6 Bacterial genetics
Key points
• The properties of a bacterial cell are defined by the information encoded in its double-stranded DNA genetic complement and its interaction with the environment. The single circular chromosome that, in most cases, comprises the genome, carries most of this, but much additional information resides within extrachromosomal elements known as plasmids.
• Plasmids and several other mobile genetic elements, including bacteriophages, transposons and other integron-based entities, render the bacterial genetic complement highly susceptible to variation by the addition of new genes. Such elements are often organized into pathogenicity islands or resistance islands and may affect the medical significance of bacterial strains when the genes they encode affect antibiotic susceptibility or virulence.
• Mobile genetic elements are transferred between related bacteria through the processes of transformation, conjugation and transduction.
• Bacterial genomes are also susceptible to change through mutation, in which the primary nucleotide sequence of one or more genes or regulatory elements is altered. Because bacterial replication produces very large cell numbers, the possibilities for generation and survival of advantageous mutations (commonly at frequencies of 10−7–10−10), such as those conferring resistance to antibiotics, are of practical significance.
• These forms of genotypic variation should be distinguished from phenotypic variation, as the former are heritable whereas the latter are not, and reflect changes in gene expression and, in the case of phase variation, genetic rearrangements leading to altered expression patterns without changing the cell’s genetic complement.
• Knowledge of the genetic complement of bacteria enables their specific detection, typing and recognition of key aspects of genotypic variation by the application of specific gene (hybridization) probes and by the use of targeted DNA amplification technologies such as the polymerase chain reaction (PCR). The possibility of rapid detection (hours) of many different targets has been enhanced by the development of real-time PCR.
Genetic organization and regulation of the bacterial cell
All properties of a bacterial cell, including those of medical importance such as virulence, pathogenicity and antibiotic resistance, are determined ultimately by the genetic information contained within the cell genome. This information is normally encoded by the specific sequence of nucleotide bases comprising the DNA of the cell. There are four common nucleotide bases in DNA: adenine, guanine, cytosine and thymine; it is the linear order in which these bases are arranged that determines the properties of the cell. With only a few exceptions, most of the genetic information required by the bacterial cell is arranged in the form of a single circular double-stranded chromosome. It is worth noting that the main bacterial chromosome is not a completely stable structure, but can exhibit quite dynamic reorganization following the insertion or deletion of transposons, integrons and genomic islands (see below) under fluctuating environmental and selective conditions. In Escherichia coli the chromosome is about 1300 µm long and occurs in an irregular coiled bundle lying free in the cytoplasm. The DNA is not associated with histone proteins as it is in eukaryotic cell chromosomes, although there are many DNA binding proteins involved in regulation of gene expression, including several proteins referred to as histone-like.
In addition to the single main chromosome, bacterial cells may also carry one or more small circular extrachromosomal elements termed plasmids. Plasmids replicate independently of the main chromosome in the cell. Although dispensable, they often carry supplementary genetic information coding for beneficial properties (e.g. resistance to antibiotics) that enable the host cell to survive under a particular set of environmental conditions.
A third source of genetic information in a bacterial cell can be provided by the presence of certain types of bacterial virus, bacteriophages. Bacteriophages consist essentially of just a protein coat enclosing the virus genome and, because they are unable to multiply in the absence of their bacterial host, generally they are lethal to their host cell. In some instances, they can enter a potentially long-term state of controlled replication, lysogeny, within the bacterial cell without causing lysis. In such a state the bacteriophage genome is referred to as a prophage and effectively becomes a temporary part of the total genetic information available to the cell and may consequently bestow additional properties on the cell.
Not all genetic material consists of double-stranded DNA. Some bacteriophages contain single-stranded molecules of either DNA or RNA, which can be either circular or linear in configuration. The genetic elements under consideration here, including bacteriophages, plasmids and members of the domain Bacteria, vary in size by more than three orders of magnitude, from about 3–5000 kilobases (kb) (Table 6.1).
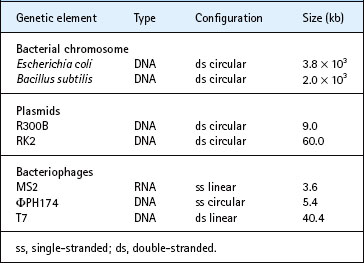
Processes leading to protein synthesis
The character of a bacterial cell is determined essentially by the specific polypeptides that comprise its enzymes and other proteins. The DNA acts as a template for the transcription of RNA by RNA polymerase for subsequent protein production within the cell. In the transcription process the specific sequence of nucleotides in the DNA determines the corresponding sequence of nucleotides in the messenger RNA (mRNA). This, in turn, is then translated into the appropriate sequence of amino acids by ribosomes. Finally, the sequence of amino acids in the resulting polypeptide chain determines the configuration into which the polypeptide chain folds itself, which in many cases determines the enzymic properties of the completed protein. A segment of DNA that specifies the production of a particular polypeptide chain is called a gene, and the processes of transcription and translation leading to protein synthesis are collectively termed the central dogma of molecular biology. These processes are illustrated schematically in Figure 6.1.
Gene regulation
Most bacteria contain enough DNA to code for the production of between 1000 and 3000 different polypeptide chains – 1000 to 3000 different genes. However, during normal bacterial life, some polypeptides will be required only at particular stages, whereas others will be needed only when the cell is provided with a new or unusual growth substrate, or is confronted with a new challenge, such as an antibiotic. Thus, many antibiotic resistance mechanisms found in bacteria are inducible (see below). Protein production is an energy-intensive process, and therefore the expression of many genes is controlled actively within the cell to prevent wasteful energy consumption.
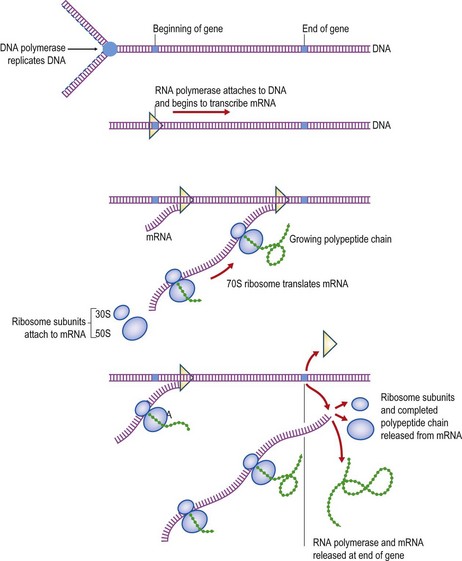
In bacteria the process of gene expression is regulated mainly at the transcriptional level, thereby conserving the energy supply and the transcription–translation apparatus. This is achieved by means of regulatory elements that either inhibit or enhance the rate of RNA chain initiation and termination for a particular gene. Numerous complex regulatory chain mechanisms are involved in coordinating the many biochemical reactions that proceed inside a cell, but related genes involved in a common regulatory system are often clustered on the bacterial chromosome. Such functional clusters are known as operons, of which the most well known example is the lactose operon of E. coli (Fig. 6.2).
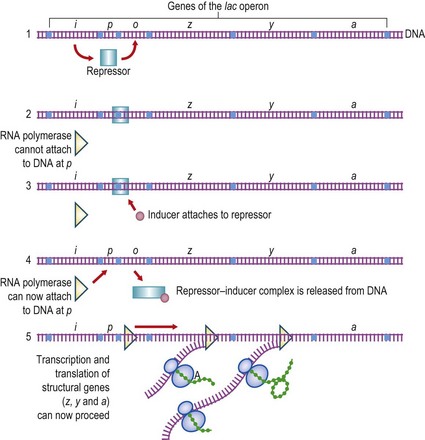
Fig. 6.2 The lac operon of Escherichia coli. (1) The lac repressor is produced from the i gene. (2) Binding of the repressor to the operator site (o) prevents transcription of the genes z (β-galactosidase), y (galactoside permease) and a (transacetylase). (3) The inducer (lactose, or a closely related derivative) can bind specifically to the repressor. (4) The repressor molecule is thereby altered at its operator-binding site and the repressor–inducer complex is released from the DNA. (5) RNA polymerase can now attach to the promoter site (p) and transcribe the structural genes of the lac operon. Note that, in bacteria, several genes may be transcribed into a single polycistronic mRNA molecule.
For transcription to occur as the first stage in protein synthesis, RNA polymerase has to attach to DNA at a specific promoter region and transcribe the DNA in a fixed direction. This process can be switched off by the attachment of a repressor molecule to a specific region of the DNA, known as the operator. This lies between the promoter and the structural gene(s) being transcribed; the repressor then blocks the movement of the RNA polymerase molecule so that the genes downstream are not transcribed.
The repressor is often an allosteric molecule with two active sites. One recognizes the operator region so that the repressor can bind to it to prevent transcription. The other recognizes an inducer molecule. When the inducer is present, it binds to the repressor and alters simultaneously the binding specificity at the other site, so that the repressor no longer binds to the operator and transcription can resume.
There are many different variations on this basic regulatory system. For example, a repressor may be normally inactive, but activated by the end-product of a biosynthetic pathway; thus, only when the end-product is present in adequate concentration will the repressor combine with the operator and switch off transcription of the operon. Alternatively, regulation of certain operons involves proteins that bind to the DNA and assist RNA polymerase to initiate transcription. These are just a few examples, and other regulatory systems display both minor and major differences. Finally, it should be stressed that prokaryotic gene regulation frequently involves interwoven regulatory circuits that respond to a variety of different stimuli. As noted in Chapter 4, these networks of genes are organized into functional units known as stimulons when they respond to a particular stimulus and regulons when they are regulated by a single protein.
Mutation
As bacteria reproduce by asexual binary fission, the genome is normally identical in all of the progeny. However, one of the fundamental requirements for evolution is that, although gene replication must normally be completely accurate to ensure stability, there must also be occasional variation to produce new or altered characters that might be of selective value to the organism. Rare inaccuracies in the DNA replication process produce a slightly altered nucleotide sequence in one of the progeny cells. Such a mutation is heritable and will be passed on stably to subsequent generations. Mutations may not produce any observable effect on the structure or function of the corresponding protein, but in a small proportion of cases an enzyme with altered specificity for substrates, inhibitors or regulatory molecules may be produced. This is the kind of mutation that is most likely to be of evolutionary value to an organism; indeed, many examples of acquired antibiotic resistance have been shown to be of this type. Other mutations may alter a gene so that a non-functional protein is formed; if this protein is essential to the cell then the mutation will be lethal.
As a mutation may occur in any one of the several thousand genes of the cell, and different mutations in the same gene may produce different effects in the cell, the number of possible mutations is very large. Particular mutations occur at fairly constant rates, normally between one per 104 and one per 1010 cell divisions. As a large bacterial colony contains at least 109 cells, even a ‘pure’ bacterial culture will contain many thousands of different mutations affecting many of the genes in the cell. Some of these mutations will be viable and could be selected by particular environmental conditions during subculture. For the same reason, in an infected patient, a variety of mutants will appear spontaneously in the population that grows from the few bacteria originally entering the body. Such mutations may enhance the ability of an organism to cause infection, for example by conferring antibiotic resistance, enhanced virulence or altered surface antigens. In such a situation, cells with the mutation will rapidly outgrow cells without the mutation, so that selection of the mutant cells occurs and they soon become the predominant type.
Phenotypic variation
The properties of a bacterial cell at a particular time are referred to as the phenotype of the cell. These properties are determined not only by its genome (genotype), but also by its environment. Phenotypic variation occurs when the expression of genes is changed in response to the environment, for instance by the induction or repression of synthesis of particular enzymes. Such changes in gene expression underpin the differentiation process involved in sporulation (see p. 19), the differing phenotypes observed during different growth phases and under different growth conditions (p. 40) and the physiological adaptive responses of bacteria (p. 46). The distinction between genotypic mutation and phenotypic variation is critical; the former is heritable and maintained through changes in environmental conditions, whereas the latter is reversible, being dependent on environmental conditions and altering when these change. Phenotypic variation is therefore not a form of mutation.
Types of mutation
Mutations can be divided conveniently into multisite mutations, involving extensive chromosomal rearrangements such as inversions, duplications and deletions, and point mutations, which are defined as only affecting one, or very few, nucleotides. The structure of DNA is such that point mutations can be divided into one of three basic types (Fig. 6.3):
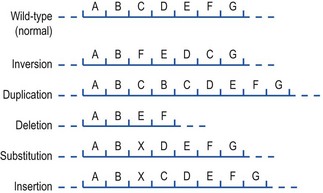
Fig. 6.3 Examples of types of mutation. The top sequence represents a portion of the wild-type chromosome from which the different mutational rearrangements shown below are derived.
Mutations occur spontaneously during replication of DNA, but most are corrected immediately by the editing apparatus of the cell. Occasional mutations, particularly those conferring a selective advantage to the cell, will be inherited stably by the progeny, but secondary mutations can occasionally restore the original nucleotide sequence. It is important to distinguish this relatively rare event of back-mutation from the separate process of phase variation, which is readily reversible and occurs with relatively high frequency in either direction, for example once per 103 cell divisions. The variation of certain Gram-negative bacteria between a fimbriate and a non-fimbriate phase, and the variation of flagellar antigens in Salmonella enterica serotypes, are examples of phase variation. These seem to involve special genetic regions that are specifically inverted to yield alternative gene products and different phenotypes. In some cases it is known that promoters initiate RNA transcription in different directions to give a flip-flop type of action. The number of such switching systems is probably quite limited, but they have value to the organism in providing a mechanism to switch to a reversible alternative, as opposed to an irreversible change.
Mutations may occur in any gene, but many individual mutations are lethal. Other mutations affect gene products that are essential only under particular cultural conditions. The detailed function and regulation of processes in the bacterial cell can often be analyzed only by searching systematically for mutations that affect each separate step in a process. Thus, mutations can be found that produce increased resistance to almost any antimicrobial agent, and the study of such mutants is an essential step in understanding the modes of action of antibiotic agents and mechanisms of resistance to them.
Gene transfer
A change in the genome of a bacterial cell may be caused either by a mutation in the DNA of the cell or result from the acquisition of additional DNA from an external source. DNA may be transferred between bacteria by three mechanisms:
Each of these mechanisms probably occurs at a low frequency in nature and may therefore be of value in bacterial evolution. It is important to note that acquisition by bacteria of new properties following gene transfer or mutation is only significant if the new genetic end-product is subject to favourable selection by the conditions under which the bacteria are growing.
Transformation
Most species of bacteria are unable to take up exogenous DNA from the environment; indeed, most bacteria produce nucleases that recognize and break down foreign DNA. However, bacteria in some genera, notably pneumococci, Haemophilus influenzae and certain Bacillus species, have been shown to be capable of taking up DNA either extracted artificially or released by lysis from cells of another strain. Cells are competent for transformation only under certain conditions of growth, usually in late log phase or, in Bacillus species, during sporulation. However, bacterial geneticists have also developed treatments by means of which organisms can be made artificially competent.
Once a piece of DNA has entered the cell by transformation, it has to become incorporated into the existing chromosome of the cell by a process of recombination in order to survive. This is a complex molecular process for which the transformed DNA must have been derived from a closely related strain, as pieces of DNA can normally recombine with the chromosome only when there is a high degree of nucleic acid similarity (homology).
Any gene may be transferred by transformation, as any fragment of a donor chromosome may be taken up by the recipient cells. However, a piece of DNA introduced into a cell by transformation will normally be relatively short, and will contain only a very small number of genes. For this reason, transformation is of limited use for studying the organization of genes in relation to one another (genetic mapping; see below).
Conjugation
Conjugation is a process by which one cell, the donor or male cell, makes contact with another, the recipient or female cell, and DNA is transferred directly from the donor into the recipient. Certain types of plasmids carry the genetic information necessary for conjugation to occur. Only cells that contain such a plasmid can act as donors; those lacking a corresponding plasmid act as recipients.
Transfer of DNA between cells by conjugation requires direct contact between donor and recipient cells. Plasmids capable of mediating conjugation carry genes coding for the production of a 1–2-µm long protein appendage, termed a pilus, on the surface of the donor cell. The tip of the pilus attaches to the surface of a recipient cell and holds the two cells together so that DNA can pass into the recipient cell. It is probable, but not absolutely certain, that transfer actually occurs through the pilus; alternatively, the pilus could act simply as a mechanism by which the donor and recipient cells are drawn together. Different types of pilus are specified by different types of plasmid and can therefore be used as an aid to plasmid classification.
In the vast majority of cases, the only DNA transferred during the conjugation process is the plasmid that mediates the process. It is thought that one strand of the circular DNA of the plasmid is nicked open at a specific site and the free end is passed into the recipient cell. The DNA is replicated during transfer so that each cell receives a copy. As donor ability is dependent upon having a copy of the plasmid, the recipient strain becomes converted into a donor, able to conjugate with further recipients and convert them in turn. In this way a plasmid may spread rapidly through a whole population of recipient cells; this process is sometimes described as infectious spread of a plasmid.
Mobilization of chromosomal genes by conjugation
Many different types of plasmid have the ability to transfer themselves. Some (but not all) plasmids also have the ability to mobilize the chromosomal genes of bacteria. The first reported plasmid of this type was the ‘F factor’ (fertility factor) of E. coli. Cells that contain the F plasmid free in the cytoplasm (F+ cells) have no unusual characteristics apart from the ability to produce F pili and to transfer the F plasmid to F− cells by conjugation. In a very small proportion of F+ cells, the F plasmid becomes inserted into the bacterial chromosome. Once inserted, the entire chromosome behaves like an enormous F plasmid, and hence chromosomal genes can be transferred in the normal manner to a recipient cell at a relatively high frequency. Such cultures are termed high-frequency recombination (Hfr) strains.
It is important to emphasize that the F plasmid system is confined to E. coli and other closely related enteric bacteria. However, many other plasmids are capable of mediating conjugation, and sometimes chromosome mobilization, not only in E. coli but also in other bacteria. For example, plasmid RP4 and its relatives have been used to mediate conjugation in a wide range of Gram-negative bacteria, and there have been reports of conjugation systems in Gram-positive bacteria, such as Enterococcus faecalis, and several Streptomyces species.
Transduction
The third known mechanism of gene transfer in bacteria involves the transfer of DNA between cells by bacteriophages. Most bacteriophages carry their genetic information (the phage genome) as a length of double-stranded DNA coiled up inside a protein coat. Other phages are known in which the phage genome consists of single-stranded DNA or RNA but, as far as is known, transducing phages all contain double-stranded DNA. Two major types of transduction are known to occur in bacteria: generalized and specialized transduction. In both types, bacterial genes are occasionally and accidentally incorporated into new phage particles. When such a phage particle subsequently infects a second bacterial cell, the DNA that enters the cell includes a short segment of chromosome from the original host. Bacterial genes have been transduced by the phage into the second cell. Genes can be transduced only between fairly closely related strains, as particular phages usually attack only a limited range of bacteria. As well as chromosomal genes, transducing bacteriophages may also pick up and transfer plasmid DNA. As an example, the penicillinase gene in staphylococci is usually located on a plasmid, and it may be transferred into other staphylococcal strains by transduction.
Lysogenic conversion
The presence of prophage DNA constitutes a genetic alteration to the host cell. Usually only the phage repressor gene is expressed, but in certain cases it can be demonstrated that other genes are also expressed by the host cell. For example, Corynebacterium diphtheriae produces diphtheria toxin only when it is lysogenized by β phage; the toxin is specified by one of the phage genes. This process is termed lysogenic conversion. It is probable that the production of many toxins by staphylococci, streptococci and clostridia is also dependent upon lysogenic conversion by specific bacteriophages. In such cases, lysogenic conversion not only gives the cell superinfection immunity, but also actively influences the virulence of the bacterium for humans.
Plasmids
Properties encoded by plasmids
As described above, plasmids are circular extrachromosomal genetic elements that may encode a variety of supplementary genetic information, including the information for self-transfer to other cells by conjugation. Not all plasmids can transfer themselves, but non-conjugative plasmids can be mobilized by other conjugative plasmids present in the same donor cell. Apart from this optional transfer ability, all bacterial plasmids contain the basic genetic information necessary for self-replication and segregation into daughter cells at cell division. Plasmids seem to be ubiquitous in bacteria; many encode genetic information for such properties as resistance to antibiotics, bacteriocin production, resistance to toxic metal ions, production of toxins and other virulence factors, reduced sensitivity to mutagens, or the ability to degrade complex organic molecules.
Plasmid classification
Because of the vast range of plasmids, it is necessary to have a means of classification so that their distribution and epidemiology can be studied. Plasmids can be grouped initially according to the properties that they encode, but other physical and genetical methods are needed to study their spread and distribution.
As all plasmids are relatively small structures that are normally separate from the bacterial cell chromosome, it is possible to isolate them from the chromosome by centrifugation and electrophoresis techniques that allow the sizes of different plasmids to be compared directly. Plasmids of similar size that confer identical phenotypes on host cells can be compared by generating restriction endonuclease fingerprints from purified plasmid DNA. A restriction endonuclease is an enzyme that cuts the DNA molecule at, or near to, a specific nucleotide sequence to produce discrete DNA fragments that can be separated by gel electrophoresis. The pattern (‘fingerprint’) of fragments produced is dependent on the distribution of the specific DNA sequences recognized by the enzyme. Closely related plasmids will produce the same, or very similar, fingerprints, whereas unrelated plasmids will produce different fingerprints. Restriction endonucleases of different specificities may be needed to generate distinctive fingerprints.
An initial genetic test will distinguish groups of plasmids that are self-transmissible from those that are not. Linked with the question of transferability is the question of host range; for example, some groups can be transferred only between members of the enterobacteria, other groups can be transferred from the enteric bacteria to the Pseudomonas family, whereas others can be transferred between almost any Gram-negative bacteria. Similar host range relationships exist among plasmids of Gram-positive bacteria.
Once the host range of a plasmid has been determined, plasmids may be classified by incompatibility testing. This method relies on the fact that closely related plasmids are unable to coexist stably in the same bacterial cell. Plasmids that are sufficiently closely related to interfere with one another’s replication in this manner are said to be incompatible and to belong to the same incompatibility group. In contrast, unrelated plasmids can coexist stably and are therefore said to belong to different incompatibility groups. Recent developments in molecular biology enable the incompatibility group of a plasmid to be determined from the DNA sequence of its replicase (rep) gene as opposed to the original more time-consuming process of testing the stability of pairs of plasmids in a bacterial cell.
A final method for plasmid classification involves the use of specific virulent bacteriophages. All members of the same plasmid incompatibility group produce the same type of pilus for conjugation. For some incompatibility groups, specific virulent bacteriophages have been isolated that will adhere only to the type of pilus produced by that particular group of plasmids. Lysis by such a phage shows that a particular type of pilus is being produced, which in turn allows the identification of the group to which the plasmid contained in the cell belongs.
Plasmid epidemiology and distribution
Some plasmid groups have been identified in many different countries of the world, whereas others have so far been found only in a single bacterial species isolated from a solitary ecological niche. There seem to be two major ways in which plasmids spread:
1. by direct transfer from one bacterium to another in a particular micro-environment
2. by being carried in a particular host from one environment, such as a hospital, to another.
The epidemiological tracing of these pathways requires identification not just of the plasmids involved but also typing of their host bacterial strains (see Ch. 3).
Transposons, integrons and genomic islands
As mentioned above, the main bacterial chromosome can exhibit quite dynamic reorganization following the insertion or deletion of transposons, integrons and genomic islands under fluctuating environmental and selective conditions. Transposons are linear pieces of DNA, often including genes for antibiotic resistance (see below), that can migrate between unrelated plasmids and/or the bacterial chromosome independently of the normal bacterial recombination processes. Integrons form an essential ‘building block’ of many transposons and allow the rapid formation and expression of new combinations of genes in response to selection pressures. A detailed description of the properties of these elements lies outside the scope of this text (see Recommended Reading), but suffice it to say that these elements seem to provide the primary mechanism for ‘foreign’ gene capture and dissemination, certainly among Gram-negative bacteria. It is also now recognized that transposons and integrons (as well as other genes) can often be found inserted into the main bacterial chromosome at specific sites where they form genomic islands of ‘foreign’ genetic material. Depending on the precise genes contained in these islands, they can also be referred to as pathogenicity islands or resistance islands. In some cases, the collection of genes contained in such islands can form a highly mobile structure. For example, highly mobile superantigen-encoding pathogenicity islands can be found in Staph. aureus; these are characterized by a specific set of phage-related functions that enable them to use the phage reproduction cycle for their own transduction across quite large phylogenetic distances.
Genetic mapping
The location of genes with reference to one another and to their respective control regions by mapping techniques is an essential part of genetic analysis. Historically, this required the transfer of genetic material between different mutants, initially by conjugation to locate the approximate position of an unknown gene on the chromosome or a plasmid, followed by fine structure mapping with generalized transduction.
A more modern approach that can be used for all bacterial genera makes use of molecular techniques allowing the sequencing of whole genomes (see Genomics, below), followed if appropriate by the isolation, cloning and nucleotide sequencing of individual genes. Once a particular gene or sequence of DNA has been selected and analyzed, it is possible to label it and use the gene as a probe in hybridization experiments. Alternatively, it is possible to synthesize a small stretch of nucleotides, termed an oligonucleotide, from within the overall gene sequence for use as a probe. Hybridization is a process in which two single strands of nucleic acid come together to form a stable double-stranded molecule. As long as the sequence of bases is complementary on each strand, the two strands will bind together by the formation of hydrogen bonds. Oligonucleotide probes are normally only 10–40 bases in length, and can be synthesized and chemically labelled by automated instruments designed specifically for this purpose. The labelled probe can then be used in hybridization experiments with relatively large fragments (‘fingerprints’) generated from the entire bacterial chromosome with rare-cutting restriction endonucleases. The probe will hybridize only to the chromosomal DNA fragment containing the original gene from which the probe was derived, and will therefore indicate the precise physical location of the original gene in relation to other known genes and control regions. Thus, it is now possible to elucidate the relationship between gene structure and function in the cell at the most fundamental molecular level.
Genetic basis of antibiotic resistance
All of the properties of a micro-organism are determined ultimately by genes located or inserted either on the main chromosome or on plasmids or on lysogenic bacteriophages. With regard to antibiotic resistance, it is important to distinguish between intrinsic and acquired resistance. Intrinsic resistance is dependent upon the natural insusceptibility of an organism. In contrast, acquired resistance involves changes in the DNA content of a cell, such that the cell acquires a phenotype (i.e. antibiotic resistance) that is not inherent in that particular species.
Intrinsic resistance
Organisms that are naturally insensitive to a particular drug will always exist. The most obvious determinant of bacterial response to an antibiotic is the presence or absence of the target for the action of the drug. Thus, polyene antibiotics such as amphotericin B kill fungi by binding tightly to the sterols in the fungal cell membrane and altering the permeability of the fungal cell. As bacterial membranes do not contain sterols they are intrinsically resistant to this class of antibiotics. Similarly, the presence of a permeability barrier provided by the cell envelopes of Gram-negative bacteria is important in determining sensitivity patterns to many antibiotics. Intrinsic resistance is usually predictable in a clinical situation and requires an informed and judicious choice of appropriate antimicrobial therapy.
Acquired resistance
An ongoing problem of antimicrobial chemotherapy has been the appearance of resistance to particular drugs in a normally sensitive microbial population. An organism may lose its sensitivity to an antibiotic during a course of treatment. In some cases the loss of sensitivity may be slight, but often organisms become resistant to clinically achievable concentrations of a drug. Once resistance has appeared, the continuing presence of an antibiotic exerts a selective pressure in favour of the resistant organisms. Three main factors affect the frequency of acquired resistance:
1. the amount of antibiotic that is being used
2. the frequency with which bacteria can undergo spontaneous mutations to resistance
3. the prevalence of plasmids able to transfer resistance from one bacterium to another.
Chromosomal mutations
Random spontaneous mutations occur continuously at a low frequency in all bacterial populations, and some mutations may confer resistance to a particular antibiotic. The rate at which these mutations occur is not normally influenced by presence of the antibiotic, but in the presence of the drug the resistant mutant can survive, grow and eventually become the predominant, or only, member of the population. The degree of resistance conferred by chromosomal mutation depends on the biological consequences of the mutation. With single large-step mutations, the drug target is altered by mutation so that it is totally unable to bind a drug, although it can still carry out its normal biological functions sufficiently well to permit the continued survival of the cell. This type of mutation occurs with nalidixic acid and rifampicin. More commonly, the target is altered so that it can no longer bind a drug as efficiently, although it still has some residual affinity. In such a case a higher concentration of antibiotic would be required to produce the same antimicrobial effect: the minimum inhibitory concentration (MIC) of the antibiotic for the organism would be increased. Once a slightly resistant organism has been produced, additional mutational events – each conferring an additional small degree of resistance – can eventually lead to the production of organisms that are highly resistant. This is called the multistep pattern of resistance. Spontaneous chromosomal mutation is of clinical importance in tuberculosis, in which mutants resistant to any single drug (e.g. streptomycin, rifampicin or isoniazid) are likely to be present in the patient before the start of treatment. If only one drug is given to the patient, the few resistant mutant bacteria will multiply and eventually cause a relapse of the disease. Combined therapy with several drugs to which the organism is sensitive is used in the treatment of tuberculosis, so that each drug kills the few mutants that are resistant to the other. The frequency with which double or triple mutations occur spontaneously in the same cell is so low as to be clinically insignificant.
There are many other examples of chromosomal mutations to antibiotic resistance that have assumed clinical importance. Bacterial enzymes called β-lactamases are commonly responsible for resistance to penicillins, cephalosporins and related antibiotics that contain a β-lactam ring (see Ch. 5). Mutations in the genes controlling the production of chromosomally encoded β-lactamases in Gram-negative bacteria can result in overproduction of these enzymes and consequent resistance to the cephalosporin antibiotics normally regarded as stable to β-lactamase.
Chromosomal mutations leading to antibiotic resistance are in many cases just as important clinically as the types of transferable resistance described in the next section.
Transferable antibiotic resistance
Of the three modes of gene transfer in bacteria, plasmid-mediated conjugation is of greatest significance in terms of drug resistance. Plasmids conferring resistance to one or more unrelated groups of antibiotics (R plasmids) can be transferred rapidly by conjugation throughout the population.
R plasmids were first demonstrated in Japan in 1959, when it was shown that resistance to several antibiotics could be transferred by conjugation between strains of Shigella and E. coli. Many surveys since then in all parts of the world have shown that R plasmids are common and widespread.
The way in which R plasmids are built up in vivo probably varies from case to case, but it is clear that simple transfer factors can pick up resistance genes and combine them with non-transmissible resistance plasmids to produce complex transmissible R plasmids that encode resistance to as many as eight or more different antimicrobial drugs. This process of plasmid evolution is accelerated considerably by the involvement of transposons and integrons (see above). R plasmids can transfer themselves into a wide range of commensal and pathogenic bacteria. Once resistance to an antibiotic appears in any one of these species, the process of transposition assists the dissemination of the responsible gene between different R plasmids and subsequent distribution to other bacterial species. As described above, integrons form an essential ‘building block’ of many transposons and allow the rapid formation and expression of new combinations of antibiotic resistance genes in response to selection pressures. These, in turn, can be assembled into resistance islands that can be found inserted into the bacterial chromosome. For example, antibiotic resistance in several Salm. enterica serovars that cause gastrointestinal disease in humans is caused by the presence in the chromosome of a set of related genomic islands carrying a class 1 integron, which carries the resistance genes. Salmonella genomic island 1 (SGI1), the first island of this type, was found in Salm. enterica serovar Typhimurium DT104 isolates, which are resistant to ampicillin, chloramphenicol, florfenicol, streptomycin, spectinomycin, sulphonamides and tetracycline. Several Salmonella serovars and Proteus mirabilis have since been shown to harbor SGI1 or related islands carrying various sets of resistance genes and some distinct groups have emerged. SGI1 is an integrative mobilizable element and can be transferred experimentally into Escherichia coli.
As the prevalence of multiple-resistance R plasmids carrying transposons and integrons continues to increase, infections caused by a wide range of pathogens become more difficult to treat. In addition, R plasmids can also carry genes, for example for toxin production, that confer increased virulence on a bacterial cell. Thus, use of antibiotics may select for bacteria carrying plasmids that confer not only multiple drug resistance but also increased pathogenicity.
Control of antibiotic resistance
The major cause of the spread of genes conferring antibiotic resistance is the selection pressure brought about by the increased, and often indiscriminate, use of antibiotics in man and animals. Plasmid-encoded drug resistance is increased by the widespread use of antibiotics in animal husbandry, where antibiotics are used as animal feed supplements and whole animal populations may be treated, rather than an individual subject as occurs in medical practice. When R plasmids are present, the mass use of antibiotics fails to prevent the spread of resistance and selects R plasmids in the gut microbiota of the whole population of animals. Such R plasmids, evolved in farm animals, have the potential to spread to human commensal bacteria, followed by transfer to more important human pathogens.
It is important to minimize the use of antibiotics as much as possible and to reduce the chance of cross-infection. Rational use of antibiotics and sensible restriction of their availability in man and animals could prevent further spread of R plasmids and perhaps reduce their incidence. Some R plasmids are unstable and tend to lose resistance genes when the selection pressure is removed. R plasmids are also lost spontaneously from a small proportion of cells in a culture, as plasmid replication and segregation are not always precisely synchronous with chromosome replication and segregation. Cells that lose an R plasmid may have a slight metabolic advantage and may slowly outgrow drug-resistant organisms. Moreover, R plasmids that evolve in one species may be unstable in another or may transfer themselves to other organisms much less efficiently. Similarly, organisms that are adapted to the gut of a calf, pig or chicken may not establish readily in human beings. Such factors may help to contain the spread of R plasmids.
Applications of molecular genetics
Specific gene probes
Every properly classified species must, by definition, have somewhere on its chromosome a unique DNA sequence that distinguishes it from every other species. If this sequence can be identified, a specific labelled DNA probe (see above) can be used in a hybridization reaction to recognize pathogen-specific DNA released from clinical samples. Initial isolation of the infecting pathogen is not necessary and, consequently, DNA probes can be used to detect pathogens that cannot be cultured easily in vitro.
DNA probes have already been used successfully to identify a wide variety of pathogens, from simple viruses to pathogenic bacteria and parasites. Probes have also been developed that can recognize specific antibiotic resistance genes, so that antimicrobial susceptibility of an infecting organism can be determined directly without primary isolation and growth. Commercial kits incorporating DNA probes are now available to detect a range of bacteria and viruses.
Nucleic acid amplification technology
In the polymerase chain reaction (PCR) a thermostable DNA polymerase and two specific oligonucleotide primers are used to produce multiple copies of specific nucleic acid regions quickly and exponentially (Fig. 6.4). The specificity of the reaction is controlled by the oligonucleotide primers that direct replication of the intervening ‘target’ region. In an exponential reaction, the target sequence is amplified a million-fold or more within a few hours. Although PCR is the most widely used method, other amplification techniques for DNA and RNA molecules are available. Once an amplification reaction has occurred, a variety of manual and automated methods is available to detect the amplified product, of which the simplest is to identify the product by size after electrophoresis and migration on an agarose gel. For many diagnostic applications, the simple visualization of an amplification product of characteristic size is a significant result, because it indicates the presence of the target DNA sequence in the original sample, but confirmation of sequence identity by specific hybridization tests is often required.
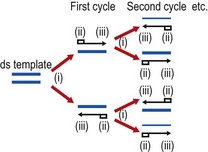
Fig. 6.4 Schematic outline of the polymerase chain reaction (PCR). Each cycle in the exponential reaction consists of three steps: (i) heat denaturation, typically at 94°C, to dissociate double-stranded (ds) DNA; (ii) annealing of primers ( ) at a temperature determined empirically for each individual PCR; (iii) elongation at an optimal temperature for thermostable polymerase activity, typically 72°C for Taq polymerase.
) at a temperature determined empirically for each individual PCR; (iii) elongation at an optimal temperature for thermostable polymerase activity, typically 72°C for Taq polymerase.
Amplification offers an exquisitely sensitive approach to the detection and identification of specific microorganisms in a variety of sample types. Potentially, a characteristic DNA or RNA sequence from a single virus particle or bacterial cell can be amplified to detectable levels within a very short period of time. The method has received particular attention for detecting the presence of low numbers of bacteria or virus particles in clinical and environmental specimens. For example, although a diagnostic antibody response may take up to 8 weeks to develop in an individual infected with human immunodeficiency virus (HIV), specific HIV sequences can be detected in a few hours by PCR, even if present at only 1 part per 100 000 human genome equivalents.
Real-time amplification
It is the detection process that discriminates real-time amplification from conventional PCR assays. The possibility of detecting the accumulation of amplification product in real time as the PCR progresses has been made possible by the labelling of primers, oligonucleotide probes (oligoprobes) or amplicons with molecules capable of fluorescing in the reaction tube. These fluorescent labels produce a change in signal at a specific wavelength following direct interaction with, or hybridization to, the amplicon. The signal produced is related to the amount of amplicon present at the end of each cycle and increases as the amount of specific amplicon increases (see Recommended Reading). Commercially available robotic nucleic acid extraction systems, combined with rapid thermal cyclers and instrumentation (e.g. LightCycler®, TaqMan®) capable of detecting and differentiating multiple amplicons, make real-time PCR an attractive and viable proposition for the routine diagnostic laboratory. Real-time PCR assays have been extremely useful for studying microbial agents of infectious disease. The greatest impact to date has been in the field of virology, where real-time assays have been used to rapidly detect a range of viruses in human specimens and to monitor quantitatively viral loads and response to antiviral therapy. Benefits to the patient can also be seen in bacteriology, where rapid detection of bacterial pathogens and/or antibiotic resistance genes can help to ensure the appropriate use of antibiotics, reduce the duration of hospital stay and minimize the potential for resistant strains of bacteria to emerge. Recent developments in real-time PCR have suggested a future in which rapid identification, quantification and typing of a range of microbial targets in single multiplex reactions will become commonplace, although it is not clear whether current resource-limited clinical microbiology services will be enabled to introduce these improved new technologies on a wide scale.
Molecular typing of micro-organisms
Typing of micro-organisms is increasingly important for studying cross-infection and epidemiological relationships, particularly during outbreaks of nosocomial infection (see Ch. 69). Molecular fingerprinting methods are now the most commonly used techniques for assessing the relatedness of individual bacterial isolates in epidemiological studies. These techniques can be used to study any organism from which DNA can be prepared and offer the possibility of a unified approach to microbial typing that can be applied immediately to a new epidemiological problem with no prior knowledge of the organisms being investigated (see Ch. 3).
A complete DNA sequence forms the ultimate reference standard for identifying micro-organisms and their sub types. Increasing numbers of micro-organisms are now being sequenced and the knowledge gained is of immense value for research purposes. However, even with rapid automated sequencing techniques, it is highly unlikely that routine diagnostic laboratories will ever have the facilities or resources to sequence all their isolates of clinical or epidemiological interest routinely. One possibility might involve the sequencing of small relatively conserved regions of the genome that can provide diagnostic information, and such techniques for sequencing genes encoding 16S ribosomal RNA are sometimes used presumptively to identify ‘unknown’ or unculturable isolates derived from clinical specimens.
Recent developments in automation and sequencing chemistries have resulted in the development of a typing technique termed multilocus sequence typing (MLST). The technique relies on the analysis of sequence variation in a relatively small set of ‘housekeeping genes’ (usually about seven) that are present in all isolates of a particular bacterial species. The genes selected for analysis should be widely separated on the chromosome and should not be adjacent to genes that may be under selective pressure. Specific primers are designed that amplify c. 500-bp fragments of these genes, which are then sequenced to determine naturally occurring variation. The sequences can be compared with those already contained in worldwide databases in order to analyse both global and local epidemiology. MLST schemes and databases are already available for more than 40 bacterial species, including Neisseria meningitidis, Streptococcus pneumoniae, Staphylococcus aureus and Haemophilus influenzae. This is a powerful new approach for the characterization of micro-organisms, as it provides unambiguous data that are electronically portable between laboratories and can be used for global epidemiological studies.
Genomics
Entire genome sequences for pathogenic micro-organisms are increasingly becoming available. It is becoming increasingly apparent that many species of bacteria exhibit significant variability among strains in terms of pathogenicity and ecological flexibility. As described above, such differences reflect the dynamic nature of the bacterial genome, which is now considered to be composed of a relatively invariable ‘core genome’ (comprising the basic gene complement forming much of the main chromosome) and a highly variable ‘accessory genome’ (comprising plasmids, bacteriophages, transposons, integrons and genomic islands, some or all of which may also be integrated into the main chromosome). Such structures may play an important role in bacterial evolution. For example, Vibrio cholerae contains a large chromosomal structure with integron characteristics that harbours several hundred gene cassettes, as well as a large second structure which has variously been referred to as a second chromosome, a megaplasmid or a chromid. This second chromosome also carries a gene capture system (termed the integron island) and host ‘addiction’ genes that are typically found on plasmids. It therefore appears that these structures represent a very efficient system by which bacteria can scavenge foreign genes that may confer increased fitness in particular environmental niches.
New methods for obtaining genome-wide mRNA expression data mean that a global view of changes in gene expression patterns in response to physiological alterations or manipulations of transcriptional regulators can also now be obtained. An alternative approach for identification and typing is already available that involves the use of DNA microarrays (sometimes called ‘DNA chips’). These consist of a very large number of evenly spaced spots of DNA fixed to a microscope slide. Each spot is a unique DNA fragment transferred by a gridding robot from multi-well plates on to the slide. Such DNA chips may become commercially available and could then be hybridized in diagnostic laboratories with DNA extracts from ‘unknown’ isolates to yield distinctive patterns of hybridization on the slide. Such patterns would be readily amenable to computerized analysis and comparison with electronic databases. More advanced chips are ‘nanolaboratories’, in that nanotechnology can be used to perform PCR, chromatography, electrophoresis and other techniques on a single chip. Some chips allow multiplexed systems, so that several techniques can be used simultaneously with several samples. Microarrays have already been used to investigate genetic expression, polymorphisms and antibiotic resistance in pathogens such as Mycobacterium tuberculosis, E. coli, S. aureus and Helicobacter pylori, not only in the context of epidemiological research, but also as a rapid diagnostic method for use with critically ill patients. Such techniques are not yet used routinely in diagnostic laboratories, but this is the beginning of a new molecular era for the diagnosis and treatment of infectious diseases, and these approaches offer the possibility of a very important shortcut to early diagnosis and treatment. The fundamental strategy of the current era of genomics is to move from studying single genes to the simultaneous study of a large number of interacting genes and their corresponding proteins. At the time of writing, this technology is still in the process of gradual development for routine use, but may ultimately revolutionize diagnostic microbiology in the twenty-first century.
Dijkshoorn L, Towner KJ, Struelens M. New Approaches for the Generation and Analysis of Microbial Typing Data. Amsterdam: Elsevier, 2001.
Erlich HA. PCR Technology – Principles and Applications of DNA Amplification. New York: Stockton Press; 1989.
Goering RV. The molecular epidemiology of nosocomial infection: past, present and future. Reviews in Medical Microbiology. 2000;11:145–152.
Hall RM. Salmonella genomic islands and antibiotic resistance in Salmonella enterica. Future Microbiology. 2010;5:1525–1538.
Hall RM, Collis CM. Antibiotic resistance in Gram-negative bacteria: the role of gene cassettes and integrons. Drug Resistance Updates. 1998;1:109–119.
Innis MA, Gelfand DH, Sninsky JJ, White T. PCR Protocols – A Guide to Methods and Applications. San Diego: Academic Press; 1990.
Kung VL. The accessory genome of Pseudomonas aeruginosa. Microbiology and Molecular Biology Reviews. 2010;74:621–641.
Mackay IM. Real-time PCR in the microbiology laboratory. Clinical Microbiology and Infection. 2004;10:190–212.
Mazel D, Davies J. Antibiotic resistance in microbes. Cellular and Molecular Life Sciences. 1999;56:742–754.
Novick RP, Christie GE, Penadés JR. The phage-related chromosomal islands of Gram-positive bacteria. Nature Reviews Microbiology. 2010;8:541–551.
Rowe-Magnus DA, Mazel D. Resistance gene capture. Current Opinion in Microbiology. 1999;2:483–488.
Rowe-Magnus DA, Guèrout A-M, Mazel D. Super-integrons. Research in Microbiology. 1999;150:641–651.
Spratt BG. Multilocus sequence typing: molecular typing of bacteria pathogens in an era of rapid DNA sequencing and the internet. Current Opinion in Microbiology. 1999;2:312–316.
Summers DK. The Biology of Plasmids. Oxford: Blackwell; 1996.
Tenover FC. Diagnostic deoxyribonucleic acid probes for infectious diseases. Clinical Microbiology Reviews. 1988;1:82–101.