 CHAPTER 173 Short Stature
CHAPTER 173 Short Stature
GROWTH
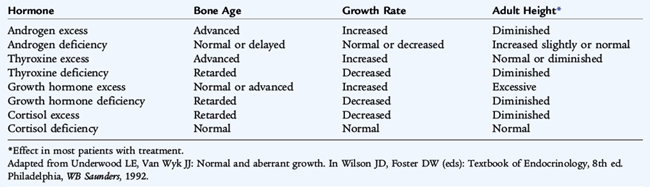
Normal growth is the final common pathway of many factors, including endocrine, environmental, nutritional, and genetic influences (see Chapter 5). A normal linear growth pattern is good evidence of overall health and can be considered a bioassay for the well-being of the whole child. The effects of certain hormones on growth and ultimate height are listed in Table 173-1. Just as various factors influence stature, stature itself influences psychological, social, and potentially economic well-being. Parental concern about the psychosocial consequences of abnormal stature often causes a family to seek medical attention. There is an increased bias toward heightism, particularly among males. Physicians must be sensitized to the overall context of a particular youngster’s attention to stature.
Growth Hormone Physiology
Growth hormone (GH) secretion is pulsatile, stimulated by hypothalamic GH-releasing factor (GRF) and inhibited by GH release inhibitory factor (somatostatin, SRIF), which interact with their individual receptors on the somatotrope in a noncompetitive manner. GH is also stimulated by ghrelin, produced in the stomach. GH circulates bound to a GH-binding protein (GHBP) which is the proteolytic product of the extracellular domain of the membrane-bound GH receptor; GHBP abundance reflects the abundance of GH receptors. GH has direct effects on tissue and also causes production and secretion of insulin-like growth factor-1 (IGF-1) in many tissues. IGF-1 is most closely associated with postnatal growth. GH stimulates IGF production in liver along with production of the acid-labile subunit (ALS) and the IGF-binding protein (IGF-BP3); this forms the complex that delivers IGF-1 to tissue. Serum concentrations of IGF-1 follow serum concentrations of GH. IGF-BP3 is measurable in clinical assays and is itself GH dependent, but less influenced by nutrition and age than is IGF-1; measuring IGF-1 and IGF-BP3 is useful in evaluating GH adequacy, particularly in infancy and early childhood. IGF-1 production is influenced by disease states such as malnutrition, chronic renal and liver disease, hypothyroidism, or obesity.
IGF-1 acts primarily as a paracrine and autocrine agent, so the IGF-1 measured in the peripheral circulation is far removed from the site of action and is an imperfect reflection of IGF-1 physiology. IGF-1 resembles insulin in structure, and the IGF-1 receptor resembles the insulin receptor, so cross-reaction of one agent, if present in excess, can cause physiologic effects usually attributed to the other agent. When IGF-1 attaches to its membrane-bound receptor, second messengers are stimulated to change the physiology of the cell and produce growth effects.
Measurement of Growth
The correct measurement of an infant’s length requires one adult to hold the infant’s head still and another adult to extend the feet with the soles perpendicular to the lower legs. A caliper-like device, such as an infantometer, or the movable plates on an infant scale are used so that the exact distance between the two calipers or plates can be determined. Marking the position of the head and feet of an infant lying on a sheet of paper on the examining table leads to inaccuracies and may miss true disorders of growth or create false concerns about a disorder of growth in a normal child. Accurate measurements of height, or length, and weight should be plotted on the Centers for Disease Control and Prevention growth charts for the timely diagnosis of growth disorders (http://www.cdc.gov/growthcharts/).
After 2 years of age, the height of a child should be measured in the standing position. Children measured in the standing position should be barefoot against a hard surface. A Harpenden stadiometer or equivalent device is optimal for the measurement of stature, but the flexible bars that extend upward on some health scales are unreliable and may be misleading. A decrease of roughly 1.25 cm in height measurement may occur when the child is measured in the standing position rather than in the lying position; many children who appear to be not growing are referred to a subspecialist, but all the only change is the position of the child at the time of measurement.
Measurement of the arm span is essential when the diagnoses of Marfan or Klinefelter syndrome, short-limbed dwarfism, or other dysmorphic conditions are considered. Arm span is measured as the distance between the tips of the fingers when the patient holds both arms outstretched horizontally while standing against a solid surface. The upper-to-lower segment ratio is the ratio of the upper segment (determined by subtraction of the measurement from the symphysis pubis to the floor [known as the lower segment] from the total height) to the lower segment. This ratio changes with age. A normal term infant has an upper-to-lower ratio of 1.7:1, a 1-year-old has a ratio of 1.4:1, and a 10-year-old has a ratio of 1:1. Conditions of hypogonadism, not commonly discerned or suspected until after the normal age for onset of puberty, lead to greatly decreased upper-to-lower ratio in an adult, whereas long-lasting and untreated hypothyroidism leads to a high upper-to-lower ratio in the child.
Endocrine Evaluation of Growth Hormone Secretion
GH, or somatotropin, is a 191–amino acid protein secreted by the pituitary gland under the control of GRF and SRIF (see Fig. 170-2). GH secretion is enhanced by α-adrenergic stimulation, hypoglycemia, starvation, exercise, early stages of sleep, and stress. GH secretion is inhibited by β-adrenergic stimulation and hyperglycemia. Because concentrations of GH are low throughout the day except for brief secretory peaks in the middle of the night or early morning, the daytime ascertainment of GH deficiency or sufficiency on the basis of a single determination of a random GH concentration is impossible. Adequacy of GH secretion may be determined by a stimulation test to measure peak GH secretion. A normal response is a vigorous secretory peak after stimulation; the lack of such a peak is consistent with GH deficiency. However, there is a high false positive rate (on any day, about 10% or more of normal children may not reach the normal GH peak after even two stimulatory tests). Indirect measurements of GH secretion, such as serum concentrations of IGF-1 and IGF-BP3, are replacing GH stimulatory tests.
The factors responsible for postnatal growth are not the same as the factors that mediate fetal growth. Thyroid hormone is essential for normal postnatal growth, although a thyroid hormone–deficient fetus achieves a normal birth length; similarly, a GH-deficient fetus has a normal birth length, although in IGF-1 deficiency resulting from GH resistance (Laron dwarfism), fetuses are shorter than control subjects. Adequate thyroid hormone is necessary to allow the secretion of GH. Hypothyroid patients may appear falsely to be GH deficient; with thyroid hormone repletion, GH secretion normalizes. Gonadal steroids are important in the pubertal growth spurt. The effects of other hormones on growth are noted in Table 173-1.
ABNORMALITIES OF GROWTH
Short Stature of Nonendocrine Causes
Short stature is defined as subnormal height relative to other children of the same gender and age, taking family heights into consideration. It can be caused by numerous conditions (Table 173-2). The Centers for Disease Control and Prevention growth charts use the 3rd percentile of the growth curve as the demarcation of the lower limit. Growth failure denotes a slow growth rate regardless of stature. Ultimately a slow growth rate leads to short stature, but a disease process is detected sooner if the decreased growth rate is noted before the stature becomes short. Height velocity may be derived and plotted on special height velocity charts. Plotted on a growth chart, growth failure appears as a curve that crosses percentiles downward and is associated with a height velocity below the 5th percentile of height velocity for age (Fig. 173-1). A corrected mid-parental, or genetic target, height helps determine if the child is growing well for the family (see Chapter 6). To determine a range of normal height for the family under consideration, the corrected mid-parental height is bracketed by 2 standard deviations (SDs), which for the United States is approximately 10 cm (4 inches). The presence of a height 3.5 SDs below the mean, a height velocity below the 5th percentile for age, or a height below the target height corrected for mid-parental height requires a diagnostic evaluation.
TABLE 173-2 Causes of Short Stature
VARIATIONS OF NORMAL
ENDOCRINE DISORDERS
SKELETAL DYSPLASIAS
LYSOSOMAL STORAGE DISEASES
SYNDROMES OF SHORT STATURE
CHRONIC DISEASE
AIDS, acquired immunodeficiency syndrome; CNS, central nervous system; GH, growth hormone; IGF, insulin-like growth factor.
* Only if caloric intake is severely diminished.
Modified from Styne DM: Growth disorder. In Fitzgerald PA (ed): Handbook of Clinical Endocrinology. Norwalk, CT, Appleton & Lange, 1986.
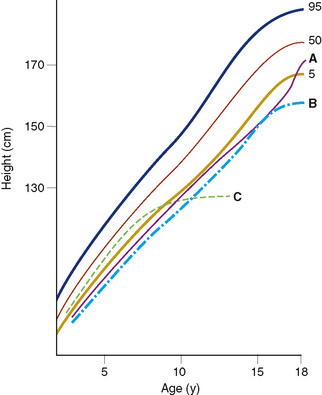
FIGURE 173-1 Patterns of linear growth. Normal growth percentiles (5th, 50th, and 95th) are shown along with typical growth curves for A, constitutional delay of growth and adolescence (short stature with normal growth rate for bone age, delayed pubertal growth spurt, and eventual achievement of normal adult stature); B, familial short stature (short stature in childhood and as an adult); and C, acquired pathologic growth failure (e.g., acquired untreated primary hypothyroidism) (see Chapter 5).
Nutrition is the most important factor affecting growth on a worldwide basis (see Chapter 28). Failure to thrive may develop in the infant as a result of maternal deprivation (nutritional deficiency or aberrant psychosocial interaction) or as a result of organic illness (anorexia, nutrient losses through a form of malabsorption, or hypermetabolism caused by hyperthyroidism) (see Chapter 21). Psychological difficulties also can affect growth, as in psychosocial or deprivation dwarfism, in which the child develops functional temporary GH deficiency and poor growth as a result of psychological abuse; when placed in a different, healthier psychosocial environment, GH physiology normalizes, and growth occurs.
The common condition known as constitutional delay in growth or puberty or both is a variation of normal growth, caused by a reduced tempo, or cadence, of physiologic development and not a disease (see Fig. 173-1). Usually a family member had delayed growth or puberty but achieved a normal final height. The bone age is delayed, but the growth rate remains mostly within the lower limits of normal. Constitutional delay usually leads to a delay in secondary sexual development. Genetic or familial short stature (Table 173-3) refers to the stature of a child of short parents, who is expected to reach a lower than average height and yet normal for these parents. If the parents were malnourished as children, grew up in a zone of war, or suffered famine, the heights of the parents are less predictive. Although there are differences in height associated with ethnicity, the most significant difference in stature between ethnic groups is the result of nutrition. Growth curves exist for Asian children and for children with some chromosomal disorders.
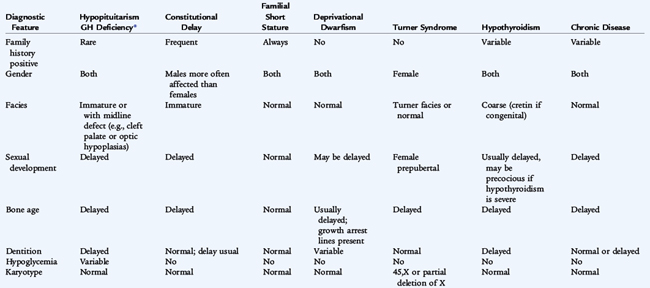

Phenotypic features suggesting an underlying chromosomal disorder can occur in a host of syndromes. These syndromes can be suspected by attending to arm spans and upper-to-lower segment ratios. Genetic syndromes often combine obesity and decreased height, whereas otherwise normal obese children are usually taller than average and have advanced skeletal development and physical maturation (see Table 173-2). The Prader-Willi syndrome includes fetal and infantile hypotonia, small hands and feet (acromicria), postnatal acquired obesity with an insatiable appetite, developmental delay, hypogonadism, almond-shaped eyes, and abnormalities of the SNRP portion of the 15th chromosome at 15q11-q13. Most cases have deletion of the paternal sequence, but about 20% to 25% have uniparental disomy, in which both chromosomes 15 derive from the mother; when both chromosomes 15 come from the father, Angelman syndrome develops. Laurence-Moon-Bardet-Biedl syndrome is characterized by retinitis pigmentosa, hypogonadism and developmental delay with an autosomal dominant inheritance pattern. Laurence-Moon syndrome is associated with spastic paraplegia, however, and Bardet-Biedl syndrome is associated with obesity and polydactyly. Pseudohypoparathyroidism leads to short stature and developmental delay with short fourth and fifth digits (Albright hereditary osteodystrophy phenotype), resistance to parathyroid hormone (PTH) and resultant hypocalcemia and elevated levels of serum phosphorus.
Short Stature Caused by Growth Hormone Deficiency
Etiology and Epidemiology
Classic congenital or idiopathic GH deficiency occurs in about 1 in 4000 to 1 in 10,000 children. Idiopathic GH deficiency, of unidentified specific etiology, is the most common cause of both congenital GH deficiency and acquired GH deficiency. Less often, GH deficiency is caused by anatomic defects of the pituitary gland, such as pituitary aplasia or other midline defects with variable degrees of deficiency of other pituitary functions. Hereditary forms of GH deficiency that affect pituitary differentiation are the result of heterogeneous defects of the gene for GH, GRF, or GH receptor. Classic GH deficiency refers to very reduced to absent secretion of GH; numerous short children may have intermediate forms of decreased GH secretion. Acquired GH deficiency causing late-onset growth failure suggests the possibility of a tumor of the hypothalamus or pituitary (Table 173-4). Other rare causes of direct injury to the pituitary gland include infection, inflammation, and autoimmunity.
TABLE 173-4 Growth Failure: Screening Tests
| Test | Rationale |
|---|---|
| CBC | |
| ESR, CRP | Inflammation of infection, inflammatory diseases, malignancy |
| Metabolic panel (electrolytes, liver enzymes, BUN) | Signs of acute or chronic hepatic, renal, adrenal dysfunction; hydration and acid-base status |
| Carotene, folate, and prothrombin time; celiac antibody panel | Assess malabsorption; detect celiac disease |
| Urinalysis with pH | Signs of renal dysfunction, hydration, water and salt homeostasis; renal tubular acidosis |
| Karyotype | Determines Turner (XO) or other syndromes |
| Cranial imaging (MRI) | Assesses hypothalamic-pituitary tumors (craniopharyngioma, glioma, germinoma) or congenital midline defects |
| Bone age | Compare with height age, and evaluate height potential |
| IGF-1, IGF-BP3 | Reflects growth hormone status or nutrition |
| Free thyroxine | Detects panhypopituitarism or isolated hypothyroidism |
| Prolactin | Elevated in hypothalamic dysfunction or destruction, suppressed in pituitary disease |
BUN, blood urea nitrogen; CBC, complete blood count; CRP, C-reactive protein; ESR, erythrocyte sedimentation rate; IGF-1, insulin-like growth factor-1; IGF-BP3, insulin-like growth factor–binding protein 3; MRI, magnetic resonance imaging.
Clinical Manifestations
Infants with congenital GH deficiency achieve a normal or near-normal birth length and weight at term, but the growth rate slows after birth, most noticeably after age 2 to 3 years, as these children become progressively shorter for age. They also tend to have an elevated weight-to-height ratio and appear chubby and short. Careful measurements in the first year of life may suggest the diagnosis, but most patients elude diagnosis until several years of age. A patient with classic GH deficiency has the appearance of a cherub (a chubby, immature appearance), with a high-pitched voice resulting from an immature larynx. Unless severe hypoglycemia occurs or dysraphism (midline defects) of the head includes a central nervous system (CNS) defect that affects mentation, the patient has normal intellectual growth and age-appropriate speech. Male neonates with isolated GH deficiency with or without gonadotropin deficiency may have a microphallus (a stretched penile length of <2 cm [normal is 3–5 cm]) and fasting hypoglycemia. Patients who lack adrenocorticotropic hormone (ACTH, cortisol) in addition to GH may have more profound hypoglycemia since cortisol also stimulates gluconeogenesis.
GH resistance or GH insensitivity is caused by abnormal number or function of GH receptors or by a postreceptor defect. Patients with the autosomal recessive Laron syndrome, involving mutations of the GH receptor, have a prominent forehead, hypoplastic nasal bridge, delayed dentition, sparse hair, blue sclerae, delayed bone maturation and osteoporosis, progressive adiposity, hypercholesterolemia, and low blood glucose. They have elevated serum GH concentrations, although serum IGF-1 and IGF-BP3 concentrations are low. The characteristic decrease in the number of GH receptors is reflected in the decreased serum concentration of GH-binding protein. The patients do not respond to the administration of GH with an increase in growth or an increase in serum concentrations of IGF-1 or IGF-BP3. Subjects with Laron syndrome have been treated experimentally with recombinant human IGF-1 with varying degrees of success. Malnutrition or severe liver disease may cause acquired GH resistance because serum GH is elevated, and IGF-1 is decreased.
Diagnosis
If family or other medical history does not provide a likely diagnosis, screening tests should include a metabolic panel to evaluate kidney and liver function, a complete blood count (CBC) to rule out anemia, a celiac panel with antibodies to tissue transglutaminase to rule out celiac disease, and carotene and folate levels to reflect nutrition and rule out malabsorption. A urinalysis aids in evaluation of renal function. Urinary pH and serum bicarbonate can assess for renal tubular acidosis. In a girl without another explanation for short stature, a karyotype may rule out Turner syndrome. A bone age will establish skeletal maturation.
If chronic disease or familial short stature are ruled out and routine laboratory testing is normal (see Table 173-4), two GH stimulatory tests are commonly performed (see Table 170-2). GH testing should be offered to a patient who is short (<5th percentile and usually >3.5 SDs below the mean), growing poorly (<5th percentile growth rate for age), or whose height projection, based on current height and skeletal maturation as assessed by bone age, is below the target height when corrected for family height.
Classic GH-deficient patients do not show an increase in serum GH levels after stimulation by various secretagogues. Some patients release GH in response to secretagogue testing, but cannot release GH spontaneously during the day or night. Measuring serum IGF-1 and IGF-BP3 is suggested but not consistently helpful in establishing a diagnosis of GH deficiency. Tests for GH secretion are insensitive, not very specific, and fairly variable. However, they do allow distinction of GH deficiency from GH insensitivity. In equivocal cases, an operational definition of GH deficiency might be that patients who require GH grow significantly faster when administered a normal dose of GH than before treatment.
Treatment
GH deficiency is treated with biosynthetic recombinant DNA–derived GH. Dosage is titrated to the growth rate. Treatment with GH carries the risk of an increased incidence of slipped capital femoral epiphysis, especially in rapidly growing adolescents, and of pseudotumor cerebri.
Administration of GH to patients with normal GH responsiveness to secretagogues is controversial, but as noted earlier, diagnostic tests are imperfect; if the patient is growing extremely slowly without alternative explanation, GH therapy is sometimes used. GH is effective in increasing the growth rate and final height in Turner syndrome and in chronic renal failure; GH also is used for treatment of short stature and muscle weakness of Prader-Willi syndrome. Other indications include children born small for gestational age who have not exhibited catch-up growth by 2 years of age and the long-term treatment of idiopathic short stature with height 2.25 SDs or less below the mean. This condition also is called non–GH-deficient short stature.
Psychological support of children with severe short stature is important because of possible ridicule. Although there is controversy, marital status, satisfaction with life, and vocational achievement may be decreased in children of short stature who are not given supportive measures.