Chapter 10 Respiratory System
The lower respiratory organs (larynx, trachea, bronchi, and lungs) begin to form during the fourth week of development.
 Respiratory Primordium
Respiratory Primordium
The respiratory system starts as a median outgrowth—the laryngotracheal groove—that appears in the floor of the caudal end of the primordial pharynx (Fig. 10-1B and C; see Fig. 10-3A). This primordium of the tracheobronchial tree develops caudal to the fourth pair of pharyngeal pouches. The endoderm lining the laryngotracheal groove gives rise to the pulmonary epithelium and glands of the larynx, trachea, and bronchi. The connective tissue, cartilage, and smooth muscle in these structures develop from the splanchnic mesoderm surrounding the foregut (see Fig. 10-4A).
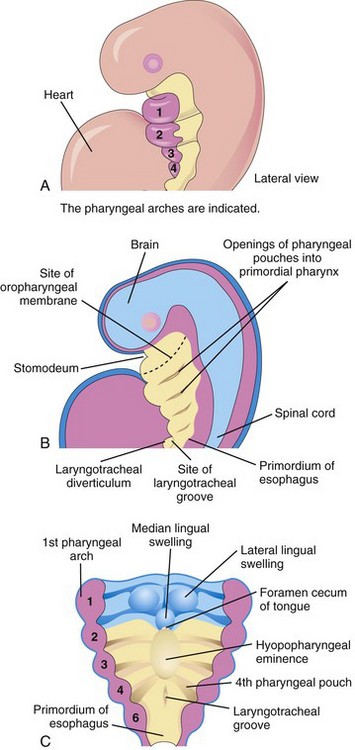
FIGURE 10–1 A, Lateral view of a 4-week embryo illustrating the relationship of the pharyngeal apparatus to the developing respiratory system. B, Sagittal section of the cranial half of the embryo. C, Horizontal section of the embryo illustrating the floor of the primordial pharynx and the location of the laryngotracheal groove.
By the end of the fourth week, the laryngotracheal groove has evaginated (protruded) to form a pouch-like laryngotracheal diverticulum (lung bud), which is located ventral to the caudal part of the foregut (Figs. 10-1B and 10-2A). As this diverticulum elongates, it is invested with splanchnic mesenchyme (primordial embryonic connective tissue), and its distal end enlarges to form a globular respiratory bud; this bud denotes the single bud from which the respiratory tree originates (Fig. 10-2B).
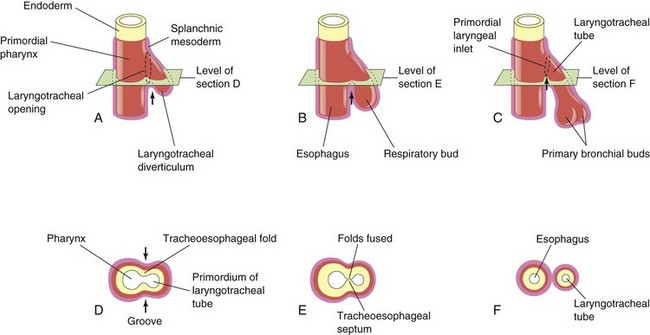
FIGURE 10–2 Successive stages in the development of the tracheoesophageal septum during the fourth and fifth weeks. A to C, Lateral views of the caudal part of the primordial pharynx showing the laryngotracheal diverticulum and partitioning of the foregut into the esophagus and laryngotracheal tube. D to F, Transverse sections illustrating formation of the tracheoesophageal septum and showing how it separates the foregut into the laryngotracheal tube and esophagus. The arrows indicate cellular changes resulting from growth.
The laryngotracheal diverticulum soon separates from the primordial pharynx; however, it maintains communication with it through the primordial laryngeal inlet (Fig. 10-2C). Longitudinal tracheoesophageal folds develop in the laryngotracheal diverticulum, approach each other, and fuse to form a partition—the tracheoesophageal septum (Fig. 10-2D and E) by the end of the fifth week. This septum divides the cranial portion of the foregut into a ventral part, the laryngotracheal tube (primordium of larynx, trachea, bronchi, and lungs), and a dorsal part (primordium of oropharynx and esophagus (Fig. 10-2F). The opening of the laryngotracheal tube into the pharynx becomes the primordial laryngeal inlet (Figs. 10-2C and 10-3B to D).
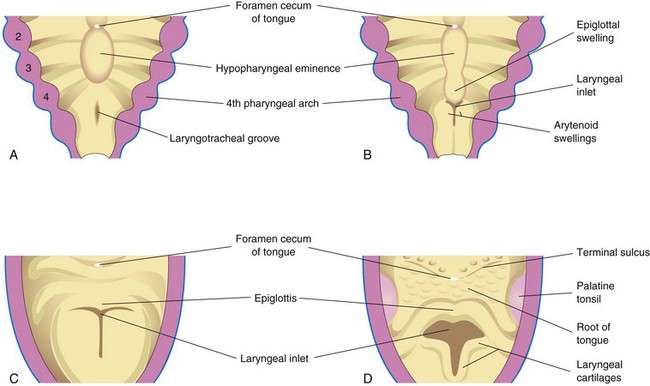
FIGURE 10–3 Successive stages in the development of the larynx. A, At 4 weeks. B, At 5 weeks. C, At 6 weeks. D, At 10 weeks. The epithelium lining of the larynx is of endodermal origin. The cartilages and muscles of the larynx arise from mesenchyme in the fourth and sixth pairs of pharyngeal arches. Note that the laryngeal inlet changes in shape from a slit-like opening to a T-shaped inlet as the mesenchyme surrounding the developing larynx proliferates.
Expression of the transcription factors NKx2.1 (ventral) and Sox2 (dorsal) are critical for dorsal-ventral patterning and trachea-esophageal separation.
Development of Larynx
The epithelial lining of the larynx develops from the endoderm of the cranial end of the laryngotracheal tube. The cartilages of the larynx develop from those of the fourth and sixth pairs of pharyngeal arches. The laryngeal cartilages develop from mesenchyme that is derived from neural crest cells. The mesenchyme at the cranial end of the laryngotracheal tube proliferates rapidly, producing paired arytenoid swellings (see Fig. 10-3B). These swellings grow toward the tongue, converting the slit-like aperture—the primordial glottis—into a T-shaped laryngeal inlet and reducing the developing laryngeal lumen to a narrow slit (Fig. 10-3C).
The laryngeal epithelium proliferates rapidly, resulting in temporary occlusion of the laryngeal lumen. Recanalization of the larynx normally occurs by the 10th week. The laryngeal ventricles form during this recanalization process. These recesses are bounded by folds of mucous membrane that become the vocal folds (cords) and vestibular folds.
The epiglottis develops from the caudal part of the hypopharyngeal eminence, a prominence produced by proliferation of mesenchyme in the ventral ends of the third and fourth pharyngeal arches (Fig. 10-3B to D). The rostral part of this eminence forms the posterior third or pharyngeal part of the tongue (see Chapter 9).
Because the laryngeal muscles develop from myoblasts in the fourth and sixth pairs of pharyngeal arches, they are innervated by the laryngeal branches of the vagus nerves (cranial nerve X) that supply these arches (see Table 9-1). Growth of the larynx and epiglottis is rapid during the first 3 years after birth. By this time, the epiglottis has reached its adult form.
Laryngeal Atresia
Laryngeal atresia, a rare birth defect, results from failure of recanalization of the larynx, which produces obstruction of the upper fetal airway—congenital high airway obstruction syndrome. Distal to the region of atresia (blockage) or stenosis (narrowing), the airways become dilated, the lungs are enlarged and filled with fluid; the diaphragm is either flattened or inverted and there is fetal ascites and/or hydrops (accumulation of serous fluid in the intracellular spaces causing severe edema). Incomplete atresia (laryngeal web) results from incomplete recanalization of the larynx during the 10th week. A membranous web forms at the level of the vocal folds, partially obstructing the airway. Treatment is by endoscopic dilation of the laryngeal web.
Development of Trachea
During its separation from the foregut, the laryngotracheal diverticulum forms the trachea and two lateral outpouchings, the primary bronchial buds (Fig. 10-2C; see Figs. 10-7A and 10-8). The endodermal lining of the laryngotracheal tube distal to the larynx differentiates into the epithelium and glands of the trachea and the pulmonary epithelium. The cartilage, connective tissue, and muscles of the trachea are derived from the splanchnic mesenchyme surrounding the laryngotracheal tube (Fig. 10-4).
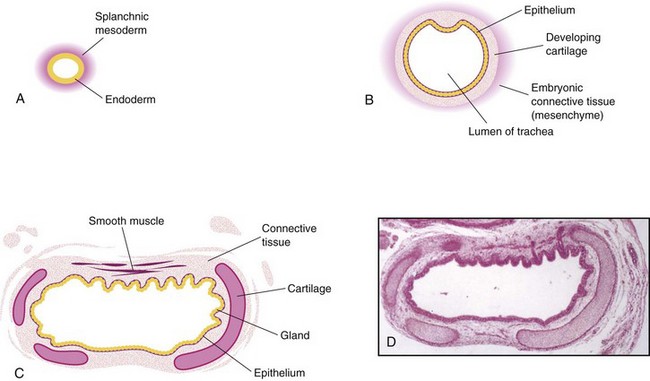
FIGURE 10–4 Transverse sections through the laryngotracheal tube illustrating progressive stages in the development of the trachea. A, 4 weeks. B, 10 weeks. C, 11 weeks (drawing of micrograph in D). Note that endoderm of the tube gives rise to the epithelium and glands of the trachea and that mesenchyme surrounding the tube forms the connective tissue, muscle, and cartilage. D, Photomicrograph of a transverse section of the developing trachea at 12 weeks.
(D, From Moore KL, Persaud TVN, Shiota K: Color Atlas of Clinical Embryology, 2nd ed. Philadelphia, WB Saunders, 2000.)
Tracheoesophageal Fistula
A fistula (abnormal passage) between the trachea and esophagus occurs once in 3000 to 4500 live births (Figs. 10-5 and 10-6); most affected infants are males. In more than 85% of cases, the tracheoesophageal fistula (TEF) is associated with esophageal atresia. A TEF results from incomplete division of the cranial part of the foregut into respiratory and esophageal parts during the fourth week. Incomplete fusion of the tracheoesophageal folds results in a defective tracheoesophageal septum and a TEF between the trachea and esophagus.
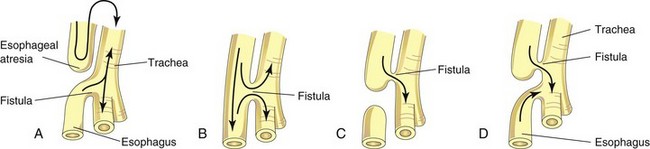
FIGURE 10–5 The four main varieties of tracheoesophageal fistula (TEF). Possible directions of the flow of the contents are indicated by arrows. Esophageal atresia, as illustrated in A, is associated with TEF in more than 85% of cases. B, Fistula between the trachea and esophagus. C, Air cannot enter the distal esophagus and stomach. D, Air can enter the distal esophagus and stomach, and the esophageal and gastric contents may enter the trachea and lungs.
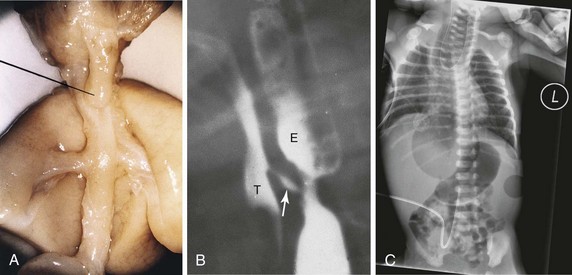
FIGURE 10–6 A, Tracheoesophageal fistula (TEF) in a 17-week male fetus. The upper esophageal segment ends blindly (pointer). B, Contrast radiograph of a neonate with TEF. Note the communication (arrow) between the esophagus (E) and trachea ( T ). C, Radiograph of esophageal atresia and trachea-esophageal fistula. The blind proximal esophageal sac is clearly visible. Note the air present in the distal GI tract indicating the presence of the tracheo-esophageal fistula. An umbilical venous catheter can also been seen.
(A, From Kalousek DK, Fitch N, Paradice B: Pathology of the Human Embryo and Previable Fetus. New York, Springer-Verlag, 1990. B, Courtesy of Dr. Prem S. Sahni, formerly of the Department of Radiology, Children’s Hospital, Winnipeg, Manitoba, Canada. C, Courtesy of Dr. J.V. Been and Dr. M.J. Schuurman, Department of Pediatrics, and Dr. S.G. Robben, Department of Radiology, Maastricht University Medical Centre, Maastricht, Netherlands.)
TEF is the most common anomaly of the lower respiratory tract. Four main varieties of TEF may develop (see Fig. 10-5). The usual anomaly is for the superior part of the esophagus to end blindly (esophageal atresia) and for the inferior part to join the trachea near its bifurcation (see Figs. 10-5A and 10-6). Other varieties of this anomaly are illustrated in Figure 10-5B to D. Infants with the common type of TEF and esophageal atresia cannot swallow, so they frequently drool saliva at rest and immediately regurgitate milk when fed. Gastric and intestinal contents may also reflux from the stomach through the fistula into the trachea and lungs. This refluxed acid, and in some cases bile, can cause pneumonitis (inflammation of the lungs) leading to respiratory compromise. Polyhydramnios (see Chapter 7) is often associated with esophageal atresia. The excess amniotic fluid develops because fluid cannot pass to the stomach and intestines for absorption and subsequent transfer through the placenta to the mother’s blood for disposal.
Laryngotracheoesophageal Cleft
Uncommonly, the larynx and upper trachea may fail to separate completely from the esophagus. This results in a persistent connection of variable lengths between these normally separated structures. Symptoms of this congenital anomaly are similar to those of tracheoesophageal fistula because of aspiration into the lungs, but aphonia (absence of voice) is a distinguishing feature.
Tracheal Stenosis and Atresia
Narrowing (stenosis) and obstruction (atresia) of the trachea are uncommon anomalies that are usually associated with one of the varieties of TEF. Stenoses and atresias probably result from unequal partitioning of the foregut into the esophagus and trachea. Sometimes there is a web of tissue obstructing airflow (incomplete tracheal atresia). Atresia or agenesis of the trachea is uniformly fatal.
Tracheal Diverticulum (Tracheal Bronchus)
This anomaly consists of a blind, bronchus-like projection from the trachea. The outgrowth may terminate in normal-appearing lung tissue, forming a tracheal lobe of the lung. A tracheal diverticulum may cause recurrent infection and respiratory distress in infants.
Development of Bronchi and Lungs
The respiratory bud develops at the caudal end of the laryngotracheal diverticulum during the fourth week (Fig. 10-2B). The bud soon divides into two outpouchings—the primary bronchial buds (Figs. 10-2C, 10-7A, and 10-8). These buds grow laterally into the pericardioperitoneal canals, the primordia of the pleural cavities (Fig. 10-7B). Secondary and tertiary bronchial buds soon develop.
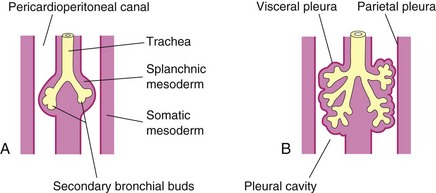
FIGURE 10–7 Illustrations of the growth of the developing lungs into the splanchnic mesenchyme adjacent to the medial walls of the pericardioperitoneal canals (primordial pleural cavities). Development of the layers of the pleura is also shown. A, 5 weeks. B, 6 weeks.
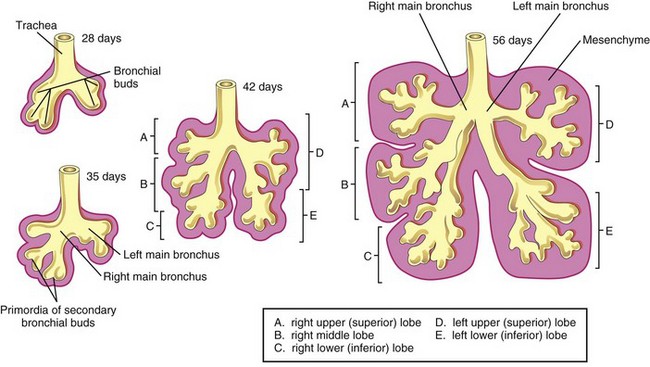
Together with the surrounding splanchnic mesenchyme, the bronchial buds differentiate into the bronchi and their ramifications in the lungs. Early in the fifth week, the connection of each bronchial bud with the trachea enlarges to form the primordia of the main bronchi (Fig. 10-8).
The embryonic right main bronchus is slightly larger than the left one and is oriented more vertically. This relationship persists in the adult; consequently, a foreign body is more likely to enter the right main bronchus than the left one.
The main bronchi subdivide into secondary bronchi that form lobar, segmental, and intrasegmental branches (Fig. 10-8). On the right, the superior lobar bronchus will supply the upper (superior) lobe of the lung, whereas the inferior bronchus subdivides into two bronchi, one to the middle lobe of the right lung and the other to the lower (inferior) lobe. On the left, the two secondary bronchi supply the upper and lower lobes of the lung. Each lobar bronchus undergoes progressive branching.
The segmental bronchi, 10 in the right lung and eight or nine in the left lung, begin to form by the seventh week. As this occurs, the surrounding mesenchyme also divides. The segmental bronchi, with the surrounding mass of mesenchyme, form the primordial of the bronchopulmonary segments. By 24 weeks, approximately 17 orders of branches have formed and respiratory bronchioles have developed (Fig. 10-9B). An additional seven orders of airways develop after birth.
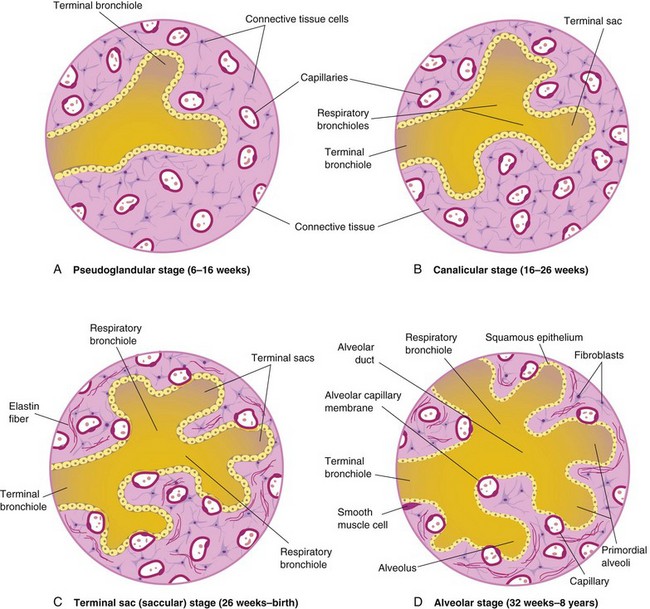
FIGURE 10–9 Diagrammatic sketches of histologic sections illustrating the stages of lung development. A and B, Early stages of lung development. C and D, Note that the alveolocapillary membrane is thin and that some capillaries bulge into the terminal sacs and alveoli.
As the bronchi develop, cartilaginous plates develop from the surrounding splanchnic mesenchyme. The bronchial smooth muscle and connective tissue and the pulmonary connective tissue and capillaries are also derived from this mesenchyme. As the lungs develop, they acquire a layer of visceral pleura from the splanchnic mesenchyme (Fig. 10-7). With expansion, the lungs and pleural cavities grow caudally into the mesenchyme of the body wall and soon lie close to the heart. The thoracic body wall becomes lined by a layer of parietal pleura, derived from the somatic mesoderm (Fig. 10-7B). The space between the parietal and visceral pleura is the pleural cavity.
Maturation of Lungs
Maturation of the lungs is divided into four stages: pseudoglandular, canalicular, terminal sac, and alveolar.
Pseudoglandular Stage (6 to 16 Weeks)
The developing lungs histologically somewhat resemble exocrine glands during this stage (Figs. 10-9A and 10-10A). By 16 weeks, all major elements of the lung have formed, except those involved with gas exchange. Fetuses born during this period are unable to survive.
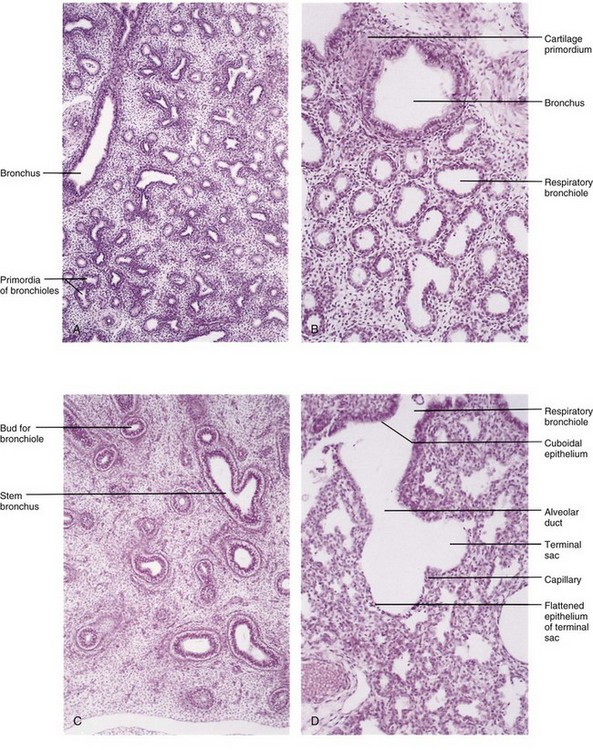
FIGURE 10–10 Photomicrographs of sections of developing embryonic and fetal lungs. A, Pseudoglandular stage, 8 weeks. Note the “glandular” appearance of the lung. B, Canalicular stage, 16 weeks. The lumina of the bronchi and terminal bronchioles are enlarging. C, Canalicular stage, 18 weeks. D, Terminal sac stage, 24 weeks. Observe the thin-walled terminal sacs (primordial alveoli) that have developed at the ends of the respiratory bronchioles. Also observe that the number of capillaries have increased and some of them are closely associated with the developing alveoli.
(From Moore KL, Persaud TVN, Shiota K: Color Atlas of Clinical Embryology, 2nd ed. Philadelphia, WB Saunders, 2000.)
Canalicular Stage (16 to 26 Weeks)
This period overlaps the pseudoglandular stage because cranial segments of the lungs mature faster than caudal ones. During the canalicular stage, the lumina of the bronchi and terminal bronchioles become larger and the lung tissue becomes highly vascular (Figs. 10-9B and 10-10B). By 24 weeks, each terminal bronchiole has given rise to two or more respiratory bronchioles, each of which then divides into three to six passages—primordial alveolar ducts.
Respiration is possible at the end of the canalicular stage because some thin-walled terminal sacs (primordial alveoli) have developed at the ends of the respiratory bronchioles, and the lung tissue is well vascularized. Although a fetus born toward the end of this period may survive if given intensive care, it often dies because its respiratory and other systems are still relatively immature.
Terminal Sac Stage (26 Weeks to Birth)
During this period, many more terminal sacs (saccules) develop (Figs. 10-9C and 10-10D), and their epithelium becomes very thin. Capillaries begin to bulge into these sacs (developing alveoli). The intimate contact between epithelial and endothelial cells establishes the blood–air barrier, which permits adequate gas exchange for survival of the fetus if it is born prematurely.
By 26 weeks, the terminal sacs are lined mainly by squamous epithelial cells of endodermal origin—type I pneumocytes—across which gas exchange occurs. The capillary network proliferates rapidly in the mesenchyme around the developing alveoli, and there is concurrent active development of lymphatic capillaries. Scattered among the squamous epithelial cells are rounded secretory epithelial cells—type II pneumocytes, which secrete pulmonary surfactant, a complex mixture of phospholipids and proteins.
Surfactant forms as a monomolecular film over the internal walls of the alveolar sacs and counteracts surface tension forces at the air-alveolar interface. This facilitates expansion of the sacs by preventing atelectasis (collapse of sacs during exhalation). The maturation of type II pneumocytes and surfactant production varies widely in fetuses of different gestational ages. The production of surfactant increases during the terminal stages of pregnancy, particularly during the last 2 weeks.
Surfactant production begins at 20 to 22 weeks, but it is present in only small amounts in premature infants; it does not reach adequate levels until the late fetal period. By 26 to 28 weeks after fertilization, the fetus usually weighs approximately 1000 g and sufficient alveolar sacs and surfactant are present to permit survival of a prematurely born infant. Before this, the lungs are usually incapable of providing adequate gas exchange, partly because the alveolar surface area is insufficient and the vascularity underdeveloped. It is not the presence of thin terminal sacs or a primordial alveolar epithelium so much as the development of an adequate pulmonary vasculature and sufficient surfactant that is critical to the survival and neurodevelopmental outcome of premature infants.
Fetuses born prematurely at 24 to 26 weeks after fertilization may survive if given intensive care; however, they may suffer from respiratory distress because of surfactant deficiency. Survival of these infants has improved with the use of antenatal corticosteroids, which induces surfactant production, and also with postnatal surfactant replacement therapy.
Alveolar Stage (32 Weeks to 8 Years)
Exactly when the terminal sac stage ends and the alveolar stage begins depends on the definition of the term alveolus. Sacs analogous to alveoli are present at 32 weeks. The epithelial lining of the terminal sacs attenuates to a thin squamous epithelial layer. The type I pneumocytes become so thin that the adjacent capillaries bulge into the alveolar sacs (Figs. 10-9D and 10-10D). By the late fetal period, the lungs are capable of respiration because the alveolocapillary membrane (pulmonary diffusion barrier or respiratory membrane) is sufficiently thin to allow gas exchange. Although the lungs do not begin to perform this vital function until birth, they are well developed so that they are capable of functioning as soon as the baby is born.
At the beginning of the alveolar stage, each respiratory bronchiole terminates in a cluster of thin-walled alveolar sacs, separated from one another by loose connective tissue. These sacs represent future alveolar ducts (Fig. 10-9D). The transition from dependence on the placenta for gas exchange to autonomous gas exchange requires the following adaptive changes in the lungs:
Approximately 95% of mature alveoli develop postnatally. Before birth, the primordial alveoli appear as small bulges on the walls of respiratory bronchioles and alveolar sacs (Fig. 10-9D). After birth, the primordial alveoli enlarge as the lungs expand, but the greatest increase in the size of the lungs results from an increase in the number of respiratory bronchioles and primordial alveoli rather than from an increase in the size of the alveoli.
Alveolar development is largely completed by 3 years of age, but new alveoli are added until approximately 8 years of age. Unlike mature alveoli, immature alveoli have the potential for forming additional primordial alveoli. As these alveoli increase in size, they become mature alveoli. The major mechanism for increasing the number of alveoli is the formation of secondary connective tissue septa that subdivide existing primordial alveoli. Initially, the septa are relatively thick, but they are soon transformed into mature thin septa that are capable of gas exchange.
Lung development during the first few months after birth is characterized by an exponential increase in the surface area of the air–blood barrier through the multiplication of alveoli and capillaries. Approximately 150 million primordial alveoli, one half of the adult number, are present in the lungs of a full-term neonate. On chest radiographs, therefore, the lungs of neonates are denser than adult lungs. Between the third and eighth years, the adult complement of 300 million alveoli is achieved.
Molecular studies indicate that lung development is controlled by a cascade of signaling pathways that are regulated by the temporal and sequential expression of highly conserved genes. The commitment and differentiation of endodermal foregut cells to form respiratory-type epithelial cells are associated with expression of several transcription factors, including thyroid transcription factor 1, hepatocyte nuclear factor (HNF) 3β, and GATA-6, as well as other Zinc-finger family members, retinoic acid receptors, and homeobox (Hox) domain-containing genes. Hox genes specify the anteroposterior axis in the embryo. Fibroblast growth factor 10 and other signals from splanchnic mesenchyme probably induce the outgrowth of the respiratory bud. Branching of this bud (branching morphogenesis) and its proliferation depend on epithelial (endodermal foregut)–mesenchymal (mesoderm) interactions. The Wnt signaling pathway plays an essential role in the inductive interactions between epithelium and mesenchyme. Recent studies suggest that Wnt7b signaling from the epithelium regulates mesenchymal proliferation and blood vessel formation in the lung. The patterning morphogen sonic hedgehog (Shh-Gli) modulates the expression of fibroblast growth factor 10, which controls the branching of the bronchial buds. Also, the morphogen retinoic acid regulates Hox a5, b5, and c4, which are expressed in the developing lung.
Fetal breathing movements (FBMs), which can be detected by real-time ultrasonography, occur before birth, exerting sufficient force to cause aspiration of some amniotic fluid into the lungs. FBMs occur intermittently (approximately 30% of them during rapid eye movement sleep) and are essential for normal lung development (Fig. 10-11). The pattern of FBMs is widely used in the monitoring of labor and as a predictor of fetal outcome in a preterm delivery. By birth, the fetus has had the advantage of several months of breathing exercise. FBMs, which increase as the time of delivery approaches, probably condition the respiratory muscles. In addition, these movements stimulate lung development, possibly by creating a pressure gradient between the lungs and the amniotic fluid.
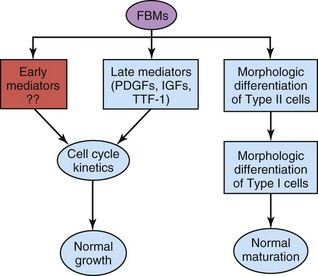
FIGURE 10–11 Fetal breathing movements (FBMs) seem to play a role in lung growth through their effects on lung cell cycle kinetics by regulating the expression of growth factors, such as platelet-derived growth factors (PDGFs) and insulin-like growth factors (IGFs), and establishing the gradient of thyroid transcription factor 1 (TTF-1) expression at the last stage of lung organogenesis (i.e., late mediators). It is also suggested that FBMs influence the expression of other unknown growth factors (i.e., early mediators) that are responsible for changes in cell cycle kinetics at earlier stages of lung development. FBMs appear to also be required for the accomplishment of the morphologic differentiation of type I and II pneumocytes.
(From Inanlou MR, Baguma-Nibasheka M, Kablar B: The role of fetal breathing-like movements in lung organogenesis. Histol Histopathol 20:1261, 2005.)
At birth, the lungs are approximately half-filled with fluid derived from the amniotic cavity, lungs, and tracheal glands. Aeration of the lungs at birth is not so much the inflation of empty collapsed organs but rather the rapid replacement of intra-alveolar fluid by air.
The fluid in the lungs is cleared at birth by three routes:
In the near-term fetus, the pulmonary lymphatic vessels are relatively larger and more numerous than in the adult. Lymph flow is rapid during the first few hours after birth and then diminishes. Three factors are important for normal lung development: adequate thoracic space for lung growth, FBMs, and adequate amniotic fluid volume (Fig. 10-12).
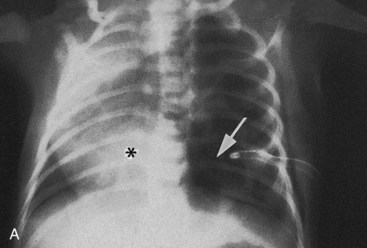
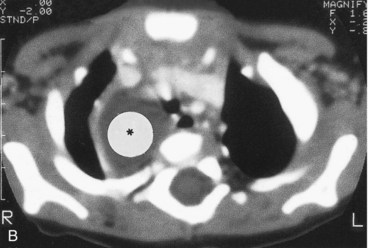
FIGURE 10–12 Congenital lung cysts. A, Chest radiograph (posteroanterior) of an infant showing a large left-sided congenital cystic adenomatoid malformation (arrow). The heart (asterisk) has shifted to the right. Note the chest tube on the left side, which was placed on the initial diagnosis of a pneumothorax (air in pleural cavity). B, Axial computed tomography image of the thorax in an infant with a large right-sided congenital bronchogenic cyst (asterisk).
(Courtesy of Dr. Prem S. Sahni, formerly of the Department of Radiology. Children’s Hospital, Winnipeg, Manitoba, Canada.)
Oligohydramnios and Lung Development
When oligohydramnios (insufficient amount of amniotic fluid) is severe and chronic because of amniotic fluid leakage or decreased production, lung development is retarded and severe pulmonary hypoplasia results from restriction of the fetal thorax.
Lungs of Neonates
Fresh healthy lungs always contain some air; consequently, pulmonary tissue removed from them will float in water. A diseased lung, partly filled with fluid, may not float. Of medicolegal significance is the fact that the lungs of a stillborn infant are firm and sink when placed in water because they contain fluid, not air.
Respiratory Distress Syndrome
Respiratory distress syndrome (RDS) affects approximately 2% of live newborn infants; those born prematurely are most susceptible. These infants develop rapid, labored breathing shortly after birth. RDS is also known as hyaline membrane disease (HMD). An estimated 30% of all neonatal disease results from RDS or its complications.
Surfactant deficiency is a major cause of RDS or HMD. The lungs are underinflated and the alveoli contain a fluid with a high protein content that resembles a glassy or hyaline membrane. This membrane is thought to be derived from a combination of substances in the circulation and from the injured pulmonary epithelium. It has been suggested that prolonged intrauterine asphyxia may produce irreversible changes in the type II alveolar cells, making them incapable of producing surfactant. There appears to be other causes for an absence or deficiency of surfactant in premature and full-term infants.
All growth factors and hormones controlling surfactant production have not been identified, but corticosteroids and thyroxine, which are involved in fetal lung maturation, are potent stimulators of surfactant production. Maternal glucocorticoid treatment during pregnancy accelerates fetal lung development and surfactant production. This finding has led to the routine clinical use of corticosteroids (betamethasone) for the prevention of RDS in preterm labor. In addition, administration of exogenous surfactant (surfactant replacement therapy) reduces the severity of RDS and neonatal mortality.
Lobe of Azygos Vein
This lobe appears in the right lung in approximately 1% of people. It develops when the apical bronchus grows superiorly, medial to the arch of the azygos vein, instead of lateral to it. As a result, the vein lies at the bottom of a fissure in the superior (upper) lobe, which produces a linear marking on a radiograph of the lungs.
Congenital Lung Cysts
Cysts (filled with fluid or air) are thought to be formed by the dilation of terminal bronchi. They probably result from a disturbance in bronchial development during late fetal life. If several cysts are present, the lungs have a honeycomb appearance on radiographs. Congenital lung cysts (Fig. 10-12) are usually located at the periphery of the lung.
Agenesis of Lungs
Absence of the lungs results from failure of the respiratory bud to develop. Agenesis of one lung is more common than bilateral agenesis, but both conditions are rare. Unilateral pulmonary agenesis is compatible with life. The heart and other mediastinal structures are shifted to the affected side, and the existing lung is hyperexpanded.
Lung Hypoplasia
In infants with congenital diaphragmatic hernia (see Chapter 8), the lung is unable to develop normally because it is compressed by the abnormally positioned abdominal viscera. Lung hypoplasia (under development) is characterized by a markedly reduced lung volume and hypertrophy of the smooth muscle in the pulmonary arteries. The pulmonary hypertension leads to decreased blood flow through the pulmonary vascular system as the blood continues to shunt through the ductus arteriosus.
Many infants with congenital diaphragmatic hernia may die of pulmonary insufficiency, despite optimal postnatal care, because their lungs are too hypoplastic for air exchange and there is too much resistance for pulmonary blood flow to support extrauterine life.
Summary of Respiratory System
Clinically Oriented Problems
Case 10–1
Choking and continuous coughing were observed in a male neonate. There was an excessive amount of mucous secretion and saliva in the infant’s mouth. He also experienced considerable difficulty in breathing. The pediatrician was unable to pass a catheter through the esophagus into the stomach.
Case 10–2
A premature infant developed rapid, shallow respiration shortly after birth. A diagnosis of RDS was made.
How do you think the infant might attempt to overcome his or her inadequate exchange of oxygen and carbon dioxide?
Discussion of these problems is given at the back of the book.
References and Suggested Reading
Abel R, Bush A, Chitty RS, et al. Congenital lung disease. In Chemick V, Boat T, Wilmott R, Bush A, editors: Kendig’s Disorders of the Respiratory Tract in Children, ed 7, Philadelphia: WB Saunders, 2006.
Brunner HG, van Bokhoven H. Genetic players in esophageal atresia and tracheoesophageal fistula. Curr Opin Genet Dev. 2005;15:341.
Domyan ET, Sun X. Patterning and plasticity in development of the respiratory lineage. Dev Dyn. 2011;240:477.
Holinger LD. Congenital anomalies of the larynx; congenital anomalies of the trachea and bronchi. In Behrman RE, Kliegman Jenson HB, editors: Nelson Textbook of Pediatrics, ed 17, Philadelphia: WB Saunders, 2004.
Ioannides AS, Massa V, Ferraro E, et al. Foregut separation and tracheo-esophageal malformations: the role of tracheal outgrowth, dorso-ventral patterning and programmed cell death. Dev Dyn. 2010;237:351.
Jobe AH. Lung development and maturation. In Martin RJ, Fanaroff AA, Walsh MC, editors: Fanaroff and Martin’s Neonatal-Perinatal Medicine: Diseases of the Fetus and Infant, ed 8, Philadelphia: Mosby, 2006.
Kays DW. Congenital diaphragmatic hernia and neonatal lung lesions. Surg Clin North Am. 2006;86:329.
Moore KL, Dalley AF, Agur AMR. Clinically Oriented Anatomy, ed 6. Baltimore: Williams & Wilkins; 2010.
Morrisey EE, Hogan BL. Preparing for the first breath: genetic and cellular mechanisms in lung development. Dev Cell. 2010;18:8.
O’Rahilly R, Boyden E. The timing and sequence of events in the development of the human respiratory system during the embryonic period proper. Z Anat Entwicklungsgesch. 1973;141:237.
Rawlins EL. The building blocks of mammalian lung development. Dev Dyn. 2011;240:463.
Shanks A, Gross G, Shim T, et al. Administration of steroids after 34 weeks of gestation enhances fetal lung maturity profiles. Am J Obstet Gynecol. 2010;203:47.
Shi W, Chen F, Cardoso WV. Mechanisms of lung development. Proc Am Thorac Soc. 2009;6:558.
Sluiter I, van de Ven CP, Wijnen RM, et al. Congenital diaphragmatic hernia. Semin Fetal Neonatal Med. 2011. [Epub ahead of print]
Turell DC. Advances with surfactant. Emerg Med Clin North Am. 2008;26:921.
Turner BS, Bradshaw W, Brandon D. Neonatal lung remodeling. J Perinat Neonat Nurs. 2006;19:362.
Warburton D, El-Hashash A, Carraro G, et al. Lung organogenesis. Curr Top Dev Biol. 2010;90:73.
Wells LJ, Boyden EA. The development of the bronchopulmonary segments in human embryos of horizons XVII and XIX. Am J Anat. 1954;95:163.
Whitsett JA, Wert SE. Molecular determinants of lung morphogenesis. In Chernick V, editor: Kendig’s Disorders of the Respiratory Tract in Children, ed 7, Philadelphia: WB Saunders, 2006.
Wladimiroff JW, Cohen-Overbeek TE, Laudy JAM. Ultrasound evaluation of the fetal thorax. In Callen PW, editor: Ultrasonography in Obstetrics and Gynecology, ed 5, Philadelphia: WB Saunders, 2008.