 CHAPTER 174 Disorders of Puberty
CHAPTER 174 Disorders of Puberty
The staging of pubertal changes and the sequence of events are discussed in Chapter 67. The onset of puberty is marked by pubarche and gonadarche. Pubarche results from adrenal maturation or adrenarche. With the appearance of pubic hair, other features include oiliness of hair and skin, acne, axillary hair, and body odor. Adrenarche does not include breast development in females or testicular enlargement in males. Gonadarche is characterized by increasing secretion of gonadal sex steroids as a result of the maturation of the hypothalamic-pituitary-gonadal axis. These sex steroids differ by gender, consisting of testosterone from the testes and estradiol and progesterone from the ovaries. In males, physical signs are pubic hair, axillary hair, facial hair, increased muscularity, deeper voice, increased penile size, and increased testicular volume. In females, the physical signs are breast development, development of the female body habitus, increased size of the uterus, and menarche with regular menstrual cycles. The third component is the growth spurt of puberty.
Hypothalamic gonadotropin-releasing hormone (GnRH), produced by cells in the arcuate nucleus, is secreted from the median eminence of the hypothalamus into the pituitary portal system and reaches the membrane receptors on the pituitary gonadotropes to cause the production and release of luteinizing hormone (LH) and follicle-stimulating hormone (FSH) into the circulation. In females, FSH stimulates the ovarian production of estrogen and, later in puberty, causes the formation and support of corpus luteum. In males, LH stimulates the production of testosterone from the Leydig cells; later in puberty, FSH stimulates the development and support of the seminiferous tubules. The gonads also produce the protein inhibin. Both sex steroids and inhibin suppress the secretion of gonadotropins. The interplay of the products of the gonads and GnRH modulates the serum concentrations of gonadotropins. GnRH is released in episodic pulses that vary during development and during the menstrual period. These pulses ensure that gonadotropins are released in a pulsatile manner. With the onset of puberty, the amplitude of the pulses of gonadotropins increases, first at night and then throughout the day. Sex steroids are secreted in response, first during the night and then throughout the day. The hypothalamic-pituitary-gonadal axis is active in the fetus and newborn, but is suppressed in the childhood years until activity increases again at the onset of puberty.
Adrenarche occurs several years earlier than gonadarche and is heralded by increasing circulating dehydroepiandrosterone (DHEA) or androstenedione. Serum DHEA increases years before the appearance of its effects, such as the development of pubic or axillary hair.
DELAYED PUBERTY
Puberty is delayed when there is no sign of pubertal development by age 13 years in girls and 14 years in boys living in the United States (Tables 174-1 and 174-2).
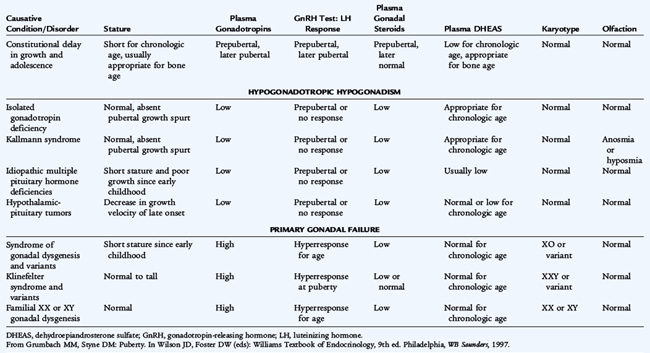
TABLE 174-1 Classification of Delayed Puberty and Sexual Infantilism
CNS, central nervous system.
Modified from Grumbach MM, Styne DM: Puberty. In Wilson JD, Foster DW (eds): Williams Textbook of Endocrinology, 9th ed. Philadelphia, WB Saunders, 1998.
Constitutional Delay in Growth and Adolescence
Patients with constitutional delay have delayed onset of pubertal development and significant bone age delay (2 standard deviations [SDs] below the mean, which is equal to a 1.5- to 2-year delay as a teenager). The height of the patient should remain close to the genetic potential, based on the parental heights, when reinterpreted for that bone age (see Chapter 173). The bone age should be consistent with the somatic maturity of the patient. Usually, height gain is below, although fairly parallel to, the normal percentiles on the growth curve. The prepubertal nadir, or deceleration before their pubertal growth spurt, is prolonged or protracted. A family history of delayed puberty in a parent or sibling is reassuring. Spontaneous puberty usually begins in these patients by the time the bone age reaches 12 years in boys and 11 years in girls. Other causes of delayed puberty must be eliminated before a diagnosis of constitutional delay in puberty is made. Observation and reassurance are appropriate. Spontaneous progression into puberty occurs when the bone age reaches 12 to 13 years, and an adult height normal for the genetic potential is attained. In some cases, boys may be treated with low-dose testosterone for a few months if the bone age is at least 11 to 12 years. Treatment is not required for longer than 4 to 8 months since endogenous hormone production usually ensues. No similar form of therapy for constitutional delay is available for girls. Young women with delayed puberty may need to be evaluated for primary amenorrhea.
Hypogonadotropic Hypogonadism
As a cause of delayed or absent puberty, hypogonadotropic hypogonadism may be difficult to distinguish from constitutional delay (see Tables 174-1 and 174-2). Hypogonadotropic hypogonadism precludes spontaneous entry into gonadarche; adrenarche usually occurs to some degree. Throughout childhood and in early puberty, patients with hypogonadotropic hypogonadism have normal proportions and growth. When these patients reach adulthood, eunuchoid proportions may ensue because their long bones grow for longer than normal, producing an upper-to-lower ratio below the lower limit of normal of 0.9 and an arm span greater than their height. If a patient has concurrent growth hormone (GH) deficiency, however, stature is exceptionally short, and the condition may have been diagnosed in infancy with a microphallus.
Isolated Gonadotropin Deficiency
Disorders that can cause hypogonadism include congenital hypopituitarism, such as midline defects, tumors, infiltrative disease (hemochromatosis), and many syndromes, including Laurence-Moon-Bardet-Biedl, Prader-Willi, and Kallmann syndromes. If there is an inability to release gonadotropins but no other pituitary abnormality, the patient has isolated gonadotropin deficiency (almost universally a result of absent GnRH). Patients grow normally until the time of the pubertal growth spurt, when they fail to experience the accelerated growth characteristic of the normal growth spurt.
Kallmann syndrome combines isolated gonadotropin deficiency with disorders of olfaction. There is genetic heterogeneity; some patients have a decreased sense of smell, others have abnormal reproduction, and some have both. Most cases are sporadic but a number of patients may have mutations in the KAL1 gene at Xp 22.3 (X chromosome) or KAL2 gene. The mutation causes the GnRH neurons to remain ineffectually located in the primitive nasal area, rather than migrating to the correct location at the medial basal hypothalamus. Olfactory bulbs and olfactory sulci are often absent on magnetic resonance imaging (MRI). Other symptoms include disorders of the hand, with one hand copying the movements of the other hand, shortened fourth metacarpal bone, and an absent kidney.
Abnormalities of the Central Nervous System
Central nervous system (CNS) tumors, including pituitary adenoma, germinoma, glioma, prolactinoma, or craniopharyngioma, are important causes of gonadotropin deficiency. Craniopharyngiomas have a peak incidence in the teenage years and may cause any type of anterior or posterior hormone deficiency. Craniopharyngiomas usually calcify, eroding the sella turcica when they expand. They may impinge on the optic chiasm, leading to bitemporal hemianopsia and optic atrophy. Germinomas are noncalcifying hypothalamic or pineal tumors that frequently produce human chorionic gonadotropin (hCG), which may cause sexual precocity in prepubertal boys (hCG cross-reacts with the LH receptor because of the similarity of structure between LH and hCG). Other tumors that may affect pubertal development include astrocytomas and gliomas.
Idiopathic Hypopituitarism
Congenital absence of various combinations of pituitary hormones may produce idiopathic hypopituitarism. Although this disorder may occur in family constellations, after X-linked or autosomal recessive patterns, sporadic types of congenital idiopathic hypopituitarism are more common. Congenital hypopituitarism may manifest in a male with GH deficiency and associated gonadotropin deficiency with a microphallus or with hypoglycemia with seizures, especially if adrenocorticotropic hormone (ACTH) and GH deficiency occurs as well.
Syndromes of Hypogonadotropic Hypogonadism
Weight loss resulting from voluntary dieting, malnutrition, or chronic disease leads to decreased gonadotropin function when weight decreases to less than 80% of ideal weight. Anorexia nervosa is characterized by striking weight loss and psychiatric disorders (see Chapter 70). Primary or secondary amenorrhea frequently is found in affected girls, and pubertal development is absent or minimal, depending on the level of weight loss and the age at onset. Regaining weight to the ideal level may not immediately reverse the condition. Increased physical activity, even without weight loss, can lead to decreased menstrual frequency and gonadotropin deficiency in athletic amenorrhea; when physical activity is interrupted, menstrual function may return. Chronic or systemic illness (e.g., cystic fibrosis, diabetes mellitus, inflammatory bowel disease, or hematologic diseases) can lead to pubertal delay or to amenorrhea from hypothalamic dysfunction. Hypothyroidism inhibits the onset of puberty and delays menstrual periods. Conversely, severe primary hypothyroidism may lead to precocious puberty.
Hypergonadotropic Hypogonadism
Hypergonadotropic hypogonadism is characterized by elevated gonadotropin and low sex steroid levels resulting from primary gonadal failure. This permanent condition is almost always diagnosed following the lack of entry into gonadarche and is not suspected throughout childhood. Gonadotropins do not increase to greater than normal until shortly before or around the normal time of puberty.
Ovarian Failure
Turner syndrome or the syndrome of gonadal dysgenesis is a common cause of ovarian failure and short stature. The karyotype is classically 45,XO, but other abnormalities of the X chromosome or mosaicism are possible. The incidence of Turner syndrome is 1 in 2000 to 1 in 3000 births. The features of a girl with Turner syndrome need not be evident on physical examination or by history. The diagnosis must be considered in any girl who is short without a contributory history. Patients with other types of gonadal dysgenesis and galactosemia as well as patients treated with radiation therapy or chemotherapy for malignancy may develop ovarian failure.
Testicular Failure
Klinefelter syndrome (seminiferous tubular dysgenesis) is the most common cause of testicular failure. The karyotype is 47,XXY, but variants with more X chromosomes are possible. The incidence is approximately 1 in 500 to 1 in 1000 in males. Testosterone levels may be close to normal, at least until mid-puberty, because Leydig cell function may be spared; however, seminiferous tubular function characteristically is lost, causing infertility. Commonly, LH levels may be normal to elevated, whereas FSH levels are usually more unequivocally elevated. The age of onset of puberty is usually normal, but secondary sexual changes may not progress because of inadequate Leydig cell function.
Primary Amenorrhea
When it is determined that no secondary sexual development is present after the upper age limits of normal pubertal development, serum gonadotropin levels should be obtained to determine whether the patient has a hypogonadotropic or hypergonadotropic hypogonadism (see Table 174-2). Based only on gonadotropin measurements, the differentiation between constitutional delay in growth and hypogonadotropic hypogonadism is difficult; the gonadotropin levels are low in both conditions. Sometimes observation for months or years is necessary before the diagnosis is confirmed.
Differential Diagnosis
Lack of menstruation in the presence of normal pubertal development might be the result of physiologic variation. The entities leading to hypogonadism include congenital hypopituitarism; tumors such as a pituitary adenoma, germinoma, glioma, prolactinoma, and craniopharyngioma; infiltrative disease; and Laurence-Moon-Biedl, Prader-Willi, and Kallmann syndromes. In females, stress, competitive athletics, or inadequate nutrition from chronic or systemic illness or from an eating disorder can lead to extreme pubertal delay, or amenorrhea on the basis of hypothalamic dysfunction. When primary amenorrhea occurs, an anatomic defect may be responsible; the Mayer-Rokitansky-Kuster-Hauser syndrome of congenital absence of the uterus occurs in 1 in 4000 to 1 in 5000 female births. Anatomic obstruction by imperforate hymen or vaginal septum also presents with normal secondary sexual development without menstruation. The complete syndrome of androgen insensitivity includes normal feminization, absence of pubic or axillary hair, and primary amenorrhea. In this syndrome, all müllerian structures, including ovaries, uterus, fallopian tubes, and upper third of the vagina, are lacking; the karyotype is 46,XY, and subjects have intra-abdominal testes. Undiagnosed Turner syndrome is another relatively common cause of amenorrhea, resulting from abnormal ovarian function. Secondary sexual development also is absent or minimal.
Treatment
If a permanent condition is apparent, replacement with sex steroids is indicated. Females are given low-dose ethinyl estradiol (5–10 μg) or conjugated estrogens (starting at 0.3 mg daily, increasing to 0.625 or 0.9 by 6–12 months later) in low daily doses until breakthrough bleeding occurs, at which time cycling is started with a dose on the first 25 days of the month; on days 20 to 25 of the month, a progestational agent, such as medroxyprogesterone acetate (5 mg), is added to mimic the normal increases in gonadal hormones and to induce a normal menstrual period. In males, testosterone enanthate or cypionate (50 mg monthly with a progressive increase to 100–200 mg) is given intramuscularly once every 4 weeks. Transdermal administration also is possible. This starting regimen is appropriate for patients with hypogonadotropic or hypergonadotropic hypogonadism, and doses are increased gradually to adult levels. Oral agents are not used for fear of hepatotoxicity. Patients with apparent constitutional delay in puberty who have, by definition, passed the upper limits of normal onset of puberty may be given a 3-month course of low-dose, sex-appropriate gonadal steroids to see if spontaneous puberty occurs. This course of therapy might be repeated once without undue advancement of bone age. All patients with any form of delayed puberty are at risk for decreased bone density; adequate calcium intake is essential. Patients with hypogonadotropic hypogonadism may be able to achieve fertility by the administration of gonadotropin therapy or pulsatile hypothalamic-releasing hormone therapy administered by a programmable pump on an appropriate schedule. Subjects with hypergonadotropic hypogonadism, whether Turner syndrome or Klinefelter syndrome, have, by definition, a primary gonadal problem and are unlikely to achieve spontaneous fertility.
In subjects with Turner syndrome, the goals of therapy include promoting growth with exogenous human GH supplementation and induction of the secondary sexual characteristics and of menses with low-dose cyclic estrogen replacement therapy, with progestagen added at the end of each cycle. Patients with Turner syndrome have had successful pregnancies after in vitro fertilization with a donor ovum and endocrine support.
SEXUAL PRECOCITY
Classification
Sexual precocity (precocious puberty) is classically defined as secondary sexual development occurring before the age of 9 years in boys or 8 years in girls (Tables 174-3 and 174-4). The lower limit of normal puberty may be 7 years in white girls and 6 years in African-American girls. At present, the mean age at which girls exhibit Tanner II breast development (thelarche) is approximately 10 years for white girls and 9 years for African-American girls (normal range 8–13 years). The mean age at which girls exhibit Tanner II pubic hair development is 9 years for white girls and 10.5 years for African-American girls. Menarche usually occurs at 12.2 and 12.9 years (range 10–15 years). Pubertal progression results from two components: gonadarche (maturation of the gonad and secretion of sex steroid, estrogen in females, testosterone in males) and adrenarche (maturation of the adrenal and secretion of adrenal sex steroid, DHEA and androstenedione). The normal developmental sequence in girls is thelarche (due to gonadarche) followed closely by pubarche (due to adrenarche) and finally menarche, 2 to 3 years later. In boys, the first normal event is enlargement of testes followed by appearance of pubic hair (long diameter of the testis greater than 2.5 cm, volume greater than 4 mL).
TABLE 174-3 Classification of Sexual Precocity
TRUE PRECOCIOUS PUBERTY OR COMPLETE ISOSEXUAL PRECOCITY
INCOMPLETE ISOSEXUAL PRECOCITY (GNRH-INDEPENDENT SEXUAL PRECOCITY)
VARIATIONS OF PUBERTAL DEVELOPMENT
CONTRASEXUAL PRECOCITY
CNS, central nervous system; GnRH, gonadotropin-releasing hormone; hCG, human chorionic gonadotropin; LH, luteinizing hormone.
From Grumbach MM, Styne DM: Puberty. In Wilson JD, Foster DW (eds): Williams Textbook of Endocrinology, 9th ed. Philadelphia, WB Saunders, 1998.
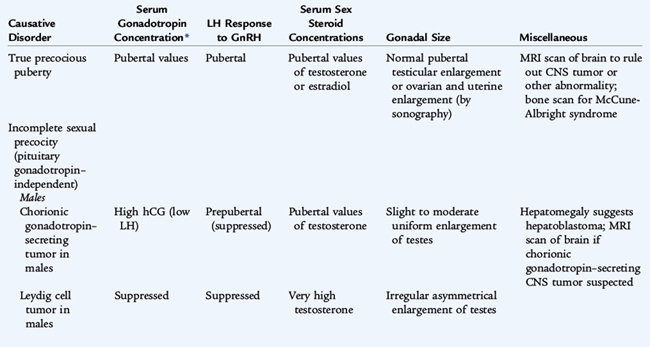
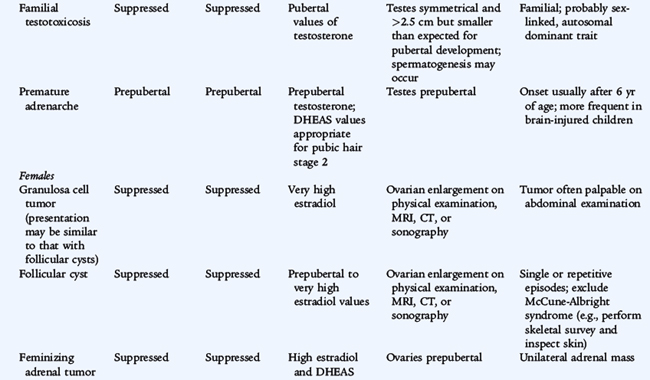

Central precocious puberty, resulting in gonadarche, emanates from premature activation of the hypothalamic-pituitary-gonadal axis (GnRH-dependent). Peripheral precocious puberty, gonadarche or adrenarche, does not involve the hypothalamic-pituitary-gonadal axis (GnRH-independent).
Central Precocious Puberty (Constitutional or Familial Precocious Puberty)
In central precocious puberty, every endocrine and physical aspect of pubertal development is normal but too early; this includes tall stature, advanced bone age, increased sex steroid and pulsatile gonadotropin secretion, and increased response of LH to GnRH. The clinical course of central precocious puberty may wax and wane. Benign precocious puberty is the presumptive diagnosis in individuals who begin puberty early on a constitutional or familial basis. If no cause can be determined, the diagnosis is idiopathic precocious puberty; this condition occurs much more often in girls than in boys. Obese girls have earlier adrenarche, and sometimes menarche as well, than normal weight girls, and the high incidence of overweight and obesity in developed countries is contributing to a shift to earlier entry into puberty. Compared with girls, boys with precocious puberty have a higher incidence of CNS disorders, such as tumors and hamartomas, precipitating the precocious puberty. Almost any condition that affects the CNS, including hydrocephalus, meningitis, encephalitis, suprasellar cysts, head trauma, epilepsy, mental retardation and irradiation, can precipitate central precocious puberty.
A CNS tumor or disease must be considered in all children with precocious puberty before the condition is diagnosed as idiopathic. Hamartomas are nonmalignant tumors of the tuber cinereum with a characteristic appearance on computed tomography (CT) or MRI; biopsy is rarely required. The mass of GnRH neurons secrete GnRH and cause precocious puberty. While hamartomas are not true neoplasms they may require neurosurgical attention, such as ventriculoperitoneal shunting, if they lead to increased intracranial pressure. The resulting precocious puberty is responsive to medical therapy with GnRH agonists, and surgery is rarely indicated.
Other masses that cause precocious puberty are not benign. Optic or hypothalamic gliomas (with or without neurofibromatosis), astrocytomas, and ependymomas may cause precocious puberty by disrupting the negative restraint of the areas of the CNS that normally inhibit pubertal development throughout childhood. These tumors may require radiotherapy, which contributes to a significant risk for hypopituitarism. Growth is greater than that found in age-matched control subjects, but less than in GH-replete patients with precocious puberty. The GH deficiency is not as obvious as in GH-deficient patients without precocious puberty. With GnRH analog treatment, precocious puberty is controlled and the growth rate decreases to that of a GH-deficient child. GH is additional therapy.
Gonadotropin-Releasing Hormone–Independent Precocious Puberty
The most common cause of GnRH-independent precocious puberty, McCune-Albright syndrome, is more frequent in girls than boys and includes precocious gonadarche, a bone disorder with polyostotic fibrous dysplasia, and hyperpigmented cutaneous macules (café au lait spots). The precocious gonadarche results from ovarian hyperfunction and sometimes cyst formation, leading to episodic estrogen secretion. This disorder results from a postconception somatic mutation in the G protein intracellular signaling system (specifically Gsα, which leads to unregulated constitutive activation of adenylate cyclase, and of cAMP in the absence of trophic hormone stimulation), in the affected cells in ovary, bone, and skin; other endocrine organs may also autonomously hyperfunction for the same reason. Hyperthyroidism, hyperadrenalism, or acromegaly may occur. Adrenal carcinomas usually secrete adrenal androgens, such as DHEA; adrenal adenomas may virilize a child as a result of the production of androgen or may feminize a child as a result of the production of estrogen.
Boys may have precocious gonadarche on the basis of a rare entity called familial GnRH-independent sexual precocity with premature Leydig cell maturation. This condition, with germ cell maturation caused by an X-limited dominant defect, produces constitutive activation of the LH receptor which leads to continuous production and secretion of testosterone without requiring the presence of LH or hCG. Outside of the pituitary, gonadotropins can originate from a tumor. hCG-secreting tumors stimulate LH receptors and increase testosterone secretion. These tumors may be found in various places, including the pineal gland (dysgerminomas, which are radiosensitive) or the liver (hepatoblastoma, which may lead to death within a few months after diagnosis).
Ovarian cysts may occur once or may be recurrent. High serum estrogen values may mimic ovarian tumors. Congenital adrenal hyperplasia is a cause of virilization in girls and is discussed in the following sections.
Evaluation of Sexual Precocity
The first step in evaluating sexual precocity is to determine which characteristics of normal puberty are apparent (Chapter 67) and whether estrogen effects, androgen effects or both are present (see Table 174-4). In girls, androgen effect manifests as adult odor, pubic and axillary hair, and facial skin oiliness and acne, whereas estrogen effect manifests as breast development, uterine increase, and eventually menarche. In boys, androgen effect manifests as adult odor, pubic and axillary hair, and facial skin oiliness and acne; it is also important to note whether the testes are enlarged more than 2.5 cm in length, which implies gonadarche. If the testes are not enlarged, but virilization is progressing, the source of the androgens may be the adrenal glands or exogenous sources. If the testes are slightly enlarged but not consistent with the stage of pubertal development, ectopic production of hCG or familial Leydig and germ cell maturation may be the cause. Most of the enlargement of testes during puberty is the result of seminiferous tubule maturation. If only Leydig cells are enlarged, as in these conditions, the testes make considerable testosterone, but show only minimal enlargement.
Laboratory evaluation includes determination of sex steroid (testosterone, estradiol, or DHEA-S or androstenedione) and baseline gonadotropin concentrations. The inherent nature of gonadotropin secretion is characterized by low secretory rates throughout childhood and pulsatile secretion in adolescents and adults. If baseline gonadotropin values are elevated into the normal pubertal range, central precocious puberty is likely. If baseline gonadotropins are low, however, no immediate conclusion may be drawn as to GnRH-dependent versus GnRH-independent precocious puberty. This distinction often requires assessment of gonadotropin responsiveness to GnRH stimulation. A prepubertal GnRH response is FSH predominant, whereas a pubertal response is more LH predominant. Thyroid hormone determination also is useful because severe primary hypothyroidism can cause incomplete precocious puberty. If there is a suggestion of a CNS anomaly or a tumor (CNS, hepatic, adrenal, ovarian, or testicular), MRI of the appropriate location is indicated. The diagnosis of central precocious puberty mandates that MRI of the CNS be performed.
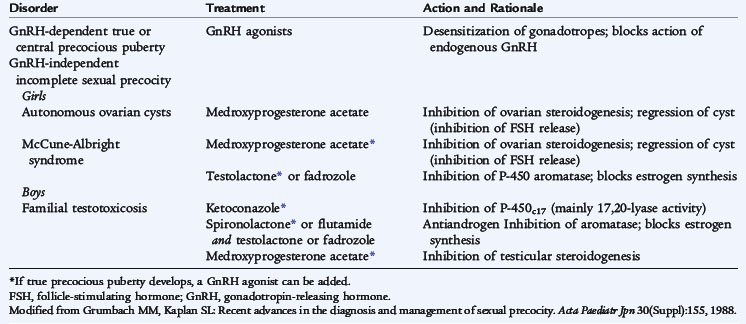
Treatment
Long-acting, superactive analogs of GnRH are the treatment of choice for central precocious puberty because they suppress gonadotropin secretion by down-regulating GnRH receptors in the pituitary gonadotropes (Table 174-5). After a brief (2–3 days) increase in gonadotropin secretion and, rarely, some withdrawal bleeding in girls, gonadotropin values decrease, and the gonadal secretion reverts to the prepubertal state. The early sexual development and increased height of a patient with precocious puberty mandate psychological support or even counseling for children and families. Boys with GnRH-independent premature Leydig cell and germ cell maturation do not respond to GnRH analogs, but require treatment with an inhibitor of testosterone synthesis (e.g., ketoconazole), an antiandrogen (e.g., spironolactone), or an aromatase inhibitor (e.g., testolactone or letrozole). Patients with precocious puberty from a hormone-secreting tumor require surgical removal, if possible. The precocious puberty of the McCune-Albright syndrome is GnRH independent and unresponsive to therapy with GnRH analog. Therapy is provided with testolactone and antiandrogens or antiestrogen, such as tamoxifen. After successful therapy for the latter conditions, central precocious puberty may develop secondarily; GnRH agonist administration is effective therapy.
VARIATIONS IN PUBERTAL DEVELOPMENT
Isolated Premature Thelarche (Premature Breast Development)
Benign premature thelarche is the isolated appearance of unilateral or bilateral breast tissue in girls, usually at ages 6 months to 3 years. There are no other signs of puberty and no evidence of excessive estrogen effect (vaginal bleeding, thickening of the vaginal secretions, increased height velocity, or bone age acceleration). Ingestion or dermal application of estrogen-containing compounds must be excluded. Laboratory investigations are not usually necessary, but a pelvic ultrasound study rarely may be indicated to exclude ovarian disease. Girls with this condition should be re-evaluated at intervals of 6 to 12 months to ensure that apparent premature thelarche is not the beginning of progression into precocious puberty. The prognosis is excellent; if no progression occurs, no treatment other than reassurance is necessary. There is no indication for a breast biopsy in a patient with established premature thelarche.
Gynecomastia
In males, breast tissue is termed gynecomastia, and it may occur to some degree in 45% to 75% of normal pubertal boys (see Chapter 67). Androgens normally are converted to estrogen by aromatization; in early puberty, only modest amounts of androgens are produced, and the estrogen effect can overwhelm the androgen effects at this stage. Later in pubertal development, the androgen production is so great that there is little effect from the estrogen produced by aromatization. Gynecomastia also can suggest the possibility of Klinefelter syndrome as puberty progresses. Prepubertal gynecomastia suggests an unusual source of estrogen either from exogenous sources (oral or dermal estrogen administration is possible from contamination of food or ointments) or from endogenous sources (from abnormal function of adrenal gland or ovary or from increased peripheral aromatization).
Isolated Premature Adrenarche (Pubarche)
The isolated appearance of pubic hair before age 6 to 7 years in girls or before age 9 years in boys is termed premature pubarche, usually results from adrenarche, and is relatively common. If the pubic hair is associated with any other feature of virilization (clitoral or penile enlargement or advanced bone age) or other signs (acne, rapid growth, or voice change), a detailed investigation for a pathologic cause of virilization is indicated to rule out a life-threatening cause, such as an adrenocortical carcinoma. Measurements of serum testosterone, 17-hydroxyprogesterone (17-OHP), and DHEA in basal and ACTH-stimulated states are indicated in heavily virilized patients to investigate the possibility of congenital adrenal hyperplasia (CAH). Ultrasound studies may reveal a hyperplastic adrenal gland or a virilizing adrenal or ovarian tumor. Most patients with isolated pubic hair do not have progressive virilization and simply have premature adrenarche (pubarche) resulting from premature activation of DHEA secretion from the adrenal gland. The skeletal maturation, as assessed by bone age, may be slightly advanced and the height slightly increased, but testosterone concentrations are normal. DHEA levels usually are high for prepubertal age, but are consistent with Tanner (sexuality maturity rating) stages II and III.