Chapter 43 Techniques in microscopy
The microscopical characters of many drugs have already been described, but it will be realized that microscopical techniques require considerable skill; years of experience are necessary to acquire a really good knowledge of the microscopy of drugs, foodstuffs and other plant materials. It is first necessary to learn how to use a microscope properly and to understand the purpose of the different reagents used in the examination of crude drugs. The preparation of systematic and illustrated reports is also important.
Mountants for specimens
Definition, particularly of colourless structures, is increased by choice of a mountant of refractive index different from that of the object. A mountant of lower refractive index is to be preferred so that the outline shadow is on the side away from the object. The ratio refractive index of object to refractive index of mountant should be of the order of 1.06. The value of this relative refractive index for cellulose (cotton) to water is 1.17, to chloral hydrate solution (5:2) 1.08 and to glycerin 1.06. This serves to emphasize the value of chloral hydrate and glycerin as mountants for plant structures.
THE MICROSCOPE
Magnification and field of view
For work in pharmacognosy, microscopes are usually fitted with two objectives, 16 mm and 4 mm, two or three eyepieces and a condenser. The procedure for making microscopical measurements is described below. Different combinations of eyepiece and objective give different magnifications and fields of view, as indicated in the table below.
When using the microscope, it is useful to know the size of the fields of view. For instance, if we know that using a 4 mm objective and a ×6 eyepiece our field of view is approximately 0.5 mm, or 500 μm, the size of objects such as the Arachnoidiscus diatom in agar (100–300 μm) or the large rosette crystals of calcium oxalate in rhubarb (up to 200 μm) may be roughly estimated. For accurate measurement, however, an eyepiece micrometer or camera lucida is used.
Apparatus for making microscopical measurements and drawings to scale
Microscopical measurements can be made using a stage micrometer in conjunction with an eyepiece micrometer, camera lucida or microprojector.
Micrometers
Two scales are required, known, respectively, as a stage micrometer and an eyepiece micrometer. The stage micrometer is a glass slide 7.6 × 2.5 cm (3 × 1 inch) with a scale engraved on it. The scale is usually 1 or 1.1 mm long and is divided into 0.1 and 0.01 parts of a millimetre. The eyepiece micrometer may be a linear scale (Fig. 43.1A and the scale 0–10 in Fig. 43.1B) or it may be ruled in squares. The value of one eyepiece division is determined for every optical combination to be used, a note being made in each case of the objective eyepiece and length of draw-tube.
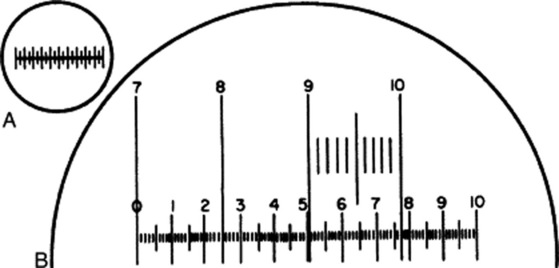
Fig. 43.1 A, Eyepiece micrometer; B, eyepiece micrometer superimposed on portion of stage micrometer scale.
To do this, unscrew the upper lens of the eyepiece, place the eyepiece micrometer on the ridge inside, and replace the lens. Put the stage micrometer on the stage and focus it in the ordinary way. The two micrometer scales now appear as in Fig. 43.1B, when the 4 mm objective is in use. In the example figured, it will be seen that when the 7 line of the stage micrometer coincides with the 0 of the eyepiece, the 10 of the stage coincides with 7.7 of the eyepiece. As the distance between 7 and 10 on the stage scale is 0.3 mm, 77 of the small eyepiece divisions equal 0.3 mm or 300 μm; therefore, 1 eyepiece division equals 300/77 or 3.9 μm.

Camera lucida
Various forms of apparatus have been designed so that a magnified image of the object under the microscope may be traced on paper. The Swift-Ives camera lucida and the Abbé drawing apparatus are examples. The former fits over the eyepiece, and when in use light from the object passes direct to the observer’s eye through an opening in the silvered surface of a prism. At the same time, light from the drawing paper and pencil is reflected by a second prism and by the silvered surface, so that the pencil appears superimposed on the object, which may thus be traced.
When the instrument is in use, the illumination of both object and paper must be suitably adjusted and the paper must be tilted at the correct angle to avoid distortion. The Abbé drawing apparatus utilizes, instead of the adjustable prism, a plane mirror carried on a side-arm; with the mirror at 45° to the bench surface, no inclined board is necessary.
To make measurements or scale drawings, the divisions of a stage micrometer are first traced on paper and then, using the same objective, eyepiece and length of draw-tube, the object to be recorded is traced.
The above camera lucidas which have served pharmacognosists well in the past do not now appear to be available commercially. This seems apparent from some of the line illustrations now submitted for publication!
Microprojection
With a suitable apparatus objects beneath a microscope can be transmitted to a screen or on to paper. Suitable particles can then be traced at the desired magnification.
Photomicrography
Modern research microscopes have built-in facilities for photo- micrography; with less expensive or with older microscopes it is necessary to fit a suitable camera to the equipment. The photographic record may seem, at first sight, to dispense with the older tracing techniques discussed above. However, although the photographic record is suitable for thin sections of plant material, rarely is it completely so for powdered drugs. A photograph contains everything visible in a particular field of view and much of this is usually uninformative; rarely does a single field of view show all the diagnostic features of the powdered drug to best advantage, and also, owing to limitations on depth of focus, structures such as large fibres, vessels, trichomes, etc. are seldom seen with all details completely in focus. Fragments of disintegrated material adhering to, or partly covering, the element photographed are also confusing to the inexperienced observer.
Polarization
The apparatus consists of a polarizer, or Nicol prism, fitting below the microscope stage, and a similar prism forming the analyser fitting above the objective. As in the case of the polarimeter, with which students will be familiar, one Nicol is kept stationary while the other is rotated. With geological microscopes, polarizers are usually permanently fitted, but for botanical work the analyser is usually fitted when required, either between the objective and nosepiece or over the eyepiece. Less cumbersome are discs cut from polaroid sheets; one of these can be of a size to fit into the filter holder of the microscope and the other to rest on the eyepiece.
When both the polarizer and analyser have their diagonal surfaces parallel, the ray of plane polarized light is transmitted by the analyser. If now the polarizer is revolved, the light diminishes in intensity until at a position 90° from the first it is entirely extinguished, the polarized light being now totally reflected by the analyser. This position, when the diagonal surfaces of the two Nicols are at right angles, is termed ‘crossed Nicols’.
Isotropic substances are characterized by having the same physical properties in all directions (e.g. gases, liquids and isometric crystals). Such substances are monorefringent (i.e. they have only one refractive index). Isotropic substances are not visible, however they be oriented, when examined between crossed Nicols. They in no way affect the polarized light passing through them from the polarizer.
Anisotropic substances exhibit different physical properties according to the direction along which they are examined. Such substances show more than one refractive index. The great majority of crystalline materials show birefringence. When a uniaxial crystal is placed with its optical axis horizontal to the stage and examined between crossed Nicols, then as the stage is rotated it will alternately shine bright (or coloured) and disappear. Through the 360° it becomes invisible (i.e. shows extinction) four times. The examination of crystals between crossed Nicols enables us to determine their crystal system (see ‘Calcium oxalate’ Chapter 42). The crystal is placed with its axis parallel to the longer diagonal of the polarizing Nicol. If the crystal belongs to the tetragonal system, the polarized light passes unchanged and on reaching the analyser is completely absorbed, the field appearing dark (i.e. extinction takes place). Conversely, monoclinic crystals show extinction only when the vertical axis makes an angle with the diagonal of the Nicol known as the extinction angle.
Many crystalline substances show brilliant colours when examined in polarized light (e.g. asbestos, sucrose, cinnamic acid). Starch grains often show a black cross, a phenomenon due to the crystalline refraction of the material. Polarized light is useful for the detection of calcium oxalate, especially when only small quantities are present in the tissues under examination. It appears bright on a black background.
Phase-contrast microscopy
This has proved particularly useful for the examination of living cells, the constituents of which normally show little differentiation. Monochromatic light is employed; light directly transmitted through the sample is reduced in intensity and the deflected light is brought half a wavelength out of phase with the transmitted light. Strong contrasts in the material under examination are thereby obtained without reduction in the resolving power of the microscope.
Ultraviolet microscopy
The limit of resolution of any microscope is governed by the wavelength of the beam employed; the shorter the wavelength, the smaller the object which can be resolved. The ultraviolet microscope with lenses of fused quartz will transmit radiation down to the wavelength of 240 nm. The images produced are recorded photographically. The instrument has been valuable in the study of cell division and differentiation.
Electron microscopy
Just as a beam of light can be focused by an optical lens, so a stream of electrons can be focused by an electromagnet acting as a lens. Objects placed in the path of the electrons produce an image which can be recorded either on a fluorescent screen or on a photographic plate. Both the focal length and the magnification can be varied by regulation of the field strength, which is controlled by the current passing through the lens. Good stabilization of the lens current is essential for the best lens performance. Because gas molecules will cause a scattering of electrons, electron images are formed only in a high vacuum (less than 10−4 mmHg). Although commercial electron microscopes were available in 1939, it was not until the 1950s that their potential could be fully exploited for biological work. The breakthrough in this field centred on the preparation of ultra-thin sections of biological tissue by the use of glass knives and on the development of suitable staining, fixation and embedding materials. To prevent complete scattering of the electrons by the tissue, sections of the order of 20–200 nm are used and a buffered solution of osmium tetroxide is commonly employed for fixation and staining. Unstained cell components of a tissue have a fairly uniform electron scattering power, similar to that of the embedding medium, so that little contrast of the image is obtainable. However, the incorporation of electron-dense atoms (osmium) into the cell organelles enables a good degree of contrast to be obtained on the electron micrographs of the sections. There is, at present, no objective way of determining how much the fine structure of cells is altered by the fixation methods employed, but indirect correlation of the results obtained with those from other techniques is reassuring.
The light microscope gives magnifications of the order of ×1000 with a resolution, set by the wavelength of the light employed, down to about 0.2 μm for visible light; no further magnification of the image can increase the detail. The theoretical limit of resolution of the electron microscope is similarly governed by the wavelength of the electrons (about 0.003 nm) and in practice electron microscopes give resolutions to about 0.4 nm. Magnifications of ×10 000 to ×24000 are commonly employed and to show all the available detail on high-quality electron micrographs, prints at magnifications of around ×500 000 may be required.
Much knowledge of the detailed structure of the living cell has only been made possible by the advent of the electron microscope. For the routine examination of vegetable drugs the light microscope with polarizing attachment is generally fully adequate, but scanning electron micrographs at a much lower magnification than the above can be extremely useful for depicting structural details not obvious with the light microscope, for example, maize starch and digitalis (Fig. 23.17).
PREPARATION OF DRUGS FOR MICROSCOPICAL EXAMINATION AND GENERAL USE OF REAGENTS
The following aims should be kept in mind for the microscopical examination of crude drugs.
Dried material often requires softening by exposing it to a moist atmosphere (leaves) or by boiling in water (roots and barks). Botanical sections of the plant material may need to be made (cut either by hand or with a freezing microtome). Sections of the dry material may be necessary for the examination of mucilage or water-soluble cell components. Disintegration serves for the isolation of specific tissues and bleaching and defatting techniques for observing deeply coloured materials and fatty seeds respectively. Almost certainly, clearing reagents will be required together with a range of suitable stains for cell walls and cell contents.
Any report should state what characters appear to be of the greatest diagnostic importance and these should be illustrated by suitable sketches.
Distribution of tissues
A general idea of the distribution of tissues can be obtained by the examination of transverse and radial and tangential longitudinal sections. Such sections should first be mounted in water or dilute glycerin. Subsequently sections should be cleared by means of chloral hydrate or other clearing agents (see below) and some stained as follows.
Phloroglucinol and hydrochloric acid
Mount the section in a 1% solution of phloroglucinol in ethanol (90%) and allow to stand for about 2 min; remove any alcohol which has not evaporated with a piece of filter paper; add concentrated hydrochloric acid, cover and examine. All lignified walls stain pink or red.
Hydrochloric acid is a powerful clearing agent and it must be remembered that it will dissolve many cell contents, including calcium oxalate. The vegetable debris of catechu contains phloroglucinol and in this case the wood stains on the simple application of hydrochloric acid.
To prevent damage to the microscope either by liquid contact or by vapours, preparations mounted in concentrated hydrochloric acid should be free of excess acid and must be removed from the microscope stage as soon as possible.
Clearing, defatting and bleaching
Structures are frequently obscured by the abundance of cell contents, the presence of colouring matters and the shrinkage or collapse of the cell walls. Therefore, reagents are used for the removal of cell contents, for bleaching and for restoring as far as possible the original shape of the cell wall. If the microscopical examination is to be made from the section mounted in the clearing agent, the refractive index of the latter is important. It may be advisable to wash the section and mount in a different medium. The commonly used mountants glycerin, alcohol, carbolic acid, lactophenol, clove oil and Canada balsam all have some clearing effect. The following clearing and bleaching agents are particularly useful.
Solution of chloral hydrate
This dissolves starch, proteins, chlorophyll, resins and volatile oils, and causes shrunken cells to expand. Chloral hydrate may be used, not only for sections but also for whole leaves, flowers, pollen grains, etc. It does not dissolve calcium oxalate and is therefore a good reagent for detection of these crystals.
Solution of potash
Solutions of potassium hydroxide, both aqueous and alcoholic, up to a strength of 50% are used for different purposes, but for use as a clearing agent a 5% aqueous solution is most generally useful. A 0.3% solution of potash may be used to dissolve aleurone grains. A 5% solution is much more powerful, and rapidly dissolves starch, protein, etc., causing the swelling of cell walls. Potash should be washed out as soon as clearing is completed, as more prolonged action is liable to cause disintegration (see below).
Ether–ethanol
A mixture of equal parts of ether and ethanol (96%) is useful for the removal of fixed oils, fats, resins, volatile oils, tannins or chlorophyll. Defatting is particularly necessary in the case of oily seeds such as linseed and strophanthus.
Solution of sodium hypochlorite
This solution is useful for bleaching dark-coloured sections such as those of many barks and for removing chlorophyll from leaves. When bleaching is complete, the sections should not be left in the reagent but should be removed and washed with water. Prolonged contact with solution of chlorinated soda causes the removal of starch and lignin, which may not be desirable.
Disintegration and isolation of tissues
The use of reagents for purposes of disintegration is based on their action on the cell wall, particularly the middle lamella. Woody tissues are usually disintegrated by means of oxidizing agents, as these oxidize away the middle lamella, which is composed mainly of lignin. Thus, dilute nitric acid has a marked disintegrating effect on wood, whereas dilute sulphuric acid has not. The middle lamella of cellulose cells is composed of pectic substances which are made soluble by dilute acids or dilute alkalis, which thus effect disintegration. Pure celluloses, however, are resistant to hydrolysing and oxidizing agents, and the stability of cellulose in boiling 5% potash is made use of for the separation of cotton from wool. Other materials such as mannans, galactans, pectin, hemicelluloses, gums and lichenin, which may occur in the cell wall, are much more readily attacked by hydrolysing agents. It will thus be seen that the composition of the ‘crude fibres’ (i.e. those tissues which remain after the material has been subjected to the action of hydrolysing agents under controlled conditions) is likely to vary in both amount and chemical nature in different drugs. However, quantitative comparisons of the crude fibres of different samples of the same drug are useful.
Potassium chlorate and nitric acid
The strength of the reagent and the time it is allowed to act must be varied according to the nature of the material. For woods (e.g. quassia) the material, in small pieces or thick sections, is immersed in 50% nitric acid. Minute quantities of potassium chlorate are added at intervals to maintain an evolution of gas. From time to time a fragment of the wood should be removed and teased with needles. When it breaks up readily, it should be washed free from acid and examined. The process should not be continued longer than is necessary, since prolonged bleaching causes more or less complete destruction of the lignin.
Chromic acid and nitric or sulphuric acid
The reagent usually consists of a mixture of equal parts of 10% chromic acid and 10% nitric or sulphuric acid. It is frequently used for the disintegration of sclerenchymatous tissues such as the testas of capsicum and colocynth seeds or for the separation of lignified hairs such as those of nux vomica and strophanthus.
Solution of potash or soda
As mentioned above, alkalis are used both for clearing and disintegrating. The material is usually digested with 5% potash on a water-bath until the more resistant cells can be teased out of the more or less completely disintegrated parenchyma. The method is useful for the separation of the heavy cuticularized epidermis of leaves and for the isolation of secretory tissue such as the vittae of umbelliferous fruits and the latex vessels of lobelia. Suberized and cutinized tissues are very resistant to the potash. Potash is also useful for the isolation of lignified elements such as are found in the veins of leaves, in senna stalks and in many barks.
Preparation of a crude fibre
For qualitative work the following procedure may be adopted. Mix about 2 g of the drug, in No. 60 powder, with 50 ml of 10% nitric acid in a casserole. Bring to the boil and maintain at the boiling point for 30 s. Dilute with water and strain through a fine filter cloth held over the mouth of a filter funnel. Transfer the washed residue to the casserole and boil for a further 30 s with 50 ml of a 2.5% solution of sodium hydroxide. Collect and wash the residue as before, mount and examine. It will be found that the tissues disintegrate readily and are in a condition well suited to microscopical examination.
Reagents
Directions for making the following reagents, if not given below, will be found in the appendices of the BP. Some of the uses of each are mentioned, but further details will be found elsewhere.
Ethanol
Different strengths are used for preserving material and for hardening. Alcohol acts as a clearing agent by dissolving oils, resins, chlorophyll, etc. It does not dissolve gums and mucilages (therefore a useful mountant for drugs containing them).
Alkanna tincture
A supply sufficient for a few months only should be made by macerating 1 part of alkanet root and 5 parts of alcohol 90% for 1 week, afterwards filtering. Stains oils and fats and suberized and cuticularized walls.
Chloral hydrate and glycerin
combines the properties of chloral hydrate and glycerin and is therefore useful for slow clearing without heat. Preparations mounted in it may be left for some days without undue evaporation.
Chloral hydrate solution BP
(chloral 80 g, water 20 ml). A valuable and widely used clearing agent. See above.
Chromic and nitric acids solution
10% Chromic acid and 10% nitric acid. See ‘Disintegration and isolation of tissues’.
Chlor-zinc-iodine solution (syn. Schulze’s solution)
Prepared by adding a solution of zinc chloride (zinc chloride 20 g; water 8.5 ml) dropwise to a solution of potassium iodide (1.0 g) and iodine (0.5 g) in water (20 ml) until a precipitate of iodine forms which does not disappear on cooling. This requires about 1.5 ml. Used as test for walls containing celluloses. Iodine solution followed by sulphuric acid gives similar results.
Copper oxide, ammoniacal solution of, BP
This solution must be freshly prepared. It causes swelling and solution of cellulose walls. The balloon-like swellings produced in raw cotton are best observed if the reagent be diluted with an equal volume of distilled water. This solution is commonly known as cuoxam.
Corallin, alkaline solution of, BP (syn. Corallin-soda)
Stains the callose of sieve-plates and some gums and mucilages.
Glycerin, dilute
One volume of glycerin is mixed with two volumes of distilled water. A useful mountant for preparations which may be left for some time, as it does not dry up. It has some clearing action, but is much inferior in this respect to chloral hydrate. It can usefully be added to a mount cleared with chloral hydrate solution to prevent the formation of crystals. It is not a good mountant for starch, as the grains tend to become transparent and striations, etc. are difficult to see; water is preferable.
Hydrochloric acid
This (density c. 1.18; c. 11.5 M) is used in testing silk and preparations containing colchicine, and with phloroglucinol as a test for lignin.
Iodine water, BP (see BP Iodine Solutions R1 and R2)
This gives a blue colour with starch and hemicelluloses. Iodine Tincture BP, followed by sulphuric acid, resembles chlor-zinc-iodine (q.v.).
Mercury–nitric acid solution BP (syn. Millon’s Reagent)
Test for protein-containing materials e.g. aleurone grains, wool and silk.
Picric acid solution
A saturated solution in water which is used to stain aleurone grains and animal fibres.
Potash solution
A 5% solution is commonly used for clearing and disintegrating (q.v.) and for the separation of cotton from wool. A 50% solution is used in testing for chitin in ergot and for eugenol in clove. A 2.5% solution is used for preparing crude fibre (q.v.).
Sodium carbonate solution BP
This is useful for the disintegration of fibres such as flax, where the use of an oxidizing agent is not required.
Sodium hypochlorite solution
The BP includes a strong and weak solution; for use see ‘Clearing, defatting and bleaching’.
Sudan III (Sudan red) solution
A solution in equal parts of glycerin and alcohol, stains oils and suberized walls, and is useful in the examination of secretory cells and ducts.
Sulphuric acid 80%
Concentrated sulphuric acid causes rapid charring, but dilutions containing 80% or less form useful reagents. The behaviour of cotton, wool, chalks, calcium oxalate and sections of strophanthus seeds should be noted. The acid dissolves cellulose and lignified walls, but has little action on suberin.
POWDERED DRUGS
The systematic approach to the identification of powdered drugs can proceed in a number of ways; for organized drugs, however, all methods depend on the microscopical recognition of characteristic cell types and cell contents. Identification can then be made by reference to tables and appropriate illustrations, by the use of punched cards, and by employing a suitable computer program. For dealing with mixtures of drugs, greater skills and practice are required and only the first of the above approaches is applicable. When a tentative identification has been made, further confirmatory observations and chemical tests can be performed; most pharmacopoeial drugs now have TLC tests for identity.
Preliminary tests
Microscopical examination
If the preliminary tests have shown that the drug dissolves or becomes mucilaginous in water, trials should be made with other liquids such as alcohol, olive oil or lactophenol until one is found in which the drug is insoluble. In most cases, however, the following procedure may be adopted.
Examination for starch
Mount in water, examine and sketch any granules observed and prove whether they are starch or not by irrigation with iodine water. Do not waste time trying to find other structures that are best seen in the following mountants.
Examination for epidermal trichomes and calcium oxalate solution
Mount in chloral hydrate solution, boil gently until clear, and examine. To ensure that calcium oxalate, if present in small quantity, is not overlooked, polarized light may be used.
Examination for lignin
Moisten the powder with an alcoholic solution of phloroglucinol and allow to stand until nearly dry; add concentrated hydrochloric acid, apply a cover-glass, and examine. Note the presence or absence of lignified vessels, fibres, parenchyma, sclereids or hairs (e.g. nux vomica). If vessels or fibres do not stain pink, suspect rhubarb or ginger.
A considerable amount of information should have been derived from the preliminary tests and the above three mounts, and the examination may be continued by the application of further microchemical tests (chlor-zinc-iodine, tincture of alkanet, ruthenium red, corallin soda, etc.). It is often advisable to defat oily powders, to bleach highly coloured ones and to prepare a crude fibre of those powders containing much starch.
Microscopical measurements of cells and cell contents should be made whenever possible and compared with those published in the literature.
The application of a knowledge of the anatomy of those plant organs occurring in drugs should make possible a recognition of the organs represented. This should be followed by reference to an atlas of vegetable powders or tables of the diagnostic characters of powdered drugs. The identity of a powder should not be regarded as established until it has been compared with one of known authenticity.
A series of tables to assist with the identification of powdered drugs, based on the presence or absence of starch, epidermal trichomes and calcium oxalate, will be found in earlier editions of this book (up to and including the 13th edition). Tables of histological characters of drugs are also given in Wallis’s Textbook of Pharmacognosy (1967); in these a morphological arrangement is employed so that in order to commence the identification process the student must first decide to which group his unknown powder belongs. For the appropriate illustrations of the elements present in powdered drugs see Jackson and Snowdon (1990) in the literature cited in Chapter 2.
Computer-assisted identification of powdered drugs
In 1976 Jolliffe and Jolliffee published details of a computer program for the identification of 174 powdered drugs of organized structure (Analyst, 101, 622). Further elaboration of the method followed as indicated in the literature quoted below. The procedure involves the examination of a single unknown powder in four different mountants for the presence or absence of eleven histological characters, namely, calcium oxalate (if present, type of crystal), aleurone, cork, lignified parenchyma, stomata (if present, which of six types), trichomes (if present, type and structure), vessels/tracheids (if present, lignified or non-lignified), sclereids, fibres, starch and pollen. The information is coded as a string of characters comprising six blocks of five digits, each of which indicates the absence or presence of the microscopical characters being examined. Processing involves a validity check of the input data and a comparison of the observed characters for the unknown with those of the 174 powdered drugs stored in the data bank. The output then gives those drugs having zero errors compared with the input followed by those having 1, 2, etc., errors. Allowance is made in the program for the fact that a particular feature of the drug might be present in such small amount that it could be missed by the inexperienced microscopist. For drugs where the computer cannot give a unique identification, simple distinguishing tests are suggested e.g. the measurement of starch grains and fibres for cassia and cinnamon. Contamination of the drug with moulds, mites, etc. does not affect the identification and the presence of additional material in the powder affording one characteristic only, e.g. starch, is accommodated.
In addition to organized single powders it should be noted that computer-aided identification programs have also been described for unorganized drugs, homoeopathic tinctures, textile and surgical dressing fibres, and food materials (see below).
Bannerman HJ, Cox BJ, Musset JH. Computer-assisted identification of unorganised drugs. Pharmaceutical Journal. 1982;228:716-717. [CAIDUD]
Jolliffe GH, Jolliffe GO. Microcomputer-aided identification of powdered vegetable drugs. Pharmaceutical Journal. 1978;221:385-386. [POWDERS]
Jolliffe GH, Jolliffe GO. ‘MICROAID’. Practical Computing. 1979;2:120-132.
Jolliffe GH, Jolliffe GO. The microcomputer as an analytical aid in drug microscopy. Trease and Evans’ Pharmacognosy. 13th edition. Baillière Tindall. London, UK, 1989;784-798
Stevens RG. Computer-aided identification of textile and surgical dressing fibres. Pharmaceutical Journal. 1980;223:293-294. [FIBRES/BAS]
QUANTITATIVE MICROSCOPY
In addition to the simple measurement of the sizes of tissues, cells and cell contents by means of the micrometer eyepiece or camera lucida, it is possible to estimate the percentage of foreign organic matter in many powdered drugs by a lycopodium spore method which was developed by T. E. Wallis (Analytical Microscopy, 3rd ed., 1965) and subsequently adopted by the Pharmacopoeia. Although the method appears now to be little used, the principles involved, as cited below, are of interest. Other microscopical determinations which may usefully be made in certain cases are vein-islet numbers, palisade ratios, stomatal numbers and stomatal indices.
Lycopodium spore methods
Wallis showed that lycopodium spores are exceptionally uniform in size (about 25 μm) and that 1 mg of lycopodium contains an average of 94 000 spores. The number of spores mg−1 was determined by direct counting and by calculation based on specific gravity and dimensions of the spores. The methods gave values in good agreement. These facts make it possible to evaluate many powdered drugs, provided that they contain one of: (1) well-defined particles which may be counted (e.g. pollen grains or starch grains); or (2) single-layered tissues or cells the area of which may be traced at a definite magnification and the actual area calculated; or (3) characteristic particles of uniform thickness, the length of which can be measured at a definite magnification and the actual length calculated. Whichever method is adopted, mounts containing a definite proportion of the powder and lycopodium are used and the lycopodium spores are counted in each of the fields in which the number or area of the particles in the powder is determined. The method is somewhat laborious and has not been subjected to statistical assessment. For powdered pharmacopoeial drugs reliance is now placed on other methods. Details can, however, be found in the BP 1973 and earlier editions of this book. Classical examples of drugs to which it was applied are senna and linseed (area measurements), nux vomica (trichome-rib lengths) and pypethrum and ginger (counts).
Leaf measurements
A number of leaf measurements are used to distinguish between some closely related species not easily characterized by general microscopy.
Palisade ratio
The average number of palisade cells beneath each upper epidermal cell is termed the palisade ratio. Quite fine powders can be used for the determination.
Pieces of leaf about 2 mm square, or powder, are cleared by boiling with chloral hydrate solution, mounted and examined with a 4 mm objective. A camera lucida or other projection apparatus is arranged so that the epidermal cells and the palisade cells lying below them may be traced. First a number of groups each of four epidermal cells are traced and their outlines inked in to make them more conspicuous. The palisade cells lying beneath each group are then focused and traced. The palisade cells in each group are counted, those being included in the count which are more than half-covered by the epidermal cells; the figure obtained divided by 4 gives the palisade ratio of that group. The range of a number of groups from different particles should be recorded.
Drugs for which palisade ratios have been utilized include belladonna, stramonium, buchu, senna, digitalis and, for recognition as adulterants, xanthium and phytolacca.
Stomatal number
The average number of stomata per square millimetre of epidermis is termed the stomatal number. In recording results the range as well as the average value should be recorded for each surface of the leaf and the ratio of values for the two surfaces.
Fragments of leaf from the middle of the lamina are cleared with chloral hydrate solution or chlorinated soda. Timmerman counted the number of stomata in 12–30 fields and from a knowledge of the area of the field was able to calculate the stomatal number; the camera lucida method described for vein-islet numbers may also be used, the position of each stoma being indicated on the paper by a small cross.
Using fresh leaves, replicas of leaf surface may be made which are satisfactory for the determination of stomatal number and stomatal index. An approximate 50% gelatin and water gel is liquefied on a water-bath and smeared on a hot slide. The fresh leaf is added, the slide is inverted and cooled under a tap and after about 15–30 min the specimen is stripped off. The imprint on the gelatin gives a clear outline of epidermal cells, stomata and trichomes.
The early investigations by Timmerman (1927) indicated that stomatal numbers are usually useless for distinguishing between closely allied species, but that in certain cases the ratio between the number of stomata on the two surfaces may be of diagnostic importance. It is possible, for example, to distinguish Datura innoxia from other species of Datura by this means.
Stomatal index
The percentage proportion of the ultimate divisions of the epidermis of a leaf which have been converted into stomata is termed the stomatal index:
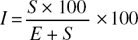
where S = number of stomata per unit area and E = number of ordinary epidermal cells in the same unit area. While stomatal number varies considerably with the age of the leaf, stomatal index is highly constant for a given species and may be determined on either entire or powdered samples. It is employed in the BP and EP to distinguish leaflets of Indian and Alexandrian sennas.
Pieces of leaf other than extreme margin or midrib are suitably cleared and mounted, and the lower surface examined by means of a microscope with a 4 mm objective and an eyepiece containing a 5 mm square micrometer disc. Counts are made of the numbers of epidermal cells and of stomata (the two guard cells and ostiole being considered as one unit) within the square grid, a cell being counted if at least half of its area lies within the grid. Successive adjacent fields are examined until about 400 cells have been counted and the stomatal index value calculated from these figures. The stomatal index may be determined for both leaf surfaces.
Rowson (1943 and 1946) found that stomatal index values may be used to distinguish between leaves of co-generic species (Table 43.1).
Table 43.1 Stomatal index values.
| Species | Stomatal index | |
|---|---|---|
| Upper surface | Lower surface | |
| Atropa acuminata | 1.7 to 4.8 to 12.2 | 16.2 to 17.5 to 1.83 |
| Atropa belladonna | 2.3 to 3.9 to 10.5 | 20.2 to 21.7 to 23.0 |
| Cassia senna | 11.4 to 12.4 to 13.3 | 10.8 to 11.8 to 12.6 |
| Cassia angustifolia | 17.1 to 19.0 to 20.7 | 17.0 to 18.3 to 19.3 |
| Datura inermis | 18.1 to 18.3 to 18.7 | 24.5 to 24.9 to 25.3 |
| Datura metel | 12.7 to 17.4 to 19.4 | 21.2 to 22.3 to 23.9 |
| Datura stramonium | 16.4 to 18.1 to 20.4 | 24.1 to 24.9 to 26.3 |
| Datura tatula | 15.6 to 20.2 to 22.3 | 28.3 to 29.8 to 31.0 |
| Digitalis lanata | 13.9 to 14.4 to 14.7 | 14.9 to 16.1 to 17.6 |
| Digitalis lutea | 2.5 to 5.5 to 8.4 | 21.6 to 22.9 to 25.2 |
| Digitalis purpurea | 1.6 to 2.7 to 4.0 | 17.9 to 19.2 to 19.5 |
| Digitalis thapsi | 5.9 to 7.0 to 7.8 | 11.9 to 12.4 to 13.5 |
| Erythroxylum coca | Nil | 12.2 to 13.2 to 14.0 |
| Erythroxylum truxillense | Nil | 8.9 to 10.1 to 10.7 |
| Phytolacca acinosa | Nil | 15.0 |
| Phytolacca americana | 2.9 to 4.2 to 5.7 | 13.0 to 13.2 to 13.4 |
Vein-islet number
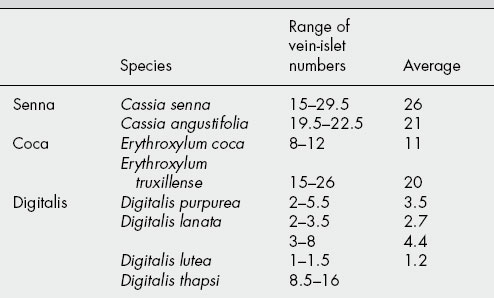
The term ‘vein-islet’ is used to denote the minute area of photosynthetic tissue encircled by the ultimate divisions of the conducting strands. The number of vein-islets mm−2 calculated from four contiguous square millimetres in the central part of the lamina, midway between the midrib and the margin, is termed the vein-islet number. When determined on whole leaves, the area examined should be from the central part of the lamina, midway between the margin and midrib.
Many leaves may be cleared by boiling in chloral hydrate solution in a test-tube placed in a boiling water-bath. Those which are difficult to clear in this way may, after soaking in water, be treated successively with sodium hypochlorite to bleach, 10% hydrochloric acid to remove calcium oxalate, and finally chloral hydrate.
A camera lucida or projection apparatus is set up and by means of a stage micrometer the paper is divided into squares of 1 mm2 using a 16 mm objective. The stage micrometer is then replaced by the cleared preparation and the veins are traced in four contiguous squares, either in a square 2 mm × 2 mm or a rectangle 1 mm × 4 mm (Fig. 43.2). When counting, it is convenient to number each vein-islet on the tracing. Each numbered area must be completely enclosed by veins, and those which are incomplete are excluded from the count if cut by the top and left-hand sides of the square or rectangle but included if cut by the other two sides. For example, the vein-islets in Fig. 43.2 total 62 and the vein-islet number is therefore 15.5.
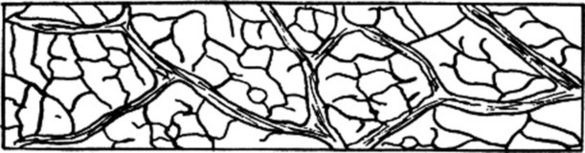
The vein-islet number frequently serves to distinguish closely related plants (Table 43.2).
Veinlet termination number
Hall and Melville (1951) determined veinlet termination number, which they define as ‘the number of veinlet terminations per mm2 of leaf surface. A vein termination is the ultimate free termination of a veinlet or branch of a veinlet’. By this character they distinguished between Peruvian and Bolivian coca leaves and between Alexandrian and Tinnevelly senna leaflets. The values are recorded in Table 43.3.
Table 43.3 Veinlet termination numbers.
| Atropa acuminata | 1.4–3.5 |
| Atropa belladonna | 6.3–10.3 |
| Cassia angustifolia | 25.9–32.8 |
| Cassia senna | 32.7–40.2 |
| Datura stramonium | 12.6–20.1 |
| Digitalis purpurea | 2.5–4.2 |
| Erythroxylum coca | 16.8–21.0 |
| Erythroxylum truxillense | 23.1–32.3 |
| Hyoscyamus niger | 12.4–19.0 |
One practical difficulty in the measurement of vein-islet and veinlet-termination numbers is deciding exactly where, and if, a veinlet terminates. This may appear to vary according to the preliminary treatment a leaf has received.
At present, of the above leaf measurements, only stomatal index is employed officially. With the increasing number of whole herbs and leaves now being introduced into the European and British Pharmacopoeias and the need for standardization of the many herbal products of interest world-wide, a further investigation of the-possible usefulness of these leaf measurements might prove rewarding. For acceptance, the results from any future measurements would necessitate a more sophisticated statistical analysis than was probably afforded the examples quoted above.