CHAPTER 43 Glomerulonephropathies
Glomerulonephritis (GN), or inflammation of the glomeruli and tubules, is the most common type of glomerulonephropathy and is usually caused by immune complexes within the glomerular capillary walls. It is thought to be one of the major causes of chronic kidney disease (CKD) in dogs, and several studies have shown that the prevalence of GN in randomly selected dogs is as high as 50%. The deposition of amyloid within the glomeruli and glomerular basement membrane structural abnormalities (e.g., hereditary X-linked nephropathy of male Samoyeds and Cocker Spaniels) are additional important, although less common, causes of glomerulonephropathy. Loss of plasma proteins, principally albumin, in the urine is the hallmark of glomerulonephropathy. In addition to its diagnostic utility, the magnitude of proteinuria is associated with progression of CKD, and therefore it has become a major focus in the treatment of patients with glomerulopathies.
Etiology and Pathophysiology
Most glomerulonephropathies in dogs and cats are mediated by immunologic mechanisms. Immune complexes present in the glomerular capillary wall are usually responsible for initiating glomerular damage and proteinuria. For example, soluble circulating antigen-antibody complexes may be deposited or trapped in the glomeruli (Fig. 43-1). In contrast to the glomerular deposition of preformed complexes, immune complexes may also form in situ in the glomerular capillary wall (see Fig. 43-1). This occurs when circulating antibodies react with endogenous glomerular antigens or “planted,” nonglomerular antigens in the glomerular capillary wall. Nonglomerular antigens may localize in the glomerular capillary wall as a result of an electrical charge interaction or a biochemical affinity with the glomerular capillary wall. Immune complexes have been shown to form in situ in dogs with glomerulonephritis associated with dirofilariasis.
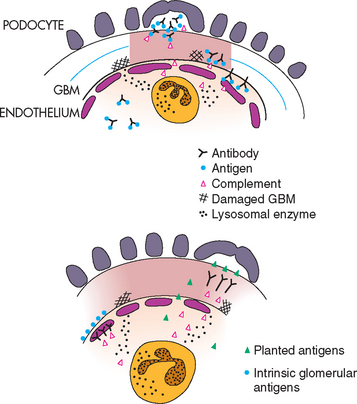
FIG 43-1 The two major types of immunologically mediated glomerular injury. Circulating soluble immune complexes have become trapped in the glomerular filter and have fixed complement. Chemotactic complement components have attracted neutrophils to the area. The release of oxygen free radicals and lysosomal enzymes from neutrophils has resulted in damage to the glomerulus (top). Damage may also result from the attachment of antibodies directed against fixed intrinsic glomerular antigens (bottom, left). Finally, damage may result from the attachment of antibodies directed against planted nonglomerular antigens (bottom, right). GBM, Glomerular basement membrane; PMN, polymorphonuclear leukocyte.
(From Chew DJ et al: Manual of small animal nephrology and urology, London, 1986, Churchill Livingstone.)
Although antibodies directed against intrinsic glomerular basement membrane material have not been found in dogs and cats with naturally occurring glomerulonephritis, several infectious and inflammatory diseases have been associated with immune-mediated glomerular disease (Box 43-1). In many cases, however, the antigen source or underlying disease is not identified; in such cases, the glomerular disease is referred to as idiopathic. It is not difficult to identify endogenous immunoglobulin or complement within glomeruli using various immunologic techniques, but the antigens associated with the immune complex within glomerular tissue are rarely identified.
 BOX 43-1 Diseases Associated with Glomerulonephritis in Dogs and Cats
BOX 43-1 Diseases Associated with Glomerulonephritis in Dogs and Cats
Despite the widespread acceptance of the term GN, in most cases glomerular lesions associated with the presence of immune complexes do not have classic evidence of neutrophilic inflammation. In very simplistic terms, the histopathologic changes observed in the glomerulus usually include one or more of the following: cellular proliferation, mesangial matrix expansion, and capillary wall thickening. Additional histopathologic subclassification of glomerular lesions associated with immune complexes that use immunohistochemical and ultrastructural studies will be necessary to improve the ability to effectively treat and accurately prognosticate this disease process.
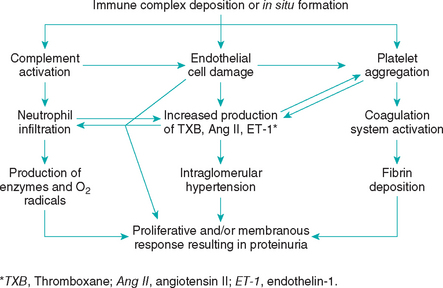
The glomerulus provides a unique environment for injurious immune complexes to stimulate production of bioactive mediators such as proinflammatory cytokines, vasoactive substances, growth factors, and extracellular matrix proteins and proteases that can contribute to the injury (see Fig. 43-1). These substances may be produced by endogenous glomerular cells or by platelets, macrophages, and neutrophils that are attracted to the immune-mediated lesion. For example, activation of the renin-angiotensin-aldosterone system (RAAS) can have hemodynamic and inflammatory/fibrotic effects on the kidney. The main hemodynamic effect is vasoconstriction of the efferent glomerular arteriole, resulting in intraglomerular hypertension. This increased hydrostatic pressure within the glomerular capillaries helps drive plasma albumin through the injured glomerular capillary wall. Angiotensin and aldosterone are also proinflammatory and can stimulate glomerular cell proliferation and fibrosis. Aldosterone also stimulates release of plasminogen activator inhibitor 1 (PAI-1), a powerful inhibitor of fibrinolysis that perpetuates glomerular thrombosis (see next paragraph).
In addition to the RAAS, several factors, including activation of the complement system, platelet aggregation, activation of the coagulation system, and fibrin deposition, also contribute to glomerular damage. Platelet activation and aggregation occur secondarily to endothelial damage or antigen-antibody interaction. Platelets, in turn, exacerbate glomerular damage by release of vasoactive and inflammatory substances and by activation of the coagulation cascade. Platelets are also capable of releasing growth-stimulating factors that promote proliferation of vascular endothelial cells. The glomerulus responds to this injury by cellular proliferation, thickening of the glomerular basement membrane, and, if the injury persists, hyalinization and sclerosis (Fig. 43-2). In those cases when identification and correction of an underlying disease process is not possible, treatment is focused on decreasing this glomerular response to the immune complexes (e.g., angiotensin and platelet antagonists).
Once a glomerulus has been irreversibly damaged by GN, the entire nephron becomes nonfunctional. Fibrosis and scarring of irreversibly damaged nephrons may resemble primary interstitial inflammation. In fact, for many years renal interstitial inflammation, or “chronic interstitial nephritis,” was thought to be the primary lesion that caused CKD in dogs. As more and more nephrons become involved, glomerular filtration in toto decreases. Remaining viable nephrons compensate for the decrease in nephron numbers with increased individual glomerular filtration rates (Fig. 43-3). This “hyperfiltration,” coupled with systemic hypertension if present, may further contribute to glomerular hyalinization and sclerosis. Although it has not been documented in dogs with naturally occurring GN, hyperfiltration and proteinuria in remnant nephrons may result in progressive nephron loss, independent of the primary disease process.
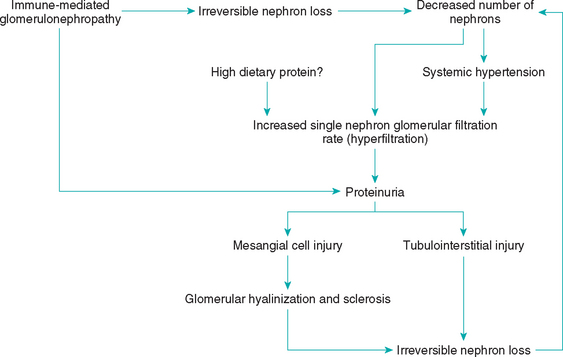
FIG 43-3 Proposed pathogenesis of progressive loss of nephrons secondary to a primary glomerulonephropathy.
Although glomerular amyloidosis is less common than GN, it is a progressive disease that also frequently leads to CKD. It is characterized by the extracellular deposition of nonbranching fibrillar proteins that stack into a specific β-pleated sheet conformation and exhibit green birefringence under polarized light when stained with Congo red (Fig. 43-4). Amyloidosis in dogs and cats is the reactive systemic form, in which amyloid may be deposited in several organs besides the kidneys. Reactive systemic amyloid deposits contain amyloid protein AA, which is an amino-terminal fragment of the acute-phase reactant protein, serum amyloid A protein (SAA), and is produced by hepatocytes in response to tissue injury. Cytokines (e.g., interleukins, tumor necrosis factor) released from macrophages after tissue injury stimulate hepatocytes to produce SAA. Amyloidosis is usually associated with an underlying inflammatory or neoplastic process; however, no predisposing factors can be identified in many dogs and cats with amyloidosis. Amyloidosis has been associated with cyclic neutropenia and with ciliary dyskinesia and recurrent respiratory tract infections in dogs. Renal amyloidosis is a familial disease in the Abyssinian cat; it results in medullary (not glomerular) amyloid deposition as a part of systemic amyloidosis. A similar form of suspected familial medullary amyloidosis resulting in renal failure has been observed in Chinese Shar-Pei dogs. Intermittent fever that occurs in association with tibiotarsal joint swelling and that resolves regardless of treatment is often observed in these dogs. The staining characteristics of the amyloid in Chinese Shar-Peis indicate that the amyloid is an inflammatory type. This amyloidosis syndrome in Chinese Shar-Peis is similar to that observed in people with familial Mediterranean fever. The medullary deposition of amyloid in Abyssinian cats and Chinese Shar-Pei dogs makes proteinuria uncommon; renal failure, however, is a common sequela.

Clinical Features
There may be no clinical signs associated with low level proteinuria; alternatively, if signs are present they are usually mild and nonspecific (e.g., weight loss and lethargy). If proteinuria is severe and results in serum albumin concentration <1.5 to 1.0 mg/dl, edema and/or ascites may occur (Table 43-1). If the glomerular disease process causes loss of more than three quarters of the nephrons, clinical signs consistent with advanced stage CKD may be present (e.g., polydipsia-polyuria, anorexia, nausea, vomiting, weight loss). Occasionally, clinical signs associated with an underlying infectious, inflammatory, or neoplastic disease may be the reason owners seek veterinary care. Rarely, dogs may be presented with acute dyspnea or severe panting caused by a pulmonary thromboembolism or may have signs associated with thromboembolism elsewhere (e.g., lameness from aortic thromboembolism).
TABLE 43-1 Signs Associated with Different Manifestations of Glomerular Disease
| MANIFESTATION | CLINICAL SIGNS | CLINICOPATHOLOGIC FINDINGS |
|---|---|---|
| Mild-to-moderate proteinuria* | Lethargy, mild weight loss, decreased muscle mass | Serum albumin 1.5-3.0 g/dl |
| Marked proteinuria (>3.5 g/day) | Severe muscle wasting, weight gain may occur, however, as result of edema or ascites | Serum albumin <1.5 g/dl, hypercholesterolemia |
| Renal failure | Depression, anorexia, nausea, vomiting, weight loss, polyuria-polydipsia | Azotemia, isosthenuria or minimally concentrated urine, hyperphosphatemia, nonregenerative anemia |
| Pulmonary thromboembolism | Acute dyspnea or severe panting | Hypoxemia; normal or low Pco2; fibrinogen >300 mg/dl; antithrombin <70% of normal |
| Retinal hemorrhage and/or detachment | Acute blindness | Systolic blood pressure >180 mm Hg |
* Microalbuminuria, as discussed in Chapter 42, may precede proteinuria and therefore be an early diagnostic tool. Pco2, Partial pressure of carbon dioxide.
Persistent proteinuria may lead to clinical signs of nephrotic syndrome, which is usually defined as a combination of proteinuria, hypoalbuminemia, ascites or edema, and hypercholesterolemia. Decreased plasma oncotic pressure and hyperaldosteronism activity causing sodium retention are thought to be the primary cause of ascites and edema. It has also been hypothesized that intrarenal mechanisms, independent of aldosterone, may contribute to sodium retention. The hypercholesterolemia associated with the nephrotic syndrome probably occurs because of a combination of decreased catabolism of proteins and lipoproteins and increased hepatic synthesis of proteins and lipoproteins. This results in the accumulation of large–molecular-weight, cholesterol-rich lipoproteins, which are not as easily lost through the damaged capillary wall as are the smaller–molecular-weight proteins, such as albumin.
In addition to the previously mentioned clinical signs, systemic hypertension and hypercoagulability are frequent complications in dogs with nephrotic syndrome. A combination of activation of the RAAS and decreased renal production of vasodilators, coupled with increased responsiveness to normal vasopressor mechanisms, are likely involved in the pathogenesis of the systemic hypertension. Systemic hypertension has been commonly associated with immunemediated GN, glomerulosclerosis, and amyloidosis, and in one study, 84% of dogs with glomerular disease were found to be hypertensive. Retinal changes, including hemorrhage, detachment, and papilledema, can be consequences of systemic hypertension; occasionally, blindness may be the presenting sign in hypertensive dogs. In most cases, the systemic hypertension is thought to occur secondary to the kidney disease rather than being a primary entitiy that causes the kidney disease. Systemic hypertension can be transmitted into the glomerular capillaries, especially as autoregulation fails, resulting in intraglomerular hypertension. This increased hydrostatic pressure within glomerular capillaries can exacerbate loss of plasma proteins across the already abnormal capillary wall or sufficiently damage the wall to induce nascent glomerular protein loss. Blood pressure measurement should be part of the evaluation and management of dogs with glomerular disease because it is likely that control of systemic hypertension may slow the progression of glomerular disease.
Hypercoagulability and thromboembolism associated with the nephrotic syndrome occur secondarily to several abnormalities in hemostasis. In addition to mild thrombocytosis, a hypoalbuminemia-related platelet hypersensitivity increases platelet adhesion and aggregation proportionally to the magnitude of hypoalbuminemia. Loss of antithrombin (AT) in urine also contributes to hypercoagulability. Antithrombin works in concert with heparin to inhibit serine proteases (clotting factors II, IX, X, XI, and XII) and normally plays a vital role in modulating thrombin and fibrin production. Finally, impaired fibrinolysis caused by aldosterone-induced production of PAI-1 further enhances blood clotting. The pulmonary arterial system is the most common location for a thromboembolic disease in dogs with glomerular lesions. Dogs with pulmonary thromboembolism are usually dyspneic and hypoxemic and have minimal pulmonary parenchymal radiographic abnormalities. Treatment of pulmonary thromboembolism is difficult, often expensive, and frequently unrewarding; therefore early prophylactic treatment to prevent thrombus formation is important.
There is increasing suspicion that proteinuria may cause glomerular and tubulointerstitial damage that can lead to progressive nephron loss in dogs and cats. Plasma proteins that have crossed the glomerular capillary wall can accumulate within the glomerular tuft and stimulate mesangial cell proliferation and increased production of mesangial matrix. In addition, excessive amounts of protein in the glomerular filtrate can damage tubular epithelial cells and lead to interstitial inflammation, fibrosis, and cell death. Mechanisms for the tubulointerstitial lesions associated with proteinuria include tubular cell lysosomal damage/rupture, peroxidative and immune-mediated damage, increased production of growth factors, cytokines and vasoactive agents, and transdifferentiation of tubular cells to myoepithelial cells that can produce collagen.
In dogs with naturally occurring CKD, proteinuria resulting in a urine protein : creatinine ratio ≥1.0 was associated with a threefold greater risk of developing uremic crises and death compared with dogs with urine protein : creatinine ratio <1.0. The relative risk of adverse outcome was approximately 1.5 times higher for every 1 unit increase in urine protein : creatinine ratio. In addition, dogs with urine protein : creatinine ratio ≥1.0 had a decrease in renal function that was greater in magnitude than that observed in dogs with urine protein : creatinine ratio <1.0. In cats with naturally occurring CKD, proteinuria appears to be very highly related to survival. The hazard ratios (95% confidence intervals) for death or euthanasia were 2.9 and 4.0 for urine protein : creatinine ratio 0.2 to 0.4 and>0.4, respectively, compared with the baseline group with a urine protein : creatinine ratio <0.2. On the basis of this evidence, it is possible that proteinuria is not only a marker of CKD in the dog and cat but also a mediator of progressive renal injury. Attenuation of proteinuria should be a major treatment objective in dogs and cats with CKD.
Diagnosis
Persistent, severe proteinuria with a normal urine sediment (hyaline casts may be observed) is the hallmark clinicopathologic sign of glomerulonephropathies. The urine protein : creatinine ratio is used to quantify the magnitude of the urine protein loss. Microalbuminuria may precede overt proteinuria in many cases (see the section on proteinuria in Chapter 42). Protein-losing nephropathies are definitively diagnosed on the basis of renal cortical histopathologic findings. (See sections on proteinuria and renal biopsy in Chapters 41 and 42.)
Treatment
Inasmuch as immune complexes usually initiate GN, primary treatment objectives include (1) identification and elimination of causative/associated antigens and (2) reduction of the glomerular response to the immune complexes.
Elimination of the source of antigenic stimulation is the treatment of choice for GN. For example, proteinuria associated with dirofilariasis in dogs often improves or resolves after successful treatment of parasitic infection. Unfortunately, elimination of the antigen source often is not possible because the antigen source or underlying disease may not be identified or may be impossible to eliminate (e.g., neoplasia). In a retrospective study by Cook (1996) of 106 dogs with GN, 43% had no identifiable concurrent disease or disorder and 19% had neoplasia. Infection, polyarthritis, hepatitis, hyperadrenocorticism, and immune-mediated hemolytic anemia are additional commonly identified concurrent medical problems (Box 43-2).
 BOX 43-2 Treatment Guidelines for Dogs and Cats with Glomerulonephritis
BOX 43-2 Treatment Guidelines for Dogs and Cats with Glomerulonephritis
PO, By mouth; ACEIs, angiotensin-converting enzyme inhibitors.
Immunosuppressive drugs have been recommended in dogs with GN, but despite these recommendations, there has been only one controlled clinical trial in veterinary medicine assessing the effects of immunosuppressive treatment. In this study by Vaden (1995) cyclosporine treatment was found to be of no benefit in reducing proteinuria associated with GN in dogs. The association between hyperadrenocorticism or long-term exogenous corticosteroid administration and GN and thromboembolism in the dog, as well as the lack of consistent therapeutic response to corticosteroids, raises questions about use of these drugs in dogs with GN. In a retrospective study of dogs with naturally occurring GN, treatment with corticosteroids appeared to be detrimental, leading to azotemia and worsening of proteinuria. Similarly, prednisone increased the urine protein : creatinine level from 1.5 to 5.6 in carrier female dogs with X-linked hereditary nephropathy. Consequently, routine use of corticosteroids to treat GN in dogs is not recommended. Treatment with corticosteroids may be indicated, however, if the underlying disease process is known to be steroid responsive (e.g., systemic lupus erythematosus). It is likely that there are specific subtypes of canine immune complex GN (e.g., minimal change GN) that are steroid responsive if they are appropriately identified and treated.
If an underlying or concurrent disease process cannot be identified and treated, or if immunosuppressive treatment is deemed inappropriate, treatment may be aimed at decreasing the glomerular response to the presence of immune complexes. Platelets appear to play an important role in the glomerular response to immune complexes, and therefore aspirin treatment is often recommended. Appropriate dosage is probably important if nonspecific cyclooxygenase inhibitors, such as aspirin, are used to decrease glomerular inflammation and platelet aggregation. An extremely low dosage of aspirin (0.5 mg/kg administered orally once a day) may selectively inhibit platelet cyclooxygenase without preventing the beneficial effects of prostacyclin formation (e.g., vasodilation, inhibition of platelet aggregation). Low-dose aspirin is easily administered on an outpatient basis and does not require extensive monitoring. Because fibrin accumulation within the glomerulus is a frequent and irreversible consequence of GN and thromboembolic disorders can complicate the management of protein-losing nephropathies, antiplatelet/anticoagulant treatment with aspirin may serve several purposes.
Treatment with angiotensin-converting enzyme inhibitors (ACEIs) can reduce proteinuria and slow disease progression. In dogs with unilateral nephrectomy and experimentally induced diabetes mellitus, ACEI administration reduced glomerular transcapillary hydraulic pressure and glomerular cell hypertrophy as well as proteinuria. In another study by Grodecki (1997) ACEI treatment of Samoyed dogs with X-linked hereditary nephritis decreased proteinuria, improved renal excretory function, decreased glomerular basement membrane splitting, and prolonged survival compared with control dogs. A double-blind, multicenter, prospective clinical trial assessed the effects of enalapril (EN) versus standard care in dogs with naturally occurring, idiopathic GN. The enalapril treatment group had decreased proteinuria, systolic blood pressure, and stable renal function compared with the placebo-treated group. In prospective randomized, con trolled clinical trials (King, 2006; Mizutani, 2006) in cats with spontaneous CKD, benazepril has been shown to reduce proteinuria, delay CKD progression, and extend survival time.
Treatment with ACEI probably decreases proteinuria and preserves renal function associated with glomerular disease by several mechanisms. In dogs administration of lisinopril decreases efferent glomerular arteriolar resistance, which results in decreased glomerular transcapillary hydraulic pressure and decreased proteinuria. In rats administration of EN prevents the loss of glomerular heparan sulfate that can occur with glomerular disease. Administration of ACEI also is thought to attenuate proteinuria by decreasing the size of glomerular capillary endothelial cell pores in people. In addition, the antiproteinuric and renal protective effects of ACEI in people may be, in part, associated with improved lipoprotein metabolism. Decreased production of angiotensin and aldosterone may also result in decreased renal fibrosis. Finally, administration of ACEI in dogs slows glomerular mesangial cell growth and proliferation that can alter the permeability of the glomerular capillary wall and lead to glomerulosclerosis.
Supportive therapy is important in the management of dogs with GN and should be aimed at alleviating systemic hypertension, decreasing edema/ascites, and reducing the risk of thromboembolism. ACEIs are recommended as the first line of treatment for proteinuric, hypertensive dogs. In those cases wherein systemic hypertension is refractory to ACEI treatment, a calcium channel blocker should be added to the antihypertensive regimen. Although similar studies have not been performed in dogs or cats, in people the combination of ACEI and an aldosterone receptor antagonist (e.g., spironolactone) have had additive effects in reducing proteinuria and renal disease progression.
Cage rest and restriction of dietary sodium should be the primary treatment considerations for patients with edema and/or ascites. Paracentesis and diuretics should be reserved for those dogs with respiratory distress or abdominal discomfort. Overzealous use of diuretics may cause dehydration and acute renal decompensation. Plasma transfusions will provide only temporary benefit in terms of increasing oncotic pressure resulting from the addition of albumin. In the past, dietary protein supplementation was recommended to offset the effects of proteinuria and reduce edema and ascites; however, recent studies in proteinuric heterozygous female dogs with X-linked nephropathy suggest that reduced dietary protein is associated with reduced proteinuria. N-3 fatty acid supplementation may also be beneficial; in dogs with surgically reduced remnant kidneys, dietary supplementation with fish oil reduced proteinuria, intraglomerular pressures, and glomerular lesion and maintained the glomerular filtration rate.
Similar to the treatment of GN, the primary treatment for amyloidosis, if possible, should be the identification and treatment of any underlying inflammatory process. Dimethylsulfoxide (DMSO) has been shown to dissolve amyloid fibrils in vitro and in vivo in mice. It has been hypothesized that DMSO has a similar amyloid-dissolving effect in domestic animals. The antiinflammatory effects of DMSO may also serve to decrease production of the acute-phase reactant SAA and the inflammation associated with an underlying disease. Decreased urinary protein excretion was observed in one dog with amyloidosis treated with DMSO; however, the effects of DMSO were difficult to determine because two potential underlying causes (interdigital pyoderma and a Sertoli cell tumor) were eliminated before the DMSO treatment. The dosage of DMSO used in that dog was 80 mg/kg administered subcutaneously three times per week; the treatment was continued for more than a year without apparent adverse effects. Other studies assessing the effects of DMSO in dogs with amyloidosis, however, have shown the treatment to be ineffective.
Colchicine is another drug that is frequently mentioned for the treatment of amyloidosis. It prevents the production of SAA by hepatocytes and has been shown to prevent amyloidosis in humans and mice if used early in the disease. Although colchicine has been recommended to prevent medullary amyloidosis in Chinese Shar-Pei dogs with fever and tibiotarsal joint swelling, no controlled studies of its use in this setting have been performed. The dosage of colchicine that has been recommended for the prophylactic treatment of amyloidosis is 0.025 mg/kg given orally q24h. Increasing the dose to 0.025 mg/kg, given orally q12h, may be considered if the animal tolerates the initial dose well for 2 weeks. However, because adverse effects of colchicine include bone marrow toxicity, the patient should be monitored closely with periodic complete blood counts.
Monitoring
It is important to monitor the urine protein : creatinine ratio after initiating treatment. Immunosuppressive treatment could alter the ratio of antigen to antibody, thus exacerbating the glomerular lesions and the proteinuria (i.e., a decrease in antibody formation leading to a mild excess of antigen or equal amounts of antigen and antibody in the immune complexes), in which case treatment should be altered or discontinued. In addition, corticosteroids can induce proteinuria owing to a number of mechanisms, so an increase in the urine protein : creatinine ratio can be iatrogenic, not necessarily a result of the progression of the disease. Lack of response to ACEI treatment may suggest the need for increasing the dosage or adding one or more drugs.
In addition, blood pressure and serum creatinine and urea nitrogen concentrations should be monitored in animals with GN. In cases in which the glomerular filtration rate depends on sodium retention and volume expansion, treatment with ACEIs can be associated with a decrease in renal excretory function. Finally, although proteinuria then occurs before the onset of azotemia, GN can lead to CKD. With the development of CKD, the glomerular filtration rate decreases and the proteinuria therefore usually also decreases. Management guidelines for CKD are presented in Chapter 44.
Prognosis
The prognosis for dogs with GN is variable and is best based on consideration of the following factors: severity of dysfunction (i.e., the magnitude of the proteinuria and the presence or absence of azotemia), the response to therapy, and the assessment of renal histopathology. Clinical experience suggests that the disease is progressive in many cases, but decreases in the urine protein : creatinine ratio and increases in albumin and AT concentration can occur in dogs with immune-mediated GN treated with diet, ACEIs, and low-dose aspirin. In selected cases, immunosuppressive treatment with corticosteroids and azathioprine may be of benefit.
Inasmuch as glomerular amyloid deposition results in severe proteinuria, with its attendant effects, the disease is relentlessly progressive, often resulting in CKD and uremia; and given that no specific treatment has proved to be effective, the prognosis for animals with renal amyloidosis is guarded to poor.
Brown S, et al. Guidelines for the identification, evaluation, and management of systemic hypertension in dogs and cats. J Vet Intern Med. 2007;21:542.
Cook AK, et al. Clinical and pathologic features of protein-losing glomerular disease in the dog: A review of 137 cases. J Am Anim Hosp Assoc. 1996;32:313-322.
Dambach DM, et al. Morphologic, immunohistochemical, and ultrastructural characterization of a distinctive renal lesion in dog putatively associated with Borrelia burgdorferi infection: 49 cases (1987–1992). Vet Pathol. 1997;34:85.
Grauer GF, et al. Effects of enalapril vs placebo as a treatment for canine idiopathic glomerulonephritis. J Vet Intern Med. 2000;14:526.
Grauer GF. Management of glomerulonephritis. In Elliott JA, Grauer GF, editors: BSAVA manual of canine and feline nephrology and urology, ed 2, Gloucester, England: British Small Animal Veterinary Association, 2007.
Grodecki K, et al. Treatment of X-linked hereditary nephritis in Samoyed dogs with angiotensin-converting enzyme (ACE) inhibitor. J Comp Pathol. 1997;117:209.
Jocob F, et al. Evaluation of the association between initial proteinuria and morbidity rate or death in dogs with naturally occurring chronic renal failure. J Am Vet Med Assoc. 2005;226:393-400.
King JN, et al. Tolerability and efficacy of benazepril in cats with chronic kidney disease. J Vet Intern Med. 2006;20:1054.
Lees GE, et al. Assessment and management of proteinuria in dogs and cats: 2004 ACVIM Forum Consensus Statement (Small Animal). J Vet Intern Med. 2005;19:377.
Mizutani H, et al. Evaluation of the clinical efficacy of benazepril in the treatment of chronic renal insufficiency in cats. J Vet Intern Med. 2006;20:1074.
Syme HM, et al. Survival of cats with naturally occurring chronic renal failure is related to severity of proteinuria. J Vet Intern Med. 2006;20:528.
Vaden SL, et al. The effects of cyclosporin versus standard care in dogs with naturally occurring glomerulonephritis. J Vet Intern Med. 1995;9:259.
Vaden SL. Glomerular diseases. In Ettinger SJ, Feldman EC, editors: Textbook of veterinary internal medicine, ed 6, St Louis: Elsevier/Saunders, 2005.