Data regarding predicted susceptibility of bacteria to specific antimicrobials can be found in Prescott JF et al: Can Vet J 25:289, 1984.
BRAIN TRAUMA
Definition and Etiology
Because of their size, behavior, and relatively thin calvarium, horses are more susceptible to head trauma than other livestock. Traumatic injuries of horses most often result from kicks, sharp blows, or falling over backward.700 Blows to the poll of the horse, particularly associated with falling over backward, are very common and result in fracture or displacement of the basisphenoid, occipital, and petrosal bones and the basioccipital and basisphenoid sutures.701-703 Young horses are particularly prone to this type of injury, probably because of their more fractious nature and tendency to react strongly to restraint, as well as to lesser strength of the immature skull.703 The basisphenoid and basioccipital bones form a part of the foramen lacerum and the jugular foramen. Fractures around these foramina may result in dysfunction of CNs IX, X, and XII.704 Hematomas form at the fracture site and extend into the membranous labyrinths and basilar areas of the brain, where they cause vestibular and occipital cortex dysfunction. Basilar region fractures carry a much more guarded prognosis than trauma at other sites.703 Blows to the forehead result in depression fractures of the dome of the calvarium and trauma to the underlying cerebrum.
Skull fractures occur in cattle from blows to the top of the calvarium. Most skull fractures are located in the center of the frontal bones, where the internal and external plates of the frontal sinus are fused into a single-layer dorsal wall of the cranial vault. This position can be located on the skull as the imaginary cross found by intersecting lines drawn between the medial canthus of the eye and the horn of the opposite side. Injuries in this area compress the frontal and parietal lobes of the cerebral cortex. The pressure changes result in loss of sensorium, sensory deficits, blindness contralaterally, or convulsions.
In young goats and horned sheep under 4 to 6 months of age, the calvarium can be inadvertently opened by removal of excessive bone during disbudding or dehorning. In goats, cerebrocortical burns can occur from overapplication of a hot iron or caustic dehorning paste. Cortical necrosis caused by bacterial infections after dehorning of calves has also been described.705
Clinical Signs
The clinical presentation of cerebral trauma depends on the area of the brain damaged, the extent of the lesion, and the duration of the injury.706Lesions of the cerebral cortex and thalamus are characterized by variably altered mentation, circling, head pressing, pacing, aimless wandering, and cortical blindness (blindness with normal eyes, pupillary light reflexes, and oculomotor reflexes). Seizures also may result; however, occurrence of a seizure does not necessarily indicate a poor prognosis.703 Compression of the midbrain results in decerebrate rigidity caused by loss of the reticulospinal tracts. In more severe cases of midbrain compression, abnormal breathing patterns may be observed, together with hyperreflexia, tetraplegia, and absence of pupillary reflexes. Compressive lesions of the mesencephalon in the region of the oculomotor nucleus result in mydriatic pupils on the ipsilateral side of unilateral brainstem lesions. Medulla oblongata compression is characterized by serial dysfunction of cranial nerves, severe disturbance of consciousness, and abnormal respiratory rhythm. Involvement of the long motor and sensory pathways to the limbs can occur with brain injuries at any level and results in ataxia and paresis, which may be worse on the side contralateral to the injury in the case of cerebral cortex, thalamic, and midbrain injury, or on the ipsilateral side in the case of traumatic damage to the medulla or cerebellum.
Basioccipital fractures of horses result in asymmetric signs of vestibular disturbance, including horizontal or rotary nystagmus, ipsilateral ventrolateral strabismus, contralateral dorsomedial strabismus, head tilt, and contralateral blindness. Horses that remain ambulatory lean or circle toward the side of the lesion. Additional signs of this syndrome include dysphagia, facial paralysis, conscious proprioceptive deficits, recumbency, depression, and coma. Horses that are recumbent struggle violently. Fracture of the petrous temporal bone may cause profuse bleeding from the ipsilateral nares, external ear canal, and guttural pouch.
Brain trauma caused by overaggressive dehorning in goats results in depressed sensorium, loss of menace response, increased extensor tonus on the contralateral side, ipsilateral mydriasis, sluggish pupillary reflex, and loss of conscious proprioceptive responses. The clinical syndrome may be delayed by several days in cases of cortical burns or trauma caused by caustic paste and may be complicated by brain abscess or bacterial meningitis.
Pathology and Pathogenesis
The pathogenetic events leading to cerebral edema and increased intracranial pressure (ICP) are complex. Trauma to the head results in a variety of abnormal physical forces exerted on brain tissue, including acceleration-deceleration, shearing, compressive, tearing, and rotational forces.706-708 The consequence of direct physical insult to brain tissue that occurs immediately on impact is considered primary traumatic brain damage.707 Such physical insult results in axonal injuries that may be immediate (primary axotomy), such as axonal tearing, or occur many hours after the initiating event (secondary axotomy).708 Trauma activates neuronal mechanoreceptors, causing cellular depolarization that spreads outward from the site of impact. Together with direct axonal injury, this may underlie initial signs of concussion, including loss of consciousness.706,708 Processes that follow the initial mechanical trauma and further exacerbate injury are considered secondary brain damage.707 Intracranial hemorrhage or loss of vascular integrity and cerebral edema occur after concussive blows to the head. Hemorrhage after head trauma may be epidural, subdural, subarachnoid, or intraparenchymal.707 Displacement of the neural tissue is caused by cerebral swelling or hematoma formation. The increased pressure is transmitted to the CSF and interferes with normal vascular flow, resulting in cerebral hypoxia and interneuronal and intraneuronal edema. Diminution of cerebral perfusion results from a combination of increased ICP, disruption of the vascular architecture, and decreased systemic blood pressure. The net result is reduced oxygen delivery to the brain, a tissue that relies on aerobic glycolysis for the production of energy. Interruption of energy production within the brain results in failure of tissue homeostasis. Breakdown of the blood-brain barrier further contributes to brain swelling and loss of intracranial homeostasis.
In addition to gross damage to tissue, brain trauma results in a complex series of biochemical events that disrupt cellular integrity.706,709 One of the most important is the depletion of adenosine triphosphate (ATP), the main energy store within neurons. This results in dysfunction of cell membrane ionic pumps, permitting influx of sodium (Na+) and calcium (Ca++) into the cell. Influx of these ions activates a number of secondary pathways within the cell, including the kinin, arachidonic, complement, and xanthine-oxidase pathways. Activation of these pathways results in the production of a variety of substances that are deleterious to cellular function, including oxygen free radicals, vasoactive mediators, cytokines, nitric oxide, excitatory neurotransmitters, and enzymes. Together, these contribute to a destructive cascade of events that further damages cell integrity.709
When brain swelling becomes severe, the respiratory centers are depressed, resulting in hypoxemia and acidosis. The extra carbon dioxide diffuses into the brain, and water follows, which further swells the CNS. Acidosis and hypoxemia also worsen the vascular leakage and hypoxemia. Extreme swelling of the cerebral cortex results in herniation through one or more anatomic sites of the calvarium. Four forms of brain herniation have been described in large animals.710 These include cingulate gyrus herniation ventral to the falx cerebri, herniation of parts of the temporal cortex ventral to the tentorium cerebelli (caudal tentorial herniation), caudal cerebellar vermis herniation through the foramen magnum, and herniation of the rostral cerebellar vermis ventral to the tentorium cerebelli (rostral tentorial herniation). Compressed tissue becomes hypoxemic and edematous. Compression of the CNS causes more hypoxia, prompting a dramatic and rapid deterioration.
Clinical Pathology
Clinical pathologic variables noted after head trauma include nonspecific changes consistent with a stress response, such as mild neutrophilia, lymphopenia, and hyperglycemia, as well as those resulting from systemic trauma, such as elevated serum CK.703 Hyperglycemia has been associated with more guarded prognosis in people with head injuries, possibly from deleterious effects on cerebral vasculature.711 In one study of horses that had head trauma, only elevated packed cell volume (PCV) was shown to be associated with a more guarded prognosis.703
Collection of CSF from the atlantooccipital cistern is generally contraindicated in head trauma, especially if signs of increased CNS pressure, uncontrolled hemorrhage from the ears or the nose, or dorsal sagittal sinus fractures are observed. Clinical signs that may suggest the presence of increased ICP include dull mentation (especially if this is worsening over time), mydriasis, blindness, or papilledema. The CSF changes that occur from traumatic injuries are characteristic. For the first 24 hours, blood is admixed evenly in the CSF. Iatrogenic hemorrhage from the tapping procedure can be differentiated from that caused by trauma because in the former case, the CSF is irregularly streaked with blood; CNS hemorrhage usually results in an even admixture of blood through the CSF. During the first 24 hours, the protein concentration and WBC count of CSF are elevated and are in the approximate ratio as that of peripheral blood. By 48 hours after the traumatic episode, the amount of blood in the CSF decreases, and when centrifuged, the cell-free CSF appears xanthochromic. WBC counts of the CSF are only marginally increased by 24 hours after hemorrhage, and the protein concentration may range from 500 to 1000 mg/mL (albuminocytologic dissociation). Thereafter, the protein concentration gradually decreases, and the xanthochromia disappears by 14 days after the acute hemorrhage. The number of mononuclear inflammatory cells gradually increases as parts of the CNS degenerate. The CK level of the CSF is elevated (10 to 100 IU/dL) for approximately 1 to 2 days after the acute traumatic episode.
Diagnostic Imaging
Radiography or more advanced imaging studies (CT, MRI) are the primary modalities for definitive diagnosis of skull fractures. It should be remembered, however, that significant brain trauma can occur in the absence of skull fracture and that this is a common situation. Radiography has the advantages of being widely available and relatively simple to perform, requiring restraint or only mild sedation in most situations. However, false-negative findings are common, especially in cases where the bony lesion may be particularly difficult to identify, as with basilar bone fractures in horses.703 While basilar fractures may be difficult to identify, other radiographic changes such as soft tissue densities in the tympanic bullae due to hemorrhage, or gas opacities adjacent to the basilar region, may support this diagnosis.711aCT is the technique of choice for the diagnosis of bony lesions and for acute intracranial hemorrhages, whereas MRI facilitates diagnosis of a variety of pathologic changes within the brain parenchyma. These advanced imaging techniques have limited availability, are costly, and usually require general anesthesia. Clinical findings and historical information may form the sole bases for diagnosis in many cases.
Treatment
The treatment of brain trauma remains one of the most controversial areas of clinical neurology in all species. Opinions differ widely and evidence in the scientific literature is often contradictory. Treatments that show promise in rodent models of head injury often are disappointing in clinical trials.711b Successful treatment depends largely on early recognition and initiation of therapies that maintain cerebral and whole body homeostasis. General medical principles for treating CNS trauma include (1) establishment of proper respiratory function, (2) support of blood pressure and maintenance of cerebral perfusion and oxygenation, (3) control of seizures, (4) nutritional and fluid support, and (5) protection from decubitus and self-inflicted damage. Many treatment modalities recommended for head-injured large animals are based on anecdotal reports and are not supported by rigorous scientific studies. It is reasonable, however, to apply the principles of head trauma management established in other species, including humans, to the management of larger mammals that sustain similar injuries. Immediate treatment involves establishment of a patent airway and administration of oxygen via mask, endotracheal tube, or nasal catheterization. In cases of severe upper airway obstruction, emergency tracheostomy may be indicated.711c Aggressive intravenous crystalloid therapy is indicated to establish and maintain a normal systemic blood pressure and to ensure that the brain receives adequate blood supply, with use of colloids or blood products as necessary. Principles of fluid therapy and treatment of shock are described in Chapter 34.
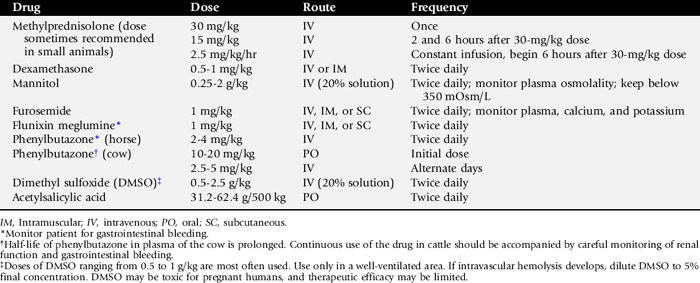
Administration of dexamethasone, methylprednisolone, mannitol, or dimethyl sulfoxide (DMSO) has been recommended for controlling CNS pressure caused by edema. The clinician should bear in mind that little or no scientific data exist to support the use of corticosteroids or DMSO in the treatment of CNS trauma, and that these drugs may have deleterious effects that outweigh any potential benefits. Doses listed below are largely anecdotal and are included because of the continued widespread tendency to use these agents, despite lack of evidence that they are effective. Studies of the use of corticosteroids in people with head injuries are ongoing, and the pendulum of opinion continues to swing between positive and negative.711d An empiric recommendation for the treatment of horses is administration of dexamethasone at 0.1 to 0.25 mg/kg by slow IV injection every 4 hours for 1 to 4 days or, for mature animals, 100 to 1000 mg of methylprednisolone by slow IV injection.716 Similar dosages could be used in ruminants.
Data supporting the use of methylprednisolone sodium succinate for treatment of acute spinal cord injuries in people comes from the National Acute Spinal Cord Injury Study (NASCIS). A dose of 30mg/kg bodyweight given by intravenous infusion within 8 hours of injury has been recommended, followed by a second and third dose (15 mg/kg each) given intravenously 2 and 6 hours later and a subsequent infusion of 2.5 mg/kg/hr for the next 48 hours.717 The use of a similar treatment for head injuries does not yet have good scientific support. In addition, the methodology and results of the NASCIS study have been called into question. The potential deleterious effects of such high doses of corticosteroids in large animal species include enhanced susceptibility to infection, muscular weakness, renal potassium and calcium loss, abortion in ruminants, and laminitis in horses. These adverse consequences of corticosteroid use probably outweigh its uncertain benefits. Table 35-8 presents a list of antiedema drugs often administered to large animals with traumatic brain disease.
The use of the osmotic diuretic mannitol has regained favor in the treatment of head trauma in humans.711d Intravenous administration of a 20% solution of mannitol (1 g/kg) or oral administration of glycerol (20 mL/kg) has been used for the treatment of increased ICP in large animal species. The physiologic activity of mannitol for lowering the CSF pressure may be related more to its vasoconstrictive effects than its activity as an osmotic diuretic. Response to the treatment may occur as early as 1 hour after administration. Mannitol is expensive and usually only economically justifiable for use in neonates. If response to the initial mannitol dosage is noted, additional treatments should be given every 4 to 6 hours for the first day. Mannitol should be administered through blood administration filter sets to minimize the occurrence of microcrystalline emboli. The drug should not be given to animals with active CNS hemorrhage, because diffusion of mannitol into the center of a newly forming hematoma exerts an osmotic effect, enlarges the size of the lesion, and further attenuates the nervous system tissues. Active CNS hemorrhage can be recognized by the presence of unclotted blood in the nose or ears, or parietal bone fractures that lacerate the dorsal sagittal sinus. Despite this provision, administration of mannitol is probably justified in an animal with rapidly worsening and potentially fatal deterioration in neurologic status, even in the likely presence of intracranial hemorrhage.
Intravenous use of DMSO has been recommended for the reduction of increased CSF pressure in large animals. The drug is administered IV at 0.5 to 4 g/kg twice daily.701,712,718,719 DMSO is diluted fivefold to tenfold in saline (10% to 20% solution) to minimize the hemolytic and hyperthermic effects. In horses, administration of 5L of 10% DMSO in a balanced electrolyte solution has been shown to have minimal deleterious clinical or clinicopathologic effects.719aHigher doses (solutions >20%) have been reported to have a number of adverse side effects such as intravascular hemolysis, colic, diarrhea, muscle tremors, and collapse.719b The use of DMSO for cerebral trauma is controversial, and benefits may be species specific. For example, anecdotal reports of benefit have been shown for horses, but controlled experiments in dogs have shown limited clinical benefit when treating experimental CNS trauma.714
DMSO has several beneficial pharmacologic actions, including free-radical scavenging, interference with neutrophil chemotaxis, prevention of microthrombi, increased penetration of corticosteroids and antibiotics into the brain, and vasodilation.720 A major effect of the drug is probably caused by its diuretic action, which is greater than that of furosemide. In experimental situations, administration of DMSO to animals with experimentally induced CNS lesions resulted in more rapid neurologic recovery than treatment with urea, corticosteroids, or mannitol.720 The adverse effects of DMSO include muscular fasciculations, intravascular hemolysis, hemoglobinuria, and sweating.719 Deaths have been reported in laboratory animals after intraperitoneal injections of 10 mg/kg and in dogs after IV dosages of 2.5 mg/kg.721-723 The median lethal dosage of DMSO in large animals is unknown. The drug is teratogenic when administered to pregnant laboratory animals.722,723 When the drug is administered IV, approximately 70% of the dosage is excreted through the respiratory tract.724 These data indicate that DMSO should be administered only in well-ventilated areas. Exposure of pregnant women and animals should be avoided. In cattle, DMSO is excreted rapidly and is essentially completely cleared from the plasma by 5 days.724 A low-level residue of DMSO may persist in the fat tissues for at least 20 days. When administered IV to horses at 1.0 and 0.1 g/kg, the biologic half-life of DMSO is 8.6 and 9.8 hours, respectively.719
Depression fractures of the frontal and parietal bones may be reduced surgically. Lacerations of cerebral tissue can be treated surgically with gentle cleaning and debridement of contaminated and devitalized tissue.725 Such procedures require general anesthesia. Prognosis in animals with exposed and contaminated brain tissue is very guarded, so expectations should be realistic before such intervention is undertaken.Surgical repair of skull fractures in horses using intrafragmentary wires or bone plates may be indicated to improve functional and cosmetic outcomes.725a,725b
Convulsions may be controlled initially by IV administration of diazepam (Valium), phenobarbital, or pentobarbital. The recommended dosages and mode of administration of these drugs are presented in Table 35-7. Good nursing care is essential. These drugs are usually highly protein bound in plasma and can be displaced or functionally altered by other drugs. All anticonvulsant treatments should begin at the lowest possible dosage, which can be increased daily or every second or third day until the seizures have been controlled. If seizures cannot be controlled without causing depression or ataxia, a second anticonvulsant is added. The dosage of the second drug is increased gradually until the seizures stop. This combination treatment is continued for 2 to 4 weeks. Thereafter, the dosage of the first anticonvulsant is tapered until it is discontinued. If seizures reappear, the dosage of this drug is increased until the seizures disappear again. The trough blood concentration of all anticonvulsants is checked monthly. The suggested therapeutic trough concentration of phenobarbital ranges from 15 to 40 μg/mL of plasma and that of diphenylhydantoin from 5 to 20 μg/mL. Any attempt to withdraw anticonvulsant therapy should be done gradually over a 4-week period.
Horses with recurrent convulsions should not be ridden or used for sporting purposes. Infrequent seizures generally do not justify anticonvulsant treatment, and economic considerations often limit the amount of drug therapy that is possible. Status epilepticus can be treated with IV diazepam in 5-mg doses until seizures are controlled or by titrated doses of phenobarbital or pentobarbital. Mares with estral-related seizures may be treated with an ovariectomy.
EQUINE PROTOZOAL MYELOENCEPHALITIS (TOXOPLASMA-LIKE AGENT; PROTOZOAL ENCEPHALOMYELITIS; SEGMENTED MYELITIS)
Definition and Etiology
Equine protozoal myeloencephalitis (EPM) is a multifocal, progressive disease of the central nervous system (CNS) that is primarily caused by infection with Sarcocystis neurona.777 Recently, another protozoan parasite, Neospora caninum/N. hughesi, has been implicated as a cause of EPM in six cases.778-783 The condition has mostly been reported from many U.S. states, Canada, Panama, Brazil, and Argentina.784-790 Several reports of the disease in countries other than those in the Western Hemisphere were primarily in horses that originated from the Americas.791-794 More recently, there have been reports of horses in France that developed neurologic deficits with positive S. neurona antibody titers that were native to France and had not resided in the United States.795,796 Young standardbred, thoroughbred, and quarter horses are most often affected, although horses of any breed may develop the disease. There does not appear to be a gender predilection for EPM, and any age may be affected.797 The risk would appear to be higher in young horses, but horses as old as 30 years have developed the condition.797
The parasite produces inflammation and necrosis of the brain, brainstem, and spinal cord. Under light microscopy, the structure of the EPM agent resembles that of Toxoplasma gondii, but comparative electron microscopic analyses of the three agents show differences. The Sarcocystis agent of horses has been grown in explant cultures of monolayered bovine monocytes.787,798 Antibodies in the sera or cerebrospinal fluid (CSF) can be detected using these specimens as probes of immunoblots of the cultured parasites. Sera from clinically affected cases recognized eight S. neurona—specific antigens,799 several of which are the basis for current diagnostic testing. DNA analysis has been very important in characterizing and classifying S. neurona. Using a random primed polymorphic DNA assay (RAPD), a unique sequence of base pairs was identified that distinguished S. neurona from eight related coccidia, specifically two Sarcocystis species, one Toxoplasma species, and five Eimeria species.800 This research demonstrated that unique DNA sequences could be successfully used as a species-specific probe for S. neurona, and that these probes permitted differentiation of S. neurona from other coccidia of equines.800
Clinical Signs
Descriptions of clinical signs of horses diagnosed with EPM may vary greatly because the organisms that cause this disease can affect any CNS tissue. Therefore, any horse exhibiting neurologic abnormality could be diagnosed with EPM.
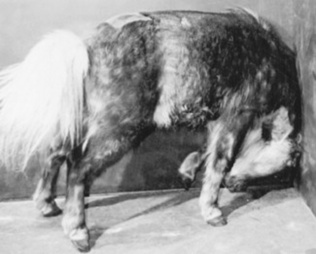
Clinical signs recognized in the earliest studies of this disease still characterize neurologic abnormalities in horses with EPM. Early workers described horses with EPM as having an asymmetric ataxia and associated muscle atrophy.801,802 Horses may have a sudden onset of clinical signs, or disease may progress slowly over several months.801 Vague, intermittent lameness that is nonresponsive to therapy may be caused by EPM, and encephalitic signs typified by asymmetric cranial nerve deficits may also be seen in affected horses.801 Gait abnormalities in horses with EPM include ataxia, tetraparesis, knuckling, circumduction, and crossing over. The abnormalities may be asymmetric. Depending on the location of the lesion in the spinal cord, areflexia, hyporeflexia, or hyperreflexia may be seen. Infections of the myelencephalon may result in head tilt, facial paralysis, circling, nystagmus, dysphagia, facial paralysis, and apparent blindness, with or without abnormal pupillary reflexes. Parasitic invasion of the ventral spinal rootlets or the radicles of the maxillary branch of the trigeminal nerve may result in neurogenic atrophy of the tongue and masticatory muscles (Fig. 35-9). This is often accompanied by focal areas of desensitization. Regional sweating (“strip sweating”) may be observed if the sympathetic tracts of the spinal cord are affected. Although EPM is typified by the presence of asymmetric, multifocal neurologic abnormalities, horses with EPM may have focal or symmetric signs.
Cerebral signs are rarely seen in horses with EPM. However, three horses with EPM presented to Ohio State University Veterinary Teaching Hospital displayed seizure activity and evidence of cortical electrical activity abnormalities on electroencephalographic (EEG) examination.803 Horses with cerebral neurologic signs often have a poor prognosis. However, seizure activity in horses with EPM may be treatable. Visual deficits and behavioral abnormalities have been reported in horses with EPM.804 Head shaking was also reported in a recent case series describing three horses diagnosed with EPM.805 Head shaking resolved in these horses after treatment for EPM. Recently, urinary incontinence and incoordination have been reported in three horses diagnosed with EPM. Resolution of the clinical signs were mixed in those cases.806
Differential diagnoses of the most common neurologic diseases of horses that resemble EPM include equine degenerative myelopathy (EDM), cervical spinal injuries, cervical vertebral stenosis or malformation (CVM), equine herpesvirus type 1 (EHV-1), and equine lower motor neuron disease (ELMND).
Another disease has become the number-one differential for EPM over the last 7 years. West Nile Virus (WNV) was first reported in the United States in 1999; however, the number of equine cases has increased consistently from 25 in 1999 to greater than 14,000 in 2002.807 Almost all the equine cases in 1999 had been diagnosed with EPM first before a definitive diagnosis of WNV was determined.808 Asymmetric neurologic deficits with profound weakness and ataxia make it difficult to differentiate WNV from EPM.
Clinical Pathology
A Western blot (WB) analysis for the diagnosis of EPM has been described and commercially marketed.799 Macrophage-cultured S. neurona is used as the antigen. After electrophoresis the blots are probed with suspect CSF or serum. Reactions are seen as bands developing on the blotted membrane. The sensitivity and specificity of WB has been reported as 89% based on 295 postmortem examinations.809 However, these figures are likely based on more severe cases. Although promising, exhaustive examinations of the sensitivity and specificity of the test in clinical cases are not yet available. Recent research suggests that the sensitivity is excellent, but the specificity in current clinical cases is much lower than originally reported.810 There is no apparent serologic cross-reactivity between the parasites of EPM (S. neurona and N. caninum) and T. gondii. Use of the WB for antibody to S. neurona differs depending on the prevalence of the disease in the population studied.811 If the test is applied in the normal horse population, where the prevalence of EPM is likely less than 1%, the predictive value of a positive test is extremely low (<8%) based on the 89% sensitivity and specificity. However, given the presence of neurologic signs, the prevalence increases dramatically (50% at Ohio State), leading to a positive predictive value of about 90%. This suggests the test should not be applied in normal horses. A recent report demonstrates that the specificity of WB was much lower than originally reported.812 Another report attempted to demonstrate increased specificity for WB for S. neurona antibody detection by blocking the reaction using Sarcocystis cruzi antibody.813 However, a double-blind investigation indicated that this increased specificity is questionable (William J.A. Saville, unpublished data).
Several new diagnostic tests are still measures of reactivity to antigens. Initially, a recombinant surface antigen (SAG1) protein was discovered, and an ELISA test was developed to test horses for S. neurona exposure.814 However, no mention of controlled environment or heat-treated feed in this study makes it difficult to interpret the results. Further development of an ELISA test using a recombinant baculovirus-expressed SAG1 antigen was completed by others.815 This test was able to detect both naturally and experimentally infected horses and other species. Another diagnostic test was developed using direct agglutination, the S. neurona agglutination test (SAT).816 In mice the sensitivity of SAT was 100% and the specificity 90%.816 In California an indirect fluorescent antibody (IFA) test was developed for detection of S. neurona antibody.817 This test was compared with two currently commercially available WB tests, and the results suggested that the IFA was better than either WB test with regard to specificity.817 Further evaluation of the IFA using both naturally and experimentally infected horses resulted in likelihood ratios that would be useful in diagnosing EPM.818 The one concern regarding this test is that it may cross-react with horses recently infected with Sarcocystis fayeri, another common parasite infecting U.S. horses.819 Further research with the IFA suggests that using this test on CSF of horses has no benefit if used on the serum in the diagnosis of EPM.820
More recently, it has been reported that surface antigens (SAGs) of S. neurona are similar to the SAGs of T. gondii.821 The SAGs of S. neurona were named SnSAG1 through SnSAG4.821 Recombinant proteins were developed to the four SnSAGs to produce a set of ELISAs for detection of antibodies to the SnSAGs.822 Testing these ELISAs with confirmed cases of EPM serum and CSF samples suggested that the SnSAG2, SnSAG3, and SnSAG4 assays would be good tests in the diagnosis of EPM.822 The ELISA to the rSnSAG1 demonstrated poor sensitivity (68.2%) and specificity (71.4%), likely because of the differences in strains of the parasite, some of which do not express antibody to SAG1.822 There was also no cross-reactivity when testing horses infected with S. fayeri or Neospora hughesi.822 The most recent test developed for diagnosis of EPM has been the S. neurona—specific IgM-capture ELISA.823 This test was developed using samples from previous experimental infection studies performed at Ohio State University.823 The IgM titers developed and peaked between weeks 2 and 3 postinoculation (PI), and the IgM response waned by 7 weeks PI.823 The IgM test is still a measure of exposure; however, it indicates an acute infection and therefore may be useful in the early diagnosis of EPM.823 Further evaluation of this diagnostic test is needed using large numbers of field tests.
Regardless of the tests developed in the past few years, they are primarily still tests of exposure, so diagnosis of EPM is problematic.824 Therefore, antibodies in serum or CSF must be accompanied by asymmetric neurologic deficits followed by rule-outs of other possible causes of the neurologic deficits.824
One of the difficulties in diagnosing EPM is caused by the large number of horses that have detectable quantities of antibody to S. neurona in the CSF for several months after therapy, or in horses that do not exhibit neurologic signs. S. neurona—specific IgG found in the CSF is assumed to be produced locally because the blood-brain-barrier (BBB) should prevent large molecules from freely entering the cerebrospinal space. However, antibody detected in the CSF could have been produced systemically if the BBB is compromised by disease or if peripheral blood contaminates the spinal fluid during sampling. To help determine whether IgG in CSF was produced locally or resulted from peripheral blood contamination, a series of tests were modified from those used in human patients with neurologic diseases. The albumin quotient (AQ) and the IgG index are calculated using concentrations of albumin and IgG found in serum and CSF of the patient.825 The AQ is a measure of BBB integrity, and the IgG index is a measure of intrathecal antibody production.825 Some reports suggest that horses with EPM usually have a normal CSF albumin concentration and a normal AQ, but the IgG index is usually elevated.826 It has been suggested that these indices, specifically an elevated AQ, can be used to distinguish true-positive WB tests in horses with EPM from false-positive tests resulting from blood contamination during sampling.826 False-positive results caused by blood contamination may alter the AQ and the IgG index by increasing the albumin and IgG concentrations in the CSF.826 It has also been recommended that the CSF indices may be used to monitor the response to therapy for EPM by identifying a decrease in the IgG index that would mark a decrease in intrathecal antibody production over time. However, small numbers of horses were used in the initial evaluation of these tests, with some inconsistencies in the reported results. Although the IgG index did decrease in 9 of 12 (75%) horses treated for EPM, 3 of 12 horses demonstrated an increase in the IgG index.827 The reliability of the CSF indices has been questioned by others.828,829 One controlled investigation suggests the CSF indices are inconsistent, therefore, if used, they should be interpreted with caution.810
Polymerase chain reaction (PCR) is available to aid in diagnosing EPM.830,831 The sensitivity and specificity of PCR testing of CSF has been reported to be 83% and 100%, respectively, when histologically confirmed cases of EPM were used to validate the assay.832 However, other research suggests that the sensitivity of PCR may be only about 40%.833 PCR requires that parasite DNA be intact. A strong inflammatory response favors enzymatic degradation of parasite DNA, and this process may affect sensitivity of the PCR test. Therefore, PCR may be useful early in the course of the disease and in chronic cases.832 In addition, PCR analysis of CSF may be insensitive because the parasite is most frequently found in tissues and not floating freely in the CSF. Therefore, parasite DNA may not be present in CSF, even when adjacent tissue is infected.833 Toxoplasma gondii infections are difficult to detect in people for the same reasons.834 A recent study of clinical cases found that PCR alone was not useful for antemortem diagnosis of EPM based on the low sensitivity (12/151).829
Sarcocystis neurona DNA has been detected in blood samples. The presence of DNA in blood samples is thought to indicate recent ingestion of S. neurona sporocysts and subsequent infection. However, it is not known how long detectable amounts of DNA remain in the blood stream after infection.832 Controlled investigations of PCR testing in horses with neurologic deficits as well as in normal horses are required to define the usefulness of this procedure.
Cerebrospinal fluid analysis has been used to aid in determining the etiology of neurologic diseases in the horse. Early studies suggested that horses with EPM had mildly elevated CSF protein concentrations, increases in numbers of mononuclear cells, and mild elevations in CSF enzyme activity (creatine kinase, CK; aspartate transaminase, AST).835 Two early studies reported marked elevations in the CSF CK activity in horses diagnosed with EPM.836,837 However, more recent studies suggest that neurologic disease of horses cannot be reliably differentiated based on CSF leukocyte counts, CK activity, AST activity, or protein concentration.810,838,839
Several serologic tests have been developed for detection of Neospora caninum antibodies in animal species.840 The three methods currently used are ELISA, IFA test, and direct agglutination test.840 However, these antibody tests are only measures of exposure to the organism. New diagnostic tests for N. hughesi used in experimentally infected equines included a whole-parasite ELISA, a recombinant-protein ELISA, a modified direct agglutination test, and an IFA test, which showed the most consistent results.841 However, a recombinant NhSAG1 ELISA has been developed that suggests, with a sensitivity of 94.4% and a specificity of 95%, it may be an excellent diagnostic test for N. hughesi infections in horses.842
Pathogenesis and Pathologic Changes
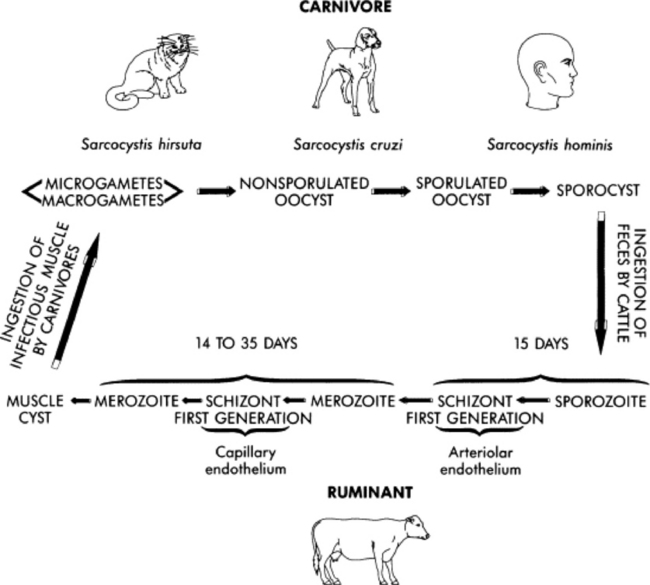
Little is known about the pathogenesis of EPM. It is assumed that the horses ingest S. neurona and that the course of infection and disease is then similar to that observed in other host species infected with Sarcocystis species. Because the sporocysts of S. neurona are passed in the feces of the opossum, infective oocysts likely are introduced into the feed and water supply of intermediate hosts. Once ingested, the sporocysts excyst and release sporozoites, which penetrate the gut and enter arterial endothelial cells of various organs. Meronts develop and rupture the host cell, releasing merozoites into the bloodstream. This is probably followed by a second round of merogony throughout the body. In most sarcocystis-like diseases, this process results in the formation of sarcocysts in the muscle. Subsequent ingestion of the infected muscle tissue by the predator or definitive host completes the life cycle. Sarcocysts of S.neurona had not been found in affected horses, indicating that the horse is likely an aberrant, dead-end host.843 However, a recent report suggested that the horse may develop S. neurona sarcocysts and therefore may be a natural intermediate host.844 The fact that this has been seen only once in the literature is troublesome, and the inability to fulfill Koch’s postulates is also a problem. Horse muscle fed to naive opossums has failed to produce sporocysts after numerous attempts (William J.A. Saville; unpublished data).
Little is known about the life cycle of N. caninum or N. hughesi in horses. Recent reports have demonstrated that the definitive host of N. caninum is likely the dog.845 At present it is not known if the dog is the definitive host of N. hughesi. The dog is the definitive host and also can be an intermediate host, which is similar to T. gondii in cats.840 Unlike EPM caused by S. neurona, tachyzoites have been found in horse tissues, as well as tissue cysts in two horses reported to have EPM caused by Neospora.840 In addition, one case of neosporosis in a foal was determined to have been congenitally infected.780 Congenital infections have not been demonstrated in horses infected with S. neurona.
Sarcocystis neurona has been recovered from CNS lesions in several horses and subsequently propagated in culture in the laboratory.846 When administered to horses parenterally or introduced through the epidural space, cultured merozoites have not induced clinical disease in the horse.846 The merozoite stage of Sarcocystis species is not known to be transmissible to other animals.846 However, nude mice have been inoculated intraperitoneally with cultured merozoites and subsequently developed evidence of S. neurona—associated encephalitis.847 These mice were immunosuppressed strains, and intraperitoneal injection would not likely be the normal route of infection with S. neurona in horses. A better mouse model has recently been developed by feeding sporocysts from feral opossums to interferon-γ knockout (IFN-γ/KO) mice.848 These procedures in IFN-γ/KO mice also help to differentiate Sarcocystis species that are excreted in opossum feces; at least three species appear to be present.848 The mechanism by which the merozoites enter the CNS is currently unknown. The organism likely enters the CNS through infected leukocytes or through the cytoplasm of endothelial cells.846 Recent research may help in confirmation of this speculation. When co-cultured with equine peripheral leukocytes, S. neurona merozoites penetrated the cells within 5 minutes after starting the culture.849 This may be the mechanism for entering the CNS. In addition, a microneme protein for host cell invasion has been documented.850
There have long been anecdotal reports that the parasite may create immune suppression in the horse. One report demonstrated a strong association between health events and development of clinical signs of EPM.797 Earlier reports suggested that nitric oxide is important to resistance to intracellular parasites, decreased nitric oxide was reported in horses with experimental and naturally occurring cases of EPM.851 Another report indicated that decreased levels of transforming growth factor beta (TGF-β) in CSF of horses may be important in development of EPM.852 Two recent reports found a decrease in IFN-γ production in lymphocytes from EPM-positive horses compared with negative horses.853,854 Other studies have corroborated these findings.855 Two reports in mice suggest that protection against S. neurona infections requires CD4 and primarily CD8 cells.856,857 This phenomenon has been demonstrated in T. gondii infections as well.856,857
Much more research is needed to elucidate the mechanisms and pathogenesis of S. neurona infections in horses.
Life Cycle of Sarcocystis neurona
Most specific details regarding the life cycle of S. neurona are currently unknown, although recent research has demonstrated that the opossum is likely the definitive host. The geographic distribution of opossums is similar to the geographic distribution of EPM, and areas with lower seroprevalence of S. neurona appear to coincide with regions outside the natural range of opossums.858 Further evidence that the opossum is the definitive host for S. neurona was obtained by experimental induction of EPM.859 When sporocysts from feral opossums were fed to horses, neurologic disease developed.859 This study has been repeated by other research groups.860-865 However, induction of clinical EPM by feeding Sarcocystis falcatula sporocysts was not successful.866 More recent work has demonstrated at least three species of Sarcocystis sporocysts in feces from the opossum.867 Other studies suggest that four species of Sarcocystis sporocysts may be present in opossum feces.868,869 Development of DNA probes that distinguish Sarcocystis species will better enable researchers to characterize sporocysts from opossum feces, using additional induction studies to help develop a reliable equine model for EPM.
Several recent studies have induced experimental infection in horses with S. neurona sporocysts. Initially, in a University of Kentucky study, naive foals were infected with 1 × 106 to 4 × 107 sporocysts orally, which resulted in mild to moderate neurologic deficits. The sporocysts were collected from wild-caught opossums, cleaned, and administered by nasogastric intubation. Unfortunately, the parasite was not cultured from the CNS tissues, resulting in the inability to fulfill Koch’s postulates.859 At the University of Florida, three studies used sporocysts detected using molecular DNA probes.869 One study administered 1 × 106 S. neurona sporocysts and another 5 × 105 sporocysts orally once daily for 7 consecutive days.864,865 Both studies resulted in mild to moderate neurologic deficits, and no parasite was detected.864,865 In another Florida study, sporocysts characterized as S. falcatula were administered to horses, with no development of neurologic signs and no seroconversion.866 This study corroborated that the opossum excreted more than one Sarcocystis species of sporocysts.866 Recently, a study at Ohio State University attempted to infect horses using 8 × 104 S. neurona sporocysts with three different treatment groups.863 All nine horses in the infected groups developed neurologic signs; however, the most severe signs (mild to moderate) were seen in horses in the transport stress group. Unfortunately, as in the Kentucky study,859 Koch’s postulates were not fulfilled in the Ohio study.863
The previous studies attempted to mimic stress using dexamethasone, but the clinical signs were less severe, and the horses’ clinical signs appeared to improve.859,863,864 In addition, regardless of the dose of sporocysts administered, some horses demonstrated an improvement in their clinical signs with no treatment. These results suggest that horses are capable of clearing large numbers of these organisms. Those findings may explain the high number of CSF antibody-positive horses with no evidence of neurologic deficits. Equally troubling, after orally inoculating horses with 1 × 108 S. neurona sporocysts, clinical signs of neurologic deficits were readily detectable; however, no parasite was found in the CNS at 7 or 14 days postinfection (PI) (W.J.A. Saville and J.P. Dubey, unpublished observations). In the natural intermediate host, the raccoon, S. neurona was readily detectable in the CNS 7 days PI.870
Additional infection studies have been attempted in horses. A horse with severe combined immunodeficiency disease (SCID) received characterized sporocysts of S. neurona orally. Although a parasitemia was detected in this horse, the first detection of the parasite in an experimentally challenged horse, the horse did not develop evidence of neurologic dysfunction. This suggests that development of clinical signs of EPM requires an intact immune system, which also supports anecdotal evidence that EPM is a neuropathologic disease in horses.860,871
Studies at Ohio State University (OSU) have further characterized S. neurona infection in horses. The infectious sporocysts were produced using laboratory-raised opossums and infected raccoons and the life cycle as previously reported.870,872 The second trial was done to determine the effect of sporocyst dose on development of clinical signs of EPM.862 Horses in the group that received 1 × 106 sporocysts seroconverted earlier and developed more consistent clinical signs than those infected with lower doses of sporocysts. Horses were transported a second time and developed worsening clinical signs after residing at the Veterinary Teaching Hospital at OSU.862 Therefore, another trial was carried out testing the effect of a second transport after infection with sporocysts immediately on arrival at the first study site.861 The results demonstrated more significant clinical signs in the horses not transported a second time, refuting the hypothesis from the 2002 study. Neither the 2002 study nor the 2004 study resulted in detection of the parasite in the tissues of infected horses. Another study, in 2005, demonstrated that S. neurona could be detected in the blood of an experimentally infected, immunocompetent horse.873 The horse had been infected daily for 98 days, and S. neurona was detected from the blood of one of six horses tested, demonstrating that the parasite could be detected in immunocompetent, experimentally challenged horses.873 Because of the lack of parasite detection in equine infection models at most sites, researchers at OSU decided to attempt culture early in the infections rather than at the end of the infection period. Eight naive horses negative for antibody to S. neurona were included; six were infected with sporocysts derived from laboratory production using the opossum-raccoon cycle, and two were control animals.874 Parasite was cultured from mesenteric lymph node, liver, and lung at 1, 2, and 7 days PI; 2, 5, and 7 days PI; and 5, 7, and 9 days PI, respectively.874 Although no parasite was detected in CNS tissue, evidence of infection was present at 7 and 9 days PI.874 This recent OSU study was able to fulfill Koch’s postulates and shows evidence that the parasite can invade the tissues very quickly after ingestion, as demonstrated in previous studies in two different species of animals.844,848,870 Further work needs to be done to examine an equine model for the disease.
Unlike most Sarcocystis species, S. neurona may aberrantly infect a large number of intermediate hosts. Although the full range of intermediate hosts for S. neurona has not yet been identified, several species of animals and birds have been reported to exhibit symptoms similar to those seen in horses with EPM. Several reports indicate that an S. neurona—like organism infected and caused neurologic disease in dogs, sheep, cats, mink, raccoons, a striped skunk, a golden hawk, Pacific harbor seals, sea otters, chickens, a Grant’s zebra, Canada lynx, and a Fisher.875-888 The harbor seals and sea otter had evidence of sarcocysts in the muscle that were S. neurona positive.878,879 This seems to be the first evidence for potential S. neurona sarcocysts to date, although the significance of this finding was not well understood. This positive reaction to anti—S. neurona antibody could be caused by cross-reactivity to other Sarcocystis species. A recent report indicates that S. neurona cycles normally between the opossum and various intermediate host species. Even though the opossum is the definitive host and does shed the parasite in its feces, the opossum does not develop antibodies to S. neurona.889 Sarcocysts are found in the muscles of five species: infected domestic cats (Felis domesticus), nine-banded armadillos (Dasypus novemcinctus), striped skunks (Mephitis mephitis), raccoons (Procyon lotor), and sea otters (Enhydra lutris nereis).872,890-893 It was thought that the domestic cat was only a laboratory intermediate host, but studies in Missouri and Ohio suggest that the cat is a natural intermediate host as well;895,894 this also seems to hold true for the striped skunk.
Recently, the life cycle of S. neurona has been completed in the laboratory.892 Previous reports of serum prevalence of antibodies to S. neurona in striped skunks suggest that they are likely a natural intermediate host as well.896 When muscle from wild-caught raccoons and road-killed armadillos was fed to laboratory-raised opossums, it resulted in shedding of sporocysts infective for ponies, horses, and IFN-γ/KO mice.872,891 High seroprevalence of S. neurona antibodies in armadillos tested (100%) from three states and raccoons (57%) tested from four states supports that these two species are natural intermediate hosts.891,897 After ingestion of the infected muscle, opossums shed sporocysts in their feces. Small numbers of sporocysts were found after feeding sea otter muscle, but the sporocysts were infective for IFN-γ/KO mice.890 The role of the sea otter as a natural intermediate host is likely limited because it is a marine mammal. Results of these life cycle studies demonstrate that many species may be potential intermediates hosts for S. neurona. This wide host range is atypical for Sarcocystis species. This host range behavior is similar to that of T. gondii, which is phylogenetically close to S. neurona.898,899
Based on the number of intermediate hosts determined so far, a number of isolates of S. neurona are postulated to exist. Recent work found that there are differences between South American and North American isolates.900,901 Also, evidence indicates that the U.S. group could be divided into northern and southern U.S. groups, which suggests geographic groupings. Other work has demonstrated differences in the SAG1 gene in different isolates of S. neurona, with 73% to 100% sequence similarity.902 This is in contrast to the SAG1 gene of Neospora species, with 96% to 98% similarity.902 These differences in the isolates was confirmed when monoclonal antibodies developed against immunodominant proteins of S. neurona failed to detect all isolates.903
Based on the estimated numbers of opossums in North America, the poor survival rate of these animals, and the small areas in which they travel, S. neurona may be transmitted by routes other than direct contact with opossum feces. Experiments performed by researchers in the 1980s indicate that some transmission may occur through birds.904 In experiments attempting to characterize the life cycle of S. falcatula, birds were apparently infected by aerosol spread.904 Vector transmission was also demonstrated by the recovery of sporocysts after budgerigars, canaries, white mice, and chickens were fed opossum feces.905 The recovered sporocysts were then fed to budgerigars to assess the viability of the sporocysts. Four of six budgerigars died, demonstrating that the sporocysts were viable. These experiments suggest that sporocysts might be transmissible between intermediate hosts. Considering the apparent wide range of natural and aberrant intermediate hosts for S. neurona and S. falcatula, transmission of infectious organisms between intermediate hosts implies that control of disease caused by these organisms may be extremely difficult. Insects such as flies and cockroaches may also be transport vectors for S. neurona. Early work demonstrated that flies and cockroaches may act as transport vectors for T. gondii.906,907 In addition, fatal pulmonary disease developed in psittacine birds fed cockroaches after the cockroaches had been fed opossum feces.908 Although this suggests that insects may play a role in transmission of S. neurona, further investigation is necessary to determine which insects are actually involved in its life cycle.
Stress may play a role in the development of EPM,843,899 but limited evidence is available to support this hypothesis. The severity of EPM may be related to the size of the infective dose, immune competency of the host, and the environmental stresses to which the horse is exposed.846 A similar association between immunosuppression and disease has been documented in other species with EPM-like symptoms. For example, recent mouse models have been developed for EPM using nude mice and IFN-γ/KO mice, both of which are immunocompromised strains.847,848 Raccoons have been identified that were concurrently infected with a Sarcocystis-like protozoan and canine distemper virus (CDV).884,909 Interestingly, CDV is known to be immunosuppressive and has often been associated with cerebral toxoplasmosis in dogs, foxes, and raccoons.909 Immunocompromised people are often infected with T. gondii, and stress plays a major role in the recrudescence of the clinical signs of T. gondii—associated encephalitis.910 Infections with either N. caninum or T. gondii can cause T-cell hyporesponsiveness to the parasite antigen. It has also been demonstrated that an intact T-cell response, specifically, appropriate interleukin-12 (IL-12) and IFN-γ production, is necessary for resistance against either N. caninum or T. gondii. The parasite may therefore facilitate further infection by compromising host immune responses.911 Recent evidence suggests that neuropeptides called neuroimmune proteins (NIPs) are released from the CNS when an animal is stressed, which may lead to suppression of lymphocyte production and function.912 Stress leads to high circulating glucocorticoid concentrations, which are also immunosuppressive.912 The combination of high resting concentrations of glucocorticoids and an increase in NIP release may result in immunosuppression and facilitate development of clinical disease in horses infected with S. neurona. Recent evidence from a controlled investigation at OSU demonstrated that health events before diagnosis of EPM were strongly associated with the disease.797 Transport stress and induction of the disease in an experimental equine model provide supporting evidence that stress may play a role in the pathogenesis of EPM.863 It has long been known that transport is a stressor in horses and other species.913-917 Horses are transported year-round to equestrian events in the United States, sometimes across the country and to other countries. Further controlled investigations are needed to examine the role of stress in the development of clinical signs of EPM in horses.
Diagnosis
The diagnosis of EPM has been difficult because of a lack of understanding regarding its pathogenesis and the variety of clinical signs. Postmortem examination was the first method used to diagnose EPM definitively, and many still consider it the “gold standard” for diagnosis. Grossly, the CNS lesions identified postmortem are described as multifocal areas of hemorrhage to light discoloration of the brain or spinal cord.843 Histology often reveals a marked mononuclear perivascular cuffing with necrosis and loss of neurons, with infiltration of monocytes, lymphocytes, some eosinophils, and rarely, neutrophils.802,835 Protozoan organisms can be seen in some of the lesions but are often difficult to detect.835 Difficulty in detecting the organisms increases if the animal has been treated with antiprotozoal medications.918 Immunohistochemical staining techniques can be used to identify parasites definitively in situ.919,920 Postmortem examination is also the definitive diagnostic test for EPM caused by N. caninum (N. hughesi).840 Inmmunohistochemistry is also a useful tool for identification of the Neospora organisms.840 However, a significant problem with this diagnostic method is that, by definition, it cannot be applied in horses antemortem and therefore cannot be applied to most clinical cases. A reliable diagnostic test that can be used for antemortem diagnosis is needed to better understand EPM and appropriately manage horses with this disease.
Epidemiology
Little is known about the epidemiology of EPM, although more and more knowledge is being accumulated about this disease.
A small study from one county in Pennsylvania indicated that the seroprevalence was approximately 45% of the horse population (95% confidence interval, 36.3% to 54.3%), and prevalence increased with age.921 Another report found an overall seroprevalence of 45% among horses in Oregon, with differences in seroprevalence among geographic regions.858 In Oregon the seroprevalence ranged from 22% in the eastern arid region of the state to 65% in the coastal region. A third study reported a 53.6% prevalence of serum antibodies to S. neurona in Ohio horses.922 The Ohio study demonstrated an increase in prevalence with age of the horse, and greater prevalence in southwestern Ohio versus northeastern Ohio. The geographic differences in Ohio may have been related to climatic differences and freezing days in various regions of the state.922 These studies suggest that in many areas of the United States, approximately 50% of the horses may have serum antibodies to S. neurona.923 Another study suggested horses are exposed to S. neurona in the eastern half of the United States at a rate 10% to 15% higher than the exposure rate in the western half of the country.832 More recently, antibodies to S. neurona were found in 33.6% of various equid serum submitted to a laboratory in Colorado. As previously reported, the prevalence increased with age; prevalence was 26% in horses age 1 to 5 years versus 37% in 10-year-old horses.924 Seroprevalence was lowest during the colder months, as reported previously. Another report on seroprevalence of S. neurona in horses demonstrated 27% in California, 28% in Florida, 54% in Missouri, 0% in Montana, and 0% from New Zealand.925 A Michigan study indicated a seroprevalence of 60%, with lower rates in colder areas, which corroborates earlier findings.926 Another study tested two populations of horses for S. neurona antibodies: the wild horse population in Wyoming and horses from western Canada.927 Using the WB test yielded 18 of 276 Wyoming horses positive and 0 of 243 Canadian horses. These results are difficult to interpret in the Wyoming horses because of the range of the opossum; the Canadian results fit with its range. Two recent reports suggest that the seroprevalence of S. neurona antibody in horses in Argentina and Brazil are 35.5% and 35.6%, respectively.784,785 Another recent study examined S. neurona exposure in Brazilian horses using an rSnSAG4 ELISA, resulting in a seroprevalence of 69.6%.928 Another study examined American-born horses exported to India for S. neurona exposure.929 Of the 86 horses tested in this study, 42 were still positive, even though some of the horses had been in India for 13 years. This work suggests that antibody to S. neurona has an extremely long half-life, or that chronic infection occurs with this parasite.
These results suggest that exposure to S. neurona is common, but that geographic differences may exist.
Little work has been performed regarding the prevalence of antibody to N. caninum/N. hughesi in horses. However, recent work found a seroprevalence of 23.3% in sera examined from two U.S. horse slaughterhouses and a lack of antibody detection in Argentina and Brazil.784,785,930 Because of the low numbers of horses involved, these studies may not reflect the true prevalence of N. caninum/N. hughesi antibody in horses. A more recent study found seroprevalence to Neospora of 2% to 3% in Missouri and California.925 Antibodies to N. caninum were found in 31.1% of Wyoming horses.927 However, testing of the Brazilian horses using the rNhSAG1 ELISA demonstrated a seroprevalence rate of 2.5% for N. hughesi.928 Based on these varying results, More work is needed regarding N. caninum/N. hughesi exposure.
No formal studies on the incidence or prevalence of EPM in the United States have been done until recently. Based on the number of cases diagnosed postmortem at the University of Kentucky, the incidence of EPM may be increasing.846 The number of samples submitted for immunoblot analysis suggests that several hundred new cases of EPM might be diagnosed in the United States each year.846 The estimated incidence of EPM based on accessions to the University of Kentucky diagnostic laboratory was 1% or less of all horses each year.843 The number of U.S. cases has only recently been enumerated by the U.S. Department of Agriculture (USDA), which provide a baseline for future reference. Based on the USDA study, the average incidence of EPM was 14 ± 6 cases per 10,000 horses per year.931 The incidence was examined based on primary use of the horse in the operation, and the lowest incidence was found in farm/ranch horses (1 ± 1 cases/10,000 horses/year). Incidence in pleasure horses increased to 6 ± 5 cases/10,000 horses/year. A marked increase followed in breeding horses (17 ± 12 cases), racing horses (38 ± 16 cases), and competition/show horses (51 ± 39 cases). The racing horses did not include horses at racetracks. These estimates reflect a similar incidence of EPM as previously reported, if not lower, and provide a baseline for the future.
Regarding the incidence of neosporosis in horses, no controlled investigations have been performed. There have been six reports of neosporosis in horses caused by N. caninum (N. hughesi).778-783 However, only four cases were in horses with neurologic signs, one was in an aborted fetus, and one was related to an intestinal problem.
Equine protozoal meningoencephalitis has been reported from a number of U.S. states, as well as from Canada, Mexico, Panama, Argentina, and Brazil.784-790 EPM has also been reported in England among horses imported from the eastern United States.794 EPM was diagnosed in an 8-month-old Arabian horse in South Africa that had been imported from the United States approximately 5 months before the onset of signs.793 The most recent report was a California horse that developed clinical signs of EPM after 10 months in Hong Kong.792 An American-born horse developing EPM has also been reported in Japan.791 Neurologic disease has been reported in horses in France, both American-born horses and horses native to France.795,796 EPM is thus primarily a disease of the Western Hemisphere.
Several authors have suggested prevalence of disease may be high among standardbred horses.801,802,918,932 However, two of these authors also suggested that this apparent predilection may be caused by the environment in which horses were kept rather than breed characteristics.801,918 Another case series reported that disease was most common in thoroughbreds.788 A controlled investigation into risk factors for development of EPM did not find a breed predilection, but occupations such as racing and showing demonstrated increased risk compared with breeding and pleasure horses.797 This finding was corroborated by the recent National Animal Health Monitoring System (NAHMS) study.931
Early reports on EPM suggested that young horses had an increased risk of disease.788,802,918 A consistent theme among reports was that at least 60% of the affected horses were 4 years old or younger. An OSU study also found increased risk in young horses, although increased risk was seen in horses older than 13 years as well.797
Historically, EPM has been reported as a “sporadic” disease; more than one case is rarely reported on farms.801,933 In reports of EPM cases from Panama, all affected horses were stabled at the same location, although this is not a common occurrence.787 Also, an outbreak was reported on a farm in Kentucky.934 An Ohio study suggested an increased risk for EPM if the disease was previously diagnosed on the farm (>2.5 times higher), which suggests clustering of cases may occur.797
Several other risk factors for development of EPM have been reported. The Ohio study found an increased risk if opossums were seen on the farm and with the presence of woods on the farm, seasonal effect, or a health event before development of clinical signs of EPM.797 The seasonal effect increased the risk of EPM as the temperature increased, with the highest risk in the fall.797 Risk decreased if a creek or river was present on the farm and if the feed was kept protected from wildlife access.797 The NAHMS study found an increased risk if opossums were seen (vs. never seen) on the premises and even higher risk if the opossums were seen frequently.931 Risk also increased with increased numbers of horses, purchased versus homegrown grain, use of wood chips or shavings as bedding, presence of rats and mice on the premises, and increased human population density. A lower risk was seen when woods were within 5 miles of the premises and when surface water was used as the primary drinking source. As in the Ohio study, the highest risk for disease was in the fall of the year. It is difficult to explain some of the findings from these studies, but management apparently plays a role in development of clinical EPM.
Recent results demonstrate that transplacental transmission of both S. neurona and N. hughesi is unlikely. After following horses at three breeding farms in California and one farm in Kentucky, investigators concluded that there was no detectable risk of transmission of either parasite.935
Treatment and Prognosis
Because of the slow development of a consistent equine model for induction of EPM, and because clinical patients require medication due to the severity of the disease, treatment regimens have evolved empirically. Until recently, recommended therapy had not changed since EPM was originally identified. However, recent use of liquid combination therapies with questionable stability resulted in a lack of response and consistently longer duration of treatment. This has led to the development of novel treatments.
The standard therapy for horses with EPM is a combination of sulfadiazine and pyrimethamine, both antifolate medications. Based on the description of the pathologic lesions of EPM and identification of organisms that resemble T. gondii, the first recommendations for treatment of EPM were extrapolated from therapy used to treat toxoplasmosis in humans.936,937 Numerous changes have since been made with regard to dosage and duration of the therapy.896,933,938,939 Most recommendations were empirically based on clinical impressions rather than controlled clinical trials.896 More recently, some therapeutic recommendations have been based on pharmacokinetic data.940,941 One study tested pyrimethamine, trimethoprim, sulfonamides, and combinations of these drugs against S. neurona merozoites in tissue culture.942 Pyrimethamine was demonstrated to be completely inhibitory and coccidiocidal at 1.0 μg/mL. The same was true for trimethoprim at 5.0 μg/mL. None of the sulfonamides alone had activity at 100 μg/mL. Sulfonamides (5.0 or 10.0 μg/mL) in combination with pyrimethamine (0.1 μg/mL) improved activity against S. neurona.942 However, these findings are based on in vitro studies, and further work is needed in controlled clinical trials in horses. Controversy surrounds the duration of therapy required to treat horses with EPM effectively; initially, recommendations were based on clearing of specific IgG from the CSF, as indicated by a negative WB. However, many horses remain CSF positive for antibody to S. neurona for months after therapy. Many clinicians have adopted the recommendation that the medications be continued at least 2 weeks after resolution of signs or 4 weeks after a plateau of the clinical signs. Current recommendations for the pyrimethamine/sulfadiazine combination is 20 mg/kg of sulfadiazine once or twice daily and pyrimethamine at 1 mg/kg daily orally for at least 150 to 180 days. Horses with EPM are often treated for long periods with medications that act by inhibiting folate metabolism. Some suggest that complete blood counts should be monitored for signs of folic acid deficiency in horses treated for EPM. Potential side effects of treatment with antifolate medications include bone marrow suppression, anemia, colitis, and even teratogenesis. Most of the anemias are mild and improve after withdrawal of the medication. One other side effect of trimethoprim-sulfamethoxazole and pyrimethamine, a commonly used combination in the past, is its effect on reproductive function in pony stallions.943 Although it may not affect semen quality, testicular volume, sperm production efficiency, erection, or libido of healthy stallions, it may induce changes in copulatory form and agility and alter the pattern and strength of ejaculation.943 Therefore, caution should be used when treating stallions for neurologic disease believed to be EPM.
Recently, triazine derivatives have been used to treat EPM. Two of these drugs, diclazuril and toltrazuril, were originally designed for use as herbicides and have been used in other countries in the prophylaxis of coccidiosis in poultry and swine. The response to therapy in horses with EPM was slightly better than the response documented for the standard therapy.944 The pharmacokinetics of both diclazuril and toltrazuril have been demonstrated.945 Currently, diclazuril is only available as a ration premix, so large volumes must be given daily. Another disadvantage is the poor palatability of diclazuril in its present form. One advantage to use of these compounds is an appreciably shorter duration of therapy. Diclazuril is administered at 5 mg/kg for a minimum of 28 days.946 Recent in vitro testing for activity of diclazuril against S. neurona has been demonstrated.947 Diclazuril may need to be administered by nasogastric tube daily.946 Toltrazuril is another coccidiostat becoming increasingly popular because of its ease of use and good absorption orally in horses.948 Toxicity studies of toltrazuril in horses at 50 mg/kg for 10 days resulted in mild anorexia and depression.949 The current recommended dose is 5 to 10 mg/kg for a minimum of 28 days.946 Further in vitro evidence indicates that ponazuril, a metabolite of toltrazuril, is effective against S. neurona.950
Nitazoxanide (NTZ) is another novel treatment recently used in the treatment of EPM. NTZ is a 5-nitrothiazole with a broad spectrum of activity against bacterial, protozoal, and helminthic parasites.946 It has been shown to kill S. neurona in cell culture.946 Toxicity studies showed that when horses were given two times the recommended dose, they became lethargic after 1 week of daily dosing.946 When horses were given NTZ at four times the recommended dose, they became significantly ill, with one death.946 At present, the suggested dose schedule is 25 mg/kg once daily for the first week and 50 mg/kg once daily for the next 23 days.946 Two of these three new EPM medications, Marquis and NTZ, have been approved by the U.S. FDA.
The prognosis for horses diagnosed with EPM is similar regardless of the treatment used. Most reports suggest an approximate improvement rate of 70% when using the standard therapy,804,951,952 but earlier work suggested the success rate of therapy was about 50%.939 Less than 25% of affected horses may return to their original function, although little objective information exists on this issue.804 A recent study with diclazuril resulted in approximately 75% improvement in horses severely affected with EPM.944 In the diclazuril study, approximately 30% of the horses (11/36) treated either returned to their original level of performance before EPM diagnosis or improved their level of performance.944 An efficacy study of 70 horses given NTZ found 63% of the horses met the criteria for success after treatment.946 A growing concern is the percentage of horses which have a relapse in clinical disease after cessation of therapy. Some horses will relapse days, weeks, or even months after cessation of therapy, but the mechanism of relapse is unknown.896 Relapse may be caused by recrudescence of a truly latent stage of the parasite, presence of a small, persistent focus of infection, or perhaps reexposure to the parasite.896 Anecdotal estimates of the relapse rates range from 10% to 28% of treated horses.804,951,952 In a controlled study performed at OSU, relapse rates were 19%, close to previous anecdotal reports.953 The relapse rate using diclazuril for the treatment of EPM was less than 5%.944
Identification of protein activity in S. neurona merozoites has demonstrated two potential targets for therapy, including serine protease and enolase.954,955 In the future, better treatments might be developed against certain proteins to remove or reduce relapse rates and improve the resolution of clinical signs of EPM.
Before recognition of EPM, corticosteroids were widely recommended for treatment of neurologic diseases in horses. However, corticosteroids should be used with caution in horses with suspected EPM because the host immune response to the organism could be adversely affected.789,794,936,937 NSAIDs and DMSO have also been routinely used in the treatment of horses with EPM since the mid 1980s.794,804
Because antibody to S. neurona persists in CSF for long periods in some horses with EPM, and because some horses may not mount a sufficient immune response to clear the organism, immune stimulants (nonspecific T-cell—stimulating compounds) have been recommended.804 Unfortunately, no controlled trials have examined the efficacy of these treatments.
Supplementation with folic acid, folinic acid, and brewer’s yeast has been recommended for treatment of presumed folic acid deficiency, particularly in pregnant mares.794,939 However, folic acid supplementation has been discouraged by other investigators because of poor absorption and the potential for toxic effects on bone marrow activity.804 Toxicity has also been reported in newborn foals born to mares that were treated for EPM with antifolate medications and concurrently supplemented with folic acid.956 These foals showed evidence of bone marrow aplasia and hypoplasia, renal nephrosis or hypoplasia, and skin lesions. Another case report involved an adult horse being treated for EPM.957 A cause-and-effect relationship between folic acid supplementation and these developmental abnormalities has not been conclusively demonstrated. At present, however, folic acid supplementation should not be used, particularly in pregnant mares, until controlled clinical trials can be performed to corroborate or refute these findings.
Use of additional supplements, such as vitamin E and thiamine, that may facilitate healing of nervous system tissue have been recommended for treating horses with EPM.804,951 However, clinical trials have not been performed to establish the efficacy of this supplementation.
Prevention
Based on the proclivity for transportation to equestrian events and the nature of the horse business, prevention of clinical cases of EPM will be difficult. A complicating factor is that the parasite is widespread throughout much of the United States. A killed—S. neurona vaccine was developed and released on a conditional license in 2000. The vaccine has been shown to induce both humoral and cell-mediated immunity in the horse.958 However, efficacy of the vaccine to prevent EPM using an equine model have not been completed to date. Because development of other parasite vaccines is extremely difficult, efficacious vaccines most likely will not be developed for many years.949-962
Based on age as a risk factor for the disease, close monitoring for evidence of neurologic disease in high-risk age-groups (young horses, old horses) may help detect EPM early. The seasonal risk for EPM should raise the index of suspicion that it may be the cause of the clinical signs when horses are presented for neurologic disease in the warmer months. This seasonal risk factor is compounded by many major horse competitions occurring in the fall. Therefore, monitoring of horses subsequent to transport and competition may be helpful. Access to feed and water by wildlife (opossums) and pests (mice, rats) should be restricted by using rodent-proof containers, which may help to prevent some cases of EPM. Forages should also be protected from wildlife access by keeping the forages in enclosed facilities to prevent access to opossums.797 Some research suggests that preventing bird access to facilities may help to prevent some cases of EPM, although it is not known what role birds may play in its pathogenesis. Ingesting sporocysts may result in passage through the intestinal tract of birds and may be infective for other species of wildlife or horses.904,905 Horses may develop EPM after some other health event, as confirmed by a recent controlled investigation.797 Therefore, monitoring of broodmares close to foaling and horses that develop a major illness or injury is important because it may assist in the early diagnosis of EPM cases.
Focusing prevention on manipulation of risk factors for the disease, such as keeping opossums out of the feed and water, is very important. However, one also must consider the intermediate host’s role in EPM. As discussed previously, several species of mammals have been reported to act as natural or laboratory intermediate hosts in the life cycle of S. neurona.872,891-893 These animals are not a threat unless they are dead. Therefore, veterinarians should encourage horse owners to pick up dead cats, armadillos, skunks, and raccoons on their property. It is important to dispose of the carcass so that opossums cannot eat them and excrete more infectious organisms to infect horses. Picking up these carcasses should be done carefully with an inverted plastic garbage bag, plastic gloves, or some other instrument.
These protozoan parasites are extremely difficult to kill in the environment. A recent study tested the most common disinfectants used in veterinary medicine, including povidone-iodine, chlorhexidine, formalin, two different strengths of NaOH, and high-temperature steam.963 The S. neurona sporocysts thrived on the disinfectants and the lower concentration of NaOH. At the higher concentration of NaOH, the parasites did not survive the treatment, however, most barn materials could not withstand this agent. The parasite could not withstand temperatures of 60°C (140°F) for 1 minute or higher temperatures for shorter periods. Therefore the only alternative for cleaning horse facilities is use of steam.
Recently, medications to prevent development of clinical signs in IFN-γ/KO mice and in horses were attempted. The first study used pyrantel tartrate, a daily anthelmintic prophylactic treatment, and concluded that it does not prevent S. neurona infections in mice.964 A similar study in horses tested for seroconversion in serum and CSF and days to seroconversion.965 There was no difference in the groups receiving and those not receiving pyrantel. Another study used the triazine-derivative anticoccidial diclazuril;* mice treated before or up to 7 days after infection did not develop clinical signs, and no parasites were recovered from the mice.966 This study suggests that anticoccidial drugs may be useful in the prevention of S. neurona infections in animals. A second triazine derivative, ponazuril, was tested in IFN-γ/KO mice.967 A single dose prevented abnormal neurologic signs and death, depending on when it was administered. This further corroborates the potential for prevention of EPM using anticoccidial drugs. In a further study on prevention of EPM in horses, two dose ranges of ponazuril were administered daily based on the recommendations for treatment; 71% of horses given 2.5 mg/kg and 40% given 5 mg/kg developed neurologic deficits.968 Seroconversion was decreased in the 5-mg/kg group compared with the controls. Therefore, this study needs to be repeated to attempt other doses of ponazuril for prevention of clinical signs of EPM, because coccidiostats may be one method of prevention useful in the future.
POLIOENCEPHALOMALACIA (CEREBROCORTICAL NECROSIS)
CHRISTOPHER CEBRA
GUY LONERAGAN
DANIEL GOULD
Definition and Etiology
Polioencephalomalacia (PEM) is a common and important neurologic disease of ruminants1064,1065 with a worldwide distribution. An animal with clinical manifestations of PEM often is referred to as having “polio” or being a “sleeper” or “brainer.”
Polioencephalomalacia is a descriptive term for histologic lesions1064,1066 that may arise from a variety of etiologies. Literally, the name means softening or necrosis (malacia) of regions of the gray matter (polio) of the brain (encephalo). Thus a definitive diagnosis of PEM requires appropriate histologic examination of brain tissue. The possible etiologies of PEM include (but are not limited to) excessive sulfur consumption,1067-1069 presumably manifested through elevated ruminal sulfide production;1070,1071 altered thiamine metabolism;1072 so-called salt poisoning or water deprivation;1073 amprolium administration;1074-1076 the molasses-urea diet;1077 and lead intoxication.1078
Clinical Signs
PEM appears to manifest both subacutely and acutely.1064,1071 In the subacute form, signs may develop within hours or over several days. In the early stages of the disease, the affected animals detach from the herd or flock, become anorectic, and stagger. They often appear blind, walk with the head held erect, and demonstrate a slight hypermetric gait. Occasionally, affected animals are excitable and charge around their enclosure, which may present a significant hazard to the veterinarian and animal handlers. Other early signs of PEM can include diarrhea, hyperesthesia, and muscle tremors, which are most obviously observed as ear flicking or facial twitching. Progression of the condition is associated with cortical blindness, head pressing, opisthotonos, dorsomedial strabismus, miosis, repetitive chewing, profuse ptyalism, and odontoprisis1064,1068,1079-1081 (Figs. 35-10 and 35-11). Despite the defective menace response, the animals usually have normal palpebral reflexes. Affected animals may also develop a variable nystagmus, strabismus (Fig. 35-12), and head tilt. The rectal temperature is normal unless excessive muscular fasciculations have developed. The pulse and respiratory rates are usually increased. An odor of hydrogen sulfide may be detected on the breath if the PEM is associated with excessive sulfur consumption.
Although most of these animals respond favorably to aggressive therapeutic intervention, clinical signs may progress to recumbency, tonic-clonic convulsions, and death. In facilities with certain types of fencing, such as cables, affected animals may push or press with such force that they die of asphyxiation.
In the acute form of PEM, animals are found recumbent and comatose.1064,1071 These animals often experience episodic tonic-clonic convulsions, and they remain recumbent and hypertonic between seizures. The prognosis is poor for acutely affected animals or those with advanced subacute manifestations. Survivors may remain irreversibly decorticated and are culled because of poor performance, chronic anorexia and ataxia, or blindness. However, mildly affected animals may remain as productive members of the herd.
Because the clinical manifestations of PEM may be subtle and nonspecific, they can be confused with other disorders. PEM often is temporarily associated with lactic acidosis that develops after consumption of excessive amounts of readily fermentable carbohydrates. Animals with lactic acidosis may appear ataxic and obtunded in addition to having a foul-smelling, watery stool and a distended, fluid-filled rumen. Concurrent PEM may not be diagnosed, or producers may confuse PEM for lactic acidosis or primary ruminal tympany in acutely affected animals that have remained laterally recumbent for some time.
The major differential diagnoses for PEM include enterotoxemia type D (focal symmetric encephalomalacia form), Haemophilus meningoencephalitis (thrombotic meningoencephalitis), coccidiosis with nervous system involvement, listeric meningoencephalitis, vitamin A deficiency, ethylene glycol poisoning, locoism, rabies, and infectious bovine rhinotracheitis encephalitis (calves only).
Clinical Pathology
Although a definitive diagnosis depends on histologic confirmation, a presumptive diagnosis may be made antemortem based on the history and clinical signs or on a definitive diagnosis in herdmates of affected animals. If a diagnosis of PEM is made, either presumptive or definitive, attention should be focused on identifying the likely causes so that exposure of herdmates to etiologic agents can be mitigated or eliminated. The investigation may proceed at the animal, herd, and environmental level to identify evidence supportive of sulfide toxicity, thiamine deficiency, lead toxicity, or water deprivation—salt toxicosis.
Sulfide concentrations in the ruminal fluid and gas cap in experimentally induced sulfur-associated PEM have been shown to be high.1082,1083 However, unpublished data supported a finding of a decrease in gas cap sulfide concentrations in naturally developing PEM associated with increased sulfur consumption.1084 This probably is caused by the rapid metabolism of sulfate to sulfide in the rumen and resultant absorption or eructation of hydrogen sulfide (H2S). Animals with naturally developing PEM are likely to have been anorectic for some time, resulting in a decrease in oxidized and reduced forms of ruminal sulfur.
Estimation of the rumen gas cap H2S concentration in clinically healthy penmates of affected cattle is an effective chute-side diagnostic procedure that can indicate excessive sulfur consumption.1071 This method provides real-time results that may aid direction of further animal and environmental investigations. In short, an area in the left paralumbar fossa is prepared for ruminocentesis. An 18-gauge, 3½-inch spinal needle is inserted through the body wall and into the rumen gas cap. A modified gas sampler is attached to the spinal needle by means of an extension set, and a known amount of gas is drawn through an H2S detector tube.1083 It is important to adjust the values to account for any dead space of the sampling instrument, such as the extension set and other modifications. H2S concentrations greater than 1000 ppm are indicative of excessive sulfur consumption.1085
Appropriate blood samples may be analyzed for lead concentration and possibly estimation of thiamine status. Thiamine status generally is evaluated using one of several available methods, including determining the total blood thiamine concentration using a thiamine-dependent Lactobacillus bioassay.1086 The erythrocyte thiamine pyrophosphate concentration may be measured by high-performance liquid chromatography (HPLC).1087 The value of estimating all phosphorylation forms of thiamine (free, diphosphate, and triphosphate) is questionable. Table 35-9 shows the reference ranges for the total thiamine concentration in normal and affected cattle.
Another method of evaluating thiamine status is determining erythrocyte transketolase activity. This is a sensitive and specific measurement of active thiamine status.1088 Transketolase catalyzes the reaction between xylulose-5-P and ribose-5-P to form sedoheptulose-7-P and 3-phosphoglyceraldehyde in the pentose phosphate pathway. Normal mean transketolase activity has been reported to range from 0.301 to 2.9 mmol pentose/hr/109 RBCs (mean, 0.782). Transketolase assays often are reported as the mean thiamine pyrophosphate effect (Table 35-10). This test compares the specific activity in the active (holoenzyme) and inactive (apoenzyme) forms with the activity of the two forms after addition of thiamine to the homogenates. A large increase in specific transketolase activity after addition of the thiamine pyrophosphate suggests thiamine deficiency. Theoretically, in animals with thiamine-associated PEM, the concentration of holoenzyme is decreased, and that of the apoenzyme is increased. Thiaminase may be detected in the rumen and feces of affected animals, but the value of this assay is questionable.1072,1087,1089,1090 If thiamine deficiency is identified in an affected animal, caution should be used in interpreting the results because a period of anorexia may result in a decrease in ruminal de novo synthesis;1092,1091 thus, affected animals may have a thiamine deficiency that develops secondary to PEM.
Table 35-10 Mean (and 95% Confidence Range) Values of Erythrocyte Transketolase as Percentage of Thiamine Pyrophosphate Effect in Erythrocytes of Normal Cattle and Sheep and in Patients with Polioencephalomalacia
| Species |
Normal (%) |
Polioencephalomalacia (%) |
| Cattle |
15 |
172 |
| (2–114) |
(120–247) |
| Sheep |
23 |
122 |
| |
(12–41) |
(96–158) |
From Edwin EE et al: Diagnostic aspects of cerebrocortical necrosis, Vet Rec 104:4, 1979.
Changes in CSF of affected animals usually are vague. They include mild pleocytosis (5 to 50 WBCs/dL) with vacuolation and increased protein concentrations (>50 mg/dL).1072,1093
Electrophysiologic studies of affected animals show a normal latency and decreased amplitude of the late peaks of the visual-evoked potentials. These changes reflect a decreased population of neurons capable of responding to the photic stimulation.1094 Electroencephalographic (EEG) changes in some animals include constant high amplitude (50 to 60 mV) and slow activity (1 to 4 Hz). Another change is diffuse lowered activity, which is consistent with diffuse necrosis.1095
Environmental investigations should include evaluation of all practical feed and water sources for sulfur concentrations or the possibility of lead contamination. The diet should also be checked for molasses and urea content.
Pathogenesis
The fundamental lesion with PEM appears to be neuronal edema with secondary compartment syndrome. In most cases the edema is postulated to result from adenosine triphosphate (ATP) depletion and insufficient function of the sodium-potassium pump. With salt intoxication, aberrant accumulation of intraneuronal osmolar substances, followed by mass movement of water into neurons, offers a different pathway to cerebral edema. In either case, the bony calvarium limits expansion, and neuronal swelling leads to pressure necrosis. Additional neuronal dysfunction independent of edema and compartment syndrome cannot be ruled out.
The brain is susceptible to PEM because of its high energy and oxygen requirements. Brain tissue is only about 2% of body weight but accounts for 20% to 30% of body glucose utilization and 20% of oxygen utilization in adults.1096 More than 85% of glucose used by the brain is energy substrate, and the blood-brain barrier prohibits the switching to many alternate energy substrates. Compared with liver or muscle, the brain has relatively small glycogen stores, only about a 10-minute supply. It is entirely dependent on circulating oxygen, and under normal conditions, the extracellular fluid contains approximately 100 times as much glucose as the oxygen required for complete aerobic glycolysis. Thus, oxygen delivery or utilization is much more rate limiting for ATP production under most circumstances than glucose availability,1097 and factors that inhibit oxygen utilization (e.g., H2S, lead) could have profound effects on neuronal ATP production.
One intriguing but controversial theory on cerebral energy metabolism could further explain the high neuronal susceptibility to ATP depletion. By this theory, glucose is reduced anaerobically to lactate in astrocytes.1098 This lactate diffuses into neurons, which are then absolutely dependent on aerobic glycolysis for ATP production. Even if this theory is false, it is unlikely that anaerobic glycolysis alone could meet neuronal ATP needs.
Much of the early work on PEM focused on the importance of thiamine (vitamin B1, thiamin, aneurin). In adult ruminants, thiamine is produced by rumen microbes at a rate that is marginally faster than the rate of consumption; very little is stored.1099 Preruminants depend on dietary thiamine. Thiamine compounds play several important roles in the glycolytic pathways. Thiamine pyrophosphate (thiamine diphosphate) is an important coenzyme for transketolase, the rate-limiting enzyme in the hexose monophosphate pathway (pentose phosphate shunt) of glycolysis, and the α-ketoacid dehydrogenases of the Krebs cycle. The role of thiamine in the hexose monophosphate pathway was long thought to be key in the development of PEM, but this pathway actually accounts for less than 3% of cerebral glycolysis.1100 It is unlikely that impairment of this pathway alone could lead to such severe disease. However, decreased function of the Krebs acid cycle through inactivity of the α-ketoacid dehydrogenases could probably cause the necessary reduction in ATP production. Thiamine triphosphate may also play a role in neuronal function independent of its enzymatic function.
The dependence of ruminants on microbial thiamine production has lead to investigation of factors that might decrease production, absorption, or function. The mechanisms proposed include ruminal production of bacterial thiaminases, production or ingestion of inactive thiamine analogs, ingestion of preformed plant thiaminases, decreased intake of preformed thiamine by preruminants, impaired absorption or phosphorylation of thiamine by rumen bacteria, increased fecal excretion of thiamine, and decreased ruminal production of thiamine diphosphate. The most frequently reported inactive thiamine analog is amprolium.1074-1076 Two types of bacterial thiaminases have been described. Thiaminase I, which is produced by Bacillus thiaminolyticus or Clostridium sporogenes,1101 catalyzes the cleavage of thiamine at the methylene bridge between the pyrimidinyl and the thiazole ring. A basic cosubstrate is required to combine with the pyrimidinyl derivative to form a new compound1102 (Fig. 35-13). Many common feedlot medications, including benzimidazoles, levamisole, and promazines, appear to be able to serve as this cosubstrate.
Several plants also appear to make a thiaminase similar to thiaminase I. These include bracken fern (Pteridium aquilinum)1103,1104 horsetail (Equisetum arvense),1104 and Nardoo fern (Marsilea drummondii).1105 Of these, only the Australian Nardoo fern has been strongly linked to outbreaks of PEM. Thiaminase II is produced by Bacillus aneurinolyticus, which proliferates in response to excessive grain intake.1106 The enzyme catalyzes the hydrolysis of the methylene bridge between the two ring structures of the thiamine molecule. The specific relationship of this thiaminase to the clinical syndrome of PEM as seen in the field is unclear.
Complete correlation has not been established among production of ruminal and fecal thiaminase, tissue and plasma concentration of thiamine, and development of clinical encephalopathy.1089,1107 Some affected animals may show normal amounts of thiamine in the plasma but have greatly decreased levels in the erythrocytes and other tissues, supporting the diversity of causes of PEM.1108
Dietary sulfur and sulfates are an important factor in the development of many cases of ruminant PEM. Beef cattle require 0.15% to 0.20% sulfur on a dry matter basis.1109 Sources of sulfur include elemental sulfur,1067 feed additives such as gypsum and ammonium sulfate,1068,1110 feedstuffs such as corn-processing by-products,1109 cruciferous crops,1111,1112 molasses,1077 and fertilizers. Water can be an important contributor to sulfur intake, usually in the form of sulfates.1070,1113,1114
There are two primary metabolic pathways of sulfur in the rumen.1114-1119 The assimilatory pathway involves reduction of sulfate to sulfides and incorporation into sulfur-containing organic compounds such as cysteine and methionine.1117 These are ultimately incorporated into microbial crude protein. The dissimilatory pathway is an energy-producing pathway in which microorganisms use sulfate as a terminal electron acceptor, similar to how mammals use oxygen.1120 The end product is liberated sulfide ion. At a ruminal pH of 5.2, 97.2% of sulfide ions are in the form of H2S and move freely to the rumen gas cap.1116,1121 H2S is readily absorbed and transported to the liver and oxidized to sulfate.1121,1122 Some H2S may be lost through eructation,1082,1121 but the significance of this route has been questioned.1123 Excess sulfur is excreted in the urine and large intestine1124,1125 or recycled to the rumen.1126,1127 A period of adaptation is required for maximum H2S production after exposure to sulfur.1118,1124,1127-1129
Because sulfur and sulfate demonstrate low cellular toxicity, it is unlikely that sulfur-associated PEM results from a sulfate or sulfur toxicity. However, sulfides are highly toxic.1130-1132 Sulfur-associated PEM is more likely to occur secondary to a sulfide toxicity.1069,1071,1082
It has been proposed that the pathogenesis of sulfur-induced PEM involves inhibition of cytochrome-c oxidase, an enzyme in the electron transport chain; this chain is important for regenerating the intermediaries of aerobic glycolysis and the final round of ATP production. With oxygen availability already limiting aerobic glycolysis in the brain, inhibiting the electron transport chain could have a profound effect on neuronal ATP production.
For highly toxic sulfide to reach the brain, it must escape hepatic oxidation. This might be achieved by two mechanisms. A surge in ruminal sulfide generation usually follows a period of adaptation to high-sulfur diets. This may overwhelm the hepatic detoxification capacity. As an alternative possibility, cattle inhale the majority of eructated ruminal gas.1133 These inhaled gases can contain high amounts of H2S, and if absorbed via the pulmonary route, the H2S would completely bypass the hepatic circulation. However, this concept has been questioned.1134
In feedlot cattle, a summer peak in PEM cases was associated with consumption of water containing 2500 mg/L of sulfate.1070 The total sulfur intake of these steers was estimated to be 0.6% on a dry matter basis during the hottest days of the year. Most PEM cases occurred between 15 and 35 days after arrival in this feedlot. In another investigation, 11% of weaners consuming a diet containing 0.9% sulfur on a dry matter basis developed clinical manifestations of PEM.1071 Lesions were confirmed in one steer that died. Addition of gypsum (calcium sulfate) to feeder steer rations at a final concentration more than 2% organic sulfate results in a significantly greater risk of developing PEM. The addition of sodium sulfate (0.6% to 0.8%) to diets may induce PEM within 11 days.1082,1128,1135,1136 High sulfur concentrations in well water in combination with accumulation in forage has been traced to an outbreak of PEM in Canada.1071 One survey described an outbreak of cerebrocortical necrosis in cattle eating diets containing 7200 mg/kg of sodium sulfate.1114
The recommended maximum tolerance level of sulfur is 0.4% of dry matter intake.1109 Other effects of excessive sulfur intake are decreases in feed intake and weight gains. Accurate diagnosis of sulfur toxicity requires measurement of sulfur in all food and water sources. Sulfur from water must be included when calculating total sulfur intake. One third of the molecular weight of sulfate is sulfur. Therefore, if an animal drinks 30 L of water a day containing 2000 mg/L of sulfate, this contributes 60,000 mg of sulfate, or 20 g of sulfur. Furthermore, if this animal consumes an average of 10 kg of dry matter daily at 0.15% sulfur, the feed or forage contributes 15 g of sulfur, making the total sulfur intake 35 g, or 0.35% on a dry matter basis. Water therefore may be a substantial source of sulfur.
Some have suggested that sulfides result in thiamine destruction, thereby directly implicating a thiamine deficiency in the pathogenesis of sulfur-associated PEM. Rumen thiamine production was slightly reduced by the inclusion of excessive sulfur in the ration.1137 However, the authors deemed the reduction clinically insignificant. Apparently, sulfur-associated PEM occurs independent of thiamine status.
PEM induced by feeding of molasses and urea is thought to be related to the high sulfur content of the molasses and to the depletion of propionate and other glucogenic precursors induced by the foodstuff.1077 It is not considered to be caused by an underlying thiamine destruction. The tissue thiamine concentrations of animals with molasses-related PEM are normal,1138 and signs are preventable by concomitant feeding of glycerol, which is converted to glucose in the rumen. Outbreaks of PEM in range cattle have been associated with ingestion of the plant Kochia scoparia.1139,1140 The pathogenesis of this condition is unknown; however, some have suggested that the plant has the capacity to accumulate sulfur in the forage.1114
Some cases of PEM cannot be linked to either problems with thiamine or sulfur toxicosis. Other compounds, such as lead, affect electron transport in a manner similar to sulfides and therefore also impair ATP production. Water intoxication creates a similar histologic lesion, but the pathogenesis of edema relates more to the generation of osmotic compounds and fluid shifts with rapid fluctuations of blood and neuronal osmolality.
Epidemiology
PEM has a worldwide distribution. The condition is seen both in individuals and as herd outbreaks. In one instance, approximately 2000 of 2200 sheep grazing were clinically affected with PEM.1067 No predilection by gender or breed is seen, although anecdotal reports suggest that heifers are less likely to develop PEM than steers in a feedlot environment. The condition affects cattle, sheep, goats, deer, camels, and camelids.1068,1074,1141-1144 Although PEM is seen predominantly in animals that eat a high-concentrate supplement, the condition also can occur in unsupplemented animals on pasture. The inciting cause of PEM can be identified in some cases.
One report from the United States indicated a predominance of cases in the summer in range cattle.1064 The age range for susceptibility to PEM has been reported as 3 weeks to 5 years in sheep, 3 weeks to 8 years in cattle, and 2 months to 2½ years in goats. The peak age of incidence is 18 months or younger in cattle and sheep, but this depends on the production system.1064,1071,1145 The incidence of PEM has been reported as high as 90% in some sheep flocks, with mortality of 1% to 10%.1067 The incidence of PEM is high in sheep exported by sea from Australia to the Middle East. In these cases the underlying disturbance was thought to be associated with a thiamine deficiency caused by a lack of rumen synthesis secondary to the shipboard conditions.1146
Necropsy Findings
In cases of sulfur-associated PEM, rumen contents may have an odor of H2S. In other cases there may be evidence of grain overload, coccidiosis, or respiratory disease. Some of these lesions relate to management conditions, but may also relate directly or indirectly to PEM. The macroscopic pathologic brain lesions of PEM include cortical swelling, softening, flattening, and yellowish discoloration of the gyri. Necrotic areas of the cerebral cortex autofluoresce under ultraviolet light (365 nm).1147,1148 Severe cases show herniation of the cerebellum through the foramen magnum or the occipital cortex under the tentorium cerebelli. Animals necropsied months after recovery may show cerebral atrophy and submeningeal cortical cysts. The major microscopic lesion is a diffuse laminar necrosis. Other changes include intracellular and intercellular edema, neuronal necrosis, gliosis, and neuronophagia.1066,1080
Treatment and Prognosis
Regardless of the underlying cause, animals with the subacute form of PEM often respond favorably to parenteral administration of thiamine hydrochloride. These animals may remain blind and may have depressed sensorium for weeks or months.1149 Thiamine should be administered at 10 to 20 mg/kg IM or subcutaneously (SC) three times daily. In severe cases, IV administration of the first dose might be warranted. If given IV, thiamine should be diluted in 5% dextrose or other isotonic fluid and administered slowly to avoid adverse reactions. If no improvement occurs initially, the treatment should be continued for at least 3 days. In some patients, recovery may take as long as 7 days, but most patients show improvement by 24 hours. A single administration of sodium dexamethasone at 0.25–1 mg/kg IM or IV or 1 g/kg of mannitol in a 20% solution IV may be beneficial in reducing cerebral edema. Anecdotal reports indicate that feedlot animals that have recovered from PEM are at increased risk of respiratory disease; therefore, prophylactic antimicrobial administration may be indicated. Convulsions may be controlled with phenobarbital, pentobarbital, or diazepam. Specific dosing regimens are listed in Tables 35-7 and 35-8.
Animals with the acute form of PEM usually have more severe cortical and deep gray matter lesions than animals with the subacute form.1071 These animals generally do not respond to therapeutic regimens. Ruminants with PEM secondary to the molasses-urea diet do not appear to respond to treatment with thiamine. They may respond more favorably to parenteral administration of glucose or enteral or parenteral administration of a glucose precursor.
Prevention and Control
Thiamine supplementation may not prevent outbreaks of PEM. Ultimately, the best way to prevent outbreaks is to manage the dietary intakes of susceptible animals appropriately. Ruminants should be allowed an adequate period of adaptation to high-concentrate rations. All feedstuffs and water sources should be carefully analyzed on a routine basis with an estimate of total sulfur intake. If excess sulfur consumption is a factor, steps should be taken to remove sources such as high-sulfur hay, ammonium sulfate, and molasses. If the excess sulfur intake is unavoidable, steps can be taken to limit its effects. Older members of the cow herd could be used to graze the high-sulfur pastures, and younger, more susceptible animals could be kept to lower-sulfur pastures or given hay supplementation. Personnel should be trained so that animals with PEM can be identified early in the disease and treated appropriately.
Thiamine may be supplemented (3 to 10 mg/kg of feed) in rations in which the concentrate/fiber ratio is high, but this has little or no effect in preventing PEM. Other recommendations for preventing PEM include addition of brewer’s yeast to the ration and gradual adaptation of ruminants (at least 2 weeks) to high-concentrate diets. If present as a feed-limiting additive, gypsum should be removed from the diet. Elimination of supplementation and rotation of pastures have been sufficient for controlling some outbreaks.1145 Supplementation with cobalt in trace-mineral salt mixes may be necessary in deficient areas.
SALT POISONING
Definition and Etiology
Salt poisoning is a common CNS disease of livestock. Salt-rich solutions ingested over time can cause production-related losses and even death. Ingestion of water containing more than 7000 mg/L of total dissolved salts is likely to result in acute salt poisoning.1152,1153 Water that contains less than 3000 mg/L of total dissolved salts is considered safe for consumption. Salt poisoning can be associated with water deprivation.1152,1154,1155 Provided that access to free water is constantly available, animals may tolerate as much as 13% dietary salt intake.1156 The total dietary salt concentration should never exceed 4%.
The acute toxic dose of oral sodium chloride (NaCl) for cattle and horses has been reported as approximately 2.2 g/kg body weight and for sheep about 6 g/kg.1156 With water restriction, the toxic dose of salt is considerably less, and poisonings have resulted from ingestion of 0.9% NaCl in water-restricted cattle.1157 Chronic toxicity can occur at lower dietary salt levels than acute toxicity. Ingestion of water with salt concentrations above 1% uniformly results in toxicosis if no other source of ion-free water is provided.1158,1159
Ingestion of water containing 0.7% salt lowers the fertility of females,1159 and water containing 0.25% salt suppresses milk production in cattle.1160 Dairy calves have been poisoned by daily feeding of 4 L of milk replacer containing 2.6% NaCl.1161 Animals are most susceptible to salt poisoning during the summer because of the increased insensitive loss of water at that time. Salt poisoning in ruminants is most frequently a syndrome of “water intoxication”: a period of restricted access to low-salt water, followed by unrestricted access to water.1152,1154,1155
Clinical Signs
Rapid ingestion of large amounts of salt causes gastrointestinal and neurologic signs.1158,1161-1166 These include mucohemorrhagic diarrhea and colic, head-neck extension (“star gazing”), blindness, aggressiveness, hyperexcitability, psychomotor seizures (paddling and loss of consciousness), vocalization, ataxia, proprioceptive deficits, head pressing, constant chewing movements, nystagmus, muscle twitching, and coma. Death occurs as a result of respiratory failure. Before the onset of neurologic signs, cattle with chronic salt toxicosis may appear to be depressed and dehydrated. Table 35-11 summarizes the spectrum of clinical effects associated with different levels of salt intake.
Table 35-11 Effects of Different Salt Concentrations in Drinking Water on Performance in Cattle
| Salt Concentration (mg/L or ppm) |
Clinical Effect |
| <1000 |
No effect |
| 1000–3000 |
Temporary diarrhea; reduced milk production |
| 3000–5000 |
May reduce milk production and feed intake; may produce reproductive failures (failure to conceive) |
| 5000–7000 |
Conception failures (abortion, infertility), reduced appetite |
| >7000 |
Unsafe, especially in hot weather; may produce encephalopathic signs, abdominal pain, mucoid diarrhea, thirst, salivation, polyuria, central nervous system signs; include knuckling, blindness, convulsions, coma, and abdominal pain |
From McCoy CP, Edwards WC: Sodium ion poisoning in livestock from oilfield wastes, Bovine Pract 15:152, 1980.
Excessive salt intake also may interfere with productivity in the absence of acute neurologic signs. In one study, cattle were given either tap water (196 ppm of dissolved salts) or saline (2500 ppm NaCl), and their milk production was measured.1160 Cows given tap water had a greater fluid intake and a significantly greater lactational persistence and daily milk production than did cows given saline. The serum concentrations of sodium and potassium were normal in the animals fed saline.
The clinical diagnosis of salt poisoning depends on the demonstration of exposure to toxic concentrations (>7000 ppm or 0.7% of sodium), the presence of water deprivation, or the determination of serum or cerebrospinal fluid (CSF) sodium concentrations greater than 160 mEq/L. CSF/serum sodium ratios greater than 1 also suggest salt poisoning. The serum sodium concentration may vary, depending on whether the patient had recently been given ion-free water before measurement. Some animals with acute neurologic lesions may be normonatremic if they have recently drunk to repletion with ion-free water, whereas others that have not had ion-free water may be hypernatremic. The CSF sodium concentration in salt-poisoned animals is consistently elevated and may exceed 200 mEq/L.1162 Ruminal sodium concentrations above 0.36% to 0.5% or brain sodium concentrations above 150 mEq/g or 1800 ppm1167 also suggest salt poisoning in cattle.1162,1163,1168 Rapid intake of low-sodium water during the rehydration phase of the disease may cause intravascular hemolysis with resultant hemoglobinuria; such findings raise the index of suspicion of salt poisoning.1153
The concentration of acetylcholinesterase in plasma and RBCs is decreased in animals that have been ingesting excessive salt (>0.49% of diet).1169 The decrease is first seen after 4 months of continuous ingestion of the high-salt diet.
Pathophysiology
The pathogenesis of salt poisoning involves the deposition of sodium ions in the CNS parenchyma and the CSF, which occurs either acutely from ingestion of a large quantity of salt or chronically after long periods of reduced water consumption. The ionic sodium accumulates in the CSF and neurons by passive diffusion. The resulting hyperosmolality reduces energy-dependent sodium transport mechanisms and the anaerobic glycolytic pathways.1170 These mechanisms normally provide energy by which the sodium ion is removed from the cell cytoplasm.1165 The thirst receptors are triggered in response to the hyperosmolality. The animal is permitted to drink ion-free water to repletion, and the fluid is absorbed from the gastrointestinal tract, resulting in expansion of the extracellular fluid and a return to normal plasma osmolality. Water then diffuses from the blood into the relatively hyperosmolar CSF and neurons, resulting in CNS edema, increased intracranial pressure, and acute encephalopathy. If the patient has had sudden access to a large quantity of salt, the hyperosmolality in the intestine results in saline catharsis and diarrhea.
Epidemiology
Animals are tolerant of high dietary salt levels if they have concomitant access to fresh drinking water. Feedstuffs that are common sources of excessive salt include whey, saline-preserved fish or fish meals, bakery by-products, and certain milk replacers.1171 Confined calves may be poisoned by improperly formulated milk replacers or oral electrolyte replacements.1162,1171 Cattle eagerly ingest large amounts of oil well sludge, which is a potential source of salt for cattle in the western and southwestern United States.1166,1172,1173 Brine is used extensively as a flush during the drilling of oil wells. Effluents from drilling rigs may contain as much as 100,000 ppm of salt. The effluents are also contaminated by heavy metals and magnesium salts, which complicate the clinical syndrome of salt poisoning. Concurrent neurologic disease may predispose animals to reduced water intake, as occurred in a group of goats with locoweed (Oxytropis species) poisoning who became water deprived because of reluctance to move to a water source. They developed clinical and pathologic evidence of salt poisoning when moved subsequently to an area where water was easily available.1154 Salt poisoning caused by water restriction may occur either inadvertently from freezing of water sources in northern climates or from intentional water restriction of veal calves.1162 Ingestion of brackish or tidal water is a cause of salt poisoning in cattle pastured on the coastal regions of the world.
Necropsy Findings
The pathologic changes of salt poisoning include cerebral edema and softening and flattening of the cortical gyri. Microscopic lesions include laminar cortical necrosis, poliomalacia, and occasionally, meningeal or perivascular infiltration of eosinophils. Perivascular infiltration of eosinophils is not as reliable an indicator of salt poisoning in ruminants as in pigs because most affected ruminants show perivascular cuffing of mononuclear cells.
Treatment
Treatment of animals affected by salt poisoning is difficult. Many animals die even after intensive medical treatment. Prognosis depends on severity of clinical signs at the start of treatment: animals that already have significant cerebral edema have a guarded prognosis. Therapy should be aimed at limiting the ingestion of nonionic water and attempting to remove intracellular solutes slowly from the brain while simultaneously controlling cerebral edema and attendant CNS signs. To prevent brain swelling and herniation of the brain through the foramen magnum or the tentorium cerebelli, slow reduction of the CSF and plasma sodium is imperative. Slow intravenous (IV) administration of normal or hypertonic saline is the mainstay of treatment.
Adult cattle should receive normal or hypertonic saline intravenously (IV) at maintenance rate (7% body weight in adult cattle, 10% in neonates).1153 Slight underestimation of fluids is preferable to excess fluid administration. Serum sodium should be monitored regularly and correction effected over 24 hours or longer. Oral fluids can be introduced gradually toward the end of the first day of treatment; salt should initially be added to oral fluids to make them isotonic to blood. When IV replacement is not feasible, isotonic to hypertonic oral fluids should be administered at a maintenance level divided into four to six feedings daily. Access to low-salt water is gradually allowed after 3 to 4 days. Any worsening of clinical signs is an indication to increase the salt level in fluids and may require IV mannitol (0.5 to 2.0 mg/kg as a 20% solution) or oral glycerin (1 mL/kg diluted 50:50 with water) to reduce cerebral edema.1153
In calves, treatment can be accomplished by administration of hypertonic saline concomitantly with feeding of 2 to 4 L of fresh milk daily. First, the plasma sodium concentration is measured, and the calf then is given 1 to 2 L of a hypertonic saline solution IV. The molar strength of the sodium ion in the IV fluid should be equal to or slightly greater than that of the plasma. If the hypotonicity of the whole milk is not counterbalanced by treatment with hypertonic saline, the CNS will rapidly expand as a result of absorption of free water. The plasma electrolyte concentration is measured twice daily. If the plasma sodium concentration declines too rapidly, 1 L of hypertonic saline solution is infused over several hours. The concentration of sodium in this fluid should be greater than the most recent plasma sodium measurement and less than the beginning plasma sodium concentration. If the calf develops nervous twitching, salivation, head-neck extension, stiff forelimbs, or convulsions, 0.5 to 1 g/kg of mannitol is immediately infused IV. A blood administration set is used to filter insoluble mannitol crystals. The calf should have no access to fresh water.
The use of solutions containing 5% glucose is dangerous and probably contraindicated because this represents ion-free extracellular fluid, which can exacerbate brain edema. Administration of corticosteroids (dexamethasone, 0.25 to 1 mg/kg by slow IV injection twice daily for 2 to 3 days) may be helpful in animals with acute cerebral edema. However, potential benefits should be weighed against the possibility of inducing extrarenal sodium retention. If the neurologic signs diminish and the plasma sodium level returns to normal, the animal may be given fresh ion-free drinking water. Thiamine (10 mg/kg by slow IV injection) may be a useful adjunctive therapy.
Control
Cattle should be fenced away from polluted ponds and oil wells. Cattle on coastal pastures should have access to fresh well water. The total daily dietary salt intake should not exceed 4% of dry matter intake. Drinking water must contain less than 7000 ppm of sodium unless the dietary sodium load is reduced correspondingly. Oral rehydrating fluids for calves should be dissolved in strict accordance with the manufacturer’s recommendations and should not be administered for longer than 3 consecutive days.
VITAMIN A DEFICIENCY
Definition and Etiology
Vitamin A (retinol) is found in green plants and can be synthesized by the small intestinal mucosal cells from plant carotenoid precursors. Precursors of vitamin A are usually fed in cattle rations as β-carotene or as retinoids (retinyl palmitate or acetate). Carotenoid in forage is converted to retinol in the liver and gut. Vitamin A deficiency occurs primarily in growing ruminants in feedlots. Deficiency develops under these conditions because the growing animal has a higher requirement for the vitamin, and feedlot-reared animals may have limited access to succulent plants. The vitamin is labile in foodstuffs and is essentially depleted after several years of storage. Diets that are naturally low in vitamin A include cereal grains, beet pulp, and cottonseed hulls. Conditions in which the immune system is challenged, as when exposure to pathogens is high, also increase the requirement for vitamin A.
The clinical signs of vitamin A deficiency in cattle are related to increased intracranial pressure and ill-thrift caused by secondary infections. Signs include intermittent convulsions, depression, and blindness. The usual dietary or management conditions that favor the vitamin deficiency include grazing on dry pastures or cereal grains other than corn, exclusive feeding of cereal grains that have been stored at high temperature and humidity, or prolonged feeding of mineral oil as a preventive for frothy bloat.
Clinical Signs
The neurologic signs of vitamin A—depleted animals are age dependent. Signs in deficient calves include anorexia, ill-thrift, blindness, diarrhea, and pneumonia. The syndrome in adults is characterized by “star-gazing” posture, blindness, diarrhea, anisocoria, nystagmus, strabismus, exophthalmos, loss of pupillary light reflexes, and intermittent tonic-clonic convulsions. The seizures last for only a few minutes and are followed by partial recovery.1174-1176 Animals may die during seizures. Stimulation of the animals frequently precipitates seizures.1177,1178 Death often is preceded by hyperesthesia and coma.1174,1175,1179 Inadequate vitamin A supplementation of calves is associated with unthriftiness, intermittent fevers, and a higher incidence of diarrhea.1180 Vitamin A—deficient adults appear to be in good body condition unless parasitism or some other nutritional deficiency is superimposed on the low vitamin A intake.1174,1181 Secondary factors that can influence the appearance of the animals include concomitant nutritional deficiencies, parasitism, and pneumonia.
The ocular changes of vitamin A deficiency are characteristic. The pupils become dilated and unresponsive. As papilledema develops, the optic disc becomes pale and its borders become indistinct, particularly in the upper quadrants,1174,1176,1182 giving the appearance of an inverted heart. The swollen disc may cast a shadow on the adjacent retina. The color of the disc becomes faded. In advanced cases the disc may become atrophic and appear dull, gray, flattened, and smaller than normal. The retinal blood vessels become tortuous or appear to be occluded as they course over the disc. Retinal detachment and subretinal hemorrhages are possible.1174 Corneal changes are an uncommon clinical finding.1174,1175
Reproductive disturbances can occur, including malformed fetuses, abortions, loss of libido, testicular degeneration, and decreased sperm counts. Calves born to vitamin A—deficient dams are blind, have domed foreheads and thickened carpal joints, and are weak at birth.1183
Vitamin A deficiency can be clinically differentiated from polioencephalomalacia (PEM) and salt poisoning by comparing the menace response with the pupillary light reflex. Calves with lead poisoning and PEM generally have intact pupillary light reflexes because of the proper functioning of the mesencephalon and optic nerves, whereas vitamin A—deficient cattle have absent pupillary light responses because of retinal degeneration and constriction of cranial nerve II at the level of the optic foramen.
Clinical Pathology
Assay of vitamin A and carotene concentrations in the plasma and feed is the most direct method of diagnosing the dietary deficiency. The concentration of plasma vitamin A and β-carotene in normal animals ranges from 25 to 85 μg/dL and 150 and 397 μg/dL, respectively.1175 Plasma concentrations of vitamin A—deficient and β-carotene—deficient animals usually are less than 7 and 70 μg/dL, respectively. Papilledema first occurs when plasma concentrations of the vitamin fall below 18 μg/dL.1184 Ataxia and blindness occur when the serum vitamin A concentration ranges from 4.87 to 8.88 μg/dL.1177 The hepatic concentration of vitamin A and carotene in normal calves ranges from 60 to 200 and from 4 to 800 μg/g of tissue, respectively. In deficient calves the hepatic concentrations of the vitamin A and carotene nutrients range from 2 to 14 and from 0.5 to 32 μg/g, respectively.1181,1184 There are no consistent changes in the blood chemistry analysis or the hemogram of deficient animals. Increased CSF pressure (>200 mm Hg) may occur; however, standardization of the measurement for all forms of anesthesia and methods of measurement is difficult.1185 Changes in the CSF of vitamin A—deficient animals include a mononuclear cell pleocytosis (40 to 50 nucleated cells/dL) and an increased protein concentration (140 mg/dL).1186
Pathophysiology
Vitamin A is responsible for the regeneration of rhodopsin in the retina and the maintenance of tissue integrity. The vitamin has effects on osteoblasts and osteoclasts, epithelial tissues, the choroid plexus, and reproductive tissues. The arachnoid villi and the retina are most sensitive to a deficiency of the vitamin. Vitamin A deficiency causes a thickening of the dura mater, resulting in diminished CSF absorption from the arachnoid granulations and the nerve rootlets. Narrowing of all the bony foramina of the skull occurs in immature animals, although bone remodeling does not occur in adults. The combined effects cause an increase in CSF pressure.1181 In severe cases the brain may herniate through the foramen magnum. Closure of the optic foramen may lead to transection of the optic nerve. The high CSF pressure is transmitted into the optic nerves and results in papilledema.
Three causes of blindness have been associated with vitamin A deficiency. Nyctalopia is presumably caused by the decreased formation of vitamin A aldehyde in the regeneration of the visual pigment rhodopsin; this type of blindness usually is reversible. Degenerative changes in the outer retinal layers also cause blindness; this is reversible if treated in the early stages. The third cause is associated with stenosis of the optic foramen and compression of the optic nerve; this condition is irreversible.1187 An experimental study has shown that humoral immune function also is impaired in sheep with vitamin A deficiency. The pathogenesis of this condition is unclear.1188
Epidemiology
The vitamin A requirement of all species ranges from 40 to 110 IU/kg daily.1189-1191 The minimum recommended daily dose of vitamin A for growing calves up to 1 year of age, for pregnant sheep, and for growing horses is 40 IU/kg. Pregnant cattle and pregnant or lactating horses require 40 to 50 IU (13.76 to 17.2 μg/kg) of vitamin A daily.1189 Lactating cattle require 80 IU (27.5 μg/kg) of vitamin A daily. Horses are susceptible to vitamin A deficiency, but the condition is rarely seen in that species. This is thought to be the result of differing conditions of management rather than an inherent resistance to the deficiency. The daily dietary requirement for carotene is 0.12 mg/kg.1189 Pasture forage, silage, and properly cured hay (<1 year old) contain large amounts of carotene. Common constituents that have low concentrations of vitamin A are sorghum, brewer’s grain, and wheat straw.
Livestock are protected from short-term deprivation of vitamin A by their ability to accumulate the vitamin in the liver; however, it is estimated that the intake required to initiate storage is at least three times the minimum daily intake.1189 Vitamin A—replete cattle fed a diet devoid of vitamin A require approximately 180 days before they begin to show clinical signs. During that time the cattle grow and fatten normally and show no adverse effects. Offspring born to these animals may show severe deficiencies. Papilledema and blindness develop rapidly after the hepatic stores are depleted.1189 An interesting sexual dimorphism in susceptibility to dietary deficiency of vitamin A was found in one study of feedlot cattle; although all cattle were fed the same deficient diet, only steers had clinical signs.1190
Vitamin A deficiency may be categorized as a primary or a secondary condition. Primary deficiencies of vitamin A develop in cattle confined in drylot corrals or pasture on dry grass forage for prolonged periods or when cattle are kept indoors and fed unsupplemented, vitamin-depleted cereals and dry forage in which the activity of carotene has been destroyed. Also, there is a seasonal difference in the concentration of vitamin A in feedstuffs. For example, cattle grazed on green pastures are consistently replete with the vitamin, whereas those grazed on dry pastures at the end of summer may become marginally deficient. Approximately 80% of the vitamin A concentration of hay is lost during field curing.1192
Destruction of carotene is hastened by many environmental and physical factors, including heat, sunlight, trace mineral supplements, and humidity. In one study, exposure of nine different supplements to trace minerals in a humidified atmosphere (60% relative humidity) at 28°C (82.2°F) resulted in depletion of 47% to 92% of the total vitamin A after 1 week of incubation.1193 Improper storage has been implicated as the cause of the depletion of vitamin A in one field case.1189 Other factors that affect the stability of vitamin A in feedstuff include pelleting and exposure to rancid fat in the feed. The addition of gelatin to vitamin premixes has been recommended to stabilize the vitamin A activity in feed.1184,1194
Secondary deficiencies of vitamin A result from interference with vitamin absorption, inhibition of the conversion of β-carotene to retinol (vitamin A) in the small intestine, or an increased requirement in the face of limited vitamin intake. The conversion of carotene to retinol is impaired in vitamin A—deficient patients.1179 Sheep may be more resistant to vitamin A deficiency because they convert β-carotene more efficiently than cattle.1179 Extensive destruction of preformed vitamin A by microflora appears to occur in the rumen and the abomasum.1194-1197 Microbial destruction, fever, lactation, high ambient temperatures, and inadequate dietary energy may increase the daily requirement for vitamin A.1198 Females are slightly more resistant to the vitamin deficiency than males, presumably because of the interconversions of estrogenic hormones into vitamin A.
Secondary deficiencies of vitamin A may be caused by impaired vitamin absorption, which may occur from long-term feeding of mineral oil. Ingestion of highly chlorinated naphthalenes (X disease) causes severe vitamin A deficiency as a result of interference with the conversion of carotene to vitamin A. Some in vitro evidence indicates that a high level of dietary nitrates inactivates intraruminal vitamin A by oxidation. This may not be clinically important, however, because studies performed in vivo failed to show a greater requirement for the vitamin when animals were fed subtoxic doses of nitrates.1197-1202 Other factors that can affect availability of vitamin A or the requirement for the vitamin include diets with low forage content, high proportion of corn silage to hay in the diet, increased exposure of animals to pathogens, and periods where immunocompetence is reduced (e.g., peripartum period).1191 Challenges to the immune system increase the requirement for vitamin A.
Necropsy Findings
The major pathologic changes in the fundus of vitamin A—deficient calves include papilledema, small flame-shaped hemorrhages around the optic disc, venous congestion in the area of the swollen optic disc, degeneration of the retinal ganglion cells, focal retinal thinning, and fusion of parts of the retina to the choroid plexus.1182 Other changes associated with vitamin A deficiency include doming of the frontal bones, enlargement of the carpi, cerebellar and cerebral compression, partial transtentorial herniation of the cerebellum, cystic dilation of the hypophyseal cleft, focal ruminal hyperkeratosis, and increased keratinization of the squamous epithelium of the penile and the preputial mucous membrane.1203,1204 Corneal ulceration and clouding have been observed in the eyes of calves with naturally occurring deficiencies.1175,1185 Vitamin A deficiency also can cause anasarca, squamous metaplasia of the salivary ducts, degeneration of the germinal testicular epithelium, degenerative changes in the intestinal epithelium in lambs, and reduction in intramuscular fat in cattle.1204-1206
Microscopic changes in the CNS include attenuation of the optic nerve with necrosis and demyelination. Focal accumulations of phagocytic cells containing lipofuscin and hemosiderin are present in the necrotic area. The optic nerve is attenuated along its entire length. Gliosis and focal vacuolization of the nerve also are seen, as is a focal loss of granular and molecular layers and Purkinje’s cells in the cerebellum. The meninges are thickened by fibrosis and mononuclear cell inflammation. The microscopic changes in the bones include wider than normal spacing of the central canals and reduction of osteoclastic lacunae.
Treatment and Control
Cattle with severe blindness caused by damage to the retina or optic nerves do not regain their vision when treated with vitamin A; however, cattle with acute encephalopathy and simple papilledema may respond favorably after a short period of vitamin supplementation.1189 Affected cattle should receive 440 IU/kg (1 IU = 0.4 μg) of vitamin A parenterally and then 6000 IU/kg parenterally every 50 to 60 days until the diet has been enriched. High-dose oral therapy is important because carotene and oil suspensions of vitamin A are not efficiently used when administered by parenteral injection.1207 Administration of large doses orally is important because conversion of β-carotene to vitamin A is inhibited in deficient calves. The recommended concentration of vitamin A in milk replacers for preruminant calves is 11,000 IU/kg dry matter.1180
Prophylactic dietary supplementation of vitamin A should be considered in all cattle that lack access to green feed. Dietary supplements could include leafy, freshly cured hay, green pasture, or 0.5 to 2 kg of alfalfa meal daily. Concentrate feeds formulated with exogenous, stabilized vitamin A are commercially available. Vitamin A powder* may be added to the drinking water at a rate of 425,000 U/50 gallons. This treatment should be continued for as long as the dietary deficiency exists. The recommended vitamin A requirements in cattle are 80 IU/kg for growing animals and 110 IU/kg for adult animals (including pregnant and lactating cows). Recommended concentrations (IU/kg dry matter) of vitamin A in feed are 2200 IU for feedlot cattle, 2800 IU for pregnant cows, and 3900 IU for lactating cows.1191
Subclinical deficiencies in ewes have been treated with a vitamin-mineral premix containing 0.3 to 0.27 kg iodine, 20.6 kg zinc, 7.9 kg copper, and 1644.5 million IU vitamin A per ton of feed. Addition of this premix to the diet of a group of sheep increased their productivity, as measured by viability, birthweight and rate of gain of lambs, and amount and quality of wool.1208