be made known to the approving regulatory authority. Since the majority of changes are made in the quality section, very small changes take lots of resources for the company as well as for the authority. The US legislation has allowed the submission of annual reports collecting those changes that have no impact on quality. This possibility has also been introduced in EU in 2010 with the purpose of saving time and resources.
Safety assessment (pharmacology and toxicology)
Next we consider how to design and integrate pharmacological and toxicological studies in order to produce adequate documentation for the first tests in humans. ICH guidelines define the information needed from animal studies in terms of doses and time of exposure, to allow clinical studies, first in healthy subjects and later in patients. The principles and methodology of animal studies are described in Chapters 11 and 15. The questions discussed here are when and why these animal studies are required for regulatory purposes.
Primary pharmacology
The primary pharmacodynamic studies provide the first evidence that the compound has the pharmacological effects required to give therapeutic benefit. It is a clear regulatory advantage to use established models and to be able at least to establish a theory for the mechanism of action. This will not always be possible and is not a firm requirement, but proof of efficacy and safety is helped by a plausible mechanistic explanation of the drug’s effects, and this knowledge will also become a very powerful tool for marketing. For example, understanding the mechanism of action of proton pump inhibitors, such as omeprazole (see Chapter 4), was important in explaining their long duration of action, allowing once-daily dosage of compounds despite their short plasma half-life.
General pharmacology
General pharmacology1 studies investigate effects other than the primary therapeutic effects. Safety pharmacology studies (see Chapter 15), which must conform to good laboratory practice (GLP) standards, are focused on identifying the effects on physiological functions that in a clinical setting are unwanted or harmful.
Although the study design will depend on the properties and intended use of the compound, general pharmacology studies are normally of short duration (i.e. acute, rather than chronic, effects are investigated), and the dosage is increased until clear adverse effects occur. The studies also include comparisons with known compounds whose pharmacological properties or clinical uses are similar.
When required, e.g. when pharmacodynamic effects occur only after prolonged treatment, or when effects seen with repeated administration give rise to safety concerns, the duration of a safety pharmacology study needs to be prolonged. The route of administration should, whenever possible, be the route intended for clinical use.
There are cases when a secondary pharmacological effect has, eventually, been developed into a new indication. Lidocaine, for example, was developed as a local anaesthetic agent and its cardiac effects after overdose were considered a hazard. Later that cardiac effect was exploited as a treatment for ventricular arrhythmia.
All relevant safety pharmacology studies must be completed before studies can be undertaken in patients. Complementary studies may still be needed to clarify unexpected findings in later development stages.
Pharmacokinetics: absorption, distribution, metabolism and excretion (ADME)
Preliminary pharmacokinetic tests to assess the absorption, plasma levels and half-life (i.e. exposure information) are performed in rodents in parallel with the preliminary pharmacology and toxicology studies (see Chapter 10).
Studies in humans normally start with limited short-term data, and only if the results are acceptable are detailed animal and human ADME studies performed.
Plasma concentrations observed in animals are used to predict the concentrations that may be efficacious/tolerated in humans, under the assumption that similar biological effects should be produced at similar plasma levels across species. This is a reasonable assumption provided the in vitro target affinity is similar.
Investigations during the toxicology programme give the bulk of the pharmacokinetic information due to the long duration of drug exposure and the wide range of doses tested in several relevant species. They also give data about tissue distribution and possible accumulation in the body, including placental transfer and exposure of the fetus, as well as excretion in milk.
Metabolic pathways differ considerably between species, often quantitatively but sometimes also qualitatively. Active metabolites can influence study results, in particular after repeated use. A toxic metabolite with a long half-life may accumulate in the body and disturb results. The characterization and evaluation of metabolites are long processes, and are generally the last studies to be completed in a development programme.
Toxicology
The principles and methodology of toxicological assessment of new compounds are described in Chapter 15. Here we consider the regulatory aspects.
In contrast to the pharmacological studies, toxicological studies generally follow standard protocols that do not depend on the compound characteristics. Active comparators are not used, but the drug substance is compared at various dose levels to a vehicle control, given, if possible, via the intended route of administration.
Single and repeated-dose studies
The acute toxicity of a new compound must be evaluated prior to the first human exposure.
This information is obtained from dose escalation studies or dose-ranging studies of short duration. Lethality is no longer an ethically accepted endpoint. The toxicology requirements for the first exploratory studies in man are described in the ICH guidline M3 which reached step 5 in June 2009 (see website reference 1). Table 20.1 shows the duration of repeated-dose studies recommended by ICH, to support clinical trials and therapeutic use for different periods.

Genotoxicity
Preliminary genotoxicity evaluation of mutations and chromosomal damage (see Chapter 15) is needed before the drug is given to humans. If results from those studies are ambiguous or positive, further testing is required. The entire standard battery of tests needs to be completed before Phase II (see Chapter 17).
Carcinogenicity
The objective of carcinogenicity studies is to identify any tumorigenic potential in animals, and they are required only when the expected duration of therapy, whether continuous or intermittent, is at least 6 months. Examples include treatments for conditions such as allergic rhinitis, anxiety or depression.
Carcinogenicity studies are also required when there is particular reason for concern, such as chemical similarities to known carcinogens, pathophysiological findings in animal toxicity studies, or positive genotoxicity results. Compounds found to be genotoxic by in vitro as well as in vivo tests are presumed to be trans-species carcinogens with hazards to humans.
Carcinogenicity studies normally run for the lifespan of the test animals. They are performed quite late in the development programme and are not necessarily completed when the application for marketing authorization is submitted. Indeed, for products for which there is a great medical need in the treatment of certain serious diseases, the regulatory authority may agree that submission of carcinogenicity data can be delayed until after marketing approval is granted.
Reproductive and developmental toxicity
These studies (see Chapter 15) are intended to reveal effects on male or female fertility, embryonic and fetal development, and peri- and postnatal development.
An evaluation of effects on the male reproductive system is performed in the repeated-dose toxicity studies, and this histopathological assessment is considered more sensitive in detecting toxic effects than are fertility studies. Men can therefore be included in Phase I–II trials before the male fertility studies are performed in animals.
Women may enter early studies before reproductive toxicity testing is completed, provided they are permanently sterilized or menopausal, and provided repeated-dose toxicity tests of adequate duration have been performed, including the evaluation of female reproductive organs.
For women of childbearing potential there is concern regarding unintentional fetal exposure, and there are regional differences (Box 20.1) in the regulations about including fertile women in clinical trials.
Box 20.1
Requirement for reproduction toxicity related to clinical studies in fertile women
| EU: | Embryo/fetal development studies are required before Phase I, and female fertility should be completed before Phase III |
| USA: | Careful monitoring and pregnancy testing may allow fertile women to take part before reproduction toxicity is available. Female fertility and embryo/fetal assessment to be completed before Phase III |
| Japan: | Embryo/fetal development studies are required before Phase I, and female fertility should be completed before Phase III. |
Local tolerance and other toxicity studies
The purpose of local tolerance studies is to ascertain whether medicinal products (both active substances and excipients) are tolerated at sites in the body that may come into contact with the product in clinical use. This could mean, for example, ocular, dermal or parenteral administration. Other studies may also be needed. These might be studies on immunotoxicity, antigenicity studies on metabolites or impurities, and so on. The drug substance and the intended use will determine the relevance of other studies.
Efficacy assessment (studies in man)
When the preclinical testing is sufficient to start studies in man, the RA department compiles a clinical trials submission, which is sent to the regulatory authority and the ethics committee (see Regulatory procedures, below).
The clinical studies, described in detail in Chapter 17, are classified according to Table 20.2.
Table 20.2 ICH classification of clinical studies
| Type of study | Study objectives | Traditional terminology |
|---|---|---|
| Human pharmacology | Assess tolerance; describe or define pharmacokinetics/pharmacodynamics; explore drug metabolism and drug interactions; estimate activity | Phase I |
| Therapeutic exploratory | Explore use for the targeted indication; estimate dosage for subsequent studies; provide basis for confirmatory study design, endpoints, methodologies | Phase II |
| Therapeutic confirmatory | Demonstrate or confirm efficacy; establish safety profile; provide an adequate basis for assessing benefit–risk relationship to support licensing (drug approval); establish dose–response relationship | Phase III (a and b) |
| Therapeutic use | Refine understanding of benefit–risk relationship in general or special populations and/or environments; identify less common adverse reactions; refine dosing recommendations | Phase IV |
Human pharmacology
Human pharmacology studies refer to the earliest human exposure in volunteers, as well as any pharmacological studies in patients and volunteers throughout the development of the drug.
The first study of a new drug substance in humans has essentially three objectives:
To investigate tolerability over a range of doses and, if possible, see the symptoms of adverse effects
To obtain information on pharmacokinetics, and to measure bioavailability and plasma concentration/effect relations
To examine the pharmacodynamic activity over a range of doses and obtain a dose–response relationship, provided a relevant effect can be measured in healthy volunteers.
Further human pharmacology studies are performed to document pharmacodynamic and pharmacokinetic effects. Examples of the data needed to support an application for trials in patients are the complete pharmacokinetic evaluation and the performance of bioavailability/bioequivalence studies during the development of new formulations or drug-delivery systems. Information is also obtained on the possible influence of food on absorption, and that of other concomitant medications, i.e. drug interaction. Exploration of metabolic pattern is also performed early in the clinical development process.
Special patient populations need particular attention because they may be unduly sensitive or resistant to treatment regimens acceptable to the normal adult population studied. One obvious category is patients with renal or hepatic impairment, who may be unable to metabolize or excrete the drug effectively enough to avoid accumulation. The metabolic pattern and elimination route are important predictors for such patients, who are not included in clinical trials until late in development.
Gender differences may also occur, and may be detected by the inclusion of women at the dose-finding stage of clinical trials.
An interaction is an alteration in the pharmacodynamic or the pharmacokinetic properties of a drug caused by factors such as concomitant drug treatment, diet, social habits (e.g. tobacco or alcohol), age, gender, ethnic origin and time of administration.
Interaction studies can be performed in healthy volunteers looking at possible metabolism changes when co-administering compounds that share the same enzymatic metabolic pathway. Also, changed pharmacokinetic behaviour can be investigated in combinations of drugs that are expected to be used together. Generally, such human volunteer studies are performed when clinical findings require clarification or if the company wishes to avoid standard warning texts which would otherwise apply to the drug class.
Therapeutic exploratory studies
After relevant information in healthy volunteers has been obtained, safety conclusions from combined animal and human exposure will be assessed internally. If these are favourable, initial patient studies can begin. To obtain the most reliable results, the patient population should be as homogeneous as possible – similar age, no other diseases than the one to be studied – and the design should, when ethically justified, be placebo controlled. For ethical reasons only a limited number of closely monitored patients take part in these studies. Their importance lies in the assumption that any placebo effect in the group treated with active drug should be eliminated by comparison with blinded inactive treatment. They are used primarily to establish efficacy measured against no treatment.
Studies in special populations: elderly, children, ethnic differences
Clinically significant differences in pharmacokinetics between the elderly and the young are due to several factors related to aging, such as impaired renal function, which can increase the variability in drug response, as well as increasing the likelihood of unwanted effects and drug interactions. Bearing in mind that the elderly are the largest group of consumers, this category should be studied as early as possible in clinical trials.
Clinical trials in children
Studies in children require experience from adult human studies and information on the pharmacokinetic profile of the substance. Because of the difficulties, and the often small commercial return, companies have seldom considered it worthwhile to test drugs in children and to seek regulatory approval for marketing drugs for use in children. Nevertheless, drugs are often prescribed ‘off-label’ for children, on the basis of clinical experience suggesting that they are safe and effective. Such off-label prescribing is undesirable, as clinical experience is less satisfactory than formal trials data as a guide to efficacy and safety, and because it leaves the clinician, rather than the pharmaceutical company, liable for any harm that may result. Recently, requirements to include a paediatric population early have forced the development of new guidance as to how to include children in clinical development. Market exclusivity prolongation has been successfully tried for some years in the USA, and in July 2003 the Federal Food Drug and Cosmetics Act was amended to request paediatric studies in a new submission unless omission is justified. In September of 2007, the Paediatric Research Equity Act replaced the 2003 Act and an assessment was made to evaluate the quality of the studies submitted. The results were positive and between September 2007 and June 2010 more than 250 clinical trials comprising more than 100 000 children have been performed. (see website reference 2).
In Europe the European Commission adopted the Paediatric Regulation in February 2007 (see website reference 3). This stipulates that no marketing approval will be granted unless there is an agreed paediatric investigation plan in place or, alternatively, there is a waiver from the requirement because of the low risk that the product will be considered for use in children. The paediatric studies may, with the agreement of the regulatory authority, be performed after approval for other populations. For new compounds or for products with a Supplementary Protection Certificate (see p. 300), paediatric applications will be given a longer market exclusivity period, and off-patent products will be given a special paediatric use marketing authorization (PUMA); funds will be available for paediatric research in these products.
To further encourage paediatric research, authority scientific advice is free. Paediatric clinical data from the EU and elsewhere are collected in a common European database to avoid repetition of trials with unnecessary exposure in children. The US FDA and the EMA in Europe exchange information on paediatric clinical research.
Ethnic differences
In order for clinical data to be accepted globally, the risk of ethnic differences must be assessed. ICH Efficacy guideline E5 defines a bridging data package that would allow extrapolation of foreign clinical data to the population in the new region. A limited programme may suffice to confirm comparable effects in different ethnic groups.
Ethnic differences may be genetic in origin, as in the example described in Chapter 17, or related to differences in environment, culture or medical practice.
Therapeutic confirmatory studies
The therapeutic confirmatory phase is intended to confirm efficacy results from controlled exploratory efficacy studies, but now in a more realistic clinical environment and with a broader population.
In order to convincingly document that the product is efficacious, there are some ‘golden rules’ to be aware of in terms of the need for statistical power, replication of the results, etc. Further, for a product intended for long-term use, its performance must usually be investigated during long-term exposure. All these aspects will, however, be influenced by what alternative treatments there are, the intended target patient population, the rarity and severity of the disease as well as other factors, and must be evaluated case by case.
Clinical safety profile
An equally important function of this largest and longest section of the clinical documentation is to capture all adverse event information to enable evaluation of the relative benefit–risk ratio of the new compound, and also to detect rare adverse reactions. To document clinical safety, the ICH E1 guideline on products intended for chronic use stipulates that a minimum of 100 patients be treated for at least 1 year, and 300–600 treated for at least 6 months. In reality, however, several thousand patients usually form the database for safety evaluation for marketing approval.
Not until several similar studies can be analysed together can a real estimate be made of the clinical safety of the product.
The collected clinical database should be analysed across a sensible selection of variables, such as sex, age, race, exposure (dose and duration), as well as concomitant diseases and concomitant pharmacotherapy This type of integrated data analysis is a rational and scientific way to obtain necessary information about the benefits and risks of new compounds, and has been required for FDA submissions for many years, though it is not yet a firm requirement in the EU or Japan.
To further emphasize the accountability for the product by the pharmaceutical company a more proactive legislation for risk evaluation has been introduced in USA and EU. The term Risk Evaluation and Mitigation Strategy (REMS) in the USA is matched by Risk Management System in EU.
The difference from the established method of reporting adverse reactions that have occurred is that any possible or potential risks for a patient being harmed should be foreseen or identified early and mitigation/minimization activities be planned in advance. The risk management document is generally part of the regulatory approval and follows the product’s entire lifecycle. The true risk profile will develop along with the increased knowledge base. The goal is at any time to be able to demonstrate how and why the treatment benefits outweigh the risks (see website references 4 and 5).
Regulatory aspects of novel types of therapy
As emphasized in earlier chapters, the therapeutic scene is moving increasingly towards biological and biopharmaceutical treatments, as well as various innovative, so-called advanced therapies. There are also broadening definitions of what constitutes medical devices. The regulatory framework established to ensure the quality, safety and efficacy of conventional synthetic drugs is not entirely appropriate for many biopharmaceutical products, and even less so for the many gene- and cell-based products currently in development. Recombinant proteins have been in use since 1982 and the regulatory process for such biopharmaceuticals is by now well established, hence this chapter will mainly focus on these. The EU also has, for example, new legislation in force since December 2008 specifically on Advanced Therapy Medicinal Products (ATMP), meaning somatic cell therapy, gene therapy and tissue engineering. This involves requirements for the applicant, as part of a marketing authorization, to establish a risk management system. In addition to the standard requirements in terms of safety follow-up for an approved product, this system should also include an evaluation of the product’s effectiveness (see website reference 6). In the USA, there are a number of FDA guidelines relating to cellular and gene therapies. The regulatory framework for even newer therapeutic modalities relating, for example, to nanotechnologies is not yet clearly defined, and the regulatory authorities face a difficult task in keeping up with the rapid pace of technological change.
Biopharmaceuticals
Compared with synthetic compounds, biopharmaceuticals are by their nature more heterogeneous, and their production methods are very diverse, including complex fermentation and recombinant techniques, as well as production via the use of transgenic animals and plants, thereby posing new challenges for quality control. This has necessarily led to a fairly pragmatic regulatory framework. Quality, safety and efficacy requirements have to be no less stringent, but procedures and standards are flexible and generally established on a case-by-case basis. Consequently, achieving regulatory approval can often be a greater challenge for the pharmaceutical company, but there are also opportunities to succeed with novel and relatively quick development programmes.
Published guidelines on the development of conventional drugs need to be considered to determine what parts are relevant for a particular biopharmaceutical product. In addition, there are, to date, seven ICH guidelines dealing exclusively with biopharmaceuticals, as well as numerous FDA and CHMP guidance documents. These mostly deal with quality aspects and, in some cases, preclinical safety aspects. The definition of what is included in the term ‘biopharmaceutical’ varies somewhat between documents, and therefore needs to be checked. The active substances include proteins and peptides, their derivatives, and products of which they are components. Examples include (but are not limited to) cytokines, recombinant plasma factors, growth factors, fusion proteins, enzymes, hormones and monoclonal antibodies (see also Chapters 12 and 13).
Quality considerations
A unique and critically important feature for biopharmaceuticals is the need to ensure and document viral safety aspects. Furthermore, there must be preparedness for potentially new hazards, such as infective prions. Therefore strict control of the origin of starting materials and expression systems is essential. The current battery of ICH quality guidance documents in this area reflects these points of particular attention (ICH Q5A-E, and Q6B).
At the time when biopharmaceuticals first appeared, the ability to analyse and exactly characterize the end-product was not possible the way it was with small molecules. Therefore, their efficacy and safety depended critically on the manufacturing process itself, and emphasis was placed on ‘process control’ rather than ‘product control’.
Since then, much experience and confidence has been gained. Bioanalytical technologies for characterizing large molecules have improved dramatically and so has the field of bioassays, which are normally required to be included in such characterizations, e.g. to determine the ‘potency’ of a product. As a result, the quality aspects of biopharmaceuticals are no longer as fundamentally different from those of synthetic products as they used to be. The quality documentation will still typically be more extensive than it is for a small-molecule product.
Today the concept of ‘comparability’ has been established and approaches for demonstrating product comparability after process changes have been outlined by regulatory authorities. Further, the increased understanding of these products has in more recent times allowed the approval of generic versions also of biopharmaceuticals, so-called follow-on biologics or biosimilars. A legal framework was established first in the EU, in 2004, and the first such approvals (somatropin products) were seen in 2006. Approvals are so far slightly behind in the USA since there was actually not a regulatory pathway for biosimilars until such provisions were signed into law in March 2010 (see website reference 7). The FDA is, at the time of writing, working on details of how to implement these provisions to approve biosimilars. Also, in Japan, the authorities have published ‘Guidelines for the Quality, Safety and Efficacy Assurance of Follow-on Biologics’, the most recent version being in March 2009.
Safety considerations
The expectations in terms of performing and documenting a non-clinical safety evaluation for biotechnology-derived pharmaceuticals are well outlined in the ICH guidance document S6. This guideline was revised in 2011 driven by scientific advances and experience gained since publication of the original guidance. It indicates a flexible, case-by-case and science-based approach, but also points out that a product needs to be sufficiently characterized to allow the appropriate design of a preclinical safety evaluation.
Generally, all toxicity studies must be performed according to GLP. However, for biopharmaceuticals it is recognized that some specialized tests may not be able to comply fully with GLP. The guidance further comments that the standard toxicity testing designs in the commonly used species (e.g. rats and dogs) are often not relevant.
To make it relevant, a safety evaluation should include a species in which the test material is pharmacologically active. Further, in certain justified cases one relevant species may suffice, at least for the long-term studies. If no relevant species at all can be identified, the use of transgenic animals expressing the human receptor or the use of homologous proteins should be considered.
Other factors of particular relevance with biopharmaceuticals are potential immunogenicity and immunotoxicity. Long-term studies may be difficult to perform, depending on the possible formation of neutralizing antibodies in the selected species. For products intended for chronic use, the duration of long-term toxicity studies must, however, always be scientifically justified. Regulatory guidance also states that standard carcinogenicity studies are generally inappropriate, but that product-specific assessments of potential risks may still be needed, and that a variety of approaches may be necessary to accomplish this.
In 2006 disastrous clinical trial events took place with an antibody (TGN 1412), where healthy volunteers suffered life-threatening adverse effects. These effects had not been anticipated by the company or the authority reviewing the clinical trial application. The events triggered intense discussions on risk identification and mitigation for so-called first-in-human clinical trials, and in particular for any medicinal product which might be considered ‘high-risk’. Since then, specific guidelines for such clinical trial applications have been issued by the regulatory authorities.
Efficacy considerations
The need to establish efficacy is in principle the same for biopharmaceuticals as for conventional drugs, but there are significant differences in practice. The establishment of a dose–response relationship can be irrelevant, as there may be an ‘all-or-none-effect’ at extremely low levels. Also, to determine a maximum tolerated dose (MTD) in humans may be impractical, as many biopharmaceuticals will not evoke any dose-limiting side effects. Measuring pharmacokinetic properties may be difficult, particularly if the substance is an endogenous mediator. Biopharmaceuticals may also have very long half-lives compared to small molecules, often in the range of weeks rather than hours.
For any biopharmaceutical intended for chronic or repeated use, there will be extra emphasis on demonstrating long-term efficacy. This is because the medical use of proteins is associated with potential immunogenicity and the possible development of neutralizing antibodies, such that the intended effect may decrease or even disappear with time. Repeated assessment of immunogenicity may be needed, particularly after any process changes.
To date, biopharmaceuticals have typically been developed for serious diseases, and certain types of treatments, such as cytotoxic agents or immunomodulators, cannot be given to healthy volunteers. In such cases, the initial dose-escalation studies will have to be carried out in a patient population rather than in normal volunteers.
Regulatory procedural considerations
In the EU, only the centralized procedure can be used for biopharmaceuticals as well as biosimilars (see Regulatory procedures, below). In the USA, biopharmaceuticals are in most cases approved by review of Biologics License Applications (BLA), rather than New Drug Applications (NDA).
Personalized therapies
It is recognized that an individual’s genetic makeup influences the effect of drugs. Incorporating this principle into therapeutic practice is still at a relatively early stage, although the concept moves rapidly towards everyday reality, and it presents a challenge for regulatory authorities who are seeking ways to incorporate personalized medicine (pharmacogenomics) into the regulatory process. How a drug product can or should be co-developed with the necessary genomic biomarkers and/or assays is not yet clear. Another regulatory uncertainty has been how the generation of these kinds of data will translate into label language for the approved product. Already in 2005 the FDA issued guidance on a specific procedure for ‘Pharmacogenomic Data Submissions’ to try and alleviate potential industry concerns and instead promote scientific progress and the gaining of experience in the field. In the EU there is, at the time of writing, draft guidance published on the use of pharmacogenetic methodologies in the pharmacokinetic evaluation of medicinal products. However, still today, there are relatively few approved products to learn from.
Orphan drugs
Orphan medicines are those intended to diagnose, prevent or treat rare diseases, in the EU further specified as life-threatening or chronically debilitating conditions. The concept also includes therapies that are unlikely to be developed under normal market conditions, where the company can show that a return on research investment will not be possible. To qualify for orphan drug status, there should be no satisfactory treatment available or, alternatively, the intended new treatment should be assumed to be of significant benefit (see website references 8 and 9)
To qualify as an orphan indication, the prevalence of the condition must be fewer than 5 in 10 000 individuals in the EU, fewer than 50 000 affected in Japan, or fewer than 200 000 affected in the USA. Financial and scientific assistance is made available for products intended for use in a given indication that obtain orphan status. Examples of financial benefits are a reduction in or exemption from fees, as well as funding provided by regulatory authorities to meet part of the development costs in some instances. Specialist groups within the regulatory authorities provide scientific help and advice on the execution of studies. Compromises may be needed owing to the scarcity of patients, although the normal requirements to demonstrate safety and efficacy still apply.
The most important benefit stimulating orphan drug development is, however, market exclusivity for 7–10 years for the product, for the designated medical use. In the EU the centralized procedure (see Regulatory procedures, below) is the compulsory procedure for orphan drugs.
The orphan drug incentives are fairly recent. In the USA the legislation dates from 1983, in Japan from 1995, and in the EU from 2000. The US experience has shown very good results; a review of the first 25 years of the Orphan Drug Act resulted in 326 marketing approvals, the majority of which are intended to treat rare cancers or metabolic/endocrinological disorders.
Environmental considerations
Environmental evaluation of the finished pharmaceutical products is required in the USA and EU, the main concern being contamination of the environment by the compound or its metabolites. The environmental impact of the manufacturing process is a separate issue that is regulated elsewhere.
The US requirement for environmental assessment (EA) applies in all cases where action is needed to minimize environmental effects. An Environmental Assessment Report is then required, and the FDA will develop an Environmental Impact Statement to direct necessary action. Drug products for human or animal use can, however, be excluded from this requirement under certain conditions (see website reference 10, p. 301), for example if the estimated concentration in the aquatic environment of the active substance is below 1 part per billion, or if the substance occurs naturally. In Europe a corresponding general guideline was implemented in December 2006 for human medicinal products (see website reference 11).
Regulatory procedures
Clinical trials
It is evident that clinical trials can pose unknown risks to humans: the earlier in the development process, the greater the risk. Regulatory and ethical approvals are based on independent evaluations, and both are required before investigations in humans can begin.
The ethical basis for all clinical research is the Declaration of Helsinki (see website reference 12, p. 301), which states that the primary obligation for the treating physician is to care for the patient. It also says that clinical research may be performed provided the goal is to improve treatment. Furthermore, the subject must be informed about the potential benefits and risks, and must consent to participate. The guardians of patients who for any reason cannot give informed consent (e.g. small children) can agree on participation.
Regulatory authorities are concerned mainly with the scientific basis of the intended study protocol and of course the safety of subjects/patients involved. Regulatory authorities require all results of clinical trials approved by them to be reported back.
All clinical research in humans should be performed according to the internationally agreed Code of Good Clinical Practice, as described in the ICH guideline E6 (see website reference 1, p. 301).
Europe
Until recently, the regulatory requirements to start and conduct clinical studies in Europe have varied widely between countries, ranging from little or no regulation to a requirement for complete assessment by the health authority of the intended study protocol and all supporting documentation.
The efforts to harmonize EU procedures led to the development of a Clinical Trial Directive implemented in May 2004 (see website reference 13, p. 301). One benefit is that both regulatory authorities and ethics committees must respond within 60 days of receiving clinical trials applications and the requirements for information to be submitted are being defined and published in a set of guidelines. Much of the information submitted to the regulatory authority and ethics committee is the same. In spite of the Clinical Trial Directive, however, some national differences in format and information requirements have remained, and upon submission of identical clinical trial protocols in several countries the national reviews may reach different conclusions. Therefore, at the time of writing, there is a pilot Voluntary Harmonization Procedure (VHP) available for applications involving at least three EU member states. This comprises two steps, where in the first round one coordinator on the regulatory authority side manages national comments on the application and provides the applicant with a consolidated list of comments or questions. In the second step the usual national applications for the clinical trial are submitted but national approval should then be gained very rapidly without any further requests for change.
USA
An Investigational New Drug (IND) application must be submitted to the FDA before a new drug is given to humans. The application is relatively simple (see website reference 14, p. 301), and to encourage early human studies it is no longer necessary to submit complete pharmacology study reports. The toxicology safety evaluation can be based on draft and unaudited reports, provided the data are sufficient for the FDA to make a reliable assessment. Complete study reports must be available in later clinical development phases.
Unless the FDA has questions, or even places the product on ‘clinical hold’, the IND is considered opened 30 days after it was submitted and from then on information (study protocol) is added to it for every new study planned. New scientific information must be submitted whenever changes are made, e.g. dose increases, new patient categories or new indications. The IND process requires an annual update report describing project progress and additional data obtained during the year.
Approval from Institutional Review Board (IRB) is needed for every institution where a clinical study is to be performed.
Japan
Traditionally, because of differences in medical culture, in treatment traditions and the possibility of significant racial differences, clinical trials in Japan have not been useful for drug applications in the Western world. Also, for a product to be approved for the Japanese market, repetition of clinical studies in Japan was necessary, often delaying the availability of products in Japan.
With the introduction of international standards under the auspices of ICH, data from Japanese patients are increasingly becoming acceptable in other countries. The guideline on bridging studies to compensate for ethnic differences (ICH E5; see website reference 1, p. 301) may allow Japanese studies to become part of global development.
The requirements for beginning a clinical study in Japan are similar to those in the USA or Europe. Scientific summary information is generally acceptable, and ethics committee approval is necessary.
Application for marketing authorization
The application for marketing authorization (MAA in Europe, NDA in the USA, JNDA in Japan) is compiled and submitted as soon as the drug development programme has been completed and judged satisfactory by the company. Different authorities have differing requirements as to the level of detail and the format of submissions, and it is the task of the RA department to collate all the data as efficiently as possible to satisfy these varying requirements with a minimum of redrafting.
The US FDA in general requires raw data to be submitted, allowing them to make their own analysis, and thus they request the most complete data of all authorities. European authorities require a condensed dossier containing critical evaluations of the data, allowing a rapid review based on conclusions drawn by named scientific experts. These may be internal or external, and are selected by the applicant.
Japanese authorities have traditionally focused predominantly on data generated in Japan; studies performed elsewhere being supportive only.
Below, the procedures adopted in these three regions are described in more detail.
Europe
Several procedures are available for marketing authorization in the EU (Figure 20.3, see website reference 13):
National procedure, in which the application is evaluated by one regulatory authority. This procedure is allowed for products intended for that country only. Also, it is the first step in a mutual recognition procedure.
Mutual recognition, in which a marketing approval application is assessed by one national authority, the Reference Member State (RMS), which subsequently defends the approval and evaluation in order to gain mutual recognition of the assessment from other European authorities. The pharmaceutical company may select countries of interest. These, the Concerned Member States, have 90 days to recognize the initial assessment. The Mutual Recognition procedure is used for harmonization and conversion of nationally approved products. After mutual recognition the final marketing authorizations are given as national decisions, but the scientific assessment is quicker and requires fewer resources from all national authorities. In the case of non-agreement, referral to EMA for arbitration is done as a last resort. But before arbitration is considered, the Coordination Group for Mutual Recognition and Decentralised Procedures (CMDh/CMDv) will try to resolve outstanding issues. This is a group composed of expert members from European regulatory authorities. The worst outcome of an arbitration would be that the marketing authorizations obtained are withdrawn in all EU countries, including the RMS.
Decentralized procedure is similar to the Mutual Recognition Procedure. It is a modernization the aim of which is to share the work among authorities earlier in the process, with the possibility of a decision being reached before 120 days have passed from receipt of a valid submission. It is the procedure of choice for a new chemical entity that for any reason is not submitted to follow the centralized procedure.
Centralized procedure is a ‘federal’ procedure carried out by the EMA, with scientists selected from CHMP to perform the review, the approval body being the European Commission. This procedure is mandatory for biotechnological products, biosimilars, orphan drugs as well as products intended to treat diabetes, AIDS, cancer, neurodegenerative disorders, autoimmune diseases and viral diseases.
The centralized procedure starts with the nomination of one CHMP member to act as rapporteur, who selects and leads the assessment team. A selected co-rapporteur and team make a parallel review. The European Commission approves the application based on a CHMP recommendation, which in turn is based on the assessment reports by the two rapporteur teams. Products approved in this way can be marketed in all EU countries with the same prescribing information, packs and labels.
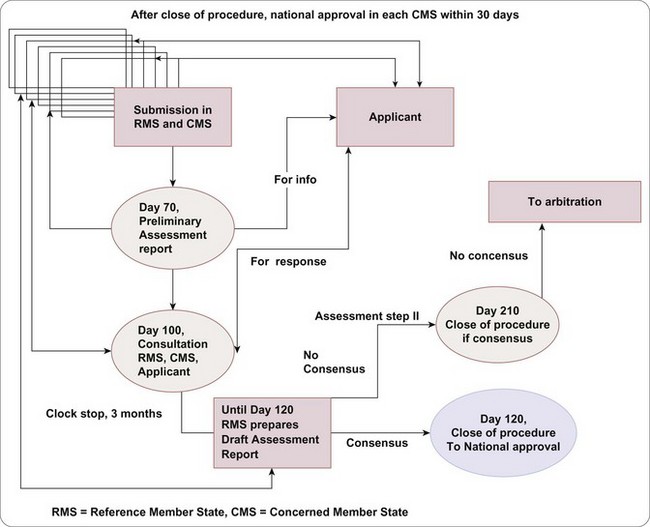
Fig. 20.3 One of the submission processes for marketing approval in EU, the Decentralized Procedure.
CHMP is prepared to give scientific advice to companies in situations where published guidance on the European position is not available, or when the company needs to discuss a possible deviation from guidelines. Such advice, as well as advice from national regulatory authorities, may be very valuable at any stage of the development programme, and may later be incorporated in new guidelines. Providing advice requires considerable effort from CHMP specialists, and fees have to be paid by the pharmaceutical company.
USA
The FDA is more willing than other large authorities to take an active part in planning the drug development process. Some meetings between the FDA and the sponsoring company are more or less compulsory. One such example is the so-called end-of-Phase II meeting. This is in most cases a critically important meeting for the company in which clinical data up until that point is presented to the FDA together with a proposed remaining phase III programme and the company’s target label. The purpose is to gain FDA feedback on the appropriateness of the intended NDA package and whether, in particular, the clinical data are likely to enable approval of a desirable product label. It is important that the advice from the FDA is followed. At the same time, these discussions may make it possible in special cases to deviate from guidelines by prior agreement with the FDA. Furthermore, these discussion meetings ensure that the authority is already familiar with the project when the dossier is submitted.
The review time for the FDA has decreased substantially in the last few years (see Chapter 22). Standard reviews should be completed within 10 months, and priority reviews of those products with a strong medical need within 6 months.
The assessment result is usually communicated either as an approval or as a complete response letter. The latter is in essence a request for additional information or data before approval.
Japan
Also in Japan the regulatory authority is today available for consultation, allowing scientific discussion and feed-back during the development phase. These meetings tend to follow a similar pattern to those in the USA and EU and have made it much easier to address potential problems well before submission for marketing approval is made.
These changes have meant shorter review times and a more transparent process. The review in Japan is performed by an Evaluation Centre, and the ultimate decision is made by the MHLW based on the Evaluation Centre’s report.
It is worth mentioning that health authorities, in particular in the ICH regions, have well-established communications and often assist and consult each other.
The common technical document
Following the good progress made by ICH in creating scientific guidelines applicable in the three large regions, discussions on standardizing document formats began in 1997. The aim was to define a standard format, called the Common Technical Document (CTD), for the application for a new drug product. It was realized from the outset that harmonization of content could not be achieved, owing to the fundamental differences in data requirements and work processes between different regulatory authorities. Adopting a common format would, nonetheless, be a worthwhile step forward.
The guideline was adopted by the three ICH regions in November 2000 and subsequently implemented, and it has generally been accepted in most other countries. This saves much time and effort in reformatting documents for submission to different regulatory authorities. The structure of the CTD (see website reference 1, p. 301) is summarized in Figure 20.4.
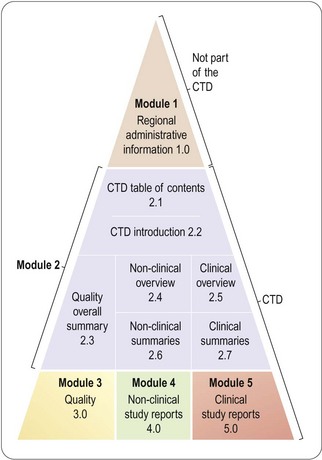
Module 1 (not part of the CTD) contains regional information such as the application form, the suggested prescribing information, the application fee, and also other information that is not considered relevant in all territories, such as environmental assessment (required in the USA and Europe, but not in Japan). Certificates of different regional needs are also to be found in Module 1, as well as patent information not yet requested in the EU.
Module 2 comprises a very brief general introduction, followed by summary information relating to quality, safety (i.e. non-clinical studies) and efficacy (i.e. clinical studies). Quality issues (purity, manufacturing process, stability, etc.) are summarized in a single document of a maximum 40 pages. The non-clinical and clinical summaries each consist of a separate overview (maximum 30 pages) and summaries of individual studies. The overviews in each area are similar to the previous EU Expert Reports in that they present critical evaluations of the programme performed. Detailed guidelines (see website references 1, 13 and 14), based on existing US, European and Japanese requirements, are available to indicate what tabulated information needs to be included in these summaries, and how the written summaries should be drafted. The non-clinical section has been fairly non-controversial. The guidance is very similar to the previous US summary, with clear instructions on how to sort the studies regarding animals, doses, durations of treatment and routes of administration.
The clinical summary is similar to what was required by the FDA, incorporating many features taken from the Integrated Summaries of Efficacy (ISE) and Safety (ISS). ISE will generally fit in the clinical summary document. The ISS document has proved very useful in drawing conclusions from the clinical studies by sensible pooling and integration, but is too large (often more than 400 pages in itself) to be accepted in the EU and Japan. This problem may be resolved by including the ISS as a separate report in Module 5.
Modules 3–5 comprise the individual study reports. Most reports are eligible for use in all three regions, possibly with the exception at present of Module 3, Quality, which may need regional content.
There is a gradual shift to fully electronic submissions – known as e-CTD – in place of the large quantities of paper which have comprised an application for marketing approval. As this gets fully implemented experimental data will be lodged mainly in databases, allowing the information to be exchanged between pharmaceutical companies and regulatory authorities much more easily. Guidelines on the structure and interface between databases in the industry setting and those at the authorities are available.
Administrative rules
Patent protection and data exclusivity
Supplementary protection certificate
During the 1980s, the time taken to develop and obtain marketing approval for a new drug increased so much that the period of market exclusivity established by the original patent could be too short to allow the company to recoup its R&D costs. To overcome this problem (see Chapter 19), the EU Council in 1992 introduced rules allowing companies to apply for a Supplementary Protection Certificate (SPC), matching similar legislation in the USA and Japan. This can prolong market protection by a maximum of 5 years to give an overall period of exclusivity of up to 15 years.
The application for an SPC has to be submitted within 6 months of first approval anywhere in Europe, within or outside the EU, and from then on the clock starts. It is thus strategically important to obtain first approval in a financially important market for best revenue.
Data exclusivity
Data exclusivity should not be confused with patent protection. It is a means to protect the originators data, meaning that generic products cannot be approved by referring to the original product documentation until the exclusivity period has ended. For a new chemical entity the protection may be up to 10 years, depending on region. The generation of paediatric data may extend it even further, between 6 and 12 months.
Pricing of pharmaceutical products – ‘the fourth hurdle’
The ‘fourth hurdle’ or ‘market access’ are expressions for the ever growing demand for cost-effectiveness in pharmaceutical prescribing. Payers, whether health insurance companies or publicly funded institutions, now require more stringent justification for the normally high price of a new pharmaceutical product. This means that, if not in the original application, studies have to demonstrate that the clinical benefit of a new compound is commensurate with the suggested price, compared to the previous therapy of choice in the country of application.
To address this concern, studies in a clinical trials programme from Phase II onwards now normally include health economic measures (see Chapter 18). In the USA, certainly, it is advantageous to have a statement of health economic benefit approved in the package insert, to justify reimbursement. Reference pricing systems are also being implemented in Europe. A favourable benefit–risk evaluation is no longer sufficient: value for money must also be demonstrated in order to have a commercially successful product (see also Chapter 21).
List of abbreviations
| CHMP | Committee for Medicinal Products for Human Use, the new name to replace CPMP early 2004 |
| CTD | Common Technical Document |
| CMDh/ | |
| CMDv | Coordination Group for Human/Veterinary Medicinal Products for Mutual Recognition and Decentralised Procedure (EU) |
| EC | Ethics Committee (EU), known as Institutional Review Board (IRB) in USA |
| eCTD | Electronic Common Technical Document |
| EMA | European Medicines Agency |
| ERA | Environmental Risk Assessment (EU) |
| EU | European Union |
| FDA | Food and Drug Administration; the US Regulatory Authority |
| GCP | Good clinical practice |
| GLP | Good laboratory practice |
| GMP | Good manufacturing practice |
| ICH | International Conference on Harmonization |
| IND | Investigational New Drug application (US) |
| IRB | Institutional Review Board (US), equivalent to Ethics Committee in Europe |
| ISS | Integrated Summary of Safety (US) |
| JNDA | Japanese New Drug Application |
| MAA | Marketing Authorization Application (EU), equivalent to NDA in USA |
| MHLW | Ministry of Health, Labor and Welfare (Jpn) |
| MTD | Maximum tolerated dose |
| NDA | New Drug Application (USA) |
| NTA | Notice to Applicants; EU set of pharmaceutical regulations, directives and guidelines |
| SPC | Supplementary Protection Certificate |
| WHO | World Health Organization |
Cartwright AC, Matthews BR. Pharmaceutical product licensing requirements for Europe. London: Ellis Horwood; 1991.
Cartwright AC, Matthews BR. International pharmaceutical product registration. London: Taylor and Francis, 1994.
Drews J. In quest of tomorrow’s medicines. New York: Springer-Verlag; 1999.
Meade TW. Subacute myelo-optic neuropathy and clioquinol. An epidemiological case-history for diagnosis. British Journal of Preventive and Social Medicine. 1975;29:157–169.
2 http://www.fda.gov/Drugs/DevelopmentApprovalProcess/DevelopmentResources/ucm049867.htm.
3 http://ec.europa.eu/health/files/eudralex/vol-1/reg_2006_1902/reg_2006_1902_en.pdf.
4 http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM184128.pdf.
5 http://www.ema.europa.eu/docs/en_GB/document_library/Regulatory_and_procedural_guideline/2009/10/WC500004888.pdf.
6 http://www.ema.europa.eu/docs/en_GB/document_library/Regulatory_and_procedural_guideline/2009/10/WC500006326.pdf.
7 http://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/ucm215031.htm.
8 http://www.ema.europa.eu/ema/index.jsp?curl=pages/special_topics/general/general_content_000034.jsp&murl=menus/special_topics/special_topics.jsp&mid=WC0b01ac058002d4eb.
9 http://www.fda.gov/ForIndustry/DevelopingProductsforRareDiseasesConditions/default.htm.
10 http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm070561.
11 http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/10/WC500003978.pdf.
12 http://www.wma.net/en/30publications/10policies/b3/index.html.
13 http://ec.europa.eu/health/documents//eudralex/index_en.htm.