Chapter 15 Assessing drug safety
Introduction
Since the thalidomide disaster, ensuring that new medicines are safe when used therapeutically has been one of the main responsibilities of regulatory agencies. Of course, there is no such thing as 100% safety, much as the public would like reassurance of this. Any medical intervention – or for that matter, any human activity – carries risks as well as benefits, and the aim of drug safety assessment is to ensure, as far as possible, that the risks are commensurate with the benefits.
Safety is addressed at all stages in the life history of a drug, from the earliest stages of design, through pre-clinical investigations (discussed in this chapter) and preregistration clinical trials (Chapter 17), to the entire post-marketing history of the drug. The ultimate test comes only after the drug has been marketed and used in a clinical setting in many thousands of patients, during the period of Phase IV clinical trials (post-marketing surveillance). It is unfortunately not uncommon for drugs to be withdrawn for safety reasons after being in clinical use for some time (for example practolol, because of a rare but dangerous oculomucocutaneous reaction, troglitazone because of liver damage, cerivastatin because of skeletal muscle damage, terfenadine because of drug interactions, rofecoxib because of increased risk of heart attacks), reflecting the fact that safety assessment is fallible. It always will be fallible, because there are no bounds to what may emerge as harmful effects. Can we be sure that drug X will not cause kidney damage in a particular inbred tribe in a remote part of the world? The answer is, of course, ‘no’, any more than we could have been sure that various antipsychotic drugs – now withdrawn – would not cause sudden cardiac deaths through a hitherto unsuspected mechanism, hERG channel block (see later). What is not hypothesized cannot be tested. For this reason, the problem of safety assessment is fundamentally different from that of efficacy assessment, where we can define exactly what we are looking for.
Here we focus on non-clinical safety assessment – often called preclinical, even though much of the work is done in parallel with clinical development. We describe the various types of in vitro and in vivo tests that are used to predict adverse and toxic effects in humans, and which form an important part of the data submitted to the regulatory authorites when approval is sought (a) for the new compound to be administered to humans for the first time (IND approval in the USA; see Chapter 20), and (b) for permission to market the drug (NDA approval in the USA, MAA approval in Europe; see Chapter 20).
The programme of preclinical safety assessment for a new synthetic compound can be divided into the following main chronological phases, linked to the clinical trials programme (Figure 15.1):
• Exploratory toxicology, aimed at giving a rough quantitative estimate of the toxicity of the compound when given acutely or repeatedly over a short period (normally 2 weeks), and providing an indication of the main organs and physiological systems involved. These studies provide information for the guidance of the project team in making further plans, but are not normally part of the regulatory package that has to be approved before the drug can be given to humans, so they do not need to be perfomed under good laboratory practice (GLP) conditions.
• Regulatory toxicology. These studies are performed to GLP standards and comprise (a) those that are required by regulatory authorities, or by ethics committees, before the compound can be given for the first time to humans; (b) studies required to support an application for marketing approval, which are normally performed in parallel with clinical trials. Full reports of all studies of this kind are included in documentation submitted to the regulatory authorities.
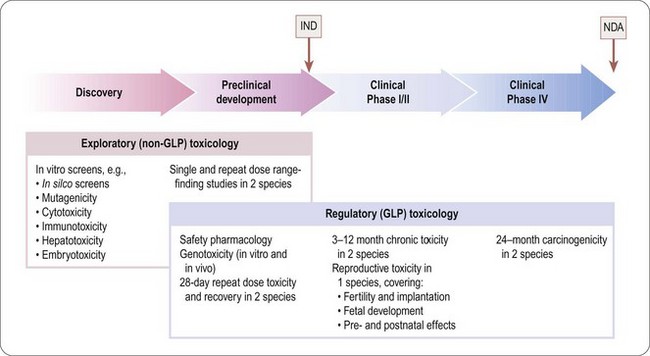
Regulatory toxicology studies in group (a) include 28-day repeated-dose toxicology studies in two species (including one non-rodent, usually dog but sometimes monkey especially if the drug is a biological), in vitro and in vivo genoxocity tests, safety pharmacology and reproductive toxicity assessment. In vitro genotoxicity tests, which are cheap and quick to perform, will often have been performed much earlier in the compound selection phase of the project, as may safety pharmacology studies.
The nature of the tests in group (b) depends greatly on the nature and intended use of the drug, but they will include chronic 3–12-month toxicological studies in two or more species, long-term (18–24 months) carcinogenicity tests and reproductive toxicology, and often interaction studies involving other drugs that are likely to be used for the same indications.
The basic procedures for safety assessment of a single new synthetic compound are fairly standard, although the regulatory authorities have avoided applying a defined checklist of tests and criteria required for regulatory approval. Instead they issue guidance notes (available on relevant websites – USA: www.fda.gov/cder/; EU: www.emea.eu.int; international harmonization guidelines: www.ich.org), but the onus is on the pharmaceutical company to anticipate and exclude any unwanted effects based on the specific chemistry, pharmacology and intended therapeutic use of the compound in question. The regulatory authorities will often ask companies developing second or third entrants in a new class to perform tests based on findings from the class leading compound.
There are many types of new drug applications that do not fall into the standard category of synthetic small molecules, where the safety assessment standards are different. These include most biopharmaceuticals (see Chapter 12), as well as vaccines, cell and gene therapy products (see below). Drug combinations, and non-standard delivery systems and routes of administration are also examples of special cases where safety assessment requirements differ from those used for conventional drugs. These special cases are not discussed in detail here; Gad (2002) gives a comprehensive account.
Types of adverse drug effect
Adverse reactions in man are of four general types:
• Exaggerated pharmacological effects – sometimes referred to as hyperpharmacology or mechanism-related effects – are dose related and in general predictable on the basis of the principal pharmacological effect of the drug. Examples include hypoglycaemia caused by antidiabetic drugs, hypokalaemia induced by diuretics, immunosuppression in response to steroids, etc.
• Pharmacological effects associated with targets other than the principal one – covered by the general term side effects or off-target effects. Examples include hypotension produced by various antipsychotic drugs which block adrenoceptors as well as dopamine receptors (their principal target), and cardiac arrythmias associated with hERG-channel inhibition (see below). Many drugs inhibit one or more forms of cytochrome P450, and hence affect the metabolism of other drugs. Provided the pharmacological profile of the compound is known in sufficient detail, effects of this kind are also predictable (see also Chapter 10).
• Dose-related toxic effects that are unrelated to the intended pharmacological effects of the drug. Commonly such effects, which include toxic effects on liver, kidney, endocrine glands, immune cells and other systems, are produced not by the parent drug, but by chemically reactive metabolites. Examples include the gum hyperplasia produced by the antiepileptic drug phenytoin, hearing loss caused by aminoglycoside antibiotics, and peripheral neuropathy caused by thalidomide1. Genotoxicity and reproductive toxicity (see below) also fall into this category. Such adverse effects are not, in general, predictable from the pharmacological profile of the compound. It is well known that certain chemical structures are associated with toxicity, and so these will generally be eliminated early in the lead identification stage, sometimes in silico before any actual compounds are synthesized. The main function of toxicological studies in drug development is to detect dose-related toxic effects of an unpredictable nature.
• Rare, and sometimes serious, adverse effects, known as idiosyncratic reactions, that occur in certain individuals and are not dose related. Many examples have come to light among drugs that have entered clinical use, e.g. aplastic anaemia produced by chloramphenicol, anaphylactic responses to penicillin, oculomucocutaneous syndrome with practolol, bone marrow depression with clozapine. Toxicological tests in animals rarely reveal such effects, and because they may occur in only one in several thousand humans they are likely to remain undetected in clinical trials, coming to light only after the drug has been registered and given to thousands of patients. (The reaction to clozapine is an exception. It affects about 1% of patients and was detected in early clinical trials. The bone marrow effect, though potentially life-threatening, is reversible, and clozapine was successfully registered, with a condition that patients receiving it must be regularly monitored.)
Safety pharmacology testing and dose range-finding studies are designed to detect pharmacological adverse effects; chronic toxicology testing is designed to detect dose-related toxic effects, as well as the long-term consequences of pharmacological side effects; idiosyncratic reactions may be revealed in Phase III clinical trials, but are likely to remain undetected until the compound enters clinical use.
Safety pharmacology
The pharmacological studies described in Chapter 11 are exploratory (i.e. surveying the effects of the compound with respect to selectivity against a wide range of possible targets) or hypothesis driven (checking whether the expected effects of the drug, based on its target selectivity, are actually produced). In contrast, safety pharmacology comprises a series of protocol-driven studies, aimed specifically at detecting possible undesirable or dangerous effects of exposure to the drug in therapeutic doses (see ICH Guideline S7A). The emphasis is on acute effects produced by single-dose administration, as distinct from toxicology studies, which focus mainly on the effects of chronic exposure. Safety pharmacology evaluation forms an important part of the dossier submitted to the regulatory authorities.
ICH Guideline S7A defines a core battery of safety pharmacology tests, and a series of follow-up and supplementary tests (Table 15.1). The core battery is normally performed on all compounds intended for systemic use. Where they are not appropriate (e.g. for preparations given topically) their omission has to be justified on the basis of information about the extent of systemic exposure that may occur when the drug is given by the intended route. Follow-up studies are required if the core battery of tests reveals effects whose mechanism needs to be determined. Supplementary tests need to be performed if the known chemistry or pharmacology of the compound gives any reason to expect that it may produce side effects (e.g. a compound with a thiazide-like structure should be tested for possible inhibition of insulin secretion, this being a known side effect of thiazide diuretics; similarly, an opioid needs to be tested for dependence liability and effects on gastrointestinal motility). Where there is a likelihood of significant drug interactions, this may also need to be tested as part of the supplementary programme.
Table 15.1 Safety pharmacology
| Type | Physiological system | Tests |
|---|---|---|
| Core battery | Central nervous system | Observations on conscious animals |
| Motor activity | ||
| Behavioural changes | ||
| Coordination | ||
| Reflex responses | ||
| Body temperature | ||
| Cardiovascular system | Measurements on anaesthetized animals | |
| Blood pressure | ||
| Heart rate | ||
| ECG changes | ||
| Tests for delayed ventricular repolarization (see text) | ||
| Respiratory system | Measurements on anaesthetized or conscious animals | |
| Respiratory rate | ||
| Tidal volume | ||
| Arterial oxygen saturation | ||
| Follow-up tests (examples) | Central nervous system | Tests on learning and memory |
| More complex test for changes in behaviour and motor function | ||
| Tests for visual and auditory function | ||
| Cardiovascular system | Cardiac output | |
| Ventricular contractility | ||
| Vascular resistance | ||
| Regional blood flow | ||
| Respiratory system | Airways resistance and compliance | |
| Pulmonary arterial pressure | ||
| Blood gases | ||
| Supplementary tests (examples) | Renal function | Urine volume, osmolality pH |
| Proteinuria | ||
| Blood urea/creatinine | ||
| Fluid/electrolyte balance | ||
| Urine cytology | ||
| Autonomic nervous system | Cardiovascular, gastrointestinal and respiratory system responses to agonists and stimulation of autonomic nerves | |
| Gastrointestinal system | Gastric secretion | |
| Gastric pH | ||
| Intestinal motility | ||
| Gastrointestinal transit time | ||
| Other systems (e.g. endocrine, blood coagulation, skeletal muscle function, etc.) | Tests designed to detect likely acute effects |
The core battery of tests listed in Table 15.1 focuses on acute effects on cardiovascular, respiratory and nervous systems, based on standard physiological measurements.
The follow-up and supplementary tests are less clearly defined, and the list given in Table 15.1 is neither prescriptive nor complete. It is the responsibility of the team to decide what tests are relevant and how the studies should be performed, and to justify these decisions in the submission to the regulatory authority.
Tests for QT interval prolongation
The ability of a number of therapeutically used drugs to cause a potentially fatal ventricular arrhythmia (’torsade de pointes’) has been a cause of major concern to clinicians and regulatory authorities (see Committee for Proprietary Medicinal Products, 1997; Haverkamp et al., 2000). The arrhythmia is associated with prolongation of the ventricular action potential (delayed ventricular repolarization), reflected in ECG recordings as prolongation of the QT interval. Drugs known to possess this serious risk, many of which have been withdrawn, include several tricyclic antidepressants, some antipsychotic drugs (e.g. thioridazine, droperidol), antidysrhythmic drugs (e.g. amiodarone, quinidine, disopyramide), antihistamines (terfenadine, astemizole) and certain antimalarial drugs (e.g. halofantrine). The main mechanism responsible appears to be inhibition of a potassium channel, termed the hERG channel, which plays a major role in terminating the ventricular action potential (Netzer et al., 2001).
Screening tests have shown that QT interval prolongation is a common property of ‘drug-like’ small molecules, and the patterns of structure–activity relationships have revealed particular chemical classes associated with this effect. Ideally, these are taken into account and avoided at an early stage in drug design, but the need remains for functional testing of all candidate drug molecules as a prelude to tests in humans.
Proposed standard tests for QT interval prolongation have been formulated as ICH Guideline S7B. They comprise (a) testing for inhibition of hERG channel currents in cell lines engineered to express the hERG gene; (b) measurements of action potential duration in myocardial cells from different parts of the heart in different species; and (c) measurements of QT interval in ECG recordings in conscious animals. These studies are usually carried out on ferrets or guinea pigs, as well as larger mammalian species, such as dog, rabbit, pig or monkey, in which hERG-like channels control ventricular repolarization, rather than in rat and mouse. In vivo tests for proarrhythmic effects in various species are being developed (De Clerck et al., 2002), but have not yet been evaluated for regulatory purposes.
Because of the importance of drug-induced QT prolongation in man, and the fact that many diverse groups of drugs appear to have this property, there is a need for high-throughput screening for hERG channel inhibition to be incorporated early in a drug discovery project. The above methods are not suitable for high-throughput screening, but alternative methods, such as inhibition of binding of labelled dofetilide (a potent hERG-channel blocker), or fluorimetric membrane potential assays on cell lines expressing these channels, can be used in high-throughput formats, as increasingly can automated patch clamp studies (see Chapter 8). It is important to note that binding and fluorescence assays are not seen as adequately predictive and cannot replace the patch clamp studies under the guidelines (ICH 7B). These assays are now becoming widely used as part of screening before selecting a clinical candidate molecule, though there is still a need to confirm presence or absence of QT prolongation in functional in vivo tests before advancing a compound into clinical development.
Exploratory (dose range-finding) toxicology studies
The first stage of toxicological evaluation usually takes the form of a dose range-finding study in a rodent and/or a non-rodent species. The species commonly used in toxicology are mice, rats, guinea pigs, hamsters, rabbits, dogs, minipigs and non-human primates. Usually two species (rat and mouse) are tested initially, but others may be used if there are reasons for thinking that the drug may exert species-specific effects. A single dose is given to each test animal, preferably by the intended route of administration in the clinic, and in a formulation shown by previous pharmacokinetic studies to produce satisfactory absorption and duration of action (see also Chapter 10). Generally, widely spaced doses (e.g. 10, 100, 1000 mg/kg) will be tested first, on groups of three to four rodents, and the animals will be observed over 14 days for obvious signs of toxicity. Alternatively, a dose escalation protocol may be used, in which each animal is treated with increasing doses of the drug at intervals (e.g. every 2 days) until signs of toxicity appear, or until a dose of 2000 mg/kg is reached. With either protocol, the animals are killed at the end of the experiment and autopsied to determine if any target organs are grossly affected. The results of such dose range-finding studies provide a rough estimate of the no toxic effect level (NTEL, see Toxicity measures, below) in the species tested, and the nature of the gross effects seen is often a useful pointer to the main target tissues and organs.
The dose range-finding study will normally be followed by a more detailed single-dose toxicological study in two or more species, the doses tested being chosen to span the estimated NTEL. Usually four or five doses will be tested, ranging from a dose in the expected therapeutic range to doses well above the estimated NTEL. A typical protocol for such an acute toxicity study is shown in Figure 15.2. The data collected consist of regular systematic assessment of the animals for a range of clinical signs on the basis of a standardized checklist, together with gross autopsy findings of animals dying during a 2-week observation period, or killed at the end. The main signs that are monitored are shown in Table 15.2.
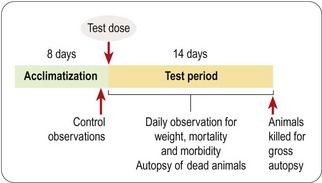
Table 15.2 Clinical observations in acute toxicity tests
| System | Observation | Signs of toxicity |
|---|---|---|
| Nervous system | Behaviour | Sedation |
| Restlessness | ||
| Aggression | ||
| Motor function | Twitch | |
| Tremor | ||
| Ataxia | ||
| Catatonia | ||
| Convulsions | ||
| Muscle rigidity or flaccidity | ||
| Sensory function | Excessive or diminished response to stimuli | |
| Respiratory | Respiration | Increased or decreased respiratory rate |
| Intermittent respiration | ||
| Dyspnoea | ||
| Cardiovascular | Cardiac palpation | Increase or decrease in rate or force |
| ?Electrocardiography | Disturbances of rhythm. | |
| Altered ECG pattern (e.g. QT prolongation) | ||
| Gastrointestinal | Faeces | Diarrhoea or constipation |
| Abnormal form or colour | ||
| Bleeding | ||
| Abdomen | Spasm or tenderness | |
| Genitourinary | Genitalia | Swelling, inflammation, discharge, bleeding |
| Skin and fur | Discoloration | |
| Lesions | ||
| Piloerection | ||
| Mouth | Discharge | |
| Congestion | ||
| Bleeding | ||
| Eye | Pupil size | Mydriasis or miosis |
| Eyelids | Ptosis, exophthalmos | |
| Movements | Nystagmus | |
| Cornea | Opacity | |
| General signs | Body weight | Weight loss |
| Body temperature | Increase or decrease |
Single-dose studies are followed by a multiple dose-ranging study in which the drug is given daily or twice daily, normally for 2 weeks, with the same observation and autopsy procedure as in the single-dose study, in order to give preliminary information about the toxicity after chronic treatment.
The results of these preliminary in vivo toxicity studies will help in the planning and design of the next steps of the development programme; they will also help to decide whether or not it is worthwhile to continue the research effort on a given chemical class.
As mentioned above, these toxicology studies are preliminary, and usually they are not sufficient for supporting the first human evaluation of the medicine. Very often, they are not conducted under good laboratory practice (GLP) conditions, nor are they conducted with test material which has been produced according to good manufacturing practice (GMP).
Subsequent work is guided by regulatory requirements elaborated by the International Conference of Harmonization or by national regulatory authorities. Their published guidelines specify/recommend the type of toxicological evaluation needed to support applications for carrying out studies in humans. These documents provide guidance, for example, on the duration of administration, on the design of the toxicological study, including the number of animals to be studied, and stipulate that the work must be carried out under GLP conditions and that the test material must be of GMP standard. The test substance for the toxicology evaluation has to be identical in terms of quality and characteristics to the substance given to humans.
Genotoxicity
Foreign substances can affect gene function in various ways, the two most important types of mechanism in relation to toxicology being:
• Mutagenicity, i.e. chemical alteration of DNA sufficient to cause abnormal gene expression in the affected cell and its offspring. Most commonly, the mutation arises as a result of covalent modification of individual bases (point mutations). The result may be the production of an abnormal protein if the mutation occurs in the coding region of the gene, or altered expression levels of a normal protein if the mutation affects control sequences. Such mutations occur continuously in everyday life, and are counteracted more or less effectively by a variety of DNA repair mechanisms. They are important particularly because certain mutations can interfere with mechanisms controlling cell division, and thereby lead to malignancy or, in the immature organism, to adverse effects on growth and development. In practice, most carcinogens are mutagens, though by no means all mutagens are carcinogens. Evidence of mutagenicity therefore sounds a warning of possible carcinogenicity, which must be tested by thorough in vivo tests.
• Chromosomal damage, for example chromosome breakage (clastogenesis), chromosome fusion, translocation of stretches of DNA within or between chromosomes, replication or deletion of chromosomes, etc. Such changes result from alterations in DNA, more extensive than point mutations and less well understood mechanistically; they have a similar propensity to cause cancerous changes and to affect growth and development.
The most important end results of genotoxicity – carcinogenesis and impairment of fetal development (teratogenicity) – can only be detected by long-term animal studies. There is, therefore, every reason to pre-screen compounds by in vitro methods, and such studies are routinely carried out before human studies begin. Because in many cases the genotoxicity is due to reactive metabolites rather than to the parent molecule, the in vitro tests generally include assays carried out in the presence of liver microsomes or other liver-derived preparations, so that metabolites are generated. Often, liver microsomes from rats treated with inducing agents (e.g. a mixture of chlorinated biphenyls known as Arochlor 1254) are used, in order to enhance drug metabolizing activity.
Selection and interpretation of tests
Many in vitro and in vivo test systems for mutagenicity have been described, based on bacteria, yeast, insect and mammalian cells (see Gad, 2002 for details). The ICH Guidelines S2A and S2B, stipulate a preliminary battery of three tests:
• Ames test: a test of mutagenicity in bacteria. The basis of the assay is that mutagenic substances increase the rate at which a histidine-dependent strain of Salmonella typhimurium reverts to a wild type that can grow in the absence of histidine. An increase in the number of colonies surviving in the absence of histidine therefore denotes significant mutagenic activity. Several histidine-dependent strains of the organism which differ in their susceptibility to particular types of mutagen are normally tested in parallel. Positive controls with known mutagens that act directly or only after metabolic activation are routinely included when such tests are performed.
• An in vitro test for chromosomal abnormalities in mammalian cells or an in vitro mouse lymphoma tk cell assay. To test chromosomal damage Chinese hamster ovary (CHO) cells are grown in culture in the presence of the test substance, with or without liver microsomes. Cell division is arrested in metaphase, and chromosomes are observed microscopically to detect structural aberrations, such as gaps, duplications, fusions or alterations in chromosome number. The mouse lymphoma cell test for mutagenicity involves a heterozygous (tk +/−) cell line that can be killed by the cytotoxic agent BrDU. Mutation to the tk −/−) form causes the cells to become resistant to BrDU, and so counts of surviving cells in cutures treated with the test compound provide an index of mutagenicity. The mouse lymphoma cell mutation assay is more sensitive than the chromosomal assay, but can give positive results with non-carcinogenic substances. These tests are not possible with compounds that are inherently toxic to mammalian cells.
• An in vivo test for chromosomal damage in rodent haemopoietic cells. The mouse micronucleus test is commonly used. Animals are treated with the test compound for 2 days, after which immature erythrocytes in bone marrow are examined for micronuclei, representing fragments of damaged chromosomes.
If these three tests prove negative, no further tests of genotoxicity are generally needed before the compound can be tested in humans. If one or more is positive, further in vitro and in vivo genotoxicity testing will usually be carried out to assess more accurately the magnitude of the risk. In a few cases, where the medical need is great and the life expectancy of the patient population is very limited, development of compounds that are clearly genotoxic – and, by inference, possibly carcinogenic – may still be justified, but in most cases genotoxic compounds will be abandoned without further ado. An updated version of the guideline is open for consultation at the time of writing.
Whereas early toxicological evaluation encompasses acute and subacute types of study, the full safety assessement is based on subchronic and chronic studies. The focus is more on the harmful effects of long-term exposure to ‘low’ doses of the agent. The chronic toxic effects can be very different from the acute toxic effects, and could accumulate over time. Three main categories of toxicological study are required according to the regulatory guidelines, namely chronic toxicity special tests and toxicokinetic analysis.
Chronic toxicology studies
The object of these studies is to look for toxicities that appear after repetitive dosing of the compound, when a steady state is achieved, i.e. when the rate of drug administration equals the rate of elimination. In long-term toxicity studies three or more dose levels are tested, in addition to a vehicle control. The doses will include one that is clearly toxic, one in the therapeutic range, and at least one in between. Ideally, the in-between doses will exceed the expected clinical dose by a factor of 10 for rodents and 5 for non-rodents, yet lack overt toxicity, this being the ‘window’ normally required by regulatory authorities. At least one recovery group is usually included, i.e. animals treated with the drug at a toxic level and then allowed to recover for 2–4 weeks so that the reversibility of the changes observed can be assessed. The aim of these studies is to determine (a) the cumulative biological effects produced by the compound, and (b) at what exposure level (see below) they appear. The initial repeated-dose studies, by revealing the overall pattern of toxic effects produced, also give pointers to particular aspects (e.g. liver toxicity, bone marrow depression) that may need to be addressed later in more detailed toxicological investigations.
The standard procedures discussed here are appropriate for the majority of conventional synthetic compounds intended for systemic use. Where drugs are intended only for topical use (skin creams, eye drops, aerosols, etc.) the test procedures are modified accordingly. Special considerations apply also to biopharmaceutical preparations, and these are discussed briefly below.
Experimental design
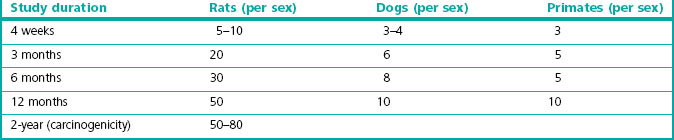
Chronic toxicity testing must be performed under GLP conditions, with the formulation and route of administration to be used in humans. Tests are normally carried out on one rodent (usually rat) and one non-rodent species (usually dog), but additional species (such as monkey or pig) may be tested if there are special reasons for suspecting that their responses may predict effects in man more accurately than those of dogs. Pharmacokinetic measurements are included, so that extrapolation to humans can be done on the basis of the concentration of the drug in blood and tissues, allowing for differences in pharmacokinetics between laboratory animals and humans (see Chapter 10). Separate male and female test groups are used. The recommended number of animals per test group (see Gad, 2002) is shown in Table 15.3. There are strong reasons for minimizing the number of animals used in such studies, and the figures given take this into account, representing the minimum shown by experience to be needed for statistically reliable conclusions to be drawn. An average study will require 200 or more rats and about 60 dogs, dosed with compound and observed regularly, including blood and urine sampling, for several months before being killed and autopsied, with tissue samples being collected for histological examination. It is a massive and costly experiment requiring large amounts of compound prepared to GMP standards, the conduct and results of which will receive detailed scrutiny by regulatory authorities, and so careful planning and scrupulous execution are essential.
Table 15.3 Recommended numbers of animals for chronic toxicity studies (Gad, 2002)
Regulatory authorities stipulate the duration of repeat-dose toxicity testing required before the start of clinical testing, the ICH recommendations being summarized in Table 20.1. These requirements can be relaxed for drugs aimed at life-threatening diseases, the spur for this change in attitude coming from the urgent need for anti-HIV drugs during the 1980s. In such special cases chronic toxicity testing is still required, but approval for clinical trials – and indeed for marketing – may be granted before they have been completed.
Evaluation of toxic effects
During the course of the experiment all animals are inspected regularly for mortality and gross morbidity, severely affected animals being killed, and all dead animals subjected to autopsy. Specific signs (e.g. diarrhoea, salivation, respiratory changes, etc.) are assessed against a detailed checklist and specific examinations (e.g. blood pressure, heart rate and ECG, ocular changes, neurological and behavioural changes, etc.) are also conducted regularly. Food intake and body weight changes are monitored, and blood and urine samples collected at intervals for biochemical and haematological analysis.
At the end of the experiment all animals are killed and examined by autopsy for gross changes, samples of all major tissues being prepared for histological examination. Tissues from the high-dose group are examined histologically and any tissues showing pathological changes are also examined in the low-dose groups to enable a dose threshold to be estimated. The reversibility of the adverse effects can be evaluated in these studies by studying animals retained after the end of the dosing period.
As part of the general toxicology screening described above, specific evaluation of possible immunotoxicity is required by the regulatory authorities, the main concern being immunosuppression. If effects on blood, spleen or thymus cells are observed in the 1-month repeated-dose studies, additional tests are required to evaluate the strength of cellular and humoral responses in immunized animals. A battery of suitable tests is included in the FDA Guidance note (2002).
The large body of data collected during a typical toxicology study should allow conclusions to be drawn about the main physiological systems and target organs that underlie the toxicity of the compound, and also about the dose levels at which critical effects are produced. In practice, the analysis and interpretation are not always straightforward, for a variety of reasons, including:
• Variability within the groups of animals
• Spontaneous occurrence of ‘toxic’ effects in control or vehicle-treated animals
• Missing data, owing to operator error, equipment failure, unexpected death of animals, etc.
• Problems of statistical analysis (qualitative data, multiple comparisons, etc.).
Overall, it is estimated that correctly performed chronic toxicity tests in animals successfully predict 70% of toxic reactions in humans (Olson et al., 2000); skin reactions in humans are the least well predicted.
Biopharmaceuticals
Biopharmaceuticals now constitute more than 25% of new drugs being approved. For the most part they are proteins made by recombinant DNA technology, or monoclonal antibodies produced in cell culture. As a rule, proteins tend to be less toxic than synthetic compounds, mainly because they are normally metabolized to smaller peptides and amino acids, rather than to the reactive compounds formed from many synthetic small molecule compounds, which are the cause of most types of drug toxicity, especially genotoxicity. Biopharmaceuticals are, therefore, generally less toxic than synthetic compounds. Their unwanted effects are associated mainly with ‘hyperpharmacology’ (see above), lack of pharmacological specificity or immunogenicity. Many protein therapeutics are highly species specific and this often means that safety studies have to be performed in monkeys or that a rodent-selective analogue has to be made. This applies to biological mediators, such as hormones, growth factors, cytokines, etc., as well as to monoclonal antibodies. The presence of impurities in biopharmaceutical preparations has an important bearing on safety assessment, as unwanted proteins or other cell constituents are often present as contaminants, and may vary from batch to batch. Quality control presents more problems than with conventional chemical products, and is particularly critical for biopharmaceuticals.
The safety assessment of new biopharmaceuticals is discussed in ICH Guidance Note S6 and the Addendum issued in June 2011. The main aspects that differ from the guidelines relating to conventional drugs are: (1) careful attention to choice of appropriate species; (2) no need for routine genotoxicity and carcinogenicity testing (though carcinogenicity testing may be required in the case of mediators, such as growth factors, which may regulate cell proliferation); and (3) specific tests for immunogenicity, expressed as sensitization, or the development of neutralizing antibodies.
Newer types of biopharmaceuticals, such as DNA-, RNA- and cell-based therapies (see Chapter 3), pose special questions in relation to safety assessment (see FDA Guidance Note, 1998). Particular concerns arise from the use of genetically engineered viral vectors in gene therapy, which can induce immune responses and in some cases retain potential infectivity. The possibility that genetically engineered foreign cells might undergo malignant transformation is a further worrisome risk. These specialized topics are not discussed further here.
Special tests
In addition to the the general toxicological risks addressed by the testing programme discussed above, the risks of carcinogenicity and effects on reproduction (particularly on fertility and on pre- and postnatal development) may be of particular concern, requiring special tests to be performed.
Carcinogenicity testing
Carcinogenicity testing is normally required before a compound can be marketed – though not before the start of clinical trials – if the drug is likely to be used in treatment continuously for 6 months or more, or intermittently for long periods. It is also required if there are special causes for concern, for example if:
• the compound belongs to a known class of carcinogens, or has chemical features associated with carcinogenicity; nowadays such compounds will normally have been eliminated at the lead identification stage (see Chapter 9)
• chronic toxicity studies show evidence of precancerous changes
• the compound or its metabolites are retained in tissues for long periods.
If a compound proves positive in tests of mutagenicity (see above), it must be assumed to be carcinogenic and its use restricted accordingly, so no purpose is served by in vivo carcinogenicity testing. Only in very exceptional cases will such a compound be chosen for development.
ICH Guidelines S1A, S1B and S1C on carcinogenicity testing stipulate one long-term test in a rodent species (usually rat), plus one other in vivo test, which may be either (a) a short-term test designed to show high sensitivity to carcinogens (e.g. transgenic mouse models) or to detect early events associated with tumour initiation or promotion; or (b) a long-term carcinogenicity test in a second rodent species (normally mouse). If positive results emerge in either study, the onus is on the pharmaceutical company to provide evidence that carcinogenicity will not be a significant risk to humans in a therapeutic setting. Until recently, the normal requirement was for long-term studies in two rodent species, but advances in the understanding of tumour biology and the availability of new models that allow quicker evaluation have brought about a change in the attitude of regulatory authorities such that only one long-term study is required, together with data from a well-validated short-term study.
Long-term rat carcinogenicity studies normally last for 2 years and are run in parallel with Phase III clinical trials. (Oral contraceptives require a 3-year test for carcinogenicity in beagles.)
Three or four dose levels are tested, plus controls. Typically, the lowest dose tested is close to the maximum recommended human dose, and the highest is the maximum tolerated dose (MTD) in rats (i.e. the largest dose that causes no obvious side effects or toxicity in the chronic toxicity tests). Between 50 and 80 animals of each sex are used in each experimental group (see Table 15.3), and so the complete study will require about 600–800 animals. Premature deaths are inevitable in such a large group and can easily ruin the study, so that housing the animals under standard disease-free conditions is essential. At the end of the experiment, samples of about 50 different tissues are prepared for histological examination, and rated for benign and malignant tumour formation by experienced pathologists. Carcinogenicity testing is therefore one of the most expensive and time-consuming components of the toxicological evaluation of a new compound. New guidelines for dose selection were adopted from 2008 which allow a more rational approach to the selection of the high dose on the basis of a 25-fold higher exposure of the rodent than that seen in human subjects on the basis of AUC measurements in plasma rather than MTD (Van der Laan, 2008).
Several transgenic mouse models have been developed which provide data more quickly (usually about 6 months) than the normal 2-year carcinogenicity study (Gad, 2002). These include animals in which human proto-oncogenes, such as hRas, are expressed, or the tumour suppressor gene P53 is inactivated. These mice show a very high incidence of spontaneous tumours after about 1 year, but at 6 months spontaneous tumours are rare. Known carcinogens cause tumours to develop in these animals within 6 months.
Advances in this area are occurring rapidly, and as they do so the methodology for carcinogenicity testing is expected to become more sophisticated and faster than the conventional long-term studies used hitherto (www.alttox.org/ttrc/toxicity-tests/carcinogenicity/).
Reproductive/developmental toxicology studies
Two incidents led to greatly increased concern about the effects of drugs on the fetus. The first was the thalidomide disaster of the 1960s. The second was the high incidence of cervical and vaginal cancers in young women whose mothers had been treated with diethylstilbestrol (DES) in early pregnancy with the aim of preventing early abortion. DES was used in this way between 1940 and 1970, and the cancer incidence was reported in 1971. These events led to the introduction of stringent tests for teratogenicity as a prerequisite for the approval of new drugs, and from this flowed concern for other aspects of reproductive toxicology which now must be fully evaluated before a drug is marketed. Current requirements are summarized in ICH Guideline S5A.
Drugs can affect reproductive performance in three main ways:
• Fertility (both sexes, fertilization and implantation), addressed by Segment 1 studies
• Embryonic and fetal development or teratology, addressed by Segment 2 studies
• Peri- and postnatal development, addressed by Segment 3 studies.
It is usually acceptable for Phase I human studies on male volunteers to begin before any reproductive toxicology data are available, so long as the drug shows no evidence of testicular damage in 2- or 4-week repeated-dose studies. The requirement for reproductive toxicology data as a prelude to clinical trials differs from country to country, but, as a general rule, clinical trials involving women of childbearing age should be preceded by relevant reproductive toxicology testing. In all but exceptional cases, such as drugs intended for treating life-threatening diseases, or for use only in the elderly, registration will require comprehensive data from relevant toxicology studies so that the reproductive risk can be assessed.
Segment 1 tests of fertility and implantation involve treating both males (for 28 days) and females (for 14 days) with the drug prior to mating, then measuring sperm count and sperm viability, numbers of implantation sites and live and dead embryos on day 6 of gestation. For drugs that either by design or by accident reduce fertility, tests for reversibility on stopping treatment are necessary.
Segment 2 tests of effects on embryonic and fetal development are usually carried out on two or three species (rat, mouse, rabbit), the drug being given to the female during the initial gestation period (day 6 to day 16 after mating in the rat). Animals are killed just before parturition, and the embryos are counted and assessed for structural abnormalities. In vitro tests involving embryos maintained in culture are also possible. The main stages of early embryogenesis can be observed in this way, and the effects of drugs added to the medium can be monitored. Such in vitro tests are routinely performed in some laboratories, but are currently not recognized by regulatory authorities as a reliable measure of possible teratogenicity.
Segment 3 tests on pre- and perinatal development entail dosing female rats with the drug throughout gestation and lactation. The offspring are observed for motility, reflex responses, etc., both during and after the weaning period, and at intervals some are killed for observations of structural abnormalities. Some are normally allowed to mature and are mated, to check for possible second-generation effects. Mature offspring are also tested for effects on learning and memory.
Reproductive and developmental toxicology is a complex field in which standards in relation to pharmaceuticals have not yet been clearly defined. The experimental studies are demanding, and the results may be complicated by species differences, individual variability and ‘spontaneous’ events in control animals.
It is obvious that any drug given in sufficient doses to cause overt maternal toxicity is very likely to impair fetal development. Non-specific effects, most commonly a reduction in birthweight, are commonly found in animal studies, but provided the margin of safety is sufficient – say 10-fold – between the expected therapeutic dose and that affecting the fetus, this will not be a bar to developing the compound. The main aim of reproductive toxicology is to assess the risk of specific effects occurring within the therapeutic dose range in humans. Many familiar drugs and chemicals are teratogenic in certain species at high doses. They include penicillin, sulfonamides, tolbutamide, diphenylhydantoin, valproate, imipramine, acetazolamide, ACE inhibitors and angiotensin antagonists, as well as many anticancer drugs and also caffeine, cannabis and ethanol. Many of these are known or suspected teratogens in humans, and their use in pregnancy is to be avoided. A classification of drugs based on their safety during pregnancy has been developed by the FDA (A, B, C, D or X). Category A is for drugs considered safe in human pregnancy, that is, adequate and well-controlled studies in pregnant women have failed to demonstrate a risk to the fetus in any trimester of pregnancy. Few drugs belong to this category. Category X is reserved for drugs (e.g. isotretinoin, warfarin) that have been proved to cause fetal abnormalities in man, and are therefore contraindicated in pregnancy. Category B covers drugs with no evidence of risk in humans; category C covers drugs in which a risk cannot be ruled out; and category D covers drugs with positive evidence of risk. Requirements for the reproductive safety testing of biologicals are laid out in the Addendum to S6 (2011) and there are specific considerations, e.g. the inability of some high-molecular-weight proteins to cross the placenta which do not apply to small molecules.
Other studies
The focus of this chapter is on the core battery of tests routinely used in assessing drug safety at the preclinical level. In practice, depending on the results obtained from these tests, and on the particular therapeutic application and route of administration intended for the drug, it is nearly always necessary to go further with experimental toxicology studies in specific areas. It must be remembered that the regulatory authorities put the onus firmly on the pharmaceutical company to present a dossier of data that covers all likely concerns about safety. Thus, where toxic effects are observed in animals, evidence must be presented to show either that these are seen only at exposure levels well outside the therapeutic range, or that they involve mechanisms that will not apply in humans. If the compound is observed to cause changes in circulating immune cells, or in lymphoid tissues, further tests for immunosuppression will be needed, as well as studies to determine the mechanism. Potential toxicological problems, not necessarily revealed in basic toxicology testing, must also be anticipated. Thus, if the compound is potentially immunogenic (i.e. it is a peptide or protein, or belongs to a known class of haptens), tests for sensitization will be required. For drugs that are intended for topical administration, local tissue reactions, including allergic sensitization, need to be investigated. For some classes of drugs skin photosensitization will need to be tested.
Interaction toxicology studies may be needed if the patients targeted for the treatment are likely to be taking another medicine whose efficacy or toxicity might be affected by the new therapeutic agent.
In summary, it is essential to plan the toxicology testing programme for each development compound on a case-by-case basis. Although the regulatory authorities stipulate the core battery of tests that need to be performed on every compound, it is up to the development team to anticipate other safety issues that are likely to be of concern, and to address them appropriately with experimental studies. In planning the safety assessment programme for a new drug, companies are strongly advised to consult the regulatory authorities, who are very open to discussion at the planning stage and can advise on a case-by-case basis.
Toxicokinetics
Toxicokinetics is defined in ICH Guideline S3A as ‘the generation of pharmacokinetic data, either as an integral component in the conduct of non-clinical toxicity studies, or in specially designed supportive studies, in order to assess systemic exposure’. In essence, this means pharmacokinetics applied to toxicological studies, and the methodology and principles are no different from those of conventional absorption, distribution, metabolism and excretion (ADME) studies. But whereas ADME studies address mainly drug and metabolite concentrations in different body compartments as a function of dose and time, toxicokinetics focuses on exposure. Although exposure is not precisely defined, the underlying idea is that toxic effects often appear to be a function of both local concentration and time, a low concentration persisting for a long time being as likely to evoke a reaction as a higher but more transient concentration. Depending on the context, exposure can be represented as peak plasma or tissue concentration, or more often as average concentrations over a fixed period. As with conventional pharmacokinetic measurement, attention must be paid to plasma and tissue binding of the drug and its metabolites, as bound material, which may comprise 98% or more of the measured concentration, will generally be pharmacologically and toxicologically inert. The principles of toxicokinetics as applied to preclinical studies are discussed in detail by Baldrick (2003).
Despite the difficulties of measuring and interpreting exposure, toxicokinetic measurements are an essential part of all in vivo toxicological studies. Interspecies comparisons and extrapolation of animal data to humans are best done on the basis of measured plasma and tissue concentrations, rather than administered dose, and regulatory authorities require this information to be provided.
As mentioned earlier, the choice of dose for toxicity tests is important, and dose-limiting toxicity should ideally be reached in toxicology studies. This is the reason why the doses administered in these studies are always high, unless a maximum limit based on technical feasibility has been reached.
Toxicity measures
Lethal dose (expressed as LD50, the estimated dose required to kill 50% of a group of experimental animals) has been largely abandoned as a useful measure of toxicity, and no longer needs to be measured for new compounds. Measures commonly used are:
• no toxic effect level (NTEL), which is the largest dose in the most sensitive species in a toxicology study of a given duration which produced no observed toxic effect
• no observed adverse effect level (NOAEL), which is the largest dose causing neither observed tissue toxicity nor undesirable physiological effects, such as sedation, seizures or weight loss
• maximum tolerated dose (MTD), which usually applies to long-term studies and represents the largest dose tested that caused no obvious signs of ill-health
• no observed effect level (NOEL), which represents the threshold for producing any observed pharmacological or toxic effect.
The estimated NTEL in the most sensitive species is normally used to determine the starting dose used in the first human trials. The safety factor applied may vary from 100 to 1000, depending on the information available, the type and severity of toxicities observed in animals, and whether these anticipated toxicities can be monitored by non-invasive techniques in man.
Variability in responses
The response to a given dose of a drug is likely to vary when it is given to different individuals or even to the same individual on different occasions. Factors such as age, sex, disease state, degree of nutrition/malnutrition, co-administration of other drugs and genetic variations may influence drug response and toxicity. As elderly people have reduced renal and hepatic function they may metabolize and excrete drugs more slowly, and, therefore, may require lower doses of medication than younger people. In addition, because of multiple illnesses elderly people often may be less able than younger adults to tolerate minor side effects. Likewise, children cannot be regarded as undersized adults, and drug dosages relative to body weight may be quite different. Drug distribution is also different between premature infants and children. The dosages of drugs for children are usually calculated on the basis of weight (mg/kg) or on the basis of body surface area (mg/m2). These important aspects of drug safety cannot be reliably assessed from preclinical data, but the major variability factors need to be identified and addressed as part of the clinical trials programme.
Conclusions and future trends
No drug is completely non-toxic or safe2. Adverse effects can range from minor reactions such as dizziness or skin reactions, to serious and even fatal effects such as anaphylactic reactions. The aims of preclinical toxicology are (a) to reduce to a minimum the risk to the healthy volunteers and the patients to whom the drug will be given in clinical trials; and (b) to ensure that the risk in patients treated with the drug once it is on the market is commensurate with the benefits. The latter is also a major concern during clinical development, and beyond in Phase IV.
It is important to understand the margin of safety that exists between the dose needed for the desired effect and the dose that produces unwanted and possibly dangerous side effects. But the extrapolation from animal toxicology to safety in man is difficult because of the differences between species in terms of physiology, pathology and drug metabolism.
Data from preclinical toxicity studies may be sufficiently discouraging that the project is stopped at that stage. If the project goes ahead, the preclinical toxicology data provide a basis for determining starting doses and dosing regimens for the initial clinical trials, and for identifying likely target organs and surrogate markers of potential toxicity in humans. Two particular trends in preclinical toxicology are noteworthy:
• Regulatory requirements tend to become increasingly stringent and toxicology testing more complex as new mechanisms of potential toxicity emerge. This has been one cause, over the years, of the steady increase in the cost and duration of drug development (see Chapter 22). Only in the last 5–10 years have serious efforts been made to counter this trend, driven by the realization that innovation and therapeutic advances are being seriously slowed down, to the detriment, rather than the benefit, of human healthcare. The urgent need for effective drugs against AIDS was the main impetus for this change.
• In an effort to reduce the time and cost of testing, and to eliminate development compounds as early as possible, early screening methods are being increasingly developed and applied during the drug discovery phase of the project, with the aim of reducing the probability of later failure. One such approach is the use of cDNA microarray methods (see Chapter 7) to monitor changes in gene expression resulting from the application of the test compound to tissues or cells in culture. Over- or underexpression of certain genes is frequently associated with the occurrence of specific toxic effects, such as liver damage (Nuwaysir et al., 1999; Pennie, 2000), so that detecting such a change produced by a novel compound makes it likely that the compound will prove toxic, thereby ruling it out as a potential development candidate. Such high-throughput genomics-based approaches enable large databases of gene expression information to be built up, covering a diverse range of chemical structures. The expectation is that the structure–activity patterns thus revealed will enable the prediction in silico of the likely toxicity of a wide range of hypothetical compounds, enabling exclusion criteria to be applied very early in the discovery process. This field of endeavour, dubbed ‘toxicogenomics’, is expected by many to revolutionize pharmaceutical toxicology (Castle et al., 2002).
At present, extensive toxicity testing in vivo is required by regulatory authorities, and data from in vitro studies carry little weight. There is no likelihood that this will change in the near future (Snodin, 2002). What is clearly changing is the increasing use of in vitro toxicology screens early in the course of a drug discovery programme to reduce the risk of toxicological failures later (see Chapter 10). New guidelines have recently (2009) been introduced to avoid delays in the evaluation of anticancer drugs (S9 ICH). The main impact of new technologies will therefore be – if the prophets are correct – to reduce the attrition rate in development, not necessarily to make drug development faster or cheaper.
Baldrick P. Toxicokinetics in preclinical evaluation. Drug Discovery Today. 2003;8:127–133.
Castle AL, Carver MP, Mendrick DL. Toxicogenomics: a new revolution in drug safety. Drug Discovery Today. 2002;7:728–736.
Committee for Proprietary Medicinal Products. Points to consider for the assessment of the potential QT prolongation by non-cardiovascular medicinal products. Publication CPMP 986/96. London: Human Medicines Evaluation Unit; 1997.
De Clerck F, Van de Water A, D’Aubiol J, et al. In vivo measurement of QT prolongation, dispersion and arrhythmogenesis: application to the preclinical cardiovascular safety pharmacology of a new chemical entity. Fundamental and Clinical Pharmacology. 2002;16:125–140.
FDA Guidance Note. Cell therapy and gene therapy products. http://www.fda.gov/cber/gdlns/somgene.pdf, 1998.
FDA Guidance Note. Immunotoxicology evaluation of investigational new drugs. www.fda.gov/guidance/index.htm, 2002.
Gad SC. Drug safety evaluation. New York: Wiley Interscience; 2002.
Haverkamp W, Breitlandt G, Comm AJ, et al. The potential for QT prolongation and proarrhythmias by non-anti-arrhythmic drugs: clinical and regulatory implications. European Heart Journal. 2000;21:1232–1237.
ICH Guideline S1A: Guideline on the need for carcinogenicity studies of pharmaceuticals. www.ich.org.
ICH Guideline S1B: Testing for carcinogenicity of pharmaceuticals. www.ich.org.
ICH Guideline S1C: Dose selection for carcinogenicity studies of pharmaceuticals and S1C(R): Addendum: addition of a limit dose and related notes. www.ich.org.
ICH Guideline S2A: Genotoxicity: guidance on specific aspects of regulatory tests for pharmaceuticals. www.ich.org.
ICH Guideline S2B: Genotoxicity: a standard battery for genotoxicity testing for pharmaceuticals. www.ich.org.
ICH Guideline S3A: Note for guidance on toxicokinetics: the assessment of systemic exposure in toxicity studies. www.ich.org.
ICH Guideline S5A: Detection of toxicity to reproduction for medicinal products. www.ich.org.
ICH Guideline S6: Preclinical safety evaluation of biotechnology-derived pharmaceuticals. www.ich.org.
2011 ICH Guideline 6 – Addendum. www.ich.org, 2011.
ICH Guideline S7A: Safety pharmacology studies for human pharmaceuticals. www.ich.org.
ICH Guideline S7B: Safety pharmacology studies for assessing the potential for delayed ventricular repolarization (QT interval prolongation) by human pharmaceuticals. www.ich.org.
ICH Guideline S9: Non clinical evaluation for anticancer pharmaceuticals. www.ich.org.
Netzer R, Ebneth E, Bischoff U, et al. Screening lead compounds for QT interval prolongation. Drug Discovery Today. 2001;6:78–84.
Nuwaysir EF, Bittner M, Trent J, et al. Microassays and toxicology; the advent of toxicogenomics. Molecular Carcinogenesis. 1999;24:152–159.
Olson H, Betton G, Robinson D, et al. Concordance of the toxicity of pharmaceuticals in humans and animals. Regulatory Toxicology and. Pharmacology. 2000;32:56–67.
Pennie WD. Use of cDNA microassays to probe and understand the toxicological consequences of altered gene expression. Toxicology Letters. 2000;112:473–477.
Snodin DJ. An EU perspective on the use of in vitro methods in regulatory pharmaceutical toxicology. Toxicology Letters. 2002;127:161–168.
Van der Laan JW. Dose selection for carcinogenicity studies of pharmaceuticals. www.ich.org, 2008.