Cytology
Exfoliative cytology is the study of cells shed from body surfaces. It refers to examination of cells present in body fluids (such as cerebrospinal, peritoneal, pleural, and synovial fluids), on mucosal surfaces (such as the trachea or vagina), or in secretions (such as semen, prostatic fluid, and milk). The primary purpose of the cytology evaluation is to differentiate inflammation and neoplasia. The types and numbers of cells present in a properly collected and prepared cytology specimen can provide rapid diagnostic information to the clinician. Samples for cytology evaluation can be collected quickly and do not generally require specialized materials or equipment for proper evaluation. With careful attention to quality control, including the use of appropriate collection, preparation, and staining techniques, a high-quality cytology sample can be obtained. Such samples yield valuable results for the clinician and often preclude the need for more invasive procedures to determine diagnosis, treatment, and prognosis for a patient.
Several different preparations are usually made from each sample. This allows for additional diagnostic testing without additional collection. Samples may be processed by a variety of techniques, including impression smears, compression or modified compression preparations, line smears, starfish smears, or wedge smears. The exact type of preparation depends on the characteristics of the sample. Some samples may also require concentration by centrifugation. Fluid samples may require anticoagulant and/or preservatives. A variety of staining techniques are also available for cytology specimens. Some samples require processing with more than one staining procedure.
Cytology provides somewhat different information than a histopathologic evaluation. Histopathology observes cells in relation to their neighboring cells. The histopathologist evaluates the cellular architecture. The preparation of a sample for histopathology involves several complex steps and some specialized equipment. To prepare a sample for histopathology, the tissue is first immersed in fixative. Several steps are involved in dehydrating the tissue before it is imbedded in paraffin. The paraffin block is then sliced and the slice mounted on a glass slide before staining (Fig. 9-1). Cytologic evaluations observe the cells individually or in small groups. The cells in a cytologic preparation are randomly distributed with no evidence of their in vitro relationship to each other.

SAMPLE COLLECTION
Cytology samples from solid masses on an animal’s body or obtained from a surgical procedure can be collected by the swab, scrape, or imprint technique. Fine needle biopsy can also be used for some solid samples as well as fluid samples. Centesis refers to fluid samples collected from body cavities.
Swabs
Swabs generally are collected only when imprints, scrapings, and aspirates cannot be made, such as with fistulous tracts and vaginal collections. The area is swabbed with a moist, sterile cotton or rayon swab (Procedure 9-1 and Fig. 9-2). Sterile isotonic fluid, such as 0.9% saline, should be used to moisten the swab. Moistening the swab helps minimize cell damage during sample collection and smear preparation. For collection of vaginal swabs, restrain the animal in a standing position with the tail elevated. Clean and rinse the vulva, then insert a lubricated speculum or smooth plastic tube to a point just cranial to the urethral orifice in the vagina. The cells collected are those exfoliated, or shed, from the vaginal wall (epithelial cells and neutrophils) and those passing through the vagina from the uterus, especially erythrocytes in proestrus and estrus in the bitch. If collecting a sample from a moist lesion, the swab need not be moistened. After sample collection, the swab is gently rolled along the flat surface of a clean glass microscope slide (Fig. 9-3). The swab should not be rubbed across the slide surface because this causes excessive cell damage.
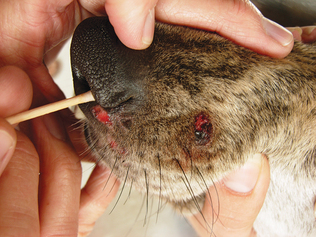

Ear swab samples may contain excess amounts of wax. This may interfere with the evaluation of the sample. To minimize this effect, gentle heating of the slide may be necessary. Passing the slide briefly through a flame or gentle heat from a warm hair dryer may be used to dissolve the wax. Excess heat must be avoided because it will destroy the cellular components of the sample. Aside from the Gram stain procedure, this is the only circumstance under which cytology samples might require heat application.
Scrapings
Smears of scrapings may be prepared from tissues collected during necropsy or surgery or from external lesions on the living animal. Scraping has the advantage of collecting many cells from the tissue and therefore is advantageous when the lesion is firm and yields few cells. The major disadvantages of scrapings are that they are more difficult to collect and they collect only superficial samples. As a result, scrapings from superficial lesions often reflect only a secondary bacterial infection and/or inflammation-induced tissue dysplasia, which markedly hinders their use in diagnosis of neoplasia.
Veterinary technicians may prepare a scraping by holding a scalpel blade perpendicular to the lesion’s cleaned and blotted surface and pulling the blade toward themselves several times (Procedure 9-2 and Fig. 9-4). The material collected on the blade is transferred to the middle of a glass microscope slide and spread by one or more of the techniques described in the following sections for preparation of smears from aspirates of solid masses.

Imprints
Imprints for cytologic evaluation may be prepared from external lesions on the living animal or from tissues removed during surgery or necropsy. They are easy to collect and require minimal restraint, but they collect fewer cells than scrapings and usually contain a greater amount of contamination (bacterial and cellular) than fine needle biopsies (FNBs). As a result, imprints from superficial lesions often reflect only a local secondary bacterial infection and/or inflammation-induced tissue dysplasia. In many instances the bacteria and tissue dysplasia markedly hinder making an accurate diagnosis of neoplasia.
The Tzanch preparation is a type of imprint collection that can be used on external lesions (Procedure 9-3). To perform this procedure, prepare four to six clean glass slides. The lesion is imprinted before it is cleaned and that first slide designated as number one. The lesion should then be cleaned with a saline-moistened surgical sponge and reimprinted with the slide marked as imprint number two. The lesion is then debrided and reimprinted with the slide, marked as imprint number three. If a scab is present, the underside of the scab should be imprinted and the slide marked as imprint number four (Fig. 9-5). Imprints from the tissue exposed from the scab removal and scrapings or swabs from that exposed tissue can both be collected.
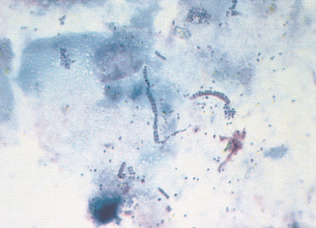
Figure 9-5 This sample was prepared with an imprint of the underside of a scab. Squamous epithelial cells and chains of coccoid organisms (Dermatophilus congolensis) are present. (Reprinted from Cowell RL, Tyler RD, Meinkoth JH: Diagnostic cytology and hematology of the dog and cat, ed 2, St Louis, 1999, Mosby.)
To collect imprints from tissues collected during surgery or necropsy, blood and tissue fluid first should be removed from the surface of the lesion being imprinted by blotting with a clean, absorbent material (Procedure 9-4 and Fig. 9-6). Excessive blood and tissue fluids inhibit tissue cells from adhering to the glass slide, producing a poorly cellular preparation. Also, excessive fluid inhibits cells from spreading and assuming the size and shape they usually have on air-dried smears. If a delay occurs from the time of sample collection until the imprint is taken, a scalpel blade should be used to expose a fresh surface before blotting and sampling the surface. The middle of a clean glass microscope slide is then touched against the blotted surface of the tissue to be imprinted. Multiple imprints generally are made on each slide (Fig. 9-7). Several slides may be imprinted so that slides are available for special stains if necessary.
Fine Needle Biopsy
Fine needle biopsies may be collected from masses, including lymph nodes, nodular lesions, and internal organs. For cutaneous lesions, they provide an advantage over other methods by avoiding superficial contamination (bacterial and cellular). However, fewer cells are usually collected than with other methods, such as scrapings. Fine needle biopsy can be performed by either an aspiration or a nonaspiration method.
Preparation of the Site for Fine Needle Biopsy
If microbiologic tests are to be performed on a portion of the sample collected or a body cavity (such as peritoneal and thoracic cavities and joints) is to be penetrated, the area of aspiration is surgically prepared. Otherwise preparation is essentially that required for a vaccination or venipuncture. An alcohol swab may be used to clean the area.
Selection of Syringe and Needle
For the aspiration method of fine needle biopsy, use a 21- to 25-gauge needle and a 3- to 20-ml syringe. The softer the aspirated tissue is, the smaller the needle and syringe used. The use of a needle larger than 21 gauge for aspiration is seldom advantageous, even for firm tissues such as fibromas. When larger needles are used, tissue cores tend to be aspirated, resulting in a poor yield of free cells suitable for cytologic preparation. Also, larger needles tend to cause greater blood contamination.
The size of the syringe used is influenced by the consistency of the tissue being aspirated. Softer tissues, such as lymph nodes, often may be aspirated successfully with a 3-ml syringe. Firm tissues, such as fibromas and squamous cell carcinomas, require a larger syringe to maintain adequate suction for sufficient collection of cells. Because the ideal size of the syringe is not known for many masses before aspiration, a 12-ml syringe is a useful size.
Aspiration Procedure
The mass to be aspirated is held firmly to aid penetration of the skin and mass and to control the direction of the needle (Procedure 9-5). The needle, with syringe attached, is introduced into the center of the mass and strong negative pres-sure is applied by withdrawing the plunger to approximately three fourths the volume of the syringe (Fig. 9-8). Several areas of the mass should be sampled, but aspiration of the sample into the barrel of the syringe and contamination of the sample by aspiration of tissue surrounding the mass must be avoided. To accomplish this, when the mass is large enough to allow the needle to be redirected and moved to several areas in the mass without danger of the needle’s leaving the mass, negative pressure is maintained during redirection and movement of the needle. However, when the mass is not large enough for the needle to be redirected and moved without danger of the needle leaving the mass, negative pressure is relieved during redirection and movement of the needle. In this situation, negative pressure is applied only when the needle is static. Often high-quality collections do not have aspirate material visible in the syringe and sometimes not even in the hub of the needle.
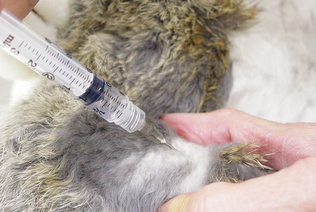
When material is observed in the hub of the needle or after several areas are sampled, the negative pressure is relieved from the syringe and the needle is withdrawn from the mass and skin. Next the needle is removed from the syringe and air is drawn into the syringe. Then the needle is replaced onto the syringe and some of the tissue in the barrel and hub of the needle is expelled onto the middle of a glass microscope slide by rapidly depressing the plunger. When possible, several preparations should be made as described in the following sections.
Nonaspirate Procedure (Capillary Technique, Stab Technique)
This technique is easier to perform than aspiration because it does not involve directing the syringe and needle and pulling the plunger with the same hand (Procedure 9-6). The mass to be sampled is held firmly to aid penetration of the skin and mass and to help direct the needle. A 22-guage needle is introduced into the mass. The needle is moved rapidly back and forth through the mass five to six times along the same tract. The cells are collected by shearing and capillary action. The needle is removed from the mass and attached to a 10-ml syringe prefilled with air. The material is expelled onto a clean glass microscope slide by rapidly depressing the plunger. The expelled material should be smeared using one of the techniques described for preparation of smears. A variation of this procedure involves attachment of the needle to an empty air-filled syringe without the plunger. The syringe can then be held by the barrel and the sample collected as described previously (Fig. 9-9).
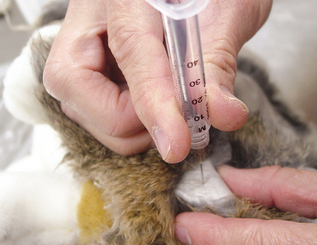
Figure 9-9 Collection of sample by the fine needle biopsy nonaspiration technique. Note that the syringe plunger has been removed.
Generally, enough material is collected to make only one smear. Therefore the procedure should be repeated two or three times in different sites of the mass to have adequate slide numbers and areas of the mass to evaluate.
Tissue Biopsy
Tissue biopsy is the sampling of a piece of tissue for cytologic and/or histopathologic examination. Many organs or tissues, including kidney, liver, lung, lymph node, prostate, skin, spleen, and thyroid, or masses (tumors) may be biopsied. Biopsy techniques include gentle abrasion with a blade, needle aspiration, and excision, including punch biopsy and endoscope-guided biopsy. The technique used varies with the tissue to be biopsied. Considerations include its location, accessibility, and nature. Prospective skin biopsy sites are clipped of hair, being careful to avoid skin irritation and inducement of an inflammatory artifact. Cleansing of the site is neither recommended nor necessary. The lesion must not be scrubbed nor any scales, crusts, or surface debris disturbed because they may offer valuable diagnostic clues.
Tissue samples for histopathologic examination often are fixed in 10% neutral phosphate-buffered formalin. To ensure adequate fixation, slabs of tis-sue no more than 1 cm wide should be placed in fluid-tight jars containing formalin at approximately 10 times the specimen’s volume. With large tissues, the sample can be removed to a smaller jar with less formalin once it has been fixed for 24 hours.
Wedge Biopsy
Elliptic wedge biopsy specimens are commonly obtained with a scalpel. The wedge biopsy offers the advantages of a large, variably sized specimen that is easily oriented by the pathology technician. Solitary lesions are often best removed with this technique. When a wedge biopsy specimen is taken, a sharp scalpel blade is used to excise the entire lesion, or the wedge is taken from an area of the lesion, through a transition zone, to normal tissue. The pathology technician then can trim the specimen on its long axis to provide the pathologist with a slide showing abnormal tissue, a transition zone, and normal tissue.
Punch Biopsy
The punch biopsy technique has a number of advantages over wedge biopsy, particularly the ease and speed of the procedure (Procedure 9-7). Keyes cutaneous biopsy punches (4-, 6-, and 8-mm disposable skin biopsy punches) are most commonly used (Fig. 9-10). Used punches may be placed in the sterilization tray and reused until dull. Most disposable punches may be reused at least three or four times.
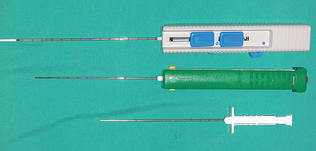
Figure 9-10 Examples of punch biopsy instruments. (From Busch SJ: Small animal surgical nursing: skills and concepts, St Louis, 2006, Mosby.)
With the biopsy punch, 4-mm specimens require no sutures and 6- or 8-mm biopsy specimens require only one or two sutures. Ideally, two or three punch biopsy specimens of various lesions should be collected. The punch is gently rotated in one direction until the punch blade has sectioned the tissue. The punch is rotated in only one direction because back-and-forth rotation increases the likelihood of specimen damage from shearing forces (Fig. 9-11).
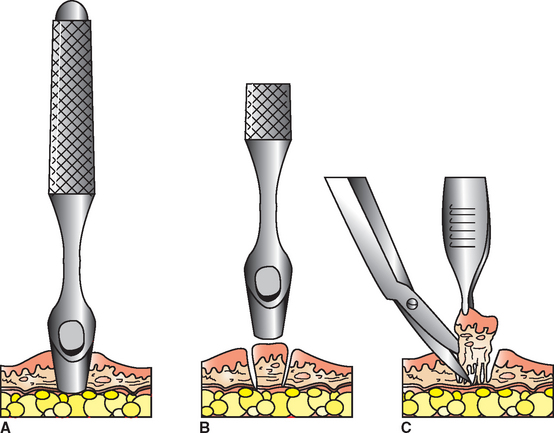
Figure 9-11 Technique for collecting a specimen with a biopsy punch. (From Busch SJ: Small animal surgical nursing: skills and concepts, St Louis, 2006, Mosby.)
Once the specimen has been collected, by either the wedge or the punch method, the specimen should be gently removed by grasping the margin of the tissue with a pair of fine forceps. Fresh, unfixed tissue is extremely fragile. Specimens collected by endoscopy can be gently flushed from the tip of the endoscope with sterile saline (Fig. 9-12). The specimen then is blotted gently on a paper towel to remove excess blood and is placed on a small piece of wooden tongue depressor or cardboard (Fig. 9-13). Skin specimens should be placed with the subcutaneous side down. Gently pressing the biopsy specimen flat facilitates adherence and allows proper anatomic orientation of the specimen in the laboratory. Allow the tissue to dry onto the “splint.”
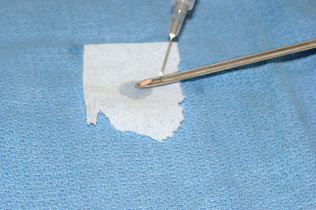
Figure 9-12 Specimens collected by endoscopy should be gently flushed from the tip of the endoscope.
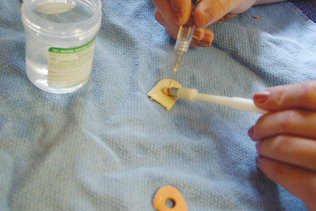
Specimens with the attached “splint” are then immersed or floated specimen side down in the fixative. Timely placement of the specimen in the fixative is critical because artifactual changes may begin to occur within 1 minute after the biopsy specimen is obtained. Adequate specimen fixation requires at least 24 hours before processing. Formalin freezes at −11° C (−24° F), and this freezing may cause substantial artifactual damage in unfixed specimens. Therefore, to ensure proper fixation without freezing artifact, specimens should remain at room temperature for at least 6 hours before exposure to possible extreme cold.
Centesis
Centesis refers to the introduction of a needle into any body cavity or organ for the purpose of removing fluid. Collection of fluid from the peritoneal cavity (abdominocentesis or paracentesis) and thoracic cavity (thoracocentesis) are commonly performed in small animal practice. Cystocentesis (percutaneous aspiration of urine from the urinary bladder) is discussed in Chapter 5. Fluid samples for cytologic evaluation may also be collected from around the spinal column, from within joints (arthrocentesis), and from the eye. General anesthesia is required for collection of cerebrospinal fluid, synovial fluid, and aqueous and vitreous humor.
Before collection of peritoneal or pleural fluid, the site should be aseptically prepared and all equipment and supplies gathered. Preparing several smears from the fluid as soon as it is collected is helpful, so a plentiful supply of glass slides should be ready. A portion of the fluid should be collected in an ethylenediamine tetraacetic acid (EDTA) tube. A 21-gauge needle is most commonly used and should be attached to a 60-ml syringe. For small animal patients, thoracocentesis is usually performed with the animal in a standing position and the needle inserted in the seventh or eighth intercostal space along the cranial aspect of the rib. Abdominocentesis may be performed in a standing animal or with the patient in lateral recumbency (Fig. 9-14). The needle is introduced into the ventral abdomen to the right of the midline approximately 1 to 2 cm caudal to the umbilicus. The procedure is slightly different for exotic and farm animals.
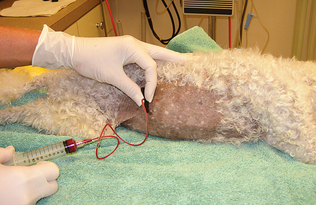
Ease of collection may reflect the volume of fluid present and/or the pressure within that body cavity. It also is influenced by the technical proficiency of the operator and, in the case of conscious animals, the cooperation of the animal. The total volume collected must be recorded at the time of collection. Certain gross characteristics of body fluids should be recorded at collection, including sample color and turbidity. Subsequently, total nucleated cell counts, cell types, and morphologic features are determined.
Color and Turbidity
Color and turbidity are influenced by protein concentration and cell numbers. Gross discoloration, with increased turbidity, may be caused by iatrogenic contamination with peripheral blood, recent or old hemorrhage, inflammation, or a combination of these conditions.
Perforation of superficial vessels during collection may result in contamination with peripheral blood. Such admixture of blood with the sample may be obvious as a streak of blood in otherwise clear fluid at some stage during collection. Blood-tinged fluid also may be the result of recent or old hemorrhage into the body cavity being sampled. Both peripheral blood contamination and recent hemorrhage result in a clear supernatant and red (erythrocyte-rich) sediment after centrifugation. Recent hemolysis imparts a reddish discoloration to the supernatant. Hemorrhage that occurred at least 2 days previously generally causes a yellowish supernatant (because of hemoglobin breakdown products), usually with little erythrocytic sediment (Fig. 9-15).

Figure 9-15 Gross appearance of various effusion (left to right): clear and colorless, yellow and slightly turbid, hemolyzed and slightly turbid, orange and turbid, sedimented fluid, bloody and turbid, brown and slightly turbid. (From Raskin RE, Meyer DJ: Atlas of canine and feline cytology, St. Louis, 2001, Saunders.)
Cytologic examination of the fluid also may assist in determining the time of hemorrhage. Clumps of platelets may be observed in recent, often iatrogenic (operator-induced), hemorrhage. These clumps are not obvious after approximately 1 hour. Blood must be present in the cavity for several hours before macrophage ingestion of erythrocytes becomes evident. If the hemorrhage occurred a day or so before collection, hemoglobin breakdown products, such as hemosiderin, may be seen in the macrophages. Inflammation also may discolor body fluids, with the degree of turbidity reflecting leukocyte numbers. Color may vary from an off-white or cream to a red-cream or dirty brown, depending on the number of erythrocytes also involved and the integrity of the cells present.
Consistent terminology must be used when describing cell types. Specific details on the morphologic features of each cell type also assist the clinician in making the diagnosis. Neutrophils and macrophages should be evaluated for the presence of vacuoles or phagocytized material. Neoplastic cells should be evaluated for malignant changes, such as mitotic figures and basophilic cytoplasm.
Transtracheal/Bronchial Wash
Cytologic evaluation of samples obtained from the trachea, bronchi, or bronchioles may assist with diagnosis of pulmonary disease in animals. Tracheal washes may be performed by passage of a catheter through an endotracheal tube in an anesthetized animal (orotracheal approach), through the nasal passages (nasotracheal approach), or through the skin and trachea (percutaneous approach) in a conscious, sedated animal. The transtracheal route minimizes pharyngeal contamination of the specimen, but it is an invasive procedure and consequently requires aseptic technique. These procedures are commonly used in both small and large animals.
Percutaneous Technique
The percutaneous method requires the use of an 18- to 20-gauge through-the-needle (jugular) catheter. The laryngeal area is clipped of hair and aseptically prepared. A small amount (usually 0.5 to 1.0 ml) of 2% lidocaine is injected into the cricothyroid membrane and surrounding skin. The needle is inserted into the trachea through the cricothyroid membrane (Fig. 9-16). Sterile physiologic saline solution is infused through the catheter at a rate of 0.5 to 1.0 ml/kg body weight. When the animal coughs, the syringe plunger should be retracted several times and the fluid collected placed into a plain sterile tube. Samples should be processed immediately.
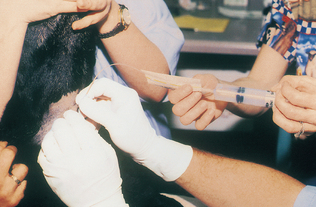
Orotracheal Technique
This technique may be preferred in very small or fractious animals. The patient must be lightly anesthetized and an appropriate-size endotracheal tube placed. A urinary or jugular catheter is then placed through the endotracheal tube and saline is infused as described for the percutaneous method. A red rubber catheter may be used but care must be taken to ensure that the catheter does not collapse upon aspiration as may occur with highly viscous samples (Fig. 9-17). Depending on the level of anesthesia, the animal will often not cough, so the saline should be withdrawn within a few seconds and evaluated. Bronchoalveolar lavage (BAL) is an orotracheal technique used to collect samples specifically from the lower respiratory tract. Bronchoscopy is the preferred method for performing a BAL, but specialized equipment (e.g., bronchoscope) is required.
With either method, only a small amount of the saline infused will be harvested with the initial collection. Subsequent coughing of the animal may also contain cells of interest, so all fluid released during coughing subsequent to the initial collection should be collected once the animal has been returned to its cage. This fluid should be placed in a sterile tube with a notation containing the site of collection. Such fluids are often contaminated but can sometimes be used for evaluation when the initial collection yields insufficient information.
Samples with little mucus (generally corresponding to small numbers of cells) should be centrifuged at low speed and smears prepared from the sediment. Samples containing much mucus (and usually numerous cells) may not need to be centrifuge concentrated before a smear is made. Total nucleated cell counts are usually not performed on tracheal wash fluids. Cell numbers are subjectively recorded from evaluation of the smear. A tracheal wash smear from a normal animal contains few cells, usually with a small amount of mucus. The mucus often appears microscopically as eosinophilic to purple strands that may enmesh the cells. Epithelial cells are the principal cell type.
Cytologic evaluation of samples obtained from the nasal cavity may be useful in investigation of diseases affecting the upper airway. Fluid (normal saline) may be infused into the nasal cavity through the nose with a syringe and tubing, then aspirated. This procedure is referred to as a nasal flush. Such specimens are processed as for a tracheal wash. Various abnormalities may be demonstrated with this procedure, such as inflammation secondary to sepsis, fungi and yeasts, and neoplasia. These should not be confused with glove powder, which may be present in some specimens.
CONCENTRATION TECHNIQUES
When a cytologic smear is to be made of fluid with a cell count less than 500/μl, concentration of cells is mandatory. Such concentration may be helpful even at higher cell counts. Four methods are described.
Low-Speed Centrifugation
To concentrate fluids by centrifugation, the fluid is centrifuged 5 minutes at 165g to 360g using a centrifuge with a radial arm length of 14.6 cm (the arm length of most urine centrifuges) at 1000 to 1500 rpm. After centrifugation, the supernatant is separated from the sediment and analyzed for total protein concentration. The sediment is resuspended in a few drops of supernatant by gently thumping the side of the tube. A drop of the resuspended sediment is placed on a slide, and a smear is made by the blood smear or compression preparation technique. When possible, several smears should be made by each technique. Addition of plasma may help cells adhere to the microscope slide. After air drying, the slide may be stained with a Romanowsky stain.
Gravitational Sedimentation
Gravitational sedimentation is another method used to concentrate cells and is most commonly used for cerebrospinal fluid (CSF) evaluations. One method uses a glass cylinder (which can be made by cutting the end off a test tube) attached to a microscope slide with paraffin wax. (The smooth tube end is dipped in melted wax and placed on a warm slide.) The cells in approximately 1 ml of CSF are allowed about 30 minutes to settle. The supernatant is then carefully removed with a pipette and the tube detached. Excess CSF may be gently removed with absorbent paper. The slide is air dried and residual paraffin carefully scraped off. The slide may be stained with a Romanowsky stain.
Membrane Filtration
Membrane filtration of alcohol-diluted CSF also may be used to concentrate cells. A membrane pore size of five is usually satisfactory. Filter holders that attach to a syringe are available. The CSF is permitted to gravity feed from the syringe barrel, or it is gently injected through the filter at no more than 1 drop/sec. The filter paper must be kept horizontal to distribute the cells evenly. Increased resistance to filtration suggests that the pores are becoming obstructed by cells or protein, and no more CSF should be forced through the filter. Filtration of another, smaller volume of CSF through fresh filter paper results in a less-crowded preparation.
After removal from the syringe holder, the filter is fixed in 95% ethanol for at least 30 minutes. Holders are available for easy handling of the filter paper during fixation and staining. A trichrometype stain must be used. Romanowsky stains are unsuitable because they stain the filter paper too intensely. A satisfactory staining procedure is as follows.
The filter paper is immersed for 2 minutes in each specific substance in the following order: 80%, 70%, 50%, and 30% ethanol and then distilled water, then 4 minutes in hematoxylin, 5 minutes in running tap water, 4 minutes in Pollak’s stain, 1 minute in 0.3% acetic acid, 1 minute in 95% ethanol, 2 minutes in n-propyl-alcohol (propanol), 2 minutes in a 1:1 mixture of propanol and xylene, and finally three rinses of 2 minutes each in xylene. At all stages the filter must be treated gently to avoid dislodging cells. Depending on the size of the filter, it may need to be cut to a suitable size before placement on a microscope slide (cell side up). The filter is then flooded with a mounting medium with a refractive index similar to that of filter paper (approximately 1.5), and a cover slip is applied.
Cytologically, the cells trapped by the membrane filter are rounder than those seen after sedimentation (and therefore may be harder to distinguish) and are in slightly different planes of focus. Furthermore, the filter produces a patterned background that may be distracting. This distraction is minimized by ensuring the sample is not overstained and by using the appropriate mounting medium. The pore size generally used is far too large to trap free bacteria. Quantitatively, more cells are collected by filtration than by the two sedimentation methods.
Cytocentrifugation
As with any fluid of low cellularity, a cytocentrifuge (e.g., Shandon Cytospin, Thermo Fisher Scientific, Inc., Waltham, MA) can be used for preparation of CSF cytologic smears. Such equipment is generally too expensive for a veterinary practice to justify purchasing. However, it often is used in a referral laboratory. This technique allows cells to be concentrated within a small circular area on the slide.
SMEAR PREPARATION
Preparation of Smears from Solid Masses
Several methods may be used to prepare smears for cytologic evaluation of solid masses including lymph nodes and internal organs. The experience of the person preparing the smears and characteristics of the sample influence the choice of smear preparation technique. A combination of slide preparation techniques is therefore sug-gested. Some cytologic preparation techniques are described in the following sections.
Compression (“Squash”) Preparation
The compression technique, sometimes referred to as the “squash prep,” can yield excellent cytologic smears (Procedure 9-8). However, in less experienced hands it often yields unreadable cytologic smears because too many cells are ruptured or the sample is not sufficiently spread. A compression preparation is made by expelling the aspirate onto the middle of one slide and then gently placing a second slide (spreader slide) over the aspirate horizontal with and at right angles to the first slide (prep slide) (Figs. 9-18 and 9-19). The spreader slide is then quickly and smoothly slid across the prep slide. Downward pressure should not be placed on the spreader slide because this may cause excessive cell rupturing, making the sample unable to be interpreted.
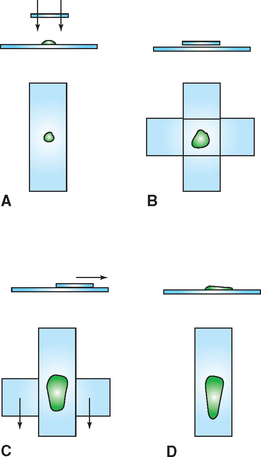
Figure 9-18 Compression preparation. A, A portion of the aspirate is expelled onto a glass microscope slide. B, Another slide is placed over the sample, spreading the sample. If the sample does not spread well, gentle digital pressure can be applied to the top slide. Care must be taken not to place excessive pressure on the slide, which could cause the cells to rupture. C, The slides are smoothly slipped apart, which usually produces well-spread smears (D) but may result in excessive cell rupture.
A modification of the compression preparation that has fewer tendencies to rupture cells is to lay the second slide over the aspirate, rotate the second slide 45 degrees, and lift it upward (Procedure 9-9 and Fig. 9-20).
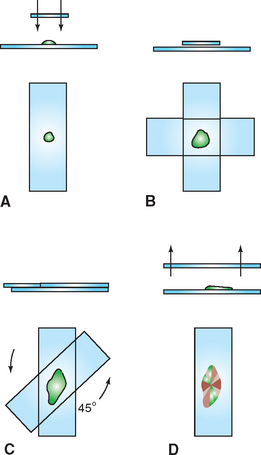
Figure 9-20 Modification of the compression preparation. A, A portion of the aspirate is expelled onto a glass microscope slide. B, Another slide is placed over the sample, causing the sample to spread. If necessary, gentle digital pressure can be applied to the top slide to spread the sample more. Care must be taken not to place excessive pressure on the slide, which could cause the cells to rupture. C, The top slide is rotated approximately 45 degrees and lifted directly upward, producing a squash preparation with subtle ridges and valleys of cells (D).
Combination Technique
One combination procedure involves spraying the sample onto the middle of a clean glass microscope slide or prep slide. Place the prep slide on a flat, solid, horizontal surface and pull another slide, called the spreader slide, backward at a 45-degree angle to the first slide until it makes contact with approximately one third of the aspirate. Then the spreader slide is slid smoothly and rapidly forward, as if making a blood smear. Next, the spreader slide is placed horizontally over the back third of the aspirate at a right angle to the prep slide. The weight of the spreader slide (top slide) is usually sufficient to spread the material. Avoid the temptation to compress the slides manually. Keep the spreader slide flat and horizontal, and use a quick, smooth motion to slide the spreader slide across the prep slide (Fig. 9-21).
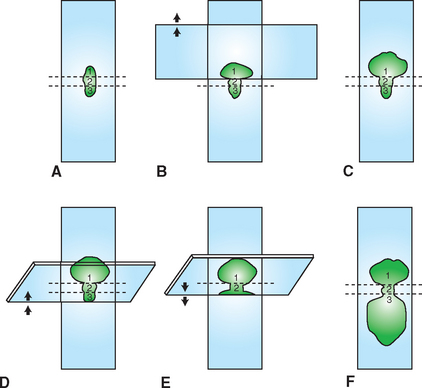
Figure 9-21 Combination cytologic preparation. A, A portion of the aspirate is expelled onto a glass microscope slide (prep slide). B, Another glass microscope slide is placed over approximately one third of the preparation. If additional spreading of the aspirate is needed, gentle digital pressure can be used. Excessive pressure should be avoided. C, The spreader slide is smoothly slid forward. This procedure makes a compression preparation of approximately one third of the aspirate (area 1). The spreader slide also contains a squash preparation (not depicted). Next, the edge of a tilted glass microscope slide (second spreader slide) is slid backward from the end opposite the compression preparation until it makes contact with approximately one third of the expelled aspirate (D and E). F, Then the second spreader slide is slid rapidly and smoothly forward. These steps produce an area (3) that is spread with mechanical forces like those of a blood smear preparation. The middle area (2) is left untouched and contains a high concentration of cells.
This procedure makes a compression preparation of the back third of the aspirate. The middle third of the aspirate is left untouched. This procedure leaves the front third of the aspirate gently spread. If the aspirate is of fragile tissue, this area should contain sufficient intact cells to evaluate. The back third of the aspirate has been spread with the shear forces of a compression preparation. If the aspirate contains clumps of cells that are difficult to spread, some clumps should be sufficiently spread in the back third of the preparation. If the aspirate is of low cellularity, the middle third remains more concentrated and is the most efficient area to study.
Starfish Smear
Another technique for spreading aspirates is to drag the aspirate peripherally in several directions with the point of a syringe needle, producing a starfish shape (Procedure 9-10 and Fig. 9-22). This technique tends not to damage fragile cells but does allow a thick layer of tissue fluid to remain around the cells. Sometimes the thick layer of fluid prevents the cells from spreading well and interferes with evaluation of cell detail. Usually, however, some acceptable areas are present. The starfish smear technique is especially useful for the preparation of highly viscous samples.
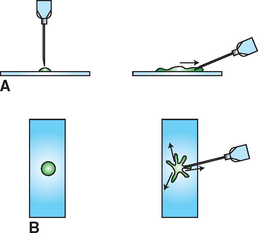
Figure 9-22 Needle spread or “starfish” preparation. A, A portion of the aspirate is expelled onto a glass microscope slide. B, The tip of a needle is placed in the aspirate and moved peripherally, pulling a tail of the sample with it. This procedure is repeated in several directions, resulting in a preparation with multiple projections.
Preparation of Smears from Fluid Samples
Cytologic smears should be prepared immediately after fluid collection. When possible, fluid samples for cytologic examination should be collected in EDTA tubes. Smears may be prepared directly from fresh, well-mixed fluid or from the sediment of a centrifuged sample by wedge (blood) smear, line smear, and/or compression preparation techniques. The cellularity, viscosity, and homogeneity of the fluid influence the selection of smear technique.
Line Smear
When the fluid cannot be concentrated by centrifugation or the centrifuged sample is of low cellularity, the line smear technique may be used to concentrate cells in the smear (Procedure 9-11 and Fig. 9-23). A drop of fluid is placed on a clean glass slide and the blood smear technique is used, except the spreading slide is raised directly upward approximately three fourths of the way through the smear, yielding a line containing a much higher concentration of cells than the rest of the slide. Unfortunately, an excessive amount of fluid also may remain in the “line” and prevent the cells from spreading well.
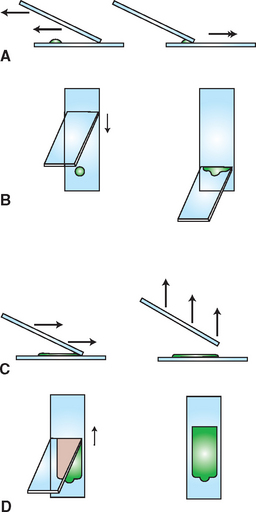
Figure 9-23 Line smear concentration technique. A, A drop of fluid sample is placed on a glass microscope slide close to one end, then another slide is slid backward to make contact with the front of the drop (B). When the drop is contacted, it rapidly spreads along the juncture between the two slides. C, The spreader slide is then smoothly and rapidly slid forward. D, After the spreader slide has been advanced approximately two thirds to three fourths the distance required to make a smear with a feathered edge, the spreader slide is raised directly upward. This procedure produces a smear with a line of concentrated cells at its end instead of a feathered edge.
The compression preparation technique often spreads viscous samples and samples with flecks of particulate material better than the blood smear and line smear techniques. The blood smear technique usually produces well-spread smears of sufficient cellularity from homogeneous fluids containing at least 5000 cells/μl but often produces smears of insufficient cellularity from fluids containing less than 5000 cells/μl. The line smear technique may be used to concentrate fluids of low cellularity but often does not sufficiently spread cells from highly cellular fluids. In general, translucent fluids are of low to moderate cellularity, whereas opaque fluids usually have high cellularity. Therefore translucent fluids often require concentration, either by centrifugation or by the line smear technique. When possible, concentration by centrifugation is preferred.
To prepare a smear by the wedge (blood smear) technique, a small drop of the fluid is placed on a glass slide approximately 1.0 to 1.5 cm from the end. Another slide is pulled backward at a 30- to 40-degree angle until it makes contact with the drop. When the fluid flows sideways along the juncture between the slides, the second slide is quickly and smoothly pushed forward until the fluid has all drained away from the second slide. This procedure makes a smear with a feathered edge.
FIXING AND STAINING THE CYTOLOGY SAMPLE
Although many stains incorporate a cellular fixative, accomplishing this as a separate step in the procedure is advantageous to ensure the highest quality preparation. The preferred fixative for cytology specimens is 95% methanol. The methanol must be fresh and not contaminated with stain or cellular debris. Methanol containers must be protected from evaporation and dilution resulting from environmental humidity, which will introduce humidity artifacts onto the slide. The prepared cytology slides should remain in the fixative for 2 to 5 minutes. Longer fixative times will improve the quality of the staining procedure and not harm the samples.
Several types of stains have been used for cytologic preparations. The two general types most commonly used are the Romanowsky-type stains (Wright’s, Giemsa, Diff-Quik) and Papanicolaou stain and its derivatives, such as Sano’s trichrome. The advantages and disadvantages of both types of stains are discussed. However, because the Romanowsky-type stains are more rewarding, practical, and readily available in practice situations, the remainder of this discussion deals predominantly with Romanowsky-stained preparations.
Romanowsky Stains
Romanowsky stains are inexpensive; readily available; and easy to prepare, maintain, and use. They stain organisms and the cytoplasm of cells excellently. Although nuclear and nucleolar detail cannot be perceived as well with Romanowsky stains as with Papanicolaou stains, nuclear and nucleolar detail usually is sufficient for differentiating neoplasia and inflammation and for evaluating neoplastic cells for cytologic evidence of malignant potential (criteria of malignancy).
Smears to be stained with Romanowsky stains are first air dried. Air drying partially preserves (fixes) the cells and causes them to adhere to the slide so that they do not fall off during the staining procedure.
Many Romanowsky stains are commercially available, including Diff-Quik (Dade Behring, Deerfield, IL.) and other quick Wright’s stains (Fig. 9-24). Most, if not all, Romanowsky stains are acceptable for staining cytologic preparations. Diff-Quik stain does not undergo the metachromatic reaction. As a result, granules of some mast cells do not stain. When mast cell granules do not stain, the mast cells may be misclassified as macrophages, which may lead to confusion in examination of some mast cell tumors. Increasing the fixative time to approximately 15 minutes may alleviate this problem. Also, in evaluation of blood smears or bone marrow aspirates, Diff-Quik does not stain polychromatophilic red blood cells well and occasionally does not stain basophils. The variations among different Romanowsky stains should not cause a problem once the evaluators become familiar with the stain they routinely use.

Each stain usually has a unique recommended staining procedure. These procedures should be followed in general but should be adapted to the type and thickness of smear being stained and to the evaluator’s preference. The thinner the smear and the lower the total protein concentration of the fluid, the less time needed in the stain. The thicker the smear is and the greater the total protein concentration of the fluid, the more time needed in the stain. As a result, fluid smears with low protein and low cellularity, such as abdominal fluid, may stain better in half or less of the recommended time. Thick smears, such as smears of neoplastic lymph nodes, may need to be stained twice the recommended time or longer. Each technician tends to have a different preferred staining technique. By trying variations in the recommended time intervals for stains, the evaluator can establish which times produce the preferred staining characteristics.
New Methylene Blue Stain
New methylene blue (NMB) stain is a useful adjunct to Romanowsky stains (Fig. 9-25). It stains cytoplasm weakly, if at all, but gives excellent nuclear and nucleolar detail. Because NMB stains cytoplasm weakly, the nuclear detail of cells in cell clumps may be better visualized. Generally, red blood cells do not stain with NMB but may develop a pale blue tint. As a result, marked red blood cell contamination of smears does not obscure nucleated cells.
Papanicolaou Stains
The delicate Papanicolaou stains give excellent nuclear detail and delicate cytoplasmic detail. They allow the viewer to see through layers of cells in cell clumps and evaluate nuclear and nucleolar changes well. They do not stain cytoplasm as strongly as Romanowsky stains and therefore do not demonstrate cytoplasmic changes as well. They also do not demonstrate bacteria and other organisms as well as Romanowsky stains do.
Papanicolaou staining requires multiple steps and considerable time. In addition, the reagents often are difficult to locate, prepare, and maintain in practice. Papanicolaou stains and their derivatives require the specimen to be wet fixed (i.e., the smear must be fixed before the cells have dried). Wet fixing requires spraying the smear with a cytologic fixative or placing it in ethanol immediately after preparation. When the smear is to be placed in ethanol, it should be made on a protein-coated slide, which prevents the cells from falling off the slide when it is immersed.
Staining Problems
Poor stain quality often perplexes the novice and experienced cytologist. Most staining problems can be avoided if the following precautions are taken:
• Always use new, clean slides. Even “precleaned” slides should be wiped with alcohol before use to remove residue.
• Fresh, well-filtered (if periodic filtration is required) stain(s) and fresh buffer solution (if a buffer is required) should be used.
• Cytologic preparations should be fixed immediately after air drying unless they are being sent to an outside laboratory. The laboratory should be consulted before fixing slides.
• The surface of the slide or smear should not be touched at any time by human hands.
Occasionally a sample may be contaminated with a foreign substance, such as lubrication jelly, that alters the specimen’s staining. Table 9-1 shows some of the problems that can occur with Romanowsky stains and some proposed solutions to these problems.


SUBMISSION OF CYTOLOGIC PREPARATIONS AND SAMPLES FOR INTERPRETATION
When in-house evaluation of a cytologic preparation does not furnish sufficient reliable information for managing a case, the preparation may be submitted to a veterinary clinical pathologist or cytologist for interpretation, or an alternative procedure, such as biopsy and histopathologic evaluation, may be performed. If possible, the person to whom the cytologic preparation is sent should be contacted, and specifics concerning sample handling should be discussed, such as the number of smears to send or whether to fix or stain the smears before mailing.
When possible, two or three air-dried unstained smears, and two or three air-dried Romanowsky-stained smears should be submitted. Pathologists may stain the air-dried unstained smears with the Romanowsky or NMB stains of their choice. The Romanowsky-stained smears are a safety factor. Some tissues stain poorly when air dried but not stained for several days. Also, slides occasionally are shattered during transport and cannot be stained on receipt. Sometimes microscopic examination of shards from the broken prestained smears allows diagnosis. If only a few smears can be prepared from the sample, one should be submitted air dried and unstained and the other submitted air dried, fixed, and stained. Smears should be well labeled with alcohol-resistant ink or another permanent labeling method. If a Papanicolaou stain is to be used, several wet-fixed smears should be submitted.
Fluid samples should have smears prepared from them immediately. Direct smears and concentrated smears should be submitted. Also, an EDTA (lavender top) and sterile serum tube (red top) fluid sample should be submitted. A total nucleated cell count and total protein concentration can be performed on the EDTA tube sample and, if necessary, chemical analyses can be performed on the serum tube sample.
Slides must be protected when mailed. Simple cardboard mailers do not provide sufficient protection to prevent slide breakage if they are mailed in unpadded envelopes. Marking the envelope with phrases such as “fragile,” “glass,” “breakable,” and “please hand cancel” has little success. Placing a pad of bubble wrap or polystyrene on each side of the slide holder usually prevents slide breakage. Slides may also be mailed in plastic slide holders or innovative holders, such as small pill bottles.
Unfixed slides should not be mailed with formalin-containing samples and should be protected against moisture. Formalin fumes alter the staining characteristics of smears, and water causes cell lysis (Fig. 9-26).
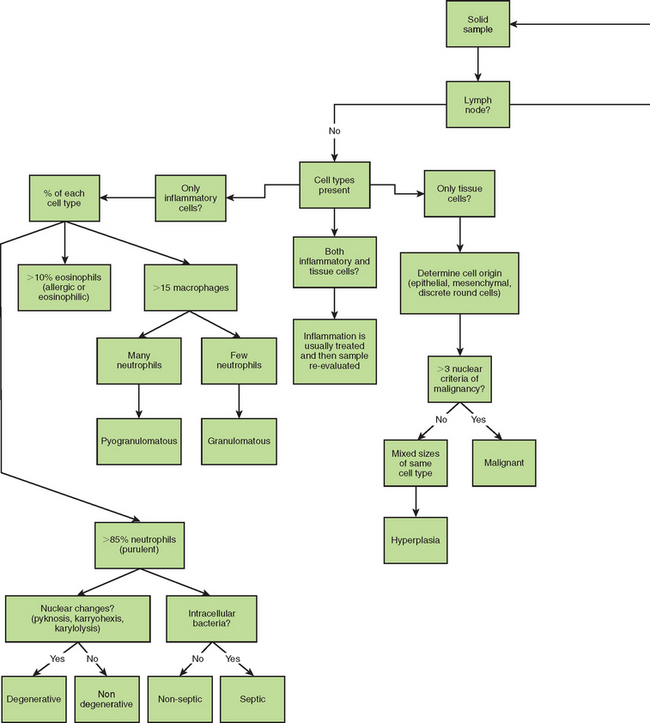
INITIAL MICROSCOPIC EVALUATION
Figure 9-27 summarizes steps in the evaluation of cytology specimens. The initial evaluation of the cytology preparation should be performed on low magnification (100×) to determine if all areas are adequately stained and to detect any localized areas of increased cellularity. To improve resolution by decreasing light refraction, a drop of immersion oil can be placed on the smear and a cover slip added. A consistent approach to examination helps ensure high-quality results. Large objects such as cell clusters, parasites, crystals, and fungal hyphae are normally evident on the low power examination. This initial evaluation should be used to characterize the cellularity and composition of the sample by recording the types of cells present and relative numbers of each type. A high power examination (400× to 450×) should then be performed to evaluate and compare individual cells and further characterize the types of cells present. Oil immersion must be used to identify specific nuclear criteria of malignancy and cytoplasmic abnormalities indicative of malignancy and various inflammatory reactions. The cytology report should indicate the cell types present, their appearance, and relative proportions.
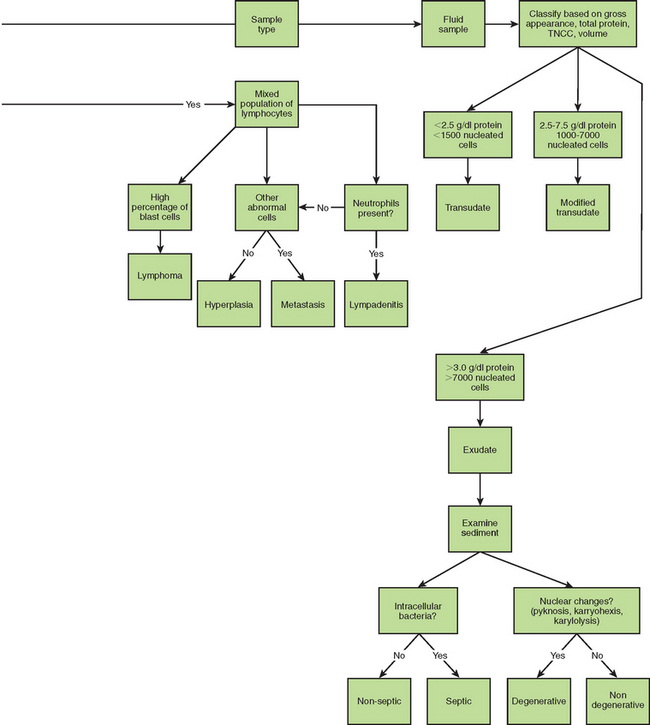
Inflammation
Inflammation is a normal physiologic response to tissue damage or invasion by microorganisms. This damage releases substances that have a chemotactic effect on certain white blood cells. These chemotactic factors are therefore involved in attracting white blood cells to the site of inflammation. The first white blood cells to arrive are the neutrophils. Neutrophils phagocytize dead tissue and microorganisms. The process of phagocytosis creates pH changes both within the neutrophils and in the site. As the pH changes, neutrophils become unable to phagocytize any further and the cells quickly die. At this point, macrophages move in to the site and pick up the phagocytic activity. Cytology samples from inflammatory sites are therefore characterized by the presence of white blood cells, particularly neutrophils and macrophages. Occasionally eosinophils or lymphocytes may also be present. In fluid samples, total nucleated cell counts of greater than 5000/μl is a common finding with inflammation. The fluid is often turbid and may be white or pale yellow. Total protein is often greater than 3 g/dl
Inflammation can be categorized as suppurative (purulent), granulomatous, pyogranulomatous, or eosinophilic based on the relative numbers of the various cell types present.
Suppurative (purulent) inflammation (Fig. 9-28) is characterized by the presence of large numbers of neutrophils, usually greater than 85% of the total nucleated cell count. When significant numbers of macrophages are present (greater than 15%), the sample is classified as granulomatous or pyogranulomatous (Fig. 9-29). Fungal and parasitic infections often manifest with this presentation. The presence of greater than 10% of eosinophils along with increased neutrophils indicates an eosinophilic inflammation (Fig. 9-30). This is usually found with parasitic infection but may also be present in some neoplastic disorders.
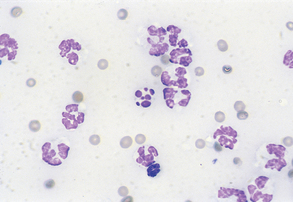
Figure 9-28 Suppurative inflammation as evidenced by the large number of neutrophils. Note the presence of karyorrhexis in the center cell. (From Raskin RE, Meyer DJ: Atlas of canine and feline cytology, St Louis, 2001, Saunders.)
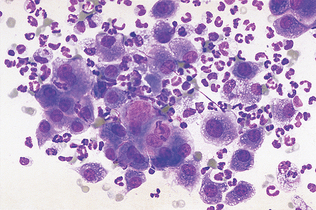
Figure 9-29 Pyogranulomatous inflammation. Macrophages represent more than 15% of the cells present. (From Cowell RL, Tyler RD, Meinkoth JH: Diagnostic cytology and hematology of the dog and cat, ed 2, St Louis, 1999, Mosby.)
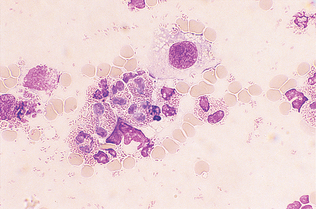
Figure 9-30 Eosinophilic inflammation. Note the single macrophage and numerous free eosinophilic granules. (From Cowell RL, Tyler RD, Meinkoth JH: Diagnostic cytology and hematology of the dog and cat, ed 2, St Louis, 1999, Mosby.)
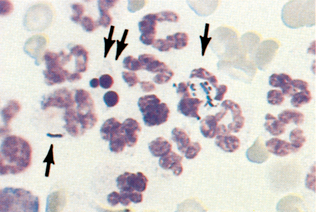
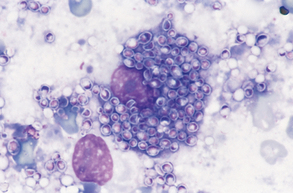
Once designated as inflammatory, the cells must also be evaluated for evidence of degeneration and presence of microorganisms. Nuclear changes that may be found in inflammatory cells (e.g., neutrophils) include karyolysis, karyorrhexis, and pyknosis, with karyolysis having the greatest significance. Pyknosis represents slow cell death (aging) and refers to a small, condensed, dark nucleus that may fragment (karyorrhexis). Karyolysis represents rapid cell death, as in some septic (bacterial) inflammatory reactions, and appears as a swollen, ragged nucleus without an intact nuclear membrane and with reduced staining intensity. Cells should also be evaluated for the presence of bacteria. Inflammatory cells that contain phagocytized microorganisms are referred to as septic (Fig. 9-31). Additional phagocytized material may include erythrocytes, parasites, and fungal organisms (Fig. 9-32).
Neoplasia
Unlike inflammation, neoplastic specimens normally contain rather homogeneous populations of a single cell type. Although mixed cells populations are sometimes seen, these usually involve a neoplastic area with a concurrent inflammation. Neoplasia is indicated when the cells present are of the same tissue origin. Once identified as neoplastic, the technician should identify the tissue origin and evaluate the cells for the presence of malignant characteristics (Table 9-2).
Neoplasia must first be differentiated as either benign or malignant. Benign neoplasia is represented by hyperplasia with no criteria of malignancy present in the nucleus of the cells. The cells are of the same type and are relatively uniform in appearance. Cells that display at least three abnormal nuclear configurations are identified as malignant. Nuclear criteria of malignancy can include any of the following:
• Anisokaryosis: Any unusual variation in overall cell size.
• Pleomorphism: Variability in the size and shape of same cell type.
• High or variable nucleus/cytoplasm ratio
• Increased mitotic activity: Mitosis is rare in normal tissue, and cells usually divide evenly in two Any increase in the present of mitotic figures or cells that are not dividing equally is considered a malignant criteria.
• Coarse chromatin pattern: The chromatin pattern is coarser than normal and may appear ropy or cordlike.
• Nuclear molding: A deformation of nuclei by other nuclei within the same cell or adjacent cells.
• Multinucleation: Multiple nuclei within a cell.
• Nucleoli that vary in size (anisonucleoliosis), shape (angular nucleoli), and number (multiple nucleoli).
In general, if three or more nuclear criteria of malignancy are present, the specimen is identified as malignant. Exceptions to this general rule are indicated if inflammation is also present or only a few cells display malignant characteristics. Histopathologic verification of cytologic findings is important for most tumors (whether cytologically benign or malignant). Also, cytologically benign cells may be obtained from malignant tumors. Histopathologic examination offers the advantage of enabling assessment of factors such as local tissue infiltration and vessel or lymphatic invasion by tumor cells. These characteristics of malignant tumors are not evident cytologically.
Specimens that have been classified as malig-nant should be further evaluated to determine the cell type involved. The primary types of tumors encountered in veterinary medicine are categorized as epithelial cell tumors, mesenchymal cell tumors, and discrete round cell tumors. The overall characteristics of samples from each of these cell types are summarized in Table 9-3. A variety of terminology is used to describe these various tumor types, and some references may differ in their classifications of specific types of tumors.

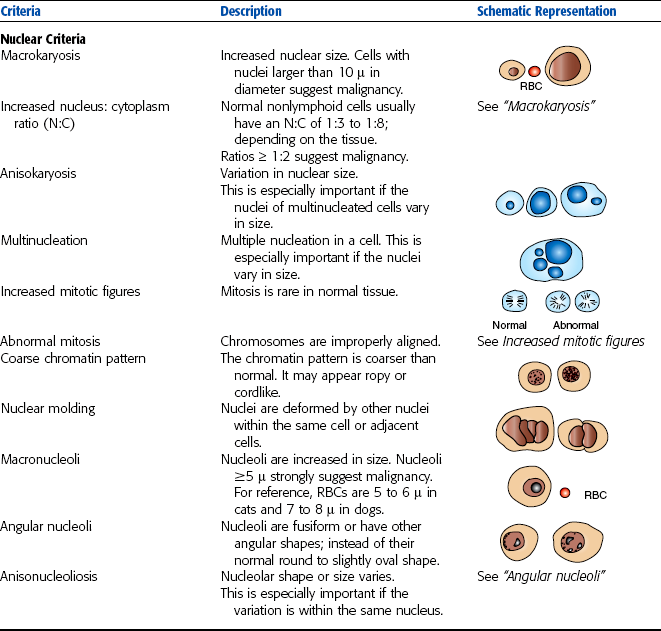
Epithelial cell tumors are also referred to as carcinoma or adenocarcinoma. The samples tend to be highly cellular and often exfoliate in clumps or sheets (Fig. 9-33). Mesenchymal cell tumors are also referred to as sarcoma and are usually less cellular. The cells tend to exfoliate singly or in wispy spindles (Fig. 9-34). Discrete round cell tumors exfoliate very well but are usually not in clumps or clusters. Round cell tumors include histiocytoma, lymphoma, mast cell tumors, plasma cell tumors, transmissible venereal tumors, and melanoma. Histiocytoma and transmissible venereal tumors appear somewhat similar except that histiocytoma is not usually highly cellular (Fig. 9-35). Plasma cell tumors can be recognized by the presence of large numbers of cells with an eccentri-cally located nucleus and prominent perinuclear clear zone (Fig. 9-36). Mast cells can be recognized by their prominent purple/black granules (Fig. 9-37). Melanoma is characterized by cells with prominent dark black granules (Fig. 9-38). Occasionally, cells from poorly differentiated tumors may contain few or no granules (amelanotic melanoma).
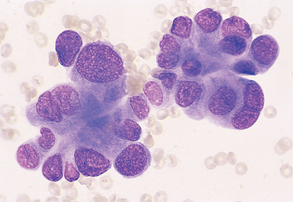
Figure 9-33 Lung carcinoma. Clusters of cells with anisokaryosis, binucleation, and high and variable nucleus/cytoplasm ratios. (From Raskin RE, Meyer DJ: Atlas of canine and feline cytology, St Louis, 2001, Saunders.)
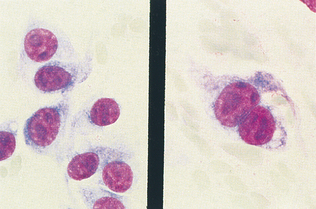
Figure 9-34 Sarcoma. Aspirate from a malignant spindle cell tumor with cells showing anisokaryosis; anisonucleiosis; and large, prominent, and occasionally angular nucleoli. (From Cowell RL, Tyler RD, Meinkoth JH: Diagnostic cytology and hematology of the dog and cat, ed 2, St Louis, 1999, Mosby.)
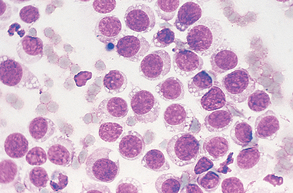
Figure 9-35 Large numbers of round cells from an imprint of a transmissible venereal tumor. (From Raskin RE, Meyer DJ: Atlas of canine and feline cytology, St Louis, 2001, Saunders.)
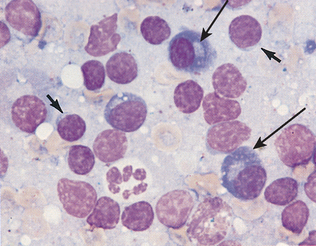
Figure 9-36 Several plasma cells are evident in this sample (long arrows) from a hyperplastic lymph node. Small lymphocytes are also present (short arrows). (From Cowell RL, Tyler RD, Meinkoth JH: Diagnostic cytology and hematology of the dog and cat, ed 2, St Louis, 1999, Mosby.)
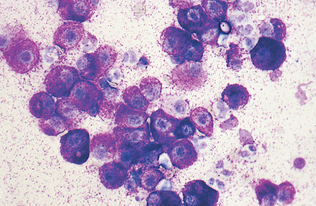
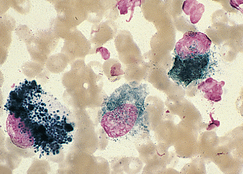
CYTOLOGY OF SPECIFIC SITES
Under normal circumstances, the peritoneal and thoracic cavities contain only enough fluid to adequately lubricate the surfaces of the organs and the cavity walls. Fluid is collected in EDTA tubes for total nucleated cell counts, cytologic examination, and refractometric protein measurements and in a plain tube for determination of the total protein concentration. Other clinical chemistry determinations are performed infrequently on peritoneal and pleural fluid.
Color, Turbidity, and Odor
Normal peritoneal and pleural fluids are colorless to straw yellow and transparent to slightly turbid. Both fluids should be odorless. Gross discoloration and increased turbidity may be the result of increased cell numbers and/or protein concentration. Collection of malodorous peritoneal fluid at abdominocentesis may indicate a necrotic segment of bowel within the peritoneal cavity, a ruptured segment of bowel with free gut contents in the cavity, or accidental enterocentesis. These conditions may be distinguishable cytologically and by reference to other clinical findings. Although not commonly seen, chylothorax may be evident as a “milky” fluid, especially if the animal has recently eaten, because of the chylous effusion’s high fat content and large number of mature lymphocytes. In fasted animals, the fluid may be tan. Unlike fluids with high leukocyte counts (which also may have a whitish color), chylous fluid does not have a clear supernatant after centrifugation. The fat in chylous fluid is present as small droplets (chylomicrons), which can be stained with Sudan III or IV. The fat in chylous fluid may be dissolved with ether after the fluid has been alkalinized with sodium hydroxide or sodium bicarbonate. If significant numbers of erythrocytes are present, the fluid may have a reddish color.
Total Nucleated Cell Count
A total nucleated cell count (TNCC) is performed using the same methods as for a complete blood count (see Chapter 2). As a cross-species generalization, normal peritoneal and pleural fluids have less than 10,000 nucleated cells/μl, usually 2000 to 6000/μl. Mononuclear cells may be visible as clusters of cells, which can make counting individual cells difficult.
A differential count of at least 100 nucleated cells should be performed, noting cell type and morphologic characteristics. Nucleated cells are categorized as neutrophils, large mononuclear cells (a collective grouping of mesothelial cells and macrophages), lymphocytes, eosinophils, and any other nucleated cells. Notes on cell morphologic characteristics should include comments on nuclear and cytoplasmic appearance. If bacteria are present, their morphologic features (bacilli, coccobacilli, cocci) and location (free or intracellular; i.e., phagocytosed) must be recorded. In such cases, another smear can be stained with Gram stain and the fluid cultured.
Cellular Elements
Normal peritoneal and pleural fluids contain few erythrocytes. The number present on a smear should be estimated. Suitable categories include rare, few, many, and large numbers. The number present varies with the method of sample preparation. Iatrogenic contamination and acute and chronic hemorrhage are distinguished grossly, as previously outlined. If erythrocytes have been present in the fluid for several hours, they may be phagocytosed by macrophages (erythrophagocytosis) (Fig. 9-39).
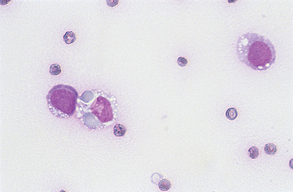
Figure 9-39 Fluid aspirate with macrophages. One cell is exhibiting erythrophagia. (From Raskin RE, Meyer DJ: Atlas of canine and feline cytology, St Louis, 2001, Saunders.)
Peritoneal and pleural fluid samples should also be evaluated for cellularity. The TNCC and total protein values for the sample allow it to be classified as transudate, modified transudate, or exudate (Table 9-4). Published normal values for peritoneal and pleural fluid cytology of dogs and cats are scarce. Normal horses generally have an average of approximately 55% to 60% neutrophils, 25% to 30% large mononuclear cells, 10% to 20% lymphocytes, and an occasional eosinophil (less than 1%). Values for cattle are somewhat similar, but normal animals often have comparable numbers of neutrophils and lymphocytes.
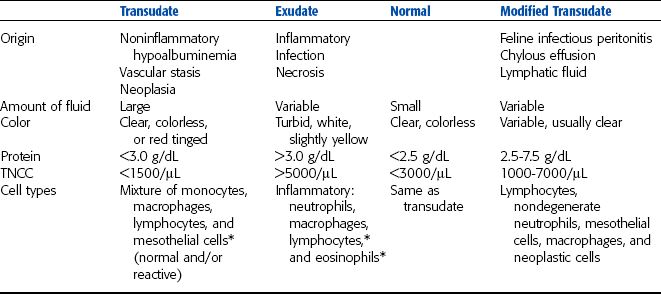
Exudates are fluids with increased cellularity and protein concentration because of inflammation. The following cross-species generalization can be made. Suppurative inflammatory reactions increase the total cell count, the percentage, and the absolute numbers of neutrophils to greater than 85% of the nucleated cells. Suppurative inflammatory reactions usually cause high-normal to elevated total nucleated cell counts, with elevated neutrophil percentages and numerous mesothelial cells and/or macrophages. Mesothelial cells line the body cavities. In the presence of increased fluid within the cavities, these cells may become reactive (i.e., multinucleate, with anisocytosis and anisokaryosis, prominent nucleoli, and basophilic cytoplasm) (Fig. 9-40). Reactive mesothelial cells may be difficult to distinguish from some neoplastic cells. Some mesothelial cells may be present as clusters or rafts of cells. These clusters result from proliferation and exfoliation of cells from the peritoneal lining, or mesothelium, in response to a decrease in contact inhibition between cells on opposing surfaces of the peritoneum, because of the effusion. Macrophages may phagocytize degenerate cells or cellular debris. Migrating parasite larvae can cause increased neutrophil and eosinophil percentages, with or without an elevated total nucleated cell count.
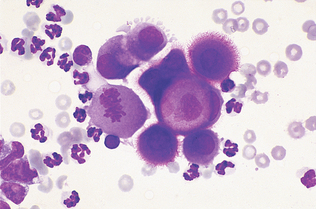
Figure 9-40 A cluster of reactive mesothelial cells. Note the mitotic figure. (From Cowell RL, Tyler RD, and Meinkoth JH: Diagnostic cytology and hematology of the dog and cat, ed 2, St Louis, 1999, Mosby.)
Cellular morphologic features depend on microorganism(s) present and may vary from cytoplasmic vacuolation with few nuclear changes evident to marked cytoplasmic vacuolation, marked nuclear swelling and disruption (karyolysis), and general cellular degeneration or fragmentation. Bacteria may be evident within the cytoplasm of neutrophils and/or macrophages (Fig. 9-41). Cases of simple peritonitis may have a single type of bacterium evident or the bacterial population may be mixed as a result of devitalization or rupture of the bowel. Accidental penetration of the bowel during abdominocentesis may also result in a mixed population of bacteria in the smear. However, in the latter case, leukocyte numbers and morphologic characteristics are usually normal, and the bacteria are frequently not phagocytosed. Large ciliated organisms also may be noted in large-bowel enterocentesis in horses.
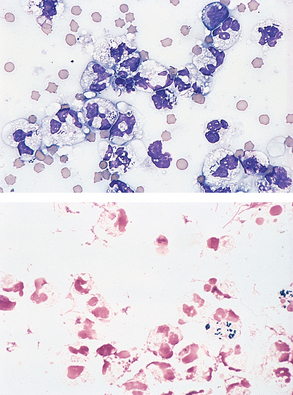
Figure 9-41 Septic exudates. Note the presence of intracellular bacterial rods. (From Raskin RE, Meyer DJ: Atlas of canine and feline cytology, St Louis, 2001, Saunders.)
Transudates (ascitic effusions) typically have low protein concentrations and low total nucleated cell counts (less than 1500/μl), with fairly normal differential counts or possibly an increase in the percentage of large mononuclear cells (Fig. 9-42). The mononuclear cells are principally mesothelial cells, which may be in clusters or rafts and may be quite reactive in appearance. Transudates are frequently secondary to congestive heart failure or may occur in animals with low blood albumin concentration.
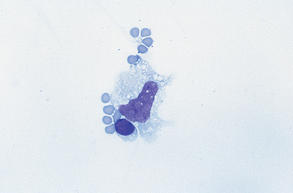
Figure 9-42 Macrophage, small lymphocyte, and several red blood cells. This sample is characteristic of normal fluid and transudates. (From Raskin RE, Meyer DJ: Atlas of canine and feline cytology, St Louis, 2001, Saunders.)
Modified transudates are characterized by relatively low to moderate total cell counts, predominantly resulting from leakage of lymphatics. This leakage is responsible for the high total protein concentration of modified transudates. Cells present include low numbers of inflammatory cells (nondegenerate), mostly small mature lymphocytes; few macrophages; and some mesothelial cells (Fig. 9-43).
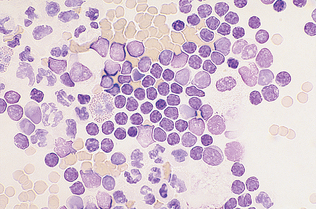
Figure 9-43 Smear from a chylous effusion. A mixture of mature lymphocytes, neutrophils, and an eosinophil are present. This is characteristic of a modified transudate. (From Cowell RL, Tyler RD, Meinkoth JH: Diagnostic cytology and hematology of the dog and cat, ed 2, St Louis, 1999, Mosby.)
Intraabdominal tumors may exfoliate cells into the peritoneal fluid. Cytologic diagnosis of such neoplasia may be difficult and is often a task for a specialist cytologist. However, the technician should be able to recognize abnormal lymphocytes by using the criteria of malignancy previously outlined and should be suspicious of clusters of pleomorphic, secretory-type cells. The presence of unexpected cells, such as mast cells, must also be noted.
Lymph Nodes
Cytologic evaluation of lymph node tissue is performed to diagnose causes of lymph node enlargement and differentiate hyperplasia, inflammation, primary neoplasia (lymphoma), and metastatic neoplasia. Lymph nodes may show evidence of inflammation (lymphadenitis), hyperplasia (benign neoplasia), mixed (both inflammatory and neoplastic cells present), neoplasia (lymph node cells with abnormal nuclear features), and metastasis (neoplastic cells from other body tissues that spread to lymph nodes). Each of these has specific cell types associated with the abnormality.
Lymph node tissue is normally collected from the periphery of an enlarged lymph node by fine needle biopsy. In patients with generalized lymphadenopathy, samples should be obtained from two lymph nodes. Because lymph nodes that drain the oral cavity and gastrointestinal tract are antigenically stimulated under normal conditions, these should be avoided. Samples are then prepared by the compression technique and stained with standard Romanowsky-type stains.
A variety of cell types may be found in lymph node aspirates. These include lymphocytes, plasma cells, white blood cells, and neoplastic cells. Microorganisms, lymphoglandular bodies, and bacteria may also be present. Lymphoglandular bodies are small cytoplasmic fragments that may be seen between cells and are not a pathologic feature. Table 9-5 contains a summary of the cell types that may be found in lymph node aspirates.
TABLE 9-5
Cell Types Found in Lymph Node Aspirates
| Cell Type | Characteristics |
| Lymphocytes, small | Similar in appearance to the small lymphocyte seen on a peripheral blood film. Slightly larger than a Scanty cytoplasm, dense nucleus. |
| Lymphocytes, intermediate | Nucleus approximately twice as large as an RBC. Abundant cytoplasm. |
| Lymphoblasts | Two to four times as large as an RBC. Usually contain a nucleolus. Diffuse nuclear chromatin. |
| Plasma cells | Eccentrically located nucleus, trailing basophilic cytoplasm, perinuclear clear zone. Vacuoles and/or Russell bodies may be present. |
| Plasmablasts | Similar to lymphoblasts with more abundant, basophilic cytoplasm. May contain vacuoles. |
| Neutrophils | May appear similar to neutrophils in peripheral blood or show degenerative changes. |
| Macrophages | Large phagocytic cell. May contain phagocytized debris, microorganisms, etc. Abundant cytoplasm. |
| Mast cells | Round cells that are usually slightly larger than lymphoblasts. Distinctive purple-staining granules may not stain adequately with Diff-Quik. |
| Carcinoma cells | Epithelial tissue origin. Usually found in clusters; pleomorphic. |
| Sarcoma cells | Connective tissue origin. Usually occur singly with spindle-shaped cytoplasm. |
| Histiocytes | Large, pleomorphic, single or multinuclear; nuclei are round to oval. |
In a normal lymph node, the predominant cell type is the small, mature lymphocyte. These tend to comprise more than three fourths of the total cells present. Smaller numbers of intermediate lymphocytes and lymphoblasts as well as macrophages are also present. Plasma cells may occasionally be seen (Fig. 9-44). Mast cells are usually rare in cytologic preparations of lymph node tissue. Lymph nodes with evidence of inflammation (lymphadenitis) will have a predominance of leukocytes, as previously described (Fig. 9-45).
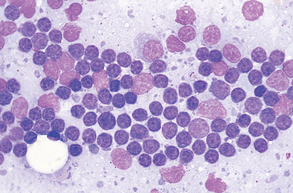
Figure 9-44 Aspirate from a normal lymph node. Small, mature lymphocytes predominate. (From Raskin RE, Meyer DJ: Atlas of canine and feline cytology, St Louis, 2001, Saunders.)
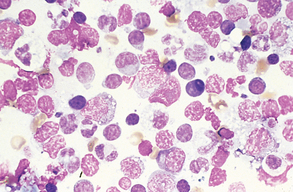
Figure 9-45 Pyogranulomatous lymphadenitis. Numerous macrophages and neutrophils are evident along with a mixture of lymphocyte types. (From Raskin RE, Meyer DJ: Atlas of canine and feline cytology, St Louis, 2001, Saunders.)
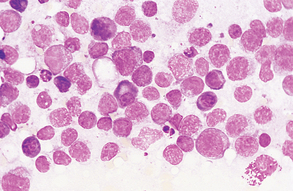
Reactive Lymph Node
Lymph nodes that are responding to antigenic stimulation also contain predominantly small, mature lymphocytes. However, plasma cells, lymphoblasts, and intermediate lymphocytes are more abundant than in a normal lymph node (Fig. 9-46). Occasional Mott cells (plasma cells containing secretory vesicles of immunoglobulin) may also be seen (Fig. 9-47). Antigenic stimulation can also cause an inflammatory response and would be characterized by the presence of neutrophils and/or macrophages.
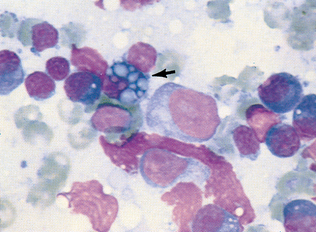
Malignant Neoplasia
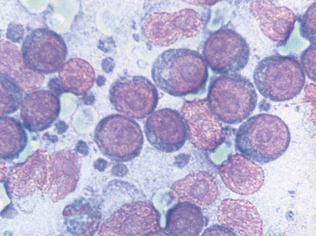
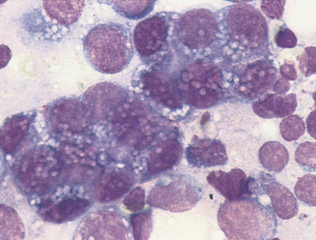
Primary lymphoid neoplasia, or lymphoma, is characterized by a predominance of lymphoblasts, and mitotic figures are common. Macrophages are also present, and plasma cells are scarce. Other neoplastic cells that may be present in lymph node aspirates include mast cells, carcinoma cells, sarcoma cells, and histiocytes. Cells that display at least three abnormal nuclear configurations usually are identified as malignant (Fig. 9-48). Lymph node samples may also contain metastatic cells from other body parts (Fig. 9-49).
Cerebrospinal Fluid
As a cross-species generalization, normal CSF contains no erythrocytes and less than 25 nucleated cells per milliliter (usually 0 to 10/L) (Fig. 9-50). Pleocytosis is an elevated CSF nucleated cell count. Normal CSF contains 95% to 100% mononuclear cells, almost all of which are lymphocytes. Bacterial infections involving CSF generally cause marked pleocytosis, mostly because of neutrophils. Inflammation associated with viruses, fungi, neoplasia, or degenerative conditions generally causes less dramatic pleocytosis, with a significant proportion of mononuclear cells (often lymphocytes). Eosinophils sometimes are seen, especially in parasitic inflammatory responses. In general, the causative agent often is not cytologically apparent. Neoplastic cells are seldom observed in CSF.
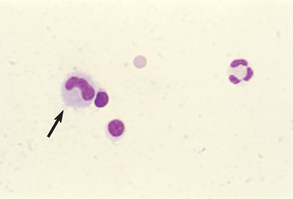
Figure 9-50 Cell types found in CSF. Two small lymphocytes, one large mononuclear cell (arrow), one neutrophil, and one erythrocyte are present. (From Raskin RE, Meyer DJ: Atlas of canine and feline cytology, St Louis, 2001, Saunders.)
Normal CSF contains virtually no erythrocytes. Erythrocytes may be counted by charging a hemacytometer with a well-mixed sample of undiluted and unstained CSF. All cells in the entire boxed area of one side are counted. With this method both erythrocytes and nucleated cells are observed. Distinguishing between these two groups of cells is usually possible, but not to subcategorize the nucleated cells. Cell counts for undiluted CSF are multiplied by 1.1 to give the total number of cells per milliliter. (If ethanol-diluted CSF is used, the cell counts are multiplied by 2.2.) If distinguishing erythrocytes from nucleated cells in unstained CSF is difficult, the total number of cells counted by the latter method can be subtracted from the number of nucleated cells counted to calculate the erythrocyte count.
Use of various correction factors has been advocated to adjust CSF nucleated cell counts (NCC) for any peripheral blood leukocytic contamination. Because normal CSF contains no erythrocytes, the observed nucleated cell count may be corrected if the number of erythrocytes per milliliter of CSF is known. The simplest approach is to consider that each 500 to 1000 erythrocytes would be accompanied by one leukocyte. A more complicated method is to use the following equation:
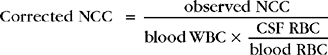
This equation requires values for blood leukocyte (white blood cells) and erythrocyte (red blood cell) counts and a CSF erythrocyte count. Despite its complexity, it is no more accurate than the preceding approximation. Ideally CSF samples should be free of iatrogenic peripheral blood contamination. Use of the previous correction factor is, at best, a rough guide to the uncontaminated CSF nucleated cell count.
In addition to the cytology evaluation, a variety of chemical and immunologic tests are performed on CSF samples. These include the Pandy test, Nonne-Apelt test, sulfosalicylic acid test, trichloroacetic acid test, fibrinogen concentration, glucose concentration, lactate concentration, electrolyte concentrations, and creatine kinase concentration. These tests are discussed further in Chapter 3.
AQUEOUS AND VITREOUS HUMOR
Fluid from the eye is similar to CSF in that it has low cellularity, being composed of mostly small mononuclear cells, essentially no erythrocytes, and a low protein concentration. Interpretation of changes in aqueous humor is similar to that for CSF.
Synovial Fluid Analysis
If only one or two drops can be obtained, as in some normal joints of cats and dogs, gross assessment of fluid color and turbidity and cytologic examination of a direct smear, possibly with concurrent subjective assessment of viscosity, may be all that is practical. If 0.5 to 1 ml is collected, a total nucleated cell count and refractometric protein measurement (on EDTA-preserved fluid) may be added to the list of tests. Collection of larger volumes permits additional tests, such as the mucin clot test.
Color and Turbidity
Normal synovial fluid is clear to straw yellow and nonturbid. Yellow synovial fluid is common in large animals, especially horses. Turbidity, when present, is caused by cells, protein (or fibrin), or cartilage.
Normal synovial fluid contains few erythrocytes. Iatrogenic contamination at arthrocentesis is common. Differentiation of contamination and recent or old hemorrhage is as previously described.
Viscosity
Viscosity reflects the quality and concentration of hyaluronic acid, which is part of the synovial fluid mucin complex. The function of mucin is joint lubrication. Viscosity may be quantitated with a viscometer; however, subjective assessment is most often used.
Normal synovial fluid is sticky. If a drop is placed between the thumb and forefinger, as the digits are separated it forms a 1- to 2-inch strand before breaking. Similarly, when gently expressed through a needle on a horizontally held syringe, it hangs in a 1- to 2-inch strand before separating from the needle tip.
In general, viscosity is not decreased in normal joints and those with degenerative problems. It frequently is decreased in joints with bacterial inflammation as a result of mucin degradation by bacterial hyaluronidase and in joints with a significant effusion (including hydrarthrosis) as a result of dilution of mucin (and hyaluronic acid).
Because EDTA may degrade hyaluronic acid, both viscosity and mucin clot formation usually are assessed on fluid to which no anticoagulant has been added. If anticoagulation is necessary because of a high fluid fibrinogen concentration, heparin is the preferred anticoagulant.
Cytologic Examination (Cell Counts)
Slides for cytologic examination may be prepared on EDTA-preserved fluid or fluid without anticoagulant, the latter especially if only a few drops are obtained and the smear is made immediately. Thin smears are made by slowly advancing the spreader slide. Because of the high viscosity of normal synovial fluid, cells usually do not accumulate at the feathered edge of the smear. Margination of cells increases as viscosity of the fluid decreases. At low cell counts, especially less than 500/μl, concentration of cells by centrifugal sedimentation and subsequent resuspension of cells in a small volume of supernatant fluid produces a more cellular smear. Slides usually are stained with a Romanowsky stain. Table 9-6 summarizes classification of synovial fluid.
TABLE 9-6
Classification of Synovial Fluid
From Raskin RE, Meyer DJ: Atlas of canine and feline cytology, St Louis, 2001, Saunders.
Normal synovial fluid generally contains at least 90% mononuclear cells and less than 10% neutrophils (Fig. 9-51). Eosinophils are rarely observed. Mononuclear cells comprise about equal numbers of lymphocytes and monocytic/macrophage-type cells, which are nonvacuolated and nonphagocytic. Large vacuolated or phagocytic mononuclear cells comprise less than 10% of the differential count of normal synovial fluid. Macrophages become vacuolated in normal synovial fluid that is not processed soon after collection; therefore prompt preparation of smears is important to prevent this artificial finding.
Figure 9-51 Normal synovial fluid has low cellularity with primarily mononuclear cells (arrow). The background material is related to the mucin content and may appear granular to ropy. (From Raskin RE, Meyer DJ: Atlas of canine and feline cytology, St Louis, 2001, Saunders.)
Cells in synovial fluid with good viscosity tend to align in a linear fashion in the direction of the smear, giving a “windrow” appearance (Fig. 9-52). Mucin precipitation produces an eosinophilic granular background in Romanowsky-stained smears, the density of which reflects smear thickness. Cells in smears from viscous fluid may not spread out well on the slide, which can make their identification difficult. Such fluid may be diluted 1:1 with saline-reconstituted hyaluronidase (150 U/ml). This decreases sample viscosity after a few minutes, allowing more accurate cell morphology when smeared.
Figure 9-52 Synovial fluid from a patient with inflammatory joint disease. These neutrophils are in the typical “windrow” arrangement commonly seen in synovial fluid. (From Raskin RE, Meyer DJ: Atlas of canine and feline cytology, St Louis, 2001, Saunders.)
As a generalization, mononuclear cells predominate in traumatic and degenerative arthropathies, usually with increased numbers of large vacuolated or phagocytic cells.
Occasionally when joint erosion has progressed through to subchondral bone, osteoclasts may be observed. In contrast, neutrophils predominate in infectious arthropathies (because of bacteria, viruses, and mycoplasmas, for example) and many noninfectious conditions, such as rheumatoid arthritis and systemic lupus erythematosus. When cells are clumped together in a smear, NMB stain may demonstrate interlocking fibrin strands. Rarely the causative organism in septic joint fluid may be observed cytologically, especially when phagocytosed. Culture is recommended when an infectious process is suspected. A granulomatous type of arthropathy is suggested when neutrophils and vacuolated/phagocytic macrophages are both increased in number. Lupus erythematosus cells, which are neutrophils or macrophages containing phagocytosed nuclear chromatin, are seen occasionally in the synovial fluid of animals with systemic lupus erythematosus.
Tracheal Wash
Total nucleated cell counts are usually not performed on tracheal wash fluids. Cell numbers are subjectively recorded from evaluation of the smear. A tracheal wash smear from a normal animal contains few cells, usually with a small amount of mucus. The mucus often appears microscopically as eosinophilic to purple strands that may enmesh the cells. Epithelial cells are the principal cell type. If the sample is collected from the level of the trachea, ciliated epithelial cells predominate. These cells are columnar to cuboidal, with a polar nucleus on the border opposite the cilia (Fig. 9-53). If the specimen is collected from the bronchi, bronchoalveolar epithelial cells are also fairly common. They are round, nonciliated cells with basophilic cytoplasm and may occur in clumps. A few goblet cells (secretory epithelial cells) also may be observed (Fig. 9-54).
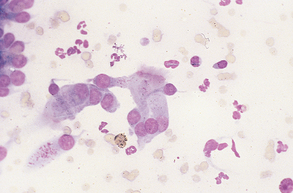
Figure 9-53 Normal ciliated columnar epithelial cells in a normal tracheal wash sample. (From Raskin RE, Meyer DJ: Atlas of canine and feline cytology, St Louis, 2001, Saunders.)
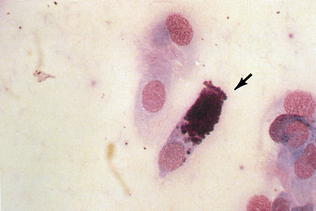
Figure 9-54 A goblet cell (arrow) and ciliated columnar cells in a tracheal wash sample. (From Cowell RL, Tyler RD, and Meinkoth JH: Diagnostic cytology and hematology of the dog and cat, ed 2, St Louis, 1999, Mosby.)
If the sample is a bronchoalveolar wash, alveolar macrophages may predominate. These are large individual cells with a large round to oval nucleus and a moderate amount of basophilic cytoplasm. If they become reactive or activated, the cytoplasm increases in volume and becomes more granular and vacuolated. Neutrophils, lymphocytes, eosinophils, plasma cells, mast cells, and erythrocytes are seen rarely in specimens from normal animals.
Abnormal tracheal washes are generally exudates. These samples contain numerous mucus strands and are cellular. Eosinophilic, spiral casts from small bronchioles (Curschmann’s spirals) suggest a chronic bronchiolar problem (Fig. 9-55). Cell morphology is highly variable among and within samples. Many cells may be unidentifiable. Neutrophils and macrophages are numerous. In acute inflammation, neutrophils are the predominant cell type and may represent more than 95% of nucleated cells (Fig. 9-56). As the process becomes more chronic, mononuclear macrophages increase in number. The causative agent, possibly bacterial or fungal, may be noted, free and/or phagocytosed, in the smear. Tracheal wash samples can be cultured by routine microbiologic procedures.
Figure 9-55 Tracheal wash sample from a dog with chronic bronchial disease. A large Curschmann’s spiral and several macrophages are present. The eosinophilic background represents mucus. (From Cowell RL, Tyler RD, Meinkoth JH: Diagnostic cytology and hematology of the dog and cat, ed 2, St Louis, 1999, Mosby.)
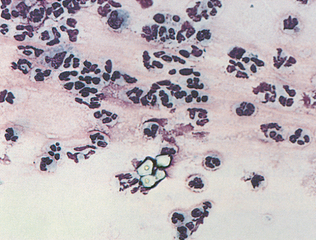
Figure 9-56 High numbers of neutrophils, an alveolar macrophage, and a cluster of four granules of cornstarch from glove powder in a tracheal wash sample. (From Cowell RL, Tyler RD, Meinkoth JH: Diagnostic cytology and hematology of the dog and cat, ed 2, St Louis, 1999, Mosby.)
The presence of bacteria or fungi in a tracheal wash does not necessarily mean the organism is pathogenic. Plant or fungal spores sometimes contaminate tracheal washes from herbivores (inhaled from feed) and may be phagocytosed by macrophages. Oral or pharyngeal contamination of the collection apparatus, or increased inspiratory effort with contamination of the upper tracheal mucosa by pharyngeal microflora, may cause inclusion of bacteria in a tracheal wash specimen. Such bacteria frequently are associated with or are adherent to squamous epithelial cells of the pharyngeal mucosa.
Eosinophils are prominent (possibly more than 10% of nucleated cells) in inflammatory reactions with an allergic or parasitic component. Because cell preservation is often only fair, free eosinophil granules rather than intact eosinophils may be noted. Rarely parasite eggs or larvae may be noted in the smear.
Erythrocytes rarely are seen in normal tracheal wash specimens. Recent hemorrhage may be evidenced by numerous intact erythrocytes in the smear. In contrast, with old hemorrhage few erythrocytes may be noted, and many of the macrophages may contain hemosiderin granules (dark-blue or red cytoplasmic granules).
Neoplastic cells may be detected in tracheal wash specimens. Criteria for malignancy are as previously described. Neoplastic cells are frequently in clusters, generally epithelial in origin, and frequently secretory in appearance (i.e., their cytoplasm is basophilic and vacuolated).
Nasal Flush
A nasal flush from a normal animal contains cornified and noncornified squamous epithelial cells, often with adherent bacteria, and negligible evidence of hemorrhage or inflammation. Various abnormalities may be demonstrated with this procedure, such as inflammation secondary to sepsis, fungi and yeasts, and neoplasia (Fig. 9-57). These should not be confused with glove powder, which may be present in some specimens (see Fig. 9-56).
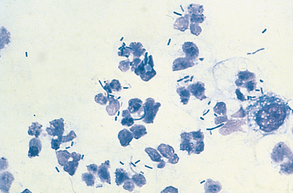
Ear Swabs
“Normal” specimens contain cornified squamous epithelial cells, with negligible evidence of inflammation and few microorganisms. Common abnormal findings are bacteria and yeasts, with or without inflammation. The organism Malassezia, a potential cause of chronic skin lesions and ear infections, will stain with Gram stain, Diff-Quik, or even NMB for a quick “wet prep.” Look for the characteristic peanut-shaped organisms (Fig. 9-58). Some controversy exists among specialists regarding whether the presence of Malassezia in small numbers is significant. Some believe that any organisms found are grounds for treatment, whereas others claim that low numbers may be found in normal ears.
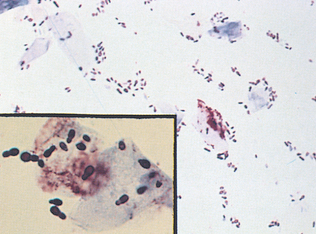
Vaginal Cytology
Exfoliative vaginal cytology is a useful adjunct to the history and clinical examination in determining the stage of the estrous cycle in bitches and queens. It assists with optimal timing of mating or artificial insemination in small animals but is not of practical value for these purposes in mares, cows, does, ewes, or sows. Cytologic findings must be interpreted in conjunction with the history and clinical signs. The findings at each stage of the canine estrous cycle are detailed subsequently. These stages are convenient divisions in a continuum of change, brought about by variations in blood estrogen and progesterone concentrations. Because determination of the stage of the estrous cycle may be difficult on the basis of a single examination, repeat examinations every few days may be necessary. Unlike the bitch, the queen (female cat) ovulates after coital stimulation. Cytologic findings at different stages of the estrous cycle are similar to those of the bitch for epithelial cells and neutrophils; however, erythrocytes are usually not present at any stage.
Cell Types Seen on Vaginal Cytology Sample Preparations
Some variation exists in the terminology used to describe the cell types commonly seen in vaginal cytology preparations. In addition to neutrophils and erythrocytes, a variety of squamous epithelial cells are also seen in vaginal cytology preparations (Fig. 9-59). These cells are further categorized on the basis of their size and degree of cornification. The epithelial cells present may include the small basal cells; the slightly larger parabasal epithelial cells (Fig. 9-60); and the largest sized noncornified squamous epithelial cells, sometimes referred to as intermediate (Fig. 9-61). At some stages of the estrous cycle, these intermediate cells may contain pyknotic nuclei (Fig. 9-62). Cornified epithelial cells are angular in appearance and usually have no nuclei (Fig. 9-63) or contain a pyknotic nuclei. Bacteria may be present in vaginal smears at any stage of the estrous cycle (but especially during estrus) and usually have no pathologic significance (i.e., they are part of the normal vaginal microflora).
Figure 9-59 Diagrams of cells from the canine vagina. A, Parabasal epithelial cell. B, Small intermediate cell. C, Large intermediate cell. D, Superficial cell with pyknotic nucleus. E, Anuclear superficial cell. (Courtesy of Kal Kan Forum.)
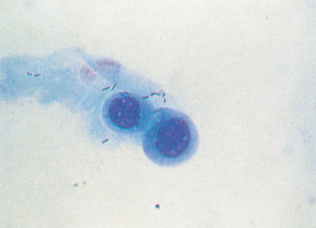
Figure 9-60 Parabasal vaginal epithelial cells from a dog. (From Cowell RL, Tyler RD, Meinkoth JH: Diagnostic cytology and hematology of the dog and cat, ed 2, St Louis, 1999, Mosby.)
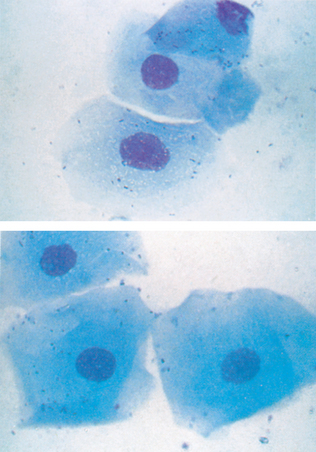
Figure 9-61 Small and large intermediate vaginal epithelial cells from a dog. (From Cowell RL, Tyler RD, Meinkoth JH: Diagnostic cytology and hematology of the dog and cat, ed 2, St Louis, 1999, Mosby.)
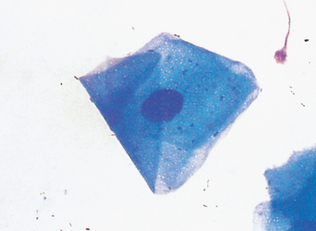
Figure 9-62 Superficial epithelial cell with a slightly pyknotic nucleus and folded angular cytoplasm. (From Cowell RL, Tyler RD, Meinkoth JH: Diagnostic cytology and hematology of the dog and cat, ed 2, St Louis, 1999, Mosby.)
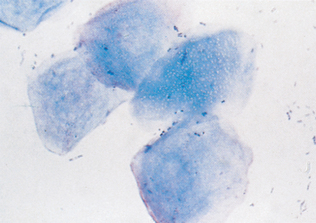
Figure 9-63 Anuclear superficial (cornified) vaginal epithelial cells from a dog. (From Cowell RL, Tyler RD, Meinkoth JH: Diagnostic cytology and hematology of the dog and cat, ed 2, St Louis, 1999, Mosby.)
Anestrus: The anestrual bitch has no vulvar swelling and does not attract male dogs. A vaginal smear reveals predominantly noncornified squamous epithelial cells (large cells with a rounded border; abundant basophilic cytoplasm; and a large, round nucleus) (Fig. 9-64). On the basis of size, these cells may be categorized as intermediate, parabasal, or basal epithelial cells. The smear also may contain some neutrophils but no erythrocytes. Anestrus is variable in length but generally lasts less than 4.5 months.
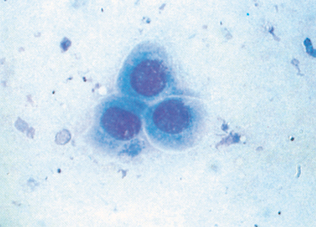
Proestrus: A bitch in proestrus has a swollen vulva, with a reddish vulvar discharge. The bitch attracts but does not accept male dogs attempting to breed. Proestrus may last 4 to 13 days, with an average of 9 days. Proestrus is often further subdivided into early proestrus and late proestrus. Gradual changes in physiology and cellular morphology are seen as the stages progress. In early proestrus, high numbers of erythrocytes are present along with basal and parabasal epithelial cells (Fig. 9-65). As proestrus continues, the numbers of erythrocytes gradually decrease and the epithelial cells begin to show signs of cornification, such as pyknotic nuclei. In late proestrus, nearly all epithelial cells present are intermediate cells with pyknotic nuclei. Small numbers of neutrophils are sometimes seen in proestrus samples, especially in the earlier stage.
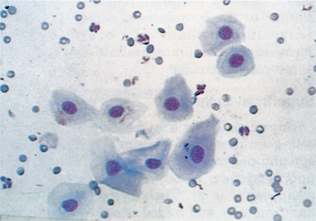
Estrus: The estrual bitch has a history of recent proestrus and a swollen vulva, with possibly a pinkish to straw-colored discharge, becoming whiter as diestrus approaches. Bitches in estrus accept male dogs attempting to mate. A vaginal smear reveals that all squamous epithelial cells are cornified and usually anuclear (Fig. 9-66), neutrophils are absent, and small numbers of erythrocytes may be present. Erythrocyte numbers decrease further and neutrophil numbers increase rapidly at the end of estrus. Estrus generally lasts 4 to 13 days, with an average of 9 days.
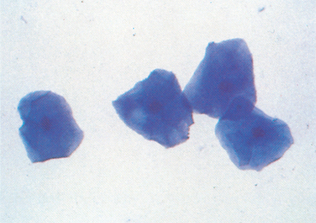
Diestrus: A bitch in diestrus has a history of recent estrus. The vulvar swelling and discharge have decreased, and she no longer attracts or is receptive to male dogs. Cornified squamous epithelial cells are replaced by noncornified squamous epithelial cells and abundant cytologic debris. By approximately the tenth day after estrus, all epithelial cells are noncornified. Neutrophils increase in number until approximately the third day of diestrus and then decrease to few by about the tenth day. Erythrocytes are generally absent throughout diestrus (Fig. 9-67). Diestrus may last 2 to 3 months. Cytologically, diestrus and anestrus are often difficult to differentiate. Pregnancy is not cytologically distinguishable from diestrus or anestrus. Some references may refer to diestrus as metestrus.
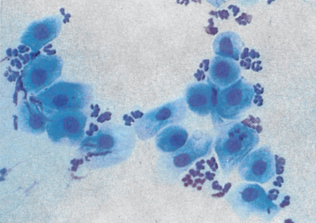
Vaginitis and Metritis: Inflammation of the vagina or uterus results in a pinkish-white vulvar discharge, usually without vulvar swelling or clinical signs of proestrus or estrus. A vaginal swab reveals noncornified squamous epithelial cells and massive numbers of neutrophils, possibly with free and/or phagocytosed bacteria (Fig. 9-68).
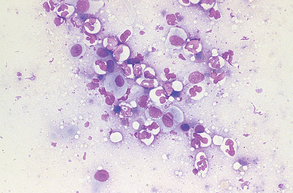
SEMEN EVALUATION
The American Society of Theriogenologists has published some guidelines for examination of male animals for breeding soundness. Evaluation of semen is an important part of such an assessment. Techniques for semen collection in common domestic animals are described in standard theriogenology texts.
Avoid exposing semen samples to marked changes in temperature (especially cold), water, disinfectants, or variations in pH. All laboratory equipment used in semen collection and examination should be clean and dry and warmed to approximately 37° C (98.6° F). This equipment includes microscope slides, cover slips, and pipettes. Stains and diluents also should be warmed to approximately 37° C. Samples should be processed in a warm room as soon as possible after collection.
The following characteristics are readily determined in the laboratory: volume of ejaculate, gross appearance, wave motion, microscopic motility, spermatozoal concentration, ratio of live/dead spermatozoa, assessment of morphologic features, and presence of foreign cells or material. Recording the animal’s species, breed, age, brief history with salient clinical findings, suspected abnormalities, and the method of semen collection (e.g., artificial vagina, electroejaculation, massage) is important.
Volume of Ejaculate
The volume of ejaculate is measured with a volumetric flask, which may be incorporated into the collection receptacle. Marked species variations occur, and the method of collection greatly influences the volume obtained, its gross appearance, and spermatozoal concentration. As a generalization, ejaculate volume is larger but spermatozoal concentration lower (and the specimen apparently more dilute) when collected by electroejaculation than when collected by artificial vagina. Further, repeated ejaculation, whether associated with semen collection or sexual activity, decreases the volume and concentration of semen obtained at subsequent collections. Semen volume tends to be greater if collection is preceded by a period of sexual arousal (“teasing”).
The ejaculate is composed of three portions: a sperm-free watery secretion, a sperm-rich fraction, and a sperm-poor fraction. The first and third fractions are derived from accessory sex glands. In bucks, bulls, rams, and toms, all three fractions are collected together. However, with boars, dogs, and stallions, the third fraction conveniently may be collected separately, which is advisable because the third fraction is voluminous in these three animals and is therefore an unnecessary encumbrance in subsequent evaluation of the semen sample. In these three species, the first two fractions (collected together) are used in the other procedures that follow.
Approximate average total ejaculate volumes (all three fractions) are as follows: boar, 250 ml; buck and ram, 1 ml; bull, 5 ml; dog, 10 ml; stallion, 65 ml; and tom, 0.04 ml. Ejaculate volume does not necessarily correlate with fertility. In general, spermatozoal number, motility, and morphologic characteristics are better guides to fertility. However, small ejaculates may be of concern in species that should have voluminous ejaculates. Knowledge of the ejaculate volume is necessary to determine total spermatozoal numbers if the sample is to be divided (and possibly diluted) for artificial insemination procedures.
Gross Appearance of Ejaculate
The opacity and color of the sample should be recorded. Opacity subjectively reflects the concentration of spermatozoa. Categories used include thick, creamy, opaque; milky opaque; opalescent milky; and watery white. This rule of thumb works best for semen from bucks, bulls, and rams, which normally have opaque, creamy-white semen because of high spermatozoal concentration. As the density of spermatozoa decreases, the specimen becomes more translucent and milkier in appearance. Semen from boars, dogs, and stallions is normally fairly translucent and white to gray. Contaminants, especially intact or degenerate erythrocytes, cause discoloration of semen.
Sperm Motility
Sperm motility (movement) is subjectively assessed and depends on careful handling of the sample for meaningful results. Variations in temperature and exposure to nonisotonic fluids or destructive chemicals (including detergents) must be avoided. Motility is correlated with fertility; however, improper specimen handling adversely affects its assessment. If other tests, especially sperm morphology, suggest the semen is normal but sperm motility is poor, another sample should be examined to ensure that technical errors were not responsible for the poor motility. Motility may be conveniently assessed in two ways.
Wave Motion: Wave motion is a subjective assessment of the gross motility of sperm. Four general classifications are used—very good, good, fair, and poor—on the basis of the amount of swirling activity observed in a drop of semen on a microscope slide at low power (40×) magnification. These categories respectively correspond to distinct vigorous swirling, moderate slow swirling, barely discernible swirling, and lack of actual swirling but with motile sperm present, which may cause the sample to have an irregular oscillating appearance. Wave motion depends on high sperm density and is therefore best in samples from bucks, bulls, and rams, which normally have high sperm concentrations. Wave motion decreases as sperm concentration decreases. Consequently, normal boars, dogs, stallions, and toms may have fair or poor wave motion. As a guide, if wave motion is very good or good, the sample should be diluted for evaluation of the percentage of motile sperm and their rate of motility.
Motility: Progressive motility of individual spermatozoa is determined on a relatively dilute drop of semen under a cover slip examined at 100× magnification. Because the motility of individual spermatozoa is difficult to appreciate in dense samples, such concentrated samples should be diluted before examination. Warm physiologic saline or fresh, buffered 2.9% sodium citrate solutions are suitable diluents.
A drop of semen is placed on a slide and diluted until a satisfactory concentration of spermatozoa is observed. A cover slip is placed on top to produce a monolayer of cells. Excessive dilution of the sample makes evaluation of motility difficult. The rate of motility is generally subjectively classified as very good, good, fair, or poor, corresponding to rapid linear activity, moderate linear activity, slow linear or erratic activity, and very slow erratic activity, respectively. The percentage of motile spermatozoa is broadly categorized as very good, good, fair, or poor, corresponding to approximately 80% to 100%, 60% to 80%, 40% to 60%, and 20% to 40% motile cells, respectively. As a generalization, to be considered satisfactory a sample should have at least 60% moderately active spermatozoa.
Sperm Concentration
Several solutions are satisfactory for semen dilution before counting sperm numbers, including 5 g of sodium bicarbonate or 9 g of sodium chloride with 1 ml of formalin in 1 L of distilled water; 3% chlorazene; or 12.5 g of sodium sulfate with 33.3 ml of glacial acetic acid in 200 ml of distilled water (Gower’s solution). The sample is diluted to a 1:200 dilution and thoroughly mixed. A Neubauer hemacytometer is charged with fluid. The sample is allowed to settle for a few minutes and is then checked for homogeneous distribution of spermatozoa. If this is not present, the sample should be thoroughly mixed again and the cleaned hemacytometer recharged.
The number of spermatozoa in the central grid area of one side of the chamber is counted at 400× magnification. The number of spermatozoa per milliliter of semen is calculated by multiplying the number observed by 2 million. If spermatozoal concentration is high (e.g., in bucks, bulls, and toms), fewer squares may be counted and the multiplication factor adjusted accordingly. Spermatozoal concentration also may be determined by colorimetric and electronic particle counter techniques.
As previously outlined, sperm concentration varies with the method of collection. As a generalization, average sperm concentrations (in millions per milliliter) are approximately 150 for boars and stallions, 3000 for bucks and rams, 1200 for bulls, 300 for dogs, and 1700 for toms.
Live/Dead Sperm Ratio
Staining with a vital dye permits discrimination between live and dead spermatozoa. An eosin/nigrosin mixture is popular for this purpose and also permits examination of sperm morphologic features. The stain is prepared by adding 1 g of eosin B and 5 g of nigrosin to a 3% solution of sodium citrate dihydrate. This solution is stable for at least 1 year.
A small drop of warm stain is gently mixed with a small drop of semen on a warm microscope slide. After several seconds of contact between specimen and dye, the mixture is smeared, as when making a blood smear, and then rapidly dried. Once the smear is dried, microscopic examination may be delayed. Live sperm resist staining and appear white (clear) against the blue-black nigrosin background. In contrast, dead sperm passively take up the eosin and are stained a pinkish red. The ratio of live/dead sperm, expressed as a percentage, is determined by examination at 400× or 1000× magnification, preferably by observation of 200 cells.
Unfortunately, this procedure is susceptible to technical problems. Conditions that kill sperm, especially temperature changes, produce misleading results. Findings should always be interpreted with regard to other results, such as sperm concentration, motility, and morphology.
Sperm Morphology
Sperm morphology is readily assessed on a nigrosin and eosin stained smear, prepared as outlined previously. Other stains, such as India ink, hematoxylin and eosin, Wright’s stain, Giemsa stain, and Cavarett’s stain (a mixture of eosin B and phenol), have been used but offer no distinct advantages over nigrosin and eosin.
Species differences exist regarding the fine points of sperm morphology, but all sperm have the same basic structure (Fig. 9-69). The percentage of abnormal spermatozoa and their types are recorded after observing 100 to 500 cells. Counting the lower number of cells (100) is usually adequate once the technician has become proficient. Abnormalities are conveniently divided into head, midpiece, and tail problems. Abnormalities often are categorized as primary or secondary.
Primary defects occur during spermatozoal production and include heads that are double, too large, too small, or oddly shaped (e.g., pearlike, round, twisted, knobby) (Fig. 9-70); midpieces that are swollen, kinked, twisted, double, or eccentrically attached to the head (abaxial); and tails that are coiled (Fig. 9-71). Primary abnormalities generally are considered more serious than secondary ones. Their percentage is fairly consistent if another semen sample is collected within several days. Slightly abaxial midpiece attachment in boars is probably not as significant as in other species because it may be found in numerous spermatozoa of apparently normal boars.
Figure 9-70 Diagrammatic representation of primary spermatozoal abnormalities involving the head. Abnormalities depicted are double head (bicephaly) (A), small head (microcephaly) (B), large head (macrocephaly) (C), pear-shaped head (pyriform) (D), elongated head (E), and round head (F).
Figure 9-71 Diagrammatic representation of primary spermatozoal abnormalities involving the midpiece and tail. Abnormalities depicted are swollen midpiece (A), coiled midpiece and coiled tail (B), bent midpiece (C), double midpiece (D), and abaxial midpiece (E).
Secondary defects may occur at any time from storage in the epididymis until the smear is made. Therefore, because secondary abnormalities may be artifactual, careful specimen handling is mandatory. Minimization of technique-induced secondary abnormalities permits easier sample interpretation. Secondary defects include tailless heads, protoplasmic droplets on the midpiece, and bent or broken tails (Fig. 9-72). Obviously, for every tailless head is a headless tail, so the latter need not be counted. Protoplasmic droplets are distinct from swollen midpieces. Protoplasmic droplets are normally present while spermatozoa are in the epididymis. The droplets migrate caudally along the midpiece while the sperm cells mature in the epididymis. The droplets usually are shed before the spermatozoa leave the epididymis.
Figure 9-72 Diagrammatic representation of secondary spermatozoal abnormalities involving the midpiece and tail. Abnormalities depicted are tailless head (A), Headless tail (B), distal protoplasmic droplets (C), proximal protoplasmic droplets (D), and bent tail (E).
As a broad generalization, less than 20%—and usually less than 10%—of spermatozoa are abnormal in a normal animal. Higher percentages of abnormal spermatozoa may compromise fertility. Obviously, however, the total number of normal spermatozoa is important, not the percentage of abnormal sperm.
Other Cells in Semen
Normal semen contains few (if any) leukocytes, erythrocytes, or epithelial cells and no bacteria or fungi. If present, their approximate quantity should be noted. If bacteria or fungi are observed without an inflammatory response, sample contamination by the normal preputial microflora should be suspected. Attention to preputial sanitation before sample collection should remedy the problem. If indicated, a semen sample may be submitted for microbiologic examination.
Cells from the germinal layers of the testes are an unusual finding in semen and represent severe testicular damage. Such cells include spermatids, spermatocytes, and large ciliated cells (often called medusa heads). Precise categorization is unimportant as long as these cells are classified as immature sperm cells.
EVALUATION OF PROSTATIC SECRETIONS
Disorders of the prostate are not uncommon in dogs but are rare in other domestic animals. Cells of prostatic origin may be collected by urethral catheterization combined with prostatic massage (or penile massage) performed per rectum to stimulate prostatic secretion. Prostatic tissue may be aspirated by transcutaneous needle biopsy. Cytologic smears are prepared as previously described.
An enlarged prostate may be the result of prostatic hypertrophy, hyperplasia, metaplasia, neoplasia, or inflammation. Prostatic cells may occur singly or in clusters. Spermatozoa may be present in some fluid samples, especially those collected by penile massage.
Normal prostatic cells have a uniform size and shape, with a fairly high nucleus/cytoplasm ratio, transparent to gray cytoplasm, homogeneous nuclear chromatin, and no obvious nucleoli. Normal prostatic fluid or tissue contains few leukocytes. Prostatic hypertrophy is gland enlargement because of increased size of individual cells, without increased cell numbers. A hypertrophic prostate is not cytologically distinguishable from a normal prostate. The distinction is made on the basis of gland size at palpation or radiography. Prostatic hyperplasia is gland enlargement because of increased numbers of cells. The cells are uniform in size and appearance and have a high nucleus/cytoplasm ratio; basophilic cytoplasm that is often vacuolated; and a nucleus with a “roughened” chromatin pattern and a uniform, small, single nucleolus. Few leukocytes are present. Metaplasia is a change (from normal) in the population of prostatic cells. Exfoliated or biopsied cells have the appearance of noncornified squamous epithelial cells. Consequently, they have a low nucleus/cytoplasm ratio and a somewhat pyknotic nucleus. Prostatic neoplasia is characterized by a pleomorphic population of cells with a high nucleus/cytoplasm ratio and very basophilic cytoplasm. Nuclear size varies among cells and contain variable numbers of large, irregular (angular), and pleomorphic nucleoli. A cytologic diagnosis of prostatic abcessation is based on finding large numbers of neutrophils in the fluid or tissue sample. Macrophages and lymphocytes also may be present in variable numbers.
EXAMINATION OF MILK
Subclinical and clinical bovine mastitis (mammary gland infection) is an important economic concern for dairy farmers. Mastitis may be detected by several laboratory procedures. Those most frequently used indirectly or directly reflect milk nucleated cell counts and/or bacterial counts. Tests are performed on milk samples from individual quarters, all four quarters pooled together, or bulk milk (several cows together, usually the whole herd).
When individual animals are being screened, foremilk (sample obtained before milking begins) is generally used. It has more cells than a sample obtained in the middle of milking but fewer cells than one collected at the end of milking. Cell counts also vary with the stage of lactation. Milk samples from normal cows within the first week and at the end of lactation have higher cell counts than throughout the interim.
Nucleated cell counts of normal milk are generally less than 300,000 to 500,000 cells/ml. Counts greater than 500,000 cells/ml indicate mastitis.
Differential cell counts sometimes are performed. Nucleated cells are categorized as neutrophils or mononuclear cells. Normal milk in midlactation generally has less than 10% neutrophils, whereas in severe acute mastitis the milk may have up to 95% neutrophils.
Recommended Reading
Anthony, A, Sirois, M. Microbiology, cytology and urinalysis. In Sirois M, ed.: Principles and practice of veterinary technology, ed 2, St Louis: Mosby, 2004.
Cowell, R, Tyler, R, Meinkoth, J. Diagnostic cytology and hematology of the dog and cat, ed 2. St Louis: Mosby, 1999.
Latimer, KS, Prasse, KW, Mahaffey, EA. Duncan and Prasse’s veterinary laboratory medicine: clinical pathology. Ames, IA: Blackwell, 2003.
Raskin, R, Meyer, D. Atlas of canine and feline cytology. St Louis: WB Saunders, 2001.