Chemotherapy of malignancy
Approximately 20–25% of people in the Western world die from cancer. Surgery and radiotherapy are valuable for treating localised cancers but are less effective in prolonging life once the tumour has spread to produce metastases. However, the introduction of cytotoxic chemotherapy to kill rapidly proliferating neoplastic cells has had a major impact on the treatment of malignant disease, especially diffuse tumours. The successful treatment of cancer frequently involves a multidisciplinary approach, which also includes psychological and social support.
A wide range of different drugs to treat cancer has been introduced into clinical practice since the 1970s with a variety of mechanisms and sites of action within cancer cells. Although the drugs differ in their specific cellular targets, the majority rely on the rapid rate of growth and division of cancer cells to provide a degree of selectivity between normal and malignant tissue. Recent developments in molecular biology are resulting in the discovery of new potential targets for drug action and a resurgence of cytotoxic drug development. The ability of molecular biological approaches to define the mechanisms of cell–cell communication, apoptosis and angiogenesis will undoubtedly prove a major stimulus for the production of new drugs with greater selectivity for cancer cells. In addition, there is growing understanding of the potential for immunotherapy in the treatment of cancer.
There are a number of in vivo animal tests for detecting antineoplastic activity, but these frequently over-predict the likely effectiveness of a compound in clinical use because the animal tumours used as models have a much higher growth fraction. A placebo-controlled clinical trial of a new antineoplastic drug given as sole treatment is now unethical if an effective drug is already available. Therefore, the efficacy of any new drug is usually assessed by adding it to the best available current therapy. A successful new drug would have to show a clinically significant benefit above that of the optimal current treatment. As a result, many recent advances in cancer chemotherapy have arisen from the more effective use of existing drugs, by optimising drug combinations and regimens, and by minimising toxicity, rather than the introduction of novel compounds.
Cytotoxic drugs share a number of generic properties and characteristics, both beneficial and adverse, which will be discussed first. The major classes of drug will then be discussed with information on mechanisms of action and general toxic effects.
Molecular origins of cancer
Most cancers probably arise from multiple genetic mutations in a cell, with sequential gene defects resulting in progressive changes through initial metaplastic and dysplastic phases, and then to invasive and ultimately metastatic cancer. Normal cells are regulated by numerous external factors that control their growth and death. These include growth factors, cytokines and hormones that activate or suppress the genes controlling cell division. Cancers develop as a result of abnormalities in the control of cell function.
About 85% of cancers arise from exposure to environmental factors such as viruses and chemicals, with inherited genetic mutations accounting for the remainder. The environmental factors produce DNA damage, which in the normal cell can be repaired before the cell completes its cycle of division. A second protective mechanism is apoptosis (programmed cell death) of severely damaged cells. Cancer may arise by various mechanisms that influence gene expression and cell cycle control:
 inactivation of tumour suppressor genes or changes in microRNA genes (encoding RNA molecules which regulate gene expression) will promote unregulated cell proliferation,
inactivation of tumour suppressor genes or changes in microRNA genes (encoding RNA molecules which regulate gene expression) will promote unregulated cell proliferation,
inactivation of genes that repair DNA,
activation of proto-oncogenes to growth-promoting oncogenes: proto-oncogenes are normal gene sequences that control cell proliferation and differentiation. They are capable of being activated to oncogenes, the expression of which leads to tumour development. Oncogene activation occurs as a result of chromosomal rearrangement, gene mutations or gene amplification. Products of oncogene activation include transcription factors, chromatin remodellers, growth factors, altered growth factor receptors, intracellular signal transducers and apoptosis regulators. Gene mutations that activate oncogenes allow cell growth in the absence of stimulation by an external regulator,
suppression of apoptosis: cells with damaged genetic material fail to undergo programmed cell death if oncogenes are activated and tumour suppressor genes are not functional. Defective function of the tumour suppressor protein p53 may be a factor in reducing apoptosis and permitting cell proliferation.
Activation of telomerase may also be important. Telomeres are found at the ends of chromosomes and their progressive shortening as healthy cells divide ultimately arrests further division. In cancer cells, telomerase activation extends the telomeres and promotes cell growth and division. Defective function of the tumour suppressor protein p53 may be a factor in reducing apoptosis and permitting cell proliferation. Growth factors secreted by cancer cells promote angiogenesis and increase blood supply to the tumour. Other secreted factors can impede the host's immune response to the cancer cells. Genetic changes in cancer cells can give rise to metabolic changes or expression of transporters such as P-glycoprotein that confer resistance to chemotherapy (Ch. 2).
Antineoplastic drugs
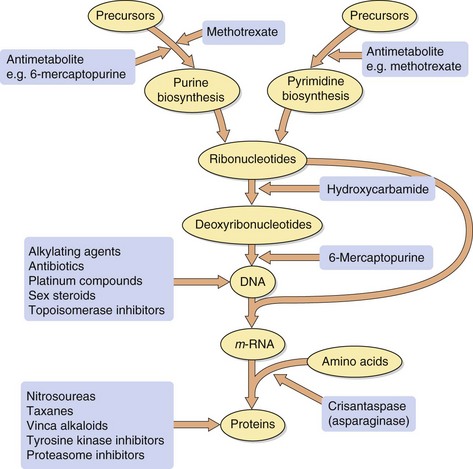
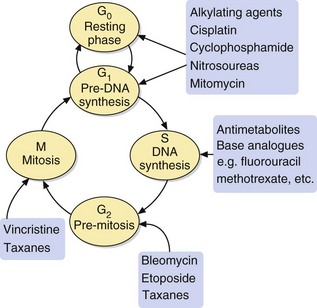
The majority of antineoplastic drugs act on the process of DNA synthesis within the cancer cell, as summarised in Figure 52.1. Selectivity of these drugs for cancer cells compared with normal tissues is determined by the rate of DNA synthesis and cell division. Resting cells in the G0 phase (Fig. 52.2) are resistant to many antineoplastic drugs. Cell cycle-specific antineoplastic drugs, such as the antimetabolites (Fig. 52.2), work effectively only when the cells are in the appropriate phase of the cell cycle at the time of treatment. Non-cell cycle-specific antineoplastic drugs, such as the alkylating drugs, nitrosoureas and cisplatin, have a ‘hit-and-run’ action on DNA, and it is not critical when the cell is exposed because the drug effect becomes apparent when the cells attempt to divide.
The sensitivity of a cancer to treatment depends on its growth fraction, which is the fraction of cells undergoing mitosis at any time. For example, in Burkitt's lymphoma almost 100% of neoplastic cells are undergoing division simultaneously and are they are very sensitive to chemotherapy, showing a dramatic response to a single dose of cyclophosphamide. In contrast, the growth fraction in a carcinoma of the colon is less than 5% of cells, resulting in its relative resistance to chemotherapy. However, metastases from colonic carcinoma deposited in the liver and elsewhere initially have a high growth fraction and are more sensitive to anti-cancer drugs.
Using in vitro cancer cell lines, it has been shown that:
antineoplastic drugs produce a proportional cell kill; in other words, a proportion such as 95% of the cells present may be eliminated during a single course of treatment; consequently, multiple treatments may be necessary to eradicate the cancer, with successive treatments producing an exponential decrease in the number of residual viable cancer cells,
essentially complete eradication of tumour cells is necessary to prevent regrowth,
efficacy of chemotherapy in vitro is increased if treatment with cell cycle-specific drugs is timed to coincide with the appropriate phase of cell division within the cell population.
In vivo, the immune system probably contributes to the final removal of residual malignant cells; however, most antineoplastic drugs compromise immunoresponsiveness, which will reduce this removal process. The periodicity of doses is probably less critical in vivo because cancer cell cycles are not synchronised within the target cell population between treatments. In clinical practice, dose intervals are often established to allow recovery of healthy cells from toxic effects of the treatment. Therefore, while these concepts apply to in vivo cancer treatment, risk–benefit considerations may change with successive treatments and preclude complete eradication of the tumour.
Resistance
Resistance to chemotherapeutic drugs may develop in a number of ways. These are explained later in the text for individual drugs, but include:
reduced drug uptake into cancer cells, e.g. methotrexate enters cells by the high-affinity transport system (the reduced folate carrier) for tetrahydrofolic acid, and downregulation of the transporter limits the uptake of methotrexate and confers resistance to the drug,
use of alternative metabolic pathways and salvage mechanisms to circumvent a blocked biochemical process; such mechanisms are usually drug-specific, e.g. induction of asparagine synthesis in cells exposed to crisantaspase (asparaginase),
alteration of intracellular drug targets, e.g. production of topoisomerase II with reduced sensitivity to the inhibitory effects of anthracyclines,
increased inactivation of the compound within the cancer cell, e.g. high intracellular levels of glutathione S-transferase isozymes inactivate cisplatin and alkylating drugs,
reduced activation of prodrugs, e.g. low intracellular levels of deoxycytidine kinase reduces activation of cytarabine (cytosine arabinoside); increased activity of thiopurine S-methyltransferase increases the metabolism of mercaptopurine and tioguanine,
increased removal of the drug from the cancer cell. This involves the possibility of increased transcription of the gene for proteins which act as carriers for the elimination from the cell of complex foreign chemicals (Fig. 2.1), including a number of cytotoxic compounds. There are several such proteins (see Table 2.1), including P-glycoprotein and the multidrug resistance-related proteins (MRPs). Increased production of the carrier protein confers multidrug resistance to a number of structurally unrelated natural compounds or their derivatives, including vinca alkaloids, etoposide, taxanes, anthracyclines, dactinomycin (actinomycin D), mitomycin C and mitoxantrone. The carrier can be inhibited by calcium channel blockers, such as nifedipine or verapamil, by ciclosporin, or by tamoxifen. These drugs may be added to cytotoxic drug regimens to prevent resistance.
Unwanted effects
Cytotoxic antineoplastic drugs are among the most toxic compounds given to humans. Many have a therapeutic index of approximately 1, as the therapeutic dose is essentially the same as the toxic dose. Because drug action is usually greater in tissues with a high growth fraction, a number of normal, rapidly dividing non-malignant tissues are also affected. In addition to effects that occur in all rapidly dividing tissues, many chemotherapeutic drugs also have specific toxic effects on other tissues. Dosage regimens are usually designed so that normal tissues, especially bone marrow and gut, can recover between doses (Fig. 52.3).
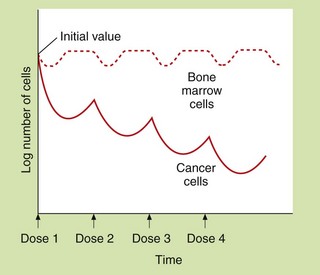
Fig. 52.3 Hypothetical dosing schedule of anti-cancer drugs to allow recovery of normal tissues.
At least 109 tumour cells are usually present when tumours are first detectable. The malignant cells show a greater proportional kill than normal cells because a larger fraction is in division at any time. Theoretically, the response of the malignant cells to dose 2 would be greater than for dose 1 if cell cycles became synchronised and dose 2 was given during the correct phase of the growth cycle. A typical dose interval is 3–4 weeks.
Gastrointestinal tract
Mucosal cells have a rapid turnover. Toxicity can produce anorexia, mucosal ulceration or diarrhoea. A sore mouth is most common with fluorouracil, methotrexate and the anthracyclines. Nausea and vomiting are common, especially with alkylating drugs and cisplatin, and this may limit an individual's ability to tolerate an optimal dosage regimen (see Ch. 32).
Bone marrow
Myelosuppression is a serious consequence of treatment and can lead to severe neutropenia, thrombocytopenia and sometimes anaemia. It often occurs 7–10 days after a cycle of chemotherapy, but is delayed with drugs such as melphalan and lomustine. These haematological consequences may limit the drug dosage that the person is able to tolerate. There is a high risk of both infection (neutropenic sepsis) and haemorrhage following cytotoxic chemotherapy.
Reproductive organs
Both sexes are affected and sterility can result, particularly after therapy with cyclophosphamide or cytarabine. Because of the mechanisms of action of cytotoxic drugs, most have teratogenic activity. Pregnant women should not be exposed to cytotoxic drugs for treatment or as members of the healthcare team. Alkylating agents or procarbazine can cause permanent male infertility. Drugs that mimic or affect the activity of sex hormones are frequently used for the treatment of breast or prostate cancer, and these inevitably produce adverse effects on sexual function.
Growing tissues in children
Of particular concern in children is the possibility that intensive cytotoxic chemotherapy can impair growth. Children treated with cytotoxic drugs for malignancy also have an increased risk of the subsequent development of a second malignancy (about 10%), which is often leukaemia.
Extravasation of intravenous drug
If anti-cancer drugs leak from a vein into the surrounding tissues, they can cause severe local tissue necrosis.
Tumour lysis syndrome
Rapid breakdown of malignant cells can produce hyperuricaemia, hyperkalaemia, hypophosphataemia, hypocalcaemia with consequent renal damage or arrhythmias. The syndrome is most common with treatment of non-Hodgkin's lymphoma, Burkitt's lymphoma and acute leukaemias. Tumour lysis syndrome can be ameliorated by good hydration and the prophylactic use of allopurinol (Ch. 31).
Drug combinations
It is common practice to treat many cancers with a combination of different antineoplastic drugs simultaneously. Potential combinations of drugs are investigated using in vitro and in vivo experiments before they are subjected to clinical evaluation in humans. The most successful combinations are those that show synergism in their actions on cancer cells, rather than a simple additive effect, while showing no increase in their systemic toxicity. Criteria for selecting ideal combinations are:
each drug should be an active antineoplastic drug in its own right; a second drug would not be given simply to increase the formation of an active metabolite of the first, although sometimes drugs are given to reduce the development of toxicity or resistance to another drug,
each drug should have a different mechanism of action and target site within the cancer cell; this will increase efficacy while reducing the likelihood of resistance,
each drug should have a different site for any organ-specific toxicity; some common toxicity is almost inevitable because nearly all drugs affect tissues with a high growth fraction.
Specific antineoplastic drugs
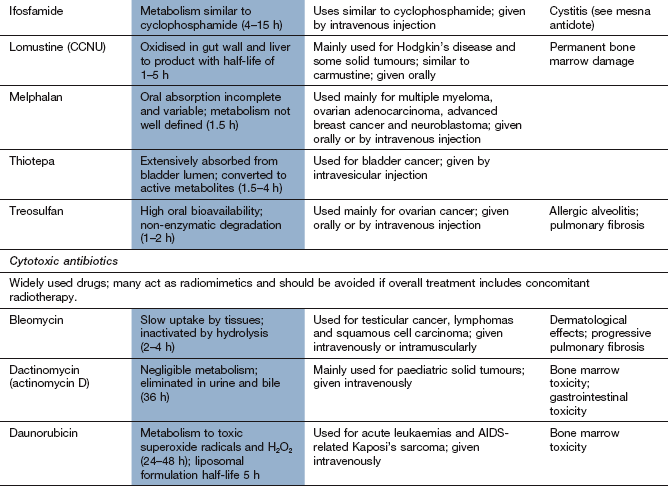
The drug compendium at the end of this chapter outlines the licensed uses of individual drugs and notes any unusual or limiting toxicity.
Drugs affecting nucleic acid function
Mechanism of action and uses: The nitrogen mustards were developed from the sulphur mustard gases used as chemical warfare agents in World War I. These gases caused bone marrow suppression in addition to the respiratory toxicity for which they were developed. Replacement of the divalent sulphur atom by trivalent nitrogen allowed the introduction of a complex side chain, which resulted in a range of more stable nonvolatile drugs that could be given therapeutically under controlled conditions. Alkylating drugs contain side chains which undergo a metabolic activation step that involves loss of part of the molecule (for example the Cl is lost from –CH2CH2Cl) and yields a highly reactive product which binds to DNA or proteins. Many alkylating drugs are bifunctional (i.e. have two reactive groups). The reactive alkylating group(s) in the molecule may be:
nitrogen mustard N–CH2CH2Cl (with Cl being the leaving group), e.g. carmustine (BCNU), chlorambucil, cyclophosphamide, ifosfamide, lomustine (CCNU), melphalan,
sulphonate ester –CH2OSO2CH3 (with SO2CH3 being the leaving group), e.g. busulfan, treosulfan,
The mechanism of action is by covalent binding to DNA (nitrogen mustards, sulphonate esters and cyclic nitrogen compounds), which prevents DNA and RNA synthesis, or by covalent binding to proteins (nitrosoureas), which blocks DNA repair processes.
When alkylating drugs bind to DNA nucleotides, such as to guanine, the alkylated nucleotide may either be repaired in which case the cell survives, or it may interfere with DNA replication by:
undergoing further metabolism via ring opening,
crosslinking to another guanine molecule via a second reactive group (with bifunctional drugs).
Because of the covalent nature of the product, these effects are not cell cycle-specific (Fig. 52.2). Alkylating agents are used to treat a wide variety of leukaemias, lymphomas and solid tumours.
Pharmacokinetics: The pharmacokinetic characteristics of the alkylating drugs depend on the nature of the reactive group(s) and the third non-reactive substituent on the N atom. Cyclophosphamide is an orally active prodrug that undergoes metabolic activation to produce two toxic metabolites, acrolein and phosphoramide mustard. Ifosfamide metabolism is similar to that of cyclophosphamide, and toxic metabolites of both cyclophosphamide and ifosfamide are excreted in the urine. Melphalan and chlorambucil, which have an aromatic substituent, undergo rapid metabolism. Most alkylating agents have half-lives of less than 6 h, but the duration of action on DNA is very long.
Alkylating drugs are highly cytotoxic and cause bone marrow suppression and neutropenia. Amifostine is a compound used to reduce the severity of neutropenia induced by cyclophosphamide or cisplatin (see cisplatin below). It is a prodrug that is metabolised in neutrophils by alkaline phosphatase to a free thiol metabolite that binds to the reactive metabolites of these cytotoxic drugs.
Fertility is reduced through impaired gametogenesis.
A particular problem with the long-term use of alkylating drugs is the development of acute myeloid leukaemia, especially if combined with radiotherapy.
Busulfan, carmustine and treosulfan can cause pulmonary fibrosis.
Busulfan and treosulfan commonly cause skin pigmentation.
Cyclophosphamide and ifosfamide cause bladder toxicity with haemorrhagic cystitis due to formation of acrolein; it can be prevented by prior treatment with mesna (mercaptoethane sulphonic acid; Ch. 53), which provides free thiol groups in the urinary bladder to detoxify acrolein. Bladder cancer can develop years after cyclophosphamide therapy.
Cytotoxic antibiotics
Mechanisms of action and uses: The cytotoxic antibiotics have diverse chemical structures.
The anthracyclines are all quinone-containing planar four-ringed structures that contain an amino sugar group.
Mitoxantrone has a three-ringed planar quinone structure with amino-containing side chains (an anthracycline derivative), and mitomycin is a non-planar tricyclic quinone.
Bleomycin and dactinomycin are complex peptide or glycopeptide derivatives.
Cytotoxic antibiotics have several possible mechanisms of action.
Intercalation: this is shown particularly by the anthracyclines, with the planar ring system intercalating between DNA bases and the amino sugar part binding to the deoxyribose phosphate groups. Intercalation blocks reading of the DNA template and also inhibits repair of DNA double-strand breaks by topoisomerase II.
Free radical attack: the metabolism of the drugs gives rise to superoxide and hydroxyl radicals and hydrogen peroxide, which cause DNA damage and cytotoxicity.
Membrane effects: interference with membrane function can occur either directly or via oxidative damage.
In general, the mechanisms of action are not cell cycle-specific, although some members of the class show greatest activity at certain phases of the cycle, for example S phase (doxorubicin, mitoxantrone), G1 and early S phase (mitomycin), and G2 phase and mitosis (bleomycin).
Cytotoxic antibiotics have a wide spectrum of activity and are used for treatment of several leukaemias and lymphomas, as well as some solid tumours.
Pharmacokinetics: The cytotoxic antibiotics are poorly absorbed from the gut and are given intravenously. They are eliminated by metabolism and some have very long half-lives (mostly 12 h or longer).
Unwanted effects: Many of these drugs have radiomimetic properties. They should not be used at the same time as radiotherapy, since toxicity can be greatly increased.
Doxorubicin, epirubicin and mitoxantrone produce dose-related irreversible myocardial damage leading to cardiomyopathy through free radical release and oxidative stress, as well as nuclear cytotoxicity. The cardiomyopathy may not become apparent until several years after treatment. Liposomal formulations of doxorubicin and longer infusion times may reduce the cardiac toxicity, as does concurrent infusion of the iron chelator dexrazoxane.
Painful skin eruptions with liposomal doxorubicin.
Bleomycin often causes skin pigmentation.
Bleomycin and mitomycin produce dose-related pulmonary fibrosis.
Tissue extravasation during infusion of anthracyclines produces severe necrosis, which can be minimised by subsequent intravenous infusion of dexrazoxane.
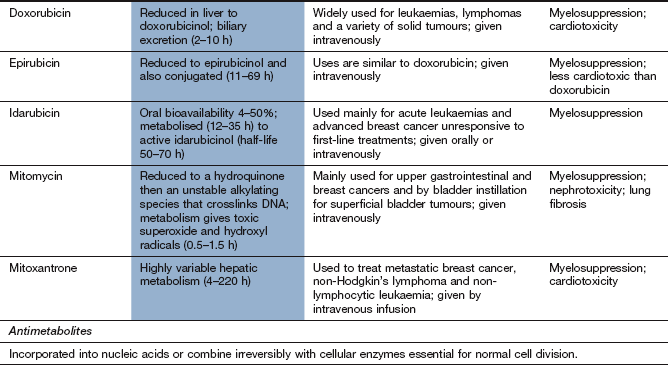
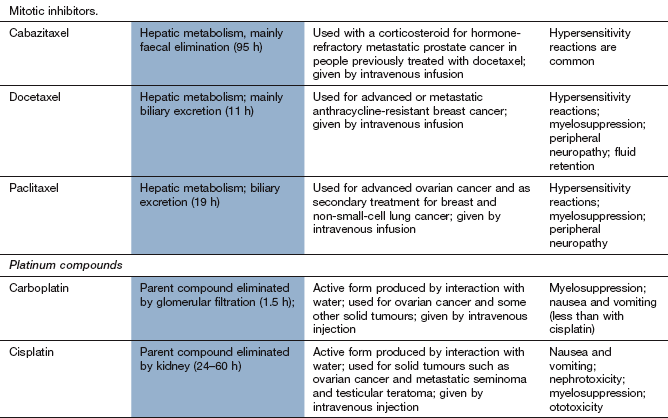
Platinum compounds
Mechanism of action and uses
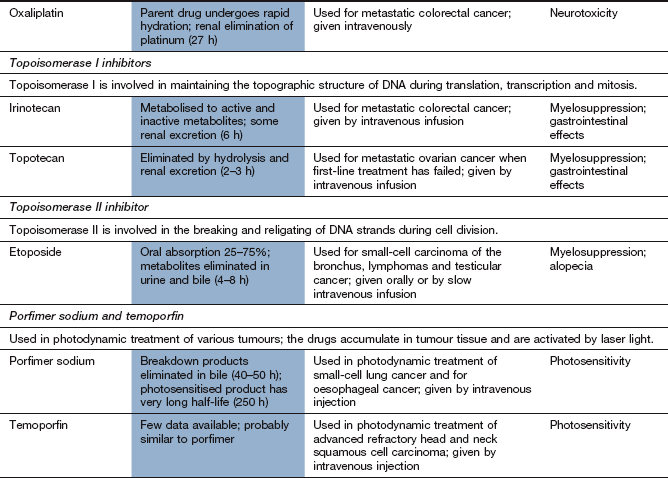
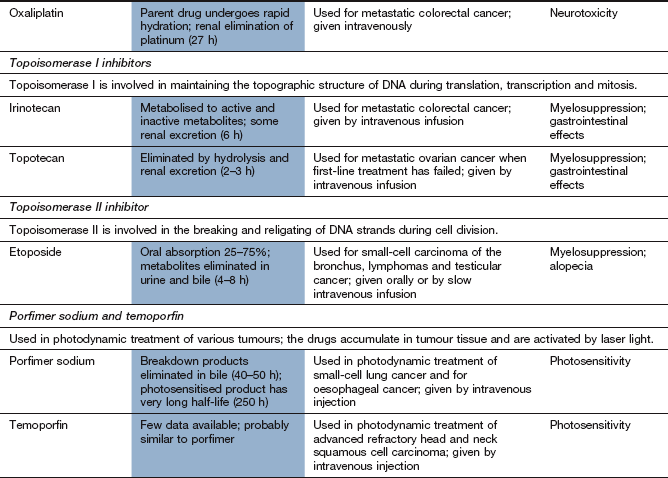
The platinum drugs enter cells and generate a reactive complex that crosslink guanine units in DNA. The result is similar to the effect of alkylating drugs by breaking the DNA chain. Cisplatin and carboplatin are used for ovarian and lung tumours. Cisplatin is also used for several other solid tumours. Oxaliplatin is used for advanced colorectal cancer.
Pharmacokinetics
These drugs are given by intravenous infusion and are mainly excreted by the kidney as platinum compounds. Cisplatin and oxaliplatin have long half-lives (24–60 h), largely owing to extensive protein binding.
Unwanted effects
Nephrotoxicity with irreversible renal impairment; hydration is important to minimise the risk.
Ototoxicity with hearing loss and tinnitus.
Most effects are more marked for cisplatin than for carboplatin. Amifostine (see above) is used to reduce the severity of cisplatin-induced neutropenia in advanced ovarian cancer. It also reduces the nephrotoxicity of cisplatin.
Antimetabolites
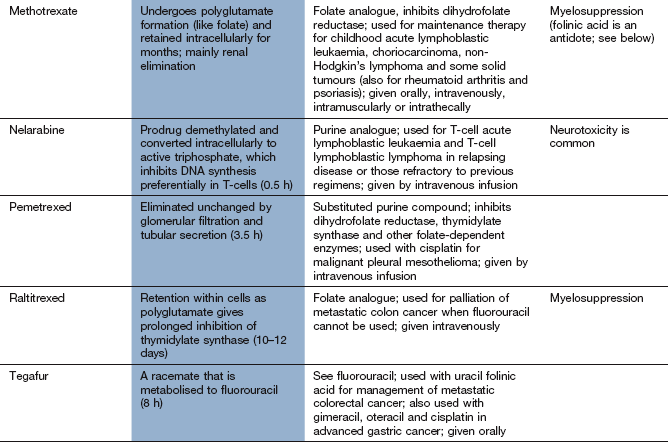
Mechanism of action and uses: An astute clinical observation that the administration of folic acid to children with leukaemia exacerbated their condition led to the development of a folate antagonist, methotrexate. This represented an important landmark in cancer chemotherapy.
Folic acid in its reduced form (tetrahydrofolic acid, THF) is an important biochemical intermediate. It is essential for synthetic reactions that involve the addition of a single carbon atom during a biochemical reaction, such as the introduction of the methyl group into thymidylate and the synthesis of the purine ring system. During such reactions, THF is oxidised to dihydrofolic acid (DHF), which has to be reduced by dihydrofolate reductase back to THF before it can accept a further one-carbon group and be reused.
Methotrexate has a very high affinity for mammalian dihydrofolate reductase and inhibits its active site. This blocks purine and thymidylate synthesis and inhibits the synthesis of DNA, RNA and protein. It may show selectivity for cancer cells because these rely more on de novo synthesis of purines and pyrimidines, whereas normal tissues use salvage pathways that reutilise preformed purines and pyrimidines to a greater extent. Methotrexate is specific for S phase and slows G1 to S phase.
Methotrexate is given for acute lymphoblastic leukaemia, non-Hodgkin's lymphomas and various solid tumours. It is also used an immunosuppressant at lower doses in non-malignant conditions such as inflammatory joint diseases and psoriasis. The mechanism of its immunosuppressant effect is different to its anti-cancer actions (Ch. 38).
Pharmacokinetics: Methotrexate is well absorbed from the gut but can also be given intravenously or intrathecally. It is eliminated by renal excretion, but a small amount may be retained for longer periods both strongly bound to dihydrofolate reductase and intracellularly as polyglutamate conjugates.
Toxicity to normal rapidly dividing tissues, especially the bone marrow.
Hepatotoxicity can follow chronic therapy as an immunosuppressant (see Ch.38).
Toxicity is increased in the presence of reduced renal excretion, and methotrexate should be avoided if there is significant renal impairment. Folinic acid (leucovorin) is frequently administered shortly after high-dose methotrexate to reduce mucositis and myelosuppression. Non-steroidal anti-inflammatory drugs such as aspirin can reduce the renal excretion of methotrexate and increase its toxicity.
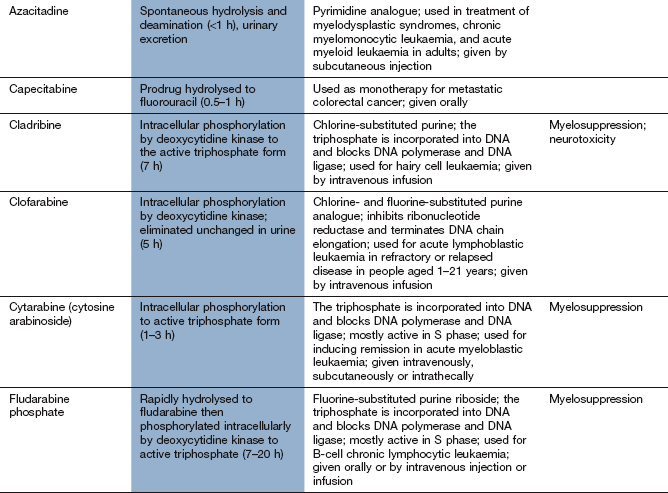
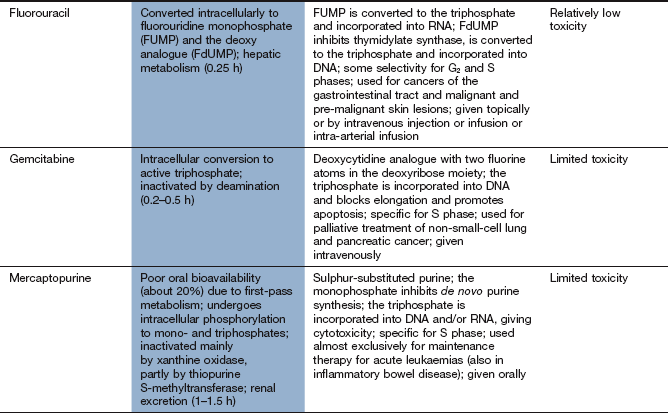
Base analogue antimetabolites
Mechanism of action and uses: A number of useful chemotherapeutic drugs have been produced by simple modifications to the structures of normal purine and pyrimidine bases (Fig. 52.4). These act in a number of ways to interfere with DNA synthesis, typically following intracellular phosphorylation and the incorporation of the triphosphate product into DNA or RNA. Detailed mechanisms are given for each drug in the Compendium at the end of the chapter. Base analogue antimetabolites are used for a wide variety of leukaemias, lymphomas and solid tumours.
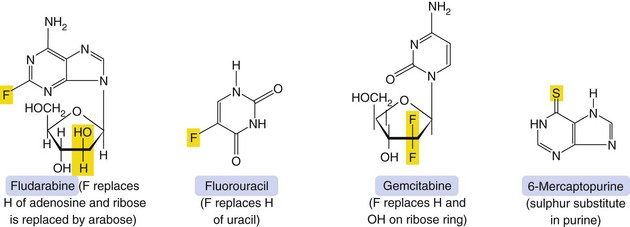
Pharmacokinetics: Base analogues are mainly absorbed and metabolised by the pathways involved in absorption and metabolism of the corresponding unmodified base. Oral absorption is often erratic and most are given intravenously. The urine is a minor route of elimination (up to 1% of the parent drug) and most half-lives are in the range 1–8 h. Tegafur is a prodrug of fluorouracil and is given in combination with uracil, or with gimeracil and oteracil, which inhibit the breakdown of fluorouracil.
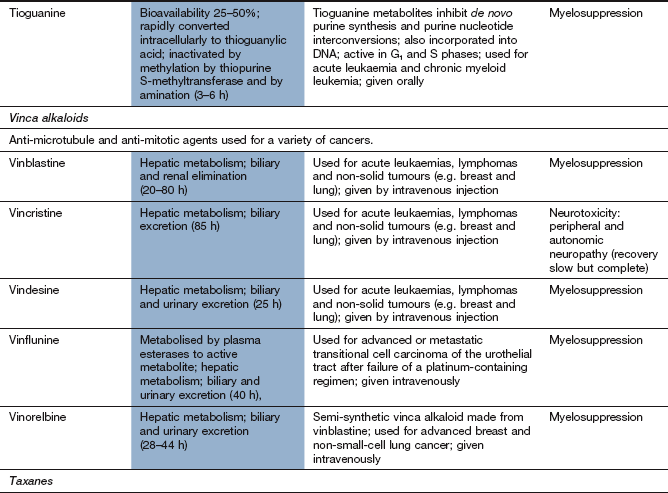
Typical cytotoxic effects are common; myelosuppression, in particular, can be severe and prolonged after cladribine, cytarabine, fludarabine and tioguanine.
Drug interaction: allopurinol (Ch. 31) interferes with the metabolism of 6-mercaptopurine, and the dose should be reduced if these drugs are used concurrently.
Mitotic inhibitors
Mechanism of action and uses: The vinca alkaloids are complex natural chemicals isolated from the periwinkle plant (Vinca rosea). Vinca alkaloids bind to tubulin and inhibit polymerisation and therefore assembly of microtubules, thus producing M-phase arrest of mitosis. They are therefore cycle-specific. Microtubules are essential for numerous cellular functions, including maintenance of cell shape, motility, transport between organelles and cell division.
The vinca alkaloids are used for various lymphomas and for acute leukaemia. They are also effective in some solid tumours.
Pharmacokinetics: Vinca alkaloids are usually given intravenously. Elimination is largely by metabolism with little renal excretion. They have very long half-lives.
Unwanted effects: The spectrum of unwanted effects differs between various drugs, despite their close structural similarities.
General cytotoxicity. Myelosuppression is dose-limiting for vinblastine, vindesine and vinorelbine, but unusual with vincristine.
Neurotoxicity, usually a sensory neuropathy, is dose-limiting with vincristine. It causes peripheral paraesthesiae, loss of tendon reflexes, abdominal pain and constipation. Motor weakness occasionally accompanies the sensory neuropathy.
Severe tissue damage if the drugs extravasate from the infusion site.
Camptothecin analogues (topoisomerase I inhibitors)
Mechanism of action and uses: These drugs are semi-synthetic derivatives of a cytotoxic alkaloid isolated from the Chinese tree Camptotheca acuminata. The drugs inhibit topoisomerase I, which is important in DNA transcription and translation. The enzyme relieves the torsional strain in DNA by producing single-strand breaks that, under normal cell conditions, are then re-ligated. The drugs bind to the DNA–topoisomerase I complex and prevent re-ligation. Although this binding is readily reversible, the consequences are irreversible, because cell death occurs when a double-strand break is produced at the DNA replication fork during S phase. Inhibition of DNA repair increases the sensitivity of the cell to ionising radiation.
Irinotecan is given as second-line treatment for metastatic colorectal cancer. Topotecan is used for lung, cervical or ovarian cancer.
Epipodophyllotoxins (topoisomerase II inhibitors)
Mechanism of action and uses: Etoposide is a synthetic derivative of a compound extracted from the mandrake root (Podophyllum peltatum). It is active during the G2 phase and binds to the complex of DNA and topoisomerase II (an enzyme involved in the breaking and rejoining of DNA strands during cell division). The etoposide-bound complex prevents DNA replication by preventing re-ligation of DNA, and leads to cell apoptosis.
Etoposide is used for small cell lung carcinoma, lymphomas and testicular cancer.
Taxanes
Mechanism of action and uses: The clinically used drugs are produced from taxane, a diterpenoid extracted from the bark of the Pacific yew tree (Taxus brevifolia). Taxanes promote the assembly of microtubules and inhibit their depolymerisation, leading to the formation of stable and non-functional microtubular bundles in the cell. They bind to a different site to that targeted by vinca alkaloids. The cell is inhibited during the G2 and M phases of the cell cycle. Taxanes are also radiosensitisers, since cells in the G2 and M phases are more sensitive to radiation.
Taxanes are used for ovarian, breast and prostate cancer and a variety of other solid tumours.
Pharmacokinetics: These drugs are given intravenously because of poor oral absorption. They are extensively metabolised in the liver and have half-lives of 10–20 h.
Severe hypersensitivity reactions can occur, with hypotension, angioedema and bronchospasm. Routine premedication with histamine (H1 and H2) receptor antagonists (Chs 33 and 39) combined with a corticosteroid (Ch. 44) is recommended for paclitaxel, and premedication with a corticosteroid for docetaxel.
Paclitaxel causes peripheral sensory neuropathy, with motor neuropathy at high dosages.
Docetaxel causes persistent leg oedema due to fluid retention.
Drugs affecting tyrosine kinase function
The products of activated oncogenes include various cell surface receptors that activate a range of intracellular tyrosine kinases which autophosphorylate the receptor (see Ch. 1). There are about 20 families of receptors, including vascular endothelial growth factor receptors (VEGFRs; of which there are three types), the human epidermal growth factor (EGF) receptors HER1 (often called EGFR), HER2, HER3 and HER4, and platelet-derived growth factor receptors (PDGFRs). Autophosphorylation of the receptor triggers a series of intracellular pathways that stimulate cancer cell proliferation and block apoptosis.
Drugs acting on these processes can be subdivided into monoclonal antibodies that inhibit the cell surface receptor and small organic molecules that act intracellularly on the tyrosine kinase enzyme.
Tyrosine kinase receptor inhibitors
Mechanism of action and uses: Bevacizumab is a monoclonal antibody that inhibits VEGFR and reduces tumour angiogenesis. It is given by intravenous infusion as part of the first-line treatment of metastatic colorectal cancer, lung cancer and renal cell carcinoma. Unwanted effects include mucocutaneous bleeding, gastrointestinal perforation and impaired wound healing.
Cetuximab is a monoclonal antibody that binds to the extracellular domain of the EGFR (HER1) and blocks ligand-induced activation of tyrosine kinase. It is given by intravenous infusion in cases of colorectal tumour expressing EGFR and in combination with radiotherapy for locally advanced squamous cell cancer of the head and neck. Unwanted effects may occur with the infusion, including chills, fever and hypersensitivity reactions. Severe keratitis leading to blindness has also been reported.
Trastuzumab is a monoclonal antibody used for metastatic breast cancer when the tumour overexpresses HER2. It binds to HER2 and prevents activation of tyrosine kinase. Unwanted effects may occur with the infusion, including chills, fever and hypersensitivity reactions. Cardiotoxicity occurs if trastuzumab is used with anthracyclines, leading to heart failure.
Tyrosine kinase inhibitors
These drugs block tyrosine kinase enzyme activity intracellularly, preventing transduction of signals from a variety of tyrosine kinase-linked cell surface receptors, and inhibiting cell growth and enhancing apoptosis. The drugs compete with ATP for binding to the enzyme and inhibit autophosphorylation of the receptor (see Ch. 1).
Dasatinib inhibits multiple tyrosine kinases, including those associated with VEGFR and PDGFR families and with Bcr-Abl (a fusion protein found in leukaemias associated with Philadelphia chromosome). Dasatinib is used for chronic myeloid leukaemia and acute lymphoblastic leukaemia.
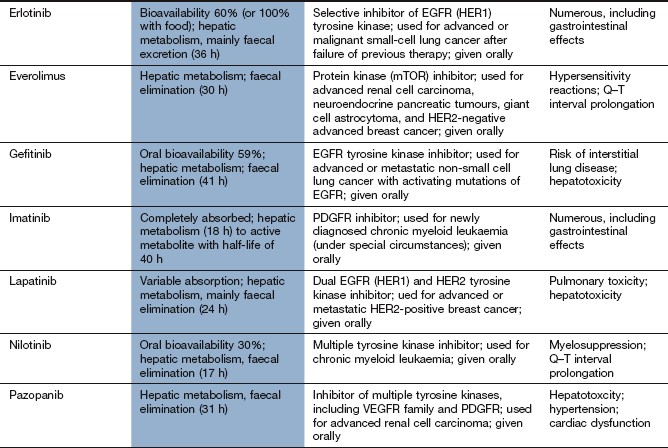
Erlotinib inhibits signals from the EGFR (HER1). It is used for lung and pancreatic cancers.
Imatinib inhibits signals from the PDGFR family and Bcr-Abl. It is used for chronic myeloid leukaemia, acute lymphoblastic leukaemia and a variety of rare tumours.
Sunitinib and sorafenib inhibit signals from several receptors, including VEGFR and PDGFR families. They are used for renal cell carcinoma; sunitinib is also used for gastrointestinal stromal tumours and sorafenib also for hepatocellular cancer.
Proteasome inhibitors
Mechanism of action: Proteasomes are large protein complexes that degrade ubiquitinated proteins (proteins conjugated to ubiquitin, which directs them to parts of the cell for destruction). Inhibition of the 26S proteasome by bortezomib interferes with degradation of pro-apoptotic proteins, leading to cell death.
Hormonal agents
Some drugs used in cancer therapy suppress cell division by actions at intracellular steroid receptors or by influencing the metabolism of steroidal hormones; examples include corticosteroids and drugs that control the division of cells sensitive to sex hormones. Cancers that arise from cell lines possessing steroid receptors that promote their growth and cell division are frequently susceptible to inhibitory steroids.
Glucocorticoids
Glucocorticoids (Ch. 44) suppress lymphocyte mitosis and are used in leukaemia and lymphoma; they are also helpful in reducing oedema around a tumour.
Oestrogens
Oestrogens such as ethinylestradiol (Ch. 45) suppress prostate cancer cells, both locally and in metastases, and provide symptomatic improvement; gynaecomastia is a common unwanted effect.
Progestogens
Progestogens such as hydroxyprogesterone acetate (Ch. 45) suppress endometrial cancer cells and kidney cancer metastases.
Oestrogen receptor antagonists
Breast cancer can be suppressed by oestrogen receptor antagonists such as tamoxifen. Tamoxifen is active orally and binds competitively to oestrogen receptors. It shows both oestrogenic effects (on bone) and anti-oestrogenic effects (on breast tissue). Tamoxifen inhibits oestrogen-regulated genes and reduces the secretion of growth factors by tumour cells. Tumour cells are affected mainly in the G2 phase of the cell cycle. Tamoxifen is extensively metabolised in the liver and has active metabolites with long half-lives, so several weeks of treatment are necessary to achieve steady-state concentrations. Unwanted effects include hot flushes and amenorrhoea in premenopausal women and vaginal bleeding in postmenopausal women. Tamoxifen inhibits CYP3A4 and, therefore, reduces the metabolism of other substrates, such as warfarin.
Gonadorelin analogues
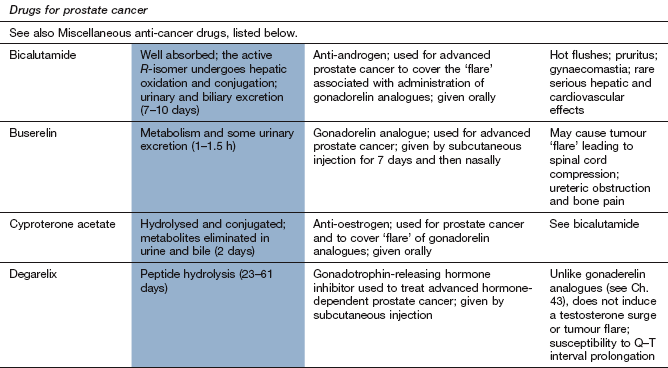
Gonadorelin is synthetic gonadotrophin-releasing hormone (GnRH) (see Ch. 43). Gonaldorelin analogues (e.g. buserelin) suppress prostate cancer cells.
Aromatase inhibitors
Aromatase is the enzyme that converts androgens to oestrogens. Inhibitors of aromatase (e.g. anastrazole and letrozole, which are non-steroidal, or exemestane, which is a steroid) reduce oestrogen production in postmenopausal women, who produce oestrogen mainly from androstenedione and testosterone in many tissues such as adipose tissue, skin, muscle and liver. Aromatase is also present in the cells of two-thirds of breast cancers. Aromatase inhibitors are used in post-menopausal women to treat breast cancers that are oestrogen-dependent.
Miscellaneous anti-cancer drugs
There are many individual anti-cancer drugs with a variety of mechanisms of action. Further details (including therapeutic uses and adverse effects) are given in the drug compendium at the end of this chapter.
Mechanisms of action and uses
Most potential biochemical sites within cells have been investigated as targets for anti-cancer drugs. Actions of different drugs include the following:
removal of asparagine required for protein synthesis (crisantaspase),
inhibition of incorporation of thymidine and adenine into DNA (procarbazine),
inhibition of adenosine deaminase, which causes a build-up of deoxyadenosine triphosphate (dATP) that inhibits the formation of other deoxyribonucleotide triphosphates (pentostatin),
inhibition of reduction of ribonucleotides to deoxyribonucleotides (hydroxycarbamide),
intercalation between DNA base pairs (amsacrine),
alkylating action, especially on thiol groups, to inhibit DNA repair (dacarbazine and temozolomide),
superoxide production causing DNA backbone cleavage and cell apoptosis (trabectedin),
increased cell differentiation and inhibition of proliferation by action on retinoid receptors (retinoic acid receptors, RARs, and retinoid X receptors, RXRs) (bexarotene, tretinoin) (Ch. 49),
photodynamic activation in superficial tumours by laser light to produce cytotoxic oxygen free radicals (porfimer sodium, temoporfin),
immunomodulation (by inhibition of tumour necrosis factor α and several other pro-inflammatory chemokines) and inhibition of angiogenesis (lenalidomide and thalidomide),
activation of cytotoxic killer cells (Ch. 38); interleukin-2 (aldesleukin) is a cytokine produced by T-lymphocytes that is made by recombinant DNA technology,
monoclonal antibodies: several monoclonal antibodies have been developed that have anti-cancer activity; for example:
alemtuzumab (which produces lysis of B-lymphocytes in treatment-resistant or rapidly relapsing chronic lymphocytic leukaemia),
rituximab (which produces lysis of B-lymphocytes in chemotherapy-resistant advanced follicular lymphoma) (see also Ch. 49),
catumaxomab (a trifunctional molecule that binds to the epithelial cell adhesion molecule (EpCAM), lymphocytes and several accessory immune cells such as macrophages, natural killer cells or dendritic cells and triggers an immune reaction against the cancer cell) is active against epithelial tumours that express EpCAM,
ipilimumab (binds to cytotoxic T-lymphocyte antigen 4 and prevents inhibition of the cytotoxic action) used for metastatic melanoma.
Clinical uses of antineoplastic drugs
Different forms of cancer vary in their sensitivity to chemotherapy. The most responsive include lymphomas, leukaemias, choriocarcinoma and testicular carcinoma, while solid tumours such as sarcomas, adrenocortical and squamous cell bronchial carcinomas generally have a poor response. An intermediate response is shown by other cancers, for example those of the bowel, bladder, head and neck, small cell bronchogenic tumours and hormone-related cancers (breast, ovary, endometrium and prostate). In addition, the sensitivity of an individual tumour can change during treatment with antineoplastic drugs, because of the development of resistance.
Chemotherapy can be used alone to treat cancer, or in combination with surgery or with radiation (chemoradiation). Chemotherapy may be given as a curative or a palliative treatment, or to reduce the risk of relapse after tumour removal. Adjuvant chemotherapy refers specifically to treatment following a surgical procedure that appears to have removed all tumour, with the intention of preventing relapse from occult disease. Neoadjuvant chemotherapy is given before surgery to reduce tumour size.
Chemotherapy is being supplemented in many cancers types by the use of orally effective small molecule inhibitors, which often require the presence of a particular gene mutation in the cancer for effect (see sections below). These are excellent examples of the increasing personalization of cancer treatments and such drugs have converted some solid cancers from rapidly fatal conditions to diseases that can remain well controlled or radiologically resolved for sometimes many years.
A major new development in systemic therapy over recent years is immunotherapy, which aims to target the cancer by immune attack. Examples for drugs that have become mainstream treatments are a vaccine in advanced prostate cancer and the immunostimulatory antibody ipilimumab in melanoma.
Anti-cancer drug therapy for specific malignancies
The following discussion selects some important cancers and outlines the role of chemotherapeutic drugs in their management. The choice of specific regimens is a complex process involving an assessment of prognosis, frailty, toxicity and the wishes of the individual. Clinical trials are producing a continuing flow of improved therapeutic options, and this is a field of medicine that changes rapidly.
Oesophageal cancer
Oesophageal cancer usually presents with advanced disease, with 50% being unresectable or having radiological metastases at presentation. Two common forms are recognized: squamous cell carcinoma is more common in older smokers, and adenocarcinoma which is associated with gastro-oesophageal reflux, obesity and Barrett's metaplasia and accounts for 75% of cases in the UK. For squamous cell cancers, if the disease is localized chemoradiation followed by surgical resection or radical chemoradiation is used. For adenocarcinoma of the lower oesophagus tumours, combination chemotherapy with oxaliplatin, epirubicin and capecitabine is commonly given before and after surgery to improve cure rates.
Gastric cancer
Surgery alone can be curative for early disease, but the majority present with more advanced disease. For those considered suitable for radical surgical resection, neoadjuvant and adjuvant chemotherapy using a combination of epirubicin, oxaliplatin and capecitabine improves survival. Fluorouracil or capecitabine combined with oxaliplatin and epirubicin can be palliative in advanced disease (response rate of 65%). Aproximately 15% of tumours are HER-2 positive, and cisplatin, fluoropyrimidine and trastuzumab can be considered for palliative treatment.
Pancreatic cancer
Most pancreatic cancers present late and 5-year survival is rare because of metastatic progression. For operable tumours, surgery followed by adjuvant chemotherapy with gemcitabine or fluorouracil is the treatment of choice. For inoperable locally advanced tumours, chemoradiation with capecitabine is used. Metastatic disease can be treated with gemcitabine alone or a combination of fluorouracil with oxaliplatin and irinotecan.
Colorectal cancer
Surgery is the mainstay of treatment for people with colorectal cancer without metastatic disease. If there is a high risk of relapse (Duke's grade C or some grade B), a combination of oxaliplatin and fluoropyrimidine improves survival by 10–15%. Pre-operative chemoradiation reduces metastatic spread and for rectal cancer reduces local recurrence. Once a person has survived for 5 years, life expectancy is similar to that in the general population.
In advanced and metastatic colorectal cancer, fluorouracil or capecitabine, with or without oxapliplatin or irinotecan, improves survival and quality of life. The EGFR-targeted monoclonal antibodies cetuximab or panitumumab can be added to oxapliplatin or irinotecan-containing regimens in KRAS wild-type colorectal tumours, leading to increased response rates. Bevacizumab, a monoclonal antibody targetting VEGF, can also enhance the effect of standard chemotherapy.
Lung cancer
There are four principal types of lung cancer. Non-small-cell cancers (adenocarcinoma, squamous cell cancer and large-cell cancer) account for about three-quarters of cases, with small-cell cancer responsible for the remainder.
For non-small-cell lung cancer, superficial lesions are amenable to several treatments, including photodynamic therapy with porfimer sodium. Surgical resection can be curative in the early stages. Neoadjuvant or adjuvant chemotherapy with cisplatin combined with one of several other drugs improves survival if the tumour is resectable. Radiotherapy or chemoradiotherapy is used after surgery when the tumour is not fully resectable, or for palliation of metastases. Chemotherapy has a limited place for advanced or recurrent disease and is mainly palliative. The current gold standard is the combination of a platinum compound (carboplatin or cisplatin) with one of several newer generation drugs (pemetrexed, a taxane, vinorelbine, gemcitabine). More recently treatment has become stratified according to histological type: squamous cell carcinomas are best treated using a platinum compound with gemcitabine, and non-sqamous cell carcinomas using a platinum compound with pemetrexed.
About 15% of non-small cell lung cancers, mainly adenocarcinomas, carry an activating mutation in the epidermal growth factor receptor (EGFR) gene. These tumours are responsive to treatment with tyrosine kinase inibitors such as erlotinib and gefitinib, which have revolutionized the management of this subgroup of tumours and maintained remissions for years in some cases. Other treatment targets have been identified, such as the cMET/ALK translocation, which accounts for about 20% of non-squamous cell carcinomas and can be treated with crizotinib. A major area of progress has been immunotherapy with anti-PD1, with previously unheard-of disease control rates and survival in people with advanced lung cancer; it is expected that immunotherapy will play a major role in lung cancer management in the future.
Small-cell lung cancer is more sensitive to chemotherapy, and has an initial response rate of 60–70%, with complete remission in 20–30% of cases. Examples of regimens are cisplatin combined with etoposide or cyclophosphamide with doxorubicin and vincristine. Radiotherapy is also given for limited-stage disease.
Melanoma
Survival in melanoma is related to tumour thickness, with 5-year survival falling from more than 95% with superficial tumours to less than 50% survival if the depth is greater than 4 mm. Wide surgical excision is the treatment of choice. Postsurgical adjuvant chemotherapy does not improve survival or disease-free outcome. Currently, interferon alfa is the only drug shown to increase disease-free survival, but it has only a small impact (3–5%) on overall survival.
The management of advanced (inoperable) disease is determined by the presence or absence of a mutation in the BRAF gene (mutation V600E). Tumours with this mutation (40–50%) can be treated with a B-Raf tyrosine kinase inhibitor, vemurafenib, with initial disease control in about 80% of cases. However the effect is sometimes short-lived with a median progression-free survival of 6 months. This appears to be mainly due to escape mutations in the BRAF gene. To overcome this, combinations with inhibitors of another intracellular signalling protein (MEK protein) appear promising. Tumours with BRAF wild-type gene or which progress after B-Raf inhibition can be treated with the immunotherapeutic antibody ipilimumab, which produces responses over 5 years in just over 20% of cases. Chemotherapy with dacarbazine, temozolamide or the vinca alkaloids (vincristine or vinblastine) produces tumour responses in 10–20% of people, but without any clear effect on survival. Combination chemotherapy increases toxicity with no improvement in response. Immunotherapy with interferon alfa has produced similar response rates to chemotherapy, but the addition of chemotherapy does not further increase survival.
Renal cancer
Nephrectomy is the treatment of choice for early stage renal cancer, but up to one-third of people have metastases at the time of diagnosis. Prior to the introduction of tyrosine kinase inhibitors for the treatment of advanced disease, interferon alfa was commonly used with response rates in the region of 10–15%. In people with significant primary tumours and modest volume metastatic disease, cytoreductive nephrectomy was generally offered and improves survival, with a small proportion of metastases spontaneously regressing after surgery. With the advent of more active systemic agents the role of nephrectomy in the presence of metastatic disease is less clear. The majority of people with renal cell cancer have the clear cell variant which is associated with high expression of VEGF. Agents that target this pathway are effective in the treatment of metastatic disease.
Drugs that inibit multiple intracellular tyrosine kinases such as sorafenib, sunitinib or pazopanib are used as the first-line treatment for advanced disease. They produce high responses rates, with average duration of response of around 1 year.
Both the mammalian target of rapamycin (mTOR) inhibitor everolimus and the VEGF-targeted kinase inhibitor axitinib are used after failure of tyrosine kinase inhibitors. By sequencing first-, second- and sometimes third-line treatment for those with advanced kidney cancer, the disease can now be controlled for several years.
Immunotherapy with interferon alfa combined with bevacizumab or aldesleukin (interleukin-2) produces responses in about 15% of cases. The toxicity of treatment can be high, but for a small proportion of those treated extremely durable response can be achieved. More recently a peptide vaccine has shown encouraging effects on survival in those vaccinated at progression after first-line treatment with sunitinib or sorafenib.
Bladder cancer
Superficial bladder tumours are removed surgically, but recurrence rates are high. Intravesical immunotherapy with Bacillus Calmette–Guérin (BCG) vaccine is used to limit recurrence in superficial disease. For more advanced disease, neoadjuvant chemotherapy with gemcitabine and cisplatin improves survival. A bladder-sparing approach using transurethral resection followed by concurrent chemotherapy (cisplatin, methotrexate and vinblastine) and irradiation has given promising results for those who do not want cystectomy.
Prostate cancer
Treatment is largely determined by the extent of spread of the cancer. There are several options:
‘watchful waiting’ for localised disease confined to the prostate. This is usually used for individuals with a life expectancy under 10 years, since many tumours do not progress in this time,
radical prostatectomy for localised disease, usually in men under 70 years, in whom the risk of subsequent metastases is reduced from 25 to 15%. Impotence is a common sequel, occurring in 35–60% of cases,
radiotherapy for localised disease or locally advanced disease in older men. Impotence follows therapy in 40–60% of cases,
interstitial implantation of radioactive pellets for localised disease or locally advanced disease (brachytherapy),
hormonal therapy for lymph node involvement or distant metastases. Prostate cancer is hormone-dependent for growth. Testosterone reduction can be achieved by bilateral orchidectomy or the use of GnRH analogues such as leuprolide or goserelin (Ch. 43). Tumour flare reactions are prevented by the use of anti-androgen therapy (e.g. with flutamide or cyproterone acetate; Ch. 46) for the first few weeks to block adrenal androgen activity,
castration-resistant disease can be treated by chemotherapy with drugs such as docetaxel and cabazitaxel. Response rates of about 50% can be achieved. Painful metastatic deposits can be treated with radiotherapy or with strontium-89, which is taken up by sclerotic metastases,
a new class of hormonal therapies has shown promising activity in advanced prostate cancer. Abiraterone is a 17-α hydroxylase inhibitor which prevents extra-testicular synthesis of androgens. It has low toxicity and increases survival when used either before or after chemotherapy in those who have become resistant to castration levels of testosterone inhibition. New androgen receptor antagonists with enhanced potency such as enzalutamide also increase survival in people with castration-resistant disease.
Testicular cancer
Testicular tumours are either seminomas or non-seminomatous germ cell tumours, depending on the tissue of origin. Cure rates are now greater than 95%. For seminomas, treatment choice includes:
orchidectomy then follow-up for recurrence or chemotherapy with carboplatin if the recurrence risk is high,
for locally advanced disease, surgery is followed by radiotherapy, perhaps combined with carboplatin,
for metastatic disease, chemotherapy with bleomycin, etoposide and cisplatin (BEP) is used.
For non-seminomatous germ cell tumours, treatment choice includes:
Ovarian cancer
Initial surgery for ovarian cancer is followed by chemotherapy for all disease that is not localised to the ovary (which occurs in 80% of cases). About 70% of these women respond to chemotherapy, with complete remission in 10–20%. Carboplatin or cisplatin with paclitaxel is often used. Liposomal doxorubicin, radiotherapy and octreotide are among the options for more advanced disease.
Cervical cancer
Surgery is the mainstay for local disease, but chemoradiation is used if there are poor prognostic predictors or advanced disease. Cisplatin is most frequently used and improves survival by 30%. For recurrent disease the combination of cisplatin and paclitaxel has a small advantage over cisplatin alone.
Endometrial cancer
Surgery is the usual initial treatment for endometrial cancer. Adjuvant radiotherapy is given to the pelvis, and radiotherapy is also used for extrauterine metastases. Disseminated disease can be treated by hormone therapy with progestogens, but responses are low (less than one-third of those treated) and depend on the presence of progesterone receptors on the tumour cells. Adjuvant chemotherapy with drugs such as carboplatin plus paclitaxel has a palliative role in advanced disease.
Breast cancer
Breast-conserving surgery is the treatment of choice for very early disease and for oestrogen receptor-positive tumours; it is usually followed by local radiotherapy. The risk of invasive recurrence is low; if this occurs, it is treated by mastectomy followed by chemotherapy. Chemotherapy or hormonal therapy is used for larger locally invasive tumours or distant spread, or as neoadjuvant treatment for recurrence.
Determination of the hormone receptor status of the tumour is an important guide to the most appropriate hormonal therapy or chemotherapy.
Oestrogen receptor-positive tumours
Options for adjuvant hormonal therapy for postmenopausal women with oestrogen receptor-positive tumours include the following.
Non-steroidal aromatase inhibitors such as anastrazole or letrozole or the steroidal aromatase inhibitor exemestane are more effective than tamoxifen, which was long considered the treatment of choice. They can also be used as neoadjuvant therapy to reduce the extent of surgical resection.
Anti-oestrogen therapy, e.g. with tamoxifen, is often considered second-line treatment for hormone-responsive cancer. If tamoxifen is used, then switching to an aromatase inhibitor after 2–3 years further improves disease-free survival.
The selective oestrogen receptor downregulator (SERD) fulvestrant is an alternative second-line treatment for locally advanced or metastatic disease.
Progestogens such as megestrol acetate are used as a third-line treatment.
GnRH analogues such as goserelin (Ch. 43) are a fourth-line treatment.
For premenopausal women with oestrogen receptor-positive tumours:
tamoxifen remains the cornerstone of treatment, with or without chemotherapy. Aromatase inhibitors are ineffective before the menopause,
tamoxifen can be combined with ovarian ablation using a GnRH analogue such as goserelin.
Hormonal treatment for early disease is usually given for 5 years and reduces mortality by 30%, with continuing benefit after stopping treatment for 15 years. The benefit of extending treatment beyond 5 years is unproven. The 10–20% of women who become unresponsive to one hormonal treatment may still respond to the use of an alternative class of drug.
Oestrogen receptor-negative tumours
Chemotherapy (treatment which does not involve hormonal manipulation) is used for oestrogen receptor-negative tumours, HER2-positive tumours, and younger women (especially under 35 years but also up to 70 years of age) or for hormonally unresponsive disease. An example of a current regimen is doxorubicin or epirubicin with cyclophosphamide, combined with docetaxel for node-positive disease, which produces response rates of up to 40%. Trastuzumab can be added to chemotherapy for cancers that express HER2; it reduces early recurrence by 50% and improves survival by 25%. Trastuzumab can also be used as first-line therapy without cytotoxic drugs.
Acute myeloid leukaemias
The acute myeloid leukaemias are a heterogeneous group of disorders (Box 52.1) that are differentiated on morphological grounds. Acute myeloid leukaemia is responsible for up to 15% of childhood leukaemias and is the commonest leukaemia of adult life. Complications usually result from bone marrow failure, and management of serious infection or bleeding are important issues in supportive care. The risk of infection is amplified by chemotherapy. The initial aim of chemotherapy is to reduce ‘blast’ cells in the marrow to below 5% of the total cell population (remission) with induction therapy and then to eradicate the leukaemic cells with consolidation therapy, usually involving at least two or three cycles of additional treatment.
Intravenous chemotherapy with two or more drugs is used in the induction phase to reduce the development of resistance. A typical regimen consists of daunorubicin with cytarabine, which produces remission in 65–70% of individuals under 60 years old; older people have a less favourable response. Consolidation is achieved with further courses of similar therapy for three to four cycles. Haematopoietic stem cell transplantation may be considered after remission is achieved. In children, treatment for the central nervous system is also given with intrathecal methotrexate. Salvage treatment is used for failure to enter remission or for relapse, with high-dose cytarabine alone or combined with fludarabine.
For acute promyelocytic leukaemia, the best initial response is obtained with tretinoin (all-trans retinoic acid), a vitamin A derivative (see Ch. 49) and consolidation achieved by the addition of an anthracycline such as idarubicin or daunorubicin.
Acute lymphoblastic leukaemia
Acute lymphoblastic leukaemia is most common in children under 10 years of age, with a few cases occurring after age 40 years. Supportive therapy is similar to that for acute myeloid leukaemia.
Remission induction (eradication of 99% of leukaemic cell burden) is achieved with combinations of three or more drugs. In children, vincristine and prednisolone or dexamethasone with crisantaspase, doxorubicin or daunorubicin is often used. Four or more drugs are used for children with high-risk disease and for most adults. Cyclophosphamide is often used for T-cell leukaemias, and imatinib or desatinib if the cells are Philadelphia chromosome-positive. Consolidation therapy is initially with at least two multidrug intensification modules, using various combinations of corticosteroid with vincristine, crisantaspase, methotrexate and mercaptopurine. Continuation therapy is used after the first 5 months with mercaptopurine and methotrexate for at least 2 years. Eradication of cranial disease is important, using intrathecal methotrexate, cytarabine and hydrocortisone; cranial irradiation is less commonly used. Selective use of haematopoietic stem cell transplantation can further improve outcome.
The results of treatment in childhood are excellent, with about 80% survival at 5 years, compared to 40% if the disease occurs in adult life.
Chronic myeloid leukaemia
Chronic myeloid leukaemia occurs in all age groups but is rare in children. Most disease follows an initial chronic course, lasting 3–4 years, with subsequent transformation to an accelerated phase, when survival is just 3–6 months. Imatinib is standard treatment for the chronic phase, and achieves cytogenetic remission in up to 87% of people. Interferon alfa, usually given with cytarabine or hydroxycarbamide, is an alternative for those who do not tolerate imatinib. In younger people, allogeneic stem cell transplantation is the treatment of choice after failed chemotherapy.
For advanced disease with blast crisis, combination chemotherapy can be considered, such as the regimen used for acute myeloid leukaemia or acute lymphoblastic leukaemia depending on the type of transformation.
Chronic lymphocytic leukaemia
Chronic lymphocytic leukaemia is predominantly a disease of the elderly. Cure is unusual and median survival is 5–8 years, but treatment is given to control symptoms due to the disease. Therefore, treatment may not be necessary if the disease is causing few problems, but oral chlorambucil, often combined with prednisolone, can be given for up to 6 months to regress the disease. Transformation of the disease to a more aggressive form can occur after several years, with increasing disease bulk, lymphoma-related symptoms or bone marrow failure. Standard therapy in these situations is either oral or intravenous fludarabine or oral chlorambucil, with the goal of reducing leukaemic cells in the marrow to below 30%. Rituximab and cyclophosphamide both enhance the efficacy of fludarabine.
Malignant lymphomas
The malignant lymphomas are a diverse group of disorders comprising Hodgkin's disease and a variety of non-Hodgkin's lymphomas, which are classified by histopathological and cytochemical techniques. Low-grade non-Hodgkin's lymphomas are managed in a similar way to chronic lymphocytic leukaemia and have a similar prognosis. Non-Hodgkin's lymphomas of intermediate grade are curable in about 40% of cases, using courses of combination chemotherapy with cyclophosphamide, doxorubicin, vincristine and prednisolone (‘CHOP’ therapy, an acronym based on the generic and proprietary names of the drugs). Rituximab may improve survival when added to standard chemotherapy, and radiotherapy is sometimes used as adjunctive treatment, or for relapsed disease. More frequent, intensive therapy is required for high-grade, aggressive non-Hodgkin's lymphomas.
For Hodgkin's disease, radiotherapy is curative if the tumour is localised; combination chemotherapy is the usual approach for more extensive disease. The most frequently used regimen is doxorubicin, bleomycin, vinblastine and dacarbazine (ABVD).
Multiple myeloma
Multiple myeloma is mainly a disorder of the elderly. Treatment is aimed at suppression of the monoclonal protein in the blood. Supportive therapy is often required to treat hypercalcaemia, renal impairment and infection. Rehydration and analgesia for bone pain are often required. Radiotherapy may be used to treat localized areas of disease for pain control, lytic bone lesions or fractures.
Autologous stem cell transplantation is increasingly used as primary therapy after intensive chemotherapy and can produce 30–50% complete remission. Induction chemotherapy is with either two or three drugs, using regimens such as dexamethasone in combination with cyclophosphamide, bortezomib, thalidomide or lenalidomide.
For those who are not eligible for transplantation, chemotherapy is usually with oral melphalan and prednisolone combined with either thalidomide or bortezomib. This reduces the myeloma protein in blood by more than 50% in half of those treated. Median survival with this treatment is 3 years.
True/false questions
1. Cancer cells are not subject to the normal feedback mechanisms which restrict cell multiplication.
2. Resting cells in G0 phase are most susceptible to antineoplastic drugs.
3. Adverse effects of some anti-cancer drugs include secondary carcinogenesis.
4. Alkylating agents interfere with normal DNA synthesis.
5. Methotrexate competitively inhibits deoxythymidylate kinase.
6. Folinic acid (leucovorin) reverses the action of methotrexate.
7. Fluorouracil is a purine antagonist.
8. The effects of cytotoxic antibiotics are due to their intercalation between DNA bases.
9. Most base analogue antimetabolites are prodrugs activated by dephosphorylation.
10. Vinca alkaloids and taxanes share a common mechanism of action.
11. Tyrosine kinase inhibitors block signalling by growth factors.
12. Tamoxifen blocks oestrogen receptors on bone cells, causing osteoporosis.
Case-based questions
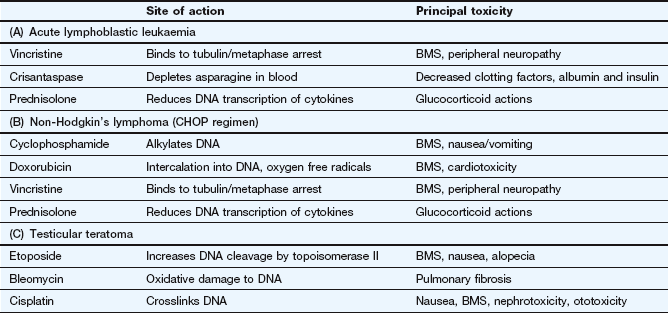
1. What are the criteria for combination chemotherapy of cancer? How well do the following treatment regimens meet the criteria?
A Acute lymphoblastic leukaemia (initial phase for induction of remission): intravenous vincristine, subcutaneous crisantaspase (asparaginase) and oral prednisolone.
B Non-Hodgkin's lymphoma: cyclophosphamide, doxorubicin, vincristine and prednisolone (CHOP regimen).
C Testicular teratoma in an adult: intravenous etoposide, bleomycin and cisplatin.
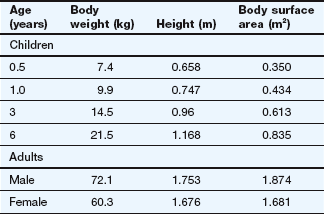
2. Why are doses of anti-cancer drugs corrected to surface area rather than body weight? Does the use of surface area correction result in higher or lower doses for children compared with simple correction for body weight? Taking the example in Table 52.1 of an adult male (body weight 72.1 kg) given 100 mg of a drug, what doses would a 1-year-old child of body weight 9.9 kg be given if corrected for body weight or for surface area?
1. True. Dysfunction in the mechanisms that normally regulate cell multiplication is a characteristic of cancer cells.
2. False. Although some anti-cancer drugs affect the resting phase, cancer cells are typically most susceptible when they are actively dividing; the sensitivity of a tumour therefore depends on its growth fraction.
3. True. Secondary cancers can occur such as bladder cancer with cyclophosphamide (due to urinary excretion of toxic metabolites) and lymphoma with alkylating agents.
4. True. The alkylated bases produce various effects on DNA function including misreading.
5. False. Methotrexate inhibits dihydrofolate reductase, a key enzyme in the folate pathway required for synthesis of purines and thymidine.
6. True. Leucovorin is used to rescue normal tissues when given after methotrexate; methotrexate has a preferential effect on cancer cells, and giving leucovorin 24 h afterwards overcomes its unwanted actions in normal tissues such as bone marrow and gut mucosa.
7. False. Fluorouracil is a pyrimidine analogue (fluorinated uracil).
8. True. As well as DNA intercalation, shown particularly by anthracyclines, cytotoxic antibiotics such as bleomycin also generate superoxide and hydrogen peroxide that cleave DNA.
9. False. Many base analogues such as cytarabine and gemcitabine are activated by intracellular phosphorylation to triphosphate derivatives, which are incorporated into DNA.
10. False. Vinca alkaloids prevent mitosis by inducing microtubule disassembly, whereas taxanes inhibit mitosis by promoting formation of stable but non-functional microtubules.
11. True. Drugs such as erlotinib, imatinib and pazopanib inhibit the receptor tyrosine kinase families associated with growth factors including epidermal growth factor (EGF), vascular endothelial growth factor (VEGF) and platelet-derived growth factor (PDGF); some also block kinases in downstream signalling pathways.
12. False. Tamoxifen is a selective oestrogen receptor modulator (SERM) that blocks oestrogen receptors on breast cancer cells but is an agonist at oestrogen receptors on bone cells, preventing osteoporosis.
Case-based answers
1. The criteria for combination therapy in cancer treatment are:
each drug should be active as a single agent; the ethics of clinical trials means that new drugs are not usually tested for this criterion in clinical studies,
each drug should have a different target within the cell; this increases cell kill and decreases drug resistance,
each drug should show different unwanted effects; ideally this will produce additive efficacy but not toxicity, and hence an increase in therapeutic index.
For each of the three drug regimens, the first criterion can be assumed to be met because all the agents are well-used drugs. The sites of action (second criterion) and side effects (third criterion) of the drugs in each regimen are shown in Table 52.2. All three regimens use drugs with different mechanisms of action, although regimen C is targeted only at DNA function. Regimen B contains three drugs that produce bone marrow toxicity and this will need careful monitoring during therapy.
2. Because many of the drugs used in cancer therapy are toxic at therapeutic doses, it is important to tailor the dosage to the individual. Children have a higher cardiac output and greater hepatic and renal blood flows than adults on a body weight basis. The clearance of drugs therefore tends to be faster in children than in adults and a proportionally higher dose is necessary to give the same blood levels. The liver and kidneys are essentially mature as the main organs of elimination by about 6–9 months of age. Hepatic and renal blood flows are related to body weight to the power of approximately 0.7. Since body surface area is also proportional to body weight0.7, it is usual to correct the drug doses by surface area to take better account of clearance. Surface area is calculated from a nomogram, or estimated using the approximation that it correlates with weight0.7, or by using the more accurate Du Bois equation:
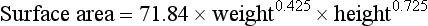
where surface area is in metres squared, weight is in kilograms and height is in metres. In the example, the adult male (body weight 72.1 kg) is given 100 mg of a drug. Simple correction for body weight of the 1-year-old child (9.9 kg) would suggest 13.7 mg (100 mg × 9.9 kg/72.1 kg) is the appropriate dose. However, using the surface area values in Table 52.1 derived from the Du Bois formula, the dose would be 100 mg × 0.434 m2/1.874 m2 = 23.2 mg. An approximation using surface area estimated as weight0.7 gives a calculated dose of 100 mg × 9.90.7/72.10.7 = 100 × 4.98/19.98 = 24.9 mg, close to that obtained using the Du Bois formula. These results appear counter-intuitive if children are assumed to be ‘more sensitive’ to drugs.
Compendium: drugs used in the treatment of cancer
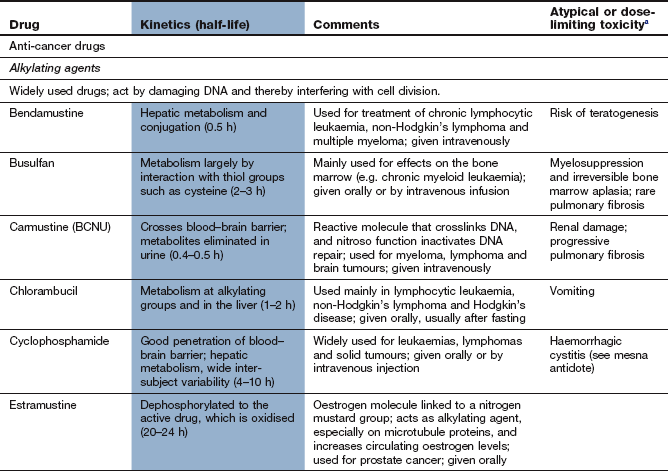

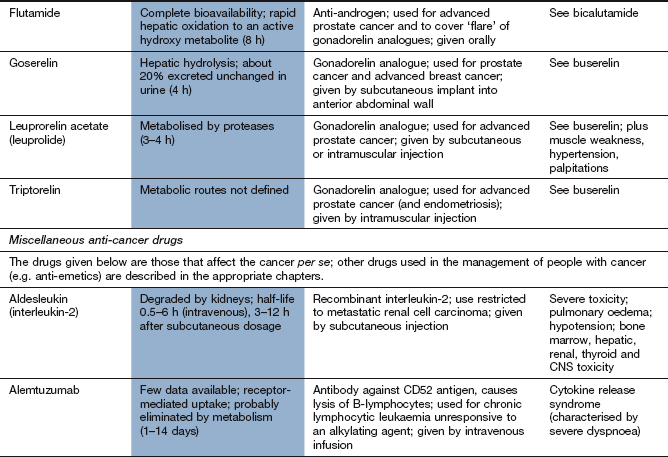
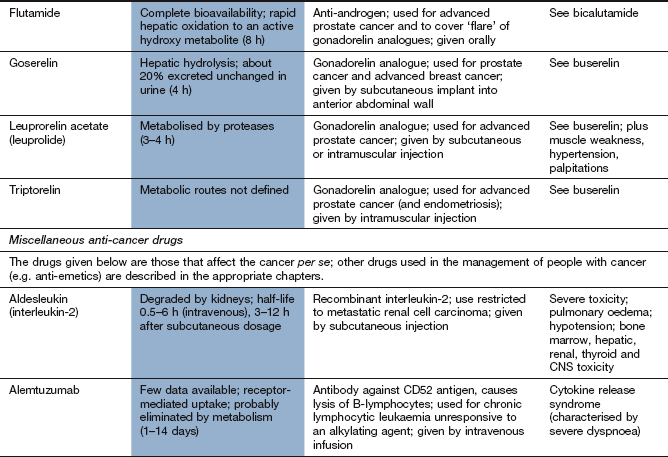
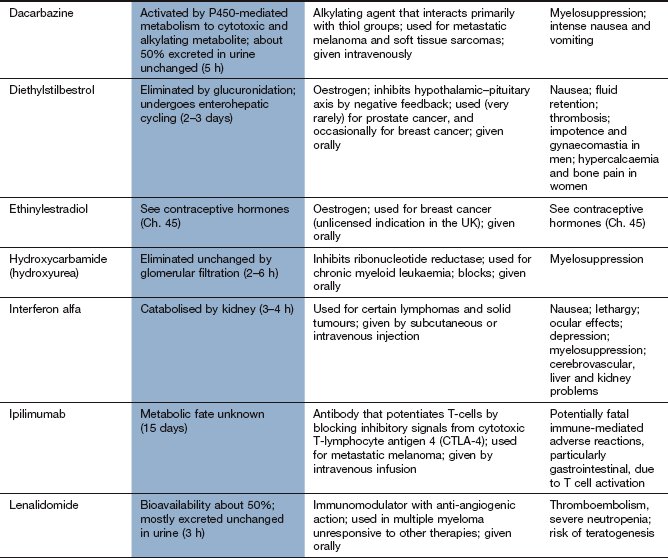
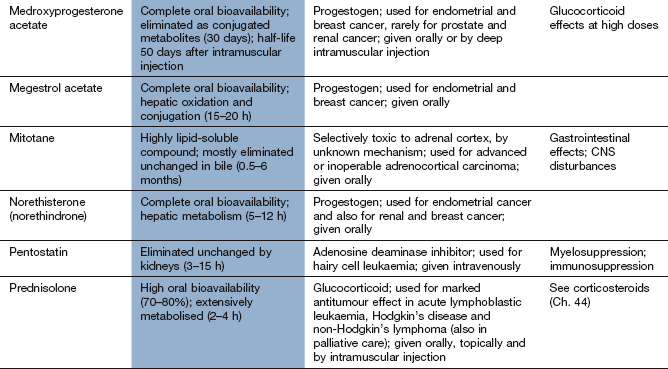
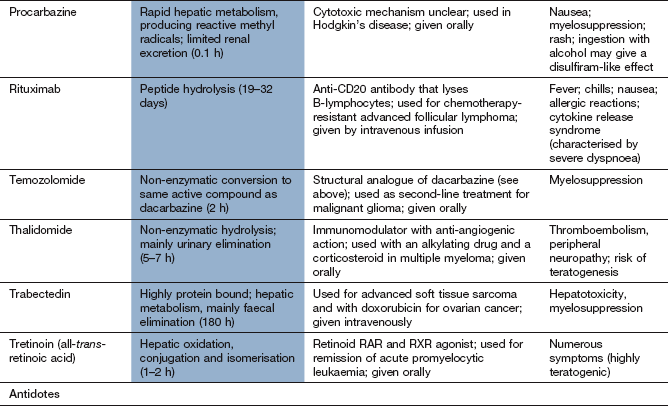
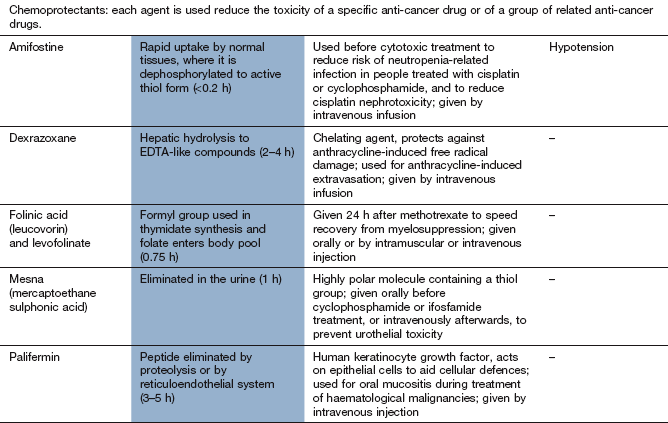
EGFR, epidermal growth factor receptor; mTOR, mammalian target of rapamycin; PDGFR, platelet-derived growth factor receptor; RAR, retinoic acid receptor; RXR, retinoid X receptors; VEGFR, vascular endothelial growth factor receptor.
aTypical toxicity for the class of drug is described in the general text; toxicity given in this column represents ‘non-class’ effects and/or severe dose-limiting toxicity.
Ambudkar, SV, Dey, S, Hrycyna, CA, Ramachandra, M, Pastan, I, Gottesman, MM. Biochemical, cellular, and pharmacological aspects of the multidrug transporter. Annu Rev Pharmacol Toxicol. 1999;39:361–398.
Ciardiello, F, Tortora, G. EGFR antagonists in cancer treatment. N Engl J Med. 2008;358:1160–1174.
Croce, CM. Oncogenes and cancer. N Engl J Med. 2008;358:502–511.
Dubowchik, GM, Walker, MA. Receptor-mediated and enzyme-dependent targeting of cytotoxic anticancer drugs. Pharmacol Ther. 1999;83:67–123.
Eccles, SA, Welch, DR. Metastasis: recent discoveries and novel treatment strategies. Lancet. 2007;369:1742–1757.
Efferth, T, Volm, M. Pharmacogenetics for individualized cancer chemotherapy. Pharmacol Ther. 2005;107:155–176.
Gottesman, MM. Mechanisms of cancer drug resistance. Annu Rev Med. 2002;53:615–627.
Griffioen, AW, Molema, G. Angiogenesis: potential for pharmacologic intervention in the treatment of cancer, cardiovascular diseases and chronic inflammation. Pharmacol Rev. 2000;52:237–268.
Links, M, Lewis, C. Chemoprotectants: a review of their clinical pharmacology and therapeutic efficacy. Drugs. 1999;57:293–308.
Marsh, S, McLeod, HL. Cancer pharmacogenetics. Br J Cancer. 2004;90:8–11.
Ballinger, AB, Anggiansah, C. Colorectal cancer. BMJ. 2007;335:715–718.
Hartgrink, HH, Jansen, EPM, van Grieken, NCT, et al. Gastric cancer. Lancet. 2009;374:477–490.
Lagergren, J, Lagergren, P. Oesophageal cancer. BMJ. 2010;341:c6280.
Meyerhardt, JA, Mayer, RJ. Systemic therapy for colorectal cancer. N Engl J Med. 2005;352:476–487.
Hidalgo, M. Pancreatic cancer. New Engl J Med. 2010;362:1605–1617.
Booton, R, Jones, M, Thatcher, N. Lung cancer 7: management of lung cancer in elderly patients. Thorax. 2003;58:711–720.
Cullen, M. Lung cancer 4: chemotherapy for non-small cell lung cancer: the end of the beginning. Thorax. 2003;58:352–356.
Jackman, DM, Johnson, BE. Small-cell lung cancer. Lancet. 2005;366:1385–1396.
Spira, A, Ettinger, DS. Multidisciplinary management of lung cancer. N Engl J Med. 2004;350:379–392.
Dahut, N, Gulley, JL, Dahut, WL. Androgen deprivation therapy for prostate cancer. JAMA. 2005;294:238–244.
Feldman, DR, Bosl, GJ, Sheinfeld, J, et al. Medical treatment of advanced testicular cancer. JAMA. 2008;299:672–684.
Hennessy, BT, Coleman, RL, Markman, M. Ovarian cancer. Lancet. 2009;374:1371–1382.
Hernandez, J, Thompson, IM. Diagnosis and treatment of prostate cancer. Med Clin North Am. 2004;88:267–279.
Horwich, A, Shipley, J, Huddart, R. Testicular germ-cell cancer. Lancet. 2006;367:754–765.
Kaufman, DS, Shipley, WU, Feldman, AS. Bladder cancer. Lancet. 2009;374:239–249.
Petignat, P, Roy, M. Diagnosis and management of cervical cancer. BMJ. 2007;335:765–768.
Rini, BI, Campbell, SC, Escudier, B. Renal cell carcinoma. Lancet. 2009;373:1119–1132.
Saso, S, Chatterjee, J, Georgiou, E, et al. Endometrial cancer. BMJ. 2011:d3954.
Waggoner, SE. Cervical cancer. Lancet. 2003;361:2217–2225.
Walsh, PC, DeWeese, TL, Eisenberger, MA. Localized prostate cancer. N Engl J Med. 2007;357:2696–2705.
Wilt, TJ, Thompson, IM. Clinically localized prostate cancer. BMJ. 2006;333:1102–1106.
Benson, JR, Jatoi, I, Keisch, M, et al. Early breast cancer. Lancet. 2009;373:1463–1479.
Turner, NC, Jones, AL. Management of breast cancer – Part 1. BMJ. 2008;337:107–110.
Turner, NC, Jones, AL. Management of breast cancer – Part 2. BMJ. 2008;337:164–169.
Eggermont, AMM. European approach to the treatment of malignant melanoma. Curr Opin Oncol. 2002;14:205–211.
Estey, E, Döhner, H. Acute myeloid leukaemia. Lancet. 2006;368:1894–1907.
Pui, C-H, Robinson, LL, Look, AT. Acute lymphoblastic leukaemia. Lancet. 2008;371:1030–1043.
Ravandi, F, Kantarajian, H, Giles, F, et al. New drugs in acute leukemia and other myeloid disorders. Cancer. 2004;100:441–454.
Dighiero, G, Hamblin, TJ. Chronic lymphocytic leukaemia. Lancet. 2008;371:1017–1029.
Hehlmann, R, Hochhaus, A, Baccarani, M, et al. Chronic myeloid leukaemia. Lancet. 2007;370:342–350.
Shanafelt, TD, Byrd, JC, Call, TG, et al. Narrative review: initial management of newly diagnosed, early-stage chronic lymphocytic leukaemia. Ann Intern Med. 2006;145:435–447.