Chapter 5 Control of breathing
 The respiratory centre in the medulla generates the respiratory rhythm using an oscillating network of six groups of interconnecting neurones. Many other diverse areas of the central nervous system influence respiratory control, these connections being coordinated by the pons. Irritant and stretch receptors in the lungs and diaphragm are involved in a series of reflex actions on the respiratory centre to influence respiratory activity.
The respiratory centre in the medulla generates the respiratory rhythm using an oscillating network of six groups of interconnecting neurones. Many other diverse areas of the central nervous system influence respiratory control, these connections being coordinated by the pons. Irritant and stretch receptors in the lungs and diaphragm are involved in a series of reflex actions on the respiratory centre to influence respiratory activity.Early in pregnancy the fetal brainstem develops a ‘respiratory centre’, which produces uninterrupted rhythmic breathing activity for many years.1 Throughout life the subject is mostly unaware of this action, which is closely controlled by a combination of chemical and physical reflexes. In addition, when required, breathing may (within limits) be completely over-ridden by voluntary control or interrupted by swallowing and involuntary non-rhythmic acts such as sneezing, vomiting, hiccupping or coughing. The control system is highly complex, with its automatic ability to adapt the action of the respiratory muscles to the changing demands of posture, speech, voluntary movement, exercise and innumerable other circumstances which alter the respiratory requirement or influence the performance of the respiratory muscles.
The Origin of the Respiratory Rhythm2
Early attempts to find the site of respiratory control used an anatomical approach involving the removal or stimulation of specific areas of the brainstem in animals (page 244). Subsequent development of precise imaging techniques allowed localisation of respiratory regions in normal human subjects, and these studies confirm that much of the historical animal work does apply to humans.3 The anatomical approach to understanding respiratory control has also been succeeded by a biochemical approach as new research methods and the possibility of therapeutic intervention have led to an explosion of interest in the chemical interactions between and within respiratory neurones.
Anatomical Location of the ‘Respiratory Centre’
The medulla is accepted as the area of brain where the respiratory pattern is generated and where the various voluntary and involuntary demands on respiratory activity are coordinated. There are many neuronal connections both into and out of the medulla, as summarised in Figure 5.1, the functions of which are described below.
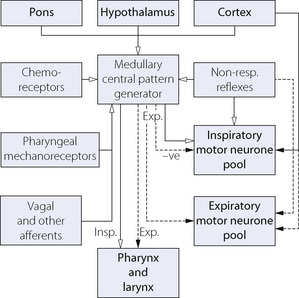
Fig. 5.1 Afferent and efferent connections to and from the medullary central pattern generator. The broken lines are expiratory pathways, which normally remain silent during quiet breathing.
Respiratory neurones in the medulla are mainly concentrated in two anatomical areas, the ventral and dorsal respiratory groups, which have numerous interconnections (Figure 5.2).
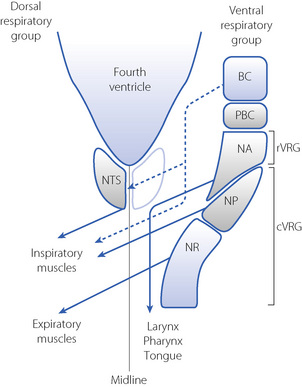
Fig. 5.2 Dorsal view of the organisation of respiratory neurones in the medulla. For clarity, the dorsal respiratory group (nucleus tractus solitarius, NTS) is shown only on the left and the ventral respiratory group (VRG) is shown only on the right. The VRG consists of the Bötzinger (BC) and Pre-Bötzinger (PBC) complexes, the rostral VRG area (rVRG) including the nucleus ambiguous (NA), and the caudal VRG area including the nucleus para-ambigualis (NP) and nucleus retroambigualis (NR). Areas with predominantly expiratory activity are shaded blue, and those with inspiratory activity shaded grey. Fibres that decussate are shown crossing the midline. The broken lines are expiratory pathways that inhibit inspiratory neurones.
The dorsal respiratory group lies in close relation to the nucleus tractus solitarius, where visceral afferents from cranial nerves IX and X terminate (Figure 5.2). It is predominantly composed of inspiratory neurones with upper motor neurones passing to the inspiratory anterior horn cells of the opposite side. The dorsal group is primarily concerned with timing of the respiratory cycle.
The ventral respiratory group comprises a column of respiratory neurones:4
 caudal ventral respiratory group, including the nucleus retroambigualis, which is predominantly expiratory with upper motor neurones passing to the contralateral expiratory muscles, and the mainly inspiratory nucleus para-ambigualis that controls the force of contraction of the contralateral inspiratory muscles. rostral ventral respiratory group, mostly made up of the nucleus ambiguous, which is involved in airway dilator functions of the larynx, pharynx and tongue.
caudal ventral respiratory group, including the nucleus retroambigualis, which is predominantly expiratory with upper motor neurones passing to the contralateral expiratory muscles, and the mainly inspiratory nucleus para-ambigualis that controls the force of contraction of the contralateral inspiratory muscles. rostral ventral respiratory group, mostly made up of the nucleus ambiguous, which is involved in airway dilator functions of the larynx, pharynx and tongue.Central Pattern Generator2,5
Unlike in the heart, there is no single ‘pacemaker’ neurone responsible for initiating breathing. Instead, a group-pacemaker hypothesis is proposed in which groups of associated neurones generate regular bursts of neuronal activity.6 For breathing, the group-pacemaker involves a complex interaction of at least six groups of neurones with identifiable firing patterns spread throughout the medulla, though concentrated in the region of the pre-Bötzinger complex. Groups of neurones include early inspiratory, inspiratory augmenting (Iaug), late-inspiratory interneurones (putative ‘off-switch’ neurones), early expiratory decrementing, expiratory augmenting, and late expiratory pre-inspiratory neurones. Typical firing patterns and the resulting muscle group activity are shown schematically in Figure 5.3. The resultant respiratory cycle may be divided into three phases:
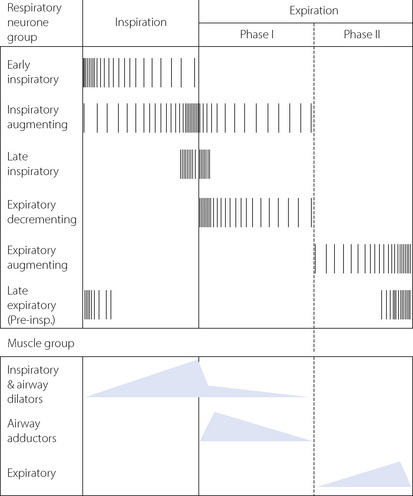
Fig. 5.3 Firing patterns of the respiratory neurone groups involved in central pattern generation and the corresponding respiratory muscle group activity. Note that expiration is divided into two phases representing passive (phase I) and active (phase II) expiration. See text for details.
Alterations in the rate at which spontaneous neuronal activity increases or decreases and the point at which the next group of neurones are activated allow an infinite variation of respiratory patterns. For example, during quiet breathing in the supine position, early expiratory neurones will reduce activity slowly and expiratory augmenting neurones will be active only briefly, resulting in almost totally passive exhalation. The converse situation will arise following exercise or at a minute volume in excess of about 40 l.min−1 when expiration will be immediately and almost totally active.
In practice, many such rhythm-generating networks are represented in parallel, so it is difficult to abolish the respiratory rhythm even with extensive brainstem damage.
Cellular mechanisms of central pattern generation.2 Respiratory neurones that exhibit spontaneous activity achieve this by a combination of intrinsic membrane properties and excitatory and inhibitory feedback mechanisms requiring neurotransmitters. In practice, neurotransmitters (both inhibitory and excitatory) have a dual effect – they recruit other cells by direct activation and modulate the spontaneous activity of a single cell by effects on its own membrane ion channels, for example slowing the rate at which an action potential travels along a dendrite.
In a similar fashion to rhythm generation in cardiac tissue, a combination of potassium and calcium ion channels are involved. For instance, in a single Iaug neurone slow membrane depolarisation occurs so producing a spontaneous discharge. These cells then ‘recruit’ other Iaug cells by excitatory postsynaptic potentials (EPSPs) and a crescendo of Iaug activity develops. Calcium-dependent potassium channels then begin to be activated and repolarise the cells so ‘switching off’ the Iaug respiratory group. Activation of other cell groups, for instance expiratory augmenting neurones, will result in activation of inhibitory postsynaptic potentials (IPSPs) on the Iaug neurones to hyperpolarise the neurone and inhibit the next wave of inspiratory activity. Similar membrane effects occur in all the respiratory neurone groups shown in Figure 5.3.
Neurotransmitters Involved in CPG and Respiratory Control2,7
These are summarised in Figure 5.4. Central pattern generation requires a combination of excitatory and inhibitory neurotransmitters. Excitatory amino acids (usually glutamate) activate several different receptors. These are divided into two groups; N-methyl-d-aspartate (NMDA) receptors, which are fast acting ion channels and non-NMDA receptors, which are slower reacting receptors involving G-protein mediated effects. Inhibitory neurotransmitters include glycine and γ-aminobutyric acid (GABA) acting via specific glycine receptors and GABAA receptors respectively to hyperpolarise the neurone and thereby inhibit its activity. These two inhibitory transmitters act quite independently during different phases of CPG.
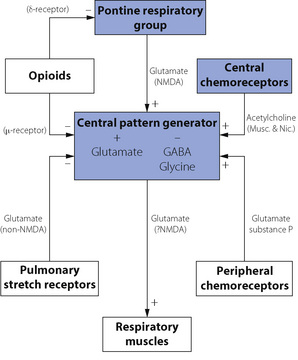
Fig. 5.4 Neurotransmitters and neuromodulators in the respiratory centre. Boxes indicate functional neuronal groups and bold type represents other influences on the respiratory centre. Substances involved in neurotransmission are shown with the most likely receptor subtype, in parentheses, if known. + indicates excitatory effect increasing respiratory activity; − indicates inhibitory activity decreasing respiration. Many of the connections shown may not be active during normal resting conditions.
Neuromodulators are substances that can influence the CPG output, but are not themselves involved in rhythm generation. There are numerous neuromodulators of respiration, many of which have several subtypes of receptors. Their exact role in normal human respiration remains unclear, but they are of undoubted relevance in both normal and abnormal breathing. For example, exogenous opioids are known to have a profound depressant effect on respiratory activity in humans (page 77), indicating the presence of opioid receptors in the respiratory centre, but administration of the opioid antagonist naloxone has no effect on respiration in resting normal subjects. Other neuromodulators include acetylcholine, which acts via both muscarinic and nicotinic receptors to mediate the effect of central chemoreceptors on respiration. Serotonin (5-hydroxytryptamine, 5HT) has many conflicting effects on respiration as a result of the numerous receptor subtypes present. Glutamate acts as a neuromodulator via both NMDA and non-NMDA receptors to mediate the pontine influence on CPG and is also involved in the influence of pulmonary stretch receptors and peripheral chemoreceptors on the respiratory pattern. Substance P also has an excitatory influence resulting in an increase in tidal volume in response to peripheral chemoreceptor activity. This diverse collection of neuromodulators probably all ultimately act via a common intra-cellular signalling pathway within CPG neurones, involving protein kinases A and C that in turn influence the activity of GABA, glycine and glutamate linked potassium and chloride channels.8
Efferent Pathways from the Respiratory Centre
Respiratory motor neurones in the brainstem are pooled into two separate areas, corresponding to inspiratory and expiratory muscle activity (see Figure 5.1). The complex integration of respiratory control seen in the CPG neurones continues to take place at the junction of the upper motor neurone with the anterior horn cell of the lower motor neurone. Three groups of upper motor neurones converge on the anterior horn cells supplying the respiratory muscles. The first group of upper motor neurones is from the dorsal and ventral respiratory groups of the medulla and are concerned with both inspiratory and expiratory output from the CPG. The second group is concerned with voluntary control of breathing (speech, respiratory gymnastics, etc.) and the third group with involuntary non-rhythmic respiratory control (swallowing, cough, hiccup, etc.). Each group of upper motor neurones occupies a specific anatomical location within the spinal cord. Neuronal control of the respiratory muscles is described in Chapter 6.
Central Nervous System Connections to the Respiratory Centre
The Pons2
There is no doubt of the existence of pontine neurones firing in synchrony with different phases of respiration, now referred to as the pontine respiratory group (PRG). Previously known as the pneumotaxic centre, three groups of neurones were identified (inspiratory, expiratory and phase-spanning) that were believed to be involved in controlling the timing of the respiratory cycle. The PRG is no longer considered to be essential for the generation of the respiratory rhythm, but does nevertheless influence the medullary respiratory neurones via a multisynaptic pathway contributing to fine control of the respiratory rhythm as, for example, in setting the lung volume at which inspiration is terminated. There are many central afferent pathways into the PRG, including connections to the hypothalamus, the cortex and the nucleus tractus solitarius. These connections suggest that the pons coordinates the respiratory effects of numerous CNS activities including cortical control, peripheral sensory information (odour, temperature), and visceral/cardiovascular inputs.
Cerebral Cortex9
Breathing can be voluntarily interrupted and the pattern of respiratory movements altered within limits determined mainly by changes in arterial blood gas tensions. This is essential for such acts as speech, singing, sniffing, coughing, expulsive efforts and the performance of tests of ventilatory function. The neurones involved in this cortical ‘over-ride’ of respiration may completely bypass the respiratory centre and act directly on the respiratory muscle lower motoneurones.10
Volitional changes in respiration are common, and under some circumstances overcome the usual chemical control of respiration. For example, conscious respiratory drive may well maintain breathing in subjects following voluntary hyperventilation when the Pco2 is below the apnoeic threshold (page 70). There are usually minor changes in the respiratory pattern when subjects focus their attention on their breathing as when physiological mouth pieces or breathing masks are used.11
In addition to volitional changes in the pattern of breathing, there are numerous other suprapontine reflex interferences with respiration such as sneezing, mastication, swallowing and coughing.12 Reflex control of respiration during speech is complex.13 During prolonged conversation, respiratory rate and tidal volume must be maintained approximately normal to prevent biochemical disturbance. In addition, for speech to be easily understood, pauses to allow inspiration must occur at appropriate boundaries in the text – for example between sentences. To achieve this, the brain performs complex assessments of the forthcoming speech to select appropriate size breaths to prevent cumbersome interruptions. This is easier to achieve during reading aloud when 88% of breaths are taken at appropriate boundaries in the text, compared with a figure of only 63% during spontaneous speech.13
Ondine’s Curse (Primary Alveolar Hypoventilation Syndrome)
In 1962 Severinghaus & Mitchell14 described three patients who exhibited long periods of apnoea, even when awake, but who breathed on command. They termed the condition ‘Ondine’s curse’ from its first description in German legend. The water nymph, Ondine, having been jilted by her mortal husband, took from him all automatic functions, requiring him to remember to breathe. When he finally fell asleep, he died. The condition is seen in adults with primary alveolar hypoventilation occurring as a feature of many different diseases, including chronic poliomyelitis and stroke.15 Characteristics include a raised Pco2 in the absence of pulmonary pathology, a flat CO2/ventilation response curve and periods of apnoea which may be central or obstructive. A similar condition is also produced by overdosage with opioids.
Ondine’s curse is also used to describe the rare condition of congenital central hypoventilation syndrome in which babies are born with a permanent defect in automatic respiratory control, leading to apnoea and hypoventilation during sleep.16 In addition, these children have abnormal respiratory responses to exercise and in keeping with the German legend also have abnormalities of cardiac control.17 In spite of such severe abnormalities, non-invasive methods of nocturnal ventilation and diaphragmatic pacing have led to almost normal lives for many of these children.16
Peripheral Input to the Respiratory Centre and Non-Chemical Reflexes
Reflexes Arising from the Upper Respiratory Tract18,19
Nose. Water and stimulants such as ammonia or cigarette smoke may cause apnoea as part of the diving reflex (page 313). Irritants can initiate sneezing which, unlike coughing, cannot be undertaken voluntarily.
Pharynx. Mechanoreceptors that respond to pressure play a major role in activation of the pharyngeal dilator muscles (page 83). There is ample evidence that local anaesthesia of the pharynx impairs their action. Irritants may cause bronchodilatation, hypertension, tachycardia, and secretion of mucus in the lower airway.
Larynx. The larynx has a dense sensory innervation with fibres from the subglottic region in the recurrent laryngeal nerve and those from the supraglottic region in the internal branch of the superior laryngeal nerve. Most reflexes arise from the supraglottic area, as section of the latter nerve abolishes almost all reflex activity. There are three groups of receptors. Mechanoreceptors respond to changes in transmural pressure or laryngeal motion and result in increased pharyngeal dilator muscle activity, particularly during airway obstruction. Cold receptors are found superficially on the vocal folds and activation generally results in depression of ventilation. The importance of this reflex in adult humans is uncertain, but these receptors may also produce bronchoconstriction in susceptible individuals (Chapter 28). Irritant receptors respond to many substances such as distilled water, cigarette smoke and inhaled anaesthetics, and, in a similar fashion to direct mechanical stimulation of the larynx, cause cough, laryngeal closure and bronchoconstriction.
The cough reflex.20 This may be elicited by chemical or mechanical stimuli arising in the larynx, trachea, carina or main bronchi. Which of these sites is responsible for the initiation of a cough is difficult to determine. For chemical stimuli the larynx may be of less importance as superior laryngeal nerve block has little effect on cough stimulated by citric acid inhalation,21 and in patients following heart–lung transplant inhalation of the normally potent stimulant distilled water results in little or no cough (page 505). Coughing can be initiated or partially inhibited voluntarily but the reflex is complex and comprises three main stages:
Expiration reflex.20,22 Similar to a cough, this reflex originates in the larynx and is believed to exist in order to prevent material being aspirated into the upper airway. It differs from a cough by the absence of an inspiratory phase, the compressive and expulsive phases occurring immediately and from the lung volume present at the time the larynx is irritated. The distinction between the cough and expiration reflexes is important – a large inspiration as seen at the start of a cough would not be helpful in the presence of solid or liquid at the laryngeal inlet.
Reflexes Arising in the Lung
Pulmonary stretch receptors and their associated reflexes.23 There are many different types of receptors in the lungs sensitive to inflation, deflation, mechanical and chemical stimulation, afferents from which are mostly conducted by the vagus, although some fibres may be carried in the sympathetic nerves. Slowly adapting stretch receptors (SARs) are found predominantly in the airways rather than in the alveoli, and are closely associated with the tracheo-bronchial smooth muscle. Lung inflation stimulates the SARs, which are named ‘slowly adapting’ due to their ability to maintain their firing rate when lung inflation is maintained, thus acting as a form of lung volume sensor. Conversely, rapidly adapting stretch receptors (RARs) are located in the superficial mucosal layer,18 and are stimulated by changes in tidal volume, respiratory frequency or changes in lung compliance.23 The RARs also differ from SARs in being nociceptive and chemosensitive, responding to a wide range of chemical irritants, mechanical stimuli and inflammatory mediators.
How these receptors transduce a mechanical change in the tissue into an action potential is unknown. Hypotheses include the release of mediators from nearby associated cells that activate a receptor on the neurone, or ion channels may exist that respond directly to an alteration in their physical shape.24 Afferent nerves from all these receptors converge on the nucleus tractus solitarius (NTS) of the medulla, where their signals are modulated and coordinated before further polysynaptic pathways communicate with the other regions of the respiratory centre. This processing of the afferent inputs by the NTS is believed to be capable of neuronal plasticity, which means the modulation can be altered by prolonged changes in external environment that influence the afferent inputs.25
The reflexes associated with pulmonary stretch receptors have attracted much attention since the associated inflation and deflation reflexes were described by Hering & Breuer in 1868.26 Breuer was a clinical assistant to Professor Hering but apparently the work was at his own instigation. However, Hering, who was a corresponding member of the Vienna Academy of Science, published Breuer’s work under his own name, in accord with the custom of the time. Breuer’s role was clearly stated in Hering’s paper but he was not a co-author. Later the same year, Breuer published a much fuller account of his work under his own name.
The inflation reflex consists of inhibition of inspiration in response to an increased pulmonary transmural pressure gradient (as in sustained inflation of the lung). An exactly similar effect may be obtained by obstructing expiration so that an inspiration is retained in the lungs.
The significance of the Hering–Breuer reflex in humans is controversial.27,28 There appears to be an important species difference between laboratory animals, in which the reflex is easy to demonstrate, and humans in whom the reflex is very weak.29 Pragmatic evidence that the Hering–Breuer reflex is unimportant in awake humans comes from studies showing normal breathing patterns in volunteers following bilateral vagal nerve block30 and in patients who have had bilateral lung transplants, when both lungs must be totally denervated (Chapter 33). Conversely, studies in which conscious perception of chest wall position is suppressed by applying imperceptible amounts of assisted ventilation have demonstrated that respiratory pattern is altered within the physiological range, demonstrating the presence of a vagal feedback mechanism.28,31 Although the Hering–Breuer inflation reflex therefore appears to exist but have minimal functional significance in adults, it is widely accepted as being present in neonates and infants.32
The deflation reflex consists of an augmentation of inspiration in response to deflation of the lung and can be demonstrated in man.33 These results are consistent with the hypothesis that lung deflation has a reflex excitatory effect on breathing, but that the threshold is higher in man than for other mammalian species.
Head’s paradoxical reflex. Head, working in Professor Hering’s laboratory, described a reversal of the inflation reflex.34 Many authors have reported that, with normal vagal conduction, sudden inflation of the lungs of many species may cause a transient inspiratory effort before the onset of apnoea due to the inflation reflex.29 A similar response may also be elicited in new-born infants,35 but it has not been established whether this ‘gasp reflex’ is analogous to Head’s paradoxical reflex. All anaesthetists are aware that, after administration of respiratory depressants, transient increases in airway pressure often cause an immediate deep gasping type of inspiration.
Other Pulmonary Afferents
C-fibre endings lie in close relationship to the capillaries, one group is in relation to the bronchial circulation and the other to the pulmonary microcirculation. The latter correspond to Paintal’s juxta-pulmonary capillary receptors (J receptors, for short).18,36
These receptors are relatively silent during normal breathing but are stimulated under various pathological conditions. They are similar to RARs described above, being nociceptive and activated by reactive oxygen species,37 tissue damage, accumulation of interstitial fluid and release of various mediators. In the laboratory they can be activated by intravascular injection of capsaicin to produce the so-called pulmonary chemoreflex which comprises bradycardia, hypotension, apnoea or shallow breathing, bronchoconstriction and increased mucus secretion.18,28 They may well be concerned in the dyspnoea of pulmonary vascular congestion and the ventilatory response to exercise and pulmonary embolisation. C-fibre endings have been characterised in physiological studies but have never been identified histologically, although non-myelinated nerve fibres are seen in the alveolar walls.
Reflexes Arising from Outside the Airway and Lungs
Phrenic nerve afferents.38 Approximately one-third of neurones in the phrenic nerve are afferent, with the majority arising from muscle spindles and tendon organs forming the spinal reflex arc described on page 89. However, some afferent neurones continue through the ipsilateral spinal cord to the brainstem and somatosensory cortex. Experimental stimulation of phrenic afferent fibres results in a reduction of respiratory efferent activity, but stimulation of some smaller afferent fibres has the opposite effect. Thus the physiological role of phrenic afferents remains obscure, but it is unlikely that they have any influence on normal breathing. The sensory information provided by phrenic afferents is believed to be important in the perception of, and compensation for, increased inspiratory loads and these afferents are important in the ‘breaking point’ following a breath hold (page 76).
Baroreceptor reflexes. The most important groups of arterial baroreceptors are in the carotid sinus and around the aortic arch. These receptors are primarily concerned with regulation of the circulation, but a large decrease in arterial pressure produces hyperventilation, while in animals a substantial rise in arterial pressure causes respiratory depression and, ultimately, apnoea.
Afferents from the musculoskeletal system. These probably do not contribute to normal resting ventilation but have an important role in the hyperventilation of exercise (Chapter 15).
The Influence of Carbon Dioxide on Respiratory Control39,40
For many years it was believed that the respiratory centre itself was sensitive to carbon dioxide. However, it is now known that both central and peripheral chemoreceptors are responsible for the effect of carbon dioxide on breathing, the latter accounting for about 80% of the total ventilatory response.39 Because of their reliance on extra-cellular pH (see below) the central chemoreceptors are regarded as monitors of steady-state arterial Pco2 and tissue perfusion in the brain, while the peripheral chemoreceptors respond more to short term and rapid changes in arterial Pco2.41
Localisation of the Central Chemoreceptors
Studies in animals indicate that central chemo-sensitive areas are located within 0.2 mm of the ventrolateral surface of the medulla, in a region now referred to as the retrotrapezoid nucleus (RTN).42 Neurones of the RTN are glutaminergic, and have selective connections to the nearby CPG.40 Many other areas of the CNS display increased neural activity with carbon dioxide stimulation including other areas of the medulla, the midline pons, small areas in the cerebellum, and the limbic system,3 though the contribution of these areas to respiratory control is unclear.
Mechanism of Action
An elevation of arterial Pco2 causes an approximately equal rise of extracellular fluid, CSF, cerebral tissue and jugular venous Pco2, which are all about 1.3 kPa (10 mmHg) more than the arterial Pco2. Over the short term, and without change in CSF bicarbonate, a rise in CSF Pco2 causes a fall in CSF pH. The blood–brain barrier (operative between blood and CSF) is permeable to carbon dioxide but not hydrogen ions, and in this respect resembles the membrane of a Pco2-sensitive electrode (page 174). In both cases, carbon dioxide crosses the barrier and hydrates to carbonic acid, which then ionises to give a pH inversely proportional to the log of the Pco2. A hydrogen ion sensor is thus made to respond to Pco2.
The mechanism by which a change in pH causes stimulation of chemoreceptor neurones remains disputed: the RTN may contain pH sensitive potassium channels, and release of adenosine triphosphate (ATP) has been proposed.39
Compensatory bicarbonate shift in the CSF. If the Pco2 of CSF is maintained at an abnormal level, the CSF pH gradually returns towards normal over the course of many hours as a result of changes in the CSF bicarbonate concentration. This is analogous to, and proceeds in parallel with, the partial restoration of blood pH in patients with chronic hyper- or hypocapnia. Compensatory changes in bicarbonate concentrations are similar in both CSF and blood, suggesting a common mechanism.43 Bicarbonate shift in CSF could therefore result simply from passive ion distribution, although the possibility of active ion transfer cannot be completely excluded. Examples of situations when this normalisation of CSF pH may occur include prolonged periods of hypocapnic artificial ventilation and the hypocapnia that occurs in response to hypoxia at altitude (page 282). Once the hypocapnia is reversed, for example when the artificial ventilation is no longer required, hyperventilation may follow for several hours. Compensatory changes in CSF pH are not confined to respiratory alkalosis, but are also found in chronic respiratory acidosis and metabolic acidosis and alkalosis. In a study of patients with a variety of pathological acid-base disturbances, values of CSF pH did not differ by more than 0.011 units from the normal value (7.326) in spite of mean arterial pH values ranging from 7.334 to 7.523.44 If the bicarbonate concentration in CSF is itself altered by pathological factors, the pH is changed and ventilatory disturbances follow. For example, after intra-cranial haemorrhage patients may spontaneously hyperventilate,45 and in these patients the CSF pH and bicarbonate have been shown to be below the normal values.
The Pco2/Ventilation Response Curve
Following a rise in arterial Pco2, respiratory depth and rate increase until a steady state of hyperventilation is achieved after a few minutes. The response is linear over the range that is usually studied and may therefore be defined in terms of two parameters, slope and intercept (see Appendix E):
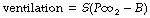
where S is the slope (l.min−1.kPa−1 or l.min−1.mmHg−1), and B is the intercept at zero ventilation (kPa or mmHg). The blue line in Figure 5.5 is a typical normal curve with an intercept (B) of about 4.8 kPa (36 mmHg) and a slope (S) of about 15 l.min−1.kPa−1 (2 l.min−1.mmHg−1). There is in fact a very wide individual variation in Pco2/ventilation response curves, including a circadian variation within individuals,46 and the response may be decreased by normal hormonal changes, disease or drugs. The dashed curve in Figure 5.5 shows the effect of changing ventilation on arterial Pco2 when the inspired carbon dioxide concentration is negligible, and is a section of a rectangular hyperbola. The normal resting Pco2 and ventilation are indicated by the intersection of this curve with the normal Pco2/ventilation response curve, which is usually obtained by varying the carbon dioxide concentration in the inspired gas.
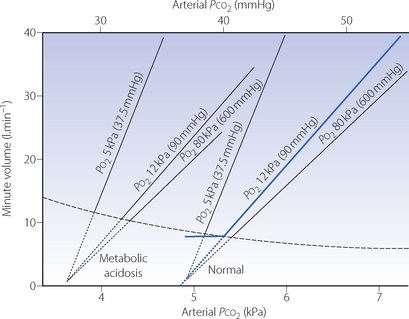
Fig. 5.5 Two fans of Pco2/ventilation response curves at different values of Po2. The right-hand fan is at normal metabolic acid-base state (zero base excess). The left-hand fan represents metabolic acidosis. The broken line represents the Pco2 produced by the indicated ventilation for zero inspired Pco2, at basal metabolic rate. The intersection of the broken curve and any response curve indicates the resting Pco2 and ventilation for the relevant metabolic acid–base state and Po2. The blue curve is the normal response. See text for details.
When subjects hyperventilate voluntarily and reduce their Pco2 below the threshold for CO2 stimulation of respiration a variety of responses are seen, varying from apnoea to normal respiration or even hyperventilation.47Figure 5.5 shows two possible extensions to the normal response curve (in blue) below the threshold for CO2 stimulation (dashed line). The first is an extrapolation of the curve to intersect the x axis (zero ventilation) at a Pco2 known as the apnoeic threshold (dotted lines in Figure 5.5). If Pco2 is depressed below this point, apnoea may result, and this is seen in some subjects. The second type of extension (shown on the blue line) is horizontal and to the left, like a hockey stick, representing the response of a subject who continues to breathe regardless of the fact that his Pco2 has been reduced. The resting arterial point at resting ventilation is normally approximately 0.3 kPa to the left of the extrapolated response curve,48 supporting the idea of a hockey stick shaped response curve. When breathing below this threshold for the onset of CO2 stimulated ventilation (the angle of the hockey stick) hypoxia seems to have no influence.47 This variable ventilatory response to low Pco2 almost certainly arises from the cortical control of respiration maintaining breathing despite a lack of chemical drive, particularly when awake.
As Pco2 is raised, a point of maximal ventilatory stimulation is reached, probably within the range 13.3–26.7 kPa (100–200 mmHg), beyond which respiratory fatigue and CO2 narcosis intervene (Chapter 23). The ventilatory stimulation is reduced until, at very high Pco2, the ventilation is actually depressed below the control value and finally apnoea results, at least in animals and almost certainly in humans as well.
The Pco2/ventilation response curve is the response of the entire respiratory system to the challenge of a raised Pco2. Apart from reduced sensitivity of the central chemoreceptors, the overall response may be blunted by partial neuromuscular blockade or by obstructive or restrictive lung disease. These factors must be taken into account in drawing conclusions from a reduced response, and diffuse airway obstruction is a most important consideration. Nevertheless the slope of the Pco2/ventilation response curve remains one of the most valuable parameters in the assessment of the responsiveness of the respiratory system to carbon dioxide and its depression by drugs.
Time course of Pco2/ventilation response.49 As described above, the initial ventilatory response to elevated Pco2 is extremely rapid occurring within just a few minutes, at which time about 75% of the final ventilatory response has occurred. With sustained hypercapnia, the minute ventilation continues to increase for a further hour before reaching a plateau, which is sustained for at least 8 hours in healthy subjects.
The Influence of Oxygen on Respiratory Control
As for carbon dioxide, it was initially thought that hypoxia stimulated respiration by a direct effect on the respiratory centre. However, around 1930 the histological studies of de Castro50 led him to suggest a chemoreceptor function for the carotid bodies, and the respiratory role of the peripheral chemoreceptors was established by Heymans51 who received a Nobel prize for his work.
Peripheral Chemoreceptors52
The peripheral chemoreceptors are fast-responding monitors of the arterial blood, responding to a fall in Pao2, a rise in Paco2 or H+ concentration, or a fall in their perfusion rate. An increase in ventilation is the result of stimulation. The bilaterally paired carotid bodies, rather than the aortic bodies, are almost exclusively responsible for the respiratory response. Each is only about 6 mm3 in volume and they are located close to the bifurcation of the common carotid artery. The carotid bodies undergo hypertrophy and hyperplasia under conditions of chronic hypoxia and are usually lost in the operation of carotid endarterectomy (see below).
Histology. The carotid bodies contain large sinusoids with a very high rate of perfusion – about 10 times the level that would be proportional to their metabolic rate, which is itself very high. Therefore the arterial/venous Po2 difference is small. This accords with their role as a sensor of arterial blood gas tensions, and their rapid response, which is within the range 1–3 seconds.
At the cellular level, the main feature is the glomus or type I cell, which is in synaptic contact with nerve endings derived from an axon with its cell body in the petrosal ganglion of the glossopharyngeal nerve. Type I cells are partly encircled by type II cells whose function is still uncertain, but they may be dormant stem cells that can be activated by hypoxia to generate new type I cells.52 Efferent nerves, which are known to modulate receptor afferent discharge, include preganglionic sympathetic fibres from the superior cervical ganglion, amounting to 5% of the nerve endings on the glomus cell.
Discharge rate in the afferent nerves from the carotid body increases in response to the following forms of stimulation:
Decrease of arterial Po2. Stimulation is by decreased Po2 and not by reduced oxygen content (at least down to about half the normal value). Thus there is little stimulation in anaemia, carboxyhaemoglobinaemia or methaemoglobinaemia. Quantitative aspects of the hypoxic ventilatory response are described in detail below.
Decrease of arterial pH. Acidaemia of perfusing blood causes stimulation, the magnitude of which is the same whether it is due to respiratory or metabolic acidosis. Quantitatively, the change produced by elevated Pco2 on the peripheral chemoreceptors is only about one-sixth of that caused by the action on the central chemosensitive areas (see below). This response does however occur very rapidly,41,49 and only develops when a ‘threshold’ value of arterial Pco2 is exceeded.47
Hypoperfusion of peripheral chemoreceptors causes stimulation, possibly by causing a ‘stagnant hypoxia’ of the chemoreceptor cells (see below). Hypoperfusion may result from severe systemic hypotension.
Blood temperature elevation causes stimulation of breathing via the peripheral chemoreceptors. In addition, the ventilatory responses to both hypoxia and CO2 are enhanced by a modest (1.4°C) rise in body temperature.
Chemical stimulation by a wide range of substances is known to cause increased ventilation through the medium of the peripheral chemoreceptors. These substances fall into two groups. The first comprises agents such as nicotine and acetylcholine that stimulate sympathetic ganglia. The second group of chemical stimulants comprises substances such as cyanide and carbon monoxide which block the cytochrome system and so prevent oxidative metabolism. Drugs which stimulate respiration via the peripheral chemoreceptors are described below.
Mechanism of Action of Peripheral Chemoreceptors52,53,54
There is now general agreement that oxygen-sensitive potassium channels are responsible for the hypoxic response of Type I cells, similar channels being found in most cells of the body that respond to hypoxia.54 Many different oxygen-sensitive potassium channels exist, with varying types occurring in different species, different tissues and under different circumstances within a species. Hypoxia inhibits the activity of the potassium channel, which alters the membrane potential of the cell and stimulates calcium channels to open, allowing an influx of extracellular calcium which stimulates transmitter release. The molecular mechanism by which potassium channels respond to Po2 is unknown, including whether or not there is a direct effect on the channel itself or whether other, hypoxia induced, molecules are responsible. Contenders for this role include reactive oxygen species (Chapter 26) produced either from mitochondria or from reduced nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, or possibly even carbon monoxide produced by haemoxygenase, an antioxidant enzyme constitutively expressed in most cells and closely associated with the potassium channels in Type I cells. These potential molecular messengers of hypoxia are probably not essential for the glomus cells to respond to low Po2, but they may be important in modulating the response, for example when perfusion of the carotid body is poor.
Stimulation of the chemoreceptors by an increased arterial Pco2 is dependent on carbonic anhydrase (present in the Type I cell) and there is therefore the possibility of both raised Pco2 and decreased arterial pH acting through an increase in intracellular hydrogen ion concentration, as in the central chemoreceptors.
Various neurotransmitters have been identified within the carotid body with dopamine, acetylcholine and adenosine triphosphate (ATP) being the most prominent, though noradrenaline, angio-tensin II, substance P and enkephalins have also been identified, though the role of each is uncertain. Acetylcholine and ATP are most likely to be the neurotransmitters involved between the Type I cell and the afferent nerves.55 The other molecules seem to have an autocrine rather than neurotransmitter role, in that their release into the carotid body tissues modulates the response of the cells to the various stimuli.55 For example, dopamine is abundant in Type I cells and released in response to hypoxia, its presence causing inhibition of calcium channels so effectively ‘damping’ the acute response. Similarly, the α2 adrenoceptor agonist clonidine reduces the ventilatory response to acute hypoxia indicating that noradrenaline also has an inhibitory effect.56 Angiotensin II increases the sensitivity of the potassium channels to hypoxia, and may be produced locally within the carotid body in response to long term hypoxia or poor carotid body perfusion, as seen in heart failure.57
The gain of the carotid bodies is under nervous control. There is an efferent pathway in the sinus nerve which, on excitation, decreases chemoreceptor activity. Excitation of the sympathetic nerve supply to the carotid body causes an increase in activity.
Other effects of stimulation. Apart from the well known increase in depth and rate of breathing, peripheral chemoreceptor stimulation causes a number of other effects, including bradycardia, hypertension, increase in bronchiolar tone and adrenal secretion. Stimulation of the carotid bodies has predominantly respiratory effects, whilst the aortic bodies have a greater influence on the circulation.
Time Course of the Ventilatory Response to Sustained Hypoxia58
By controlling the concentration of inhaled oxygen, arterial oxygen saturation can be reduced and then maintained at a constant level of hypoxia, usually with a Sao2 of about 80%. In order to separate the effects on ventilation of hypoxia and Pco2 most studies use isocapnic conditions, where the subject’s alveolar Pco2 is maintained at their control (resting ventilation) level by addition of CO2 to the inspired gas. The interaction of Pco2 and hypoxia in ventilatory control is discussed below. With a moderate degree of sustained hypoxia the ventilatory response is triphasic as shown in Figure 5.6. The three phases are described separately.
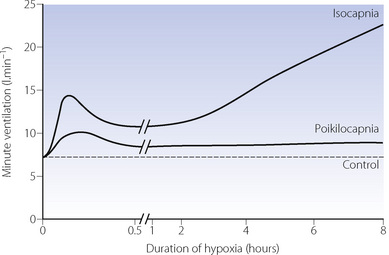
Fig. 5.6 Time course of the ventilatory response to hypoxia (Sao2 ≈ 80%). Practical problems prevent the continuous and rapid measurement of minute volume and respiratory gases for 8 hours, so the curves are produced from combining the data from three studies. When arterial Pco2 is maintained at normal levels (isocapnia) the response is triphasic. When arterial Pco2 is not controlled (poikilocapnia) the magnitude of the response is damped because the hypoxia induced hyperventilation reduces Pco2 and therefore respiratory drive. See Figure 17.3 for respiratory effects of prolonged hypoxia.
(After references 59, 60 and 61.)
Acute hypoxic response. This is the first immediate and rapid increase in ventilation. Sudden imposition of hypoxia results in stimulation of ventilation within the lung-to-carotid body circulation time (about 6 s), but in most studies the response appears slower due to the delay between reducing inspired oxygen and the reduction in alveolar and then arterial Po2. Ventilation continues to increase for between 5 and 10 minutes, rapidly reaching high levels.
Many factors affect the acute ventilatory response. There are wide variations between individuals, within an individual on different days, between male and female subjects, and with the hormonal changes of the menstrual cycle. A small number of otherwise normal subjects lack a measurable ventilatory response to hypoxia when studied at normal Pco2. This is of little importance under normal circumstances, because the Pco2 drive from the central chemoreceptors will normally ensure a safe level of Po2. However, in certain therapeutic and abnormal environmental circumstances, such as at high altitude, it could be dangerous.
Hypoxic ventilatory decline (HVD). Shortly after the acute hypoxic response reaches a peak, minute ventilation begins to decline reaching a plateau level, still above the resting ventilation, after 20–30 minutes (Figure 5.6). The degree of HVD in an individual correlates with the acute hypoxic response – the greater the initial increase in ventilation the greater the subsequent decline.60 Though not completely elucidated yet, the mechanism of HVD appears to have a significant centrally mediated component,62 and represents a change in ventilatory drive rather than a decline in the sensitivity of the receptors to hypoxia.63 In animals, central glutamate release is involved in the acute hypoxic response, whilst GABA is implicated in producing HVD.64
Ventilatory response to sustained hypoxia. Once HVD is complete, continued isocapnic hypoxia results in a second, more slow, rise in ventilation over several hours (Figure 5.6). Ventilation continues to increase for at least 8 hours,62 and reaches a plateau by 24 hours.65 Species differences in this response again make elucidation of the mechanism in humans difficult, but the most likely explanation is a direct effect of hypoxia on the carotid bodies, possibly mediated by angiotensin II (page 223).
Hypoxia for more than 2–3 days only occurs following ascent to altitude, and the effects of this are described in Chapter 17.
Ventilatory Response to Progressive Hypoxia
Instead of maintaining a constant degree of hypoxia, ventilation may be measured during a progressive reduction in Po2. Once again, by controlling inspired gas concentrations, alveolar Po2 may be reduced from 16 to 5 kPa (120 to 40 mmHg) over 15 minutes,66 and ventilation increases progressively throughout this period. The response under these circumstances probably equates to the acute hypoxic response. If alveolar Po2 is plotted against minute ventilation a Po2/ventilation response curve is produced (Figure 5.7). A Po2/ventilation response curve approximates to a rectangular hyperbola (see Appendix E), asymptotic to the ventilation at high Pao2 (zero hypoxic drive) and to the Pao2 at which ventilation theoretically becomes infinite (known as ‘C’ and about 4.3 kPa). Figure 5.7 shows a typical example but there are very wide individual variations. Note that there is a small but measurable difference in ventilation between normal and very high Po2.
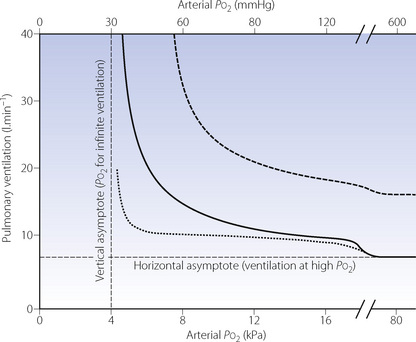
Fig. 5.7 Ventilatory response to progressive hypoxia. The heavy curve represents the normal Po2/ventilation response under isocapnic conditions, that is with Pco2 maintained at the resting value. It has the form of a rectangular hyperbola asymptotic to the ventilation at high Po2 and the Po2 at which ventilation becomes infinite. The curve is displaced upwards by both hypercapnia and exercise at normal Pco2 (dashed line). Hypocapnia displaces the curve downwards (dotted line) regardless of whether the hypocapnia results from not controlling Pco2 (poikilocapnia) or by deliberately reducing Pco2.
(Data from references 66, 67 and 68.)
The initial ventilatory response to Po2 may be expressed as:
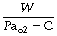
where W is a multiplier (i.e. the gain of the system) and partly dependent upon the Pco2. The ventilatory response here is the difference between the actual ventilation and the ventilation at high Po2, Pco2 being unchanged.
The inconvenience of the non-linear relationship between ventilation and Po2 may be overcome by plotting ventilation against oxygen saturation. The relationship is then linear with a negative slope, at least down to a saturation of 70%.69 This approach is the basis of a simple non-invasive method of measurement of the hypoxic ventilatory response (see below).
Iatrogenic Loss of Peripheral Chemoreceptor Sensitivity70
Nerves from the carotid bodies are usually divided during bilateral carotid endarterectomy, which provides evidence that the carotid bodies are not essential for the maintenance of reasonably normal breathing under conditions of rest and mild exercise. Indeed, there is some evidence that the common finding of atheromatous disease at the carotid bifurcation may itself reduce chemoreceptor function and that a careful, ‘nerve sparing’, carotid endarterectomy can increase the ventilatory response to hypoxia.71 Deliberate abolition of the hypoxic ventilatory response by carotid endarterectomy has been advocated as a treatment for incapacitating dyspnoea in severe respiratory disease.72
Central Hypoxic Depression of Breathing
In addition to its effects on peripheral chemoreceptors, hypoxia also has a direct effect on the respiratory centre. Central respiratory neurone activity is depressed by hypoxia, and apnoea follows severe medullary hypoxia whether due to ischaemia or to hypoxaemia. With denervated peripheral chemoreceptors, phrenic motor activity becomes silent when the medullary Po2 falls to about 1.7 kPa (13 mmHg).73 More intense hypoxia causes a resumption of breathing with an abnormal pattern, possibly driven by a ‘gasping’ centre. This pattern of central hypoxic depression appears to be particularly marked in neonates and may be the relic of a mechanism to prevent the fetus from attempting to breathe in utero.
Mechanisms of hypoxic depression of ventilation. Medullary Pco2, and therefore ventilation, may be reduced by increased cerebral blood flow induced by hypoxia, and severe hypoxia causes depletion of high-energy phosphates. However, it has also been shown that neonatal hypoxia results in decreased levels of excitatory neurotransmitters (glutamate and aspartate) and increased levels of inhibitory substances, particularly GABA and endogenous opioids, both powerful respiratory depressants.
Integration of the Chemical Control of Breathing
The two main systems contributing to chemical control of breathing have been described quite separately, but in the intact subject this is not possible. For example, the peripheral chemoreceptors respond (slightly) to changes in Pco2, and hypoxia affects the respiratory centre directly as well as via the carotid body receptors. An overall view of the chemical control of breathing is shown schematically in Figure 5.8.
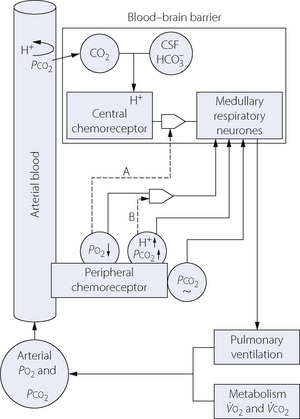
Fig. 5.8 Scheme of connections between individual aspects of chemical control of breathing. See text for details.
It was originally thought that the various factors interacted according to the algebraic sum of the individual effects caused by changes of Pco2, Po2, pH, etc. Hypoxia and hypercapnia were, for example, thought to be simply additive in their effects, but it is now realised that this was a very simplistic view of a complex system.
Effects of Pco2 and pH on the Hypoxic Ventilatory Response66
The acute hypoxic response is enhanced at elevated Pco2 as shown by the upper dashed curve in Figure 5.7, the mechanism being indicated by broken line B in Figure 5.8. This interaction contributes to the ventilatory response in asphyxia being greater than the sum of the response to be expected from the rise in Pco2 and the fall in Po2 if considered separately.
Responses to both acute and prolonged hypoxia are depressed by hypocapnia, as shown in the lower dotted curve in Figure 5.7. This results from opposing effects on the CPG of increased chemoreceptor input and decreased central chemoreceptor drive. On prolonged exposure to hypoxia at altitude, this effect continues until acclimatisation takes place (page 282).
Poikilocapnic conditions occur when no attempt is made to control Pco2 during hypoxic ventilation, and the hypoxia induced hyperventilation immediately gives rise to hypocapnia. Though rarely studied, this situation is important as poikilocapnia will occur in clinical situations. Early studies of the effects of Pco2 on hypoxic ventilation showed that without control of Pco2 the hypoxia driven increase in ventilation is almost exactly counteracted by the Pco2 driven depression of ventilation resulting in no change in minute volume until breathing less than 10% oxygen.67,68 Many earlier studies were, however, performed before technology allowed elucidation of the time course of hypoxic ventilation, and may have been measuring the plateau of ventilation after hypoxic ventilatory decline rather than the acute hypoxic response. More recent studies have shown that poikilocapnic conditions attenuate, but do not abolish, the first two phases of the ventilatory response to constant hypoxia (Figures 5.7 and 5.8).59,60 Increased ventilation with sustained (over 1 hour) hypoxia is abolished during poikilocapnia but the minute volume does remain above resting levels (Figure 5.6).61
Exercise enhances the response to hypoxia even if the Pco2 is not raised,74 possibly due to lactic acidosis, oscillations of arterial Pco2, afferent input from muscle, or perhaps to catecholamine secretion. The upper broken curve in Figure 5.7 would also correspond to the response during exercise at an oxygen consumption of about 800 ml.min−1. It is important to note that the slope of the curve at normal Po2 is considerably increased in both these circumstances, so there will then be an appreciable ‘hypoxic’ drive to ventilation at normal Po2. Enhanced response to Po2 during exercise seems to be an important component in the overall ventilatory response to exercise (Chapter 15).
Effects of Pao2 and pH on Central Chemoreceptor Response47
The broken line (A) in Figure 5.8 shows the influence of the peripheral chemoreceptor drive on the gain of the central ventilatory response to Pco2. Typical quantitative relationships are shown in Figure 5.5, with hypoxia at the left of each fan and hyperoxia on the right. The curve marked Po2 80 kPa represents total abolition of chemoreceptor drive obtained by the inhalation of 100% oxygen.
Metabolic acidosis displaces the whole fan of curves to the left as shown in Figure 5.5. The intercept (B) is reduced but the slope of the curves at each value of Po2 is virtually unaltered. Display of the fan of Pco2/ventilation response curves at different Po2 is a particularly complete method of representing the state of respiratory control in a patient, but is impractical to determine.
Periodic Breathing
This term describes a respiratory pattern in which ventilation waxes and wanes in a regular pattern. It is normal in neonates (page 254), but seen only during sleep in adults, occurring more frequently in the elderly75 and in all ages when sleeping at altitude (page 287). The cause of periodic breathing is unknown, but is likely to involve an abnormality of the chemical control of breathing, possibly a poorly responsive or over-damped control system.74 Cheyne-Stokes respiration is an extreme form of periodic breathing in which apnoea occurs during the hypoventilation phase, and is seen most commonly in patients with heart failure. In the case of Cheyne-Stokes respiration abnormalities of respiratory control and lung function contribute, but the slow circulation time seen in patients with heart failure introduces further periodicity into the breathing pattern.76
Breath Holding
Influence of Pco2 and Po2
When the breath is held after air breathing, the arterial and alveolar Pco2 are remarkably constant at the breaking point and values are normally close to 6.7 kPa (50 mmHg). This does not mean that Pco2 is the sole or dominant factor and concomitant hypoxia is probably more important. Preliminary oxygen breathing delays the onset of hypoxia, and breath-holding times may be greatly prolonged with consequent elevation of Pco2 at the breaking point. The relationship between Pco2 and Po2 at breaking point, after starting from different levels of oxygenation, is shown in Figure 5.9.
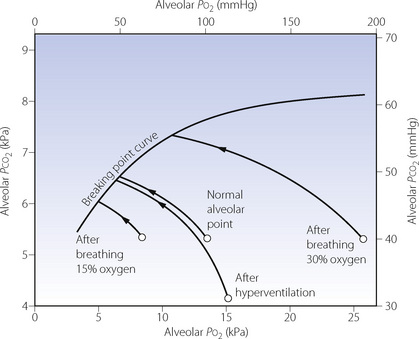
Fig. 5.9 The ‘breaking point’ curve defines the coexisting values of alveolar Po2 and Pco2, at the breaking point of breath holding, starting from various states. The normal alveolar point is shown and the curved arrow shows the changes in alveolar gas tensions that occur during breath holding. Starting points are displaced to the right by preliminary breathing of oxygen enriched gases, and to the left by breathing mixtures containing less than 21% oxygen. Hyperventilation displaces the point representing alveolar gas to the right and downwards. The length of arrows from starting point to the breaking point curve gives an approximate indication of the duration of breath hold. This can clearly be prolonged by oxygen breathing or by hyperventilation, maximal duration occurring after hyperventilation with 100% oxygen.
On the basis of changing blood gas tensions and the great variability of individuals’ responses it might be predicted that subjects with ‘flat’ ventilatory responses to oxygen and carbon dioxide would be able to hold their breath longer. Elite breath-hold divers (page 297) have been shown to have a blunted response to carbon dioxide but not to hypoxia.77
Effect of Lung Volume
Breath-holding time is directly proportional to the lung volume at the onset of breath holding, partly because this has a major influence on oxygen stores. There are, however, other effects of lung volume and its change, which are mediated by afferents arising from the chest wall, diaphragm and the lung itself. Prolongation of breath-holding times are seen after bilateral vagal and glossopharyngeal nerve block,78 and following complete muscular paralysis of conscious subjects.79 These studies suggest that much of the distress leading to the termination of breath-holding is caused by frustration of the involuntary contractions of the respiratory muscles, which increase progressively during breath-holding. Fowler’s experiment in 1954 easily demonstrates the importance of frustration of involuntary respiratory movements.80 After normal air breathing, the breath is held until breaking point. If the expirate is then exhaled into a bag and immediately reinhaled, there is a marked sense of relief although it may be shown that the rise of Pco2 and fall of Po2 are uninfluenced.
Extreme durations of breath holding may be attained after hyperventilation and preoxygenation. Times of 14 minutes have been reached and the limiting factor is then reduction of lung volume to residual volume, as oxygen is removed from the alveolar gas.
Drug Effects on the Control of Breathing
Considering the therapeutic potential of drugs that could specifically influence respiratory drive, it is surprising that so few drugs affecting respiratory control have been developed. Elucidation of the mechanisms for the control of breathing described in this chapter is a relatively recent event, particularly when considering the neurotransmitters and neuromodulators involved. The large number of different receptors involved in normal respiratory control (Figure 5.4) means that drugs affecting a single receptor may have little effect, or unpredictable effects, on respiration and so be of little clinical use. In addition, the neurotransmitters and neuromodulators involved are widely distributed throughout the central nervous system (CNS), so agonists or antagonists of their receptors are likely to have diverse effects resulting in unacceptable adverse effects.
Many other factors apart from the drug itself affect respiratory activity, so the effect that a drug exerts on the respiration of an individual patient is complex and unpredictable. For example in a healthy patient recovering from surgery under general anaesthesia, pain, anxiety, stress and changes in blood chemistry will be stimulating breathing whilst sedation, sleep and residual anaesthetic or analgesic agents will all be tending to depress respiration.
Respiratory Depressants
Any drug that depresses CNS activity may depress respiration, either individually or in combination with other CNS depressants such as alcohol. Almost all general anaesthetic agents reduce ventilation in a dose-dependent fashion, and are described in detail on page 327. Two specific groups of drugs that have well documented depressant effects on ventilation are opioid analgesics and benzodiazepines.
Opioids81,82,83 Figure 5.4 shows that both μ- and δ-opioid receptors are present in the respiratory centre. As indicated above, the role of these receptors in normal respiratory control is unknown. Animal studies suggest that μ-receptors in the pre-Bötzinger complex (Figure 5.2) may be involved in normal respiratory control.83 In humans, the evidence is less clear. In healthy subjects, administration of the non-specific opioid receptor antagonist naloxone has no effect on respiration.81
Agonists of μ-opioid receptors, such as morphine, cause dose-dependent depression of respiration normally characterised by a slow respiratory rate, but tidal volume is also commonly reduced. Female subjects show a greater susceptibility to the respiratory depression seen with opioids.83 Ventilatory responses to hypoxia and hypercapnia are also severely impaired, removing the physiological safety mechanism for patients. Partial agonists at the μ-receptor, such as nalbuphine and buprenorphine, have a ceiling effect for their analgesic efficacy that is associated with a lesser effect on ventilation than full agonists. Most of the analgesic effects of clinically used opioids are also mediated by the μ-receptor, so the respiratory depressant effect of opioid drugs is currently inseparable from their therapeutic effect. Equi-analgesic doses of different opioids show similar degrees of respiratory depression, but the speed of onset of the drug does affect the clinical pattern of respiratory depression that occurs. With rapidly acting opioids such as fentanyl, apnoea normally follows its intravenous administration, but when an equi-analgesic dose of the slower acting morphine is administered, apnoea is unusual, because hypercapnia develops to counteract the respiratory depression.84
Benzodiazepines. Benzodiazepines exert their effect by binding directly to GABAA receptors, and increasing the inhibitory effect of endogenous GABA. Figure 5.4 shows that GABA is involved in respiratory central pattern generation so it is unsurprising that benzodiazepines affect respiration. Parenterally administered benzodiazepine drugs, such as midazolam or diazepam, cause a dose-dependent reduction in resting ventilation and reduce the ventilatory response to hypoxia85 and hypercapnia.81 The degree of respiratory impairment seen correlates well with their effect on consciousness. Reduced resting ventilation with midazolam can be reversed with the benzodiazepine antagonist flumazenil, though the responses to hypoxia and hypercapnia may still be abnormal despite the subjects no longer being sedated.86 Unlike for opioids, the respiratory depressant effects of benzodiazepines seem to have a ceiling effect, with massive overdoses of these drugs rarely causing life-threatening respiratory depression unless other CNS depressants, commonly alcohol, are ingested simultaneously.
Respiratory Stimulants
Non-specific CNS stimulant drugs have existed for many years, and, as part of their general stimulant effects, also increase respiratory drive. Early drugs of this type such as nikethamide and almitrine were used as respiratory stimulants, but at doses effective for stimulating respiration they had an unacceptably high incidence of CNS toxicity such as headache, agitation, muscle spasms or convulsions.
Doxapram is the only currently used respiratory stimulant, and seems to be more specific for respiratory stimulation than its predecessors, though still has a high incidence of CNS side effects. Doxapram works by stimulating the peripheral chemoreceptors to increase respiratory drive,87 this effect occurring at lower doses than those causing more generalised CNS stimulation. In healthy subjects, infusion of a standard dose of doxapram approximately doubles resting minute volume, and also substantially increases the ventilatory responses to hypoxia and hypercapnia.88 Despite this impressive action on respiratory control, when used to treat patients with type 2 ventilatory failure (page 393) generalised CNS stimulation undoubtedly contributes to the therapeutic effect by reversing the sedative effects of hypercapnia (page 357) and increasing the patient’s perception of their breathlessness.89
Methods for Assessment of Breathing Control
In assessing the control of breathing under ideal conditions, arterial blood gas tensions would be measured continuously. In practice, this is invasive and rapid measurements are impossible, so in almost all cases end-tidal gas concentration is measured and converted to partial pressure. In normal healthy subjects with reasonable slow respiratory rates, these measurements will equate well to alveolar and therefore arterial tension, but this may not be the case in patients.
Sensitivity to Carbon Dioxide
A lack of ventilatory response to carbon dioxide may result from impaired function of the respiratory system anywhere between the medullary neurones and the mechanical properties of the lung (see Figure 27.2). Thus it cannot be assumed that a decreased ventilation/Pco2 response is necessarily due to failure of the central chemoreceptor mechanism.
Steady state method. This technique requires the simultaneous measurement of minute volume and Pco2 after Pco2 has been raised by increasing the concentration of carbon dioxide in the inspired gas. The ventilation is usually reasonably stable after 5 minutes of inhaling a fixed concentration of carbon dioxide. Severinghaus’s pseudo steady state method90 measures ventilation after 4 minutes and is a useful compromise giving highly repeatable results.48 Several points are needed to define the Pco2/ventilation response curve and it is a time-consuming process, which may be distressing to some patients.
Rebreathing method. Introduced by Read in 1967, this technique greatly simplified determination of the slope of the Pco2/ventilation response curve.91 The subject rebreathes for up to four minutes from a 6-litre bag originally containing 7% carbon dioxide and about 50% oxygen, the remainder being nitrogen. The carbon dioxide concentration rises steadily during rebreathing while the oxygen concentration should remain above 30%. Thus there should be no appreciable hypoxic drive and ventilation is driven solely by the rising arterial Pco2, which should be very close to the Pco2 of the gas in the bag. Ventilation is measured by any convenient means and plotted against the Pco2 of the gas in the bag. The Pco2/ventilation response curve measured by the rebreathing technique is displaced to the right by about 0.7 kPa (5 mmHg) compared with the steady state method, but the slope agrees closely with the steady state method,48,91 and is much easier to perform.
Sensitivity to Hypoxia92
There is often some reluctance to test sensitivity to hypoxia because of the reduced Po2 to which the patient is exposed. Various approaches to the problem have been described, of which three are used (albeit rarely) in practice.
Steady state method. This is the classical technique and is best undertaken by preparing Pco2/ventilation response curves at different levels of Po2, which are presented as a fan (Figure 5.5). The spread of the fan is an indication of peripheral chemoreceptor sensitivity but it is also possible to present the data in the form of the rectangular hyperbola (see Figure 5.7) by plotting the ventilatory response for different values of Po2 at the same Pco2.
Rebreathing method. Read’s rebreathing method is described above and has been adapted to measure the response to hypoxia.69 The oxygen concentration of the rebreathed gas is reduced by the oxygen consumption of the subject, but active steps have to be taken to maintain the Pco2 at a constant level. Calculation of the response is greatly simplified by measuring the oxygen saturation (usually non-invasively by means of a pulse oximeter) and plotting the response as ventilation against saturation. This normally approximates to a straight line and the slope is a function of the chemoreceptor sensitivity. However, even if Pco2 is held constant, the response is directly influenced by the patient’s sensitivity to Pco2.
Intermittent inhalation of high oxygen concentration. This method avoids exposing subjects to hypoxia. Temporary withdrawal of peripheral chemoreceptor drive by inhalation of oxygen should reduce ventilation by about 15%. This may be used as an indication of the existence of carotid body activity but clearly it is much less sensitive than the steady state method.
References
1. Blanco CE. Maturation of fetal breathing activity. Biol Neonate. 1994;65:182-188.
2. Bianchi AL, Denavit-Saubie M, Champagnat J. Central control of breathing in mammals: Neuronal circuitry, membrane properties, and neurotransmitters. Physiol Rev. 1995;75:1-31.
3. Corfield DR, Fink GR, Ramsay SC, et al. Evidence for limbic system activation during CO2-stimulated breathing in man. J Physiol. 1995;488:77-84.
4. Rekling JC, Feldman JL. Pre-Bötzinger complex and pacemaker neurones: hypothesised site and kernel for respiratory rhythm generation. Annu Rev Physiol. 1998;60:385-405.
5. Richter DW, Ballanyi K, Schwarzacher S. Mechanisms of respiratory rhythm generation. Curr Opin Neurobiol. 1992;2:788-793.
6. Del Negro CA, Hayes JA. A ‘group pacemaker’ mechanism for respiratory rhythm generation. J Physiol. 2008;586:2245-2246.
*7. Ramirez JM, Telgkamp P, Elsen FP, Quellmalz UJA, Richter DW. Respiratory rhythm generation in mammals: synaptic and membrane properties. Respir Physiol. 1997;110:71-85.
8. Richter DW, Lalley PM, Pierrefiche O, et al. Intracellular signal pathways controlling respiratory neurons. Respir Physiol. 1997;110:113-123.
9. Horn EM, Waldrop TG. Supra-pontine control of respiration. Respir Physiol. 1998;114:201-211.
10. Corfield DR, Murphy K, Guz A. Does the motor cortical control of the diaphragm ‘bypass’ the brain stem respiratory centres in man? Respir Physiol. 1998;114:109-117.
11. Western PJ, Patrick JM. Effects of focussing attention on breathing with and without apparatus on the face. Respir Physiol. 1988;72:123-130.
12. Matsuo K, Hiiemae KM, Gonzalez-Fernandez M, Palmer JB. Respiration during feeding on solid food: alterations in breathing during mastication, pharyngeal bolus aggregation, and swallowing. J Appl Physiol. 2008;104:674-681.
13. Winkworth AL, Davis PJ, Adams RD, Ellis E. Breathing patterns during spontaneous speech. J Speech Hear Res. 1995;38:124-144.
14. Severinghaus JW, Mitchell RA. Ondine’s curse: failure of respiratory centre automaticity while asleep. Clin Res. 1962;10:122.
15. Vingerhoets F, Bogousslavsky J. Respiratory dysfunction in stroke. Clin Chest Med. 1994;15:729-737.
16. Weese-Mayer DE, Silvestri JM, Menzies LJ, Morrow-Kenny AS, Hunt CE, Hauptman SA. Congenital central hypoventilation syndrome: diagnosis, management, and long-term outcome in thirty-two children. J Pediatr. 1992;120:381-387.
17. Woo MS, Woo MA, Gozal D, Jansen MT, Keens TG, Harper RM. Heart rate variability in congenital central hypoventilation syndrome. Pediatr Res. 1992;31:291-296.
*18. Widdicombe JG. Afferent receptors in the airways and cough. Respir Physiol. 1998;114:5-15.
19. Sant’Ambrogio G, Tsubone H, Sant’Ambrogio FB. Sensory information from the upper airway: Role in the control of breathing. Respir Physiol. 1995;102:1-16.
20. Widdicombe J, Fontana G. Cough: what’s in a name? Eur Respir J. 2006;28:10-15.
21. Stockwell M, Lang S, Yip R, Zintel T, White C, Gallagher CG. Lack of importance of the superior laryngeal nerves in citric acid cough in humans. J Appl Physiol. 1993;75:613-617.
22. Tatar M, Hanacek J, Widdicombe J. The expiration reflex from the trachea and bronchi. Eur Respir J. 2008;31:385-390.
23. Widdicombe J. Airway receptors. Respir Physiol. 2001;125:3-15.
24. Taylor-Clark T, Undem BJ. Transduction mechanisms in airway sensory nerves. J Appl Physiol. 2006;101:950-959.
25. Bonham AC, Chen C-Y, Sekizawa S, Joad JP. Plasticity in the nucleus tractus solitarius and its influence on lung and airway reflexes. J Appl Physiol. 2006;101:322-327.
26. Ullman E. About Hering and Breuer. In: Porter R, editor. Breathing: Hering–Breuer Centenary Symposium. Edinburgh and London: Churchill Livingstone; 1970:3.
27. Gaudy JH. The Hering–Breuer reflex in man? Br J Anaesth. 1991;66:627-628.
28. Kubin L, Alheid GF, Zuperku EJ, McCrimmon DR. Central pathways of pulmonary and lower airway vagal afferents. J Appl Physiol. 2006;101:618-627.
29. Widdicombe JG. Respiratory reflexes in man and other mammalian species. Clin Sci. 1961;21:163-170.
30. Guz A, Noble MIM, Trenchard D, Cochrane HL, Makey AR. Studies on the vagus nerves in man: their role in respiratory and circulatory control. Clin Sci. 1964;27:293-304.
31. BuSha BF, Stella MH, Manning HL, Leiter JC. Termination of inspiration by phase dependent respiratory vagal feedback in awake normal humans. J Appl Physiol. 2002;93:903-910.
32. Rabbette PS, Fletcher ME, Dezateux CA, Soriano-Brucher H, Stocks J. Hering–Breuer reflex and respiratory system compliance in the first year of life: a longitudinal study. J Appl Physiol. 1994;76:650-656.
33. Guz A, Noble MIM, Eisle JH, Trenchard D. The effect of lung deflation on breathing in man. Clin Sci. 1971;40:451-461.
34. Head H. On the regulation of respiration. J Physiol (Lond). 1889;10:1-70.
35. Cross KW, Klaus M, Tooley WH, Weisser K. The response of the new-born baby to inflation of the lungs. J Physiol (Lond). 1960;151:551-565.
36. Paintal AS. Some recent advances in studies on J receptors. Adv Exp Med Biol. 1995;381:15-25.
37. Gerhold KA, Bautista DM. TRPA1: irritant detector of the airways. J Physiol. 2008;586:14.
38. Frazier DT, Revelette WR. Role of phrenic nerve afferents in the control of breathing. J Appl Physiol. 1991;70:491-496.
39. Gourine AV. On the peripheral and central chemoreception and control of breathing: an emerging role of ATP. J Physiol. 2005;568:715-724.
40. Guyenet PG, Stornetta RL, Bayliss DA. Retrotrapezoid nucleus and central chemoreception. J Physiol. 2008;586:2043-2048.
41. Nattie E. Why do we have both peripheral and central chemoreceptors? J Appl Physiol. 2006;100:9-10.
42. Sato M, Severinghaus JW, Basbaum AI. Medullary CO2 chemoreceptor neuron identification by c-fos immunochemistry. FASEB J. 1991;5:A1120.
43. Forster HV, Dempsey JA, Chosy LW. Incomplete compensation of CSF [H+] in man during acclimatisation to high altitude. J Appl Physiol. 1975;38:1067-1072.
44. Mitchell RA, Carman CT, Severinghaus JW, Richardson BW, Singer MM, Snider S. Stability of cerebrospinal fluid pH in chronic acid-base disturbances in blood. J Appl Physiol. 1965;20:443-452.
45. Froman C, Crampton-Smith A. Hyperventilation associated with low pH of cerebrospinal fluid after intracranial haemorrhage. Lancet. 1966;1:780-782.
46. Spengler CM, Czeisler CA, Shea SA. An endogenous circadian rhythm of respiratory control in humans. J Physiol. 2000;526:683-694.
*47. Mohan R, Duffin J. The effect of hypoxia on the ventilatory response to carbon dioxide in man. Respir Physiol. 1997;108:101-115.
48. Lumb AB, Nunn JF. Ribcage contributions to CO2 response during rebreathing and steady state methods. Respir Physiol. 1991;85:97-110.
49. Tansley JG, Pedersen MEF, Clar C, Robbins PA. Human ventilatory response to 8 h of euoxic hypercapnia. J Appl Physiol. 1998;84:431-434.
50. de Castro F. Sur la structure et l’innervation de la glande intercarotidienne. Trab Lab Invest biol Univ Madrid. 1926;26:365.
51. Heymans C, Bouckaert JJ, Dautrebande L. Sinus carotidien et réflexes respiratoire. Arch Int Pharmacodyn Ther. 1930;39:400.
*52. López-Barneo J, Ortega-Sáenz P, Pardal R, Pascual A, Piruat JI. Carotid body oxygen sensing. Eur Respir J. 2008;32:1386-1398.
53. Prabhakar NR. Oxygen sensing by the carotid body chemoreceptors. J Appl Physiol. 2000;88:2287-2295.
54. Weir K, López-Barneo J, Buckler KJ, Archer SL. Acute oxygen-sensing mechanisms. N Engl J Med. 2005;353:2042-2055.
55. Nurse CA. Neurotransmission and neuromodulation in the chemosensory carotid body. Auton Neurosci. 2005;120:1-99.
56. Foo IT, Warren PM, Drummond GB. Influence of oral clonidine on the ventilatory response to acute and sustained isocapnic hypoxia in human males. Br J Anaesth. 1996;76:214-220.
57. Leung PS. Novel roles of a local angiotensin-generating system in the carotid body. J Physiol. 2006;575:4.
58. Powell FL, Milsom WK, Mitchell GS. Time domains of the hypoxic ventilatory response. Respir Physiol. 1998;112:123-134.
59. Huang SY, Alexander JK, Grover RF, et al. Hypocapnia and sustained hypoxia blunt ventilation on arrival at high altitude. J Appl Physiol. 1984;56:602-606.
60. Easton PA, Slykerman LJ, Anthonisen NR. Ventilatory response to sustained hypoxia in normal adults. J Appl Physiol. 1986;61:906-911.
61. Howard LSGE, Robbins PA. Ventilatory response to 8 h of isocapnic and poikilocapnic hypoxia in humans. J Appl Physiol. 1995;78:1092-1097.
62. Robbins PA. Hypoxic ventilatory decline: site of action. J Appl Physiol. 1995;78:373-374.
63. Garcia N, Hopkins SR, Elliott AR, Aaron EA, Weinger MB, Powell FL. Ventilatory response to 2-h sustained hypoxia in humans. Respir Physiol. 2000;124:11-22.
64. Soto-Arape I, Burton MD, Kazemi H. Central amino acid neurotransmitters and the hypoxic ventilatory response. Am J Respir Crit Care Med. 1995;151:1113-1120.
65. Cruz JC, Reeves JT, Grover RF, et al. Ventilatory acclimatisation to high altitude is prevented by CO2 breathing. Respiration. 1980;39:121-130.
66. Weil JV, Byrne-Quinn E, Sodal IE, et al. Hypoxic ventilatory drive in normal man. J Clin Invest. 1970;49:1061-1072.
67. Dripps RD, Comroe JH. The effect of the inhalation of high and low oxygen concentrations on respiration, pulse rate, ballistocardiogram and arterial oxygen saturation (oximeter) of normal individuals. Am J Physiol. 1947;149:277-291.
68. Cormack RS, Cunningham DJC, Gee JBL. The effect of carbon dioxide on the respiratory response to want of oxygen in man. Q J Exp Physiol. 1957;42:303-316.
69. Rebuck AS, Campbell EJM. A clinical method for assessing the ventilatory response to hypoxia. Am Rev Respir Dis. 1974;109:345-350.
*70. Timmers HJLM, Wieling W, Karemaker JM, Lenders JWM. Denervation of carotid and baro- chemoreceptors in humans. J Physiol. 2003;553:3-11.
71. Vanmaele RG, De Backer WA, Willeman MJ, et al. Hypoxic ventilatory response to carotid endarterectomy. Eur J Vasc Surg. 1992;6:241-244.
72. Vanmaele RG, De Leersnijder, Bal J, Van Kerkhoven W, Bongaerts P, Vaerenberg C. One year follow-up after bilateral carotid body resection for COPD. Eur J Respir Dis. 1983;64(suppl 126):470.
73. Edelman NH, Neubauer JA. Hypoxic depression of breathing. In: Crystal RG, West JB, editors. The lung, scientific foundations. New York: Raven; 1991:1341.
74. Weil JVW, Byrne-Quinn E, Sodal IE, Kline JS, McCullough RE, Filley GF. Augmentation of chemosensitivity during mild exercise in normal man. J Appl Physiol. 1972;33:813-819.
75. Wellman A, Malhotra A, Jordan AS, Schory K, Gautam S, White DP. Chemical control stability in the elderly. J Physiol. 2007;581:291-298.
76. Lorenzi-Filho G, Genta PR. A new straw in the genesis of Cheyne-Stokes respiration. Chest. 2008;134:7-9.
77. Grassi B, Ferretti G, Costa M, et al. Ventilatory responses to hypercapnia and hypoxia in elite breath-hold divers. Respir Physiol. 1994;97:323-332.
78. Guz A, Noble MIM, Widdicombe JG, Trenchard D, Mushin WW, Makey AR. The role of the vagal and glossopharyngeal afferent nerves in respiratory sensation, control of breathing and arterial pressure regulation in conscious man. Clin Sci. 1966;30:161-170.
79. Campbell EJM, Godfrey S, Clark TJH, Freedman S, Norman J. The effect of muscular paralysis induced by tubocurarine on the duration and sensation of breath holding during hypercapnia. Clin Sci. 1969;36:323-328.
80. Fowler WS. Breaking point of breath-holding. J Appl Physiol. 1954;6:539-545.
81. Shook J, Watkins WD, Camporesi EM. Differential roles of opioid receptors in respiration, respiratory disease, and opiate-induced respiratory depression. Am Rev Respir Dis. 1990;142:895-909.
82. Dahan A, Teppema LJ. Influence of anaesthesia and analgesia on the control of breathing. Br J Anaesth. 2003;91:40-49.
*83. Pattinson KTS. Opioids and the control of respiration. Br J Anaesth. 2008;100:747-758.
84. Gross JB. When you breathe in you inspire, when you don’t breathe, you … expire. Anesthesiol. 2003;99:767-770.
85. Alexander CM, Gross JB. Sedative doses of midazolam depress hypoxic ventilatory response in humans. Anesth Analg. 1988;67:377-382.
86. Gross JB, Blouin RT, Zandsberg S, Conard PF, Häussler J. Effect of flumazenil on ventilatory drive during sedation with midazolam and alfentanil. Anesthesiology. 1996;85:713-720.
87. Scott RM, Whitwam JG, Chakrabarti MK. Evidence of a role for the peripheral chemoreceptors in the ventilatory response to doxapram in man. Br J Anaesth. 1977;49:227-231.
88. Burki NK. Ventilatory effects of doxapram in conscious human subjects. Chest. 1984;85:600-604.
89. Ebihara S, Ogawa H, Sasaki H, Hida W, Kikuchi Y. Doxapram and perception of dyspnea. Chest. 2002;121:1380-1381.
90. Severinghaus JW. Proposed standard determination of ventilatory responses to hypoxia and hypercapnia in man. Chest. 1976;70:129s.
91. Read DJC. A clinical method for assessing the ventilatory response to carbon dioxide. Australas Ann Med. 1967;16:20-32.
92. Duffin J. Measuring the ventilatory response to hypoxia. J Physiol. 2007;584:285-293.