Drug toxicity and overdose
Most therapeutic drugs produce their beneficial response by altering human homeostatic mechanisms; only antimicrobial agents and parasiticides have the theoretical possibility of a therapeutic response without some direct action on human metabolic or physiological processes. Some therapeutic agents, for example atropine (belladonna), tubocurarine (curare), ergot alkaloids (causing St. Anthony's fire), digoxin (digitalis) and dicoumarol (causing haemorrhagic disease in cattle), have pharmacological properties that were first recognised as a result of either accidental or intentional poisonings. It is hardly surprising that all drugs are capable of producing adverse effects if the dosage is high enough. The relationship between a potentially beneficial drug and a poison was recognised five centuries ago when Paracelsus stated: ‘All things are toxic and it is only the dose which makes something a poison.’
Some of the medicines prescribed today were first used as relatively crude plant extracts, for example digitalis glycosides and opium extracts. It was the identification and isolation of the active chemical entities in plant extracts that allowed the dose and purity of the active ingredient to be controlled sufficiently to optimise the ratio between benefit and risk. In this respect, the current vogue for herbal remedies could be considered to represent a backward step in controlling the safety and efficacy of drugs.
Drug toxicity can develop at normal therapeutic doses of a drug or as a result of an acute overdose. In some cases, toxicity occurs in the majority of treated individuals because of the nature of the drug, for example cytotoxic agents used for cancer chemotherapy, but significant toxicity is rare with the majority of commonly prescribed drugs when used at recommended dosages. There is considerable inter-individual variability in both the nature and severity of adverse reactions, and toxicity can be reduced by taking into account factors that are known to increase susceptibility, such as age, concurrent disease or body weight, when selecting both the drug and the dosage. Usually, a reduction in dosage or a change of drug during chronic treatment will reduce the severity of adverse effects (but see immunological mechanisms discussed below).
Toxicity following an acute overdose usually produces predictable adverse reactions, which may be life-threatening and/or prejudice long-term health. Rapid treatment is then required and this may be aimed at preventing further drug absorption, increasing drug elimination/inactivation and managing the adverse effects produced.
This chapter is, therefore, divided into two main sections:
 drug toxicity and adverse effects, which discusses mechanisms for adverse effects produced both during normal drug therapy and after an overdose,
drug toxicity and adverse effects, which discusses mechanisms for adverse effects produced both during normal drug therapy and after an overdose,
self-poisoning and drug overdose, which is concerned with the management of drug overdose.
Drug toxicity and adverse effects
There is no consistent use of terminology in describing effects of drugs that were not intended when the drug was prescribed. Adverse effects of a drug are those that can occur when the drug is used at therapeutic doses; these have also been referred to as collateral effects. Drug toxicity is more commonly applied to effects that occur at supratherapeutic doses. This section provides a framework for classifying adverse and toxic effects of drugs which are referred to in the individual drug monographs as unwanted effects. The term side effect is a widely used alternative.
The beneficial effects of a drug in one situation (e.g. the antidiarrhoeal effect of opioids) may be an adverse effect in other circumstances (e.g. constipation, when an opioid is used for pain relief). Therefore, even classification of the nature of effect into beneficial or adverse may depend on the condition being treated. The adverse effects caused by different drugs are listed in the British National Formulary (BNF), and it is apparent that for most drugs' potential adverse effects are more numerous than beneficial properties. It is important to remember that such lists are not exhaustive, and prescribers should be alert to both predicted and unexpected reactions to medicines. The prescriber should consider the risk–benefit ratio for each individual and the suitability of alternative drugs and/or treatments and people who are prescribed drugs should be informed of the risk–benefit balance inherent in their treatment. The patient information leaflet (PIL) included with the dispensed medicine represents a useful way of providing such advice. Prescribers should also be aware of the risks of adverse and toxic effects arising from drug interactions.
It should be appreciated that all drugs are associated with some risk of adverse effects and toxicity, although both the severity and incidence differ widely among drugs. In general, the acceptability of a risk of an unwanted effect is related to the severity of the disease being treated; for example, serious idiosyncratic reactions with incidences of 1 in 10 000 have led to the withdrawal of some non-steroidal anti-inflammatory drugs (NSAIDs), whereas some cancer chemotherapeutic agents can cause significant toxicity in nearly all individuals.
A useful indication of the safety margin available for a drug is given by the therapeutic index (TI):
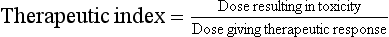
Drugs such as diazepam have a TI of about 50 and it is difficult for even the most inept doctor to cause serious toxicity with diazepam. In contrast, digoxin has a TI of only about 2, and for such drugs toxicity may be precipitated by relatively small changes in dosage regimen or the clearance of the drug from the body. The TI relates to serious toxicity and does not indicate the potential for minor adverse effects, which may inconvenience the person enough for him or her to stop treatment but are not considered to represent drug toxicity.
Types of unwanted effect
Adverse drug reactions are usually divided into two main types:
Type A (Augmented): these effects are usually dose-related and largely predictable from the known pharmacological and biochemical effects of the drug or its metabolites,
Type B (Bizarre): these effects are not obviously dose-related and are idiosyncratic and unpredictable; they are often immunological in nature.
This classification makes no allowance for time-dependency of some adverse events, or for the underlying susceptibility of the individual. Other types of adverse reactions have been defined as: continuing (type C), delayed (type D), end-of-use (type E), failure of therapy (type F) or genetic/genomic (type G). These types are not mutually exclusive, and classification of an adverse event along multiple axes, such as dose-relatedness, time-relatedness and susceptibility may be more meaningful.
Pharmacological toxicity
In ‘pharmacological toxicity’, the toxic reaction is a predictable extension of the known pharmacology of the drug at its site(s) of action (Table 53.1), and should be recognised readily when monitoring the individual's response to treatment. There are numerous examples in this book where the adverse effect is really an excessive therapeutic action.
Table 53.1
Examples of drugs with adverse effects related to their primary therapeutic properties
| Drug | Adverse effect |
| Acetylcholinesterase inhibitors | Muscle weakness |
| Beta-adrenoceptor antagonists | Heart block when used as an antiarrhythmic |
| Insulin | Hypoglycaemia |
| General anaesthetics | Medullary depression |
| Loop diuretics | Hypokalaemia |
| Warfarin | Haemorrhage |
For many drug effects, the response increases with increase in dose, with low sub-therapeutic doses giving an inadequate response, therapeutic doses giving the desired response, but very high doses giving an excessive response that can be regarded as a form of toxicity (response 1 in Fig. 53.1). A good example is warfarin (Ch. 11), inadequate doses of which are associated with a lack of the desired anti-thrombotic effect, whereas at excessive doses there is a risk of haemorrhage. This has given rise to the concept of a ‘therapeutic window’, which is a range of doses or blood/plasma concentrations within which most individuals should show a beneficial response with minimal risk of adverse effects (illustrated by response 1 in Fig. 53.1). The concept is particularly valuable in the interpretation of measurements of drug concentrations in plasma, which can be used to monitor adherence to treatment and to assess likely response (Table 53.2).
Table 53.2
Examples of therapeutic windows based on plasma concentrations
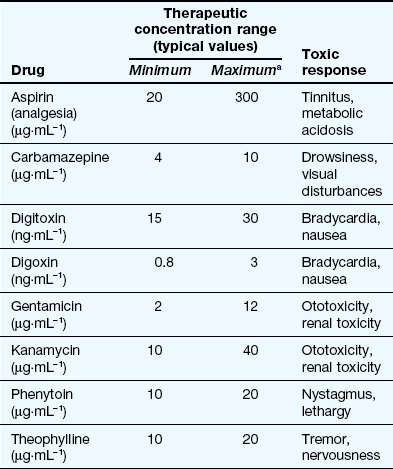
aThe maximum concentration may be limited by toxicity related to the primary therapeutic response (e.g. carbamazepine) or to an unrelated effect (e.g. gentamicin).
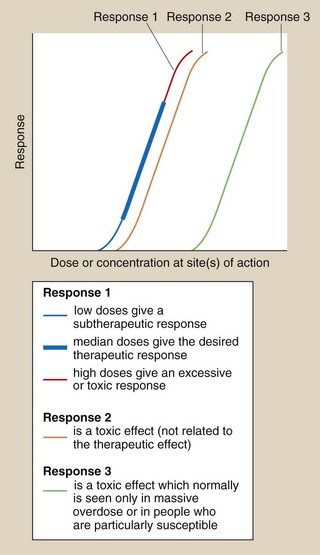
Fig. 53.1 Dose–response relationships in relation to toxicity.
Response 1 is the primary therapeutic effect, the magnitude of which increases with dose from sub-therapeutic, through therapeutic to potentially toxic. Response 2 is an undesired effect seen at a dose only slightly greater than those producing the therapeutic effect. Response 3 is an adverse effect normally seen only in overdose.
In many other cases, the toxic reaction may be unrelated to the primary therapeutic effect (Table 53.3), and may be caused by a secondary effect that is not the primary aim of the treatment given (response 2 in Fig. 53.1). This toxicity will often be present to a limited extent at appropriate therapeutic doses.
Table 53.3
Examples of drugs with adverse effects unrelated to their primary therapeutic use
| Drug | Adverse effects |
| Beta-adrenoceptor agonists | Increase in heart rate when used in asthma |
| Beta-adrenoceptor antagonists | Reduction in heart rate when used for hypertension |
| Aminoglycosides | Deafness |
| Anti-cancer drugs | Myelosuppression |
| Anticonvulsants | Sedation when used for epilepsy |
| Antipsychotics | Dystonias or parkinsonism |
| Corticosteroids | Glaucoma |
| Drugs for Parkinson's disease | Hallucinations and confusion |
| Opioid analgesics | Respiratory depression |
| Paracetamol | Liver failure |
| Statins | Rhabdomyolysis |
| Thalidomide | Birth defects |
| Thiazides | Glucose intolerance |
The separation of therapeutic and toxic dose–response curves is a measure of the TI. If these are very close (e.g. responses 1 and 2 in Fig. 53.1), then there is a low safety margin (low TI) and most individuals will show some degree of toxicity at normal therapeutic doses, for example myelosuppression with cytotoxic anti-cancer drugs. For drugs with widely separated therapeutic and toxic dose–response curves (e.g. responses 1 and 3 in Fig. 53.1) – that is, those with high TIs – toxicity would not be seen at normal doses; for example a β-adrenoceptor antagonist would be very unlikely to cause myocardial depression and heart failure in people with normal left ventricular function. However, the toxic effect may occur in individuals who are unusually sensitive because of their genetics or their physical condition; for example, standard doses of a β-adrenoceptor antagonist can precipitate heart failure in people with pre-existing impaired left ventricular function.
Pharmacological toxicity is the most common cause of adverse effects. Such toxicity can be minimised by an assessment of the risk–benefit balance for the individual to be treated. This should take into account factors that may influence both pharmacokinetics and target-organ sensitivity, including age, physiological status (e.g. renal function), concurrent medication, disease processes, environmental factors (e.g. smoking), etc.
Because of the predictable nature of pharmacological toxicity, it is usual for some treatments to be co-prescribed with drugs that will reduce the possibility of toxic effects; examples include anti-emetics given with cancer chemotherapy, vitamin B6 given with isoniazid and leucovorin (folinic acid) given after high-dose methotrexate.
Biochemical toxicity
In ‘biochemical toxicity’, the toxicity or tissue damage is caused by an interaction of the drug, or an active metabolite, with cell components, especially macromolecules such as structural proteins and enzymes. A generalised scheme is given in Figure 53.2. For most licensed drugs this form of toxicity is identified and characterised during preclinical studies in animals and monitored in clinical trials (Ch. 3), for example by measuring changes in serum levels of liver or muscle enzymes.
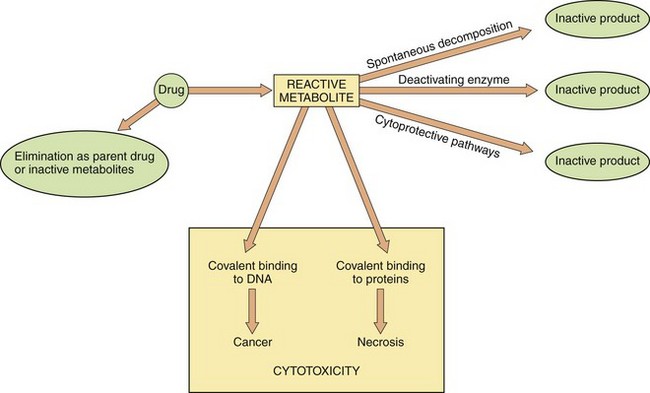
Fig. 53.2 Metabolism and cytotoxicity.
In this scheme, the propensity of the reactive metabolite of a cytotoxic drug to cause adverse biochemical effects on cellular DNA and proteins depends on the balance between formation of the reactive metabolite from the parent drug, and the rate of elimination of both the reactive metabolite and the parent drug by alternative metabolic routes. Therapeutic interventions are aimed at either increasing elimination of the parent drug or enhancing cytoprotective pathways to reduce the cytotoxic effects.
In some situations, an understanding of the mechanism of toxicity has allowed the development of appropriate treatments or antidotes. An example is the key observation that the thiol (-SH) group of the endogenous tripeptide glutathione can prevent cell damage caused by highly reactive chemical species, such as the toxic metabolite of paracetamol (see below and Fig. 53.3). The nature of the cell damage is related to the stability of the toxic reactive chemical; extremely unstable metabolites may bind covalently to and inactivate the enzyme that forms them, whereas more stable species may be able to diffuse to a distant site, for example DNA, and initiate changes such as cancer. Important examples of biochemical toxicity are given below.
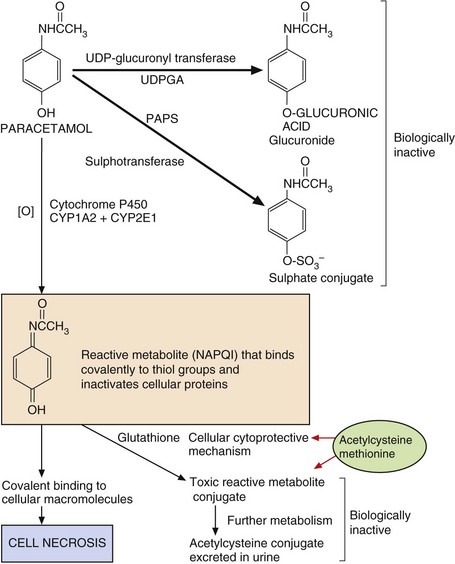
Fig. 53.3 Pathways of paracetamol metabolism.
In overdose, the concentrations of 3′-phosphoadenosine 5′-phosphosulphate (PAPS) (for sulphation) and glutathione (for cytoprotection) are depleted, and extensive macromolecular binding leads to hepatocellular necrosis. N-Acetylcysteine and methionine replenish glutathione to conjugate the toxic metabolite. NAPQI, N-acetyl-p-benzoquinone imine; UDPGA, uridine diphosphate glucuronic acid.
Paracetamol
Paracetamol-induced hepatotoxicity represents the result of an imbalance between metabolic detoxication of paracetamol, via conjugation with glucuronic acid and sulphate, and metabolic activation to an unstable toxic metabolite (N-acetyl-p-benzoquinone imine, NAPQI) via oxidation by cytochrome P450. This toxic metabolite can bind covalently to proteins and cause cell necrosis. Low doses of paracetamol are safe because they are eliminated mainly by conjugation, and any NAPQI created by cytochrome P450 oxidation is inactivated by a cytoprotective pathway involving glutathione. However, in overdose, the sulphate conjugation reaction is saturated and there is increased cytochrome P450-mediated oxidation of paracetamol to NAPQI (Fig. 53.3). Once the hepatic glutathione is depleted this toxic metabolite binds covalently to proteins and causes oxidative stress and cell necrosis. This biochemical mechanism explains:
the site of toxicity (centrilobular necrosis in the liver because of the large amounts of cytochrome P450 present),
the increased toxicity seen in individuals treated with inducers of cytochrome P450 (especially alcohol-related induction of CYP2E1),
the increased toxicity seen in individuals with low hepatic stores of glutathione due to poor nutrition,
the successful treatment of paracetamol overdose with acetylcysteine, which provides an additional source of thiol groups for inactivation of NAPQI (see treatment of drug overdose, below).
Cyclophosphamide
Cyclophosphamide is an anti-cancer drug. Its highly toxic metabolites bind to DNA as part of their mechanism of action, but they are eliminated in the urine and cause haemorrhagic cystitis due to cell damage in the bladder (Ch. 52). This adverse effect can be prevented by prior treatment with mesna (mercaptoethane sulphonic acid), which possesses both a thiol group for cytoprotection and a highly polar sulphonic acid group, and results in high renal excretion and delivery of this cytoprotective molecule to the bladder epithelium. It is usually given either orally 2 h before the cyclophosphamide or intravenously at the same time to cover the period of maximum urinary excretion of toxic cyclophosphamide metabolites. It is not yet known whether mesna also protects against bladder cancer, which can arise about 10–20 years after initial treatment with cyclophosphamide.
Isoniazid
Isoniazid, which is used for the treatment of tuberculosis (Ch. 51), causes hepatitis in about 0.5% of treated individuals. This is believed to result from the formation of a reactive metabolite, N-acetylhydrazine, which is produced by acetylation followed by oxidative metabolism. The biochemical basis for the susceptibility of some individuals to the hepatotoxic metabolite is not known. Fast acetylators (see Ch. 2) form more N-acetylhydrazine than do slow acetylators, but, unexpectedly, they are not more sensitive to isoniazid toxicity. Susceptibility may be related to the balance between further activation of N-acetylhydrazine (by oxidation) and its detoxification (by further acetylation); if this is so, then fast acetylators may produce more active metabolite but also inactivate it more rapidly.
Spironolactone
Spironolactone (Ch. 14) is oxidised by cytochrome P450. The metabolite formed in the testes binds to and destroys testicular cytochrome P450 and this causes a decrease in the metabolism of progesterone to testosterone (a step which is also catalysed by a cytochrome P450). This effect, combined with an anti-androgenic action at receptor sites (pharmacological toxicity), can result in gynaecomastia and decreased libido.
Aromatic amines and nitrites
Aromatic amines, such as the anti-leprosy drug dapsone and some antimalarials (e.g. primaquine), are oxidised in the liver to active metabolites, which are released into the circulation, where they can affect erythrocytes, causing methaemoglobinaemia and/or haemolysis.
Methaemoglobinaemia: In the erythrocyte, the active metabolite interacts with molecular oxygen (O2), which then oxidises haemoglobin (Fe2+) to methaemoglobin (Fe3+) and also oxidises the active metabolite itself (Fig. 53.4A). Because of the large amounts of haemoglobin compared with the amount of drug given this would be inconsequential, were it not for the fact that the oxidised active metabolite can be recycled back to the active metabolite by reduction with NADPH (reduced nicotinamide adenine dinucleotide phosphate) in the erythrocyte. Consequently, each molecule of the metabolite undergoes repeated redox cycling and is able to oxidise many molecules of haemoglobin. NADPH is formed during the metabolism of glucose 6-phosphate by glucose-6-phosphate dehydrogenase (G6PD) (Fig. 53.4A). The activity of G6PD, and hence the amounts of NADPH, are determined genetically. There is a high incidence of G6PD deficiency in people of African ancestry and very high in those of Mediterranean ancestry, such as Kurdish people. Such individuals have limited NADPH reserves and a low ability to reduce the oxidised active drug metabolite (Fig. 53.4A) back to the active metabolite. In consequence, there is limited redox cycling of the active drug metabolite and they are less susceptible to drug-induced methaemoglobinaemia, but they are more susceptible to haemolysis (see below).
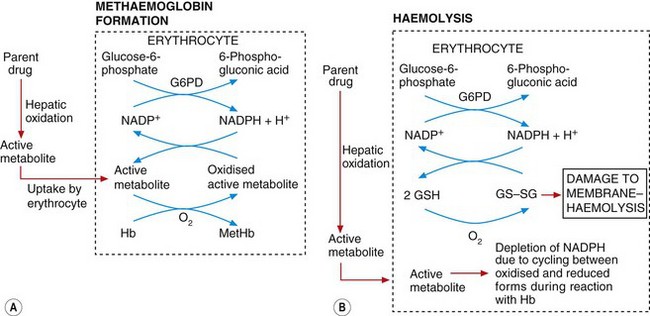
Fig. 53.4 Mechanisms of methaemoglobinaemia and haemolysis.
In methaemoglobinamia (A), the active drug metabolite oxidises haemoglobin (Hb) to methaemoglobin (MetHb). The oxidised active metabolite is repeatedly recycled to the active metabolite by reduced nicotinamide adenine dinucleotide phosphate (NADPH) generated by glucose-6-phosphate dehydrogenase (G6PD). In (B), the depletion of NADPH as a result of methaemoglobinaemia depletes levels of the reduced glutathione (GSH) necessary for maintaining erythrocyte cell membrane integrity; the build-up of oxidised glutathione dimers (GS–SG) causes membrane damage and haemolysis. The active metabolite may also react directly with glutathione to lower GSH concentrations.
Haemolysis: This arises from an increase in erythrocyte membrane permeability associated with accumulation of oxidised glutathione (GS–SG in Fig. 53.4B) in the erythrocyte. Oxidised glutathione accumulates because redox cycling of the drug metabolite linked to the formation of methaemoglobin (Fig. 53.4A) (see above) results in depletion of NADPH, which is the cofactor essential for maintaining glutathione in the reduced state. Individuals with G6PD deficiency are very susceptible to haemolysis caused by aromatic amines and nitrites because the low endogenous levels of NADPH are depleted rapidly and oxidised glutathione cannot then be reduced. Given the geographical distribution of G6PD deficiency, it is ironic that the amino groups associated with this form of toxicity are often present in drugs used to treat tropical infections (such as primaquine for the treatment of malaria; see Ch. 51 and Box 47.6).
Immunological toxicity
Immunological toxicity is frequently referred to as ‘drug allergy’ and is the form of toxicity with which people may be most familiar, for example penicillin allergy. Immunological mechanisms are implicated in a number of common adverse effects, such as rashes and fever, but may also be involved in organ-directed toxicity. Although the term ‘allergy’ may not be strictly correct for all forms of immunologically mediated toxicity, it is probably better than ‘hypersensitivity’, which has also been used to describe an elevated sensitivity to any mechanism or effect.
Low-molecular-weight compounds (<1100 Da) are not able to elicit an allergic response directly but can do so after the compound, or a metabolite, has formed a stable or covalent bond with a macromolecule. Covalent binding to a normal protein produces a hapten–protein conjugate that is recognised as foreign by the immune system and can act as an antigen (Fig. 53.5).
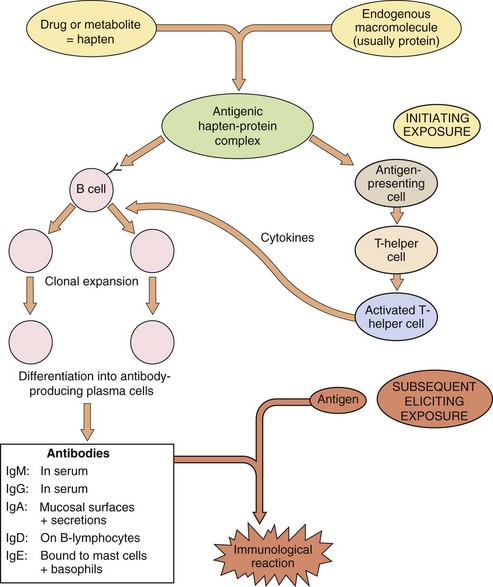
Fig. 53.5 Mechanisms of drug allergy.
The drug or drug metabolite binds as a hapten to an endogenous macromolecule. The initial exposure produces an antigenic hapten–protein complex, which results in the production of antibodies via B-cell clonal expansion and differentiation, regulated by cytokines from activated T-helper cells. The eliciting exposure occurs later (usually at least 3 days later, during which time therapy may or may not be continuing); antigen–antibody interaction then exposes a complement-binding site, which triggers the reaction. The nature of the immunological reaction depends on the nature of the antibody and/or localisation of the antigen. Treatment is with immunosuppressant drugs (Ch. 38).
Immunologically mediated toxicity can show a wide range of characteristics.
Toxicity is unrelated to dose: once the antibody has been produced, even very small amounts of antigen can trigger a reaction.
There is normally a lag of at least 3 days between initial exposure and the development of symptoms; however, the first dose of a subsequent treatment may give an immediate reaction.
Cross-reactivity is possible among different compounds that share the same antigenic determinant or structural component involved in antibody recognition, such as the penicilloyl group of the penicillin family.
The incidence varies widely between different drugs; for example, from about 1 in 10 000 people for phenylbutazone-induced agranulocytosis to 1 in 20 for ampicillin-related skin rashes.
The response is idiosyncratic but genetically controlled. Individual responsiveness cannot be predicted, but individuals who have a history of atopic disease are more likely to develop a ‘drug allergy’.
The effects produced may be subdivided into the classic four types of allergic reaction (see also Ch. 38).
Type 1: immediate or anaphylactic reactions. These are mediated via IgE antibodies attached to the surface of basophils and mast cells; the release of numerous mediators, for example histamine, serotonin and leukotrienes, produces effects that include urticaria, bronchoconstriction, hypotension, oedema and shock. A skin-prick challenge test usually produces an acute inflammatory response. Examples of drugs having this type of effect are penicillins and peptide drugs, such as crisantaspase (asparaginase).
Type 2: cytotoxic reactions. The antigen is formed by the drug binding to a cell membrane; subsequent interaction of this antigen with circulating IgG, IgM or IgA antibodies activates complement and initiates cell lysis. Depending on the carrier cell to which the drug is bound, cell lysis can result in thrombocytopenia (e.g. cephalosporins, quinine), neutropenia (e.g. metronidazole) or haemolytic anaemia (e.g. penicillins, rifampicin and possibly methyldopa).
Type 3: immune-complex reactions. The antigen–antibody interaction occurs in serum and the complex formed is deposited on endothelial cells, basement membranes, etc., to initiate a more localised inflammatory reaction, such as arteritis or nephritis. Examples include serum sickness (urticaria, angioedema, fever) with penicillins, lupus erythematosus-like syndrome with hydralazine and procainamide (especially in slow acetylators) and possibly NSAID-related nephropathy.
Type 4: cell-mediated delayed-type reactions. Reaction to the eliciting exposure is delayed. The reactions occur mostly in skin through the formation of an antigen between the drug (hapten) and skin proteins. This is followed by an infiltration of sensitised T-lymphocytes, which recognise the antigen and release cytokines to produce local inflammation, oedema and irritation; for example, contact dermatitis.
In addition to true immunologically mediated toxicity, as described above, there are examples of ‘pseudoallergic’ reactions, such as aspirin-intolerant asthma which shows many of the characteristics given above (e.g. induction of asthma only in susceptible individuals), but for which a true immunological basis has not been demonstrated. In aspirin intolerance, cross-reactivity with other NSAIDs in fact suggests a common pharmacological mechanism based on cyclo-oxygenase inhibition (Ch. 29).
It has been estimated that ‘drug allergy’ accounts for about 10% of adverse drug reactions but that severe reactions are rare. For example, only about 5 in 10 000 individuals develop an anaphylactic reaction to penicillins, but about a half of these are sufficiently serious to warrant hospital treatment (see Ch. 39). However, given the large numbers of people taking drugs such as penicillins, ‘drug allergy’ is an important source of iatrogenic morbidity.
Self-poisoning and drug overdose
Self-poisoning can be either accidental or deliberate. Approximately a quarter of a million episodes are believed to occur each year in England and Wales, although fewer than 40% of these reach hospital. Deaths from intentional self-poisoning and accidental overdose still average about 2500 each year in England and Wales, with about two-thirds being male. Accidental poisoning is common in children under 5 years of age, when it often involves household products as well as medicines. A second peak of self-poisoning occurs in the teens and early twenties, when it is more frequent in girls. The incidence then progressively falls with increasing age. Most deliberate self-poisoning represents ‘parasuicide’ or attention-seeking behaviour. True suicide attempts account for about 1000 deaths a year, occurring most frequently in those over 45 years. About 30% of the deaths from deliberate overdose are in those over 65 years of age: self-poisoning at this age occurs most often in response to depression or specific life events such as bereavement. It is important to recognise that at any age the severity of poisoning bears little relationship to suicidal intent.
Heroin and morphine overdoses are the most common single cause of death from drug overdose, accounting for almost 25% of the total. The drugs most frequently used for intentional self-poisoning are benzodiazepines, analgesics and antidepressants, often taken together with alcohol. It is important to attempt to identify the cause of the poisoning because it may determine the most suitable treatment. However, it should be remembered that information from the person, about which drug was taken, how much and the time of overdosing, is frequently unreliable. TICTAC (www.tictac.org.uk) is a computerised database which enables registered users to identify tablets and capsules.
Management principles
The emergency treatment of poisoning is described in the BNF. Additional sources of information include the UK National Poisons Information Service (www.npis.org) and its computer database TOXBASE (www.toxbase.co.uk), which has information on household products and industrial and agricultural chemicals as well as drugs, and is available to registered users.
The management of drug overdose outlined below has a number of principal aims (Fig. 53.6).
Managing adverse effects
There are certain immediate measures required when someone presents with a possible drug overdose or poisoning:
remove the person from contact with the poison if appropriate; for example, gases, corrosives,
assess vital signs, i.e. pulse, respiration and pupil size; inspect the person for injury,
ensure a clear airway; if breathing but unconscious, place in the coma position,
obtain a clear history if possible,
preserve any evidence; for example, bottles, written notes, etc.
Supportive measures
Examples of unwanted effects seen in drug overdose are shown in Table 53.4. A number of the effects will require supportive measures (Fig. 53.7).
Table 53.4
Complications of acute poisonings
| Complication | Cause | Examples of poisons |
| Cardiac arrest | Direct cardiotoxicity | Many |
| Hypoxia | Many | |
| Electrolyte/metabolic disturbance | Many | |
| Central nervous system depression | Many | |
| Seizures | Direct neurotoxicity | Tricyclic antidepressants, theophylline |
| Hypoxia | Many | |
| Hypotension | Myocardial depression | Beta-adrenoceptor antagonists, tricyclic antidepressants |
| Peripheral vasodilation | Many | |
| Arrhythmia | Direct cardiotoxicity | Beta-adrenoceptor antagonists, tricyclic antidepressants, verapamil, digoxin |
| Hypoxia | Many | |
| Electrolyte/metabolic disturbance | Many | |
| Renal failure | Hypotension | Many |
| Rhabdomyolysis | Opioids, hypnotics, ethanol, carbon monoxide | |
| Direct nephrotoxicity | Paracetamol, heavy metals | |
| Hepatic failure | Direct hepatotoxicity | Paracetamol, carbon tetrachloride |
| Respiratory depression | Direct neurotoxicity | Sedatives, hypnotics, opioids |
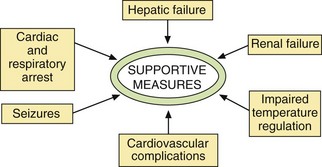
Cardiac or respiratory arrest: This may result from a toxic effect of the drug on the heart, from depression of the respiratory centre or from metabolic disturbance. Assisted ventilation, ranging from mouth-to-mouth, or Ambu-bag inflation, to the use of a ventilator, may be required. In some circumstances, recovery is possible even after prolonged resuscitation.
Hypotension: A low blood pressure is common in severe poisoning with central nervous system depressants. A systolic blood pressure below 70 mmHg can cause irreversible brain damage, but any degree of low blood pressure should be treated if accompanied by poor tissue perfusion or low urine output. Depression of the vasomotor centre can cause arterial dilation and peripheral venous pooling, producing a low central venous pressure (CVP). The CVP should be raised to 10–15 cmH2O (measured from the midaxillary line) by intravenous infusion of a crystalloid solution, such as 0.9% saline. A vasoconstrictor such as noradrenaline (norepinephrine; Ch. 4) is rarely required. If hypotension occurs in association with a normal or raised central venous pressure, this suggests myocardial depression. A positive inotropic drug such as the β1-adrenoceptor agonist dobutamine (Ch. 7) should then be used.
Arrhythmias: Disturbances of cardiac rhythm should only be treated if they are severe. Ventricular arrhythmias causing hypotension often require intervention, but caution should be exercised if there is a long Q–T interval on the electrocardiogram, since the tachycardia often fails to respond to standard antiarrhythmic drugs. It is essential to correct metabolic derangements that predispose to arrhythmias, for example hypothermia, hypoxia, hypercapnia, hypokalaemia, hyperkalaemia and acidosis. If there is widening of the QRS complex after overdose of a tricyclic antidepressant, then intravenous sodium bicarbonate should be given to reduce the risk of serious arrhythmias.
Seizures: These may be caused by a treatable underlying cause such as hypoxia, hypoglycaemia or hypocalcaemia, or they may be a direct toxic effect of the drug on neuronal function. The treatment of choice is lorazepam or diazepam intravenously, or rectal diazepam if the intravenous route is unavailable (Ch. 20). Artificial ventilation with neuromuscular blockade (Ch. 27) is used if the seizures cannot be controlled.
Renal failure: Kidney damage is usually a consequence of prolonged hypotension. Other causes include a direct nephrotoxic effect of the drug and renal damage produced by the products of toxic muscle necrosis (rhabdomyolysis).
Hepatic failure: This usually results from the direct toxic effects of specific agents, such as paracetamol.
Impaired temperature regulation: Hypothermia is common, and can be caused by depression of the metabolic rate with reduced heat production and by increased heat loss from cutaneous vasodilation. It is common with phenothiazines and barbiturates, but is seen with any prolonged coma. Rewarming, preferably by wrapping in a ‘space blanket’, reduces the risk of serious ventricular arrhythmias. By contrast, CNS stimulants such as ecstasy can produce hyperthermia, as can aspirin, which uncouples cellular oxidative phosphorylation.
Reducing toxicity
The adverse effects can be reduced by:
Prevention of absorption of poisons
There are two principal methods of preventing further absorption of the drug: gastric aspiration/lavage and activated charcoal. Inducing emesis with an irritant such as ipecacuanha is not recommended, since it has little effect on drug absorption and increases the risk of aspiration of gastric contents.
Gastric aspiration and lavage: Gastric lavage is rarely required, and should not be considered in unconscious or drowsy persons without protection of the airways by a cuffed endotracheal tube to prevent aspiration of gastric contents into the lungs. It should never be used after ingestion of corrosives or petroleum products. A large-bore orogastric tube is used to aspirate gastric contents initially and then to lavage with doses of water at body temperature. Its effectiveness is unproven. Gastric lavage can be considered for up to 1 h after ingestion of a drug that is not absorbed by activated charcoal, such as lithium or iron salts.
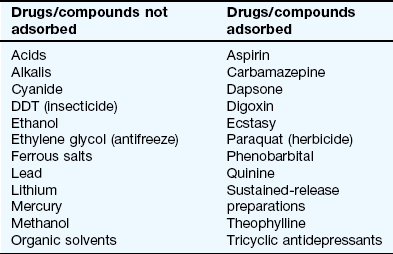
Activated charcoal: This formulation of charcoal has a large adsorbent area and is given as a suspension in water. Activated charcoal adsorbs or binds the drug and retains it in the gastrointestinal lumen. Not all drugs are adsorbed onto charcoal (see Table 53.5). About 10 g of charcoal is required for every 1 g of poison, which makes it impractical for poisons that are usually ingested in large quantities. For suitable drugs, an initial dose of 50 g of charcoal for adults can prevent absorption if given within 1 h of drug ingestion (later after poisoning with modified-release preparations, or drugs with antimuscarinic properties that delay gastric emptying). Charcoal should not be given to drowsy or comatose persons because of the risk of aspiration into the lungs. Constipation is the major unwanted effect of charcoal; charcoal should not be given in the absence of bowel sounds, because of the risk of bowel obstruction.
Elimination of poisons
There are three principal methods of enhancing elimination of the drug: activated charcoal, renal elimination and haemodialysis/haemoperfusion.
Activated charcoal: Repeated administration of 50 g of activated charcoal every 4 h for up to 24–36 h achieves further retention of adsorbed drug in the small intestine. Drug is continuously being transferred in both directions across the gut wall, with the concentration gradient normally favouring net absorption, owing to the high concentration free in solution within the gut lumen. If drug in the bowel is bound onto the charcoal, this lowers the free concentration and can result in net transfer from the body into the gut and thereby enhance elimination of the compound. Repeated-dose activated charcoal is useful for overdose with phenobarbital, carbamazepine, dapsone, quinine and theophylline.
Renal elimination: Altering urine pH, while maintaining normal urine flow, can be effective in increasing the renal elimination of drugs that are weak electrolytes. Modification of urine pH to increase the extent of ionisation of the drug will reduce reabsorption from the renal tubule (Ch. 2). Only a modest increase in urinary flow rate is required. Weak acids, such as salicylates, are excreted more readily when urine is alkalinised (alkaline diuresis, achieved by giving sodium bicarbonate),
Forced diuresis with intravenous infusion of large quantities of fluid was advocated in the past for drugs or toxic metabolites that are mostly eliminated unchanged by the kidney. However, serious disturbances of fluid or electrolyte balance can occur, and therefore it is no longer recommended.
Haemodialysis: This is reserved for the most severely poisoned individuals. The technique is only successful if a large proportion of the body burden of the drug is retained in the plasma and available for removal (i.e. the drug has a low apparent volume of distribution). Haemodialysis relies on diffusion of the drug from the blood across a semi-permeable membrane into the dialysis fluid; it is used for salicylates, phenobarbital, methanol, ethylene glycol, sodium valproate and lithium.
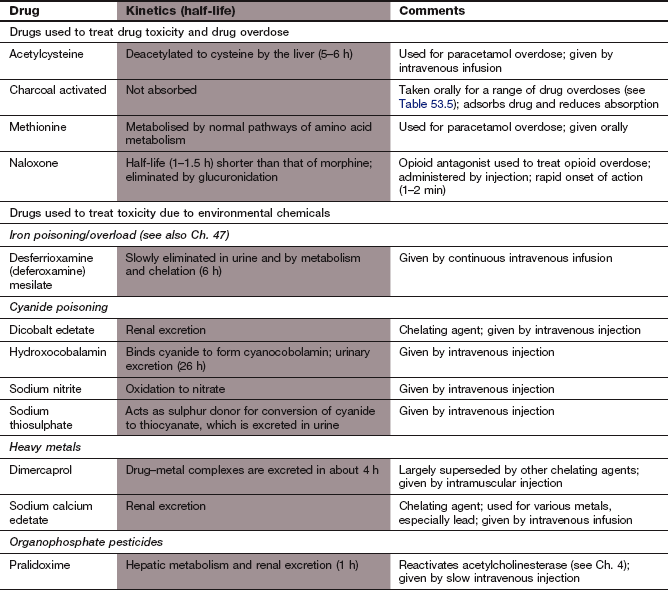
Specific antidotes
Antidotes are available for only a minority of the drugs commonly involved in poisoning cases. Some important examples are given below.
Competitive receptor antagonists:
Atropine acts at muscarinic receptors to block the parasympathetic effects of excess acetylcholine caused when organophosphate insecticides irreversibly inhibit acetylcholinesterase. It is given by intravenous or intramuscular injection.
Naloxone acts at opioid receptors to reverse the effects of opioid analgesics. Its short half-life, compared with those of most opioids, means that repeated injections or preferably an infusion is usually needed. Naloxone can precipitate acute withdrawal in someone who is dependent on opioids.
Flumazenil is an antagonist of benzodiazepine action on γ-aminobutyric acid type A (GABAA) receptors. It is given intravenously, but is rarely needed for the treatment of intentional benzodiazepine overdose because fatalities are uncommon with this class of drug. Flumazenil can cause seizures in benzodiazepine-dependent individuals. It is used to reverse the effects of benzodiazepines when toxicity occurs in people with chronic liver disease.
Chelating agents: Chelating agents act by forming a complex with the drug or chemical, thereby reducing its free (active) concentration:
Compounds that affect drug metabolism:
Fomepizole (available by special order) is a competitive inhibitor of alcohol dehydrogenase given by intravenous injection as the treatment of choice in methanol or ethylene glycol poisoning; it blocks the formation of their toxic metabolites formaldehyde and glycoaldehyde respectively. Alternatively, with caution, their formation can be slowed with oral or intravenous ethanol, which is a preferential substrate for alcohol dehyrogenase with less toxic metabolites.
Acetylcysteine provides a substrate for conjugation of the cytotoxic metabolite of paracetamol (NAPQI) when the natural conjugating ligand, glutathione, is depleted (see below).
Some specific common poisonings
Paracetamol overdose can be fatal, with about 150 deaths occurring each year in England and Wales. Metabolism of paracetamol takes place in the liver, mainly producing non-toxic conjugates (Fig. 53.3). A small amount is oxidised by the cytochrome P450 system to the reactive intermediate NAPQI, which is normally inactivated by conjugation with the thiol group of glutathione. When hepatic glutathione is depleted by paracetamol overdose, oxidative stress coupled with NAPQI-mediated denaturation of proteins produces hepatic necrosis. Similar processes in the kidney can cause renal tubular necrosis.
In the first 24 h there are few symptoms apart from nausea, vomiting, abdominal pain and sweating, which usually resolve. Liver damage begins within 24 h of a large overdose, producing right upper quadrant pain and tenderness. Jaundice is apparent by 36–48 h and liver damage is maximal by 3–4 days. Severe liver failure, requiring transplantation for survival, can ensue. The most sensitive measures of liver damage are the prothrombin time, or the international normalised ratio (INR), and the plasma unconjugated bilirubin. Renal failure is seen in about a quarter of cases with severe liver damage.
Activated charcoal in large doses is recommended within 1 h of a potentially serious paracetamol overdose. Because antidotes are most effective when given early, blood should be analysed for paracetamol if there is any suspicion of poisoning. Blood should be taken at 4 h or more after the suspected overdose. Earlier sampling is not informative because a low plasma level at that time could reflect incomplete absorption of a large overdose, rather than ingestion of a small overdose. Antidotes used in paracetamol poisoning, such as acetylcysteine and methionine, are used to replace glutathione as a thiol donor in the liver; glutathione itself is not used, because it cannot enter liver cells from the blood.
Intravenous acetylcysteine is the preferred treatment for potentially serious poisoning, and should be started prior to the analysis of a plasma paracetamol concentration. Methionine can be given orally if acetylcysteine is not available, but not with or after activated charcoal, because methionine can compete with paracetamol for adsorption. Methionine should not be used if there is vomiting, or started more than 10–12 h after ingestion of paracetamol, since the efficacy of methionine in late poisoning is unknown.
A graph is available (Fig. 53.8) to indicate the risk of liver damage for a given plasma paracetamol concentration related to the time after ingestion. Plasma concentrations after 15 h must be interpreted by extrapolation of the graph. Treatment is necessary if potentially toxic paracetamol concentrations are detected. However, after a staggered overdose or when the time of the overdose is unknown the plasma concentration is uninterpretable and treatment should always be given.
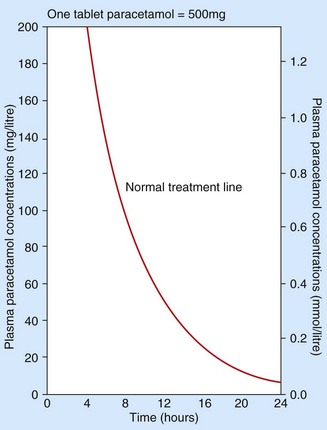
Treatment used to be confined to the first 15 h after overdose, but liver damage can be reduced even when the antidote is delayed for up to 36 h.
Salicylates
Salicylate poisoning is now uncommon in the UK. Aspirin is hydrolysed rapidly to salicylic acid after absorption, but further metabolism by conjugation with glycine is rate-limited. Symptoms of toxicity are nausea, vomiting, abdominal pain, tinnitus, deafness, hyperventilation and sweating. Agitation frequently occurs in adults, but children become comatose. The chain of metabolic events produced by aspirin is shown in Figure 53.9.
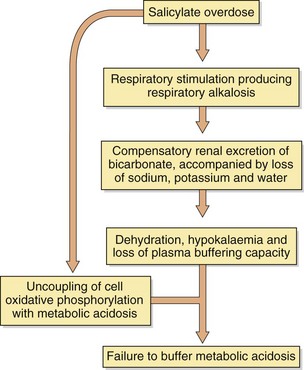
Activated charcoal is recommended for reducing absorption if given early. Correction of fluid, electrolyte and acid–base balance is fundamental to successful management; a fluid deficit of 3–4 L is not unusual in severe poisoning. Forced alkaline diuresis is no longer advocated to enhance salicylate elimination (see above); simple alkalinisation of the urine with 1.26% oral sodium bicarbonate solution to raise the pH above 7.5 is effective and safer. Haemodialysis is the treatment of choice in severe poisoning, especially if there is severe metabolic acidosis.
Tricyclic antidepressants
Approximately 400 deaths per year occur in England and Wales from overdose with tricyclic antidepressants. The antimuscarinic effects of tricyclic antidepressants delay gastric emptying and oral activated charcoal is used routinely for up to 4 h after the overdose. Drowsiness and confusion are followed by seizures and coma in more severe poisoning. Cardiac depression can produce hypotension. Serious arrhythmias, such as ventricular tachycardia, can occur, and ECG monitoring is recommended for at least 24 h. If there is widening of the QRS complex on the ECG, then intravenous infusion of sodium bicarbonate can prevent or treat arrhythmias. Antiarrhythmic drugs should usually be avoided.
Opioid analgesics
The triad of signs characteristic of opioid overdose are:
They can be reversed rapidly by administration of naloxone (Ch. 19), which is a competitive antagonist at opioid µ-receptors. After an initial intravenous bolus dose of naloxone it is often necessary to give repeated boluses or a continuous infusion, because the half-life of naloxone is very short compared with those of most opioids. In poisoning with buprenorphine the effect of naloxone is often incomplete, and assisted ventilation may also be needed. Acute poisoning with organophosphorus insecticides can produce signs that are similar to those with opioids, but naloxone will have no effect.
Beta-adrenoceptor antagonists
Poisoning by β-adrenoceptor antagonists usually presents with bradycardia and hypotension. More severe effects, including coma and convulsions, can occur with some drugs in this class. Treatment of the bradycardia is with intravenous atropine, which may increase the blood pressure. Cardiogenic shock that does not respond to atropine should initially be treated with intravenous glucagon, which has a positive inotropic effect. A temporary cardiac pacemaker may be necessary.
Ecstasy
Ecstasy (3,4-methylenedioxymetamfetamine, MDMA) toxicity is characterised by tachycardia, hyperreflexia, hyperpyrexia and initial hypertension followed by hypotension. In severe cases, delirium, seizures, coma and cardiac dysrhythmias may occur. MDMA is metabolised by CYP2D6 and genetic differences in this enzyme may result in wide inter-individual differences in susceptibility to the toxic effects of MDMA. Some people may present with hyponatraemia, possibly as a result of drinking excessive water as a precaution to prevent dehydration. Treatments include activated charcoal, but only for up to 2 h post-ingestion, since MDMA is absorbed rapidly, and diazepam for agitation or seizures.
True/false questions
1. The therapeutic index (TI) is the ratio between the dose of a drug that results in significant toxicity and the dose that provides a therapeutic response.
2. Mesna reduces the cytotoxic effects of cyclophosphamide on the bladder.
3. Acidification of the urine (acid diuresis) enhances excretion of aspirin.
Case-based questions
A 70-year-old man with a history of depressive illness and alcohol abuse was prescribed a compound analgesic containing 30 mg codeine phosphate and 500 mg paracetamol (co-codamol 30/500) for back pain. Eight hours after collecting his prescription he was seen as an emergency by his GP, who considered the man had taken an overdose and admitted him to hospital.
1. True. A high TI is a desirable property of a drug as it indicates that significant toxicity.
2. True. Mesna (mercaptoethane sulphonic acid) is a thiol donor which protects against haemorrhagic cystitis when administered with the cytotoxic drug cyclophosphamide.
3. False. Acidification of the urine would enhance excretion of weak bases. Weak acids such as aspirin are ionised by alkalinisation of the urine, so less is reabsorbed from the renal tubule.
4. True. Flumazenil is a competitive GABAA receptor antagonist that can be given intravenously in benzodiazepine overdose.
5. False. Fomepizole inhibits the metabolism of methanol by alcohol dehydrogenase, slowing the formation of its toxic metabolites formaldehyde and formic acid.
Case-based answers
A Initial features would be those of opioid overdosage caused by the codeine, with possible symptoms of respiratory depression, pinpoint pupils, coma and cardiovascular collapse. Later symptoms of nausea, abdominal pain and sweating and, if untreated, jaundice, are due to liver damage caused by the toxic metabolite of paracetamol.
B Naloxone is a rapid reversible antagonist of opioids at µ-, κ- and δ-receptors. It has a short half-life and may have to be given repeatedly. Acetylcysteine conjugates with the hepatotoxic metabolite of paracetamol (N-acetyl-p-benzoquinone imine, NAPQI) and is most effective when given early after an overdosage; the risk of liver damage is related to the time of ingestion before treatment and the plasma paracetamol concentrations. Chronic alcohol consumption would increase the toxic effects of codeine and paracetamol; alcohol enhances the central depressant actions of the opioid and also induces cytochrome P450 enzymes, increasing formation of NAPQI and causing toxicity at lower paracetamol doses. The toxicity of paracetamol would also be increased by other cytochrome P450-inducing drugs.
C Codydramol contains paracetamol with the opioid dihydrocodeine and is likely to produce similar unwanted effects to co-codamol in overdose.
Adam, J, Pichler, WJ, Yerly, D. Delayed hypersensitivity: models of T-cell stimulation. Br J Clin Pharmacol. 2011;71:701–707.
Aronson, JK. Medication errors: definitions and classification. Br J Clin Pharmacol. 2009;67:599–604.
Ardern–Jones, MR, Friedmann, PS. Skin manifestations of drug allergy. Br J Clin Pharmacol. 2011;71:672–683.
Buckley, NA, Dawson, AH, Whyte, IM, O'Connell, DL. Relative toxicity of benzodiazepines in overdose. BMJ. 1995;310:219–221.
Dawson, AH, Whyte, IM. Therapeutic drug monitoring in drug overdose. Br J Clin Pharmacol. 2001;52(suppl 1):97S–102S.
Edwards, IR, Aronson, JK. Adverse drug reactions: definitions, diagnosis and management. Lancet. 2000;356:1255–1259.
Ferner, RE, Dear, JW, Bateman, DN. Management of paracetamol poisoning. BMJ. 2011;342:d2218.
Frew, A. General principles of investigating and managing drug allergy. Br J Clin Pharmacol. 2011;71:642–646.
Heard, KJ. Acetylcysteine for acetominophen poisoning. N Engl J Med. 2008;359:285–292.
Jick, SS, Dean, AD, Jick, H. Antidepressants and suicide. BMJ. 1995;310:215–218.
Lee, WM. Drug-induced hepatotoxicity. N Engl J Med. 1995;333:1118–1127.
Park, BK, Kitteringham, NR, Pirmohamed, M, Tucker, GT. Relevance of induction of human drug-metabolising enzymes: pharmacological and toxicological implications. Br J Clin Pharmacol. 1996;41:477–491.
Park, BK, Kitteringham, NR, Powell, H, Pirmohamed, M. Advances in molecular toxicology–towards understanding idiosyncratic drug toxicity. Toxicology. 2000;153:39–60.
Thong, BY-H, Tan, T-C. Epidemiology and risk factors for drug allergy. Br J Clin Pharmacol. 2011;71:684–700.
Vale, JA, Proudfoot, AT. Paracetamol (acetaminophen) poisoning. Lancet. 1995;346:547–552.
Waring, RH, Emery, P. The genetic origin of responses to drugs. Br Med Bull. 1995;51:449–461.