Heart failure
There is no universally accepted definition of heart failure. The heart failure syndrome is usually said to exist when there is inadequate oxygen delivery to peripheral tissues, either at rest or during exercise, due to dysfunction of the heart or when adequate oxygen delivery can only be maintained with an elevated left ventricular filling pressure.
Maintenance of cardiac output
There are four major determinants of cardiac output:
 preload: this is governed by the ventricular end-diastolic volume, which in turn is related to ventricular filling pressure and therefore to venous return of blood to the heart,
preload: this is governed by the ventricular end-diastolic volume, which in turn is related to ventricular filling pressure and therefore to venous return of blood to the heart,
afterload: the systolic wall tension in the ventricle; this reflects the resistance to ventricular emptying within both the heart (during isovolumic contraction) and the peripheral circulation.
The output from both right and left sides of the heart is normally balanced. In the healthy heart, cardiac output is regulated mainly by changes in heart rate and preload. Heart rate is modulated by the autonomic nervous system, with sympathetic nervous stimulation increasing heart rate and parasympathetic stimulation via the vagus nerve slowing the rate.
The relationship between preload and stroke volume (the amount of blood ejected from the ventricle during systole with each contraction) is shown in Figure 7.1. The degree of stretch of the ventricular muscle (preload) determines the force of cardiac contraction (the Frank–Starling phenomenon). The curve describing this relationship is governed by intrinsic myocardial contractility: thus, the curve is shifted upwards and to the left when contractility is augmented, for example by sympathetic nervous stimulation. In a healthy heart with normal myocardial contractility, the left ventricular filling pressure lies on the steep part of the curve, making stroke volume very sensitive to small changes in preload.
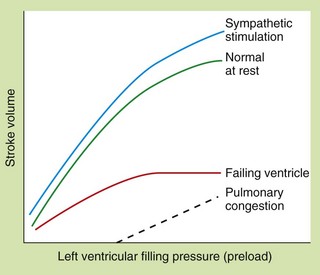
Fig. 7.1 The Frank–Starling relationship between preload (left ventricular filling pressure) and stroke volume in healthy and failing hearts.
In the severely failing heart, increases in filling pressure and heart rate are insufficient to restore cardiac output, and pulmonary congestion will occur.
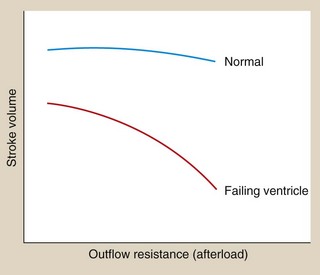
The relationship between afterload and stroke volume is shown in Figure 7.2. Afterload is determined largely by peripheral resistance but also by the size of the ventricle. Enlargement of the left ventricular cavity (e.g. as a result of increased venous return or preload) increases wall tension, and the heart must generate greater pressure both to initiate and to maintain contraction. Preload and afterload are therefore interrelated. In the healthy ventricle a rise in afterload will cause a fall in stroke volume, but the consequent sympathetic stimulation will increase myocardial contractility and maintain stroke volume.
Pathophysiology of heart failure
Heart failure is a syndrome that has several underlying causes (Box 7.1). Occasionally it arises suddenly such as after acute myocardial infarction or acute mitral regurgitation from rupture of the chordae tendineae. More commonly the onset is gradual from progressive loss of myocardial function or slow degenerative change in valve function.
The underlying problem in heart failure is reduced cardiac output and therefore low blood pressure, but the syndrome of heart failure arises largely from neurohumoral counter-regulation in response to low blood pressure and reduced renal perfusion. This is principally due to the sympathetic nervous system and the renin–angiotensin–aldosterone system (Fig. 7.3). The consequences of these compensatory mechanisms are vasoconstriction of both arteries and veins and excessive salt and water retention by the kidneys. Although these are the normal physiological responses to reduced blood pressure, in the setting of a failing heart they can create additional problems.
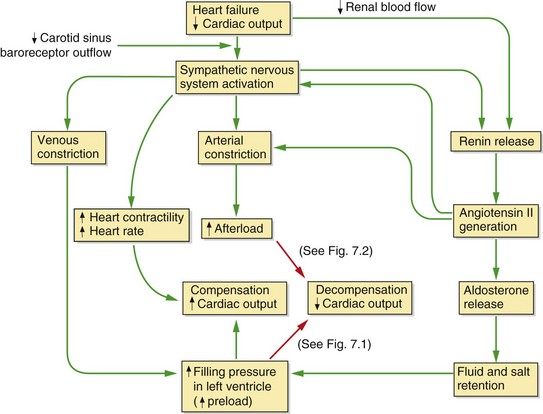
Fig. 7.3 Neurohumoral consequences of heart failure.
In the mildly impaired heart a fall in cardiac output results in a cascade of compensatory events (green arrows), including sympathetic stimulation of heart rate and contractility, constriction of arteries and veins, and activation of the renin–angiotensin–aldosterone system; overall these compensatory mechanisms restore cardiac output. If cardiac function is significantly impaired (red arrows), an increased preload cannot restore an adequate stroke volume (decompensation) (see Fig. 7.1) and the increased afterload will put additional strain on the failing heart and further decrease cardiac output (see Fig. 7.2). In chronic heart failure these effects are compounded by cardiac remodelling and downregulation of cardiac β1-adrenoceptors.
In the failing ventricle, the Frank–Starling curve is shifted downwards and to the right (failing-ventricle curve, Fig. 7.1) and the maximum achievable stroke volume is reduced. The curve is also flatter, indicating that stroke volume has become less responsive to changes in preload. As a result of activation of the compensatory mechanisms, salt and water retention expands plasma volume and venoconstriction enhances venous return to the heart. These factors increase the filling pressure of the left ventricle in an attempt to restore the resting stroke volume. Heart rate will also increase, which will raise cardiac output despite a lower stroke volume. If these responses are successful in restoring a normal resting cardiac output the heart failure is said to be compensated. However, the cardiac output may be unable to rise to meet the needs of the body during exertion.
Decompensation occurs when the combination of the increases in preload and heart rate fail to restore a normal resting cardiac output (Fig. 7.3). A persistent high level of symapathetic tone results in downregulation of β1-adrenoceptors and therefore less ability to maintain cardiac output. In most cases of heart failure, the impairment of function initially affects the left ventricle. As the central blood volume continues to increase in an attempt to raise the stroke volume, the hydrostatic pressure in the pulmonary veins will rise. When the hydrostatic pressure in the pulmonary circulation exceeds the plasma colloid osmotic (oncotic) pressure that holds fluid in the blood vessel, fluid leaves the capillaries into the interstitium of the alveoli and then into the alveolar spaces, producing pulmonary oedema (Fig. 7.2). Eventually, the raised pulmonary vascular pressure leads to right heart failure (producing biventricular failure, or congestive cardiac failure), and oedema develops in the peripheral and splanchnic tissues.
Peripheral arterial resistance (afterload) will also rise as a result of the compensatory mechanisms (Fig. 7.2). The failing ventricle cannot meet this with an increase in myocardial contractility so stroke volume will fall (Fig. 7.2) with further cardiac decompensation.
Heart failure arising from myocyte loss (such as occurs with myocardial infarction or cardiomyopathies) leads to adaptive changes in the surviving cells and extracellular matrix, known as remodelling. Remodelling is driven by several factors including local effects of catecholamines, angiotensin II, aldosterone and pro-inflammatory cytokines. This eventually produces a more globular, dysfunctional left ventricle. This type of heart failure is characterized by a reduced ejection fraction (heart failure with reduced ejection fraction or systolic heart failure).
In aortic or mitral valve regurgitation, heart failure arises because the left ventricle must accommodate the normal forward stroke volume and also the regurgitant volume (the volume leaking back into the left ventricle or left atrium respectively). Eventually, the left ventricle cannot enlarge sufficiently to maintain an effective stroke volume.
Heart failure can also arise from impaired diastolic relaxation even when contractile function in systole is normal. Stroke volume is reduced, but ejection fraction is normal. This is known as heart failure with preserved ejection fraction or diastolic heart failure. If the left ventricle fails to relax adequately, it will not accommodate the venous return, leading to pulmonary venous congestion and a low cardiac output, activating the same compensatory neurohumoral responses. Heart failure with preserved ejection fraction characteristically occurs in older people in association with left ventricular hypertrophy, but it also contributes to heart failure in ischaemic left ventricular dysfunction (see Ch. 5).
Symptoms in heart failure are caused by a reduced cardiac output (‘forward failure’) or venous congestion (‘backward failure’). The most common complaints are breathlessness from increased pulmonary venous pressure, and fatigue resulting from the reduced cardiac output and impaired skeletal muscle perfusion. In response to the reduced perfusion, biochemical changes also occur in skeletal muscle, making it less efficient. Other symptoms, such as the discomfort of peripheral oedema and anorexia due to bowel congestion, are attributable to a high systemic venous pressure. Increased stimulation of β-adrenoceptors in the heart can lead to life-threatening ventricular arrhythmias.
Acute left ventricular failure
Acute left ventricular failure usually results from a sudden inability of the heart to maintain an adequate cardiac output and blood pressure. It can follow acute myocardial infarction, acute mitral or aortic valvular regurgitation, or arise from the onset of a brady- or tachyarrhythmia if there is pre-existing poor left ventricular function. The sudden fall in cardiac output leads to reflex arterial and venous constriction (Fig. 7.3). There is a rapid rise in filling pressure of the left ventricle as a result of increased venous return. If the heart is unable to expel the extra blood, the hydrostatic pressure in the pulmonary veins rises until it exceeds the plasma oncotic pressure and produces acute pulmonary oedema. The principal symptom is breathlessness, usually at rest with orthopnoea.
Cardiogenic shock
The syndrome of cardiogenic shock arises when the systolic function of the left ventricle is suddenly impaired to such a degree that there is insufficient blood flow to meet resting metabolic requirements of the tissues. This definition excludes shock caused by hypovolaemia. The clinical hallmarks are a low systolic blood pressure (usually <90 mmHg), with a reduced cardiac output and an elevated left ventricular filling pressure. Cardiogenic shock can follow acute myocardial infarction, and in this situation usually indicates loss of at least 40% of the left ventricular myocardium. Other mechanical disturbances, such as acute mitral regurgitation or ventricular septal rupture, can produce cardiogenic shock in association with a lesser degree of myocardial damage. Less commonly, the syndrome is associated with right ventricular infarction. The mortality of cardiogenic shock, even with intensive treatment, is in excess of 70%.
Chronic heart failure
Myocardial damage from ischaemic heart disease is the most common cause of chronic heart failure, but potentially correctable causes such as valvular lesions, as well as treatable exacerbating factors such as anaemia or arrhythmias, may be identified. In most people with heart failure there are signs of both right and left ventricular failure (biventricular or congestive heart failure). Chronic heart failure is not a trivial complaint. People with left ventricular systolic dysfunction who have symptoms only on exertion have a 2-year mortality risk of about 20%, whereas the 1-year mortality is 80% if there are symptoms at rest. In heart failure with reduced ejection fraction the degree of left ventricular dysfunction is a guide to prognosis. Death is from either progressive heart failure or ventricular arrhythmias.
Positive inotropic drugs in the treatment of heart failure
Myocardial contractility can be improved by increasing the availability of free intracellular Ca2+ to interact with contractile proteins, or by increasing the sensitivity of the myofibrils to Ca2+. Only drugs that increase myocardial intracellular Ca2+ are established in clinical use; they work by one of two distinct mechanisms:
an action on the cell membrane Na+/K+-ATPase pump (e.g. digitalis glycosides),
by increasing intracellular cAMP (e.g. sympathomimetic inotropes, phosphodiesterase inhibitors).
An additional advantage of the positive inotropic drugs that increase myocardial cAMP is their ability to enhance the reuptake of Ca2+ by the sarcoplasmic reticulum in diastole. This improves diastolic relaxation in addition to augmenting systolic contractility.
Digitalis glycosides
Mechanism of action and effects
Effect on myocardial contractility: Digitalis glycosides are compounds with a steroid nucleus that were originally isolated from a species of foxglove (Digitalis purpura). Digoxin is the drug that is most widely used in clinical practice. Digoxin binds to the energy-dependent Na+ pump (Na+/K+-ATPase) in the myocyte membrane. This pump establishes and maintains the Na+ and K+ gradients across the cell (Fig. 7.4), producing low intracellular Na+ and high intracellular K+ concentrations. A separate passive transmembrane exchange of Na+ and Ca2+ occurs down their concentration gradients, with Na+ entering the cell while Ca2+ is translocated out. The rate of this exchange is dependent on the intracellular Na+ concentration. Digoxin partially inhibits the Na+/K+-ATPase, which increases the intracellular Na+ concentration. This lowers the concentration gradient for Na+ across the cell membrane, which in turn reduces the Na+/Ca2+ exchange so that Ca2+ is retained in the cell. The excess intracellular Ca2+ is stored in the sarcoplasmic reticulum during diastole and released during cell membrane excitation, leading to enhanced myocardial contraction.
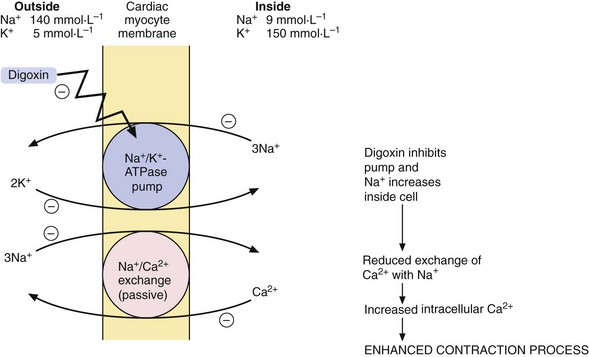
Effects on cardiac action potential and intracardiac conduction: Digoxin can be arrhythmogenic, but also has actions that are useful for treating certain arrhythmias.
Direct actions of digoxin on the heart can provoke arrhythmias by increasing myocardial cell excitability (Ch. 8), as follows.
Reduction of the resting membrane potential. The cell membrane Na+/K+-ATPase pump extrudes three Na+ out of the cell for every two K+ that enter, which increases the negative intracellular electrical potential and hyperpolarises the cell, making it less excitable (see Ch. 8). Inhibition of this membrane pump by digoxin leads to the cell membrane potential becoming less negative and closer to the threshold potential for depolarisation. Arrhythmias are therefore more readily initiated.
Triggering of spontaneous release of Ca2+ from the sarcoplasmic reticulum. This leads to transient depolarisation of the cell immediately following an action potential (‘after-potentials’), which can initiate arrhythmias (Ch. 8).
Digoxin also has useful indirect actions on the heart that arise from stimulation of the central vagal nucleus and enable it to be used for treating arrhythmias (Ch. 8). The vagal effects on the heart are:
decreased automaticity of the sinoatrial node which slightly slows heart rate in sinus rhythm,
increased refractory period of the atrioventricular node, which slows impulse transmission to the ventricles and is useful in the management of the fast ventricular rates that result from atrial flutter and fibrillation (Ch. 8).
Pharmacokinetics
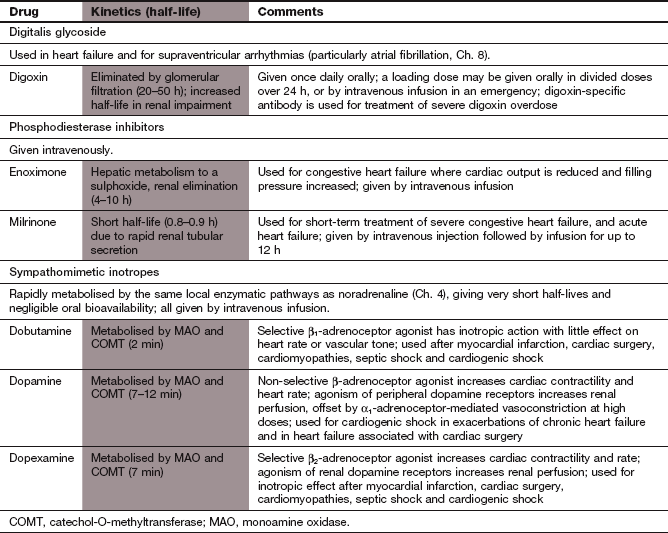
Digoxin is well absorbed from the gut and the kidney is the main route of elimination, by filtration at the glomerulus and by active tubular secretion. The half-life of digoxin is very long (about 1.5 days) and is increased if renal function is impaired. The dose must be reduced in the presence of renal impairment to avoid toxicity (see below). If an early onset of action is required, loading doses should be given over the first 24 or 36 h (Ch. 2). If a rapid response is essential, digoxin can be given by slow intravenous injection.
Unwanted effects
Digitalis glycosides have a narrow therapeutic index, and toxicity is mostly dose-related.
Gastrointestinal disturbances: anorexia, nausea and vomiting (largely a central effect at the chemoreceptor trigger zone; Ch. 32), and diarrhoea.
Neurological disturbances: fatigue, malaise, confusion, vertigo, coloured vision (especially yellow halos around lights, possibly from inhibition of Na+/K+-ATPase in the cones of the retina).
Distinctive changes on the ECG: this includes non-specific T-wave changes and sagging of the S–T segment with an upright T-wave (‘reverse tick’) often referred to as ‘digoxin effect’. These ECG effects do not indicate toxicity, but can be mistaken for myocardial ischaemia.
Consequences of intracellular Ca2+ overload: increased excitability of the atrioventricular node and Purkinje fibres produces atrial or nodal ectopic beats, atrial or nodal tachycardia, ventricular ectopic beats or (less commonly) ventricular tachycardia.
Consequences of increased vagal activity: excessive atrioventricular nodal block (‘heart block’) can occur. When associated with increased atrial excitability, this produces atrial tachycardia with atrioventricular nodal block, a rhythm characteristic of digitalis toxicity.
Gynaecomastia: during long-term treatment the steroid structure allows digitalis glycosides to bind to, and stimulate oestrogen receptors in breast tissue.
Exacerbating factors for digitalis glycoside toxicity
Hypokalaemia: reduced extracellular K+ concentration increases the effects of digitalis glycosides on the Na+/K+-ATPase pump. Care must be taken if potassium-losing diuretics, such as furosemide (Ch. 14), are used with digitalis glycosides.
Renal impairment: reduces the excretion of digoxin.
Hypoxaemia: this sensitises the heart to digitalis glycoside-induced arrhythmias.
Hypothyroidism: the renal elimination of digoxin is decreased because of a reduced glomerular filtration rate.
Drugs that displace digoxin from tissue binding sites and interfere with its renal excretion: these include verapamil (Ch. 5) and quinidine (Ch. 8), which can double the plasma concentration of digoxin. Amiodarone (Ch. 8) produces a less marked effect.
Treatment of digitalis toxicity
Digitalis glycoside toxicity can be treated by:
withholding further doses of digitalis glycoside,
using K+ supplementation (Ch. 14) for hypokalaemia. This is usually given orally, but should be given by slow intravenous infusion if there are dangerous arrhythmias,
using atropine (Ch. 8) for sinus bradycardia or atrioventricular block. Temporary transvenous pacing is used for marked bradycardia unresponsive to atropine,
digoxin-specific antibody fragments for serious digoxin toxicity (Ch. 53).
Sympathomimetic inotropes
Mechanisms of action and effects
The mechanisms of action of the inotropic sympathomimetic drugs are also considered in Chapter 4. Noradrenaline and adrenaline are not used for their inotropic action because they produce marked vasoconstriction through effects on α-adrenoceptors.
Isoprenaline is a non-selective β-adrenoceptor agonist that is now only available in the UK on special order. It is sometimes used to increase heart rate in the emergency treatment of bradycardias through its action on both β1- and β2-adrenoceptors (Ch. 8). Dobutamine, a synthetic dopamine analogue, is a selective β1-adrenoceptor agonist that produces a powerful inotropic response, with relatively less increase in heart rate than isoprenaline and little direct effect on vascular tone, even at high concentrations. Dopexamine acts on β2-adrenoceptors to increase heart rate and to vasodilate, and, to a lesser extent, on β1-adrenoceptors, giving a weak direct positive inotropic effect. It also acts on peripheral dopamine receptors and produces some increase in renal blood flow, but, unlike dopamine, it does not cause peripheral vasoconstriction with high doses.
Dopamine has dose-related actions at several receptors.
At low doses, it selectively stimulates peripheral dopamine receptors, which are structurally distinct from those in the central nervous system. This produces renal arterial vasodilation and diuresis (D1 receptors) and peripheral arterial vasodilation (D2 presynaptic receptors, which inhibit noradrenaline release from sympathetic nerves).
At moderate doses, non-selective β-adrenoceptor stimulation produces a positive inotropic response (Fig. 7.5). Tachycardia is more marked than with dobutamine, because of stimulation of both cardiac β1- and β2-adrenoceptors and the reflex response to β2-adrenoceptor-mediated peripheral arterial dilation.
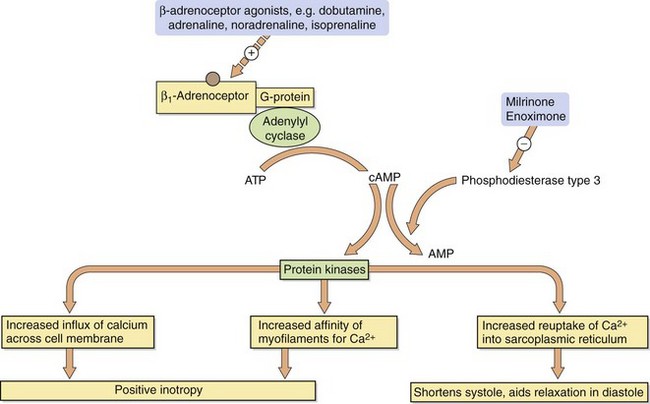
Fig. 7.5 Mechanisms by which sympathomimetics and phosphodiesterase inhibitors exert their positive inotropic effects.
At high doses, α1-adrenoceptor stimulation produces peripheral vasoconstriction, which also affects the renal arteries and overcomes D1-receptor-mediated renal vasodilation.
The doses of dopamine that produce these different effects differ widely among individuals. Unfortunately there is no dose that can be relied upon to act selectively at dopamine receptors without stimulating adrenoceptors.
Pharmacokinetics
All sympathomimetic inotropes are administered by intravenous infusion because of their very short half-lives (<12 min). Metabolic inactivation is by the same monoamine oxidase (MAO) and catechol-O-methyltransferase (COMT) pathways as for noradrenaline (Ch. 4). Desensitisation and downregulation of β-adrenoceptors (Ch. 1) rapidly reduce the response to sustained infusions over 48–72 h. Because of its unpredictable vasoconstrictor actions, dopamine is usually given into a large central vein.
Phosphodiesterase inhibitors
Mechanism of action and effects
Milrinone and enoximone are specific inhibitors of the isoenzyme of phosphodiesterase (type 3) found in cardiac and smooth muscle. Their inotropic action on the heart results from an increase in intracellular cAMP with increased mobilisation of intracellular Ca2+ (Fig. 7.5). Unlike β-adrenoceptor agonists, the activity of phosphodiesterase inhibitors is not limited by desensitisation of cell surface receptors, because they act at a site beyond the receptor. Since they have complementary sites of action, phosphodiesterase inhibitors and β-adrenoceptor agonists will have additive effects on the heart. Phosphodiesterase type 3 inhibition in vascular smooth muscle produces peripheral arterial vasodilation.
Pharmacokinetics
Phosphodiesterase inhibitors are only given for short-term treatment by intravenous infusion. Milrinone is eliminated by the kidney and enoximone by hepatic metabolism. They have elimination half-lives of about 1 h.
Management of heart failure
Acute left ventricular failure
The immediate aim of pharmacological treatment in acute left ventricular failure is to reduce the excessive venous return to the heart. Treatment includes:
oxygen in high concentration via a facemask,
intravenous opioid analgesic such as morphine (Ch. 19), often given to relieve both distress and breathlessness,
intravenous injection of a loop diuretic such as furosemide (Ch. 14). This initially produces venodilation, which increases peripheral venous pooling. Symptoms are therefore improved even before the onset of a diuresis that reduces plasma volume and further decreases preload,
sublingual glyceryl trinitrate (GTN; Ch. 5). This dilates venous capacitance vessels and is a useful alternative or additional emergency treatment to diuretics.
Whenever possible, a precipitating or exacerbating cause should be treated; for example, arrhythmias, anaemia, thyrotoxicosis, acute mitral regurgitation or critical aortic stenosis. However, if the primary cause is impairment of left ventricular systolic function, then management as for chronic heart failure is subsequently necessary.
Cardiogenic shock
The immediate aim of treatment is resuscitation, while looking for a remediable cause. If appropriate, early coronary revascularisation is crucial to increase the probability of survival. Supportive measures include the following:
Give oxygen in high concentration via a facemask.
Correct any acid–base imbalance (especially acidosis) and electrolyte abnormalities (particularly hypokalaemia).
Relieve pain, usually with an intravenous opioid analgesic such as morphine (Ch. 19).
Correct any cardiac rhythm disturbance (Ch. 8).
Ensure an adequate left ventricular filling pressure. This can be low after right ventricular infarction, despite a high central venous pressure (right ventricular filling pressure). If intravenous volume is adequate but tissue perfusion remains impaired, dobutamine is the inotropic drug of choice. Dobutamine is often given in combination with low-dose dopamine, with the intention that the dopamine will improve renal perfusion; however, there is little evidence that the addition of dopamine is beneficial.
Phosphodiesterase inhibitors are sometimes given to improve myocardial contractility and to produce peripheral vasodilation. They are usually reserved for those who fail to improve with maximum tolerated doses of dobutamine.
When there is profound hypotension, noradrenaline (norepinephrine) (Ch. 4) can be infused intravenously to produce α1-adrenoceptor-mediated peripheral vasoconstriction and maintain vital organ perfusion. The potential disadvantage is that the increase in peripheral resistance will further impair cardiac output. Vasopressin (Ch. 43) is sometimes given to raise blood pressure, as vascular sensitivity to noradrenaline is impaired in shock. Vasopressin is a vasoconstrictor that also increases vascular sensitivity to noradrenaline.
Vasodilators can be given to ‘offload’ the heart once an adequate blood pressure has been established. This strategy is particularly helpful if there is significant mitral regurgitation, since reduced resistance to left ventricular emptying will diminish the regurgitant volume. Either glyceryl trinitrate (Ch. 5) or nitroprusside (Ch. 6) is used.
Chronic heart failure with reduced ejection fraction
Much of the treatment of chronic heart failure is directed towards counteracting the compensatory mechanisms for the reduced cardiac output and low blood pressure generated by a failing heart; that is, arterial and venous vasoconstriction and fluid retention. When there is a reduced ejection fraction, a further desirable action is to reduce or reverse the shape change (remodelling) that occurs in the failing ventricle and makes contraction less efficient. Overall, treatment of heart failure with reduced ejection fraction has two main aims: symptom relief and improved prognosis.
Non-pharmacological treatment
A number of lifestyle changes can be helpful.
Weight reduction should be encouraged for an obese person; this improves exercise tolerance.
Bed rest may be appropriate to rest the heart during acute episodes of fluid retention.
Modest salt restriction is desirable (severe salt restriction is unpleasant and unnecessary).
Fluid restriction is rarely required unless profound hyponatraemia accompanies severe oedema. In this situation, diuretics may be ineffective until the plasma Na+ concentration is corrected.
If possible, drugs that exacerbate heart failure by producing myocardial depression (e.g. most calcium channel blockers) or by promoting fluid retention (e.g. non-steroidal anti-inflammatory drugs) should be withdrawn. Beta-adrenoceptor antagonists can cause myocardial depression, but should not be stopped, although a high dose may need to be reduced. Alcohol intake should be moderate at most, since alcohol depresses myocardial contractility and can be arrhythmogenic.
A graded exercise programme for people with stable heart failure can improve symptoms.
Diuretics
Diuretics remain the mainstay of treatment for chronic heart failure with fluid retention, and are very effective for relief of symptoms (Ch. 14). A loop diuretic (usually furosemide) is typically used, taken once daily in the morning. There is no evidence that the use of a loop or thiazide diuretic alters prognosis in heart failure. Hypokalaemia is unusual when loop diuretics are used in chronic heart failure, especially as an angiotensin-converting enzyme (ACE) inhibitor or angiotensin II receptor antagonist is usually taken concurrently (see below). Nevertheless, the use of a potassium-sparing diuretic is advisable if the plasma K+ falls below 3.5 mmol⋅L–1, especially if digoxin or anti-arrhythmic therapy is given concurrently (because of an increased risk of generating cardiac rhythm disturbances). Spironolactone is preferred in this situation since it improves symptoms and prognosis (at least in severe heart failure) if a low dose is added to maximal therapy with other drugs. In more severe heart failure the fluid retention may not respond to usual doses of a loop diuretic. Strategies for the management of diuretic-resistant fluid retention are considered in Chapter 14.
Angiotensin-converting enzyme inhibitors and angiotensin receptor antagonists
An ACE inhibitor (Ch. 6) is now considered to be essential in the treatment of heart failure with reduced ejection fraction, and is usually started at the same time as a diuretic. By reducing angiotensin II synthesis, ACE inhibitors produce arterial and venous dilation, which improves cardiac function by decreasing ventricular end-diastolic volume and increasing cardiac output (Figs 7.1 and 7.2). ACE inhibitors usually improve breathlessness and fatigue, and exercise tolerance increases. The full symptomatic response is often delayed for 4–6 weeks after the start of treatment, despite early haemodynamic changes. A further benefit of ACE inhibitors is improved survival, which may be due to a reversal of the remodelling of the left ventricle. High doses of an ACE inhibitor are more effective than low doses, and reduce mortality by 20–25% in heart failure with reduced ejection fraction.
There is a small risk of symptomatic hypotension after administration of the first dose of an ACE inhibitor, but the use of a small initial dose reduces the duration of any hypotension. ACE inhibitors promote K+ retention by the kidney; the combination of an ACE inhibitor with spironolactone is used to improve prognosis in severe heart failure, but carries a small additive risk of hyperkalaemia.
If an ACE inhibitor is not tolerated, usually because of cough, an angiotensin II receptor antagonist (Ch. 6) can be substituted. These agents have similar efficacy to ACE inhibitors.
Beta-adrenoceptor antagonists
Beta-adrenoceptor antagonists (Ch. 5) are highly effective for the treatment of heart failure with reduced ejection fraction, usually after stabilising the condition with an ACE inhibitor (or an angiotensin II receptor antagonist) and a diuretic. Beta-adrenoceptor antagonists were once considered to be contraindicated in heart failure because of their negative inotropic properties. However, if introduced very gradually, and starting with a low dose, they improve both symptoms and survival. The survival advantage is additive to that produced by an ACE inhibitor, with a further reduction of 30–35% in mortality at all classes of severity of heart failure. Possible explanations for the benefit of β-adrenoceptor antagonists are numerous (Box 7.2), but a reduction in cardiac remodelling is probably important. Unless there are contraindications, all people who have heart failure with reduced ejection fraction should be treated with a β-adrenoceptor antagonist once they are clinically stable. The only compounds licensed for this use in the UK are bisoprolol, carvedilol and nebivolol, although there are also data to show the efficacy of a modified-release formulation of metoprolol.
Digoxin
Digoxin is widely used to control heart rate when heart failure is associated with atrial fibrillation and a rapid ventricular rate. The use of digoxin for heart failure associated with sinus rhythm has been more controversial, but there is now conclusive evidence that its positive inotropic effect can be useful as a supplement to diuretic and ACE inhibitor therapy when there is severe left ventricular systolic dysfunction and persisting symptoms. Digoxin improves symptoms and the need for hospitalisation, and survival may be improved if the serum digoxin concentration is kept low. Importantly, the effective dose of digoxin for those in heart failure who are in sinus rhythm is smaller than the dose required for control of atrial fibrillation.
Other vasodilators
Treatment with a combination of hydralazine (Ch. 6) and isosorbide dinitrate or mononitrate (Ch. 5), in addition to a diuretic and digoxin, provides balanced arterial and venous dilation. This combination improves exercise tolerance in heart failure but produces only a modest reduction in mortality, although there may be greater benefits in people of Afro-Caribbean origin. The combination can be tried for people who cannot tolerate an ACE inhibitor or angiotensin II receptor antagonist (Ch. 6).
Cardiac pacing and defibrillation
Cardiac resynchronisation therapy (CRT; biventricular electrical pacing) is helpful in severe heart failure when the ECG shows left bundle branch block and the ventricles display marked dyssynchronous contraction on echocardiography. Pacing both ventricles simultaneously restores synchronous contraction and improves cardiac output.
About half of all people with heart failure die suddenly of ventricular arrhythmias, and an implantable cardioverter ventricular defibrillator (ICD) can improve prognosis when there is severe left ventricular impairment and a propensity to ventricular arrhythmias. Combined cardiac resynchronisation–defibrillator devices (CRT-Ds) are also available.
Anti-arrhythmic drugs do not improve survival in heart failure.
Heart failure with preserved ejection fraction
The optimal management of heart failure with preserved ejection fraction is not well established. Most of the interventions that improve prognosis in heart failure with reduced ejection fraction have shown little impact on survival when ejection fraction is preserved, and therefore treatment is mainly directed at symptom relief using the drugs discussed above (with the exception of positive inotropic agents such as digoxin).
True/false questions
1. When blood pressure falls, sympathetic outflow increases because of an increase in the sensory input from the baroreceptors in the carotid sinus.
2. In the healthy heart, a rise in afterload increases myocardial contractility.
3. Breathlessness and fatigue are common symptoms of heart failure.
4. Pulmonary oedema occurs when the hydrostatic pressure in the pulmonary veins is less than the plasma osmotic pressure.
5. In severe heart failure the attempts of the body to compensate for the cardiac dysfunction are detrimental.
6. Digoxin is the mainstay of the treatment of heart failure.
7. Digoxin inhibits the Na+/K+-ATPase pump on the cardiac myocyte membrane.
8. Hypokalaemia reduces the action of digoxin on the Na+/K+-ATPase pump.
9. Digoxin inhibits the vagus, decreasing the refractory period of the atrioventricular node.
10. Dobutamine produces peripheral vasodilation by its effect on β2-adrenoceptors.
11. Sustained infusion of dobutamine desensitises the receptor response.
12. Milrinone is given intravenously to improve tissue perfusion in cardiogenic shock.
13. Angiotensin-converting enzyme (ACE) inhibitors should not be given together with K+-sparing diuretics.
14. ACE inhibitors may cause cough by reducing the synthesis of bradykinin.
15. β-Adrenoceptor antagonists improve survival in chronic heart failure.
16. Hydralazine is a vasodilator with a predominant action on arteries.
One-best-answer (OBA) question
Which of the following is the least accurate statement about dopamine?
Case-based questions
Mr DY is 78 years of age and had a large anterior myocardial infarction 3 years ago. Echocardiography revealed significant left ventricular systolic dysfunction with an ejection fraction of 30%. He presented with several symptoms, including fatigue, decreased exercise ability, shortness of breath and peripheral oedema. Examination demonstrated cardiomegaly, a raised jugular venous pressure and crackles in the lungs. An ECG showed that he is in sinus rhythm.
A What are the choices of diuretic open to you in treating Mr DY?
B Potassium loss produced by diuretics may lead to hypokalaemia. What is an effective way of reducing urinary K+ loss?
C Mr DY was then started on an ACE inhibitor. What precautionary measures should be taken in starting this new medication?
D Could long-term oral digoxin be used as part of the treatment for Mr DY?
E Could the use of a β-adrenoceptor antagonist make Mr DY's condition worse?
1. False. A falling blood pressure reduces the baroreceptor reflex input to the vasomotor centre, resulting in increased sympathetic outflow.
2. True. A rise in afterload decreases stroke volume initially but the consequent sympathetic stimulation increases contractility in the healthy heart and restores the stroke volume. In the failing heart, contractility increases less readily and stroke volume falls.
3. True. The breathlessness is caused by increased pulmonary venous pressure leading to pulmonary oedema; fatigue is caused by reduced cardiac output and impaired perfusion of skeletal muscle.
4. False. Oedema occurs when the net hydrostatic pressure, which moves fluid out of the vessel, is greater than the the plasma osmotic pressure, which moves interstitial fluid into the vessel.
5. True. The fall in cardiac output activates the sympathetic nervous system and the renin–angiotensin-aldosterone system; these changes are appropriate to restore blood pressure in the event of haemorrhage, but are unhelpful in severe heart failure as they increase preload, afterload and heart rate, hence increasing the workload of the failing heart.
6. False. Digoxin may be of benefit but the mainstay of treatment is a diuretic such as furosemide and an ACE inhibitor (or angiotensin II receptor antagonist). If diuretics are given concurrently with digoxin, a K+-sparing diuretic such as spironolactone may also be required to prevent hypokalaemia; hypokalaemia increases the risk of digoxin-induced rhythm disturbances.
7. True. By blocking the Na+/K+-ATPase pump, cellular export of Na+ is reduced and intracellular Na+ concentrations rise. The reduced Na+ concentration gradient across the cell membrane reduces the linked export of Ca2+ ions. Increased intracellular Ca2+ concentrations enhance contractility.
8. False. Potassium ions and digoxin compete for the pump, so hypokalaemia can increase the activity and pro-arrhythmic risk of digoxin.
9. False. As well as its direct effects on the heart, digoxin increases vagal outflow from the vasomotor centre. This increases the refractory period of the atrioventricular node and is the reason why digoxin is useful in some arrhythmias, such as atrial fibrillation.
10. False. Dobutamine is a selective β1-adrenoceptor agonist and does not produce peripheral vasodilation.
11. True. Stimulation of β1-adrenoceptors by dobutamine results in cAMP synthesis and increased cardiac contractility. Prolonged stimulation (48–72 h) desensitises the receptors; this does not occur with phosphodiesterase type 3 inhibitors, such as milrinone, which bypass the receptor and raise levels of cAMP by blocking its breakdown.
12. True. Milrinone inhibits the breakdown of intracellular cAMP by phosphodiesterase type 3 in cardiac and vascular smooth muscle, resulting in an inotropic effect and peripheral vasodilation; it is used in patients with cardiogenic shock not responding to full doses of dobutamine.
13. False. The combination of an ACE inhibitor and spironolactone provides additive clinical benefit but care must be taken to avoid dangerous hyperkalaemia, with regular monitoring of the plasma K+ concentration. This is because ACE inhibitors reduce aldosterone-dependent reabsorption of Na+ in the collecting duct. As Na+ reabsorption at this site occurs in exchange for K+ efflux, ACE inhibitors may increase K+ retention, particularly in combination with potassium-sparing diuretics.
14. False. ACE inhibitors reduce the breakdown of bradykinin by ACE (also known as kininase II); increased bradykinin levels in the lungs are thought to be responsible for the cough seen in some patients receiving ACE inhibitors. An alternative strategy is to use an angiotensin II receptor antagonist.
15. True. Beta-adrenoceptor antagonists, ACE inhibitors and spironolactone improve survival in chronic heart failure, possibly by reducing the cardiac remodelling effects of catecholamines, angiotensin II and aldosterone respectively.
16. True. Hydralazine is predominantly an arterial vasodilator, while isosorbide mononitrate mainly acts as a venodilator; their combined use may be effective in chronic heart failure in people intolerant to ACE inhibitors or angiotensin II receptor antagonists.
OBA answer
Answer D is the least accurate statement as dopamine is a non-selective β-adrenoceptor agonist at moderate doses, resulting in a positive inotropic effect. Its action on β2-adrenoceptors causes peripheral vasodilation leading to marked tachycardia (answer C). At low doses, dopamine causes renal arterial vasodilation via D1 receptors, leading to diuresis (answer A), but at high doses this is counteracted by renal arterial vasoconstriction mediated by α1-adrenoceptors (answer E). Interindividual variation in the response to dopamine is high (answer B).
Case-based answers
A The treatment of first choice for fluid retention in chronic heart failure is a diuretic. For mild symptoms, a thiazide diuretic may be adequate, but in most people a loop diuretic such as furosemide is used. The loss of renal function in the elderly and renal underperfusion in heart failure means that thiazide diuretics are less effective in older people with this condition, such as Mr DY.
B Hypokalaemia is arrhythmogenic and should be avoided in people with heart failure, particularly those taking digoxin. Urinary K+ loss caused by a loop diuretic or thiazide can be reduced by combination with a K+-sparing diuretic such as amiloride or spironolactone. Amiloride directly blocks epithelial Na+/K+ exchange in the collecting duct. In severe heart failure, spironolactone, which competes with aldosterone for the mineralocorticoid receptor, also improves prognosis, possibly by blocking aldosterone-dependent cardiac remodelling.
C ACE inhibitors reduce preload and afterload by reducing the production of angiotensin II, a powerful vasoconstrictor, and by reducing blood volume (via reduced aldosterone production). They also slow the progression of heart failure and improve survival. There is a small risk of severe hypotension following the first dose, and omission of the diuretic immediately prior to this may be helpful.
D The place of digoxin is well established in heart failure associated with atrial fibrillation and a rapid ventricular rate, but the benefit of a low dose of digoxin is now established in heart failure with severe left ventricular systolic dysfunction and sinus rhythm, when combined with a diuretic and an ACE inhibitor.
E β-Adrenoceptor antagonists used injudiciously may worsen heart failure by reducing cardiac output, but low doses are beneficial (see Box 7.2), provided the patient's condition has been stabilised with diuretics and an ACE inhibitor.
Ahmed, A, Rich, MW, Love, TE, et al. Digoxin and reduction in mortality and hospitalization in heart failure: a comprehensive post hoc analysis of the DIG trial. Eur Heart J. 2006;27:178–186.
Amabile, CM, Spencer, AP. Keeping your patient with heart failure safe. A review of potentially dangerous medications. Arch Intern Med. 2004;164:709–720.
Arroll, B, Doughty, R, Andersen, V. Investigation and management of congestive heart failure. BM J. 2008;341:c3657.
Bangash, MN, Kong, M-L, Pearse, RM. Use of inotropes and vasopressor agents in critically ill patients. Br J Pharmacol. 2011;165:2015–2033.
Cleland, JGF, Coletta, A, Witte, K. Practical applications of intravenous diuretic therapy in decompensated heart failure. Am J Med. 2006;119(12A):S26–S36.
Eichhorn, EJ, Gheorghiade, M. Digoxin. Prog Cardiovasc Dis. 2002;44:251–266.
Friedrich, JO, Adhikari, N, Herridge, MS, et al. Meta-analysis: low-dose dopamine increases urine output but does not prevent renal dysfunction or death. Ann Intern Med. 2005;142:510–521.
Gowda, RM, Fox, JT, Khan, IA. Cardiogenic shock: basics and clinical considerations. Int J Cardiol. 2008;123:221–228.
Krum, H, Teerlink, JR. Medical therapy for chronic heart failure. Lancet. 2011;378:713–721.
McAlister, FA, Wiebe, N, Ezekovitz, JA, et al. β-Blocker dose, heart rate reduction, and death in patients with heart failure. Ann Intern Med. 2009;150:784–794.
McMurray, JJV. Systolic heart failure. N Engl J Med. 2010;362:228–238.
Metra, M, Felker, GM, Zaca, V, et al. Acute heart failure: multiple clinical profiles and mechanisms require tailored therapy. Int J Cardiol. 2010;144:175–179.
Molenaar, P, Parsonage, WA. Fundamental considerations of β-adrenoceptor subtypes in human heart failure. Trends Pharmacol Sci. 2005;26:368–374.
Rajagopolan, S, Arora, A, Shafiq, N, et al. Pharmacotherapy of heart failure with normal ejection fraction (HFNEF) – a systematic review. Br J Clin Pharmacol. 2011;72:369–380.
Rathore, SS, Curtis, JP, Jeptha, P, et al. Association of serum digoxin concentration and outcomes in patients with heart failure. JAMA. 2003;289:871–878.
Shah, AM, Mann, DL. In search of new therapeutic targets and strategies for heart failure: recent advances in basic science. Lancet. 2011;378:704–712.
Struthers, AD. Angiotensin blockade or aldosterone blockade as the third neuroendocrine-blocking drug in mild but symptomatic heart failure. Heart. 2006;92:1728–1731.
Waller, JR, Waller, DG. Beta-blockers for heart failure with reduced ejection fraction. BMJ. 2011;343:c5603.
Yang, EH, Shah, S, Criley, JM. Digoxin toxicity: a fading but crucial complication to recognize. Am J Med. 2012;125:337–343.