Chapter 26 Hyperoxia and oxygen toxicity
 Breathing oxygen at increased atmospheric pressure achieves very high arterial Po2 values but venous Po2, and therefore minimum tissue Po2, only increases at three atmospheres absolute pressure. Hyperbaric oxygen is used to treat a variety of conditions such as tissue infections, carbon monoxide poisoning and sports injuries, but its use remains controversial. Normal metabolic processes, particularly in the mitochondria, produce a range of powerful oxidising derivatives of oxygen, collectively referred to as reactive oxygen species.
Breathing oxygen at increased atmospheric pressure achieves very high arterial Po2 values but venous Po2, and therefore minimum tissue Po2, only increases at three atmospheres absolute pressure. Hyperbaric oxygen is used to treat a variety of conditions such as tissue infections, carbon monoxide poisoning and sports injuries, but its use remains controversial. Normal metabolic processes, particularly in the mitochondria, produce a range of powerful oxidising derivatives of oxygen, collectively referred to as reactive oxygen species.Chapter 24 described the disastrous consequences of lack of oxygen for life forms that depend on it, but for most organisms hypoxia is an infrequent event. However, oxygen itself also has toxic effects at the cellular level, which organisms have had to oppose by the development of complex antioxidant systems. The activity of toxic oxygen derivatives and antioxidant systems is perfectly balanced for most of the time. Nevertheless, there is a strengthening opinion that over many years oxidative mechanisms predominate, and may be responsible for the generalised deterioration in function associated with ageing.1 In a variety of diseases, or when exposed to extra oxygen, the balance is radically disturbed and oxidative tissue damage results.
Hyperoxia
Hyperventilation, while breathing air, can raise the arterial Po2 to about 16 kPa (120 mmHg). Higher levels can be obtained only by oxygen enrichment of the inspired gas and/or by elevation of the ambient pressure. Although the arterial Po2 can be raised to very high levels, the increase in arterial oxygen content is usually relatively small (Table 26.1). The arterial oxygen saturation is normally close to 95% and, apart from raising saturation to 100%, additional oxygen can be carried only in physical solution. Provided that the arterial/mixed venous oxygen content difference remains constant, it follows that venous oxygen content will rise by the same value as the arterial oxygen content. The consequences in terms of venous Po2 (Table 26.1) are important because minimum tissue Po2 approximates more closely to venous than to arterial Po2. The rise in venous Po2 is trivial when breathing 100% oxygen at normal barometric pressure, and it is necessary to breathe oxygen at 3 atmospheres absolute (ATA) pressure before there is a large increase in venous and therefore tissue Po2. This is because most of the body requirement can then be met by dissolved oxygen, and the saturation of capillary and venous blood remains close to 100%.
Table 26.1 Oxygen levels attained in the normal subject by changes in the oxygen tension of the inspired gas
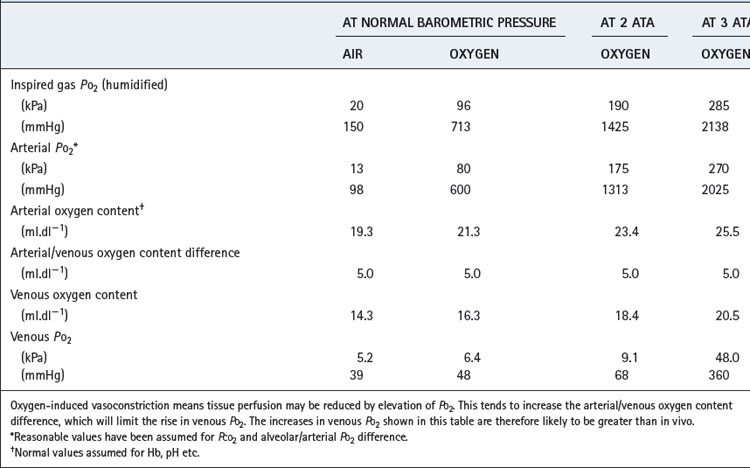
It is convenient to consider two degrees of hyperoxia. The first applies to the inhalation of oxygen-enriched gas at normal pressure, while the second involves inhaling oxygen at raised pressure and is termed hyperbaric oxygenation.
Hyperoxia at Normal Atmospheric Pressure
The commonest indications for oxygen enrichment of the inspired gas are the prevention of arterial hypoxaemia (‘anoxic anoxia’) caused either by hypoventilation (page 399) or by venous admixture (page 133). Oxygen enrichment of the inspired gas may also be used to mitigate the effects of hypoperfusion (‘stagnant hypoxia’). The data in Table 26.1 show that there will be only marginal improvement in oxygen delivery (page 202), but it may be critical in certain situations. ‘Anaemic anoxia’ will be only partially relieved by oxygen therapy but, because the combined oxygen is less than in a subject with normal haemoglobin concentration, the effect of additional oxygen carried in solution will be relatively more important.
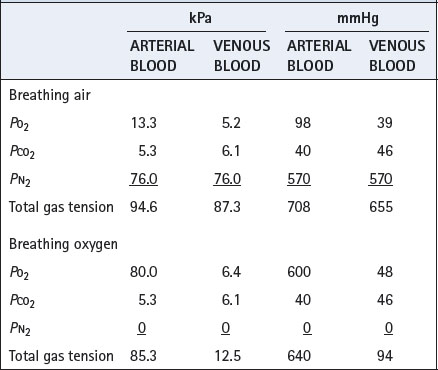
Clearance of gas loculi in the body may be greatly accelerated by the inhalation of oxygen, which greatly reduces the total tension of the dissolved gases in the venous blood (Table 26.2). This results in the capillary blood having additional capacity to carry away gas dissolved from the loculi. Total gas tensions in venous blood are always slightly less than atmospheric, and this is of critical importance in preventing the accumulation of air in potential spaces such as the pleural cavity, where the pressure is subatmospheric. Oxygen is therefore useful in the treatment of air embolism (page 426) and pneumothorax (page 446).
Hyperbaric Oxygenation
Mechanisms of Benefit
Effect on Po2. Hyperbaric oxygenation is the only means by which arterial Po2 values in excess of 90 kPa (675 mmHg) may be obtained. However, it is easy to be deluded into thinking that the tissues will be exposed to a similar Po2 as found in the chamber. Terms such as ‘drenching the tissues with oxygen’ have been used but are meaningless. In fact, the simple calculations shown in Table 26.1, supported by experimental observations, show that large increases in venous and presumably therefore minimum tissue Po2 do not occur until the Po2 of the arterial blood is of the order of 270 kPa (2025 mmHg), when the whole of the tissue oxygen requirements can be met from the dissolved oxygen. However, the relationship between arterial and tissue Po2 is highly variable (page 155), and hyperoxia-induced vasoconstriction in the brain and other tissues limits the rise in venous and tissue Po2. Direct access of ambient oxygen will increase Po2 in superficial tissues, particularly when the skin is breached.
Effect on Pco2. An increased haemoglobin saturation of venous blood reduces its buffering power and carbamino carriage of carbon dioxide, possibly resulting in carbon dioxide retention. In fact, the increase in tissue Pco2 from this cause is unlikely to exceed 1 kPa (7.5 mmHg). However, in the brain this might result in a significant increase in cerebral blood flow, causing a secondary rise in tissue Po2.
Vasoconstriction. An increase in Po2 causes vasoconstriction, which may be valuable for reduction of oedema in the reperfusion of ischaemic limbs and in burns (see below).
Angiogenesis. The growth of new blood vessels is improved when oxygen is increased to more than 1 ATA pressure.2 There seems to be no effect with 100% oxygen at 1 ATA,3 and the mechanism by which angiogenesis is promoted is uncertain. When normoxia follows a period of hypoxia, reactive oxygen species (see below) are produced, and these are known to stimulate the production of a variety of growth factors that initiate angiogenesis.4 The same mechanism may occur during hyperbaric oxygenation.5
Anti-bacterial effect. For many years oxygen was believed to play a role in bacterial killing by the formation of reactive oxygen species, particularly in polymorphs and macrophages, though this has recently been refuted (see below). However, oxygen will still have a direct toxic effect on micro-organisms, particularly on anaerobic bacteria, and relief of hypoxia improves the performance of polymorphs.6
Boyle’s Law effect. The volume of gas spaces within the body is reduced inversely to the absolute pressure according to Boyle’s Law (page 515). This effect is additional to that resulting from reduction of the total tension of gases in venous blood (see above).
Clinical Applications of Hyperbaric Oxygenation7
In practice, hyperbaric oxygen therapy means placing a patient into a chamber at 2–3 ATA and providing apparatus to allow them to breathe 100% oxygen, normally a tight fitting facemask. Treatment is usually for about 1–2 hours, and repeated daily for up to 30 days. Since its first use in 1960 enthusiasm for hyperbaric oxygenation has waxed and waned, but its use is still confined to relatively few centres. Clear indications of its therapeutic value have been slow to emerge from controlled trials, which are admittedly very difficult to conduct in the conditions for which benefit is claimed. In particular, a proper ‘control’ group of patients must undergo a sham treatment in a hyperbaric chamber, which has been used in very few trials. The most commonly accepted indications are as follows.
Infection is the most enduring field of application of hyperbaric oxygenation, particularly anaerobic bacterial infections. High partial pressures of oxygen increase the production of reactive oxygen species, which are cidal not only to anaerobes but also to aerobes. The strongest indications are for clostridial myonecrosis (gas gangrene), refractory osteomyelitis and necrotising soft tissue infections, including cutaneous ulcers.
Gas embolus and decompression sickness are unequivocal indications for hyperbaric therapy and the rationale of treatment is considered above and in Chapter 18.
Carbon monoxide poisoning. In spite of the exploitation of natural gas, there remains a high incidence of carbon monoxide poisoning from automobile exhausts, fires and defective domestic heating appliances. Indications for hyperbaric oxygenation following carbon monoxide poisoning include age over 36 years, loss of consciousness or carboxyhaemoglobin (COHb) levels of more than 25%,8 and it is now accepted that therapy improves delayed neurological sequelae.7,9,10 The rationale of therapy – increased rate of dissociation of COHb – seems simple when the half-life of COHb is approximately 4–5 hours whilst breathing air and only 20 minutes with hyperbaric oxygen. However, breathing 100% oxygen at normal pressure reduces the half life of COHb to just 40 minutes, and therefore in many cases, by the time transport to a hyperbaric chamber is achieved, COHb levels will already be considerably reduced. Other potential benefits of hyperbaric oxygen are believed to derive from minimising the effects of carbon monoxide on cytochrome-c oxidase and reducing lipid peroxidation and neutrophil adherence.9
Wound healing is improved by hyperbaric oxygenation, even when used intermittently. It is particularly useful when ischaemia contributes to the ineffective healing – for example, in diabetes mellitus or peripheral vascular disease. The mechanisms are similar to those for burns, and, in both cases, improved tissue oxygen levels probably result from direct diffusion of oxygen into the affected superficial tissues and increased release of growth factors.5
Sports injuries. Hyperbaric oxygen is believed to expedite recovery from soft-tissue injuries and fractures incurred during competitive sports.11 Early treatment (within 8 hours) is most effective indicating a probable effect on neutrophil activity at the site of injury.12
Multiple sclerosis. In the early 1980s there was great interest in the therapeutic value of hyperbaric oxygenation in multiple sclerosis. A small study reported a favourable response in a double-blind controlled trial in which the treated group received 2 ATA oxygen, while the placebo group inhaled 10% oxygen in nitrogen, also at 2 atmospheres.13 Unfortunately, these findings were not confirmed in subsequent studies, and a review of 14 controlled trials concluded that hyperbaric oxygen cannot be recommended for the treatment of multiple sclerosis.14
Oxygen Toxicity
The Oxygen Molecule and Reactive Oxygen Species (ROS)15,16
Although ground state oxygen (dioxygen) is a powerful oxidising agent, the molecule is stable and has an indefinite half-life. However, the oxygen molecule can be transformed into a range of ROS and other highly toxic substances, most of which are far more reactive than oxygen itself.
The dioxygen molecule (Figure 26.1) is unusual in having two unpaired electrons in the outer (2P) shell. Thus dioxygen itself qualifies as a ‘double’ free radical, but stability is conferred by the fact that the orbits of the two unpaired electrons are parallel. The two unpaired electrons also confer the property of paramagnetism, which has been exploited as a method of gas analysis that is almost specific for oxygen (page 208).
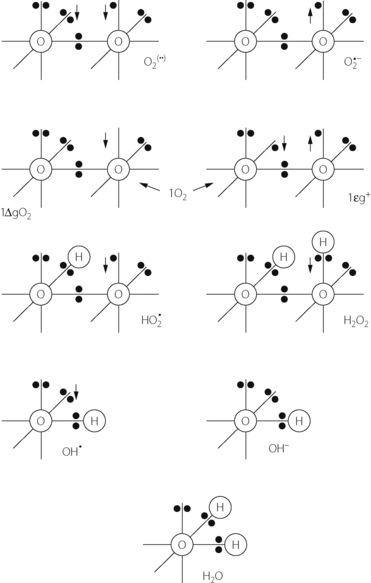
Fig. 26.1 Outer orbital ring of electrons in (from the top left): ground state oxygen or dioxygen (O2); superoxide anion (O2 −); two forms of singlet oxygen (1O2); hydroperoxyl radical (HO2); hydrogen peroxide (H2O2); hydroxyl radical (OH); hydroxyl ion (OH−); and water. The arrows indicate the direction of rotation of unpaired electrons. See text for properties and interrelationships.
−); two forms of singlet oxygen (1O2); hydroperoxyl radical (HO2); hydrogen peroxide (H2O2); hydroxyl radical (OH); hydroxyl ion (OH−); and water. The arrows indicate the direction of rotation of unpaired electrons. See text for properties and interrelationships.
Singlet oxygen. Internal rearrangements of the unpaired electrons of dioxygen result in the formation of two highly reactive species, both known as singlet oxygen (1O2). In 1ΔgO2 one unpaired electron is transferred to the orbit of the other (Figure 26.1), imparting an energy level 22.4 kcal.mol−1 above the ground state. There being no remaining unpaired electron, 1ΔgO2 is not a ROS. In 1Σg+, the rotation of one unpaired electron is reversed, which imparts an energy level 37.5 kcal.mol−1 above the ground state and this molecule is a ROS. 1Σg+ is extremely reactive, and rapidly decays to the 1ΔgO2 form, which is particularly relevant in biological systems and especially to lipid peroxidation.
Superoxide anion. Under a wide range of circumstances, considered below, the oxygen molecule may be partially reduced by receiving a single electron, which pairs with one of the unpaired electrons forming the superoxide anion (O2− in Figure 26.1), which is both an anion and a ROS. It is the first and crucial stage in the production of a series of toxic oxygen-derived ROS and other compounds. The superoxide anion is relatively stable in aqueous solution at body pH, but has a rapid biological decay due to the ubiquitous presence of superoxide dismutase (see below). Being charged, superoxide anion does not readily cross cell membranes.
Hydroperoxyl radical. Superoxide anion may acquire a hydrogen ion to form the hydroperoxyl radical thus:
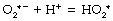
The reaction is pH dependent with a pK of 4.8, so the equilibrium is far to the left in biological systems.
Hydrogen peroxide. Superoxide dismutase (SOD) catalyses the transfer of an electron from one molecule of the superoxide anion to another. The donor molecule becomes dioxygen while the recipient rapidly combines with two hydrogen ions to form hydrogen peroxide (Figure 26.1). Although hydrogen peroxide is not a ROS, it is a powerful and toxic oxidising agent that plays an important role in oxygen toxicity. The overall reaction is as follows:
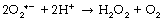
Hydrogen peroxide is continuously generated in the body. Two enzymes ensure its rapid removal. Catalase is a highly specific enzyme active against only hydrogen, methyl and ethyl peroxides. Hydrogen peroxide is reduced to water thus:
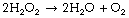
Glutathione peroxidase acts against a much wider range of peroxides (R-OOH), which react with glutathione (G-SH) thus:
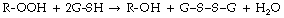
Catalase and glutathione peroxidase are discussed further below. Obligatory anaerobic bacteria are normally without catalase.
Three stage reduction of oxygen. Figure 26.2 summarises the three-stage reduction of oxygen to water, which is the fully reduced and stable state. This contrasts with the more familiar single-stage reduction of oxygen to water that occurs in the terminal cytochrome (page 200). Unlike the single-stage reduction of oxygen, the three-stage reaction shown in Figure 26.2 is not inhibited by cyanide.
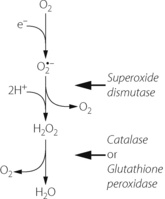
Fig. 26.2 Three-stage reduction of oxygen to water. The first reaction is a single electron reduction to form the superoxide anion reactive oxygen species. In the second stage the first products of the dismutation reaction are dioxygen and a short-lived intermediate, which then receives two protons to form hydrogen peroxide. The final stage forms water, the fully reduced form of oxygen.
Secondary Derivatives of the Products of Dioxygen Reduction
The Fenton reaction. Although both the superoxide anion and hydrogen peroxide have direct toxic effects, they interact to produce even more dangerous species. To the right of Figure 26.3 is shown the Fenton or Haber-Weiss reaction, which results in the formation of the harmless hydroxyl ion together with two extremely reactive species, the hydroxyl free radical (OH) and singlet oxygen (1O2).
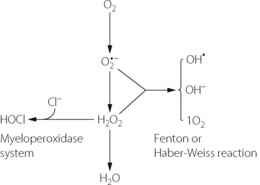
Fig. 26.3 Interaction of superoxide anion and hydrogen peroxide in the Fenton or Haber-Weiss reaction to form hydroxyl free radical, hydroxyl ion and singlet oxygen. Hypochlorous acid is formed from hydrogen peroxide by the myeloperoxidase system.
(After reference 17 by courtesy of the Editor of the Journal of the Royal Society of Medicine.)
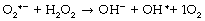
The hydroxyl free radical is much the most dangerous ROS derived from oxygen.
The myeloperoxidase reaction.18 To the left of Figure 26.3 is shown the reaction of hydrogen peroxide with chloride ion to form hypochlorous acid. This occurs in the phagocytic vesicle of the neutrophil and plays a role in bacterial killing. The reaction is accelerated by the enzyme myeloperoxidase, which comprises some 7% of the dried weight of a neutrophil. The myeloperoxidase reaction also occurs immediately after fertilisation of the ovum, and hypochlorous acid so formed causes polymerisation of proteins to form the membrane that prevents the further entry of spermatozoa.
Relationship to ionising radiation. The changes described above have many features in common with those caused by ionising radiation, the hydroxyl radical (OH) being the most dangerous product in both cases. It is, therefore, hardly surprising that the effect of radiation is increased by high partial pressures of oxygen. As tissue Po2 is reduced below about 2 kPa (15 mmHg), there is progressively increased resistance to radiation damage until, at zero Po2, resistance is increased three-fold. This unfortunate effect promotes resistance to radiotherapy of malignant cells in hypoxic areas of tumours (page 444).
Nitric oxide may behave as a ROS by reacting with the superoxide anion to produce peroxynitrite (ONOO−).15 This molecule can either rearrange itself into relatively harmless nitrite or nitrate (page 194), or give rise to derivatives with similar biological activity to the hydroxyl radical. Conversely, nitric oxide may act as an antioxidant, binding to ferrous iron molecules and preventing them from contributing to the formation of superoxide anion (see below) or the Fenton reaction. The in vivo role of nitric oxide as a ROS or antioxidant therefore remains unclear.19
Sources of Electrons for the Reduction of Oxygen to Superoxide Anion
Figure 26.3 shows the superoxide anion as the starting point for the production of many other ROS. The first stage reduction of dioxygen to the superoxide anion is therefore critically important in oxygen toxicity.
Mitochondrial enzymes. NADH dehydrogenase and a variety of other mitochondrial enzymes may ‘leak’ electrons to molecular oxygen and so produce superoxide anions during normal oxidative respiration.15,20 Animal studies indicate that this may account for almost 8% of total oxygen consumption, indicating the importance of the highly efficient mitochondrial form of SOD (see below).
The NADPH oxidase system is the major electron donor in neutrophils and macrophages. The electron is donated from NADPH by the enzyme NADPH oxidase, which is located within the membrane of the phagocytic vesicle. This mechanism is activated during phagocytosis and is accompanied by a transient increase in the oxygen consumption of the cells, a process known to be cyanide resistant. This is the so-called respiratory burst, and occurs in all phagocytic cells in response to a wide range of stimuli including bacterial endotoxin, immunoglobulins and interleukins. Superoxide anion is released into the phagocytic vesicle, where it is reduced to hydrogen peroxide which then reacts with chloride ions to form hypochlorous acid in the myeloperoxidase reaction (Figure 26.3). For many years the release of ROS into the phagocyte was believed to be the main way in which bacteria were killed by phagocytes. Recent work on pulmonary neutrophils in mice with pneumococcal infection has refuted this claim, finding no evidence of bacterial killing by neutrophil-generated ROS, though ROS were involved in neutrophil regulation.21 Powerful protease enzymes released into the phagosome by the neutrophil may be the most important bactericidal mechanism.
Although the NADPH oxidase system has extremely important biological functions, there seems little doubt that its inappropriate activation in marginated neutrophils can damage the endothelium of the lung, and it may well play a part in the production of acute lung injury (Chapter 31).
Xanthine oxidoreductase (XOR) and reperfusion injury.22 The enzyme XOR is responsible for the conversion of hypoxanthine and xanthine to uric acid (Figure 26.4). XOR is a large (300 kDa) protein involving two separate substrate binding sites, one including flavine adenine dinucleotide cofactor and the other a molybdenum molecule. In vivo, XOR exists in two interchangeable forms, with about 80% existing as xanthine dehydrogenase and the remainder as xanthine oxidase. In both forms XOR catalyses the conversion of both hypoxanthine to xanthine and of xanthine to uric acid, and under normal conditions uses NADH as a cofactor. In ischaemic or hypoxic tissue large quantities of hypoxanthine accumulate (page 363), the availability of NADH declines, and the ratio of the oxidase and dehydrogenase forms of XOR may be reversed. As a result of these changes, when oxygen is restored to the cell, the XOR catalysis of xanthine and hypoxanthine is altered with NAD+ and dioxygen now being used as cofactors, resulting in the production of hydrogen peroxide and superoxide anions (Figure 26.4). Thus during reperfusion there may be extensive production of oxygen-derived free radicals. It seems probable that, under certain circumstances, this mechanism may play a role in reperfusion tissue damage or postischaemic shock.23
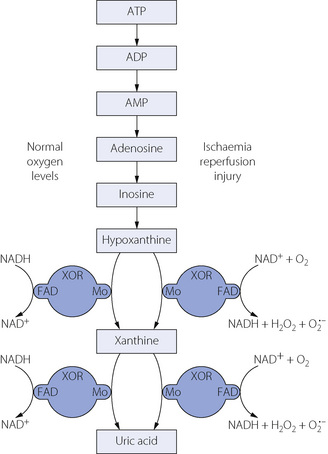
Fig. 26.4 Generation of superoxide anion from oxygen by the activity of xanthine oxidoreductase (XOR). With normal cellular oxygen levels (left side) NADH is the cofactor, binding at the flavine adenine dinucleotide (FAD) site whilst the substrate reacts with the molybdenum binding site at the opposite side of the XOR molecule. Following a period of ischaemia (right side), reperfusion causes NAD+ and oxygen rather than NADH to react at the FAD binding site of XOR, resulting in the production of hydrogen peroxide or superoxide anion.
Ferrous iron (Fe2+) loses an electron during conversion to the ferric (Fe3+) state. This is an important component of the toxicity of ferrous iron. A similar reaction also occurs during the spontaneous oxidation of haemoglobin to methaemoglobin (page 196). It is for this reason that large quantities of SOD, catalase and other protective agents are present in the young red blood cell. Their depletion may well determine the lifespan of the cell. Apart from ferrous iron acting as an electron donor, it is a catalyst in the Fenton reaction (see above).
High Po2. Whatever other factors may apply, the production of ROS is increased at high levels of Po2 by the law of mass action. It would seem that the normal tissue defences against ROS (discussed below) are usually effective only up to a tissue Po2 of about 60 kPa (450 mmHg). This accords with the development of clinical oxygen toxicity as discussed below. There is also evidence that generation of ROS is increased when normal oxygen usage is increased, for example during exercise.24
Exogenous compounds. Various drugs and toxic substances can act as an analogue of NADPH oxidase and transfer an electron from NADPH to molecular oxygen. The best example of this is paraquat which can, in effect, insert itself into an electron transport chain, alternating between its singly and doubly ionised form. This process is accelerated at high levels of Po2 and so there is a synergistic effect between paraquat and oxygen. Paraquat is concentrated in the alveolar epithelial type II cell where the Po2 is as high as anywhere in the body. Due to the very short half-life of the oxygen-derived free radicals, damage is confined to the lung. Bleomycin and some antibiotics (e.g. nitrofurantoin) can act in a similar manner. Reactions usually occur at high dose levels, are again potentiated by increased oxygen levels or radiation, and eventually lead to pulmonary fibrosis.
Biological Effects of ROS
Their use in the regulation of phagocytes, and possibly in the killing of microorganisms is a beneficial role for ROS. Elsewhere within cells, the balance between the detrimental effects of ROS and the antioxidants that counter these (see below) is described as the redox state of the cell. Cellular redox state is believed to be part of an essential, and poorly understood, cell signalling system,25,26 being involved, for example, in the sensing of oxygen levels in the carotid body. Otherwise, most effects of ROS on biological systems are harmful, and alterations in redox state are linked to a diverse range of diseases.
The three main biochemical targets for ROS damage are deoxyribonucleic acid (DNA), lipids and sulphydryl-containing proteins. All three are also sensitive to ionising radiation. The mechanisms of both forms of damage have much in common and synergism occurs.
DNA. Breakage of chromosomes in cultures of animal lung fibroblasts by high concentrations of oxygen was first demonstrated in 197827 (Figure 26.5). In-vivo studies of therapeutic hyperbaric oxygen in humans have also shown DNA damage. However, adverse clinical outcomes from hyperbaric oxygen have not been demonstrated, though susceptible subgroups, who have less effective cellular antioxidant or DNA repair systems, may exist.28
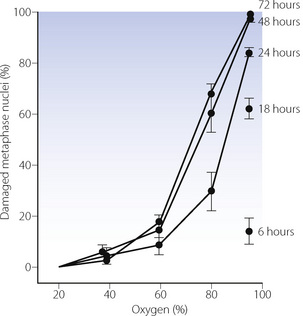
Fig. 26.5 Breakage of chromosomes in a culture of Chinese hamster lung fibroblasts by oxygen at various concentrations and for varying durations of exposure.
(Reproduced from reference 27 by courtesy of the Editors of Mutation Research.)
Lipids. There is little doubt that lipid peroxidation is a major mechanism of tissue damage by ROS. The interaction of a ROS with an unsaturated fatty acid not only disrupts that particular lipid molecule but also generates another ROS so that a chain reaction ensues until stopped by an antioxidant.16 Lipid peroxidation disrupts cell membranes and accounts for the loss of integrity of the alveolar/capillary barrier in pulmonary oxygen toxicity.
Proteins. Damage to sulphydryl-containing proteins results in formation of disulphide bridges, which inactivates a range of proteins.
Interference with these fundamental cellular molecules has widespread physiological implications. Superoxide anion, and the peroxynitrite formed from nitric oxide, initiate a wide range of pathological processes, including the inactivation of neurotransmitters, inhibition of proteins, release of cytokines and direct cytotoxic effects (Figure 26.6).29 Inevitably, cell dysfunction will rapidly occur, followed over the long term by the occurrence of inflammation, malignancy or cell death. Over an animal’s lifetime, ROS-induced damage is now closely linked with cardiovascular30 and neurological disease, cancer31 and the degenerative changes of ageing.1
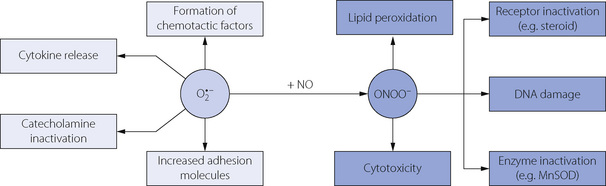
Fig. 26.6 Biochemical effects of superoxide anion and peroxynitrite. These potent cellular effects initiate numerous pathological processes including inflammation, malignancy or cell death. MnSOD, manganese superoxide dismutase.
(After reference 29 with permission of the authors.)
Defences against Reactive Oxygen Species
Life in an oxidising environment is possible only because of powerful anti-oxidant defences, which all aerobes have developed (Chapter 1). The defensive systems are freely duplicated and operate in depth.
Antioxidant Enzymes
These enzymes are widely distributed in different organs and different species but are deficient in most obligatory anaerobic bacteria. Young animals normally have increased levels of SOD and catalase, which confers greater resistance to oxygen toxicity. The reactions catalysed by antioxidant enzymes have been described above.
Superoxide dismutase.32,33 Three types of SOD exist, each derived from a separate gene; extracellular SOD, cytoplasmic SOD containing manganese (MnSOD) and mitochondrial SOD containing both copper and zinc (CuZnSOD). Extra production of SOD may be induced by several mechanisms, of which hyperoxia is the most notable, but inflammatory cytokines such as interferon, TNF, interleukins and lipopolysaccharide are important stimulants of SOD production in the intact animal.
Animal studies have consistently shown that induction of SOD confers some protection against the toxic effects of oxygen,15 and, by implication, enhanced SOD activity may be protective against the wide range of pathological processes described above. There are difficulties in the therapeutic use of SOD because the most important forms are intracellular or mitochondrial enzymes which have very short half-lives in plasma. There is therefore little scope for their use by direct intravenous injection. It is possible for SOD to enter cells if it is administered in liposomes, and extracellular SOD has been used by direct instillation into the lungs.34 Recent attempts to enhance SOD activity for therapeutic purposes have switched to the development of SOD mimetics.29 A number of small polycyclic compounds, mostly containing a central manganese molecule, have been found to catalyse the same reactions as SOD, but because of their small size and non-peptide nature they can freely enter the intracellular environment. SOD mimetics have yet to begin clinical trials, but their therapeutic potential for the future looks promising.
Catalase has a cellular and extracellular distribution similar to SOD, with which it is closely linked in disposing of superoxide anion (Figure 26.2). Although studied less extensively, catalase production is believed to be induced by the same factors as SOD. Similarly, trials of exogenous antioxidant enzymes have usually given better results when both SOD and catalase are administered.
Glutathione peroxidase system scavenges not only the ROS themselves but also reactive species formed during lipid peroxidation. Two molecules of the tripeptide (glycine-cysteine-glutamic acid) glutathione (GSH) are oxidised to one molecule of reduced glutathione (GSSG) by the formation of a disulphide bridge linking the cysteine residues. GSH is reformed from GSSG by the enzyme glutathione reductase, protons being supplied by NADPH.
Endogenous Antioxidants
Ascorbic acid is a small molecule with significant antioxidant properties, being particularly important for removal of the hydroxyl free radical. Humans, along with guinea-pigs and bats, lack the enzyme required for the production of ascorbate, and so must ingest sufficient vitamin C to compensate. In these mammals, SOD activity is markedly higher than in those able to produce endogenous ascorbate.35
Vitamin E (α-tocopherol) is a highly fat-soluble compound and is therefore found in high concentrations in cell membranes. Predictably, its main antioxidant role is in the prevention of lipid peroxidation chain reactions described above.
Glutathione is found in high concentrations in the airway lining fluid as part of the glutathione peroxidase system described above. Widespread use of paracetamol, which at high doses can reduce glutathione levels in the lung, may attenuate the antioxidant activity provided by glutathione so increasing oxidative stress and possibly contributing to the increasing incidences of asthma and COPD (Chapter 28).36
Surfactant may act as an antioxidant in the lung. Animal studies have shown that administration of exogenous surfactant prolongs the duration of oxygen exposure required to cause lung damage.37
Exogenous Antioxidants
Allopurinol. Because XOR plays a pivotal role in the reactions shown in Figure 26.4 it seemed logical to explore the use of allopurinol, which inhibits a range of enzymes including XOR. As may be expected, benefit was seen mainly following ischaemia-reperfusion injury, but under these conditions allopurinol has multiple effects on purine metabolism and may not be acting as a XOR inhibitor at all.22
Iron-chelating agents. Since ferrous iron is both a potent source of electrons for conversion of oxygen to the superoxide anion and a catalyst in the Fenton reaction, desferrioxamine has antioxidant properties in vitro.38
These compounds, along with other in vitro antioxidants such as n-acetyl cysteine, β-carotene and dimethylsulphoxide have generally failed to live up to their expectations in human disease. There are three possible explanations. First, studies of ROS production and antioxidants in human cells are relatively rare, and there is known to be considerable species variability.15 Secondly, penetration of the exogenous antioxidant to the site of ROS generation (e.g. mitochondria) or damage (e.g. nuclear DNA) is likely to be poor. Finally, ROS involvement in physiological systems such as neutrophil regulation is crucial, so any non-specific antioxidant activity may be detrimental. Their therapeutic role in oxygen toxicity or diseases known to involve ROS is therefore far from fully clarified.
Clinical Oxygen Toxicity
The most important clinical conditions in which oxygen has been identified as the sole precipitating cause are oxygen convulsions, pulmonary oxygen toxicity and retrolental fibroplasia.
Oxygen Convulsions (The Paul Bert Effect)
It is well established that exposure to oxygen at a partial pressure in excess of 2 atmospheres absolute (2 ATA) may result in convulsions, usually preceded by a variety of non-specific neurological symptoms such as headache, and visual disturbances.39 This limits the depth to which closed-circuit oxygen apparatus can be used. It is interesting that the threshold for oxygen convulsions is close to that at which brain tissue Po2 is likely to be sharply increased (Table 26.1). The relationship to cerebral tissue Po2 is supported by the observation that an elevation of Pco2 lowers the threshold for convulsions. High Pco2 increases cerebral blood flow and therefore raises the tissue Po2 relative to the arterial Po2. Hyperventilation and anaesthesia each provide limited protection.
Convulsions result from poorly understood changes in cellular interactions between gamma-aminobutyric acid (GABA) and nitric oxide. GABA concentrations decrease in the brain prior to convulsion and the change correlates with the severity of the convulsion.40 As GABA is an inhibitory neurotransmitter, it is not unreasonable to suggest that a reduced level might result in convulsions. Nitric oxide is known to sensitise neurones to the toxic effects of GABA in hypoxia, and is also involved in hyperoxic convulsions. Nitric oxide inhibitors delay the onset of convulsions in hyperoxia,41 but paradoxically, the same effect is seen with some NO donors.42 Whatever the role of NO, the final common pathway seems to be mediated by disturbed calcium fluxes and increased cyclic-GMP concentration.41
Incidence. Hyperbaric oxygen used therapeutically as described above – that is, intermittent exposure to less than 3 ATA – carries little risk of oxygen convulsions. At 2 ATA, a large series reported no convulsions in over 12 000 treatments.43 Treatment for CO poisoning is associated with a greater incidence of convulsions because of the higher pressures used (normally 2.8–3.0 ATA) and the toxic effects of CO on the brain itself. In this case, 1–2% of patients experience convulsions.44
Pulmonary Oxygen Toxicity
Pulmonary tissue Po2 is the highest in the body. In addition, a whole range of other oxidising substances may be inhaled, including common air pollutants and the constituents of cigarette smoke (Chapter 21). The lung is therefore the organ most vulnerable to oxygen toxicity and a range of defence mechanisms have developed. Overall antioxidant activity from both enzymes and other endogenous antioxidants is very high in the fluid lining the respiratory tract. Extracellular SOD is abundant in pulmonary airway tissues, and abnormalities in its regulation may contribute to some lung diseases.45 Type II alveolar epithelial cells, which produce surfactant (page 23), are believed to also incorporate vitamin E into the surfactant lipids.46
Pulmonary oxygen toxicity is unequivocal and lethal in laboratory animals such as the rat. Humans seem to be far less sensitive, but there are formidable obstacles to investigation of both human volunteers and patients. Study of oxygen toxicity in the clinical environment is complicated by the presence of the pulmonary pathology that necessitated the use of oxygen.
Symptoms.47 High concentrations of oxygen cause irritation of the tracheobronchial tree, which gives rise initially to a sensation of retrosternal tightness. Continued exposure leads to chest pain, cough, and an urge to take deep breaths. Reduced vital capacity is the first measurable change in lung function, occurring after about 24 hours of normobaric 100% oxygen. Oxygen exposure beyond this point leads to the widespread structural changes described below, which ultimately give rise to acute lung injury and possibly irreversible changes in lung function.
Cellular changes.48 Electron microscopy has shown that, in rats exposed to 1 atmosphere of oxygen, the primary change is in the capillary endothelium, which becomes vacuolated and thin. Permeability is increased and fluid accumulates in the interstitial space. At a later stage, in monkeys, the epithelial lining is lost over large areas of the alveoli. This process affects the type I cell (page 22) and is accompanied by proliferation of the type II cell, which is relatively resistant to oxygen. The alveolar/capillary membrane is greatly thickened, partly because of the substitution of type II for type I cells and partly because of interstitial accumulation of fluid.
Limits of survival. Pulmonary effects of oxygen vary greatly between different species, probably because of different levels of provision of defences against free radicals. Most strains of rat will not survive for much more than 3 days in 1 atmosphere of oxygen. Monkeys generally survive oxygen breathing for about 2 weeks, and humans are probably even more resistant. Oxygen tolerance in humans has been investigated,49 but these studies are based on reduction in vital capacity etc., which is a very early stage of oxygen toxicity. There is an approximately inverse relationship between Po2 and duration of tolerable exposure. Thus 20 hours of 1 atmosphere had a similar effect to 10 hours of 2 atmospheres or 5 hours of 4 atmospheres.
Pulmonary oxygen toxicity seems to be related to Po2 rather than inspired concentration. Early American astronauts breathed 100% oxygen at a pressure of about one-third of an atmosphere for many days (Table 19.1) with no apparent ill effects. There is abundant evidence that prolonged exposure to this environment does not result in demonstrable pulmonary oxygen toxicity, thus establishing a Po2 of 34 kPa (255 mmHg) as a safe level. It also confirms that the significant factor is partial pressure and not concentration. In contrast, the concentration of oxygen rather than its partial pressure is the important factor in absorption collapse of the lung (see below).
Clinical studies. Some limited information on human pulmonary oxygen toxicity has been obtained from patients in the course of therapeutic administration of oxygen. A study in 1967 of patients who died after prolonged artificial ventilation found more structural pulmonary abnormalities (fibrin membranes, oedema and fibrosis) in those who had received 100% oxygen.50 However, the higher concentrations of oxygen would probably have been used in the patients with more severe defects in gas exchange, and it is therefore difficult to distinguish between the effects of oxygen itself and the conditions which required its use. A similar group of patients ventilated for long periods with high concentrations of oxygen were reviewed in 1980,51 and these authors concluded that adverse effects of oxygen on the alveolar epithelium were rarely of practical importance in hypoxaemic patients. An attempt to avoid the complicating factor of pre-existing pulmonary disease was made in 197052 by ventilating a group of patients with 100% oxygen for 24 hours after cardiac surgery. Various indices of pulmonary function (Vd/Vt ratio, shunt and compliance) were not significantly different from a control group receiving less than 42% oxygen.
In contrast to these essentially negative findings, a study in 1987 obtained positive findings in a randomised trial involving patients ventilated after cardiac surgery.53 Venous admixture was significantly greater and arterial Po2 less in patients receiving 50% oxygen compared with the group receiving less than 30%. There are many possible causes for these changes but the authors concluded that unnecessary elevation of inspired oxygen concentration should be avoided, a view from which few would dissent at present.
Pulmonary absorption collapse. Whatever the uncertainties about the susceptibility of humans to pulmonary oxygen toxicity, there is no doubt that high concentrations of oxygen in zones of the lung with low ventilation/perfusion ratios will result in collapse. This occurs routinely during anaesthesia (page 334), and may be demonstrated in the healthy volunteers. A few minutes of breathing oxygen at residual lung volume results in radiological evidence of collapse, a reduced arterial Po2 and substernal pain on attempting a maximal inspiration.54
Balancing the risks. Prevention of dangerous hypoxia is always the first priority and must be treated in spite of the various hazards associated with the use of oxygen. Recent guidelines have suggested that emergency oxygen therapy should always by targeted at a pre-determined oxygen saturation, suggested values being 94–98% in most acutely ill patients or 88–92% for those at risk of hypercapnia.55 When these targets are not achievable by increasing inspired oxygen alone, it is important to remember that oxygen delivery can also be increased by improving cardiac output and haemoglobin levels.
The cornerstone of avoiding the potentially harmful effects of oxygen in the clinical environment is prevention. Although, brief periods of exposure to 100% oxygen appear safe, inspired oxygen concentrations should be titrated against arterial Po2. This is particularly important in patients exposed to paraquat or bleomycin.
Retrolental Fibroplasia (RLF)56
Shortly after RLF was first described in 1942, it became established that hyperoxia was the major aetiological factor and led to the use of oxygen being strictly curtailed in the management of neonates. This resulted in an increase in morbidity and mortality attributable to hypoxia and thereafter oxygen was carefully monitored and titrated in the hope of steering the narrow course between the Scylla of hypoxia and the Charybdis of RLF. This policy has not eradicated the condition, and there is some evidence that RLF may occur in infants who have never received additional oxygen. Vitamin E has been used in the attempt to prevent RLF but it is currently believed that hyperoxia is but one of a variety of factors that may cause RLF by changes in the retinal oxygen supply. RLF is increasingly likely to occur with greater degrees of prematurity, and there is a well-established inverse relationship between birth weight and its incidence.
References
1. Ershler WB. A gripping reality: oxidative stress, inflammation, and the pathway to frailty. J Appl Physiol.. 2007;103:3-5.
2. Thom SR. Hyperbaric oxygen therapy. Intensive Care Med.. 1989;4:58-63.
3. Marx RE, Ehler WJ, Tayapongsak P, Pierce LW. Relationship of oxygen dose to angiogenesis induction in irradiated tissue. Am J Surg.. 1990;160:519-524.
4. Maulik N, Das DK. Redox signalling in vascular angiogenesis. Free Radic Biol Med.. 2002;33:1047-1060.
5. Thom SR. Oxidative stress is fundamental to hyperbaric oxygen therapy. J Appl Physiol.. 2009;106:988-995.
6. Mandell G. Bactericidal activity of aerobic and anaerobic polymorphonuclear neutrophils. Infect Immun.. 1974;9:337-341.
7. Tibbles PM, Edelsberg JS. Hyperbaric-oxygen therapy. N Engl J Med.. 1996;334:1642-1648.
8. Weaver LK, Valentine KJ, Hopkins RO. Carbon monoxide poisoning. Risk factors for cognitive sequelae and the role of hyperbaric oxygen. Am J Respir Crit Care Med.. 2007;176:491-497.
*9. Stoller KP. Hyperbaric oxygen and carbon monoxide poisoning: a critical review. Neurol Res.. 2007;29:146-155.
10. Thom SR. Hyperbaric-oxygen therapy for acute carbon monoxide poisoning. N Engl J Med.. 2002;347:1105-1106.
11. Babul S, Rhodes EC. The role of hyperbaric oxygen therapy in sports medicine. Sports Med.. 2000;30:395-403.
12. Staples J, Clement D. Hyperbaric oxygen chambers and the treatment of sports injuries. Sports Med.. 1996;22:219-227.
13. Fischer BH, Marks M, Reich T. Hyperbaric-oxygen treatment of multiple sclerosis. A randomized, placebo controlled, double blind study. N Engl J Med.. 1983;308:181-184.
14. Kleijnen J, Knipschild P. Hyperbaric oxygen for multiple sclerosis. Review of controlled trials. Acta Neurol Scand.. 1995;91:330-334.
15. Kinnula VL, Crapo JD, Raivio KO. Generation and disposal of reactive oxygen metabolites in the lung. Lab Invest.. 1995;73:3-19.
16. Webster NR, Nunn JF. Molecular structure of free radicals and their importance in biological reactions. Br J Anaesth.. 1988;60:98-108.
17. Nunn JF. Oxygen – friend or foe. J Roy Soc Med.. 1985;78:618-622.
18. Fantone JC, Ward PA. Role of oxygen-derived free radicals and metabolites in leukocyte-dependent inflammatory reactions. Am J Pathol.. 1982;107:397-418.
*19. Dweik RA. Nitric oxide, hypoxia, and superoxide: the good, the bad, and the ugly!. Thorax. 2005;60:265-267.
20. Turrens JF. Mitochondrial formation of reactive oxygen species. J Physiol.. 2003;552:335-344.
21. Marriott HM, Jackson LE, Wilkinson TS, et al. Reactive oxygen species regulate neutrophil recruitment and survival in pneumococcal pneumonia. Am J Respir Crit Care Med.. 2008;177:887-895.
*22. Harrison R. Structure and function of xanthine oxidoreductase: Where are we now? Free Radic Biol Med.. 2002;33:774-797.
23. Traystman RJ, Kirsch JR, Koehler RC. Oxygen radical mechanisms of brain injury following ischaemia and reperfusion. J Appl Physiol.. 1991;71:1185-1195.
24. Kantner M. Free radicals, exercise and antioxidant supplementation. Proc Nutrit Soc.. 1998;57:9-13.
25. Forman HJ, Torres M. Reactive oxygen species and cell signalling. Respiratory burst in macrophage signaling. Am J Respir Crit Care Med.. 2002;166:S4-S8.
26. Clanton T. Yet another oxygen paradox. J Appl Physiol.. 2005;99:1245-1246.
27. Sturrock JE, Nunn JF. Chromosomal damage and mutations after exposure of Chinese hamster cells to high concentrations of oxygen. Mutat Res.. 1978;57:27-31.
*28. Speit G, Dennog C, Radermacher P, Rothfuss A. Genotoxicity of hyperbaric oxygen. Mutat Res.. 2002;512:111-119.
29. Salvemini D, Riley DP, Cuzzocrea S. SOD mimetics are coming of age. Nat Rev Drug Discov.. 2002;1:367-374.
30. Rapola JM. Should we prescribe antioxidants to patients with coronary heart disease. Eur Heart J. 1998;19:530-532.
31. Hennekens CH, Buring JE, Manson JE, et al. Lack of effect of long-term supplementation with beta carotene on the incidence of malignant neoplasms and cardiovascular disease. N Engl J Med.. 1996;334:1145-1149.
32. Zelko IN, Mariani TJ, Folz RJ. Superoxide dismutase multigene family: A comparison of the CuZn-SOD (SOD1), Mn-SOD (SOD2), and EC-SOD (SOD3) gene structures, evolution, and expression. Free Radic Biol Med.. 2002;33:337-349.
*33. Kinnula VL, Crapo JD. Superoxide dismutases in the lung and human lung diseases. Am J Respir Crit Care Med.. 2003;167:1600-1619.
34. Barnard ML, Baker RR, Matalon S. Mitigation of oxidant injury to lung microvasculature by intratracheal instillation of antioxidant enzymes. Am J Physiol.. 1995;265:L340-L367.
35. Nandi A, Mukhopadhyay CK, Ghosh MK, Chattopadhyay DJ, Chatterjee IB. Evolutionary significance of vitamin C biosynthesis in terrestrial vertebrates. Free Radic Biol Med.. 1997;22:1047-1054.
36. McKeever TM, Lewis SA, Smit HA, Burney P, Britton JR, Cassano PA. The association of acetaminophen, aspirin, and ibuprofen with respiratory disease and lung function. Am J Respir Crit Care Med.. 2005;171:966-971.
37. Ghio AJ, Fracica PJ, Young SL, Piantadosi CA. Synthetic surfactant scavenges oxidants and protects against hyperoxic lung injury. J Appl Physiol.. 1994;77:1217-1223.
38. Gutteridge JMC, Rowley DA, Griffiths E, Halliwell B. Low-molecular-weight iron complexes and oxygen radical reactions in idiopathix haemochromatosis. Clin Sci.. 1985;68:463-467.
39. Arieli R, Arieli Y, Daskalovic Y, Eynan M, Abramovich A. CNS oxygen toxicity in closed circuit diving: signs and symptoms before loss of consciousness. Aviat Space Environ Med.. 2006;77:1153-1157.
40. Wood JD, Watson WJ. Gamma-aminobutyric acid levels in the brain of rats exposed to oxygen at high pressures. Can J Biochem Physiol.. 1963;41:1907-1913.
41. Wang WJ, Ho XP, Yan YL, Yan TH, Li CL. Intrasynaptosomal free calcium and nitric oxide metabolism in central nervous system toxicity. Aviat Space Environ Med.. 1998;69:551-555.
42. Bitterman N, Bitterman H. L-arginine-NO pathway and CNS oxygen toxicity. J Appl Physiol.. 1998;84:1633-1638.
43. Hill RK. Is more better? A comparison of different clinical hyperbaric treatment pressures – a preliminary report. Undersea Hyperb Med.. 1993;20(suppl):12.
44. Hampson NB, Simonson SG, Kramer CC, Piantadosi CA. Central nervous system oxygen toxicity during hyperbaric treatment of patients with carbon monoxide poisoning. Undersea Hyperb Med.. 1996;23:215-219.
45. Bowler RP, Crapo JD. Oxidative stress in airways. Is there a role for extracellular superoxide dismutase. Am J Respir Crit Care Med.. 2002;166:S38-S43.
46. Kolleck I, Sinha P, Rüstow B. Vitamin E as an antioxidant of the lung. Mechanisms of vitamin E delivery to alveolar Type II cells. Am J Respir Crit Care Med.. 2002;166:S62-S66.
47. Montgomery AB, Luce JM, Murray JF. Retrosternal pain is an early indicator of oxygen toxicity. Am Rev Respir Dis.. 1989;139:1548-1550.
48. Weibel ER. Oxygen effect on lung cells. Arch Intern Med.. 1971;128:54-56.
49. Clark JM, Lambertsen CJ, Gelfand R, et al. Effects of prolonged oxygen exposure at 1.5, 2.0, or 2.5 ATA on pulmonary function in men (Predictive studies V). J Appl Physiol.. 1999;86:243-259.
50. Nash G, Blennerhassett JB, Pontoppidan H. Pulmonary lesions associated with oxygen therapy and artificial ventilation. N Engl J Med.. 1967;276:368-374.
51. Gilbe CE, Salt JC, Branthwaite MA. Pulmonary function after prolonged mechanical ventilation with high concentrations of oxygen. Thorax. 1980;35:907-911.
52. Singer MM, Wright F, Stanley LK, Roe BB, Hamilton WK. Oxygen toxicity in man. A prospective study in patients after open-heart surgery. N Engl J Med.. 1970;283:1473-1478.
53. Register SD, Downs JB, Stock MC, Kirby RR. Is 50% oxygen harmful? Crit Care Med.. 1987;15:598-601.
54. Nunn JF, Williams IP, Jones JG, Hewlett AM, Hulands GH, Minty BD. Detection and reversal of pulmonary absorption collapse. Br J Anaesth.. 1978;50:91-100.
55. O’Driscoll BR, Howard LS, Davison AG. BTS guideline for emergency oxygen use in adult patients. Thorax. 2008;63(suppl VI):1-68.
56. Lucey JF, Dangman B. A reexamination of the role of oxygen in retrolental fibroplasia. Pediatrics. 1984;73:82-96.