Chapter 16 Sleep
 During normal sleep tidal volume is reduced, with maximal reduction in ventilation occurring during rapid eye movement sleep when breathing also becomes irregular.
During normal sleep tidal volume is reduced, with maximal reduction in ventilation occurring during rapid eye movement sleep when breathing also becomes irregular.Normal Sleep
Sleep is classified on the basis of the electroencephalogram (EEG) and electro-oculogram (EOG) into rapid eye movement (REM) and non-REM (stages 1–4) sleep.
Stage 1 is dozing from which arousal easily takes place. The EEG is low voltage and the frequency is mixed but predominantly fast. This progresses to stage 2 in which the background EEG is similar to stage 1 but with episodic sleep spindles (frequency 12–14 Hz) and K complexes (large biphasic waves of characteristic appearance). Slow, large amplitude (delta) waves start to appear in stage 2 but become more dominant in stage 3 in which spindles are less conspicuous and K complexes become difficult to distinguish. In stage 4, which is often referred to as deep sleep, the EEG is mainly high voltage (more than 75 μV) and more than 50% slow (delta) frequency.
REM sleep has quite different characteristics. The EEG pattern is the same as in stage 1 but the EOG shows frequent rapid eye movements that are easily distinguished from the rolling eye movements of non-REM sleep. Dreaming occurs during REM sleep.
The stage of sleep changes frequently during the night, and the pattern varies between different individuals and on different nights for the same individual (Figure 16.1). Sleep is entered in stage 1 and usually progresses through 2 to 3 and sometimes into 4. Episodes of REM sleep alternate with non-REM sleep throughout the night. On average there are four or five episodes of REM sleep per night, with a tendency for the duration of the episodes to increase towards morning. Conversely, stages 3 and 4 predominate in the early part of the night.
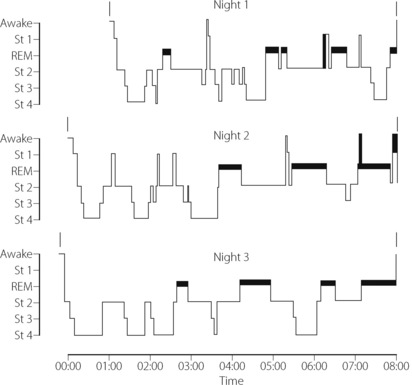
Fig. 16.1 Patterns of sleep on three consecutive nights in a young fit man aged 20. The thick horizontal bars indicate rapid eye movement (REM) sleep.
(Record kindly supplied by Dr C Thornton)
Respiratory Changes
Ventilation.1 Tidal volume decreases with deepening levels of non-REM sleep and is minimal in REM sleep, when it is about 25% less than in the awake state. Respiratory frequency is generally unchanged, though breathing is normally irregular during REM sleep. Minute volume is progressively reduced in parallel with the tidal volume. These changes in ventilation are brought about by the same neurochemical changes that cause sleep. Increased activity of GABA secreting neurones during sleep has a direct depressant effect on the respiratory centre (see Figure 5.4) and activation of cholinergic neurones is thought to be responsible for the respiratory patterns seen during non-REM sleep.1
Arterial Pco2 is usually slightly elevated by about 0.4 kPa (3 mmHg). In the young healthy adult, arterial Po2 decreases by about the same amount as the Pco2 is increased, and therefore the oxygen saturation remains reasonably normal. Mean value for rib cage contribution to breathing (page 87) was found to be 54% in stage 1–2, decreasing slightly in stages 3–4.2 However, in REM sleep, the value was reduced to 29%, which is close to the normal awake value in the supine position.
Chemosensitivity. In humans, the slope of the hypercapnic and hypoxic ventilatory responses are markedly reduced during sleep.3,4 In both cases, the slope is reduced by approximately one-third during non-REM sleep, and even further reduced during REM sleep, but fortunately the responses are never abolished completely.
Effect of age. Compared with young subjects, the elderly have more variable ventilatory patterns when awake, which seems to result in more episodes of periodic breathing (page 76) and apnoea when asleep. Elderly subjects also have significant oscillations in upper airway resistance during sleep (see below),5 which may contribute to the observed variations in ventilation. Thus as age advances, episodes of transient hypoxaemia occur in subjects who are otherwise healthy, with saturations commonly falling as low as 75% during sleep. Such changes must be regarded as a normal part of the ageing process.
Pharyngeal airway resistance. Air flow through the sharp bends of the upper airway is normally laminar, but is believed to be very close to becoming turbulent even in normal subjects.6 Pharyngeal muscles may play a crucial role in maintaining the optimum shape of the airway to maintain laminar flow, and the speed at which these control mechanisms can respond to changes in pharyngeal pressure (page 83) may be more critical than previously thought.7 Any condition that attenuates or delays these reflexes even slightly, such as sleep or alcohol ingestion, will then have a major effect on airflow in the pharynx causing breakdown of the normally laminar flow.
The nasal airway is normally used during sleep, and upper airway resistance is consistently increased, especially during inspiration and in REM sleep. The main sites of increase are across the soft palate and in the hypopharynx.8 Changes in pharyngeal muscle activity with sleep are complex. Muscles with predominantly tonic activity, such as tensor palati, show a progressive decrease in activity with deepening non-REM sleep,9 reaching only 20–30% of awake activity in stage 4 sleep. This loss of tonic activity correlates very well with increased upper airway resistance.9 Unlike in the awake state, tensor palati also fails to respond to an inspiratory resistive load. The activity of muscles with predominantly phasic inspiratory activity (e.g. geniohyoid and genioglossus) are influenced little by non-REM sleep. In spite of maintained phasic activity during non-REM sleep, tonic activity of geniohyoid is reduced whilst that of genioglossus is well preserved, and responds appropriately to resistive loading.10 During REM sleep genioglossus activity is reduced.11 It thus appears that the major effect is upon the tonic activity of nasopharyngeal muscles and the increase in hypopharyngeal resistance seems to be due to secondary downstream collapse.
The ventilatory response to increased airway resistance is important in normal sleep because of the increased pharyngeal resistance, and is generally well preserved. There are substantial and rapid increases in both diaphragmatic and genioglossal inspiratory activity following nasal occlusion in normal sleeping adults.12
Snoring
Snoring may occur at any age, but the incidence is bimodal, peaking in the first and the fifth to sixth decades of life. It is commoner in males than females, and linked to obesity. It may occur in any stage of sleep, becoming more pronounced as non-REM sleep deepens, though usually attenuated in REM sleep. As may be expected, snoring is less severe when sleeping in the lateral rather than supine position.13 About one-quarter of the population are habitual snorers, but these vary from the occasional snorer (e.g. after alcohol or with an upper respiratory tract infection) to the habitual persistent and heavy snorer.
Snoring originates in the oropharynx and in its mildest form is due to vibration of the soft palate and posterior pillars of the fauces. However, in its more severe forms, during inspiration the walls of the oropharynx collapse and the tongue is drawn back as a result of the subatmospheric pressure generated during inspiration against more upstream airway obstruction. This may be at the level of the palate as described above or may be the result of nasal polyps, nasal infection or enlarged adenoids, which are the commonest cause of snoring in children.14 As obstruction develops, the inspiratory muscles greatly augment their action and intrathoracic pressure may fall as low as −7 kPa (−70 cmH2O).
‘Normal’ snoring is not associated with either frequent arousal from sleep or apnoea, but is believed to precede the development of more serious sleep-related breathing disorders, with both increasing age and obesity making this progression more likely.15
Sleep-Disordered Breathing16
This term is used to describe a continuum of respiratory abnormalities seen during sleep, which affect around 20% of the population16 and range from simple snoring to life-threatening obstructive sleep apnoea.17,18,19 All are characterised by periods of apnoea, with or without episodes of airway narrowing or obstruction, that lead to repeated episodes of arterial hypoxia and arousal from sleep. Repeated arousals throughout the night give rise to excessive daytime sleepiness. Four syndromes are described, but there is considerable overlap between them:
Upper airway resistance syndrome14 in which tidal volume and arterial oxygen saturation 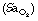 remain normal, but at the expense of extensive respiratory effort, which causes over 15 arousals per hour.
remain normal, but at the expense of extensive respiratory effort, which causes over 15 arousals per hour.
Obstructive sleep hypopnoea involves frequent (>15 per hour) episodes of airway obstruction of sufficient severity to reduce tidal volume to less than 50% of normal for over 10 seconds. There may be small decreases in 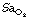 .
.
Obstructive sleep apnoea is characterised by more than 5 episodes per hour of obstructive apnoeas lasting over 10 seconds and associated with severe decreases in 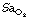 . In fact, durations of apnoea may be as long as 90 seconds and the frequency of the episodes as high as 160/hour. In severe cases, 50% of sleep time may be spent without tidal exchange.
. In fact, durations of apnoea may be as long as 90 seconds and the frequency of the episodes as high as 160/hour. In severe cases, 50% of sleep time may be spent without tidal exchange.
The last two syndromes are commonly grouped together as sleep apnoea/hypopnoea syndrome (SAHS). Severity is quantified by recording the apnoea/hypopnoea index (AHI), which is simply the number of occurrences per hour of apnoeas or hypopnoeas lasting longer than 10 seconds. Milder forms of sleep-disordered breathing tend to progress to more severe forms as patients grow older and fatter. The prevalence of SAHS, defined as an AHI of over 5, is between 3.5% and 24% in men and between 1.5% and 9% in women depending on the population studied.16,20,21
Apnoea or hypopnoea may be central or obstructive. Differentiation between central and obstructive apnoea is conveniently made by recording rib cage and abdominal movements continuously during sleep (Figure 16.2). If, as a result of upper airway obstruction, abdominal and ribcage movements become uncoordinated (Figure 16.2C), hypopnoea results. When these movements are equal but opposite in phase, there is obstructive apnoea (Figure 16.2D). Obstructive apnoea may occur in REM or non-REM sleep but the longest periods of apnoea tend to occur in REM sleep. As for snoring, airway obstruction is less frequent when sleeping in the lateral, rather than supine, position.13,22 Central apnoeas are more common in elderly patients.
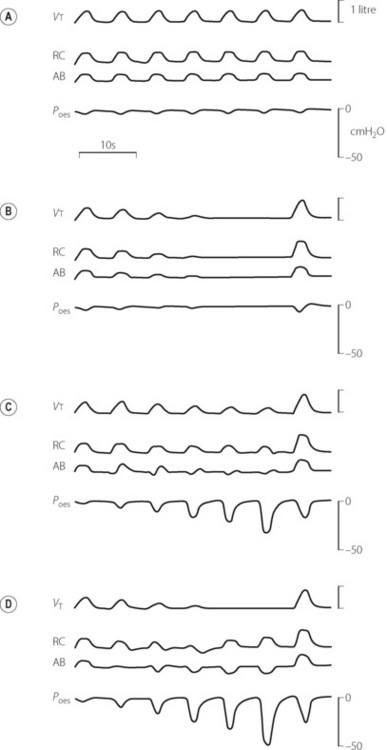
Fig. 16.2 Continuous records of breathing during differing types of apnoea and hypopnoea showing tidal volume (Vt), ribcage (RC) and abdominal (AB) contributions to breathing, and oesophageal pressure (Poes). (A) normal; (B) central apnoea; (C) obstructive hypopnoea; (D) obstructive apnoea.
Obesity hypoventilation syndrome (Pickwickian Syndrome) describes a combination of obesity, chronic hypercapnia (both nocturnal and daytime) and sleep-disordered breathing, usually severe OSA.23,24 With increasing levels of obesity in the developing world this syndrome is likely to become more common. The syndrome has a poor prognosis without treatment, which usually involves non-invasive ventilation.
The Mechanism of Airway Obstruction17,25,26
Anatomical factors. There is now widespread agreement that, on average, patients with SAHS have anatomically narrower airways than controls, and that the airway shape differs. Anatomical airway narrowing is believed to relate to two main factors.
First, obesity influences pharyngeal airway size. A central pattern of obesity, commonly seen in males, includes extensive fat deposition in the neck tissues. This accounts for the association between SAHS and neck circumference.24 Adipose tissue is best visualised using magnetic resonance imaging (MRI), and in patients with SAHS collections of fat are invariably seen lateral to the pharynx, between the pterygoid muscles and the carotid artery (Figure 16.3). Pharyngeal fat is increased above normal levels even in non-obese patients with SAHS (Figure 16.3C).27 In addition, the quantity of adipose tissue seen correlates with the AHI, and weight loss predictably reduces both.
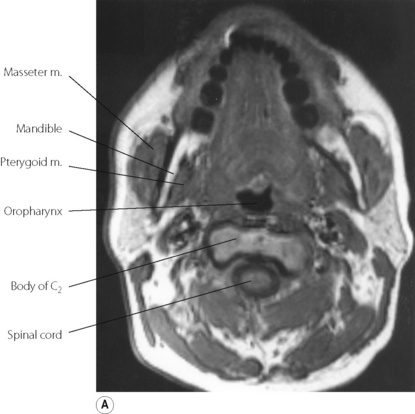
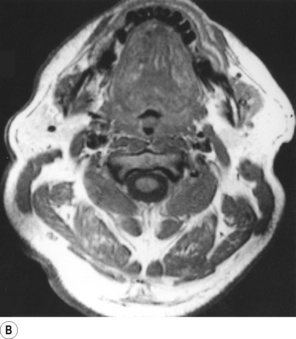
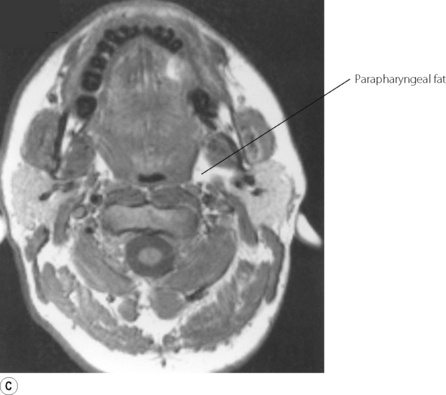
Fig. 16.3 Magnetic resonance imaging scan of the neck at the level of the oropharynx. In this type of scan fat tissue appears white. (A) Normal, non-obese, subject. (B) Obese patient with obstructive sleep apnoea, showing deposits of adipose tissue throughout the neck (the uvula is seen in the pharynx). (C) Non-obese patient with sleep apnoea, showing fat deposits lateral to the pharynx with normal amounts of adipose tissue elsewhere.
(I am indebted to Dr I Mortimore for providing the scans. Parts (A) and (C) reproduced from reference 27 by permission of the publishers of American Journal of Respiratory and Critical Care Medicine.)
Secondly, facial structure may be different in some patients with SAHS, including micrognathia (small mandible) or retrognathia (posterior positioned mandible), both of which will tend to displace the tongue backwards, requiring extra genioglossus activity to maintain a normal-sized airway. This hypothesis raises the interesting possibility that SAHS may begin in early childhood, when enlarged adenoids and tonsils can influence facial bone development, and may also go some way to explaining familial ‘aggregations’ of SAHS and snoring.14
Pharyngeal dilator muscles are more active in awake subjects with SAHS when compared with controls, presumably as a physiological response to the anatomically abnormal airway. The activity is believed to originate from the usual reflex, stimulated by a negative pharyngeal pressure (page 83), which may be present to a greater extent in SAHS subjects even when awake. This requirement for increased pharyngeal muscle activity to maintain airway size may become impossible to maintain during sleep. Coupled with the normal loss of tonic activity of pharyngeal muscles (see above), sleep quickly results in airway obstruction.
Airway collapse occurs only in obstructive sleep apnoea, and normally results from increased upstream resistance behind the soft palate leading to secondary downstream collapse. The ease with which this collapse occurs is a function of the compliance (collapsibility) of the hypopharyngeal walls, opposed by the action of the pharyngeal dilator muscles. Collapse is more likely to occur when pharyngeal compliance is high and particularly when there is increased sub-mucosal fat in the pharynx, a situation that seems to occur more commonly in men than women.28 Severe collapse of the hypopharynx occurs with the combination of enhanced diaphragmatic contraction, depressed pharyngeal dilator muscle activity and upstream obstruction.
Respiratory Control and Arousal in SAHS29
Instability of respiratory control systems also contributes to the pattern of respiration seen with OSA.17,30 Multiple feedback loops are involved in controlling breathing (Chapter 5) such as the responses to Pco2, Po2 and mechanical pharyngeal reflexes. A small alteration in the rate at which a feedback loop detects a physiological change, or responds to that change, will lead to instability of the overall system. Sleep is believed to cause sufficient disturbance of the feedback loops to cause this type of instability, and repetitive respiratory cycles are established, the simplest example being the normal periodic breathing (page 76) seen in old age, a more dramatic example being severe SAHS with long periods of apnoea, hypoxia and hypercarbia.
Apnoea or hypopnoea are terminated when the patient is aroused from sleep, though this arousal is normally subcortical; that is, the patient does not return to full consciousness. Arousal is followed by clearance of the pharyngeal airway, and this is crucially important for survival. In spite of the depressed ventilatory response curves, hypoxia and hypercapnia do contribute to arousal, probably alongside afferent input from pressure-sensitive pharyngeal receptors. Current opinion supports the view that a combination of all these factors results in increased respiratory drive, which brings about arousal.31 Whatever the mechanism, arousal is often accompanied by significant sympathetic discharge.
Medical Effects of SAHS32
The effects of the SAHS are not trivial and, over a period of years, morbidity and mortality in patients with SAHS is considerably higher than controls. There has been difficulty proving that this observation relates to the SAHS itself rather than the associated smoking, obesity and alcohol consumption, though the link with cardiovascular disease is now believed to be independent of these confounding variables, particularly with more severe forms of sleep disordered breathing.32,33,34 There are two main causes of increased mortality:
Sleep deprivation. A night’s sleep that is disturbed hundreds of times, even subconsciously, leaves the individual with severe daytime somnolence, with decrement of performance in many fields. The ability to drive is impaired, such that patients with SAHS have a much greater incidence of accidents than control subjects, with some studies finding a direct association between the AHI and likelihood of an accident.35 Treatment with nasal continuous positive airways pressure (nCPAP, see below) reverses this observation.36
Cardiovascular effects. Each arousal is associated with significant secretion of catecholamines, and often an episode of hypoxia. These events, occurring many times each night, cause multiple adverse effects on the cardiovascular system acting via excessive production of reactive oxygen species (Chapter 26) and by amplification of inflammatory processes.37 It is therefore unsurprising that SAHS is strongly implicated in the development of hypertension,34 and also believed to contribute to the development of heart failure, ischaemic strokes, diabetes mellitus and hypercholesterolaemia.33,38,39
Principles of Therapy40
Conservative treatment. Avoidance of alcohol, sedative drugs and the supine position during sleep will all improve the AHI. Weight loss is effective at reducing the AHI in obese patients with SAHS, and is believed to act by reducing peri-pharyngeal fat, so increasing airway diameter and reducing the tendency of the airway to collapse. There is some evidence that small amounts of weight loss are associated with large reductions in AHI. Many drug therapies for SAHS have been tried, including using respiratory stimulants, or drugs to reduce REM sleep or sympathetic tone, but none are currently recommended for routine use.40
Nasal continuous positive airway pressure (nCPAP)41 aims to avoid the development of a sub-atmospheric pharyngeal pressure sufficient to cause downstream pharyngeal collapse. It requires a well-fitting nasal mask or soft plastic tubes that fit inside the external nares. Compressed air must then be provided at the requisite gas flow, preferably with humidification. nCPAP serves no useful purpose during expiration and systems have been developed to return airway pressure to atmospheric during expiration. In effect this provides a low level of intermittent positive pressure ventilation. Compliance with nCPAP is the only major limitation to its use, and the technique is now widely accepted as the most effective treatment for SAHS, including for the reduction of the daytime somnolence that has such a detrimental effect on the patient’s life.
Oral appliances are available that can be maintained in the mouth at night to move either the tongue or the mandible forward so increasing the size of the airway. They are a non-invasive form of SAHS treatment that is less intrusive than nCPAP, but are also a less effective treatment, so are used mostly for patients who cannot use nCPAP.42
Surgical relief of obstruction. For snoring alone, the first approach is the removal of any pathological obstruction such as nasal polyps that cause downstream collapse, though this may not improve patients with SAHS. A variety of more radical operations are available, including uvulo-palato-pharyngoplasty, which reduces the size of the soft palate and so dampens palatal oscillations and reduces pharyngeal collapse at this level. Non-obese patients with SAHS who have facial bone abnormalities may benefit from maxillofacial corrective surgery, usually involving advancement of the anterior mandible and/or maxilla. Tracheotomy (opened only at night) has been used in some cases as a last resort. The benefits of surgical treatment of SAHS remain uncertain and are now usually reserved for patients who have a specific anatomical abnormality of their airway as part of their SAHS.40
References
*1. Joseph V, Pequignot JM, Van Reeth O. Neurochemical perspectives on the control of breathing during sleep. Respir Physiol Neurobiol.. 2002;130:253-263.
2. Millman RP, Knight H, Kline LR, Shore ET, Chung DC, Pack AI. Changes in compartmental ventilation in association with eye movements during REM sleep. J Appl Physiol.. 1988;65:1196-1202.
3. Douglas NJ, White DP, Weil JV, Pickett CK, Zwillich CW. Hypercapnic ventilatory response in sleeping adults. Am Rev Respir Dis.. 1982;126:758-762.
4. Douglas NJ, White DP, Weil JV, et al. Hypoxic ventilatory response decreases during sleep in normal men. Am Rev Respir Dis.. 1982;125:286-289.
5. Hudgel DW, Devadatta P, Hamilton H. Pattern of breathing and upper airway mechanics during wakefulness and sleep in healthy elderly humans. J Appl Physiol.. 1993;74:2198-2204.
6. Shome B, Wang L-P, Prasad AK, Santere MH, Szeri AZ, Roberts D. Modeling of airflow in the nasopharynx with applications to sleep apnea. J Biomech Eng.. 1998;120:416-422.
7. Roberts D. Invited editorial on ‘Neuromechanical interaction in human snoring and upper airway obstruction’. J Appl Physiol.. 1999;86:1757-1758.
8. Hudgel DW, Hendricks C. Palate and hypopharynx – sites of inspiratory narrowing of the upper airway during sleep. Am Rev Respir Dis.. 1988;138:1542-1547.
9. Tangel DJ, Mezzanotte WS, White DP. Influence of sleep on tensor palatini EMG and upper airway resistance in normal men. J Appl Physiol.. 1991;70:2574-2581.
10. Henke KG. Upper airway muscle activity and upper airway resistance in young adults during sleep. J Appl Physiol.. 1998;84:486-491.
11. Eckert DJ, Malhotra A, Lo YL, White DP, Jordan AS. The influence of obstructive sleep apnea and gender on genioglossus activity during rapid eye movement sleep. Chest. 2009;135:957-964.
12. Kuna ST, Smickley J. Response of genioglossus muscle activity to nasal airway occlusion in normal sleeping adults. J Appl Physiol.. 1988;64:347-353.
13. Nakano H, Ikeda T, Hayashi M, Ohshima E, Onizuka A. Effects of body position on snoring in apneic and nonapneic snorers. Sleep. 2003;2:169-172.
14. Rappai M, Colop N, Kemp S, deShazo R. The nose and sleep-disordered breathing. What we know and what we do not know. Chest. 2003;124:2309-2323.
15. Berger G, Berger R, Oksenberg A. Progression of snoring and obstructive sleep apnoea: the role of increasing weight and time. Eur Respir J. 2009;33:338-345.
16. Jennum P, Riha RL. Epidemiology of sleep apnoea/hypopnoea syndrome and sleep-disordered breathing. Eur Respir J. 2009;33:907-914.
*17. Patil SP, Schneider H, Schwartz AR, Smith PL. Adult obstructive sleep apnea: pathophysiology and diagnosis. Chest. 2007;132:325-337.
18. Bradley TD. Respiratory sleep medicine. A coming of age. Am J Respir Crit Care Med.. 2008;177:363-364.
19. Stradling JR, Davies RJO. Obstructive sleep apnoea/ hypopnoea syndrome: definitions, epidemiology, and natural history. Thorax. 2004;59:73-78.
20. Strohl KP, Redline S. Recognition of obstructive sleep apnea. Am J Respir Crit Care Med.. 1996;154:279-289.
21. Ohayon MM, Guilleminault C, Priest RG, Caulet M. Snoring and breathing pauses during sleep: telephone interview survey of United Kingdom population sample. BMJ. 1997;314:860-863.
22. Akita Y, Kawakatsu K, Hattori C, Hattori H, Suzuki K, Nishimura T. Posture of patients with sleep apnea during sleep. Acta Otolaryngol Supp.. 2003;550:41-45.
23. Mokhlesi B, Tulaimat A. Recent advances in obesity hypoventilation syndrome. Chest. 2007;132:1322-1336.
*24. Crummy F, Piper AJ, Naughton MT. Obesity and the lung: 2. Obesity and sleep disordered breathing. Thorax. 2008;63:738-746.
25. Ryan CM, Bradley TD. Pathogenesis of obstructive sleep apnea. J Appl Physiol.. 2005;99:2440-2450.
26. Sanders MH. The upper airway and sleep-disordered breathing. Getting the big picture. Am J Respir Crit Care Med.. 2003;168:509-515.
27. Mortimore IL, Marshall I, Wraith PK, Sellar RJ, Douglas NJ. Neck and total body fat deposition in nonobese and obese patients with sleep apnea compared with that in control subjects. Am J Respir Crit Care Med.. 1998;157:280-283.
28. O’Donnell CP, Schwartz AR, Smith PL. Upper airway collapsibility. The importance of gender and adiposity. Am J Respir Crit Care Med.. 2000;162:1606-1607.
29. Younes M. Role of respiratory control mechanisms in the pathogenesis of obstructive sleep disorders. J Appl Physiol.. 2008;105:1389-1405.
30. White DP. Pathogenesis of obstructive and central sleep apnea. Am J Respir Crit Care Med.. 2005;172:1363-1370.
31. Cherniack NS. If I die before I wake: not a worry for sleep apnea patients. J Appl Physiol.. 2007;103:1919-1920.
32. McNicholas WT, Bonsignore MR. Sleep apnoea as an independent risk factor for cardiovascular disease: current evidence, basic mechanisms and research priorities, and the Management Committee of EU COST ACTION B26. Eur Respir J, 29. 2007: 156-178.
33. Bradley TD, Floras JS. Obstructive sleep apnoea and its cardiovascular consequences. Lancet. 2009;373:82-93.
34. Logan A. Sleep-disordered breathing and hypertension. Am J Respir Crit Care Med.. 2009;179:1082-1083.
35. Stradling J. Driving and obstructive sleep apnoea. Thorax. 2008;63:481-483.
36. Mazza S, Pépin J-L, Naëgelé B, et al. Driving ability in sleep apnoea patients before and after CPAP treatment: evaluation on a road safety platform. Eur Respir J. 2006;28:1020-1028.
37. Gozal D, Kheirandish-Gozal L. Cardiovascular morbidity in obstructive sleep apnea: oxidative stress, inflammation, and much more. Am J Respir Crit Care Med.. 2008;177:369-375.
38. Murray BJ. Brain death by a thousand hypoxic cuts in sleep. Am J Respir Crit Care Med.. 2007;175:528-529.
39. Lorenzi-Filho G, Drager LF. Obstructive sleep apnea and atherosclerosis: A new paradigm. Am J Respir Crit Care Med.. 2007;175:1219-1221.
*40. Ryan CF. Sleep 9: An approach to treatment of obstructive sleep apnoea/hypopnoea syndrome including upper airway surgery. Thorax. 2005;60:595-604.
41. Gordon P, Sanders MH. Sleep 7: Positive airway pressure therapy for obstructive sleep apnoea/hypopnoea syndrome. Thorax. 2005;60:68-75.
42. Chan ASL, Lee RWW, Cistulli PA. Dental appliance treatment for obstructive sleep apnea. Chest. 2007;132:693-699.