5 Antimicrobial agents
Key points
• Most antimicrobial agents are active only against bacteria; smaller numbers of antifungal, antiviral, antiprotozoal and anthelminthic agents are available.
• Antimicrobial agents used in clinical medicine do not affect spores of bacteria or fungi, or latent viruses.
• The largest group of antibacterial agents are β-lactam compounds (penicillins, cephalosporins, etc.), most of which are semi-synthetic derivatives of naturally occurring antibiotics. Members of this group have widely different properties.
• Other widely used antibacterial agents include aminoglycosides, tetracyclines, macrolides, glycopeptides and quinolones.
• Most antifungal agents are suitable only for topical application. Agents used for systemic therapy of fungal infection include amphotericin B, certain azole derivatives, griseofulvin and terbinafine.
• The largest groups of antiviral compounds are the antiretroviral (anti-HIV) and the anti-herpes agents, such as aciclovir, that act on nucleic acid synthesis.
• Combinations of three or more antiretroviral drugs can effectively keep HIV levels in the circulation down below the limit of detection.
• Most antiparasitic agents have specific activity against particular protozoa or helminths, although some have broader activity.
• Routine antibiotic sensitivity testing is useful in the case of common bacterial pathogens. The disc diffusion method is commonly used.
Antimicrobial agents are used not only to treat bacterial diseases, but also infections with viruses, fungi, protozoa and helminths. These drugs have transformed the management of infectious disease, but none is free from unwanted side effects, and microbial resistance is a constant threat. Consequently, they must be used with discretion and understanding of their individual properties. The treatment of individual infections is dealt with in the appropriate chapters. The general strategy of antimicrobial chemotherapy, which is crucial to the control of antimicrobial drug resistance, is covered in Chapter 67.
Antibiotics are naturally occurring microbial products; synthetic compounds such as sulphonamides, quinolones, nitrofurans and imidazoles should strictly be referred to as chemotherapeutic agents. However, as some antibiotics can be manufactured synthetically whereas others are the products of chemical manipulation of naturally occurring compounds (semi-synthetic antibiotics), the distinction is ill defined. Nowadays the term antibiotic is used loosely to describe agents (mainly, but not exclusively, antibacterial agents) employed to treat systemic infection. Antimicrobial substances that are too toxic to be used other than in topical therapy or for environmental decontamination are referred to as antiseptics or disinfectants (see Ch. 4).
Antibacterial agents
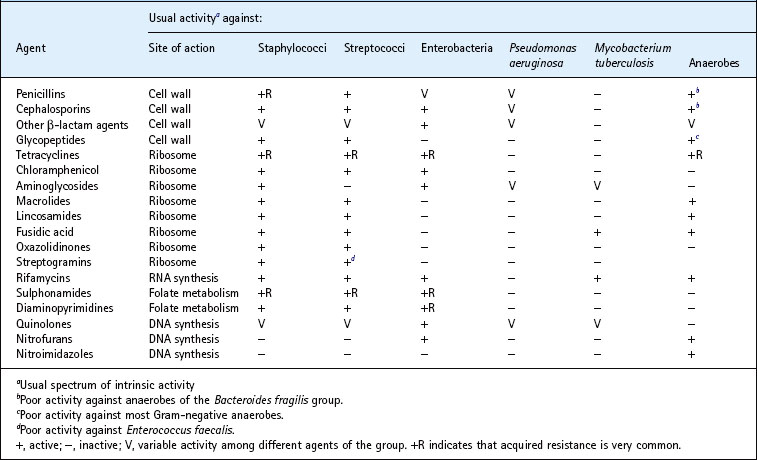
The principal types of antibacterial agent are listed in Table 5.1. Because there are so many, it is convenient to group them according to their site of action.
Table 5.1 Principal types of antibacterial agent (other than agents used exclusively in mycobacterial infection)
Inhibitors of bacterial cell wall synthesis
As most bacteria possess a rigid cell wall that is lacking in mammalian cells (see Ch. 2, p. 13), this structure is a prime target for agents that exhibit selective toxicity, the ability to inhibit or destroy the microbe without harming the host. However, the bacterial cell wall can also prevent access of agents that would otherwise be effective. Thus, the complex outer envelope of Gram-negative bacteria is impermeable to large hydrophilic molecules, which may be prevented from reaching an otherwise susceptible target.
Inhibitors of bacterial cell wall synthesis act on the formation of the peptidoglycan layer (Fig. 5.1). Bacteria that lack peptidoglycan, such as mycoplasmas, are resistant to these agents.
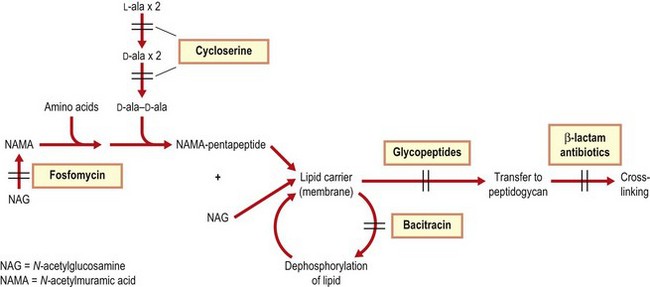
Fig. 5.1 Formation of bacterial cell wall peptidoglycan, showing the sites of action of inhibitors of the process.
β-Lactam agents
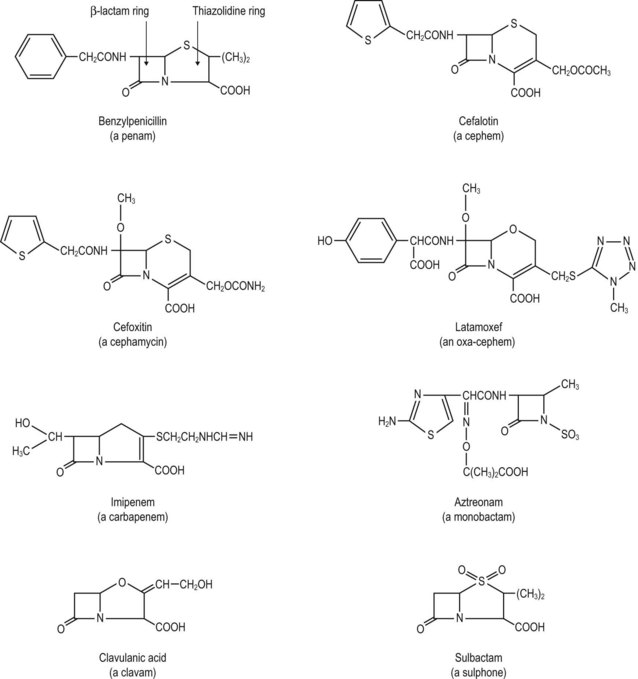
Penicillins, cephalosporins and other compounds that feature a β-lactam ring in their structure fall into this group (Fig. 5.2). All of these compounds bind to proteins situated at the cell wall–cell membrane interface. These penicillin-binding proteins are involved in cell wall construction, including the cross-linking of the peptidoglycan strands that gives the wall its strength. Opening of the β-lactam ring by hydrolytic enzymes, collectively called β-lactamases, abolishes antibacterial activity. Many such enzymes are found in bacteria. Those elaborated by Gram-negative enteric bacilli are particularly diverse in their activity and properties. Most prevalent are the so-called TEM β-lactamases (TEM-1, TEM-2, etc.), numerous forms of which have evolved under selective pressure of β-lactam antibiotic use. Gram-negative bacteria able to produce enzymes that inactivate many different β-lactam antibiotics – so-called extended-spectrum β-lactamases (ESBLs) – sometimes become endemic in hospitals, causing serious problems, especially in patients in high-dependency units.
Penicillins
The original penicillin, benzylpenicillin (penicillin G; often called simply ‘penicillin’) exhibits unrivalled activity against staphylococci, streptococci, neisseriae, spirochaetes and certain other organisms. However, resistance, normally due to the production of β-lactamase, has undermined its activity against staphylococci and, to a lesser extent, gonococci. Bacteria, including staphylococci and pneumococci, that exhibit reduced susceptibility to penicillin by a non-enzymic mechanism are also encountered. Benzylpenicillin revolutionized the treatment of infection caused by some of the most virulent bacterial pathogens, but it also suffers from several shortcomings:
• breakdown by gastric acidity when given orally
• very rapid excretion by the kidney
Further development of the penicillin family has been directed towards improving these properties. Crucial to this was the discovery that removal of the phenylacetic acid side-chain left intact the core structure, 6-aminopenicillanic acid, the starting point for the numerous semi-synthetic penicillins that have been produced. Among the most important penicillins that followed the introduction of benzylpenicillin are:
• phenoxymethylpenicillin (penicillin V), which can be given orally
• procaine penicillin, a long-acting salt of benzylpenicillin
• flucloxacillin, dicloxacillin and oxacillin, compounds resistant to staphylococcal β-lactamase
• ampicillin and amoxicillin, which are active against some enterobacteria
• ticarcillin, azlocillin and piperacillin, which are active against Pseudomonas aeruginosa.
Penicillins other than flucloxacillin and its relatives are inactivated by staphylococcal β-lactamase. Methicillin-resistant Staphylococcus aureus (MRSA; a term that has persisted although methicillin is now virtually obsolete) owe their resistance to alterations in the target penicillin-binding proteins; they are resistant to all penicillins and to all other β-lactam antibiotics with the exception of certain new cephalosporins (see below).
Ampicillin is sometimes administered as an esterified prodrug, a formulation providing improved absorption in the gut, where it releases the active ingredient into the circulation.
Cephalosporins
Cephalosporins are close cousins of the penicillins, but the β-lactam ring is fused to a six-membered dihydrothiazine ring rather than the five-membered thiazolidine ring of penicillins. The additional carbon carries substitutions that may alter the pharmacological behaviour of the molecule, and sometimes its antibacterial activity. Some cephalosporins (e.g. cefalotin [formerly called cephalothin] and cefotaxime) carry an acetoxymethyl group on the extra carbon. This can be removed by hepatic enzymes to yield a less active derivative, but it is doubtful whether this has any therapeutic significance. Other cephalosporins (e.g. cefamandole, cefoperazone and the oxa-cephem, latamoxef) possess a methyltetrazole substituent. Use of compounds with this feature has been associated with hypoprothrombinaemia and bleeding in some patients.
Cephalosporins are generally stable to staphylococcal penicillinase (though they are differentially susceptible to hydrolysis by the various types of enterobacterial β-lactamase), but they lack activity against enterococci. They exhibit a broader spectrum than most penicillins and are less prone to cause hypersensitivity reactions. Among the most important cephalosporins in therapeutic use are:
• cefalexin, cefradine and cefaclor, which can be given orally
• cefuroxime and cefoxitin, which are stable to many β-lactamases
• cefotaxime and ceftriaxone, which combine β-lactamase stability with high intrinsic activity
• cefpodoxine (as the proxetil prodrug) and cefixime, which combine stability to enterobacterial β-lactamases with oral absorption
• ceftazidime and cefpirome, which additionally exhibit good activity against Ps. aeruginosa
• ceftobiprole and ceftaroline, which are active against MRSA.
Other β-lactam agents
Various agents with diverse properties share the structural feature of a β-lactam ring with penicillins and cephalosporins (see Fig. 5.2):
• Carbapenems (e.g. imipenem, meropenem and ertapenem) have a broad spectrum of activity, embracing most Gram-positive and Gram-negative aerobic and anaerobic bacteria. Imipenem is inactivated by a dehydropeptidase in the human kidney, and is co-administered with a dehydropeptidase inhibitor, cilastatin.
• The oxa-cephem latamoxef is a broad-spectrum β-lactamase-stable compound.
• The monobactam, aztreonam, is a monocyclic compound with a spectrum that is restricted to aerobic Gram-negative bacteria.
• The clavam, clavulanic acid, exhibits poor antibacterial activity, but has proved useful as a β-lactamase inhibitor when used in combination with β-lactamase-susceptible compounds such as amoxicillin (co-amoxiclav) and ticarcillin.
• The sulphones, sulbactam and tazobactam, also act as β-lactamase inhibitors and are marketed in combination with ampicillin (or cefoperazone) and piperacillin, respectively.
Glycopeptides
Vancomycin and teicoplanin are large molecules that are unable to penetrate the outer membrane of Gram-negative bacteria, and the spectrum is consequently restricted to Gram-positive organisms. Their chief importance resides in their action against Gram-positive cocci with multiple resistance to other drugs. Enterococci and staphylococci, including MRSA, that exhibit resistance or reduced sensitivity to glycopeptides are being reported more frequently. Other glycopeptides are under development.
Other inhibitors of bacterial cell wall synthesis
• Fosfomycin is a simple phosphonic acid antibiotic used mainly for the treatment of urinary tract infection. Resistance arises readily in vitro.
• Bacitracin is active against Gram-positive bacteria, but is too toxic for systemic use. It is found in many topical preparations, and is also used in the laboratory in the presumptive identification of haemolytic streptococci of Lancefield group A (see pp. 184 and 195).
• Isoniazid and some other compounds used to treat tuberculosis interfere with the formation of the mycolic acids of the mycobacterial cell wall.
• Cycloserine is an analogue of D-alanine used occasionally against multiresistant strains of Mycobacterium tuberculosis.
Inhibitors of bacterial protein synthesis
Bacterial ribosomes are sufficiently different from those of mammalian cells to allow selective inhibition of protein synthesis. Most agents that act at this level are true antibiotics (or derivatives thereof) produced by Streptomyces species or other soil organisms. Some have an effect in eukaryotic cells, which have mitochondrial ribosomes similar to those of bacteria (see p. 13).
Tetracyclines
These are broad-spectrum agents with important activity against chlamydiae, rickettsiae, mycoplasmas and, surprisingly, malaria parasites, as well as most conventional Gram-positive and Gram-negative bacteria. They prevent binding of amino-acyl transfer RNA (tRNA) to the ribosome and inhibit, but do not kill, susceptible bacteria. The various members of the tetracycline group are closely related and differ more in their pharmacological behaviour than in antibacterial activity. Doxycycline and minocycline are in most common use. Resistance has limited the value of tetracyclines against many Gram-positive and Gram-negative bacteria, but rickettsiae, chlamydiae and mycoplasmas are usually susceptible. Tigecycline (a glycylcycline) retains activity against many bacteria resistant to other tetracyclines.
Chloramphenicol
Chloramphenicol also possesses a very broad antibacterial spectrum. It acts by blocking the growth of the peptide chain. Use of chloramphenicol has been limited because of the occurrence of a rare but fatal side effect, aplastic anaemia.
Aminoglycosides
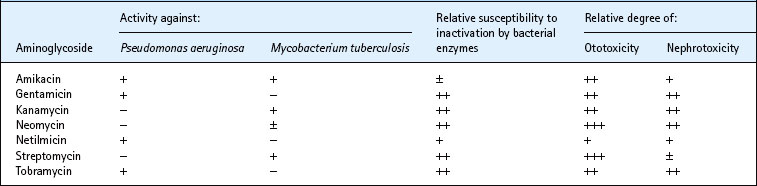
Streptomycin, the first antibiotic to be discovered by random screening of soil organisms, is predominantly active against enterobacteria and M. tuberculosis. Like other members of the aminoglycoside family it has no useful activity against streptococci, anaerobes or intracellular bacteria. The group also has in common a tendency to damage the eighth cranial nerve (ototoxicity) and the kidney (nephrotoxicity).
The chief properties of aminoglycosides are shown in Table 5.2. They inhibit formation of the ribosomal initiation complex and also cause misreading of messenger RNA (mRNA). They are bactericidal compounds, and some, notably gentamicin and tobramycin, exhibit good activity against Ps. aeruginosa. Such compounds have been used widely, often in combination with β-lactam antibiotics, with which they interact synergistically, in the ‘blind’ treatment of sepsis in immunocompromised patients. Resistance may arise from ribosomal changes (streptomycin) or alterations in drug uptake. However, it is more often caused by bacterial enzymes that phosphorylate, acetylate or adenylate exposed amino or hydroxyl groups. Enzymic resistance consequently affects the various aminoglycosides differentially, depending on the possession of exposed groups that can be attacked by the enzyme involved. Amikacin is resistant to most of the common enzymes.
Macrolides
Macrolide antibiotics have a large macrocyclic lactone ring substituted with some unusual sugars. They act by interfering with the translocation of mRNA on the bacterial ribosome. They are used mainly as antistaphylococcal and antistreptococcal agents, though some have wider applications. They have no useful activity against enteric Gram-negative bacilli. The original macrolide, erythromycin, is unstable in gastric acid and is usually administered orally as the stearate salt or as an esterified pro-drug (pharmacological preparations that improve absorption and deliver the active drug into the circulation). Salts suitable for intravenous administration are also available. Certain other macrolides, notably clarithromycin, offer improved pharmacological properties.
The macrolactone ring of most macrolides is composed of 14 atoms, but others have a 16-membered structure. These include spiramycin, which has some useful activity against the protozoan parasite, Toxoplasma gondii.
Some macrolides feature structural changes in the macrocyclic ring: in azithromycin, a compound distinguished by good tissue penetration and a long terminal half-life, the ring has been expanded by inclusion of a nitrogen atom to form an azalide; in telithromycin, a compound that retains activity against macrolide-resistant Gram-positive cocci, a keto function has been introduced to produce a ketolide.
Lincosamides
The original lincosamide antibiotic, lincomycin, has been superseded by a derivative, clindamycin, that is better absorbed after oral administration and is more active against the organisms within its spectrum. These include staphylococci, streptococci and most anaerobic bacteria, against which clindamycin exhibits outstanding activity. Enthusiasm for the use of clindamycin has been tempered by an association with the occasional development of severe diarrhoea, which sometimes progresses to a life-threatening pseudomembranous colitis (see Clostridium difficile, p. 254).
Lincosamides bind to the 50S ribosomal subunit at a site closely related to that at which macrolides act. Inducible resistance to macrolides caused by enzymic modification of the ribosomal binding site also renders the cells resistant to lincosamides (and streptogramins; see below), but only in the presence of macrolides, which alone are able to act as inducers.
Fusidic acid
The structure of fusidic acid is related to that of steroids, but the antibiotic is devoid of steroid-like activity. It blocks factor G, which is involved in peptide elongation. Fusidic acid has an unusual spectrum of activity that includes corynebacteria, nocardia and M. tuberculosis, but the antibiotic is usually regarded simply as an antistaphylococcal agent. It penetrates well into bone and has been used widely (generally in combination with a β-lactam antibiotic to prevent the selection of resistant variants) in the treatment of staphylococcal osteomyelitis.
Linezolid
Linezolid is an oxazolidinone. It is a narrow-spectrum anti-Gram-positive agent which acts by preventing the formation of the ribosomal initiation complex. It is used exclusively against MRSA and other Gram-positive cocci resistant to older agents. There is some evidence that it may be of use in drug-resistant tuberculosis.
Streptogramins
This is the collective name for a family of antibiotics that occur naturally as two synergistic components. They were formerly used mainly in animal husbandry, although one member of the group, pristinamycin, is available in some countries as an antistaphylococcal agent. Use was limited by poor solubility, but derivatives suitable for parenteral administration, quinupristin and dalfopristin, have been developed. The combination exhibits bactericidal activity against most Gram-positive cocci, but has poor activity against Enterococcus faecalis.
Inhibitors of nucleic acid synthesis
A number of important antibacterial agents act directly or indirectly on DNA or RNA synthesis.
Sulphonamides and diaminopyrimidines
These agents affect DNA synthesis because of their role in folic acid metabolism. Folic acid is used in many one-carbon transfers in living cells, including the conversion of deoxyuridine to thymidine. During this process the active form of the vitamin, tetrahydrofolate, is oxidized to dihydrofolate, and this must be reduced before it can function in further reactions.
Sulphonamides are analogues of para-aminobenzoic acid, and prevent the condensation of this compound with dihydropteridine during the formation of folic acid. Diaminopyrimidines, which include the broad-spectrum antibacterial agent trimethoprim and the antimalarial compounds pyrimethamine and cycloguanil (the metabolic product of proguanil), prevent the reduction of dihydrofolate to tetrahydrofolate. Sulphonamides and diaminopyrimidines thus act at sequential stages of the same metabolic pathway and interact synergistically, although in bacterial infections trimethoprim is generally sufficiently effective, and less toxic, when used alone.
Sulphonamides are broad-spectrum antibacterial agents, but resistance is common and the group also suffers from problems of toxicity. The numerous sulphonamides exhibit similar antibacterial activity, but differ widely in their pharmacokinetic behaviour. They have largely been replaced by safer and more active agents, although the combination of sulfamethoxazole with trimethoprim (co-trimoxazole) is still used. Sulfadoxine or sulfadiazine combined with pyrimethamine are used in malaria and toxoplasmosis, respectively.
Quinolones
These drugs act on the α subunit of DNA gyrase. Their properties allow them to be categorized roughly into three groups (Table 5.3). Nalidixic acid and its early congeners are narrow-spectrum agents active only against Gram-negative bacteria. Their use is virtually restricted to urinary tract infection, although they have also been used in enteric infections. Later quinolones, such as ciprofloxacin and ofloxacin, which are 6-fluoro derivatives and are described as fluoroquinolones, display much enhanced activity and a broader spectrum, although activity against some Gram-positive cocci, notably Streptococcus pneumoniae, is unreliable. Continued development has produced compounds that lack the latter defect and, in some cases, exhibit further broadening of the spectrum and improved pharmacokinetic properties.
Table 5.3 Principal types of quinolone antibacterial agent
| Narrow-spectrum compoundsa | Broad-spectrum compoundsb | Compounds with further enhanced spectrumc |
|---|---|---|
a Spectrum restricted to enteric Gram-negative bacilli.
b Improved activity against Pseudomonas aeruginosa and Gram-positive cocci.
c Further improved activity against Gram-positive cocci and some anaerobes.
Quinolones are quite well absorbed when given orally and are widely distributed throughout the body. Extensive metabolization may occur, particularly with nalidixic acid and the older derivatives. Ciprofloxacin and other fluoroquinolones are used widely despite certain problems of toxicity, and resistance is becoming more prevalent.
Nitroimidazoles
Azole derivatives feature prominently among antifungal, antiprotozoal and anthelminthic agents. Those that exhibit antibacterial activity are 5-nitroimidazoles. At low redox (Eh) values they are reduced to a short-lived intermediate that causes DNA strand breakage. Because of the requirement for low Eh values, 5-nitroimidazoles are active only against anaerobic (and certain micro-aerophilic) bacteria and anaerobic protozoa. The representative of the group most commonly used clinically is metronidazole; similar derivatives include tinidazole, and nimorazole.
Nitrofurans
The most familiar nitrofuran is nitrofurantoin, an agent used exclusively in urinary tract infection. Other derivatives, including furazolidone, which is used in enteric infections, are marketed for a variety of purposes in some parts of the world. These compounds probably act on DNA through a reduced metabolite, in a manner analogous to that of the nitroimidazoles.
Rifamycins
This group of antibiotics is characterized by excellent activity against mycobacteria, although other bacteria are also susceptible; staphylococci in particular are exquisitely sensitive. These compounds act by inhibiting transcription of RNA from DNA. Rifampicin, the best known member of the group, is used in tuberculosis and leprosy. Wider use has been discouraged on the grounds that it might inadvertently foster the emergence of resistance in mycobacteria. Rifapentine has similar properties, but exhibits a longer plasma half-life. Rifabutin (ansamycin) is used in infections caused by atypical mycobacteria of the avium-intracellulare group (see Ch. 19).
Miscellaneous antibacterial agents
Polymyxins
Polymyxin B and colistin (polymyxin E) act like cationic detergents to disrupt cell membranes. They exhibit potent antipseudomonal activity, but toxicity has limited their usefulness, except in topical preparations and bowel decontamination regimens. If systemic use is contemplated, a sulphomethylated derivative, colistin sulfomethate, is preferred.
Daptomycin
Daptomycin is a semi-synthetic lipopeptide antibiotic with activity against Gram-positive cocci. It has a role in the treatment of infections caused by multiresistant organisms.
Antimycobacterial agents
As well as streptomycin and rifampicin (see above), various agents are used exclusively for the treatment of mycobacterial infection. These include isoniazid, ethambutol and pyrazinamide, which are commonly found in antituberculosis regimens, and diaminodiphenylsulfone (dapsone) and clofazimine, which are used in leprosy. Cycloserine and p-aminosalicylic acid (PAS), which were formerly used in tuberculosis, have now been largely abandoned except for drug-resistant tuberculosis. Some fluoroquinolones and macrolides exhibit activity against certain mycobacteria, and may have a role in treatment.
Antifungal agents
Although fungi cause a wide variety of infections, relatively few agents are available for treatment, especially for the systemic therapy of serious mycoses. Superficial fungal infections of the skin and mucous membranes can often be treated with topical agents, including polyenes, such as nystatin, or azole derivatives, of which many (clotrimazole, miconazole, econazole, etc.) are marketed as vaginal pessaries and creams. For dermatophyte infections of the nails, oral therapy with griseofulvin or the allylamine derivative terbinafine is peculiarly suitable, as these agents are deposited in newly formed keratin.
Serious systemic disease caused by yeasts and other fungi is often treated with the polyene, amphotericin B, which is extremely toxic. Newer formulations of the drug, in which it is complexed with liposomes or lipids, are better tolerated. The pyrimidine analogue 5-fluorocytosine is active against many types of yeast, and is used in combination with amphotericin B in severe systemic yeast infections. Anidulafungin, caspofungin and micafungin are members of the echinocandin class of agents They are active against various fungi, including Candida, Aspergillus and Histoplasma spp. and Pneumocystis jirovecii (formerly P. carinii), a fungus long thought to be a protozoon.
Azole derivatives exhibit the broadest spectrum of activity, embracing yeasts, filamentous fungi and dimorphic fungi, but few are suitable for systemic use. Those that are include the 2-nitroimidazole ketoconazole, and the triazoles fluconazole, itraconazole, posaconazole and voriconazole. The latter three exhibit useful activity against Aspergillus fumigatus. Fluconazole is well distributed after oral administration, and has been used successfully in systemic yeast infections, including cryptococcal meningitis.
The spectrum of activity of the common antifungal compounds is shown in Table 5.4. Most act by interfering with the integrity of the fungal cell membrane, either by binding to membrane sterols (polyenes) or by preventing the synthesis of ergosterol (azoles and allylamines). Echinocandins interfere with β-glucan synthesis in the fungal cell wall. Use in individual fungal diseases is considered in Chapter 61.

Antiviral agents
Compared with the number of agents available for the treatment of bacterial infection, there are relatively few antiviral agents. However, an increasing number are effective in the treatment and prophylaxis of a range of viral diseases. Those antiviral agents in clinical use are presented in Table 5.5 for agents used for diseases other than human immunodeficiency virus (HIV) infection, and in Table 5.6 for the equally long list of antiretroviral agents.
Table 5.5 Antiviral agents in clinical use for infections other than human immunodeficiency virus
| Compound | Mode of action | Indication |
|---|---|---|
| Anti-herpesvirus agents | ||
| Aciclovir | Nucleoside analogue | Herpes simplex; varicella-zoster |
| Cidofovir | Nucleotide analogue | Cytomegalovirus retinitis |
| Famciclovir | Pro-drug of penciclovir | Herpes simplex; varicella-zoster |
| Fomivirsen | Antisense oligonucleotide | Cytomegalovirus retinitis |
| Foscarnet | Inhibition of DNA polymerase | Cytomegalovirus; aciclovir-resistant herpes simplex or varicella-zoster |
| Ganciclovir | Nucleoside analogue | Cytomegalovirus |
| Penciclovir | Nucleoside analogue | Herpes simplex; varicella-zoster |
| Valaciclovir | Pro-drug of aciclovir | Herpes simplex; varicella-zoster |
| Valganciclovir | Pro-drug of ganciclovir | Cytomegalovirus |
| Anti-influenza agents | ||
| Amantadine (and rimantadine) | Viral uncoating | Influenza A |
| Oseltamivir | Neuraminidase inhibitor | Influenza A and B |
| Zanamivir | Neuraminidase inhibitor | Influenza A and B |
| Anti-hepatitis agents | ||
| Adefovir | Nucleotide analogue | Chronic hepatitis B |
| Boceprevir | NS3 Protease inhibitor | Chronic hepatitis C |
| Entecavir | Nucleoside analogue | Chronic hepatitis B |
| Interferon-α | Immunomodulator | Chronic hepatitis B and C |
| Lamivudine | Nucleoside analogue | Chronic hepatitis B |
| Telaprevir | NS3 Protease inhibitor | Chronic hepatitis C |
| Telbivudine | Nucleoside analogue | Chronic hepatitis B |
| Tenofovir | Nucleotide analogue | Chronic hepatitis B |
| Other antiviral agents | ||
| Ribavirin | Nucleoside analogue | Respiratory syncytial virus; hepatitis C |
Table 5.6 Antiretroviral agents in clinical use
| Type of agent | Drug names |
|---|---|
| Nucleoside analogue reverse transcriptase inhibitor | Abacavir, didanosine, emtricitabine, lamivudine, stavudine, zalcitabine, zidovudine |
| Nucleotide analogue reverse transcriptase inhibitor | Tenofovir |
| Non-nucleoside reverse transcriptase inhibitora | Delavirdine, efavirenz, etravirine, nevirapine |
| Protease inhibitor | Atazanavir, amprenavir (and fosamprenavir), darunavir, indinavir, lopinavir (formulated with ritonavir), nelfinavir, ritonavir, saquinavir, tipranavir |
| Fusion inhibitor | Enfuvirtidea |
| Entry inhibitor | Maravirocb |
| Integrase inhibitor | Raltegravir |
b Active against only those HIV strains which use CCR5 co-receptors.
About half of all the antiviral agents presently available are nucleoside (or nucleotide) analogues, which are phosphorylated within cells to an active triphosphate and inhibit viral DNA synthesis. Some important antiviral compounds, representing a range of molecular structures, are illustrated in Figure 5.3.
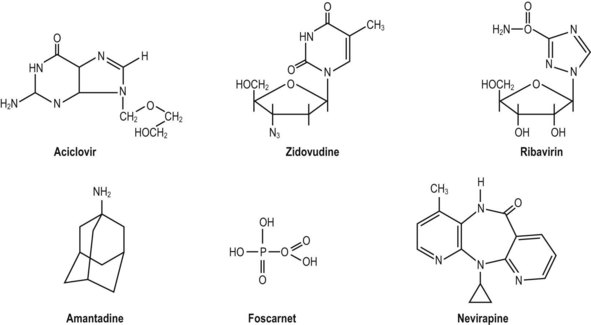
Nucleoside analogues
Inhibitors of herpesvirus DNA polymerases
The most widely used antiviral agent, aciclovir (and the related penciclovir), is first phosphorylated to the monophosphate by a virus-encoded thymidine kinase produced in cells infected by the herpes simplex virus (HSV) or by varicella-zoster virus (VZV). Subsequent phosphorylation steps are completed by cellular kinases to form aciclovir triphosphate. This competes with the natural substrate for the viral DNA polymerase, becomes incorporated into the viral DNA chain and inhibits further DNA polymerase activity (Fig. 5.4). Aciclovir lacks a 3′-hydroxyl group on its acyclic side-chain, and therefore it cannot form a phosphodiester bond with the next nucleotide due to be added to the growing herpesvirus DNA chain, which is terminated prematurely. The lack of cellular toxicity of aciclovir is a consequence of three selective features:
1. Initial phosphorylation takes place only in virus-infected cells.
2. As phosphorylation within virally-infected cells reduces the intracellular concentration of free aciclovir, more drug will diffuse into the cell from the extracellular space, thereby resulting in concentration of drug specifically within virally-infected cells.
3. Aciclovir triphosphate inhibits viral (not cellular) DNA polymerase.
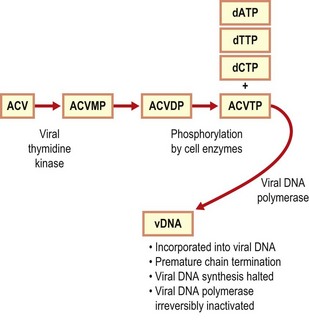
Fig. 5.4 Mode of activation and action of aciclovir (ACV). ACVMP, aciclovir monophosphate; ACVDP, aciclovir diphosphate; ACVTP, aciclovir triphosphate; dATP, deoxyadenosine triphosphate; dTTP, deoxythymidine triphosphate; dCTP, deoxycytidine triphosphate; vDNA, viral deoxyribonucleic acid.
Aciclovir has an established record in the treatment of HSV and VZV disease, as prophylaxis against HSV reactivation after transplantation, and in the long-term suppression of recurrent genital herpes. Valaciclovir and famciclovir are oral pro-drug formulations of aciclovir and penciclovir, respectively. They provide improved systemic drug levels and require less frequent administration.
Ganciclovir, which exhibits preferential activity against another herpesvirus, cytomegalovirus (CMV), is activated by a CMV protein kinase encoded by the viral gene UL97 in CMV-infected cells, but there is also considerable phosphorylation in uninfected cells, and ganciclovir is much more toxic than aciclovir, particularly to the bone marrow. Ganciclovir also exhibits antiviral effects against human herpesviruses HHV-6 and HHV-7, which do not respond to aciclovir. Valganciclovir is an oral pro-drug formulation providing a much improved systemic concentration of ganciclovir after oral administration.
Cidofovir is a nucleoside phosphonate and therefore does not require activation. It is converted in cells into the diphosphate, which inhibits CMV DNA polymerase. It shows activity against a range of DNA viruses, but its main clinical use is for CMV retinitis. It is not absorbed orally, has a very-long half-life (2 weeks), and is nephrotoxic.
These antiviral agents inhibit only replicating herpesvirus and do not eliminate the latent virus. Reduced susceptibility is occasionally found in isolates of HSV, VZV or CMV, usually derived from severely immunocompromised hosts who have been receiving prolonged antiviral therapy. Clinically significant antiviral resistance arises from mutations in the viral DNA polymerases which no longer bind the antiviral triphosphate derivatives, or, in the case of CMV, mutations in the UL97 gene such that the virus cannot phosphorylate ganciclovir.
Other inhibitors of viral RNA or DNA synthesis
Ribavirin is a nucleoside analogue that has no useful anti-herpes activity, but is used in a nebulized form in the treatment of respiratory syncytial virus infection and intravenously for Lassa fever. It is also used orally in combination with interferon for the treatment of chronic hepatitis C. Its mechanism of action is unclear, but the wide antiviral spectrum it exhibits, particularly in vitro, suggests that it may interfere with the processing of virally-derived mRNA.
The nucleoside analogues lamivudine, entecavir and telbivudine, and the nucleotide analogues adefovir and tenofovir (used as pro-drugs, adefovir dipivoxil and tenofovir disoproxil, respectively) all inhibit the reverse transcriptase activity of hepatitis B virus (HBV), thereby reducing virus replication. These drugs differ in their potency and also in the genetic barrier to development of resistance within the viral enzyme, with entecavir and tenofovir being classified as highly potent drugs with the least likelihood of emergence of resistance.
There is great interest in the development of new antiviral drugs directed against various targets within hepatitis C virus, including the NS5b RNA-dependent RNA polymerase enzyme. However, the first directly acting agents, licensed in late 2011, for the treatment of HCV infection are the NS3 protease inhibitors telaprevir and boceprevir.
Non-nucleoside anti-herpes agents
Foscarnet, a pyrophosphate analogue, inhibits nucleic acid synthesis without requiring any activation, and is an important agent for treatment of cytomegalovirus or of other herpesvirus infections that have become resistant to aciclovir or ganciclovir through mutations in the viral DNA polymerase or protein kinase genes. It is poorly absorbed orally, and exhibits nephrotoxicity. Fomivirsen is an antisense oligonucleotide that inhibits translation of mRNA into proteins. It has limited use as a local treatment for CMV retinitis.
Agents that block viral uncoating
Amantadine, a symmetrical amine compound, has long been known to inhibit influenza A virus replication. Amantadine (and its derivative, rimantadine) blocks an ion channel formed by the integral membrane protein (M2) of influenza A (but not influenza B) virus, preventing uncoating of the virus within cells. Resistance arises rapidly through mutations in the M2 gene.
Neuraminidase inhibitors
Zanamivir and oseltamivir are agents specifically designed to block the action of the influenza virus enzyme neuraminidase by occupying the catalytic site. They act on all influenza viruses. Neuraminidase is found on the surface of influenza virus, and these compounds act to prevent release of newly-formed viral particles from the cell surface, reducing the spread of influenza viruses locally. Zanamivir is taken by inhalation into the oropharynx; oseltamivir is the oral pro-drug form of a similar agent.
Interferons
Interferons, naturally occurring antiviral compounds produced by mammalian cells in response to viral infection, are now manufactured by genetic recombination. Interferon-α (given by either subcutaneous or intramuscular injection) is used to treat chronic infection with hepatitis B and C viruses. The mode of action of interferon-α is complex, producing an antiviral state in cells to which it binds, and interfering with the production of virus in those cells (see Ch. 10). However, interferons also act through immune modulation, upregulating the expression of major histocompatibility complex (MHC) molecules on cell surfaces. This is a significant part of the action in clearing chronic hepatitis B.
Modification of interferon by addition of polyethylene glycol – peginterferon – results in sustained effects after just one dose per week. This agent appears to offer an effective treatment for chronic hepatitis C, a common form of chronic hepatitis, when combined with oral ribavirin.
Antiretroviral agents
The HIV pandemic has generated an enormous interest in potential antiviral agents. Seven classes of drugs are now generally available, acting variously to inhibit viral nucleic acid synthesis, viral protein synthesis, viral entry, and integration of the DNA provirus into the host genome (see Table 5.6). All inhibit only replicating HIV and do not eliminate the integrated proviral DNA.
The use of multiple drugs in combination antiretroviral therapy (cART, previously known as ‘highly active antiretroviral therapy’ or HAART; see Ch. 55) may result in such suppression of HIV replication that virus is undetectable in the circulation. The use of cART prevents the development of immune deficiency in infected patients, and may also result in immune reconstitution in patients who have already developed the acquired immunodeficiency syndrome (AIDS).
Emergence of resistant mutants has been a problem with all classes of anti-HIV agent, and has stimulated developments in the field of antiviral susceptibility testing.
Nucleoside and nucleotide reverse transcriptase inhibitors
The earliest anti-HIV agent, zidovudine (azidothymidine), and several other compounds, including didanosine (dideoxyinosine), zalcitabine (dideoxycytidine; now rarely used), lamivudine and stavudine, are nucleoside analogues. New additions include abacavir and emtricitabine, and a nucleotide analogue, tenofovir. They are activated (phosphorylated) by cellular enzymes and inhibit the reverse transcriptase function of the viral polymerase of HIV-1 or HIV-2, with many terminating the chain of proviral DNA. Phosphorylation rates vary in different cell types, and between resting and replicating cells.
Unlike aciclovir, these nucleoside analogues are associated with some toxicity as there is less selectivity in their activation and action:
Non-nucleoside reverse transcriptase inhibitors
These compounds, which include nevirapine, delavirdine (no longer widely available), efavirenz and etravirine inhibit only HIV-1. They bind directly, without activation, away from the catalytic site of reverse transcriptase, but exert a structural change that inhibits its action. They are not incorporated into the DNA chain.
HIV protease inhibitors
These compounds act at a late stage in the viral cycle by interfering with the cleavage of essential polyprotein precursors. The numerous derivatives now available are listed in Table 5.6.
HIV fusion inhibitors
Only one example of this class of antiretroviral agents is presently available: enfuvirtide. This drug is a homologue of the short peptide sequence active in fusion of the HIV-1 envelope to the cell membrane after attachment, and it inhibits that final stage of the entry process. The drug has to be given by subcutaneous injection, and its use is reserved for patients who have limited options for treatment. It is not active against HIV-2.
Other inhibitors of the HIV entry process are under development, including drugs that block the co-receptors for attachment. Cellular as well as viral factors will influence the success of this approach.
HIV entry inhibitors
Initial binding of HIV to CD4 molecules present on the target cell surface is followed by binding to one of a number of secondary receptors, the most commonly used of which is CCR5, a chemokine receptor. Maraviroc is a drug which binds specifically to CCR5, thereby preventing access of viral particles to this molecule.
HIV integrase inhibitors
Once viral RNA has been reverse transcribed into a DNA form (known as the provirus), integration of the provirus into the host cell genome is mediated by a viral integrase enzyme. The latest antiretroviral drugs to have been developed are integrase inhibitors, acting to prevent this step in the viral life cycle.
Antiparasitic agents
The choice of agents for the treatment of protozoal and helminthic infections remains extremely limited. Part of the problem is that protozoa and helminths are very varied, reflecting diverse solutions to the problems of their specialized parasitic existence. Consequently, there are few ‘broad-spectrum’ antiprotozoal or anthelminthic agents, although some compounds exhibit a surprising range of activity. Thus the antiprotozoal nitroimidazoles, such as metronidazole, are active against Entamoeba histolytica, Trichomonas vaginalis and Giardia lamblia (see Ch. 62); benzimidazoles, such as mebendazole and albendazole, act against most intestinal nematodes; praziquantel not only exhibits good activity against all human schistosomes but also includes other trematodes and tapeworms in its spectrum (see Ch. 63). Most remarkably of all, ivermectin is active not only against many filarial worms and some intestinal roundworms but also against ectoparasites such as the scabies mite.
Antimicrobial agents commonly used against pathogenic protozoa and helminths are shown in Tables 5.7 and 5.8, respectively.
Table 5.7 Principal agents used against the major protozoan parasites of man
| Species | Agent |
|---|---|
| Cryptosporidium parvum | Nitazoxanide |
| Entamoeba histolytica | Metronidazole |
| Diloxanide furoate (Emetine) |
|
| Giardia lamblia | Metronidazole (Mepacrine) |
| Leishmania spp. | Sodium stibogluconate |
| Amphotericin B | |
| Miltefosine | |
| Plasmodium spp. | Chloroquine |
| Quinine | |
| Pyrimethamine | |
| Proguanil | |
| Mefloquine | |
| Halofantrine | |
| Artemisinin and its derivatives | |
| Toxoplasma gondii | Pyrimethamine + sulfadiazine |
| Trichomonas vaginalis | Metronidazole |
| Trypanosoma brucei ssp. rhodesiense and gambiense | Eflornithinea |
| Melarsoprol | |
| (Suramin) | |
| (Pentamidine) | |
| Trypanosoma cruzi | (Benznidazole) |
Compounds in parentheses are of limited value.
a Not active against T. brucei rhodesiense.
Table 5.8 Spectrum of activity of the principal anthelminthic agents
| Agent | Active against |
|---|---|
| Benzimidazolesa | Intestinal nematodes |
| Diethylcarbamazine | Filarial worms |
| Ivermectin | Onchocerca volvulus; other filariae |
| Levamisole | Hookworms; Ascaris lumbricoides |
| Niclosamide | Tapeworms |
| Metrifonate | Schistosoma haematobium |
| Oxamniquine | Schistosoma mansoni |
| Piperazine | Ascaris lumbricoides; Enterobius vermicularis |
| Praziquantel | Schistosoma spp.; other trematodes; tapeworms |
| Pyrantel pamoate | A. lumbricoides; E. vermicularis; hookworms |
| Trivalent antimonials | Schistosoma spp. |
Antimicrobial sensitivity tests
To establish the activity of an antimicrobial agent, micro-organisms are tested for their ability to grow in the presence of suitable concentrations of the drug. Bacteria are the simplest to test and, in practice, routine sensitivity testing is usually reserved for the common, easily grown, bacterial pathogens. Tests of mycobacteria, fungi, viruses and some other organisms are available in certain laboratories and reference units. Genotypic assays for mutations associated with antiviral resistance are becoming more widely available in specialist virology centres. Similarly, genotypic assays that detect resistance mutations affecting antituberculosis therapy are available through reference centres.
Antibacterial agents
Potency of antibacterial agents is often expressed as the minimum inhibitory concentration (MIC): the lowest concentration of the agent that prevents the development of visible growth of the test organism during overnight incubation. Serial dilutions of the agent are prepared in a suitable broth or agar medium, and a standard inoculum of the test organism is added. If agar is used, many different isolates can be tested at the same time by spot inoculation of the plate. Broth dilution MIC titrations have the advantage that the minimum bactericidal concentration (MBC) can additionally be estimated by subculture of dilutions of the antibiotic above that in which inhibition has occurred overnight. The MBC is usually taken as the lowest concentration capable of reducing the original inoculum by a factor of 1000, for example from 105 colony-forming units (cfu)/mL to 102 cfu/mL or below. To establish the rate of killing, the number of viable organisms in broth cultures is measured at timed intervals after addition of appropriate concentrations of the antibiotic.
Antibiotic titrations are too laborious for the routine assessment of antibiotic activity in clinical practice, although a truncated form of the method, in which isolates are tested at agreed break points of sensitivity or resistance, is sometimes used. A common alternative method is the disc diffusion test. The culture to be examined is seeded confluently, or semi-confluently, over the surface of an agar plate, and paper discs individually impregnated with different antibiotics are spaced evenly over the inoculated plate. Antibiotic diffuses outwards from each disc into the surrounding agar and produces a diminishing gradient of concentration. On incubation, the bacteria grow on areas of the plate except those around the drugs to which they are sensitive. The width of the zone of inhibition is a rough measure of the degree of sensitivity to the drug. A highly standardized version of the disc diffusion method, the Bauer–Kirby test, is favoured in the USA and some other countries.
Characteristics of the growth medium (such as pH or the presence of antagonizing substances), the size of the bacterial inoculum and the conditions of the test may influence the results of sensitivity tests. In disc methods, the size of the inhibition zone is additionally affected by the diffusion characteristics of the antibiotic. It is therefore important that suitable control organisms are tested in parallel in all tests of antibiotic susceptibility. In Stokes’ method, control is achieved by placing antibiotic discs at the interface between inocula of the test and control bacteria on the same plate. In this way the zone of inhibition obtained with the test organism can be compared directly with that of a known control, and variations in the cultural conditions or in disc content can be avoided.
Antiviral agents
Tests for antiviral susceptibility are still far from routine, being readily available only through reference or specialist laboratories. Experience with these assays is steadily increasing, and the clinical benefits of measuring antiviral susceptibility are gradually being established.
The detection of mutations associated with drug resistance is now a routine procedure in the management of HIV infection. Some reference laboratories have built up a great deal of experience with in-house sequencing of HIV genes and have generated large databases, which are shared on an international basis to allow for prediction of reduced susceptibility. Expert opinion on the significance of the mutations may usefully guide therapeutic decisions and is invaluable for selection of therapeutic combinations.
Phenotypic assays
Phenotypic assays measure the inhibitory effect of the antiviral agent on the clinical virus isolate. The plaque reduction assay for inhibition of HSV by aciclovir is one example. An effect is usually considered to be significant when virus replication or product formation is reduced by 50% compared to that found with no drug (50% inhibitory concentration: IC50).
For HIV assays, inhibition of replication in cultures of peripheral blood mononuclear cells is preferred. There are also recombinant virus assays in which HIV genes of interest from the test isolate are put into a laboratory strain to provide a cheaper, faster assay.
Genotypic assays
These tests detect the presence of known resistance-associated mutations in viral genes (e.g. HIV reverse transcriptase or protease, or HBV polymerase), from which reduced susceptibility is predicted. Some of these assays are available in commercial kit format, for example:
• a line probe assay by reverse hybridization of amplified viral fragments with a panel of oligonucleotides
• an automated sequencing programme detecting mutations in reverse transcriptase and protease genes.
Genotypic assays are more commonly used than phenotypic ones, being simpler, safer and less costly for routine use.
Assay of antimicrobial drugs
In most clinical circumstances in which antimicrobial agents are used it is not usually necessary to measure the concentrations achieved in blood or other body fluids (see Ch. 67). Such measurements are needed, however, during the development of a new agent to establish the pharmacokinetic behaviour of the drug.
Various techniques are available, including microbiological methods in which the antibiotic-containing material and known dilutions of the drug are titrated in parallel against a susceptible indicator organism. Analytical techniques such as high-pressure liquid chromatography may also be used.
In the few cases in which assays are needed in clinical practice, notably during treatment with aminoglycosides, immunochemical methods are often used with commercially available instrumentation.
With regard to antiviral drug assays, concerns over toxic levels of aciclovir or ganciclovir arise on occasion, and these can be measured. Therapeutic drug monitoring is also practised in relation to HIV therapy, mainly to test for compliance with the onerous regimens necessary in the management of this chronic condition.
De Clercq E. In search of a selective therapy of viral infections. Antiviral Research. 2009;85:19–24.
Finch RG, Davey PG, Wilcox MH, Irving WL. Antimicrobial Chemotherapy, ed 6. Oxford: Oxford University Press; 2012.
Finch RG, Greenwood D, Norrby SR, Whitley RJ. Antibiotic and Chemotherapy: Anti-infective Agents and their Use in Therapy, ed 9. London: Saunders, Elsevier; 2010.
Franklin TJ, Snow GA. Biochemistry and Molecular Biology of Antimicrobial Drug Action, ed 6. New York: Springer; 2005.
Grayson ML, Crowe SM, McCarthy J, et al. Kucers’ The Use of Antibiotics: A Clinical Review of Antibacterial, Antifungal and Antiviral Drugs, ed 6. London: Hodder Arnold; 2010.
Lorian V, ed. Antibiotics in Laboratory Medicine, ed 5, Baltimore: Lippincott, Williams & Wilkins, 2005.
Volberding P, Deeks SG. Antiretroviral therapy and management of HIV infection. Lancet. 2010;376:49–62.
National electronic library of infection. Bugs & drugs on the web http://www.antibioticresistance.org.uk/
United States National Library of Medicine and National Institutes of Health. Antibiotics http://www.nlm.nih.gov/medlineplus/antibiotics.html
World Health Organization. Essential Medicines and Pharmaceutical Policies http://www.who.int/medicines/en/