 CHAPTER 151 Hemostatic Disorders
CHAPTER 151 Hemostatic Disorders
NORMAL HEMOSTASIS
Hemostasis is the dynamic process of coagulation as it occurs in areas of vascular injury. This process involves the carefully modulated interaction of platelets, vascular wall, and procoagulant and anticoagulant proteins. After an injury to the vascular endothelium, subendothelial collagen induces a conformational change in von Willebrand factor (vWF), an adhesive protein to which platelets bind via their glycoprotein Ib receptor. After adhesion, platelets undergo activation and release numerous intracellular contents, including adenosine diphosphate (ADP). These activated platelets subsequently induce aggregation of additional platelets. Simultaneously, tissue factor, collagen, and other matrix proteins in the tissue activate the coagulation cascade, leading to the formation of the enzyme thrombin (Fig. 151-1). Thrombin has multiple effects on the coagulation mechanism, such as further aggregation of platelets, a positive feedback activation of factors 5 and 8, the conversion of fibrinogen to fibrin, and the activation of factor 11. A platelet plug forms, and bleeding ceases, usually within 3 to 7 minutes. The generation of thrombin leads to formation of a permanent clot by the activation of factor 13, which cross-links fibrin forming a stable thrombus. As a final element to this process, contractile elements within the platelet mediate clot retraction. Thrombin also contributes to the eventual limitation of clot size by binding to the protein thrombomodulin on intact endothelial cells, converting protein C into activated protein C. Thrombin contributes to the eventual lysis of the thrombus by activating plasminogen to plasmin. All of the hemostatic processes are closely interwoven and occur on biologic surfaces that mediate coagulation by bringing the critical players: platelets, endothelial cells, and subendothelium into close proximity with pro- and anticoagulant proteins.
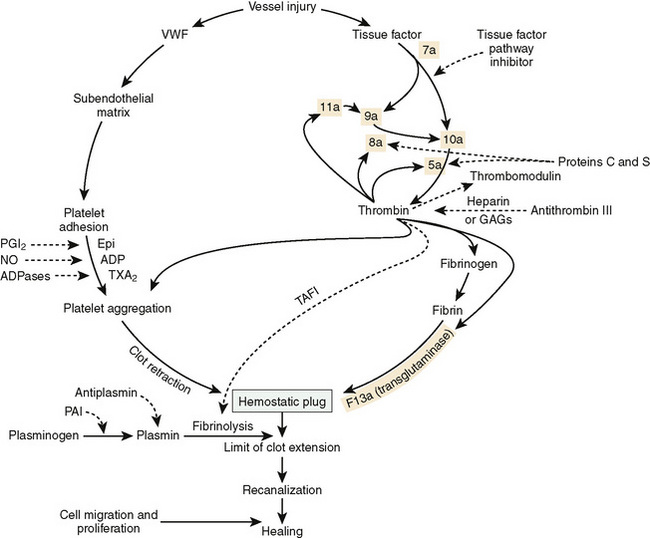
FIGURE 151-1 Diagram of the multiple interactions of the hemostatic mechanism. Solid lines indicate reactions that favor coagulation, and dashed lines indicate reactions that inhibit clotting. Epi, epinephrine; GAGs, glycosaminoglycans; NO, nitric oxide; PAI, plasminogen activator inhibitor; PGI2, prostaglandin I2 (prostacyclin); TAFI, thrombin-activated fibrinolytic inhibitor; TXA2, thromboxane A2; VWF, von Willebrand factor.
(Modified from, Scott JP, Montgomery RR: Hemorrhagic and thrombotic diseases. In Kliegman RM, Behrman RE, Jenson HB, Stanton BF [eds]: Nelson Textbook of Pediatrics, 18th ed. Philadelphia, WB Saunders, 2007, p 2062.)
Although it is convenient to think of coagulation as having intrinsic and extrinsic pathways, the reality is that these pathways are closely interactive and do not react independently (Fig. 151-2). For ease of use all coagulation factors are denoted using Arabic rather than Roman numerals to prevent misreading factor VII (7) as factor VIII (8). In vivo, factor 7 autocatalyzes to form small amounts of factor 7a. When tissue is injured, tissue factor is released and causes a burst of factor 7a generation. In vivo, tissue factor, in combination with calcium and factor 7a, activates factor 9 and factor 10. The major physiologic pathway is the activation of factor 9 by factor 7a, with eventual generation of thrombin. Thrombin then feeds back on factor 11, generating factor 11a and accelerating thrombin formation. This process explains why deficiency of factor 8 or factor 9 leads to severe bleeding disorders, whereas deficiency of factor 11 is usually mild, and deficiency of factor 12 is asymptomatic.
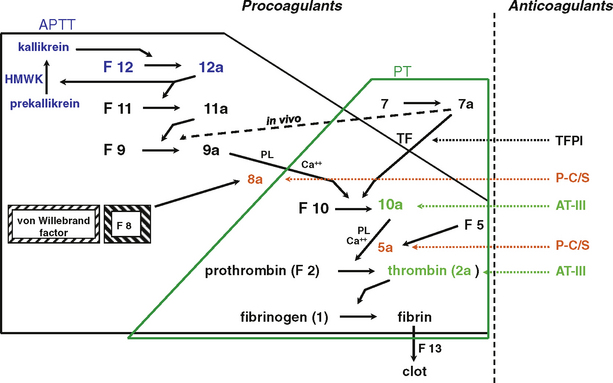
FIGURE 151-2 Simplified pathways of blood coagulation. The area inside the solid black line is the intrinsic pathway measured by the activated partial thromboplastin time (APTT). The area inside the green line is the extrinsic pathway, measured by the prothrombin time (PT). The area encompassed by both lines is the common pathway. AT-III, antithrombin III; F, factor; HMWK, high-molecular-weight kininogen; P-C/S, protein C/S; PL, phospholipid; TFPI, tissue factor pathway inhibitor.
(Modified from Scott JP, Montgomery RR: Hemorrhagic and thrombotic diseases. In Kliegman RM, Behrman RE, Jenson HB, Stanton, BF [eds]: Nelson Textbook of Pediatrics, 18th ed. Philadelphia, WB Saunders, 2007, p 2061.)
As the procoagulant proteins are activated, a series of inhibitory factors serve to tightly regulate the activation of coagulation. Antithrombin III inactivates thrombin and factors 10a, 9a, and 11a. The protein C and protein S system inactivates the activated factors 5 and 8, which are cofactors localized in the “tenase” and “prothrombinase” complexes. The tissue factor pathway inhibitor, an anticoagulant protein, limits the activation of the coagulation cascade by factor 7a and factor 10a. Fibrinolysis is initiated by the action of tissue plasminogen activator on plasminogen, producing plasmin, the active enzyme that degrades fibrin into split products. Fibrinolysis eventually dissolves the clot and allows normal flow to resume. Deficiencies of anticoagulant proteins may lead to thrombosis.
DEVELOPMENTAL HEMOSTASIS
In the fetus, fibrinogen, factor 5, factor 8, and platelets approach normal levels during the second trimester. Levels of other clotting factors and anticoagulant proteins increase gradually throughout gestation. The premature infant is simultaneously at increased risk of bleeding or clotting complications that are exacerbated by many of the medical interventions needed for care and monitoring, especially indwelling arterial or venous catheters. Most children attain normal levels of procoagulant and anticoagulant proteins by 1 year of age, although levels of protein C lag and normalize in adolescence.
HEMOSTATIC DISORDERS
Etiology and Epidemiology
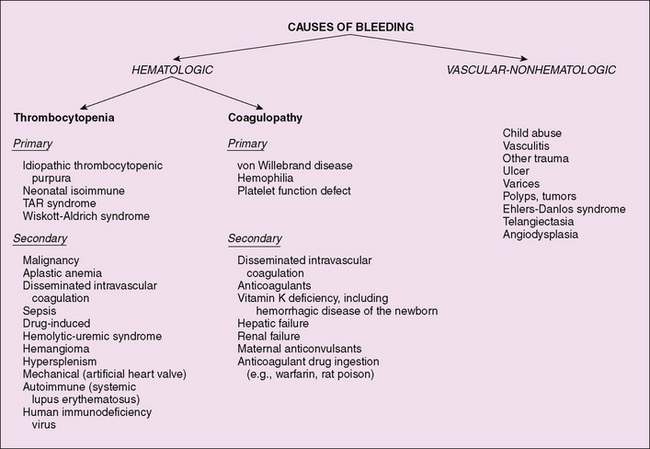
A detailed family history is crucial for bleeding and thrombotic disorders. Hemophilia is X-linked, and almost all affected children are boys. von Willebrand disease usually is inherited in an autosomal dominant fashion. In the investigation of thrombotic disorders, a personal or family history of blood clots in the legs or lungs, early-onset stroke, or heart attack suggests a hereditary predisposition to thrombosis. The causes of bleeding may be hematologic in origin or due to vascular, nonhematologic causes (Fig. 151-3). Thrombotic disorders can be congenital or acquired (Table 151-1) and frequently present after an initial event (central catheter, trauma, malignancy, infection, pregnancy, treatment with estrogens) provides a nidus for clot formation or a procoagulant stimulus.
Clinical Manifestations
Patients with hemostatic disorders may have complaints of either bleeding or clotting. Age at onset of bleeding indicates whether the problem is congenital or acquired. The sites of bleeding (mucocutaneous or deep) and degree of trauma (spontaneous or significant) required to induce injury suggest the type and severity of the disorder. Certain medications (aspirin and valproic acid) are known to exacerbate preexisting bleeding disorders by interfering with platelet function.
The physical examination should characterize the presence of skin or mucous membrane bleeding and deeper sites of hemorrhage into the muscles and joints or internal bleeding sites. The term petechia refers to a nonblanching lesion less than 2 mm in size. Purpura is a group of adjoining petechiae, ecchymoses (bruises) are isolated lesions larger than petechiae, and hematomas are raised, palpable ecchymoses.
Because systemic disorders may induce either hemorrhagic or thrombotic disorders, the physical examination should search for manifestations of an underlying disease, lymphadenopathy, hepatosplenomegaly, vasculitic rash, or chronic hepatic or renal disease. Deep venous thrombi may cause warm, swollen (distended), tender, purplish discolored extremities or organs or no findings. Arterial clots cause acute, painful, pale, and poorly perfused extremities. Arterial thrombi of the internal organs present with signs and symptoms of infarction.
Laboratory Testing
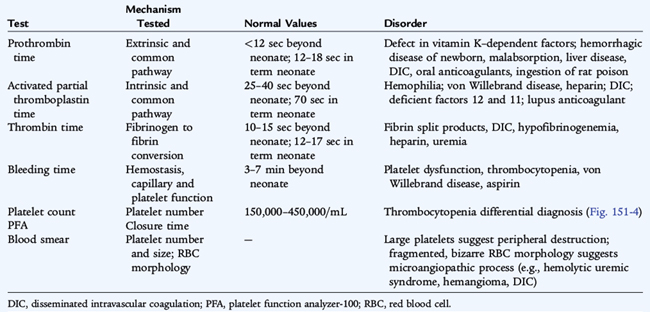
Screening laboratory studies for bleeding patients include a platelet count, prothrombin time, partial thromboplastin time, fibrinogen, and bleeding time or other screening test of platelet function. Many laboratories have adopted the platelet function analyzer (PFA) to replace the bleeding time as a screening test for platelet function abnormalities and von Willebrand disease. The PFA has variable sensitivity and specificity for common bleeding disorders. No single laboratory test can screen for all bleeding disorders. The findings on screening tests for bleeding vary with the specific disorder (Table 151-2).
Differential Diagnosis
Disorders of Platelets
Platelet counts less than 150,000/mm3 constitute thrombocytopenia. Mucocutaneous bleeding is the hallmark of platelet disorders, including thrombocytopenia. The risk of bleeding correlates imperfectly with the platelet count. Children with platelet counts greater than 80,000/mm3 are able to withstand all but the most extreme hemostatic challenges, such as surgery or major trauma. Children with platelet counts less than 20,000/mm3 are at risk for spontaneous bleeding. These generalizations are modified by factors such as the age of the platelets (young, large platelets usually function better than old ones) and the presence of inhibitors of platelet function, such as antibodies, drugs (especially aspirin), fibrin degradation products, and toxins formed in the presence of hepatic or renal disease. The size of platelets is routinely measured as the mean platelet volume (MPV). The etiology of thrombocytopenia (Fig. 151-4) may be organized into three mechanisms:
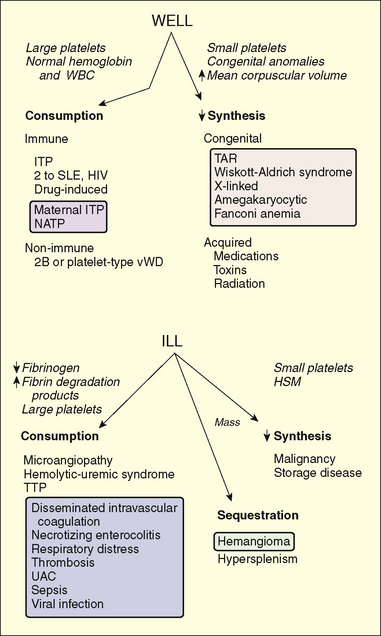
FIGURE 151-4 Differential diagnosis of childhood thrombocytopenic syndromes. The syndromes initially are separated by their clinical appearances. Clues leading to the diagnosis are presented in italics. The mechanisms and common disorders leading to these findings are shown in the lower part of the figure. Disorders that commonly affect neonates are listed in the shaded boxes. HSM, hepatosplenomegaly; ITP, idiopathic immune thrombocytopenic purpura; NATP, neonatal alloimmune thrombocytopenic purpura; SLE, systemic lupus erythematosus; TAR, thrombocytopenia with absence of radius (syndrome); TTP, thrombotic thrombocytopenic purpura; UAC, umbilical artery catheter; WBC, white blood cell.
(From Scott JP: Bleeding and thrombosis. In Kliegman RM, Greenbaum LA, Lye PS [eds]: Practical Strategies in Pediatric Diagnosis and Therapy. Philadelphia, WB Saunders, 2004, p 920.)
Sequestration Thrombocytopenia Resulting from Decreased Platelet Production
Primary disorders of megakaryopoiesis (platelet production) are rare in childhood, other than as part of an aplastic syndrome. Thrombocytopenia with absent radii syndrome is characterized by severe thrombocytopenia in association with orthopedic abnormalities, especially of the upper extremity. The thrombocytopenia usually improves over time. Amegakaryocytic thrombocytopenia presents at birth or shortly thereafter with findings of severe thrombocytopenia, but no other congenital anomalies. The marrow is devoid of megakaryocytes and usually progresses to aplasia of all hematopoietic cell lines.
Acquired thrombocytopenia as a result of decreased production is rarely an isolated finding. It is seen more often in the context of pancytopenia resulting from bone marrow failure caused by infiltrative or aplastic processes. Certain chemotherapeutic agents may affect megakaryocytes selectively more than other marrow elements. Cyanotic congenital heart disease with polycythemia often is associated with thrombocytopenia, but this is rarely severe or associated with significant clinical bleeding. Congenital (TORCH [toxoplasmosis, other agents, rubella, cytomegalovirus, herpes simplex]) and acquired viral infections (human immunodeficiency virus [HIV], Epstein-Barr virus, and measles) and some drugs (anticonvulsants, antibiotics, cytotoxic agents, heparin, and quinidine) may induce thrombocytopenia. Postnatal infections and drug reactions usually cause transient thrombocytopenia, whereas congenital infections may produce prolonged suppression of bone marrow function.
Thrombocytopenia Resulting from Peripheral Destruction
Etiology
In a child who appears well, immune-mediated mechanisms are the most common cause of thrombocytopenia resulting from rapid peripheral destruction of antibody-coated platelets by reticuloendothelial cells. Neonatal alloimmune thrombocytopenic purpura (NATP) occurs as a result of sensitization of the mother to antigens present on fetal platelets. Antibodies cross the placenta and attack the fetal platelet (see Chapter 59). Many platelet alloantigens have been identified and sequenced, permitting prenatal diagnosis of the condition in an at-risk fetus. Mothers with idiopathic thrombocytopenic purpura (maternal ITP) or with a history of ITP may have passive transfer of antiplatelet antibodies that bind to fetal platelets, with resultant neonatal thrombocytopenia (see Chapter 59). The maternal platelet count is sometimes a useful indicator of the probability that the infant will be affected. If the mother has had a splenectomy, her platelet count may be normal and is a poor predictor of the likelihood of severe neonatal thrombocytopenia because maternal antibody triggers destruction of the fetal platelets in the fetal spleen.
Clinical Manifestations
The infant with NATP is at risk for intracranial hemorrhage in utero and during the immediate delivery process. In ITP, the greatest risk seems to be present during passage through the birth canal, during which molding of the head may induce intracranial hemorrhage. Fetal scalp sampling or percutaneous umbilical blood sampling may be performed to measure the fetal platelet count.
Treatment
Administration of intravenous (IV) immunoglobulin before delivery increases fetal platelet counts and may alleviate thrombocytopenia in the infant in cases of NATP and ITP. Delivery by cesarean section is recommended to prevent central nervous system (CNS) bleeding (see Chapter 59). Neonates with severe thrombocytopenia (platelet counts < 20,000/mm3) may be treated with IV immunoglobulin or corticosteroids or both until the period of thrombocytopenia remits. If necessary, infants with NATP may receive washed maternal platelets.
Idiopathic Thrombocytopenic Purpura
Etiology
Autoimmune thrombocytopenic purpura of childhood (childhood ITP) is a common disorder in children that usually follows an acute viral infection. Childhood ITP is caused by an antibody (IgG or IgM) that binds to the platelet membrane. The condition results in Fc receptor–mediated splenic destruction of antibody-coated platelets. Rarely, ITP may be the presenting symptom of an autoimmune disease, such as systemic lupus erythematosus.
Clinical Manifestations
Young children typically exhibit ITP 1 to 4 weeks after viral illness, with abrupt onset of petechiae, purpura, and epistaxis. The thrombocytopenia usually is severe. Significant adenopathy or hepatosplenomegaly is unusual, and the red blood cell (RBC) and white blood cell (WBC) counts are normal.
Diagnosis
The diagnosis of ITP usually is based on clinical presentation and the platelet count and does not often require a bone marrow examination. If atypical findings are noted, however, marrow examination is indicated to rule out an infiltrative disorder (leukemia) or an aplastic process (aplastic anemia). In ITP, an examination of the bone marrow reveals increased megakaryocytes and normal erythroid and myeloid elements.
Treatment and Prognosis
Therapy is seldom indicated for platelet counts greater than 30,000/mm3. Therapy does not affect the long-term outcome of ITP but is intended to increase the platelet count acutely. For moderate and severe clinical bleeding with severe thrombocytopenia (platelet count < 10,000/mm3), therapeutic options include prednisone, 2 to 4 mg/kg/24 hours for 2 weeks; IV immunoglobulin, 1 g/kg/24 hours for 1 to 2 days; or IV anti-D (WinRho SD), 50 to 75 μg/kg/dose for Rh-positive individuals. All of these approaches seem to decrease the rate of clearance of sensitized platelets, rather than decreasing production of antibody. The optimal choice for therapy (if any) is controversial. Splenectomy is indicated in acute ITP only for life-threatening bleeding. Approximately 80% of children have a spontaneous resolution of ITP within 6 months after diagnosis. Serious bleeding, especially intracranial bleeding, occurs in less than 1% of patients with ITP. There is no evidence that treatment prevents intracranial bleeding.
ITP that persists for 6 to 12 months is classified as chronic ITP. Repeated treatments with IV immunoglobulin, IV anti-D, or high-dose pulse steroids are effective in delaying the need for splenectomy. Secondary causes of chronic ITP, especially systemic lupus erythematosus and HIV infection, should be ruled out. Splenectomy induces a remission in 70% to 80% of childhood chronic ITP cases. The risks of splenectomy (surgery, sepsis from encapsulated bacteria, pulmonary hypertension) must be weighed against the risk of severe bleeding.
Other Disorders
Wiskott-Aldrich syndrome is an X-linked disorder characterized by hypogammaglobinemia, eczema, and thrombocytopenia caused by a molecular defect in a cytoskeletal protein common to lymphocytes and platelets (see Chapter 74). Small platelets are seen on a peripheral blood smear. Nevertheless, thrombocytopenia often is improved by splenectomy. Hematopoietic stem cell transplantation cures the immunodeficiency and thrombocytopenia. Familial X-linked thrombocytopenia can be seen as a variant of Wiskott-Aldrich syndrome or a mutation in the GATA1 gene. Autosomal macrothrombocytopenia is due to deletions in chromosomes 22q11 or mutations in 22q12.
Thrombotic microangiopathy causes thrombocytopenia, anemia secondary to intravascular RBC destruction, and, in some cases, depletion of clotting factors. Children with thrombotic microangiopathy usually are quite ill. In a child with DIC, the deposition of fibrin strands within the vasculature and activation of thrombin and plasmin result in a wide-ranging hemostatic disorder with activation and clearance of platelets. Hemolytic uremic syndrome occurs as a result of exposure to a toxin that induces endothelial injury, fibrin deposition, and platelet activation and clearance (see Chapter 164). In thrombotic thrombocytopenic purpura, platelet consumption, precipitated by a congenital or acquired deficiency of a metalloproteinase that cleaves von Willebrand factor, seems to be the primary process, with a modest deposition of fibrin and RBC destruction.
Disorders of Platelet Function
Etiology
Primary disorders of platelet function may involve receptors on platelet membranes for adhesive proteins. Deficiency of glycoprotein Ib complex (vWF receptor) causes Bernard-Soulier syndrome. A deficiency of glycoprotein IIb-IIIa (the fibrinogen receptor) causes Glanzmann thrombasthenia. Mild abnormalities of platelet aggregation and release, detectable by platelet aggregometry, are far more common. Secondary disorders caused by toxins and drugs (uremia, valproic acid, aspirin, nonsteroidal anti-inflammatory drugs, and infections) may cause a broad spectrum of platelet dysfunction.
Clinical Manifestations
Disorders of platelet function present with mucocutaneous bleeding and a prolonged bleeding time or long PFA closure time and may be primary or secondary. The bleeding time is an insensitive screen for mild and moderate platelet function disorders, but is usually prolonged in severe platelet function disorders, such as Bernard-Soulier syndrome or Glanzmann thrombasthenia.
Disorders of Clotting Factors
Etiology
Hereditary deficiencies of most procoagulant proteins lead to bleeding. The genes for factor 8 and factor 9 are on the X chromosome, whereas virtually all the other clotting factors are coded on autosomal chromosomes. Factor 8 and factor 9 deficiencies are the most common severe inherited bleeding disorders. von Willebrand disease is the most common congenital bleeding disorder. Of the procoagulant proteins, low levels of the so-called contact factors (prekallikrein, high-molecular-weight kininogen, and Hageman factor [factor 12]) cause a prolonged activated partial thromboplastin time (aPTT) but are not associated with a predisposition to bleeding.
Hemophilia
Etiology
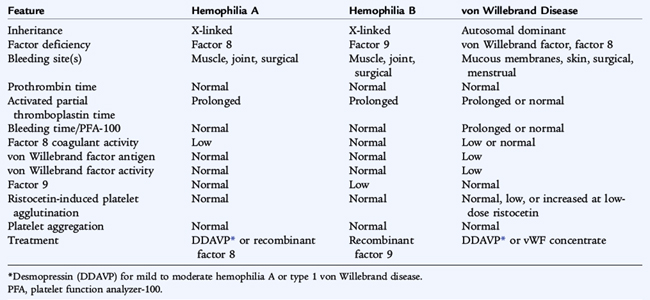
Hemophilia A (factor 8 deficiency) occurs in 1 in 5000 males. Hemophilia B (factor 9 deficiency) occurs in approximately 1 in 25,000. Clinically the two disorders are indistinguishable other than by their therapy (Table 151-3). The lack of factor 8 or factor 9 delays the generation of thrombin, which is crucial to forming a normal, functional fibrin clot and solidifying the platelet plug that has formed in areas of vascular injury. The severity of the disorder is determined by the degree of clotting factor deficiency.
Clinical Manifestations
Patients with less than 1% (severe hemophilia) factor 8 or factor 9 may have spontaneous bleeding or bleeding with minor trauma. Patients with 1% to 5% (moderate hemophilia) factor 8 or factor 9 usually require moderate trauma to induce bleeding episodes. In mild hemophilia (>5% factor 8 or factor 9), significant trauma is necessary to induce bleeding; spontaneous bleeding does not occur. Mild hemophilia may go undiagnosed for many years, whereas severe hemophilia manifests in infancy when the child reaches the toddler stage. In severe hemophilia, spontaneous bleeding occurs, usually in the muscles or joints (hemarthroses).
Laboratory Studies
The diagnosis of hemophilia is based on a prolonged activated partial thromboplastin time (aPTT). In the aPTT, a surface-active agent activates the intrinsic system of coagulation, of which factors 8 and 9 are crucial components. In factor 8 or factor 9 deficiency, the aPTT is quite prolonged, but should correct to normal when the patient’s plasma is mixed 1:1 with normal plasma. When an abnormal aPTT is obtained, specific factor assays are needed to make a precise diagnosis (see Table 151-2) to determine the appropriate factor replacement therapy. Prenatal diagnosis and carrier diagnosis are possible using molecular techniques.
Treatment
Early, appropriate replacement therapy is the hallmark of excellent hemophilia care. Acute bleeding episodes are best treated in the home when the patient has attained the appropriate age, and the parents have learned home treatment. Bleeding associated with surgery, trauma, or dental extraction often can be anticipated, and excessive bleeding can be prevented with appropriate replacement therapy. For life-threatening bleeding, levels of 80% to 100% of normal factor 8 or factor 9 are necessary. For mild to moderate bleeding episodes (hemarthroses), a 40% level for factor 8 or a 30 to 40% level for factor 9 is appropriate. The dose can be calculated using the knowledge that 1 U/kg body weight of factor 8 increases the plasma level 2%, whereas 1.5 U/kg of recombinant factor 9 increases the plasma level 1%:
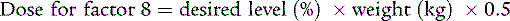
or
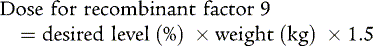
Desmopressin acetate is a synthetic vasopressin analog with minimal vasopressor effect. Desmopressin triples or quadruples the initial factor 8 level of a patient with mild or moderate (not severe) hemophilia A, but has no effect on factor 9 levels. When adequate hemostatic levels can be attained, desmopressin is the treatment of choice for individuals with mild and moderate hemophilia A. Aminocaproic acid is an inhibitor of fibrinolysis that may be useful for oral bleeding.
Patients treated with older factor 8 or 9 concentrates derived from large pools of plasma donors were at high risk for hepatitis B, C, and D and HIV. Recombinant factor 8 and factor 9 concentrates are safe from virally transmitted illnesses. Older patients who were exposed to factor concentrates, cryoprecipitate, or both before modern viral testing have a high prevalence of HIV infection. Acquired immunodeficiency syndrome (AIDS) is the most common cause of death in older hemophilia patients. Many older patients also have chronic hepatitis C.
Inhibitors are IgG antibodies directed against transfused factor 8 or factor 9 in congenitally deficient patients. Inhibitors arise in 15% of severe factor 8 hemophiliacs and less commonly in factor 9 hemophiliacs. They may be high or low titer and show an anamnestic response to treatment. The treatment of bleeding patients with an inhibitor is difficult. For low titer inhibitors, options include continuous factor 8 infusions. For high titer inhibitors, it is usually necessary to administer a product that bypasses the inhibitor, preferably recombinant factor 7a. Activated prothrombin complex concentrates, used in the past to treat inhibitor patients, paradoxically increased the risks of thrombosis, resulting in fatal complications, such as myocardial infarction. For long-term treatment of inhibitor patients, induction of immune tolerance by repeated infusion of the deficient factor with or without immunosuppression may be beneficial.
Prevention of long-term crippling orthopedic abnormalities is a major goal of hemophilic care. Early institution of factor replacement and continuous prophylaxis beginning in early childhood should prevent the chronic joint disease associated with hemophilia.
von Willebrand disease
Etiology
Von Willebrand disease is a common disorder (found in 1% of the population) caused by a deficiency of vWF. vWF is an adhesive protein that serves two functions: vWF acts as a bridge between subendothelial collagen and platelets and it binds and protects circulating factor 8 from rapid clearance from circulation. Von Willebrand disease usually is inherited as an autosomal dominant trait and rarely as an autosomal recessive trait. vWF may be either quantitatively deficient (partial = type 1 or absolute = type 3) or qualitatively abnormal (type 2 = dysproteinemia). Approximately 80% of patients with von Willebrand disease have classic (type 1) disease (i.e., a mild to moderate deficiency of vWF). Several other subtypes are clinically important, each requiring different therapy.
Clinical Manifestations
Mucocutaneous bleeding, epistaxis, gingival bleeding, cutaneous bruising, and menorrhagia occur in patients with von Willebrand disease. In severe disease, factor 8 deficiency may be profound, and the patient may also have manifestations similar to hemophilia A (hemarthrosis). Findings in classic von Willebrand disease differ from findings in hemophilia A and B (see Table 151-3).
Laboratory Testing
vWF testing involves measurement of the amount of protein, usually measured immunologically as the vWF antigen (vWF:Ag). vWF activity (vWF:Act) is measured functionally in the ristocetin cofactor assay (vWFR:Co), which uses the antibiotic ristocetin to induce vWF to bind to platelets.
Treatment
The treatment of von Willebrand disease depends on the severity of bleeding. Desmopressin is the treatment of choice for most bleeding episodes in patients with type 1 disease and some patients with type 2 disease. When high levels of vWF are needed but cannot be achieved satisfactorily with desmopressin, treatment with a virally attenuated, vWF-containing concentrate (Humate P) may be appropriate. The dosage can be calculated as for factor 8 in hemophilia. Cryoprecipitate should not be used because it is not virally attenuated. Hepatitis B vaccine should be given before the patient is exposed to plasma-derived products. As in all bleeding disorders, aspirin should be avoided.
Disseminated Intravascular Coagulation
Etiology
Disseminated intravascular coagulation (DIC) is a disorder in which widespread activation of the coagulation mechanism is usually associated with shock. Bleeding and clotting manifestations may be present. Normal hemostasis is a balance between hemorrhage and thrombosis. In DIC, this balance is altered by the severe illness, so the patient has activation of coagulation mediated by thrombin and fibrinolysis mediated by plasmin. Coagulation factors—especially platelets, fibrinogen, and factors 2, 5, and 8—are consumed, as are the anticoagulant proteins, especially antithrombin, protein C, and plasminogen. Endothelial injury, tissue release of thromboplastic procoagulant factors, or, rarely, exogenous factors (snake venoms) directly activate the coagulation mechanism (Table 151-4).
TABLE 151-4 Causes of Disseminated Intravascular Coagulation
INFECTIOUS
TISSUE INJURY
MALIGNANCY
VENOM OR TOXIN
MICROANGIOPATHIC DISORDERS
GASTROINTESTINAL DISORDERS
HEREDITARY THROMBOTIC DISORDERS
NEONATAL PERIOD
MISCELLANEOUS CONDITIONS/DISORDERS
From Scott JP: Bleeding and thrombosis. In Kliegman RM (ed): Practical Strategies in Pediatric Diagnosis and Therapy. Philadelphia, WB Saunders, 1996.
Clinical Manifestations
The diagnosis of DIC usually is suspected clinically and is confirmed by laboratory findings of a decline in platelets and fibrinogen associated with elevated prothrombin time, partial thromboplastin time, and elevated levels of D-dimer, formed when fibrinogen is clotted and then degraded by plasmin (Table 151-5). In some patients, DIC may evolve more slowly and there may be a degree of compensation. In a severely ill patient, the sudden occurrence of bleeding from a venipuncture or incision site, gastrointestinal or pulmonary hemorrhage, petechiae, or ecchymosis or evidence of peripheral gangrene or thrombosis suggests the diagnosis of DIC.
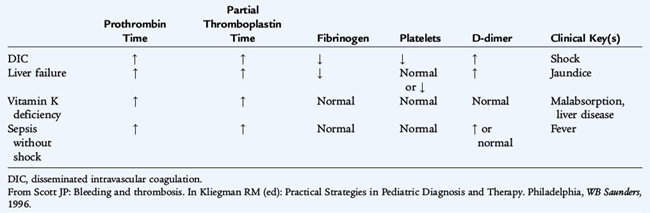
Treatment
The treatment of DIC is challenging. General guidelines include the following: treat the disorder inducing the DIC first; support the patient by correcting hypoxia, acidosis, and poor perfusion; and replace depleted blood-clotting factors, platelets, and anticoagulant proteins by transfusion. Heparin may be used to treat significant arterial or venous thrombotic disease unless sites of life-threatening bleeding coexist. Drotrecogin alfa (recombinant activated protein C) reduces mortality rate in adults with DIC and sepsis, but these results have not been confirmed in children.
Thrombosis
Etiology
A hereditary predisposition to thrombosis (see Table 151-1) may be caused by a deficiency of an anticoagulant protein (protein C or S, antithrombin, or plasminogen) (Fig. 151-5), by an abnormality of a procoagulant protein making it resistant to proteolysis by its respective inhibitor (factor 5 Leiden), by a mutation resulting in an increased level of a procoagulant protein (prothrombin 20210), or by damage to endothelial cells (homocysteinemia). Neonates with deficiency syndromes may be particularly vulnerable to thrombosis. Neonates with homozygous protein C deficiency present with purpura fulminans or thrombosis of the major arteries and veins or both. Many individuals with an inherited predisposition to thrombosis exhibit symptoms in adolescence or early adulthood. Protein C deficiency presenting in adulthood usually is inherited as an autosomal dominant trait, whereas the homozygous form usually is autosomal recessive. Protein S and antithrombin III deficiencies are inherited as autosomal dominant traits. Factor 5 Leiden is the most common hereditary cause of a predisposition to thrombosis, appearing in 3% to 5% of whites. Acquired antiphospholipid antibodies (anticardiolipin and lupus anticoagulant) also predispose to thrombosis.
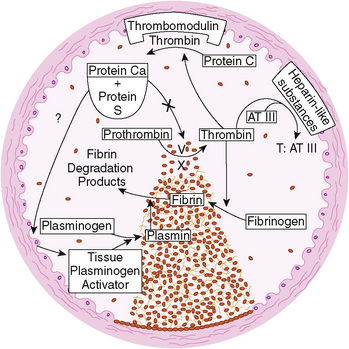
FIGURE 151-5 Formation of the hemostatic plug at the site of vascular injury. Three major physiologic anticoagulant mechanisms—antithrombin III (AT III), protein C, and the fibrinolytic system—are activated to limit clot formation to the site of damage and to prevent generalized thrombosis. T, thrombin.
(From Schafer A: The hypercoaguable state. Ann Intern Med 102:814–828, 1985.)
Clinical Manifestations
Neonates and adolescents are the most likely pediatric patients to present with thromboembolic disease. Indwelling catheters, vasculitis, sepsis, immobilization, nephrotic syndrome, coagulopathy, trauma, infection, surgery, inflammatory bowel disease, oral contraceptive agents, pregnancy, and abortion all predispose to thrombosis. The manifestations of pulmonary emboli vary from no findings to chest pain, diminished breath sounds, increased S2, cyanosis, tachypnea, and hypoxemia.
Diagnostic and Imaging Studies
Venous thrombosis can be detected noninvasively by ultrasound Doppler flow compression studies. The gold standard for diagnosis is the venogram. An abnormal (high probability) ventilation-perfusion scan or detection of an intravascular thrombus on helical computed tomography (CT) is diagnostic of pulmonary emboli. There are no appropriate screening studies for thrombotic disorders. Diagnosis of a congenital or acquired predisposition to thrombosis requires a battery of specific assays.
Treatment
Therapy of thrombotic disorders depends on the underlying condition and usually involves standard or low-molecular-weight heparin and then longer term anticoagulation with warfarin. Major vessel thrombosis or life-threatening thrombosis may necessitate treatment with fibrinolytic agents (recombinant tissue plasminogen activator). In newborns, inherited deficiency syndromes may present as emergencies and necessitate replacement with plasma, antithrombin III concentrates, or protein C concentrates.