Drug discovery, safety and efficacy
Historically, most medicines were of botanical or zoological origin and most had dubious therapeutic value. During the 20th century there were major advances in chemistry allowing the synthesis and purification of huge arrays of small organic molecules to be screened for pharmacological activity. Advances in drug development for the treatment of disease are illustrated most dramatically with antimicrobial chemotherapy, which revolutionised the chances of people surviving severe infections such as lobar pneumonia, the mortality from which was 27% in the pre-antimicrobial era but fell to 8% (and subsequently lower) following the introduction of sulphonamides and then penicillins. Latterly, advances in molecular biology have enabled the development of a number of ‘biologic’ drugs based on the structures of antibodies, receptors and other human proteins. Meanwhile, the Human Genome Project, and the growth of technologies that allow systematic study (‘omics’) of the entire range of cellular RNAs, proteins and small molecules, known as transcriptomics, proteomics and metabolonomics, respectively, have expanded knowledge of the range of gene products and processes that might present targets for novel drugs.
Early medicines consisting of crude extracts of plants or animal tissues usually contained a mixture of many organic compounds, of which one, more than one or none may have had useful biological activity. The active constituents of some plant-derived preparations are bitter-tasting organic molecules known as alkaloids. For example, opium from the opium poppy contains high concentrations of the alkaloid morphine, and various preparations of opium have been used more or less successfully for the treatment of pain and diarrhoea (e.g. dysentery) for thousands of years. Promising therapeutic approaches also included the use of foxglove extracts (which contain cardiac glycosides) for the treatment of ‘dropsy’ (fluid retention); however, there was also considerable toxicity, because the plant preparations contained variable amounts of the active glycoside and other compounds which have a narrow therapeutic index (Ch. 7). Similarly, for centuries, extracts of white willow bark have been used to ease joint pain and reduce fever, although their active ingredient, salicylic acid, also carries substantial toxicity.
A major advance in the safety of plant-derived medicines was the isolation, purification and chemical characterisation of the active component. This had three main advantages, as follows.
 The administration of controlled amounts of the purified active compound removed biological variability in the potency of the crude plant preparation.
The administration of controlled amounts of the purified active compound removed biological variability in the potency of the crude plant preparation.
The administration of the active component removed the unwanted and potentially toxic effects of contaminating substances in the crude preparations.
The identification and isolation of the active component allowed the mechanism of action to be defined, leading to the synthesis and development of chemically related compounds based on the structure of the active component but with greater potency, higher selectivity, fewer unwanted effects, altered duration of action and better bioavailability. For example, chemical modification of salicylic acid by acetylation produced acetylsalicylic acid, or aspirin, first marketed in 1899, with greater analgesic and antipyretic activity and lower toxicity than the parent compound.
Thus, although drug therapy has natural and humble origins, it is the application of scientific principles, particularly the use of controlled experiments and clinical trials to generate reliable knowledge of drug actions, which has given rise to the clinical safety and efficacy of modern medicines. In the age of ‘scientific reason’ it is surprising that so many people believe that ‘natural’ medicinal products offer equivalent therapeutic effectiveness with fewer unwanted effects.
A major advantage of modern drugs is their ability to act selectively; that is, to affect only certain body systems or processes. For example, a drug that both lowers blood glucose and reduces blood pressure may not be suitable for the treatment of someone with diabetes mellitus (because of unwanted hypotensive effects) or a person with hypertension (because of unwanted hypoglycaemic effects), or even of those with both conditions (because different doses may be needed for each effect).
Drug discovery
The discovery of a new drug can be achieved in several different ways (Fig. 3.1). The simplest method is to subject new chemical entities (novel chemicals not previously synthesised) to a battery of screening tests that are designed to detect different types of biological activity. These include in vitro studies on isolated tissues, as well as in vivo studies of complex and integrated systems, such as animal behaviour. Novel chemicals for screening may be produced by direct chemical synthesis or isolated from biological sources, such as plants, and then purified and characterised. This approach has been revolutionised in recent years by developments in high-throughput screening (or HTS), which takes advantage of laboratory robotics for liquid handling combined with in vitro cell lines expressing cloned target proteins in tiny reaction volumes in microplates containing hundreds or thousands of reaction wells. Active compounds, which may be small-molecule libraries derived from bacterial or fungal sources, or proteins derived from solid-phase peptide synthesis, can then be selected based on interactions with cells that express a range of possible sites of action, such as G-protein-coupled or nuclear receptors or enzymes important in drug metabolism. Such methods allow the screening of many hundreds of compounds each day and the selection of suitable ‘lead compounds’, which are then subjected to more labour-intensive and detailed tests.
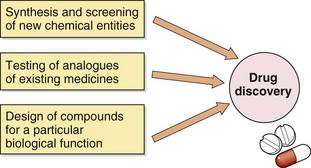
A second approach involves the synthesis and testing of chemical analogues and modifications of existing medicines; generally, the products of this approach show incremental advances in potency, selectivity and bioavailability (structure–activity relationships). However, additional or even new properties may become evident when the compound is tried in animals or humans; for example, minor modifications of the sulphanilamide antimicrobial molecule gave rise to the thiazide diuretics and the sulfonylurea hypoglycaemics.
More recently, attempts have been made to design substances to fulfil a particular biological role, which may entail the synthesis of a naturally occurring substance (or a structural analogue), its precursor or an antagonist. Good examples include levodopa, used in the treatment of Parkinson's disease, the histamine H2 receptor antagonists and omeprazole, the first proton pump inhibitor. Logical drug development of this type depends on a detailed understanding of human physiology both in health and disease. High-throughput screening is particularly useful in such a focused approach. In silico (computer-based) approaches to the modelling of receptor binding sites have facilitated the development of ligands with high binding affinities and, often, high selectivity.
The recent phenomenal advances in molecular biology have led to the increasing use of genomic techniques, both to identify genes associated with pathological conditions and subsequently to develop compounds that can either mimic or interfere with the activity of the gene product. Such compounds are often proteins, which gives rise to problems of drug delivery to the relevant tissue and to the site of action, which may be intracellular, and also raises issues related to safety testing (see below). A good example of the potential of genomic research is the drug imatinib (Ch. 52), which was developed to inhibit the Bcr-Abl receptor tyrosine kinase, which was implicated in chronic myeloid leukaemia cells by molecular biological methods; imatinib is a non-protein organic molecule with a high oral bioavailability.
Irrespective of the approach, drug development is a long and costly process, with estimates of approximately 14 years and over GB£500 million to bring one new drug to market. Much of this cost lies in gaining the preclinical and clinical evidence required for approval of a new drug by regulatory bodies.
Drug approval
Each year, many thousands of new chemical entities and also compounds purified from plant and microbial sources are screened for useful and novel pharmacological activities. Potentially valuable compounds are then subjected to a sequence of in vitro and in vivo animal studies and clinical trials in humans, which provide essential information on safety and therapeutic benefit (Fig. 3.2).
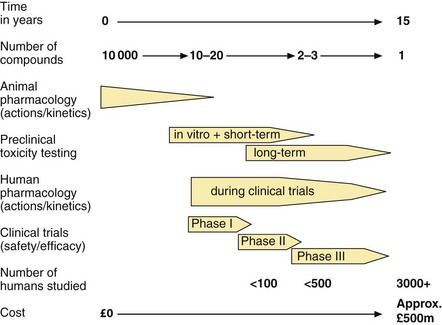
Fig. 3.2 The development of a new drug to the point at which a licence is approved.
Post-marketing surveillance will continue to add data on safety and efficacy.
All drugs and formulations licensed for sale in the UK have to pass a rigorous evaluation of:
In the European Union (EU), new drugs are approved under a harmonised procedure of drug regulation. The European Medicines Agency (EMA; www.ema.europa.eu/ema) is a decentralised body of the EU with headquarters in London and is responsible for the regulation of medicines within the EU. It is broadly comparable to the Food and Drug Adminstration (FDA; www.fda.gov) in the USA. The EMA receives advice from the Committee for Medicinal Products for Human Use (CHMP), which is a body of international experts who evaluate data on the safety, quality and efficacy of medicines. Other EMA committees are involved in evaluating paediatric medicines, herbal medicines and advanced therapies such as gene therapy. Under the current EU system, new drugs are evaluated by the CHMP and national advisory bodies have an opportunity to assess the data before a final CHMP conclusion is reached.
The UK Commission on Human Medicines (CHM) was established in 2005 to replace both the Medicines Commission (MC) and the Committee on Safety of Medicines (CSM), which previously had evaluated medicines regulated in the UK under the Medicines Act (1968). The CHM is one of a number of committees established under the Medicines and Healthcare products Regulatory Agency (MHRA; www.mhra.gov.uk). The MHRA provides advice to the Secretary of State for Health.
Safety
Historically, the introduction of new drugs has been bought at a price of significant toxicity, and regulatory systems have arisen as much to protect patients from drug toxicity as to ensure benefit. In the USA, the FDA was established in 1937, following a dramatic incident in which 76 people died of renal failure after taking an elixir of sulphanilamide which contained the solvent diethylene glycol. Similarly, some 30 years later, the occurrence of limb malformations (phocomelia) and cardiac defects in infants born to mothers who had taken thalidomide for the treatment of nausea in the first trimester of pregnancy led to the establishment of the precursor of the CSM in the UK.
Today, major tragedies are avoided by a combination of in vitro studies and animal toxicity tests (preclinical testing) and by careful observation during clinical studies on new drugs (see below). The development and continuing refinement of preclinical toxicity testing has increased the likelihood of identifying chemicals with direct organ toxicity. During clinical trials, immunologically mediated effects are likely to be seen at the lower end of the dose ranges that are used in such trials (see Ch. 53).
Quality
An important function of regulatory bodies is to ensure the consistency of prescribed medicines and their manufacturing processes. Drugs have to comply with defined criteria for purity and limits are set on the content of any potentially toxic impurities. The stability and, if necessary, sterility of the drug also have to be established. Similarly, licensed formulations must contain a defined and approved amount of the active drug, released at a specified rate. There have been a number of cases in the past in which a simple change to the manufactured formulation affected tablet disintegration, the release of drug and the therapeutic response. The quality of drugs for human use is defined by the specifications in the European Pharmacopoeia (Ph.Eur.) and the British Pharmacopoeia (BP).
Efficacy
All medicines, apart from homeopathic products, must have evidence of efficacy for their licensed indications. Efficacy, i.e. the ability to produce a predefined level of clinical response, can be established only by trials in people with the disease, for whom the medicine is intended, and therefore the demonstration of efficacy is a major aim of the later phases of clinical research (Fig. 3.2).
Establishing safety and efficacy
Regulatory bodies such as the CHMP and CHM require supporting data from in vitro studies, animal studies and clinical investigations before a new drug is approved. Although there is some overlap, the basic aims and goals are:
preclinical studies: to establish the basic pharmacology, pharmacokinetics and toxicological profile of the drug and its metabolites, using animals and in vitro systems,
phase I clinical studies: to establish the human pharmacology and pharmacokinetics, together with a simple safety profile,
phase II clinical studies: to establish the dose–response relationship and to develop the dosage protocol for clinical use, together with more extensive safety data,
phase III clinical studies: to establish the efficacy and safety profile of the drug in people with the proposed disease for which the drug will be indicated,
pharmacovigilance: to monitor adverse events following approval and marketing of the drug.
Preclinical studies
Preclinical studies must be carried out before a compound can be administered to humans. These studies investigate three areas:
pharmacological effects: in vitro effects using isolated cells, tissues or organs; receptor-binding characteristics; in vivo effects in animals and/or animal models of human diseases; prediction of potential therapeutic use,
pharmacokinetics: identification of metabolites (since these may be the active form of the compound); evidence of bioavailability (to assist with the design of both clinical trials and in vivo animal toxicity studies); establishment of principal route and rate of elimination,
toxicological effects: a battery of in vitro and in vivo studies undertaken with the aim of identifying toxicity as early as possible, and before there is extensive in vivo exposure of animals or, subsequently, humans.
Toxicity testing
Toxicity testing has two primary goals: identification of hazards and prediction of the likely risk of that hazard occurring in humans receiving therapeutic doses of the new medicine. A wide range of doses is studied; high doses are required to increase the ability to detect hazards and lower doses are needed to analyse dose–response relationships to predict the risk at doses producing the anticipated therapeutic effect. Toxicity tests include the following (see also www.emea.europa.eu/htms/human/humanguidelines/nonclinical.htm).
Mutagenicity: a variety of in vitro tests using bacterial cells (such as the Ames test) and in cell lines from rodents are employed at an early stage to define any damage to DNA or chromosomal structures that may be linked to carcinogenicity or teratogenicity; in vivo studies may be undertaken to investigate the mechanism of genotoxicity.
Acute toxicity: a single dose is given to animals by the route proposed for human use; this may reveal a likely site for toxicity and is essential in defining the initial dose for human studies. Acute toxicity data, including information on the doses causing lethality, are essential for safe manufacture; the LD50 (a precise estimate of the dose required to kill 50% of an animal population) has been replaced by simpler and more humane methods that define the dose range associated with acute toxicity.
Subacute toxicity: repeated doses are given to animals for 14 or 28 days; this will usually reveal the target for toxic effects, and comparison with single-dose data may indicate the potential for accumulation.
Chronic toxicity: repeated doses are given to animals for up to 6 months; this reveals the target for toxicity (except cancer). The aim is to define dose regimens associated with adverse effects and a ‘no observed adverse effect level’ (NOAEL; the ‘safe’ dose).
Carcinogenicity: repeated doses are given throughout the lifetime of the animal (usually 2 years in a rodent).
Reproductive toxicity: repeated doses are given to animals from before mating and throughout gestation to assess any effect on fertility, implantation, fetal growth, the production of fetal abnormalities (teratogenicity) or neonatal growth.
The extent of animal toxicity testing required prior to the first administration to humans is related to the proposed duration of human exposure and the population to be treated. All drugs are subjected to an initial in vitro screen for mutagenic potential: if satisfactory, this is followed by acute and subacute studies for up to 14 days of administration to two animal species. Doses studied are usually a low dose sufficient to cause the pharmacological/therapeutic effect, a high dose sufficient to cause target organ toxicity and an intermediate dose, together with a control group of untreated animals. Teratogenicity and reproductive toxicity studies are required if the drug is to be given to women of childbearing age; since the thalidomide tragedy, rabbits have been used for teratogenicity studies because, unlike rodents, they show fetal abnormalities when treated with thalidomide. Carcinogenicity testing is necessary for drugs that may be used for long periods, for example over 1 year.
An international review of the extent of in vivo animal testing necessary prior to phase I and phase II clinical trials concluded that the duration of animal toxicity tests should be the same as proposed human exposure (Table 3.1). In Japan and the USA, the same advice applies for phase III studies, but the EU recommends more extensive animal toxicity studies to support phase III trials. Dogs are the ‘non-rodent species’ usually studied.
Table 3.1
EMA guidelines for the length of animal toxicity studies necessary to support phase I and phase II studies in humans
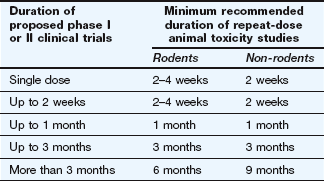
Support of phase III clinical studies may require longer animal toxicity studies than shown here.
The use of animals for the establishment of chemical safety is an emotive issue, and there is extensive current research to replace in vivo animal studies with in vitro tests based on known mechanisms of toxicity. However, toxicology as a predictive science is still in its infancy and at present it is impossible to replicate the complexity of mammalian physiology and biochemistry by in vitro systems. In vivo studies remain essential to investigate interference with either integrative functions or complex homeostatic mechanisms. Carefully controlled safety studies in animals are an essential part of the current procedures adopted to prevent extensive human toxicity, which would inevitably result from the use of untested compounds. Although toxicology has failed in the past to prevent some tragedies (see above), these have led to improvements in methods and current tests provide an effective predictive screen. Nevertheless, there have been examples of approved drugs being withdrawn because of reactions that were not detected in preclinical studies; for example, the high rate of rhabdomyolysis produced by cerivastatin, which was withdrawn worldwide in 2001. This may be increasingly important in the future because drugs developed using molecular biological methods to act specifically at human proteins may show limited or no activity at the analogous rodent receptors; however, animal studies will still provide a useful screen for non-specific, non-receptor-mediated effects.
Students should recognise that not all hazards detected at high doses in experimental animals are of relevance to human health. An important function of expert advisory bodies such as the CHMP is to assess the relevance to human health of effects detected in experimental animals at doses that may be two orders of magnitude (or more) above human exposures. Many drug ‘scare stories’ in the media are based on a hazard detected at experimental doses in animals much higher than the relevant doses for humans.
Clinical trials: phases I–III
The purposes of pre-marketing clinical studies are:
to establish that the drug has a useful action in humans,
to define any toxicity at therapeutic doses in humans,
to establish the nature of common (type A) unwanted effects (see Ch. 53).
Subjects in clinical studies give informed consent to participate and the trials are approved by ethical committees and regulatory agencies. Traditionally, pre-marketing clinical studies have been subdivided into three phases. Although the distinction between these is blurred, the following classification system provides a useful framework
Phase I studies
Phase I is the term used to describe the first trials of a new drug in humans, with typically between 20 and 100 volunteers. A principal aim of these studies is to define basic properties, such as route of administration, pharmacokinetics and tolerability. The studies are usually carried out by the pharmaceutical company, often using a specialised contract research organisation. Subjects taking part in phase I studies are often healthy volunteers recruited by open advertisement, especially when the compound is of low predicted toxicity and has wide potential use; for example, an antihistamine. In some cases, people suffering from the condition in which the drug will be used may be studied, such as cytotoxic agents used for cancer chemotherapy.
The first few administrations are usually in a very small number of subjects (n < 10) who receive an oral dose that may be as low as one-fiftieth or one-hundredth of the minimum required to produce a pharmacological effect in animals (after scaling for differences in body weight). Such a ‘microdose’ study may be termed a phase 0 trial. The dose may be then built up incrementally in larger subject groups until a pharmacological effect is observed or an unwanted action occurs. During these studies toxic effects are sought by routine haematology and biochemical investigations of liver and renal function; other tests, including an electrocardiogram, will be performed as appropriate.
It is also usual to study the disposition, metabolism and main pathways of elimination of the proposed new drug in humans at this stage. Such studies help to identify the most suitable dose and route of administration for future clinical studies. Investigations of drug metabolism and pharmacokinetics often necessitate the use of radioactively labelled compounds containing carbon-14 or tritium (3H) as part of the drug molecule.
Peptide drugs have to be given intravenously in clinical trials to mimic the route of proposed clinical use. Very low doses are studied in the first instance, especially with peptides that are designed to interact specifically with human homeostatic or signalling systems, because studies in animals may not reveal the full biological activities. Despite these safeguards, TGN 1412, a monoclonal antibody directed against CD28, a co-stimulatory molecule for T-cell receptors, caused multiple organ dysfunction in its first phase I trial in six human volunteers in March 2006. The severe toxicity occurred at a dose 500 times lower than the dose found to be safe in animals, including non-human primates.
Phase II studies
During phase II studies, the detailed clinical pharmacology of the new compound is determined, typically in groups of 100–300 individuals with the intended clinical condition. A principal aim of these studies is to define the relationship between dose and pharmacological/therapeutic response in humans. Evidence of a beneficial effect may emerge during phase II studies, although a placebo control is not always used. Additional studies may be undertaken at this stage in special groups such as elderly people, if it is intended that the drug will be used in that population. Other studies may investigate the mechanism of action, or test for interactions with other drugs. Phase II studies normally define the optimum dosage regimen, and this is then used in large clinical trials to demonstrate the efficacy and safety of the drug.
Phase III studies
Phase III studies are the main clinical trials, usually performed in 300–3000 people with the condition that the drug is intended to treat, in multiple centres, in comparison with a placebo that looks (and tastes) the same as the active compound. Allocation to active compound or placebo requires randomisation, which reduces selection bias in the assignment of treatments and facilitates the blinding (masking) of the identity of treatments from the investigators. The advantages and disadvantages of the new compound may also be compared with the best available treatment or the leading drug in the class. It may be difficult to justify the use of a placebo if an effective form of treatment has been established for the condition being studied, as substituting the novel drug for the established treatment in the trial could result in significant risk to those participating. The drug under evaluation may therefore be used in addition to the established treatment and compared with the established treatment plus a placebo.
Clinical trials are of two main types: within-subject and between-subject comparisons (Fig. 3.3). In within-subject trials, an individual is randomly allocated to commence treatment either with the new compound or with a placebo (or comparator drug) before ‘crossing over’ to the alternative therapy, usually with a washout period in between. In contrast, between-subject comparisons involve randomisation of subjects to receive only one of two (or more) treatments for the duration of the study.
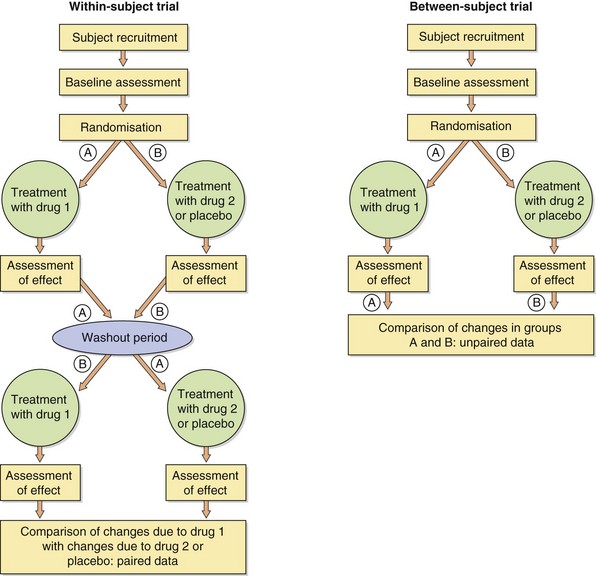
Within-subject comparisons (also called crossover trials) can usually be performed on a smaller number of subjects, since the individuals act as their own controls and most non-treatment-related variables are eliminated. However, such studies often require a longer involvement of each individual. Also, there may be carry-over effects from one treatment that affect the apparent efficacy of the second treatment, although statistical analysis should be able to deal with this problem. Studies of this type may be difficult to interpret when there is a pronounced seasonal variation in the severity of a condition, such as Raynaud's phenomenon or hay fever. Crossover studies (Fig. 3.3) cannot be used if the treatment is curative; for example, an antimicrobial for treating acute infections.
Between-subject comparisons (also called parallel-group studies) require roughly twice as many participants as within-subject trials but have the advantages that each subject will usually be studied for a shorter period and carry-over effects are avoided. Although it is not possible to provide a perfect match between subjects entering the two (or more) different treatment groups, this approach to the evaluation of new drugs is preferred by many drug regulatory authorities.
Whichever form of comparison is made, measurements of benefit (and adverse effects) are made at regular intervals using a combination of objective and subjective techniques (Table 3.2). Throughout these studies, careful attention is paid to detecting and reporting both unwanted effects (type A reactions) and unpredictable (type B) reactions (Ch. 53). Rare type B reactions are not usually seen prior to the marketing of a new drug, because they may occur only once in every 1000–10 000 or more individuals treated with the drug. It is salutary to note that by the time a new medicine is marketed only 2000–3000 people may have taken the drug, often for relatively short periods such as 6 months, amounting to only a few hundred patient-years of drug exposure.
Table 3.2
Examples of response measurements during clinical trials
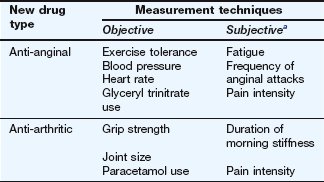
aSubjective effects are often scored on a numerical scale, e.g. 0 = no pain at all, 10 = the worst imaginable pain.
Post-marketing surveillance: phase IV (pharmacovigilance)
The full spectrum of benefits and risks of medicines may not become clear until after they are marketed. Reasons for this include the low frequency of certain adverse drug reactions, and the tendency to avoid the inclusion of children, the elderly and women of childbearing age in pre-marketing clinical trials. Another factor is the widespread use of other medicines in normal clinical practice, which could produce an unexpected interaction with the new drug. Phase IV studies involve post-marketing surveillance of efficacy and adverse reactions, sometimes in clinical indications additional to those licensed. Pharmacovigilance is the identification of risk/benefit issues for authorised medicines arising from their use in clinical practice, and includes the effective dissemination of information to optimise the safe and effective use of medicines.
Pharmacovigilance reports across the EU are coordinated by the EudraVigilance network (http://eudravigilance.ema.europa.eu/human/). Within the UK, a number of systems of post-marketing surveillance are in use. The most important is known as the Yellow Card system; it depends on doctors reporting suspected serious adverse reactions to the MHRA, either online (see MHRA website, www.mhra.gov.uk) or using postage-prepaid cards included in the British National Formulary (BNF) and the Monthly Index of Medical Specialties (MIMS). In addition to reporting suspected serious adverse effects of established drugs, doctors are asked to supply information about all unwanted effects of medicines that have been marketed recently. Each year the MHRA receives some 20 000 yellow cards/slips. In return, doctors are supplied regularly with information about current drug-related problems.
A second form of pharmacovigilance involves systematic post-marketing surveillance of recently marketed medicines. This may be organised by the pharmaceutical company responsible for the manufacture of the new drug (companies also receive information via their representatives). The MHRA administers the Clinical Practice Research Datalink (CRPD; www.cprd.eu), formed in 2012 from the General Practice Research Database, which collects the anonymised, longitudinal patient records from participating UK general practices. It is used in conjunction with the Yellow Card scheme to provide a warning system for approved medicines.
Prescription event monitoring (PEM) provides a further method for detailed study of possible associations provided by pharmacovigilance programmes. This involves:
identification of a possible health problem associated with an approved medicine,
identification by the UK Prescription Pricing Authority of individuals who have been prescribed a drug of interest,
the subsequent distribution of ‘green cards’ to those individuals' GPs, with a request that they complete all details about the person and events that occurred.
The cards are returned to the Drug Safety Research Unit (DSRU) in Southampton (www.dsru.org), which collates and analyses the data. PEM has the advantage that it does not require doctors to make a judgement concerning a possible link between the prescription of a drug and any medical event that occurs while the person is taking the drug. At first sight, a broken leg may be thought an unlikely drug-related adverse effect, but it could be the result of drug-induced hypotension, ataxia or metabolic bone disease.
Finally, detailed monitoring of adverse reactions to drug therapy takes place in some hospitals. These data contribute further to our overall knowledge.
When assessing efficacy and associated toxicity, combining the data from a number of clinical trials (meta-analysis) can provide an overview of the validity and reproducibility of clinical findings. Meta-analysis is complex and only well-designed trials should be combined. The Cochrane Library (www.thecochranelibrary.com) provides a regularly updated collection of evidence-based meta-analyses.
The UK National Institute for Clinical Excellence (NICE) was established in 1999 and became the National Institute for Health and Care Excellence (still known as NICE) in 2005 following its merger with the Health Development Agency. NICE is responsible for providing national guidance on treatments and care for people using the NHS in England and Wales. It provides advice on the clinical value and cost-effectiveness of new treatments, but also on existing treatments if there is uncertainty about their use. NICE produces guidance on:
the use of new and existing medicines and treatments in the NHS in England and Wales (technology appraisals),
the appropriate treatment and care of people with specific diseases and conditions in the NHS in England and Wales (clinical guidelines),
whether interventional procedures used for diagnosis or treatment are safe and work well enough for routine use (interventional procedures).
One-best-answer (OBA) questions
1. In which phase of drug development is a placebo most likely to be used?
2. Which of the following statements best describes a phase III crossover trial?
A Randomisation is not required in a crossover trial.
B People with the most severe disease are allocated to receive the active drug first.
C The second treatment period starts as soon as the first treatment period ends.
D Crossover trials are preferred for trials of curative drugs.
E Participants receive two or more treatments in a random sequence.
1. Answer D is correct. A drug is most likely to be compared with a placebo in large-scale (phase III) trials, although a placebo may also be used in phase II trials. The drug (and placebo) may be added to the existing best treatment, or compared with a leading drug for the same condition.
2. Answer E is correct. Randomisation is designed to produce treatment groups that match in all important characteristics, including disease severity. In a within-subject (or crossover) trial, randomisation of the sequence of treatments that each subject receives reduces the probability of confounding by an effect of one treatment persisting into the second treatment period; an interval between treatments (washout) helps to ensure this. Crossover trials are generally not used to test curative drugs for acute conditions (e.g. antimicrobials) as the condition may not persist through both treatment periods.
Adams, CP, Brantner, VV. Estimating the cost of new drug development: is it really $802 million? Health Affairs. 2006;25:420–428.
Austin, CP. The impact of the completed human genome sequence on the development of novel therapeutics for human disease. Annu Rev Med. 2004;55:1–13.
Dollery, C. The clinical pharmacologist's view; drug discovery and early development. In: Wilkins MR, ed. Experimental Therapeutics. London: Martin Dunitz, Taylor and Francis; 2003:3–24.
Kerwin, R. The National Institute for Clinical Excellence and its relevance to pharmacology. Trends Pharmacol Sci. 2004;25:346–348.
Layton, D, Hazell, L, Shakir, SA. Modified prescription-event monitoring studies: a tool for pharmacovigilance and risk management. Drug Safety. 2011;34(12):e1–e9.
Lynch, A, Connelly, J. The toxicologist's view: non-clinical safety assessment. In: Wilkins MR, ed. Experimental Therapeutics. London: Martin Dunitz, Taylor and Francis; 2003:25–50.
Marchetti, S, Schellens, JH. The impact of FDA and EMEA guidelines on drug development in relation to Phase 0 trials. Br J Cancer. 2007;97:577–581.
Persidis, A. High-throughput screening. Advances in robotics and miniaturization continue to accelerate drug lead identification. Nat Biotechnol. 1998;16:488–489.
Schneider, G, Fechner, U. Computer-based de novo design of drug-like molecules. Nat Rev Drug Discov. 2005;4:649–663.
Shah, RR, Branch, SK, Steele, C. The regulator's view; regulatory requirements for marketing authorizations for new medicinal products in the European Union. In: Wilkins MR, ed. Experimental Therapeutics. London: Martin Dunitz, Taylor and Francis; 2003:51–75.
Walker, DK. The use of pharmacokinetic and pharmacodynamic data in the assessment of drug safety in early drug development. Br J Clin Pharmacol. 2004;58:601–608.