Chapter 16 Anticholinergic agents
Introduction
In this chapter, we will discuss drugs acting on the cholinergic system. The cholinergic receptors are part of the autonomic nervous system. They are well distributed in all tissues of the body, and play major roles in the homeostasis of physiological functions. The intervention at cholinergic activity level can help in many disease states. Some of them have been extensively studied (e.g. glaucoma, myasthenia gravis and peptic ulcer) and some of them have only recently been discovered to have a cholinergic dimension (e.g. Parkinson’s disease, Alzheimer’s syndrome, psychosis, obesity and cancer).1,2,3,4 Moreover, the control of cholinergic activity is highly desirable during surgical operation, and this has had a great impact on the facilitation of surgical procedures.
Naturally occurring anticholinergic compounds have long been used in history for very different reasons. Atropa belladonna (the beautiful lady), the natural source of atropine, was used by Italian women to dilate the eye pupils in order to give a charming look. Calabar beans, the natural source of physostigmine, were used to test alleged criminals for innocence, death indicating a ‘guilty suspect’. The dry extract of curare was rubbed against the arrows used by American Indians during their fights and for hunting. Moreover, the psychological/neurological effects of these drugs have made them a good material for detective novel writers.
The botanical origin of the first receptor ligands is a common feature among all drug categories related to the cholinergic activity. Muscarine from the amanite mushroom and nicotine from tobacco leaves were the two alkaloids which exhibited agonistic activity on the muscarinic and nicotinic cholinergic receptors, respectively. Atropine from A. belladonna was the first antimuscarinic agent to be discovered. Tubocurarine (or calabash curare) was the first compound to be discovered to have nicotinic receptor antagonist activity and physostigmine was the first known anticholinesterase substance.
Drugs acting on the cholinergic systems may be divided into two main categories, each being subdivided into subgroups which will have different therapeutic indications:
The potency of these drugs makes them dangerous poisons, and it is very common throughout the chapter to mention the use of one agonist in treating the toxicity of an antagonist, and the same antagonist for treating the toxicity of that very same agonist.
Physiology of the autonomic system
The cholinergic receptors are well spread throughout the whole human body with varying abundance of one subtype of the receptors over the others in a specific tissue.
The early observation of the different responses elicited by application of muscarine and nicotine suggested two different receptors. Both receptors share the neurotransmitter acetylcholine (ACh), thus both were named cholinergic receptors, but a further nomenclature was followed to distinguish between those preferably affected by muscarine (muscarinic receptors) and those affected by nicotine (nicotinic receptors).
Nicotinic receptors (AChNR) are found in synapses between sympathetic and parasympathetic nerves, synapses between a nerve and a skeletal muscle, at the end plates of the somatic motor nerves and the adrenal medulla. Muscarinic receptors (AChMR) are distributed in the synapses between a parasympathetic nerve and a smooth muscle and cardiac muscles (Fig. 16.1).
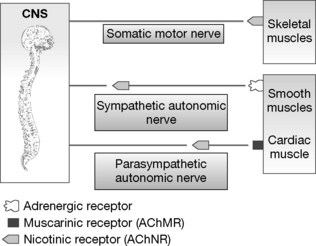
The two sets of receptors vary a great deal, but all share the neurotransmitter acetylcholine and the general structure required to accommodate it, and these differences are not detected by the natural activator ACh. However, they are sufficient for the selective acceptance/rejection of the binding of other molecules. Both types of cholinergic receptor have been isolated.
The muscarinic receptor is a seven-helix membrane-spanning G-protein-type receptor and the associated second messenger is either activation of inositol-3-phosphate (IP3) or inhibition of cyclic adenosine monophosphate (cAMP). There are five subtypes of the muscarinic receptors (M1–M5). All share the same general structure and the active site, but they differ in their tissue distribution, their second messenger mechanism and the associated physiological response.5,6
The nicotinic receptor is a channel-type receptor composed of four non-identical subunits which open an ion gate upon activation, allowing the build-up of a membrane potential which in turn transmits the signal further down the nerve or muscle. The structure of the receptors will be discussed in more detail later. The effects resulting from the activation of the muscarinic and nicotinic receptors in different tissues are summarised in Table 16.1.
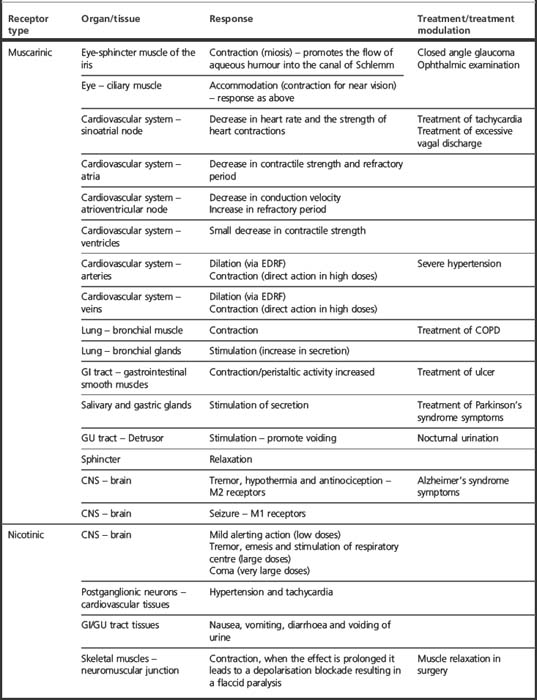
We should bear in mind that some of the above actions, especially those in the cardiovascular system, are controlled by a complex system. For example, the direct slowing effect on sinoatrial rate will result in hypotension which in turn leads to a sympathetic reflex discharge. Additionally, the muscarinic agonist can act indirectly via EDRF (endothelium-derived release factor which is nitrous oxide [NO]) or directly in high doses which will elicit opposing effects, i.e. dilation, contraction, respectively. That is why the net effect of acetylcholine agonist is dependent on its relative availability in the blood and vessels and the sympathetic reflex responsiveness.
The receptor activation process
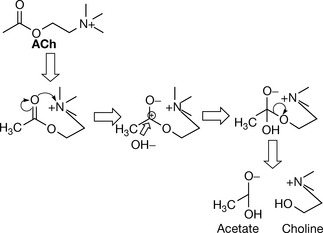
The activation of the cholinergic receptor starts with the synthesis of ACh from choline by the enzymatic action of choline acetyl transferase. ACh is then stored in vesicles pending the arrival of nerve signals interacting with the cell membrane which result in releasing ACh to the synaptic cleft. ACh then binds to its receptor and produces a physiological response and leaves the receptor. The neighbouring acetylcholinesterase captures any dissociated ACh and hydrolyses it to acetate and choline, thus inactivating it. The latter is recycled into the nerve cell by a choline carrier protein (Fig.16.2).
On the basis of the information shown in Figure 16.2, the targets for cholinergic drug action could be:
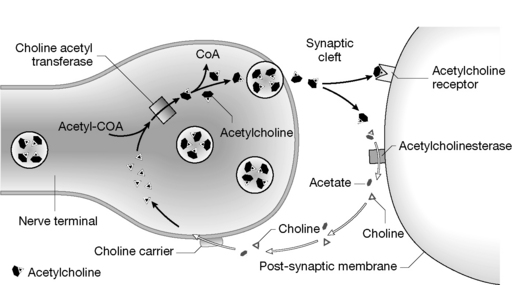
Figure 16.2 The acetylcholine (ACh) receptor activation cycle
(from Rang HP, Dale MM, Ritter JM, Flower RJ 2007 Pharmacology, 6th edn. St Louis, Elsevier, with permission).
Acetylcholine (ACh) was the first neurotransmitter to be characterised. It binds efficiently to both AChNR and AChMR despite the differences in the active sites of the receptors. The dissimilarity of the active site is good news with regards to the different responses elicited by the activation of the two receptors, as it makes possible the rational design of highly selective receptor agonists or antagonists.
ACh itself is a small molecule freely rotating on its backbone (choline). This gives rise to numerous potential freely interconvertible conformations, and ACh can switch on all cholinergic receptors, but it probably does so at each receptor with a different conformation. This is supported by the selectivity of muscarine and low-dose nicotine on one group of receptors rather than the other. ACh adopts a specific conformation at different receptors where the ‘active’ part of the molecule is locked into a limited number of conformations by incorporating a part of it into a cyclic moiety (Fig. 16.3).
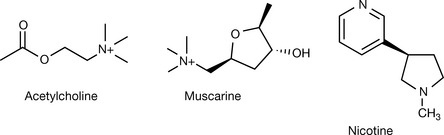
ACh contains an ionic quaternary ammonium entity and dipolar ester with accompanying electrostatic fields.7 Experiments of varying groups indicated maximum activity to be associated with the cationic nitrogen. In explorations of the role of the cationic head, the primary amine analogue, aminoethyl acetate, is inactive. However, tertiary amine counterparts, such as dimethylaminoethyl acetate, are strong enough bases to be almost totally ionised at pH 7.4, thus retaining cationic character along with lipophilic character. The importance of charge is shown by inactivity of the neutral, quaternary substance resulting from replacement of N by C. Other necessary features are the polarised carbonyl oxygen and probably the non-carbonyl oxygen.The ester group has to be in the optimum position, and extending the carbon bridge between the nitrogen and the carbonyl to more than two methylene units diminishes the activity significantly. Alkane chains with no ester group show essentially no activity. Structure–activity relationship (SAR) studies revealed that the presence of hydrogen-bond acceptor groups close to the quaternary nitrogen is detrimental for AChNR binding. (SAR results for ACh are valid for both receptors.) Box 16.1 shows the important factors in SAR at cholinergic receptors.
The active site of the receptor is thought to be an asparagine motif which interacts with the dipole of the carbonyl group in the ester side of the transmitter, while the interaction on the other quaternary ammonium side is thought to be either an ion-pair interaction with an aspartic acid residue or an interaction between the delocalised positive charge on the quaternary nitrogen+ three methyl groups and an electron-rich pocket formed by three tyrosine motifs in the receptor (cation-π-donor interaction).
Cholinergic agonists
The SAR of ACh indicates a very tight fit interaction at the receptor, resulting in a very limited space for the medicinal chemist for a rational drug design. ACh itself is rapidly hydrolysed by acetylcholinesterase (AChE). The effect of a large-bolus i.v. injection wears out in 5–20 seconds (there is large variation between individuals). ACh is very labile to hydrolysis of its ester function via either chemical or enzymatic activity. Chemical hydrolysis makes it orally inactive. This instability arises from the proximity of a quaternary ammonium function to an ester group, as it attracts the electron pairs on the oxygen to move towards its positive charge, thus withdrawing the electrons from the carbon–oxygen bond on the ester function towards the oxygen. This will make the dipole C–O greater, hence more prone to attack by a nucleophile (even weak nucleophile such as water, Fig. 16.4).
Hydrolysis by esterases (pseudocholinesterase, butyrylcholinesterase) in the circulation gives ACh a very short plasma half-life, in addition to its specific hydrolysis by AChE at the active site. Exogenous ACh has been employed therapeutically for restoring the gastrointestinal or urological movement post surgery. The minimum effective dose of ACh is 20–50 μg/min and it has no selectivity at all.
ACh as a therapeutic agent is not convenient, and modifications are needed to ensure:
Modifications of the acetylcholine structure
When designing a drug to be a cholinergic agonist, selectivity is highly desirable but not a simple term, as therapeutic candidates should be selective for the cholinergic receptor among other neurological receptors, selective for nicotinic or muscarinic, or selective for specific subtypes or tissue types. Selectivity could be achieved on the pharmacodynamic level, where the structure of the drug molecule makes it more active at one receptor than another, or on the pharmacokinetic level where the molecular structure of the drug makes it more available to specific tissues. Equally important, the new structure should retain ACh’s activity (see Box 16.1).
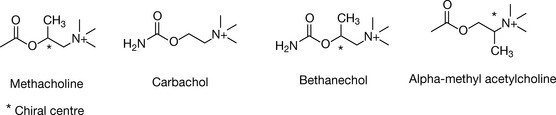
Adding an alkyl group on the beta position relative to the quaternary ammonium group will provide a shield which will inhibit nucleophilic attacks and also provide an electroinductive group which will decrease the dipole activity of the ester function. The tight fit at the receptor mentioned previously dictates this alkyl group should be only a small one; in fact, anything larger than a methyl group yields better stability but a poorer activity. Methacholine (Fig. 16.5) is mainly used via inhalation for the diagnosis of asthma (challenge test). It is a water-soluble drug and it is not active via oral absorption and does not cross the blood–brain barrier (BBB). The addition of an extra methyl group creates a chiral centre.
The extra methyl group led to enhanced selectivity at the muscarinic receptor and diminished activity at the nicotine receptor. Methacholine was designed to ensure stability against chemical hydrolysis, but the additional group appeared to provide hindrance to access of cholinesterase as well, and methacholine is three times more resistant to esterase hydrolysis than ACh.
Another approach to reduce the attacks of nucleophiles is to decrease the dipole activity by countering the positive dipole charge on the carbonyl carbon. This could be achieved by replacing the alkyl function with an amine group to create a carbamate (carbachol, Fig. 16.5). The lone pair of electrons on the nitrogen delocalises, countering the partial positive charge formed on the carbonyl carbon, hence making it less vulnerable to nucleophilic attack. Carbachol (carbamylcholine) is mainly used in eyedrops for the treatment of glaucoma, an ophthalmic disease resulting a high pressure inside the oculus which can be relieved by cholinergic agonist action. Carbachol induces miosis and increases the drainage of aqueous humour. Carbachol has a very slight increased preference for the nicotinic receptor, but is still considered a non-selective cholinergic agent just like ACh. It is introduced in 0.01% w/v sterile eyedrops (carbachol chloride) for the treatment of open-angle glaucoma.8
Bethanechol combines the two modifications described above. It is resistant to both chemical and enzymatic hydrolysis and consequently has a relatively long half-life of 60–90 minutes. Bethanechol is a chiral drug due to the methyl addition at the β-position relative to the quaternary ammonium. The muscarinic receptor is strongly stereoselective and (S)-bethanechol is 1000 times more potent than (R)-bethanechol.9 But these agents are still produced as racemates.
Bethanechol is, like methacholine, highly selective for the muscarinic receptor and shows no appreciable nicotinic action. It is reported to be selective for M3 muscarinic subtype as well. Cholinergic drugs are used to restore bowel movement following surgical manipulations or congenital megacolon, and to counteract urinary retention, for instance resulting from trauma. Bethanechol is the drug of choice for treating these conditions. It is used orally (10–25 mg three to four times per day). It shows systemic activity when administered subcutaneously and it is used in doses of 5 mg. Notice the low oral bioavailability. There is a very strict rule regarding the use of cholinergic drugs in these conditions. The depressed smooth muscles should be obstruction free; otherwise application of the drug will increase the pressure and could lead to perforation.10
Other cholinergic agonists
Demecarium (Fig.16.6) is used in veterinary medicine for the treatment of glaucoma in animals.
Pilocarpine (Fig. 16.6) is another botanical alkaloid structurally related to muscarine acting on the cholinergic system. It was first extracted from the leaves of plants from the genus Pilocarpus. Pilocarpine shows a muscarinic agonist activity, with selectivity to M3 which is found in the eye. The selectivity made it a first-line choice for the treatment of both types of glaucoma (open and closed angle). It is given as eyedrops (concentration of 4% w/v), as long-acting plastic implants in the conjunctival sac, or as eyedrops in combination with physostigmine in the case of treatment of closed-angle glaucoma. Pilocarpine is also used as an antidote for toxicity caused by scopolamine or atropine.
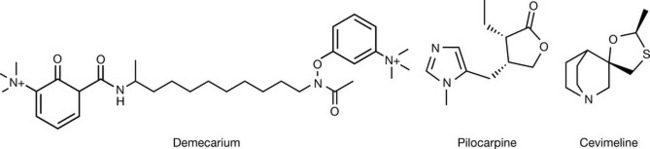
Cevimeline (Fig. 16.6) is a new direct-acting muscarinic agonist highly selective for the M3 receptor. It is used for the treatment of dry mouth associated with Sjögren’s syndrome, an autoimmune disease where atypical antibodies destroy the glands which produce tears and saliva.
Antiacetylcholinesterase drugs
Acetylcholinesterase (AChE) is situated next to any cholinergic receptor. It catches the molecule once it has departed its active site and renders it inactive by breaking the ester bond to give choline, which is taken by the presynaptic terminus, and acetate. Hydrolysis of acetylcholine and restoration of the active site of the enzyme completes in about 150 microseconds. AChE is a treelike enzyme, with the stem implemented in the cell membrane and the branches (three) protruding out of the cell membrane. Each branch holds four active sites composing an army of twelve active sites ready to capture any unbound ACh. This makes AChE activity one of the most efficient hydrolysis processes in biology. The active site was thought to include an aspartic acid residue which was supposed to hold ACh by ionic bonding, and a tyrosine residue which interacts with an oxygen ester group via a H-bond. However, recent studies have shown the active site to be buried deep in the enzyme, with no negatively charged residues. Instead, the positive charge of the quaternary ammonium interacts with a series of aromatic residues, possibly via a charge transfer interaction (see Ch. 1). AChE holds the ACh molecule in place by a peripheral site composed of a tryptophan at position 279 and tyrosine at position 121. This peripheral site fixes the ACh in the right orientation in preparation for the action of the catalytic site which consists of the catalytic triad (serine–histidine–aspartate) and two additional aromatic amino acids tryptophan 84 and phenylalanine 330 which may play a further role in orientation of the ACh. The whole arrangement holds the ester region in an orientation where it is in a proximity to a serine residue situated next to a histidine residue. This arrangement is essential for the hydrolysis efficiency, where histidine plays a vital rule in fortifying the nucleophilic activity of serine which, in the absence of the acid/base catalysis of histidine, is just an aliphatic alcohol and thus too weak a nucleophile to hydrolyse an ester by itself. Drugs which inhibit the hydrolysis action of cholinesterase will preserve endogenous ACh in the synaptic cleft, thus eliciting a drug action exactly like the injection of ACh, with a duration of action which can be modified (see below). The mechanism of the hydrolysis by AChE is shown in Figure 16.7.
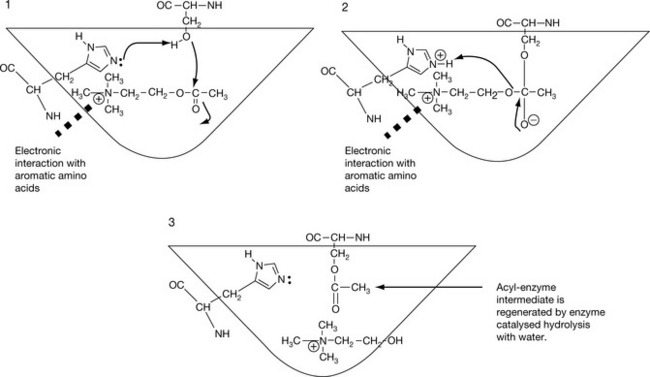
The work on anticholinesterase drugs is a good example for the impact of chemical knowledge on the outcome in medicinal chemistry. In this case, a best fit in the enzyme active site may not be the most active compound, as chemistry of hydrolysis rather than enzyme binding seems to be the determinant factor of activity. Cholinesterase inhibitors have less effect on smooth muscles, thus on blood pressure, than direct acting cholinergic agonists.
The antiacetylcholinesterase drugs
According to their mechanism of action, the anti-AChE agents can be divided into three groups.
Competitive antagonists
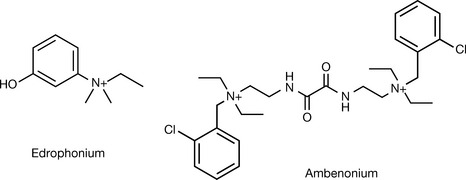
Quaternary alcohols such as edrophonium (Fig. 16.8) act as competitive antagonists with ACh to AChE. Edrophonium works without the involvement of any covalent bonds, which form in the case of other antagonists. It is used for diagnosis and dose titration purposes before and within the therapeutic regimen used for the treatment of myasthenia gravis, an autoimmune disease where the body destroys the nicotinic receptors (AChNR) (see Box 16.3). It is of vital importance to choose the right dose used for anticholinesterase treatment as low doses will keep the patient in agony, and higher doses will elicit muscarinic hyperactivity (abdominal cramps, bronchial constriction and increased secretions, among other symptoms) and an excessive dose may worsen the situation by evoking a nicotinic depolarising blockade (a clinical situation termed as cholinergic crisis). The edrophonium (Tensilon) test uses an injection of edrophonium chloride (10 mg). The activity starts 30–60 seconds after the injection and lasts for 10 minutes. Edrophonium was used to treat supraventricular arrhythmias, but has been superseded by newer agents of different pharmacological classes.
Myasthenia gravis is an autoimmune disease where antibodie to the nicotinic receptors are produced which decrease the number of functional nicotinic receptors. The syndrome is associated with general weakness in all muscles, especially the small ones such as those in the head, neck and extremities. Providing strong agonists of the nicotinic receptor or keeping a high concentration of the acetylcholine at the postsynaptic junction will improve the patient’s condition and help improve daily activities. The drug class of choice for treating the symptoms of myasthenia gravis is the short-acting anticholinesterase agents. The dose requirement in this disease changes too rapidly to allow for the use of longer-acting anticholinesterases. Any observed muscarinic activity may be treated with antimuscarinic drugs such as atropine.
Ambenonium is another competitive inhibitor of AChE. It is composed of two moieties structurally related to edrophonium connected by an oxaldiamide bridge. This is thought to provide a competitive shield of the two active sites in the AChE. It is used as its bromide salt for treating myasthenia gravis in dose intervals of 3–6 hours.
Short-acting carbamate anticholinesterase agents
The remaining groups of anti-AChE drugs all cause formation of a covalent bond with the serine residue in AChE, which is more resistant to nucleophilic attack by water than the normal acetyl group and thus slows down recycling of the enzyme. A good anticholinesterase drug should:
Physostigmine (Fig. 16.9) was discovered in 1864 and its structure was established in 1925. It is extracted from the plant Physostigma venenosum (calabar bean) and it was the first compound found to show anti-AChE activity. Physostigmine has serious side effects which limit its use, but it provided a prototype which medicinal chemists could use as a lead compound and improve it. The important structural elements within the molecule are shown in Figure 16.9.
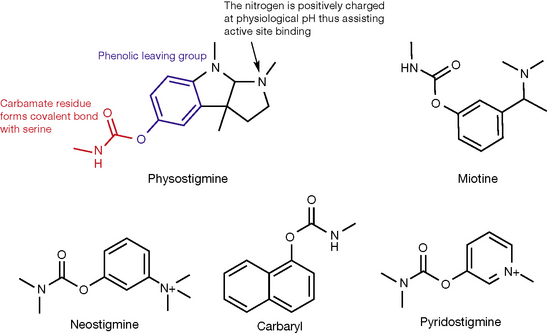
The three structural elements mentioned above with regard of the structure of physostigmine are as follows:
Physostigmine is used as a drug either locally in the treatment of glaucoma, or in the treatment of atropine poisoning. It exhibits serious side effects including psychoneurological effects, being originally used to extract the truth from criminals who were made to swallow an extract of calabar beans (ordeal beans). The drug is susceptible to chemical hydrolysis. It is available as physostigmine salicylate injection (1 mg). It is not permanently charged, and can cross the BBB (Box 16.2), causing central side effects. Antimuscarinic and tricyclic antidepressant toxicity cause severe muscarinic blockade. Physostigmine has been employed in treating toxicity due to muscarinic receptor blockers and tricyclic antidepressants via promoting an increase in ACh levels. The ability of this agent to cross the BBB makes it the chosen drug for this condition. However, it produces its own toxic effects, which restrict its use to severe cases uncontrollable with symptomatic therapy.
Box 16.2 Blood–brain barrier (BBB)
Only un-ionised drugs cross the BBB. One could ask, if the tertiary amines are all protonated, when they access their receptor, i.e. at physiological pH, how come they still can cross the BBB? This is because most, but not all, the molecules are protonated. For instance, at pH = 7.4, physostigmine (pKa = 8.2) will be only 86% protonated and 14% unprotonated. This ratio is always constant. The unprotonated lipophilic form will cross the BBB and disrupt the balanced status, leading to production of new unprotonated molecules which will, in turn, cross BBB and so on.
The task for medicinal chemists was to produce analogues of physostigmine with similar activity and fewer side effects (especially on CNS) and with better chemical stability. Miotine (Fig. 16.9) followed physostigmine as a first synthetic AChE inhibitor. It is still prone to chemical hydrolysis and its non-permanently charged tertiary amine group grants it access to the CNS, leading to undesired side effects there.
Neostigmine (Fig. 16.9) is a further modification on miotine where the methylcarbamate group is replaced with dimethylcarbamate, making the compound more resistant to chemical hydrolysis due to the added inductive effect of the second methyl group. This permanently charged quaternary nitrogen in neostigmine prevents it from crossing the BBB, making it a safe drug from a CNS perspective, and encourages binding to the enzyme active site. The quaternary ammonium is directly attached to the benzene ring, which forces nitrogen into the same plane as the ring, making a stable/fixed conformation for the whole molecule. This is thought to contribute to the enhanced activity of neostigmine. It was historically used as a pregnancy test as it induces menstrual bleeding in the absence of pregnancy, but was superseded by hormonal tests. Neostigmine has additional direct effect at nicotinic receptors, which makes it a better treatment for myasthenia gravis syndrome. The dose is titrated according to the edrophonium test, but its short duration of action requires an inconvenient frequency of dosing (every 2–3 hours). Sustained-release products are available for night usage. Neostigmine is used for treating postoperative depression of smooth muscles (paralysis of stomach or bowel, or urinary retention). It is used orally (15 mg) or subcutaneously (0.5–1 mg). Note the high oral dose which indicates its very low oral bioavailability. It is used as well in congenital megacolon and other clinical situations involving depression of smooth muscles. Neostigmine is used to promptly reverse anaesthesia-produced paralysis. The prompt effect necessitates the use of intravenous or intramuscular dosage forms. A final indication for neostigmine is its use as an antidote for atropine poisoning.
Carbaryl is another carbamate anti-AChE agent which has a high fat solubility. It was designed for rapid distribution into the CNS of insects and it was used until 2004 as a lotion for the treatment of head lice.
Pyridostigmine (Fig. 16.9) is close in structure to both neostigmine and miotine. The aromatic ring (leaving group) is preserved but attached to a methylpyridine ring rather than a benzene ring. A permanent charge is provided by the aromatic ring as well. It is used for myasthenia gravis and it leads to better patient compliance than neostigmine as the dose frequency is less (every 3–6 hours). The dose is determined by the edrophonium test.
Pyridostigmine is used to reverse anaesthesia-produced paralysis in the same fashion as neostigmine.
Long-acting organophosphate agents
Irreversible inhibitors of AChE act in a similar way as the carbamates, but the phosphate bond is stable for hundreds of hours. It is further strengthened by the ageing phenomenon, where the phosphorylated serine on the active site loses one of its oxygens, leading to a phosphine–serine bond, which is even more resistant to hydrolysis.
These compounds were first intended to be (but fortunately never used) as chemical warfare agents. The prototype compounds were dyflos and sarin (nerve gases). These compounds irreversibly phosphorylate the serine hydroxyl motif in AChE. The reaction product is very stable and can not be hydrolysed by weak nucleophiles, even with histidine catalysis. This increases the availability of endogenous ACh in the synaptic gap, leading to a constant signalling at the smooth and skeletal muscles, paralysis and death.
Dyflos and sarin (Fig. 16.10) are extremely active and have serious side effects, thus limiting their use to local ophthalmic administration for the treatment of glaucoma. The binding of these agents is irreversible and a new enzyme is needed in order for the cells to restore ACh hydrolysing capacity.
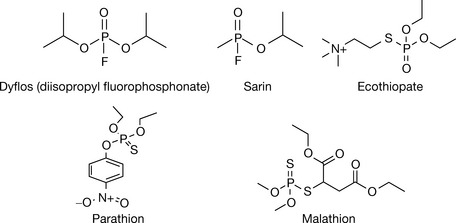
The number of organophosphates synthesised as insecticides is estimated to be 50 000. Organophosphates are generally very lipid soluble (ecothiopate is an exception, and is soluble and stable in aqueous solutions), and many of them are chemically unstable. Among the organophosphates, ecothiopate, malathion and parathion have satisfactory stability. This led to the widespread use of these agents since environmental stability determines the frequency of application. Ecothipate was developed to selectively bind to AChE. This was achieved by introducing a quaternary ammonium group at a suitable distance (the two carbon bridge rule) from the phosphorylating head group. Ecothiopate is more potent than dyflos, and its stability in aqueous solutions (up to several weeks) enables its use as eyedrops for the treatment of glaucoma. It dissociates from AChE over a few days. It is manufactured as ophthalmic drops of ecothiopate iodide (1.5–12.5 mg/5 mL) for the treatment of glaucoma and accommodative esotropia, an ocular disease in young children. Despite its toxicity, ecothiopate is considered the second line for the treatment of open-angle glaucoma uncontrollable by short-acting cholinergic agonists.
Parathion and malathion (Fig. 16.10) are examples of insecticides which function by irreversibly inhibiting AChE. These compounds have a phosphorylating head group similar to dyflos and sarin, but with one oxygen substituted by a sulphur. Surprisingly, these compounds, despite their potent insecticidal activity, are relatively safe for humans. The design of these compounds stemmed from a knowledge of the differences in biosynthetic pathways between vertebrates and insects. The compounds are inactive in both insects and humans, but they are converted to their active forms where the sulphur is changed to an oxygen by metabolism in the body. This metabolic process happens in both insects and vertebrates, but in the latter it happens in parallel with metabolic detoxification, which makes these agents rather safe.11 Parathion is prone to detoxifying metabolism with a slower rate than malathion, which makes the latter safer. Fish are not able to detoxify these agents, and fish deaths have been reported when using parathion and malathion insecticides in areas of proximity to water. In spite of the proposed safety, these compounds, owing to their high lipid solubility, can be absorbed via the skin and accumulate in fatty tissues. Thus they should be treated with extra care.
Antidote for irreversible antiacetylcholinesterases
Phosphorylated AChE, but not the phosphinylated (post aging) enzyme, is susceptible to hydrolysis when treated with a strong nucleophile such as hydroxylamine. Again, the design here is heavily based on the chemical knowledge of the designer. The serine phosphate ester created by agents such as dyflos is extremely stable, and requires a strong nucleophile to break it. Hydroxylamine seems to be the most suitable candidate for the job, but it is also very toxic. The antidote was designed as a prodrug which would release hydroxylamine, or provide a hydroxylamine moiety only at the site of action. Specific binding to the AChE enzyme is strongly needed to increase potency and reduce side effects. This could be achieved by providing an ACh-like tail separated judiciously from a hydroxylamine head. Pralidoxime (Fig. 16.11) was the outcome of this combined knowledge, with a quaternary ammonium tail which interacts with the hydrophobic pocket in AChE securing better approachability for the hydroxylamine head to attack the phosphorylated serine.
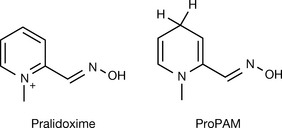
Pralidoxime is permanently charged, hence, it can not counteract the organophosphorylation in the CNS (remember, irreversible anti-AChEs are lipid soluble, thus expected to easily pass to the CNS). ProPAM (Fig. 16.11) is a prodrug of pralidoxime, and was designed as a reduced form of pralidoxime. The permanent charge (necessary for drug activity) is lost by reduction, allowing ProPAM to cross the BBB where the charge (thus the activity) can be regained by enzymatic oxidation. The permanently charged molecule cannot leave the CNS, which improves the availability of the antidote in the desired site.
Centrally acting cholinergic drugs
Acetylcholine neurotransmission was found to play an essential role in memory function, Alzheimer’s disease and parkinsonism. This has added a new (CNS) dimension to the therapeutic portfolio of cholinergic drugs, specifically muscarinic agents, as muscarinic subtype receptors are predominant in the CNS.12
Tacrine (Fig. 16.12) is an AChE inhibitor which exerts its action in the CNS as an anticholinesterase and a cholinergic agonist as well. However, it causes hepatotoxicity. It has poor absorption from the gut which necessitates an inconvenient four-times-daily dosage regimen.13 Donepezil (Fig. 16.12) is a more selective, less hepatotoxic member of this family. It has an excellent oral absorption, passes across the BBB and has a long half-life (70 hours) allowing for a once-daily (5–10 mg) treatment. Both drugs are approved to be used to ameliorate the cognitive deterioration associated with Alzheimer’s disease, and donepezil has been tested for the treatment of other types of secondary dementia. The two drugs do not seem to be structurally related, or related to other cholinesterase inhibitors.
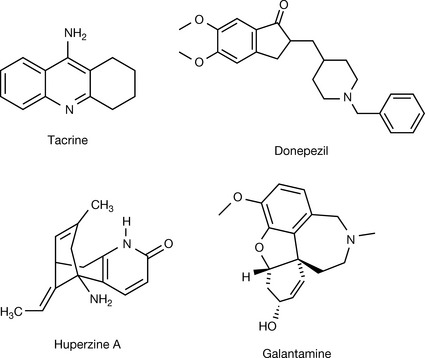
Huperzine A (Fig. 16.12) from the plant Huperzia serrata, which has a long history of use in the Chinese herb library, was found to have AChE activity. It is safer than tacrine and donepezil and has been used in clinical trials for treating Alzheimer’s disease and epilepsy.
Galanthamine (Fig. 16.12, galantamine) is another example of a drug of botanical origin. It is now chemically synthesised for large-scale production purposes, but was first isolated in 1959 from Galanthus woronowii (Voronov’s snowdrop). It is a competitive and reversible cholinesterase inhibitor with a very good oral bioavailability (80–100%) and a half-life of 7 hours. The side effects of galantamine may not be tolerable, and dose titration is necessary to improve tolerability. It is available as a tablet, extended-release capsule or oral solution in doses of 4–12 mg.
Anticholinergic drugs
Countering the action of ACh leads to several physiological responses which have been therapeutically exploited for treatment of diseases or for synergising with the action of other medications. It is of great importance for this antagonism to be selective for a specific receptor subtype or even a specific tissue. As the muscarinic (AChMR) and nicotinic (AChNR) cholinergic receptors are not structurally similar, and they do not function in the same manner, we can classify the cholinergic antagonists into two categories: muscarinic antagonists and nicotinic antagonists.
Muscarinic antagonists
The muscarinic receptors are of the G-protein family. They are all composed of seven transmembrane segments arranged in a serpentine fashion where the third cytoplasmic loop is coupled to a G-protein. Five different subtypes of muscarinic receptors have been identified by DNA cloning. Three of these subtypes M1, M2 and M3, have been assigned definite functions, where as M4 and M5 are suspected to be related to a CNS cholinergic activity.5 The activation of these subtypes results in different post-receptor second messenger signals, with M1, M3 and M5 activating the inositol-3-phosphate and diacylglycerol cascade and M2 and M4 leading to inhibition of cAMP production.14 The tissue distribution of muscarinic receptors varies as well. M1 is found in nerves, cortex, hippocampus and secretory glands. M2 is dominant in the heart and smooth muscles; hence M2 is called the cardiac M2 receptor.15 M3 is found in glands (exocrine glands), smooth muscles and endothelium.16 M4 is found in the neostriatum, and M5 in substantia nigra. It is necessary that the antimuscarinic agent has a considerable selectivity for cholinergic receptors rather than any other types of receptor and for muscarinic receptors rather than nicotinic receptors. Ideally, activity should be at a specific subtype of the muscarinic receptor in harmony with the desired action. The effect of a specific drug varies across tissues and this can be attributed to the pharmacokinetic (distribution) and pharmacodynamic (selectivity) properties of the drug.
Antimuscarinic agents
The first-discovered muscarinic antagonist was atropine which occurs as a secondary metabolite in the roots of the plant Atropa belladonna and other plants of the species Solanaceae. Atropine is an ester of tropine with hydroxymethylphenylacetic acid. The cosmetic use of extracts of belladonna for dilating the pupils to render women more attractive eventually led to its use (mydriatic) in the examination of the cornea in ophthalmology. Atropine is biosynthesised as a single enantiomer (called L-hyoscyamine), but once in solution, it easily racemises due to the carbonyl group situated in the alpha position to the chiral centre (Fig. 16.13).
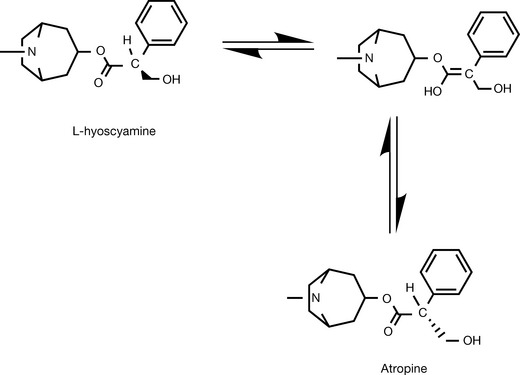
Pure atropine was isolated in 1831 and is still in use as a drug today. It is highly selective for cholinergic receptors and specifically for the muscarinic receptor. Its action at the nicotinic receptor is undetectable at subtoxic doses. It is more selective for muscarinic receptors than any of the synthetic antimuscarinic agents which may show ganglionic blocking effects or histamine blocking effects. However, it is non-selective for the subtypes of muscarinic receptors. Atropine has a half-life of 2 hours and the effect of the drug declines after this time in all organs but the eye. Certain species, e.g. rabbits, have an atropine esterase which will deactivate the drug more rapidly. It is most effective in salivary, bronchial and sweat glands, and least effective in the parietal cells in the stomach.
Atropine is used to inhibit the secretion induced by the irritant effect of inhaled anaesthetic agents at the bronchi. It is given in i.v. injection prior to anaesthesia with doses of 0.5–1 mg. Parenteral atropine has an emergency site use in countering the vagal depression of the sinoatrial node function resulting from myocardial infarction. It is used as well in myasthenia gravis for controlling the muscarinic side effects associated with prolonged anti-AChE therapy.
Atropine, owing to its long duration of action on the eye, is suitable for preventing adhesion formation in uveitis. Other ocular uses include the preparation for ophthalmic examination of non-compliant patients (e.g. children) as eyedrops (1% of atropine sulphate).
Atropine has shown a great efficacy against diarrhoea, especially when combined with a morphine receptor agonist. The commercial combination Lomotil (atropine sulphate 0.025 mg/dephenoxylate. HCl 2.5 mg) is one of the best treatments for diarrhoea. However, atropine in this case is used to produce some side effects which will discourage the abuse of diphenoxylate.
Atropine suppresses sweating, which is tolerable in adults, but can cause atropine fever in children, even at ordinary doses. Urinary retention and gastric hypomotility, usually expected post surgery, are exacerbated with antimuscarinic use.
Hyoscine (scopolamine, Fig. 16.14) is another natural antimuscarinic agent. It was isolated in 1879 from Datura stramonium (thorn apple). The structure has the addition of an oxygen bridge to the complex tropine ring system in atropine. This seems to increase the activity, especially in the CNS. This is probably because it is a much weaker base pKa 7.6 compared with atropine (pKa 9.9), this being due to the electron withdrawing effect of the oxygen reducing the availability of the lone pair on the nitrogen. Scopolamine has been used for many years as a prophylaxis and for the treatment for motion sickness. It is as effective as any other recently introduced treatment. It is given by mouth (0.3 mg three times daily), injection or transdermal batches (1.5 mg). It induces a dry mouth and sedation via all routes of administration.
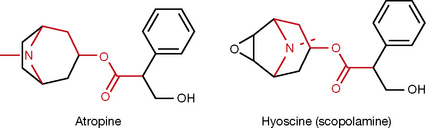
Scopolamine is interchangeable with atropine in preventing laryngospasm (by reducing bronchial secretion) in surgery. Its central effect, especially induced amnesia, is considered desirable in such traumatic events. The precautions in the use of atropine still apply to scopolamine.
Both compounds have in their skeleton an acetylcholine like section, i.e. a nitrogen, protonated at physiological pH, and an ester separated by three carbons (coloured with red in Fig. 16.14). Both compounds are tertiary amines which cross the BBB and produce CNS side effects, scopolamine being more likely to do this in view of its lower pKa value.
The two compounds occur naturally as the S (−) isomers but they are used therapeutically as racemic mixtures although with decreased activity compared to the natural form since The S (−) form is at least 100 times more potent than the R (+) form which reflects the enantioselective nature of the muscarinic receptor.
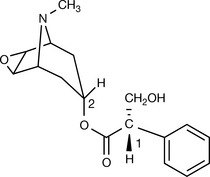
Starting from these two compounds, synthetic compounds were produced taking into consideration the need to diminish the disadvantages of the natural compounds. Note here that there is more flexibility with the length of the carbon bridge between the ester and the nitrogen, and there is no restriction with regard to alkyl substitution on the nitrogen or with regard to the acetyl motif on the ester side, as was discussed earlier in cholinergic agonists. This is because we do not need an exact fit on the receptor here to switch it on, but rather we are a seeking a mechanism to prevent endogenous ACh from accessing the active site on the receptor.
Ipratropium bromide (Fig. 16.15) is a synthetic quaternary ammonium compound made by adding an isopropyl alkyl group to the tertiary nitrogen in atropine (think of the name as three syllables ipr-atrop-ium (ipr from isopropyl, atrop from atropine, and ium) because it is a quaternary ammonIUM). Antimuscarinic activity at the muscarinic receptor M3 leads to bronchodilation and reduction of bronchial secretion. This effect is more significant in patients with chronic obstructive pulmonary diseases (COPD). The selectivity of M3/M2 for the chosen antimuscarinic agent is crucial for these effects, as blocking M2 will remove the autoinhibitory effect of ACh and trigger a stronger cholinergic discharge in postganglionic nerves, releasing more ACh to compete more with the muscarinic M3 antagonist. Ipratropium seems to be highly selective for M3, and is employed in metered-dose aerosols for the treatment of COPD (such as asthma) with a concentration of 40 μg/actuation. The dosage form provides maximum drug distribution at the desired tissue of action, and minimum systemic side effects. Ipratropium does not cross BBB due to its permanent charge; hence it shows very few CSN side effects. The dose varies widely according to the severity of the condition (2–6 actuations per dose) and the short half-life of ipratropium requires a frequent dosing (2–4 times per day).
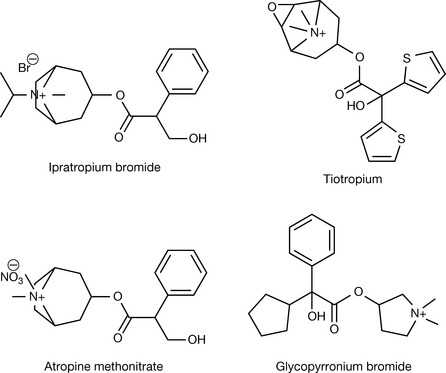
Tiotropium (Fig. 16.15) is a long-acting antimuscarinic agent with a similar selectivity profile to ipratropium. It is structurally related to scopolamine with substitution of the phenyl ring with two aromatic thiazol rings, and replacement of the hydroxymethyl group alpha to carbonyl group with a hydroxyl group. Tiotropium combines the ACh-like skeleton with the branched hydrophobic rings (discussed below). In clinical trials, tiotropium was superior to ipratropium in improving asthmatic patients, and allowed a reduction of the concomitant use of salbutamol. The long half-life of tiotropium (dissociation half-life of M3=35 hours) allows for a once-daily (18 μg) use. The dosage form is a dry powder inhalation, provided as capsules applied via an inhalation device. The capsules are ineffective when taken orally and may cause intolerable side effects.17,18,19
Atropine methonitrate (Fig. 16.15) is a methylated quaternary ammonium atropine. It was found to increase the activity of the myenteric plexus (a plexus of sympathetic and parasympathetic nerves supplying the two layers of muscles in the small intestine). Its permanent charge prevents its absorption by the gastrointestinal tract where it works locally for treating intestinal cramps.
The complex ring system is not really crucial for the antimuscarinic activity. This is illustrated by the potency of glycopyrronium bromide. The ester group seems to be important but not vital. Some compounds such as benzhexol (trihexyphenidyl (Fig. 16.16) have shown antimuscarinic activity even with no ester group at all. Glycopyrronium bromide (Fig. 16.15) is an example of an antimuscarinic compound which does not have the tropine ring system. The tropinium in atropine methonitrate was replaced by a simple pyrrolydinium ring, while the phenyl in the bulky blocking unit found in atropine was preserved, with the addition of a branched cyclopentanoyl group to the position alpha of the carbonyl. Glycopyrronium bromide is used interchangeably with atropine and scopolamine for reducing bronchial excretion and preventing laryngospasm. It is used as well to reduce or prevent the muscarinic effects of neostigmine. Glycopyrronium bromide is also used orally as an adjunctive therapy for peptic ulcer (2 mg) and topically as an antiperspirant (1% w/w and 3% w/w creams).
Recent experiments in animals have shown that glycopyrronium bromide, a quaternary amine, has conjunctival penetration and onset of action and duration of action as good as atropine when applied in ophthalmic solutions. This is contrary to the general belief which had previously prevailed that quaternary ammonium agents were not well absorbed by the eye.
Antimuscarinic compounds with no ester groups in the skeleton have a branched hydrophobic ring system, the rings are six-member or more or less, but there should be two rings which are NOT aliphatically attached to each other. These hydrophobic rings are thought to fill a hydrophobic cavity with a ‘T’ or ‘Y’ shape inside the active site, thus fixing the tail of the molecule in an orientation where the ACh-like head group blocks the access of the endogenous neurotransmitter, thus preventing the necessary conformational changes in the receptor which lead to the cholinergic activity.
The potency of molecules which lack the ester groups suggests that there are two mechanisms of binding to the receptor, one involves hydrogen bond interaction with the electron pairs on the ester oxygens and the other involves a van der Waals hydrophobic interaction with the hydrophobic rings (look at the structure of tiotropium, Fig. 16.15, which may combine the two mechanisms).
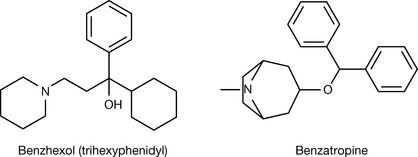
Parkinson’s disease is associated with tremor and rigidity in muscles. These symptoms seem to result from the imbalance of acetylcholine–dopamine ratio at the ganglia-stratum in favour of the first. Blocking ACh activity was the first drug treatment strategy for this disease. Latterly, dopamine treatment was deemed more important. Nevertheless, combination of both treatments yields significant synergism and is desirable when the anticholinergic treatment is tolerated by the patient. Benzhexol is used centrally to treat the symptoms of Parkinson’s disease and is added as well to the antischizophrenic therapy regimens to alleviate the extrapyramidal (Parkinson’s-like) side effects of antipsychotic drugs (see Box 16.5). It is available in tablets of 2 or 5 mg, injection (10 mg) and elixir (2 mg/5 mL). It is used in doses up to 15 mg per day. Benzhexol causes, as do other tertiary amine antimuscarinic agents, hallucinogenic effects, which puts it on the list of ‘drugs of abuse’.
Benztropine has similar uses and administration to benzhexol. It shares the tropine ring with atropine but the ester is replaced with an ether and branched benzyl groups. It has some antihistamine and local anaesthetic activity but is not indicated for such uses. Benzatropine (used as the mesilate salt) has a longer half-life than benzhexol and is used as a single daily dose (1–6 mg). It is available in oral (tablet) or injectable (i.m.) dosage forms.
Antimuscarinic agents have a remarkable effect on the gastrointestinal tract, and reduced saliva, gastric section, amount of stomach acid, pepsin and mucine are all observed. Motility is affected as well, with the walls of the viscera being relaxed and both the tone and propulsive movements being diminished upon antimuscarinic therapy. These effects make antimuscarinic drugs potential agents for the treatment of peptic ulcers, diarrhoea and intestinal cramps. Specific agents have been optimised for each of these three indications.
Pirenzepine (Fig. 16.17) makes a special antimuscarinic case. It is selective for the M1 receptor compared to its activity against M2 or M3. It is 20 and 4 times less potent at the M3 receptor than at M1 and M2, respectively. As discussed before, M1 is found in secretory glands and M2 is found in the heart. This selectivity gives pirenzepine a good activity against gastric acid secretion with no cardiac side effects. However, pirenzepine was superseded by H2-receptor antagonist and proton pump inhibitors (which are discussed in Ch. 17 and Ch. 12, respectively).
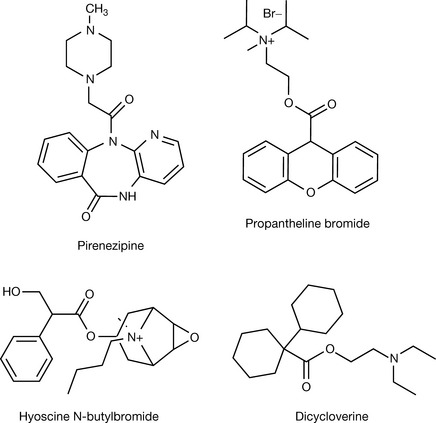
The antispasmodic agents make use of the muscle relaxation effect of the antimuscarinic agents, but seem to depend on other additional mechanisms for success in obtaining the desired action, which is directly proportionate to the ability to relax the gut muscles in the absence, as well as the presence, of cholinergic stimulation. Examples of these types of drugs are propantheline, hyoscine-N-butyl bromide and dicycloverine (Fig. 16.17).
Propantheline provides another example of the branched ring system. It is a quaternary ammonium with the bromide salt used to relieve spasms of the intestine or to treat involuntary urination in doses of 15–30 mg.
Hyoscine-N-butyl bromide is a quaternary ammonium variant of hyoscine (scopolamine) formed by adding a butyl group to the tertiary amine group. It is mainly used to relieve temporal spasms of the intestine or menstrual cramps. It produces very moderate side effects, especially in the CNS. The long-term use of hyoscine-N-butyl bromide for the treatment of irritable bowel syndrome (IBS) has been reported as well (three daily doses of 10 mg). The drug is available in oral dosage forms with doses of 10–35 mg.
Injectable hyoscine-N-butyl bromide has been successfully used as an antisecretory drug in combination with haloperidol in palliative care.20
Dicycloverine is another example of an antimuscarinic agent which binds by both hydrogen bonding and hydrophobic van der Waals forces to the receptor. It is used as a gastrointestinal spasmolytic agent for the treatment of abdominal cramps and symptoms of IBS. It is available as oral capsules (10–20 mg), syrup (20 mg/5 mL) and an injection (20 mg/2 mL) of the hydrochloride salt.
Otilonium bromide is a new generation antispasmodic. It has an ACh-like moiety (red colour) attached to an aromatic system which ends with an octyl ether. The oral bioavailability of otilonium bromide is very low, and it was found to accumulate in the lower intestine, making it a locally active agent. Otilonium bromide is highly tolerable with very few side effects due to poor absorption of the drug. Even the effect on gastric secretion, which occurs with other antimuscarinic agents, was not reported at therapeutic doses. The antimuscarinic (M3) spasmolytic activity of otilonium bromide is fortified by other mechanisms including blockade of calcium channels and binding to tachykinin neurokinin-2 receptors. Its specific indication lies in treating the symptoms of IBS, especially with associated diarrhoea, in doses of 40 mg three times daily.21
The prolongation of gastric emptying time subsequent to the diminished gastric motility makes antimuscarinic agents, even the selective agent pirenzepine which is used for peptic ulcer, contraindicated in gastric ulcer as they increase the acid–ulcer contact time. Another observation about the same effect is the interaction with drug absorption, which is affected by the emptying time, and this should be taken into consideration when adding any antimuscarinic agents to a current ongoing therapeutic regimen.
The effect of antimuscarinic agents on the eye is mydriasis, cycloplegia and reduction of lacrimal secretions. These effects are desirable in ophthalmic examination of non-compliant patients (e.g. children) but considered to be side effects (blurred vision, dry [sandy] eye) in the systemic use of these agents. Cyclopentolate and tropicamide (Fig. 16.18) are antimuscarinic agents used in ophthalmic examinations. They have shorter duration of action, 1 and 0.25 days respectively, in comparison to atropine (7–10 days). They are not structurally related, or related to any other antimuscarinic agent. However, in cyclopentolate it can be seen that there is an ester group separated by two methylene units from an amine group, as in ACh. They may also be used before or after eye surgery, such as lens replacement surgery. Both cyclopentolate and tropicamide are available in eyedrops at concentrations 0.5% and 1% w/v.
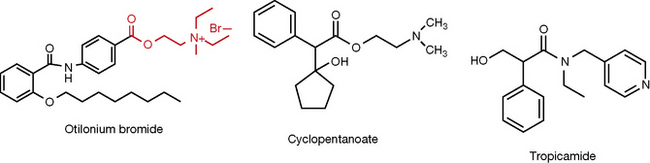
Upon antimuscarinic treatment, smooth muscles of the ureters and bladder wall are relaxed. This leads to urinary retention which is desired for patients with urinary incontinence but is contraindicated in males with prostate hyperplasia where filling the bladder increases pressure on the prostatic walls. Oxybutynin (Fig. 16.19) has shown some pharmacokinetic/pharmacodynamic selectivity towards the urinary tract. It is used orally (5–15 mg/day) in patients with overactive bladder and has been proven valuable as well in relieving bladder spasm after urological surgery. Tolterodine (Fig. 16.19) is an M3 selective antimuscarinic agent. It is used for urinary incontinence in adults.
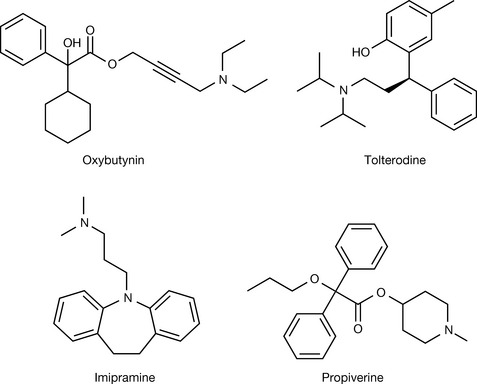
Many antihistamines, antipsychotics and antidepressants have structural similarities with muscarinic receptor antagonists and, predictably, show antimuscarinic effects which are considered in many cases the major side effects of these drug categories. Imipramine (Fig. 16.19) is a tricyclic antidepressant with a chemical structure which fits that of a muscarinic blocker. The muscarinic blockade is considered a side effect (Box 16.4) when used for treating depression, but imipramine is also employed for treating urinary incontinence in the elderly.
Box 16.4 Effects of antimuscarinic agents on the cardiovascular system
The antimuscarinic agents elicit these effects on the cardiovascular system:
Propiverine (Fig. 16.19) is a newer antimuscarinic approved for the specific use of urinary incontinence in doses of 5–75 mg. The rules for the use of antimuscarinic agents are summarised in Box 16.5.
Box 16.5 Rules for the use of quaternary and tertiary amines as antimuscarinic agents
Quaternary amines are poorly absorbed by the gut and poorly distributed to the CNS owing to their hydrophilicity and are used when a peripheral effect devoid of central effects is desirable.
Tertiary amines are well absorbed from the intestine and conjunctival membrane. They easily cross the BBB, with scopolamine showing the highest preference for central residency among all antimuscarinic agents.
All antimuscarinic agents work in a surmountable competitive fashion; reversal of their actions can be concluded easily by infusing acetylcholine or any other cholinergic agonists, but the reversing agents should be selected carefully according to a peripheral (quaternary amine) or peripheral/central (tertiary amine) toxicity that needs to be surmounted.
Antinicotinic drugs
The nicotinic receptor AChNR is quite different from the muscarinic receptor. It is a ligand-gated ion channel and does not operate with a second messenger system. Activation of the nicotinic receptor leads to a flow of Na+ and K+ across the cell membrane and a depolarising effect on the effector cell (nerve cell or neuromuscular end plate).
AChNRs are subdivided into two subtypes, which vary in structure and distribution. The muscular nicotinic receptors are distributed in the neuromuscular junctions of the skeletal muscles and the postganglionic nicotinic receptors in the ganglia of the autonomic nervous system. Both nicotinic subtypes are pentamer proteins, but the subunits are not the same.
The nicotinic receptors in muscle and neural tissues have a large N-terminal domain, consisting of four hydrophobic transmembrane domains, called TM1–TM4 regions, a large cytoplasmic loop, and a short extracellular C-terminus. The transmembrane loop is thought to form the ion pore for the nicotinic ACh receptor. Binding of ACh and AChNR agonists to the muscular AChNR takes place at the amino-terminal regions of the α/γ and γ/β interfaces, whereas binding to the neuronal AChNR takes place at the two α/β interfaces of the heteromers and to the five subunit interfaces in the homomers.22
The nicotinic receptor is found in the CNS as well. It plays a central role in nicotine (smoking) addiction. Nicotine binds strongly to the brain cholinergic receptor due to a strong cation–π donor interaction to a specific aromatic amino acid of the receptor, TrpB (tryptophan 149). The interaction is enhanced by a hydrogen bond to the carbonyl at the backbone of TrpB. This interaction is absent in the muscular AChNRs, explaining why absorption of nicotine via smoking does not cause muscle contractions. Although the amino acid sequence in the active site is similar in muscular and neurological AChNRs, a neighbouring disparity in amino acid residues (a lysine in neurologic and a glycine in muscular receptors at position153) which affects the active site shape explains the differences in nicotine-binding strength between the two receptor types. The lysine is thought to form a backbone hydrogen bond between loop B and loop C (remember, AChNRs have four transmembrane loops) whereas glycine discourages this bond.23
All AChNR have two alpha subunits, each carries one active site. To give the greatest probability of channel opening, both active sites should be occupied by agonists. Activation of only one site still can lead to channel opening but only with a moderate probability. Large amounts of ACh released by nerve impulses cause the muscle action potential. There are two types of muscle relaxation produced by manipulation of AChNR. Depolarising muscle relaxation happens when agonists occupy the active site for a prolonged period, resulting in the constant depolarisation of the effector cells (postganglionic neurons and muscular end-plate cells), which will then not fire any new signals, and this leads to muscle relaxation. This action is irreversible and is constant as long as the agonist is occupying the active site on the receptor. The non-depolarising muscle relaxants act by competitively antagonising the active site, and this is reversible by cholinergic agents.24
Non-depolarising neuromuscular blockers
Non-depolarising neuromuscular blockers (NMBs) in normal doses act by competitive inhibition of the interaction between acetylcholine and nicotinic receptors. In higher doses, they are thought to block the ion channel as well, which explains the decreased effectiveness of AChE inhibitors in treating non-depolarising neuromuscular blocker toxicity.
The non-depolarising agents vary according to their duration of action and cardiac effects. The paralysis is desirable only during surgery, and a speedy recovery is in favour of the patient. This has been an incentive to medicinal chemists to design short-acting agents.
The duration of action of non-depolarising neuromuscular blockers is basically dependent on their metabolism, with agents excreted by the kidney being the longer acting. Here, we should take into consideration that patients under surgery have different liver and kidney functions from aware patients, as do elderly and young patients.
Tubocurarine-like drugs
As with all the drug categories in this chapter, the first compound with antinicotinic activity came from a natural source. Curare was used by the American Indians as a dry extract of the plant Chondrodendron tomentosum (the curare vine) rubbed onto the tip of their arrows. The actual name, curare, is a corruption of two Tupi Indian terms meaning ‘bird’ and ‘to kill’. The name indicates the potency of this plant as a weapon. Curare extracts contain several alkaloids, the most potent of which is tubocurarine, which was isolated in 1935, but it took 35 years to determine its structure. Tubocurarine has a complex structure with two amine functions, one being quaternary and the other tertiary, and both are thought to be involved in the mechanism of action of tubocurarine (Fig. 16.20). Tubocurarine is the natural prototype of the non-depolarising neuromuscular relaxants. Its first medical use goes back to 1912 and it was introduced into anaesthesia in 1942. Tubocurarine causes a hypotension due to a moderate release of histamine and blocks the autonomic ganglionic nicotine receptors, producing a vagolytic effect (tachycardia). It is still employed in doses of 0.12–0.4 mg/kg and its action lasts for 60–90 minutes. However, its side effects make it less favoured than newer agents. Metocurine (Fig. 16.20) was introduced as an attempt to reduce the side effects of tubocurarine. It is a slight modification of the tubocurarine structure where both free hydroxyl groups were methylated, with the tertiary nitrogen being rendered quaternary by addition of a methyl group as well. The modification succeeded in decreasing histamine release, but had no impact on the vagolytic effect. Both tubocurarine and metocurine are highly selective towards the nicotinic receptor compared to the muscarinic receptors.
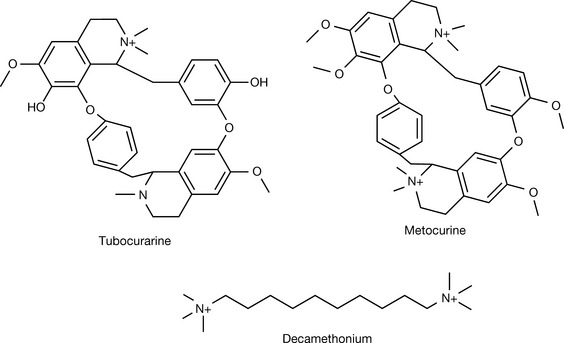
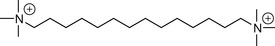
Several hypotheses have been suggested for the mechanism of binding of tubocurarine and later analogues to the nicotinic receptor, which seems puzzling considering the total absence of the ester function in these compounds. The most plausible hypothesis suggests an interaction between one of the charged nitrogens with the active site, with the other nitrogen interacting with a cysteine residue 0.9–1.2 Å away from the active site. This is supported by the distance between the two nitrogens in tubocurarine, and the fact that decamethonium (Fig. 16.20), a very simple molecule where two quaternary nitrogens are separated by an aliphatic chain of ten carbons, has shown potency comparable to tubocurarine.
Thus the active site of the receptor is blocked competitively, preventing the access of the endogenous ACh. The proposed mechanism indicates the importance of the distance between the two charged nitrogens in any antinicotinic compound to be designed, also bearing in mind that these distances change with the conformation that the molecule takes, in the case of a highly flexible molecule such as decamethonium.
Decamethonium was used as a replacement for tubocurarine but it had multiple disadvantages of its own. Decamethonium is a partial agonist giving a brief contraction of muscles which delays its onset of action, it is non-selective for the neuromuscular junction and gives cardiac side effects including tachycardia and hypotension which may persist for a considerable time given the strong interaction between the drug and the receptor. The lack of selectivity in decamethonium can be understood in terms of the flexibility of the molecule, which can take so many conformations, some of which are suitable to switch off the muscarinic receptor as well.
The design of effective analogues of tubocurarine and decamethonium took into consideration the introduction of conformationally restricted groups, as well as the mechanism governing the rate of elimination of the drug from the body while at the same time preserving the crucial distance between the two charged amines. The later non-depolarising agents have a relatively rigid structure and can be divided, from a chemical perspective, into steroid and isoquinoline derivatives.
 Self Test 16.4
Self Test 16.4
Based on the hypothesis given for NMB activity, do you think the compound below will give improved antinicotinic properties compared to decamethonium? Why?
Steroid-like neuromuscular blocking agents
Pancuronium (Fig. 16.21) was the first steroid-like neuromuscular blocking (NMB) agent employed in clinical use. It is composed of two acetylcholine motifs separated by a rigid steroid-like structure. The acetylcholine motifs use parts of the steroid as the two-carbon separator bridge, and the quaternary ammonium groups are incorporated into a piperidenium ring (outlined in red). This structure ensures a good occupancy of the active sites on both alpha subunits in the nicotinic receptor to prevent its switching on by acetylcholine. It is a potent muscle relaxant (six times more potent than tubocurarine) and has a prolonged duration of action (120–180 minutes). However, the onset of action is relatively slow (2–4 minutes) and its renal excretion (80%) makes it better avoided for patients with kidney diseases. It is inactive via the oral route and is given by i.v. injection as a bromide salt with a concentration of 1 mg/mL or 2 mg/mL. Pancuronium has been cited many times in forensic investigations of suicides and murders, and as a part of the euthanasia procedure.
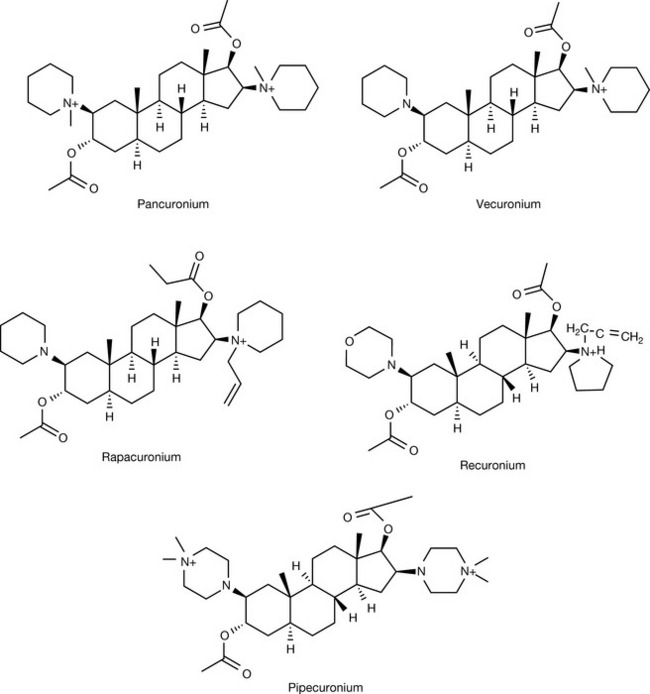
Vecuronium is an N-desmethylpancuronium, the minor change resulting in a substantial impact on the rate of elimination of the drug. Removing the methyl from one of the quaternary ammonium groups will convert it into a tertiary amine with a loss of the permanent charge. This reduces the hydrophilicity and, since the general structure is steroid-like, this facilitates elimination of the drug into the bile. Vecuronium has a shorter duration of action of 20–35 minutes (compared to 120 minutes for pancuronium), fewer cardiac side effects with comparable potency (very slightly more than pancuronium).25 The salt used is the bromide and is available in multiple-injection vials of 20 mg/20 mL concentration. The paralysing action is noticed in approximately 1 minute for a 0.08–1 mg/kg initial dose.
Pancuronium and vecuronium both have faster onset of action than tubocurarine, and they do not have significant cardiovascular side effects. Their intermediate duration of action makes them suitable for medium-sized surgical procedures (e.g. caesarean section). However, faster onset of action, fewer side effects and a shorter duration of action are still desirable features demanded by anaesthetists.
N-desmethylation on one quaternary nitrogen, and replacing the methyl with an allyl group on the other, resulted in rapacuronium (Fig. 16.21), the most rapid onset of action of non-depolarising muscle relaxants. Rapacuronium has a short duration of action as well (10–20 minutes), it is eliminated mainly by liver, and its potency is 15 times less than pancuronium. However, it was voluntarily withdrawn in 2001 by its manufacturing company because of reports of severe bronchospasm.26
Rocuronium (Fig. 16.21) preserves the vecuronium trend of tertiary versus quaternary nitrogens, but with further modifications on the nitrogen substitutes. The drug has a similar pharmacokinetic, duration of action, and toxicity profile to vecuronium but six times less potent.27 It has a fast onset of action (about 2 minutes). Recuronium is supplied as its bromide salt in multiple-dose vials containing volumes of 50 or 100 mL at a concentration of 10 mg/mL.
Pipecuronium is a modification of pancuronium where both quaternary ammonium groups were desmethylated, but quaternary nitrogens were introduced into what was a piperidine ring in pancuronium. Pipecuronium is similar to pancuronium in efficacy. It shows some elimination by liver but this is not as predominant as in recuronium or vecuronium. Pipecuronium has a half-life of 1.7 hours in normal renal function and 4 hours in renal function impairment. The onset of action is reported to be 2.5–3 minutes and the duration of action about 45 minutes. Pipecuronium is supplied in a sterile injection containing 10 mg of the bromide salt.28
The early steroid-like neuromuscular blockers show very little histamine release or ganglionic effects, but still cause cardiac side effects due to non-selective binding to the cardiac muscarinic receptor, with pancuronium showing a moderate block on that receptor. The problem was partially resolved in rocuronium and rapacuronium and diminished further in vecuronium and pipecuronium but with some loss of potency. The steroid-like agents are prone to metabolism. Of the different metabolites, the 3-hydroxy shows 40–80% activity compared to the parent compound which may cause problems upon long term accumulation of the metabolite. This should be taken into consideration in patients with liver and kidney diseases. However, this is a concern only in intensive care units (ICUs), as accumulation of the metabolite is negligible during surgery. Box 16.6 summarises the importance of muscle relaxation in anaesthesia.
Box 16.6 Importance of muscle relaxation in anaesthesia
Complications of surgeries, especially those on the abdomen or throat, necessitate skeletal muscle relaxation prior to surgery. Before the introduction of muscle relaxation, this was achieved by deep anaesthesia which itself is a hazard for patients (cardiac complications, respiratory suppression). Deep anaesthesia is no longer required by virtue of neuromuscular blocking agents providing adequate muscle relaxation for all surgical requirements.
Muscle relaxation is desirable in surgical operations and also in ICUs where paralysis is of benefit to the patient.
Isoquinoline derivative neuromuscular blocking agents
The design of these agents was based on the properties of both tubocurarine and decamethonium. The bulky ring system, which gave tubocurarine conformational restriction and thus selectivity and linear space between the two quaternary ammonium groups present in decamothenium, were combined. Isoquinoline derivatives were intended to be similar in structure to the bulky groups in tubocurarine (the natural compound) and still conserve a linear chain with a 1.1–1.2 Å distance separating two nitrogen atoms. In the atracurium structure, two exactly similar isoquinoline derivatives were connected by a 13-atom bridge. The linear (non-branched) bridge provides the necessary distance between the two quaternary ammonia. Additionally, it contains two beta-carbonyl groups which facilitate the degradation of atracurium via the Hofmann elimination. Atracurium (Fig. 16.22) was first synthesised in the Department of Pharmacy at the Strathclyde University in 1974, which was part of a remarkable double since the pharmacological screening which led to vecuronium was carried out in the Department of Pharmacology at Strathclyde at about the same time. The commercial drug is a mixture of ten stereoisomers.
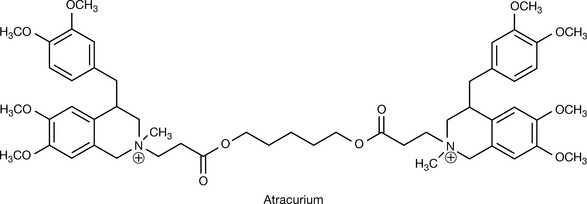
Atracurium has a duration of action similar to that of vecuronium and is eliminated by a rationally designed, purely chemical degradation mechanism via the Hofmann elimination (see Box 16.7) which occurs in an aqueous environment with the rate increasing with higher pH (Fig. 16.23). Atracurium is supplied as a sterile injection of 10 mg/mL.
Box 16.7 Hofmann elimination as a chemical degradation mechanism of isoquinoline NMBs
Hofmann elimination occurs in quaternary amines where one of the attached alkyl groups leaves the nitrogen in the form of an alkene. The reaction happens in an alkaline medium, usually with the aid of heating and vacuum. With unsymmetrical amines, the major alkene product is derived from the least substituted and generally the least stable alkyl group, an observation known as the Hofmann rule. This is in direct contrast to normal elimination reactions where the more substituted, stable product is dominant.
The conditions required for Hofmann elimination can never be achieved in vivo, but the introduction of a nearby electron withdrawing group, a carbonyl in this specific case, supported by the slightly alkaline (pH = 7.4) medium will facilitate the process. The reaction products are inactive and can no longer block the active site of the nicotinic receptor, and in the best case, the elimination products can occupy one of the two active sites on the nicotinic receptor, which, as discussed, is not sufficient for a complete blockage of ACh action.
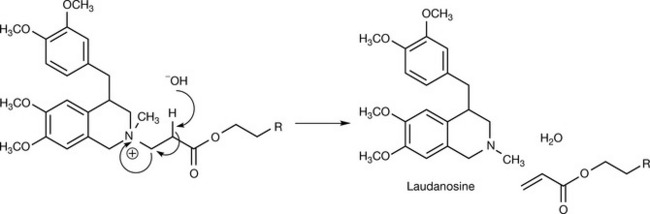
The main product of the Hofmann elimination of atracurium is laudanosine, an alpha-adrenoreceptor blocking agent which causes hypotension.29 Moreover, laudanosine crosses the blood–brain barrier (in contrast to the parent compound) and may cause excitement and seizure activity which leads to an increase in anaesthetic requirement by 30%. Accumulation and corresponding problems with laudanosine arise only in infants and with prolonged application in ICUs.30 Laudanosine-caused hypotension is enhanced by histamine released from tissue stores as a side effect of atracurium.31
To counter the problems produced by laudanosine, the stereoisomers of atracurium were isolated in order to allow a dose reduction. Cisatracurium (Fig. 16.24) is an R-cis R-cis isomer of atracurium. It is degraded by Hofmann elimination just like atracurium. However, plasma concentrations of laudanosine do not reach such high levels as in the case of atracurium, leading to fewer cardiac side effects. Cisatracurium also releases less histamine from tissue stores. The potency and duration of action are similar in both drugs. Cisatracurium besilate is provided in sterile injections with a concentration equivalent to 2 mg/mL of cisatracurium in 5- or 10-mL vials, or 10 mg/mL of cisatracurium in 20-mL vials intended for use in ICUs only.
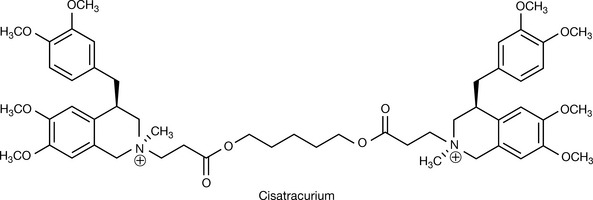
The spontaneous chemical elimination makes these agents a better option for patients with renal or hepatic diseases and allows less patient-to-patient variability. The Hofmann elimination does not occur in acidic pH, and these drugs are stable when stored at a pH = 3–4. This pH level is achieved by the addition of benzenesulphonic acid which provides some buffering capacity compared to, say, hydrochloric acid. However, even at this pH, an observed loss of potency by a rate of 6% per annum has been reported to both agents at 4°C. This rate increases to 5% per month at 25°C and thus refrigerator storage is necessary for these agents.
Mivacurium (Fig. 16.25) is another bisbenzylisoquinolinium neuromuscular blocking agent. One structural difference from atracurium is the additional methoxy group on each of the benzyl rings in the benzylisoquinoline, but the greatest change is the addition of a third carbon to separate the carbonyl group from the quaternary ammonium. This has made the elimination of the drug less dependent on chemical degradation mechanisms and more reliant on plasma cholinesterase (pseudocholinesterase). Additionally, the introduction of a double bond in the centre of the unbranched chain separating the two quaternary ammonium centres allowed multiple geometric isomers to emerge. The commercial drug is a mixture of trans-trans [52–62%], cis-trans [34–40%] and cis-cis [4–8%]. These forms, especially the first two, are easy targets for plasma cholinesterase, and the result is a very short-acting neuromuscular blocking agent (10–20 minutes). Nevertheless, the duration of action is prolonged in kidney disease patients as they have decreased pseudocholinesterase levels and in individuals with genetically variable levels of pseudocholinesterase. The separating bridge in mivacurium consists of 16 atoms (compared to 13 in atracurium) but surprisingly it is three times more potent than atracurium. Mivacurium causes histamine release comparable to that caused by atracurium. It is provided as mivacurium dichloride and the pharmaceutical dosage form is a sterile injection with a concentration of 2 mg/mL.
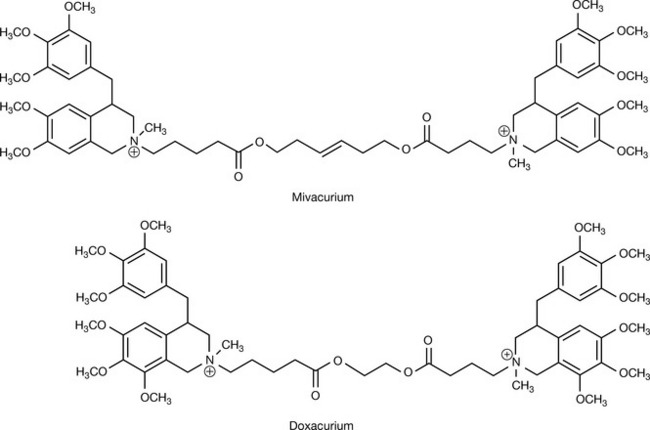
Doxacurium is a middle case between atracurium and mivacurium. The non-branched bridge was modified from atracurium by shifting the carbonyl group to the gamma position relative to the quaternary ammonium, and by eliminating three of the methylene units from the centre of the bridge. This modification (as in mivacurium) limits the Hofmann elimination and disposes doxacurium to renal elimination. Other modifications include one extra methoxy group on each of the four aromatic rings in the structure. Its duration of action is prolonged (40 minutes) and its potency is enhanced compared to atracurium (four times). The renal elimination of doxacurium makes it less optimal in patients with kidney diseases. It is supplied in sterile injections of doxacurium dichloride with a concentration equivalent to 1 mg/mL of the active compound.32
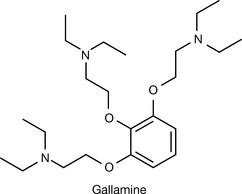
Gallamine (Fig. 16.26) was developed in 1947 as a non-depolarising neuromuscular blocker. It does not chemically belong to any of the groups above. The effect of gallamine lasts for 40–60 minutes and its potency is 20% that of tubocurarine. It is used as its tri-iodide salt and the presence of the iodide salt means that it is unsuitable for patients with thyroid problems. Gallamine blocks the cardiac muscarinic receptor very strongly, resulting in a remarkable increase in heart rate and cardiac output with occasional hypertension.
Depolarising neuromuscular blocking agents
Suxamethonium (Fig. 16.27) is the only depolarising NMB agent in clinical use. Suxamethonium (succinylcholine) was designed by introducing two ester groups in symmetric positions in the middle of the decamethonium molecule. The ester has labile functions which degrade in the circulation both chemically and enzymatically via the action of pseudocholinesterase (PAChE). The hydrolysis product, succinylmonocholine, binds only to one alpha unit of the active site of the nicotinic receptor. This produces only a mild depolarisation, not enough to cause a continuous depolarising and a subsequent neuromuscular blockade. Succinylmonocholine shows no toxicity. Plasma cholinesterase has a very large capacity, and can metabolise suxamethonium very rapidly, explaining the short duration of action (5–10 minutes). Suxamethonium is not metabolised by end-plate cholinesterase, and is removed by diffusion (Fig. 16.27). The elimination in the plasma makes the diffusion equilibrium directed towards extracellular fluid (then plasma) and prevents accumulation. This allows for pseudocholinesterase to play a great role in the potency and duration of action of suxamethonium, and makes it, as in the case of mivacurium use, highly variable in patients with kidney disease and among individuals deficient of this enzyme for genetic reasons (0.05% of humans lack PAChE).
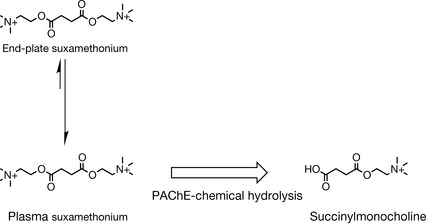
The blockade process passes through two phases. It starts with a depolarisation just like that induced by ACh, the natural ligand for the nicotinic receptor, causing random, very brief muscle contractions. Suxamethonium is not metabolised by synaptic AChE. Thus it produces a prolonged depolarisation compared to ACh. The longer depolarisation state of the membrane makes it refractory to new excitements and results in a flaccid paralysis. This phase can be augmented by AChE inhibitors (e.g. neostigmine), as they usually inhibit PAChE as well, and increase synaptic exposure to suxamethonium.33 The unsynchronised nature of the muscle contractions in this phase causes damage in the neighbouring muscle fibres, explaining the complaints of muscle pain post surgery where suxamethonium was used. In the second phase, repolarisation is achieved but the end plates are still not responsive to new depolarisation. This is described as desensitisation of the motor end-plate cells. Evidence suggests that suxamethonium acts by occupying the receptor and blocking the ion channel itself, to which the desensitisation phenomena can be attributed. Suxamethonium has the fastest onset of action (<1 minute) among all neuromuscular blocking agents. The onset of action determines how soon the patient’s trachea can be intubated, which is necessary in life-threatening emergencies. It is given in doses of 1–2 mg/kg intravenously or 3 mg/kg intramuscularly, reconstituted from a clear solution of a concentration of 50 mg/mL. Suxamethonium activates cholinergic receptors in a non-selective manner. All nicotinic (neuromuscular and ganglionic) and muscarinic receptors are affected by this drug and it causes various cardiac arrhythmias. The contradictory effect of activating cholinergic receptors in different sites (e.g. ganglionic receptors versus muscarinic receptors in the sinus node) means its effect on the cardiovascular system varies according to dose and individuals. Other side effects of suxamethonium include increased intraocular pressure and intragastric pressure. Suxamethonium occasionally causes an exaggerated release of potassium into the blood. This may lead to cardiac arrest in children. In 1993, the FDA prevented the use of suxamethonium in children, and then modified the risk level to a warning. Additionally, since suxamethonium’s basic elimination mechanism is by enzymatic hydrolysis with PAChE which varies significantly between individuals, this leads to difficulties in dose determination and recovery time prediction.
NMB agents, whether depolarising or non-depolarising, are polar compounds owing to the positive charge carried on the quaternary ammonia. This makes them biologically unavailable when given by the oral route, and less able to distribute to the central nervous system. The polarity makes their volume of distribution close to that of the blood. Box 16.8 summarises some of the drug combinations that are used in anaesthesia.
Box 16.8 Drug combinations in anaesthesia
The addition of neuromuscular blockers to the anaesthetic regimen has proven significantly valuable. However, additional therapeutic measures are necessary to combat the adverse effects of the different agents. Hence, the histamine release caused by tubocurarine, metocurine, atracurium and mivacurium are eliminated by a pre-surgery dose of an antihistamine. The muscarinic activity of suxamethonium is reversed by a dose of atropine and the post-surgery muscle pain resulting from the fasciculation caused by suxamethonium is relieved by a small dose of a non-depolarising muscle relaxant. These measures, albeit complicating the anaesthetic regimen, are in favour of the patient when measured by a risk/benefit assessment.
Ganglionic blockers
Ganglion blocking drugs are anticholinergic agents which block the nicotinic receptor at the ganglionic level. Nicotinic ion gate blockers, which work on the nicotinic receptor but at a site of interaction different from the ACh active site, are classified with this category of drugs. The lack of selectivity in the action elicited by these drugs allows for high interpersonal variability in response and limits their clinical use. These agents block the postganglionic acetylcholine signal for both sympathetic and parasympathetic nerves. For tissues innervated by both types, the net effect of this blockade is dependent on the predominant tone in the tissue.
Hexamethonium (Fig. 16.28) is a quaternary ammonium agent. It was the first successful antihypertensive treatment. However, its side effects led to discontinuation of its use. It is thought to exert its action by blocking the ion channel rather than the acetylcholine active site. Blocking the ganglionic nicotinic receptor will stop the sympathetic discharge to the veins and arteries, resulting in a vasodilation.
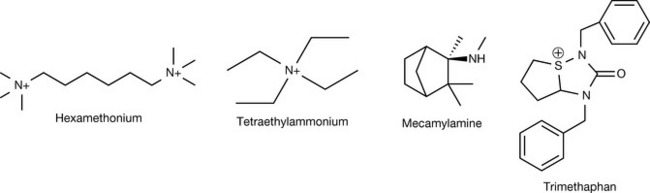
Tetraethylammonium (Fig. 16.28) is a quaternary ammonium agent with a very short duration of action. Mecamylamine is a secondary amine designed to improve the oral bioavailability of ganglionic blockers. Central side effects are not unexpected. It was used for the emergency treatment of malignant hypertension but has been superseded by safer agents such as sodium nitroprusside. Trimethaphan (Fig. 16.28) is a short-acting anticholinergic agent working at the ganglionic site. It has a complex structure compared to other agents in this class. It bears two adjacent tertiary amides, but still can not be absorbed by the gut or cross the BBB owing to the positive charge of its sulphonium ion. The structure does not seem to be related to acetylcholine. It is given i.v. for the emergency treatment of malignant hypertension.
 Self Test 16.2
Self Test 16.2
Isomer S. C – C = O > C – C – O > C – C = C. Hydrogen is in front of everything thus reverse the assignment.
Centre 2 is not chiral because the carbon 2 is attached to two identical carbon atoms due to symmetry in the tropine ring.
1. Dean ESaB. Role of the cholinergic system in the pathology and treatment of schizophrenia. Expert Rev Neurother. 2009;9:73-86.
2. Koch H.J.H.S., Jürgens T. On the physiological relevance of muscarinic acetylcholine receptors in Alzheimer’s disease. Curr Med Chem. 2005;12:2915-2921.
3. Laura Paleari A.G., Cesario A., Russo P. The cholinergic system and cancer. Semin Cancer Biol. 2008;18:211-217.
4. Maresca A.S.C. Muscarinic acetylcholine receptors as therapeutic targets for obesity. Expert Opin Ther Targets. 2008;12:1167-1175.
5. Jtirgen Wess N.B., Mutschler E., Bluml K. Muscarinic acetylcholine receptors: structural basis of ligand binding and G protein coupling. Life Sci. 1995;56:915-922.
6. Zlotos D.P., Buller S., Tränkle C., et al. Bisquaternary caracurine V derivatives as allosteric modulators of ligand binding to M2 acetylcholine receptors. Bioorg Med Chem Lett. 2000;10:2529-2532.
7. Kovacic P., Pozos R.S., Draskovich C.D. Unifying electrostatic mechanism for receptor-ligand activity. J Recept Signal Transduct Res. 2007;27:411-431.
8. Jensen A.A., Mikkelsen I., Frolund B., et al. Carbamoylcholine homologs: novel and potent agonists at neuronal nicotinic acetylcholine receptors. Mol Pharmacol. 2003;64:865-875.
9. Frank Dorje J.W., Lambrecht G., Tacke R., Mutschler E., Brann M.R. Antagonist binding profiles of five cloned human muscarinic receptor subtypes. J Pharmacol Exp Ther. 1991;256:727-733.
10. Bikádi Z.S.M. Muscarinic and nicotinic cholinergic agonists: structural analogies and discrepancies. Curr Med Chem. 2003;10:2611-2620.
11. Patrick G.L., editor. An introduction to medicinal chemistry. Oxford; New York: Oxford University Press, 2005.
12. Hanna Kaduszkiewicz T.Z., Beck-Bornholdt H.-P., van den Bussche H. Cholinesterase inhibitors for patients with Alzheimer’s disease: systematic review of randomised clinical trials. Br Med J. 2005;331:321-327.
13. Maltby N, Broe G.A., Creasey H., Jorm A.F., Christensen H., Brooks W.S. Efficacy of tacrine and lecithin in mild to moderate Alzheimer’s disease: double blind trial. Br Med J. 1994;308:879-883.
14. van Koppen C.J., Kaiser B. Regulation of muscarinic acetylcholine receptor signaling. Pharmacol Ther. 2003;98:197-220.
15. May L.T., Avlani V.A., Langmead C.J., et al. Structure-function studies of allosteric agonism at M2 muscarinic acetylcholine receptors. Mol Pharmacol. 2007;72:463-476.
16. Zhiguo Wang HSaHW. Functional M3 muscarinic acetylcholine receptors in mammalian hearts. Br J Pharmacol. 2004;142:395-408.
17. Keam S.J.K.G. Tiotropium bromide. A review of its use as maintenance therapy in patients with COPD. Treat Respir Med. 2004;3:247-268.
18. Mundy C., Kirkpatrick P. Tiotropium bromide. Nat Rev Drug Discov. 2004;3:643-644.
19. van Noord J.A., Bantje T.A., Eland M.E., et al. A randomised controlled comparison of tiotropium and ipratropium in the treatment of chronic obstructive pulmonary disease. Thorax. 2000;55:289-294.
20. Barcia E., Reyes R., Luz Azuara M., et al. Compatibility of haloperidol and hyoscine-N-butyl bromide in mixtures for subcutaneous infusion to cancer patients in palliative care. Support Care Cancer. 2003;11:107-113.
21. Susanne Lindqvist J.H., Sharp P., Johns N., et al. The colon-selective spasmolytic otilonium bromide inhibits muscarinic M3 receptor-coupled calcium signals in isolated human colonic crypts. Br J Pharmacol. 2002;137:1134-1142.
22. Villeneuve G., Cécyre D., Lejeune H., et al. Rigidified acetylcholine mimics: conformational requirements for binding to neuronal nicotinic receptors. Bioorg Med Chem Lett. 2003;13:3847-3851.
23. Xiu X., Puskar N.L., Shanata J.A.P., et al. Nicotine binding to brain receptors requires a strong cation-[π-π] interaction. Nature. 2009;458:534-537.
24. Lee C. Conformation, action, and mechanism of action of neuromuscular blocking muscle relaxants. Pharmacol Ther. 2003;98:143-169.
25. Vizi E.S., Lendvai B. Side effects of nondepolarizing muscle relaxants: Relationship to their antinicotinic and antimuscarinic actions. Pharmacol Ther. 1997;73:75-89.
26. Gyermek L. Development of ultra short-acting muscle relaxant agents: history, research strategies, and challenges. Med Res Rev. 2005;25:610-654.
27. Sluga M., Ummenhofer W., Studer W., et al. Rocuronium versus succinylcholine for rapid sequence induction of anesthesia and endotracheal intubation: a prospective, randomized trial in emergent cases. Anesth Analg. 2005;101:1356-1361.
28. Katzung B.G., editor. Basic and clinical pharmacology. New York, London: McGraw-Hill, 2001.
29. Chuliá S., Ivorra M.D., Lugnier C, Vila E., Noguera M.A., D’Ocon P. Mechanism of the cardiovascular activity of laudanosine: comparison with papaverine and other benzylisoquinolines. Br J Pharmacol. 1994;113:1377-1385.
30. Fodale V., Santamaria L.B. Laudanosine, an atracurium and cisatracurium metabolite. Eur J Anaesthesiol. 2002;19:466-473.
31. Chiodini F., Charpantier E., Muller D., et al. Blockade and activation of the human neuronal nicotinic acetylcholine receptors by atracurium and laudanosine. Anesthesiology. 2001;94:643-651.
32. Harald J. Sparr TMBaTF-B. Newer neuromuscular blocking agents 1–1 how do they compare with established agents. Drugs. 2001;61:919-942.
33. Views E. Succinylcholine: new insights into mechanisms of action of an old drug. Anesthesiology. 2006;104:633-634.