 CHAPTER 74 Neutrophil Disorders
CHAPTER 74 Neutrophil Disorders
Neutrophils play important roles in immunity and wound healing; their primary function is ingesting and killing pathogens. Neutrophil disorders can result from deficient cell numbers, defective chemotaxis, or defective function (Table 74-1). Patients with neutrophil disorders are susceptible to infections with Staphylococcus aureus, certain fungi, and gram-negative bacteria. Suggestive signs include mucous membrane infections (gingivitis), abscesses in the skin and viscera, lymphadenitis, poor wound healing, delayed umbilical cord separation, or absence of pus. Neutrophils develop in the bone marrow from hematopoietic stem cells by the action of several colony-stimulating factors, including stem cell factor, granulocyte-monocyte colony-stimulating factor (GM-CSF), granulocyte colony-stimulating factor (G-CSF), and interleukin-3 (IL-3). On leaving the bone marrow, mature neutrophils are found in the circulation or reside in the marginating pool. Chemotactic factors, including the complement fragment C5a, IL-8, and bacterial formulated peptides, mobilize neutrophils to enter tissues and sites of infections. Adhesion molecules are necessary for neutrophils to roll and adhere to vascular endothelium and extravasate from the blood into sites of infection, where they encounter, phagocytose, and kill pathogens, especially those coated by complement or antibodies. Neutrophils kill ingested pathogens using granular enzymes or by activation of oxygen radicals.
TABLE 74-1 Phagocytic Disorders
| Name | Defect | Comment |
|---|---|---|
| Chronic granulomatous disease | Bactericidal | X-linked recessive (66%), autosomal recessive (33%); eczema, osteomyelitis, granulomas, abscesses caused by Staphylococcus aureus, Burkholderia cepacia, Aspergillus fumigatus |
| Chédiak-Higashi syndrome (1q42I-44) | Bactericidal plus chemotaxis; poor natural killer function | Autosomal recessive; oculocutaneous albinism, neuropathy, giant neutrophilic cytoplasmic inclusions; malignancy, neutropenia |
| Hyperimmunoglobulin E (Job syndrome) | Chemotaxis, opsonization | Autosomal dominant (STAT3 mutation), autosomal recessive (TYK2 mutation), eczema, staphylococcal abscesses, granulocyte and monocyte chemotaxis affected |
| Myeloperoxidase deficiency | Bactericidal, fungicidal | Reduced chemiluminescence; autosomal recessive (1:4000); persistent candidiasis in diabetic patients |
| Glucose-6-phosphate dehydrogenase deficiency | Bactericidal | Phenotypically similar to chronic granulomatous disease |
| Burns, malnutrition | Bactericidal plus chemotaxis | Reversible defects |
| Lazy leukocyte syndrome | Chemotaxis | Normal bone marrow cells but poor migration; granulocytopenia |
| Leukocyte adhesion deficiency; CD18 deficiency (21q22.3) | Adherence, chemotaxis, phagocytosis; reduced lymphocyte cytotoxicity | Delayed separation or infection of umbilical cord; lethal bacterial infections without pus; autosomal recessive; neutrophilia; deficiency of LFA-1, Mac-1, CR3 |
| Shwachman-Diamond syndrome | Chemotaxis, neutropenia | Pancreatic insufficiency, metaphyseal chondrodysplasia; autosomal recessive |
ETIOLOGY AND CLINICAL MANIFESTATIONS
Disorders of Neutrophil Numbers
The normal neutrophil count varies with age. Neutropenia may be caused by decreased marrow production or peripheral neutrophil destruction and is defined as an absolute neutrophil count (ANC) less than 1500/mm3 for white children 1 year of age or older. African-American children normally have lower total white blood cell and neutrophil counts. The effect of neutropenia depends on its severity. The susceptibility to infection is minimally increased until the ANC is less than 1000/mm3. Most patients do well with an ANC greater than 500/mm3. At these levels, localized infections are more common than generalized bacteremia. Serious bacterial infections are more common with an ANC less than 200/mm3. In the absence of an adequate neutrophil count, migration of neutrophils to areas of damage in the skin and mucous membrane is delayed. Neutropenia may be congenital or acquired (Table 74-2) and may be associated with specific diseases, especially infections (Table 74-3), or result from drug reactions (Table 74-4). The major types of infections associated with neutropenia are cellulitis, pharyngitis, gingivitis, lymphadenitis, abscesses (cutaneous or perianal), enteritis (typhlitis), and pneumonia. The sites of the infection usually are colonized heavily with normal bacterial flora that becomes invasive in the presence of neutropenia.
TABLE 74-2 Mechanisms of Neutropenia
ABNORMAL BONE MARROW
Marrow Injury
Maturation Defects
PERIPHERAL CIRCULATION
Pseudoneutropenia: Shift to Bone Marrow
Intravascular
EXTRAVASCULAR MECHANISMS
Adapted from Moore JO, Oritel TL, Rosse WF: Disorders of granulocytes and monocytes. In Andreoli TE, Bennett JC, Carpenter CCJ, Plum F, editors: Cecil Essentials of Medicine, 4th ed, Philadelphia, 1997, Saunders.
There are several forms of congenital neutropenia. Severe congenital neutropenia (Kostmann syndrome) is an autosomal recessive disorder and may present in infancy. In severe congenital neutropenia, myeloid cells fail to mature beyond the early stages of the promyelocyte. The peripheral blood may show an impressive monocytosis. The genetic defect in Kostmann syndrome is a mutation in the gene for HAX-1, a protein found in mitochondria, which protects myeloid cells from apoptosis. Endogenous G-CSF levels are increased; nevertheless, exogenous G-CSF produces a rise in the neutrophil count, improving the care of these children. Acute myeloid leukemia has developed in a few patients who have survived into adolescence. Stem cell transplantation may be curative.
Severe congenital neutropenia, that may be either persistent or cyclic, also is a component of Shwachman-Diamond syndrome, an autosomal recessive syndrome of pancreatic insufficiency accompanying bone marrow dysfunction. This is a panmyeloid disorder in which neutropenia is the most prominent manifestation. Patients may have all the common complications of neutropenia, including gingivitis, which may be severe and lead to serious oral infections, alveolar bone destruction, and loss of teeth at an early age. Metaphyseal dysostosis and dwarfism may occur. Patients usually respond to G-CSF.
Other congenital neutropenias caused by deficient neutrophil production vary in severity and are poorly characterized. A gain of function mutation in the Wiskott-Aldrich syndrome protein has also been associated with an X-linked form of severe congenital neutropenia. The Wiskott-Aldrich syndrome protein demonstrates how mutations in same gene, albeit at different locations, can result in different diseases: Wiskott-Aldrich syndrome, X-linked thrombocytopenia, and X-linked congenital neutropenia. Benign congenital neutropenia is a functional diagnosis for patients with significant neutropenia in whom major infectious complications do not develop. Many patients whose ANC ranges from 100 to 500/mm3 have an increased frequency of infections, particularly respiratory infections, but the major problem is the slow resolution of infections that develop. These disorders may be sporadic or familial and, in some instances, are transmitted as an autosomal dominant disorder. Severe congenital neutropenia may be associated with SCID in reticular dysgenesis, a disorder of hematopoietic stem cells affecting all bone marrow lineages.
Cyclic neutropenia is a stem cell disorder in which all marrow elements cycle. It may be transmitted as an autosomal dominant, recessive, or sporadic disorder. Because of the short half-life of neutrophils in the blood (6 to 7 hours) compared with platelets (10 days) and red blood cells (120 days), neutropenia is the only clinically significant abnormality. The usual cycle is 21 days, with neutropenia lasting 4 to 6 days, accompanied by monocytosis and often by eosinophilia. Stomatitis or oral ulcers, pharyngitis, lymphadenopathy, fever, and cellulitis are present at the time of neutropenia. Severe, debilitating bone pain is common when the neutrophil count is low. G-CSF results in increasing neutrophil numbers and a shorter duration of neutropenia. Defects in ELA2, the gene encoding for neutrophil elastase, has been found in patients with sporadic, autosomal dominant, and autosomal recessive forms of cyclic neutropenia.
Isoimmune neutropenia occurs in neonates as the result of transplacental transfer of maternal antibodies to fetal neutrophil antigens. The mother is sensitized to specific neutrophil antigens on fetal leukocytes that are inherited from the father and are not present on maternal cells. Isoimmune neonatal neutropenia, similar to isoimmune anemia and thrombocytopenia, is a transient process (see Chapters 62 and 151). Cutaneous infections are common, and sepsis is rare. Early treatment of infection while the infant is neutropenic is the major goal of therapy. Intravenous immune globulin may decrease the duration of neutropenia.
Autoimmune neutropenia usually develops in children 5 to 24 months of age and often persists for prolonged periods. Neutrophil autoantibodies may be IgG, IgM, IgA, or a combination of these. Usually, the condition resolves in 6 months to 4 years. Clinical symptoms may dictate treatment. Although intravenous immune globulin and corticosteroids have been used, most patients respond to G-CSF. Autoimmune neutropenia rarely may be an early manifestation of systemic lupus erythematosus or rheumatoid arthritis.
Neutropenia is common in stressed neonates. Virtually any major illness, including asphyxia, may precipitate transient neonatal neutropenia. Maternal conditions, such as hypertension and eclampsia, can induce neonatal neutropenia. Significant neutropenia may develop in infected neonates, in part, due to depletion of bone marrow stores, which are far less extensive in neonates than adults.
Disorders of Neutrophil Migration
Neutrophils normally adhere to endothelium and migrate to areas of inflammation by the interaction of membrane proteins, called integrins and selectins, with endothelial cell adhesion molecules. A hallmark of defects in neutrophil migration is the absence of pus at sites of infection. In leukocyte adhesion deficiency type I (LAD-I), infants lacking the β2 integrin CD18 exhibit the condition early in infancy with failure of separation of the umbilical cord (often 2 months after birth) with attendant omphalitis and sepsis (see Table 74-1). The neutrophil count usually is greater than 20,000/mm3 because of failure of the neutrophils to adhere normally to vascular endothelium and to migrate out of blood to the tissues (Fig. 74-1). Cutaneous, respiratory, and mucosal infections occur. Children with this condition often have severe gingivitis. Sepsis usually leads to death in early childhood. This disorder is transmitted as an autosomal recessive trait. Stem cell transplantation may be lifesaving.
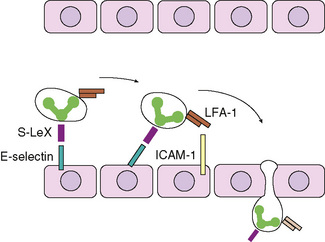
Figure 74-1 Schematic representation of neutrophil migration from the vascular space across the vascular endothelium into tissues. Neutrophils bind to selectin molecules on the surface of vascular endothelium via sialylated and fucosylated tetrasaccharides related to the sialylated Lewis X blood group (S-LeX) found on the surface of neutrophils. The bound neutrophils roll along the endothelium and become tightly bound by the interaction of the adhesion molecule leukocyte function–associated antigen type 1 (LFA-1) on the neutrophil and intercellular adhesion molecule type 1 (ICAM-1) on vascular endothelium, allowing neutrophils to move through the endothelium and into the tissue spaces.
(Adapted from Janeway CA, Travers P, Walport M, Capra JD: Immunobiology: The Immune System in Health and Disease, 4th ed, New York, 1999, Elsevier.)
LAD-II results from impairment of neutrophil rolling along the vascular wall, which is the first step in neutrophil migration into tissues and sites of infection. Rolling is mediated by sialylated and fucosylated tetrasaccharides related to the sialylated Lewis X blood group found on the surface of neutrophils, monocytes, and activated lymphocytes binding to selectin molecules on vascular endothelium (see Fig. 74-1). LAD-II results from a general defect in fucose metabolism leading to the absence of sialylated Lewis X blood group on the surface of neutrophils and other leukocytes.
A variety of conditions are associated with chemotactic defects in neutrophils. Hyper-IgE syndrome, caused by defects in the IL-6 signaling pathway, is characterized by eczema, hyperimmunoglobulinemia E, an extrinsic chemotactic defect, absent T-cell and B-cell responses to antigens, and recurrent cold boils (usually caused by S. aureus) that do not become markedly red or drain (see Chapter 73 and Table 74-1).
Disorders of Neutrophil Function
Defects in neutrophil function are relatively rare inherited disorders and tend to be associated with a marked susceptibility to bacterial and fungal infection. Chronic granulomatous disease (CGD) is a rare disorder of white blood cells that results from defective intracellular killing of bacteria and intracellular pathogens by neutrophils and macrophages because of an inability to activate the “respiratory burst,” the catalytic conversion of molecular oxygen to superoxide (O2−). Reduced nicotinamide adenine dinucleotide phosphate oxidase, the enzyme that catalyzes the respiratory burst, consists of four subunits, two subunits of cytochrome b558, gp91phox and p22phox, and two cytosolic oxidase components, p47phox and p67phox. Defects in any of these enzymes lead to an inability to kill catalase-positive pathogens such as S. aureus and enteric gram-negative bacteria and fungi (Aspergillus fumigatus, Candida albicans, Torulopsis glabrata). The gp91phox gene is located on chromosome Xp21.1 and is responsible for the more common form of the disease. Only one copy of the gene needs to be defective for the disease to be expressed in males. The other gene defects are inherited in an autosomal recessive manner. The chromosome locations for these genes are 16q24 for p22phox, 7q11.23 for p47phox, and 1q25 for p67phox. Glucose-6-phosphate dehydrogenase also is involved in the production of superoxide; severe forms of glucose-6-phosphate dehydrogenase deficiency also result in CGD. Patients characteristically have lymphadenopathy, hypergammaglobulinemia, hepatosplenomegaly, dermatitis, failure to thrive, anemia, chronic diarrhea, and abscesses. Infections occur in the lungs, the middle ear, gastrointestinal tract, skin, urinary tract, lymph nodes, liver, and bones. Granulomas may obstruct the pylorus or ureters.
Chédiak-Higashi syndrome, an abnormality of secondary granules, is an autosomal recessive disorder caused by a mutation in a cytoplasmic protein (CHS1) of unknown function (thought to be involved in organellar protein trafficking) resulting in fusion of the primary and secondary granules in neutrophils. Giant granules are present in many cells, including lymphocytes, platelets, and melanocytes. Patients usually have partial oculocutaneous albinism. The defect in Chédiak-Higashi syndrome results in defective neutrophil and natural killer cell function, leading to recurrent and sometimes fatal infections with streptococci and staphylococci. Most patients progress to an accelerated phase associated with Epstein-Barr virus infection and characterized by a lymphoproliferative syndrome with generalized lymphohistiocytic infiltrates, fever, jaundice, hepatomegaly, lymphadenopathy, and pancytopenia. The condition resembles familial erythrophagocytic lymphohistiocytosis and virus-associated hemophagocytic syndrome more than a malignant lymphoma.
LABORATORY DIAGNOSIS
The evaluation of a neutropenic child depends on clinical signs of infection, family and medication history, age of the patient, cyclic or persistent nature of the condition, signs of bone marrow infiltration (malignancy or storage disease), and evidence of involvement of other cell lines (Fig. 74-2). Neutropenia is confirmed by a complete blood count and differential. A bone marrow aspirate and biopsy may be necessary to determine whether the neutropenia is due to a failure of production in the bone marrow, infiltration of the bone marrow, or loss of neutrophils in the periphery. Antineutrophil antibodies help diagnose autoimmune neutropenia.
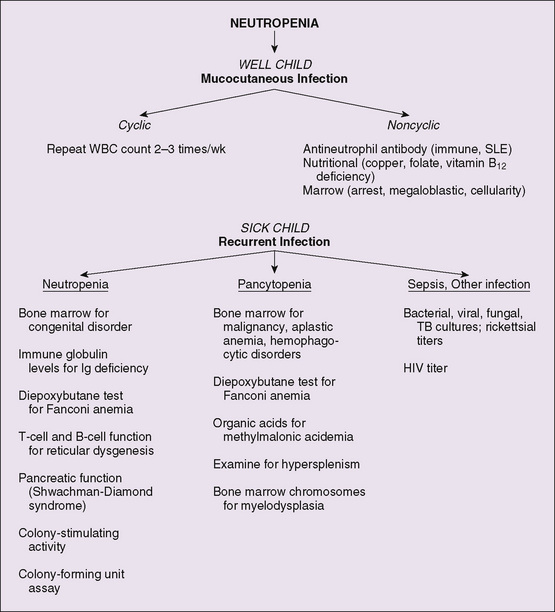
Figure 74-2 Algorithm for evaluation of neutropenia. SLE, systemic lupus erythematosus; TB, tuberculosis; WBC, white blood cell.
Neutrophil chemotactic defects can be excluded by the presence of neutrophils at the site of infection. The Rebuck skin window is a 4-hour in vivo test for neutrophil chemotaxis that is not performed routinely by most laboratories. Flow cytometry for the presence of adhesion molecules, such as CD18, can help diagnose leukocyte adhesion defects. Point mutations that affect the function of the adhesion molecule but do not alter antibody binding are missed using flow cytometry.
CGD can be diagnosed by the flow cytometry-based test using dihydrorhodamine 123 or the nitroblue tetrazolium test (see Chapter 72). Light microscopy of neutrophils for the presence of giant granules can help diagnose Chédiak-Higashi syndrome. The presence of an elevated IgE level, especially in association with poor antibody and T-cell responses to antigens, suggests hyper-IgE syndrome.
TREATMENT
Therapy for neutropenia depends on the underlying cause. Patients with severe bacterial infections require broad-spectrum antibiotics; the resolution of neutropenia is the most important prognostic factor. Most patients with severe congenital neutropenia or autoimmune neutropenia respond to therapy with G-CSF. Granulocyte transfusion should be reserved for life-threatening infection; even with transfusion, the results have been disappointing. Chronic mild neutropenia not associated with immunosuppression can be managed expectantly with prompt antimicrobial treatment of soft tissue infections, which usually are caused by S. aureus or group A streptococcus.
Frequent courses of antibiotics, including trimethoprim-sulfamethoxazole prophylaxis, and surgical débridement of infections are required in CGD. Because A. fumigatus can cause serious infection in patients with CGD, moldy hay, decomposing compost, and other nests of fungi must be avoided. The frequency of infection in CGD is lessened by treatment with recombinant interferon-γ administered subcutaneously three times a week. Stem cell transplantation (see Chapter 76) may be lifesaving in CGD, LAD-1, and Chédiak-Higashi syndrome.
PROGNOSIS AND PREVENTION
The prognosis depends on the particular defect. Milder and transient defects in neutrophil numbers have a better prognosis. Prolonged absence of neutrophils or their function has a poor prognosis, especially with the risk of bacterial and fungal sepsis. Treatment with interferon-γ has improved the prognosis of patients with CGD; however, successful stem cell transplantation is the only currently available mode of therapy that can reverse the poor prognosis of severe neutrophil defects. As in other genetic defects, prenatal diagnosis and genetic counseling are possible for all known gene mutations.