Medically compromised children
Kerrod B Hallett, Sherene Alexander, Meredith Wilson, Craig Munns, Angus C Cameron and Richard P Widmer

Introduction
Comprehensive dental care of a medically compromised child requires consideration of their underlying systemic condition and coordination of their dental treatment with their medical consultant. Although dental problems are common in this group, their oral health is overlooked frequently by the medical profession. The term used to identify this particular group, ‘medically compromised children’, has been replaced recently by the more general term ‘children with special needs’. However, the older term is still relevant because it reminds the dentist that these children often have medical conditions that can affect dental treatment or that they can present with specific oral manifestations of a systemic disease. This chapter discusses the common paediatric medical conditions that require consideration in the provision of optimal dental treatment. The prevention of oral diseases is important in children with chronic medical problems (Figure 12.1), as oral complications can severely compromise a child’s medical management and overall prognosis.

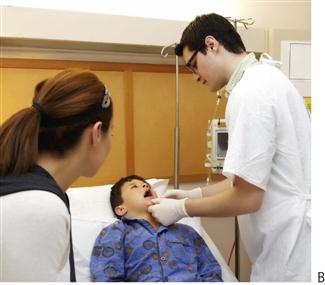
Figure 12.1 (A) Children with medical problems may have conditions with a dental manifestation. This child has osteogenesis imperfecta – or they may present with a medical comorbidity that complicates their general dental care. (B) Not all children with medical problems require hospital admission, although treatment of such patients is often challenging. Mobile dental equipment is invaluable in providing quality dental care. The majority of hospital inpatients are treated in the dental surgery and usually only those who may be in traction or in intensive care units require bedside treatment.
Cardiology
Congenital heart disease
Congenital heart disease (CHD) has an incidence of approximately 8–10 cases per 1000 live births and represents the largest group of paediatric cardiovascular diseases. Although most lesions occur individually, several form major components of syndromes or chromosomal disorders such as Down syndrome (trisomy 21) (see Figure 12.2A) and Turner syndrome (45, X chromosome) with over 40% of children being affected. However, in the majority of cases, no cause can be determined and a multifactorial aetiology is often assumed. Known risk factors associated with CHD include maternal rubella, diabetes, alcoholism, irradiation and drugs such as thalidomide, phenytoin sodium (Dilantin) and warfarin sodium (Coumadin). Turbulent blood flow is caused by structural abnormalities of the heart anatomy and presents clinically as an audible murmur. The degree of clinical morbidity is determined by the haemodynamics of the lesion. Congenital heart disease can be classified into acyanotic (shunt or stenotic) and cyanotic lesions depending on clinical presentation. Eight common conditions account for 85% of all cases.
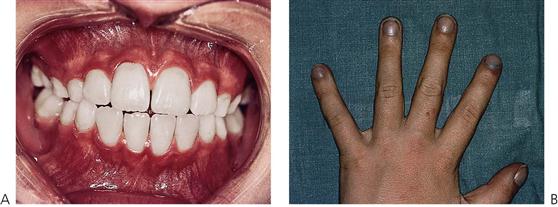
Figure 12.2 (A) Intra-oral photograph of a child with complex cyanotic cardiac disease associated with trisomy 21. Note the cyanosis of the gingival tissues. Of importance is to note that he has no dental disease. His oral hygiene is exceptional and there is no caries. Treatment would have been extremely difficult due to his intellectual disability. (B) Clubbing of the fingers associated with complex cyanotic cardiac disease. The nail beds also show cyanosis.
Acyanotic conditions
The acyanotic group of conditions is characterized by a connection between the systemic and pulmonary circulations or a stenosis (narrowing) of either circulation. Infants often present with feeding difficulties, breathlessness and failure to thrive. Shunts are from the left to right. The most common anomalies and their specific sites are:
• Atrial septal defect (ASD) – usually located near foramen ovale.
• Ventricular septal defect (VSD) – in the membranous septum of the ventricular wall.
• Patent ductus arteriosus (PDA) – caused by failure of closure of the ductus connecting the pulmonary artery with the aorta (normally closes soon after birth). If cardiac failure develops the infant is digitalized and prescribed diuretics if necessary. Hospitalization, oxygen, nasogastric tube feeding and antibiotic therapy for chest infection may also be required. If the lesion does not close spontaneously, surgery to reduce torrential pulmonary flow or repair of the defect is indicated.
Acyanotic defects with obstruction include:
• Coarctation or localized constriction of the aorta – usually in the area related to the insertion of the ductus.
• Aortic stenosis or narrowing of the aortic central orifice – due to fusion of the aortic valve cusps.
• Pulmonary stenosis – due to narrowing of the pulmonary valve which may also involve the pulmonary arteries.
Cyanotic conditions (Figure 12.2)
All cyanotic conditions exhibit right-to-left shunting of desaturated blood. Cyanotic defects become clinically evident when 50 g/L of desaturated haemoglobin is present in peripheral arterial blood. Infants with mild cyanosis may be pink at rest but become very blue during crying or physical exertion. Children with cyanotic defects are at significant risk for desaturation during general anaesthesia and preoperative consultation with the paediatric cardiologist and anaesthetist is essential.
The most common cyanotic lesions are:
• Tetralogy of Fallot – which includes a VSD, pulmonary stenosis at valve or sub-valve levels, a large overriding aorta and right ventricular hypertrophy.
• Transposition of great vessels – when the aorta exits the heart from the right side and the pulmonary artery exits from the left. Although the internal heart anatomy is normal, the systemic blood circulation cannot be resaturated with oxygen and immediate management of transposition by opening the ductus arteriosus and corrective surgery is required.
• Eisenmenger syndrome – this refers to cyanosis from any right-to-left shunt caused by increased pulmonary resistance through a VSD or PDA.
• Tricuspid atresia – due to absent tricuspid valve and may present with an absent right ventricle and pulmonary valve. The pulmonary circulation is maintained through a PDA in the neonate.
• Pulmonary atresia – which is similar to tricuspid atresia except that the tricuspid valve is patent.
Other cardiovascular diseases
Other common paediatric cardiovascular disorders include cardiomyopathies such as myocardial disease and pericardial disease, cardiac arrhythmia, infective endocarditis and rheumatic heart disease (RHD). Both CHD and RHD can predispose the internal lining of the heart to bacterial or fungal infection (infective endocarditis) and lead to the formation of friable vegetations of blood cells and organisms. Vegetations may embolize and cause renal, pulmonary or myocardial infarcts or cerebrovascular accidents. Streptococcus viridans, a common commensal organism in the oral cavity, is most frequently responsible for chronic infective endocarditis, whereas Staphylococcus aureus is often implicated in the acute fulminating form of infective endocarditis.
Dental management
Several important principles need to be followed when managing children with cardiac disease. Transient bacteraemia can occur following invasive dental procedures and potentially cause infective endocarditis in a susceptible patient. Therefore, all children with CHD or previous RHD require antibiotic prophylaxis to reduce the risk of infective endocarditis (see Appendix E). Those children who have been previously taking long-term antibiotics should be prescribed an alternative medication as per the protocol to avoid development of resistant oral organisms. In addition, a preoperative oral antiseptic mouthwash, such as 0.2% chlorhexidine gluconate, is recommended to reduce the oral bacterial counts.
Children with CHD have a higher prevalence of enamel anomalies in the primary dentition and concomitant risk of early childhood caries. Some cardiac medications may contain up to 30% sucrose and dietary prescription with high-caloric supplements (Polyjoule) further potentiate caries risk. Meticulous oral hygiene and preventive dental care, such as fissure sealants and topical fluoride therapy is recommended to reduce the risk of dental caries in susceptible children.
Dental disease in children with cardiac disorders can seriously complicate their medical management. Children with advanced cardiovascular disease should receive only palliative dental care until their medical condition has been stabilized. Aggressive treatment of pulpally involved primary teeth is recommended. Pulpotomy or pulpectomy is contraindicated in these children due to the possibility of subsequent chronic bacteraemia. Although routine treatment in the dental surgery environment is possible, it is often preferable to manage children with multiple carious teeth under general anaesthesia in the hospital environment. This protocol allows completion of treatment with one invasive procedure and negates the risk of infective endocarditis with further operative procedures. If multiple visits are planned, there is a need to prescribe alternative antibiotics or wait for a month between appointments to reduce bacterial resistance.
A thorough preoperative assessment of the child’s regular medication (including anticoagulants, antiarrhythmics, and antihypertensives) is essential to avoid any potential drug interactions during treatment. There is no contraindication to the use of vasoconstrictors in local anaesthetic solutions. If conscious sedation is used, vital signs and oxygen saturation during the procedure should be carefully monitored. Avoid the use of electrosurgery, electronic pulp testers and ultrasonic cleaning devices in children with cardiac pacemakers, in case of potential interference. Some common impediments are non-compliance with oral hygiene and dietary advice, postoperative infection and bleeding.
Haematology
Disorders of haemostasis
Primary haemostasis is initiated after injury to a blood vessel with the formation of a primary platelet plug. This process is mediated by interactions between the platelets and coagulation factors in the plasma and the vessel wall. Secondary haemostasis or coagulation is also triggered by the initial injury and reaches its greatest intensity after the primary platelet plug is formed. Fibrin deposition provides the framework for the formation of a stable blood clot.
Prolonged bleeding can occur when either phase of haemostasis is disturbed. The clinical manifestations of a haemostasis disorder vary depending on the phase affected. Defects in primary haemostasis generally result in bleeding from the skin or mucosal surfaces, with the development of petechiae and purpura (ecchymoses). These disorders include von Willebrand’s disease as well as defects in platelet function. In contrast, defects in secondary haemostasis, such as haemophilia, lead to bleeding that tends to be more deep-seated in muscles and joints. In both disorders, uncontrolled prolonged oral bleeding can occur from innocuous insults such as a tongue laceration or cheek biting.
Children with haemostasis disorders can be identified from a thorough medical history, examination and laboratory tests. Questions should reveal episodes of spontaneous bleeding or bruising; the occurrence of prolonged bleeding in other family members and prescription of anticoagulant medication. A physical examination of skin (unusual areas of bruising on the chest or back or bruising from lying on a toy), joints and oral mucosa should be undertaken for evidence of petechiae, ecchymoses and haematoma. If a haemostasis disorder is suspected, referral to a haematologist is recommended for evaluation and laboratory blood tests.
Laboratory tests
• PFA 100 (platelet function analysis) may be used as a screening test for von Willebrand’s disease and platelet dysfunction. If it is prolonged, specific testing for these disorders may be required.
• Full blood count (FBC) is required to determine platelet levels (normal range 150–400 × 109/L) and platelet function tests may be necessary in selected cases. Adequate haemostasis can usually be maintained by no less than 40 × 109 platelets/L.
• Coagulation tests include prothrombin time (PT) that is a test of the extrinsic coagulation pathway (normal range 11–17 s) and activated partial thromboplastin time (APTT), which is a test of the intrinsic coagulation pathway (normal range 24–38 s). Test results greater than 2 s compared with control values should be considered abnormal.
Classification
Vascular disorders
Vascular disorders are characterized by increased capillary fragility and include the purpuras, hereditary haemorrhagic telangiectasia, haemangiomas, vitamin C deficiency, Henoch Schönlein purpura and connective tissue disorders such as Ehlers–Danlos syndrome.
Platelet disorders
Platelet disorders can be either a deficiency (thrombocytopenia) or dysfunction.
Thrombocytopenia
Thrombocytopenia is defined as a platelet count <150 × 109/L. Clinical signs and symptoms associated with decreased platelet counts are as follows:
• <75 × 109/L – May exhibit post-surgical haemorrhage.
• <25 × 109/L – Spontaneous haemorrhage, easy bruising.
• <15 × 109/L – Petechiae appear on the skin.
• <5 × 109/L – Oral petechiae, submucosal and mucosal bleeding.
Thrombocytopenia may occur as an isolated entity of unknown cause (idiopathic thrombocytopenic purpura, ITP), as a result of marrow suppression by drugs or from other haematological diseases such as aplastic anaemia. Marrow replacement by neoplastic cells in haematological malignancies will also result in thrombocytopenia. Children undergoing chemotherapy will have decreased platelet counts.
Thrombocytosis
Thrombocytosis is an increased number of platelets (>500 × 109/L) and may be associated with prolonged bleeding due to abnormal platelet function. Myeloproliferative disorders may present with thrombocytosis.
Platelet function disorders
These may be congenital or acquired. The most common cause of acquired platelet dysfunction is the use of non-steroidal anti-inflammatory drugs (e.g. aspirin). Administration of cyclo-oxygenase inhibitors will result in blockage of the production of thromboxane A2 for the life of the platelet (7–9 days). This results in a decrease in platelet aggregation. Some metabolic diseases such as Gaucher’s disease also manifest as defects of platelet function.
A decrease in the number of platelets or platelet dysfunction will result in failure of initial clot formation. Children with thrombocytopenia will bleed immediately after trauma or surgery, unlike those with haemophilia, who usually start to bleed 4 h after the incident. The most common oral manifestations are petechiae and ecchymoses. There may also be spontaneous gingival bleeding and prolonged episodes of bleeding after minor trauma or tooth brushing.
Inherited coagulation disorders
Coagulation disorders result from a decrease in the amount of particular plasma factors in the coagulation cascade. The most common disorders are haemophilia A and von Willebrand’s disease, both manifesting a decrease in factor VIII levels. The factor VIII is produced by endothelial cells and is composed of two portions. The largest part of the molecule is the von Willebrand’s factor and is responsible for initial platelet aggregation. The factor VIII part of the complex and factor IX are responsible for activation of factor X in the intrinsic pathway of the coagulation cascade. Other disorders of coagulation include vitamin K deficiency, liver disease and disseminated intravascular coagulation usually from overwhelming (Gram-negative) infection.
Coagulation disorders are classified according to the defective plasma factor; the most common conditions are factor VIII (haemophilia A) and factor IX (haemophilia B or Christmas disease). von Willebrand’s disease occupies a unique position in that both platelet and factor VIII activity is decreased, therefore both bleeding time and APTT are prolonged.
Haemophilia A
This is inherited as an X-linked recessive disorder with deficiency of factor VIII, and occurs 1 in 10 000 live male births. Spontaneous mutation occurs in 30% of cases. The disease is classified as:
• Severe (<1% factor VIII) – with spontaneous bleeding into joints and muscles.
• Moderate (2–5% factor VIII) – with less severe bleeding usually following minor trauma.
• Mild (5–25% factor VIII) – which may not manifest until middle-age following significant trauma or surgery.
Factor VIII assay is usually performed after the initial diagnosis of a coagulopathy. Affected children and their families require considerable medical support and may have an indwelling central line for regular factor VIII concentrate infusion.
Haemophilia B or Christmas disease
This disease has clinical features similar to factor VIII deficiency. It is also inherited as an X-linked recessive gene and results in prolongation of the APTT. It is diagnosed by specific assay of factor IX.
von Willebrand’s disease
von Willebrand’s disease is inherited as an autosomal dominant trait (gene locus 12p13). The most common clinical manifestations include epistaxis and gingival and gastrointestinal bleeding. von Willebrand factor is found in the plasma, platelets, megakaryocytes and endothelial cells and circulates as a major component of the factor VIII molecule complex. This disease is divided into various subtypes, based on the platelet and plasma multimeric structure of the von Willebrand factor.
Dental management
Dental management of children with suspected haemostasis disorders should begin with screening laboratory tests. If tests are abnormal, haematological consultation is required for a definitive diagnosis. Invasive dental procedures should be performed only after the extent of the problem has been determined. Extractions must never be performed without first consulting the haematologist. It is preferable to have platelet levels >80 × 109/L before extractions. Endodontic procedures may be preferable to extractions in order to avoid the need for platelet transfusion.
Dental procedures
• Use an atraumatic technique. In the event that oral surgery is necessary, a sound surgical technique to minimize trauma and local measures to control bleeding such as careful atraumatic suturing and socket dressings are mandatory.
• Maxillary infiltration anaesthesia can generally be administered slowly without pretreatment with platelet or factor replacement. However, if the infiltration injection is into loose connective tissue or a highly vascular area, then factor replacement to achieve 40% activity levels is recommended.
• Avoid mandibular block injections as these may be complicated by dissecting haematoma and airway obstruction. In the absence of suitable factor replacement, intra-periodontal injections may be used, but with great caution. The anaesthetic solution is placed under moderate pressure along the four axial surfaces of the tooth by inserting the needle into the gingival sulcus and the periodontal ligament space.
• Nitrous oxide sedation can be effective for restorative procedures with the need for local anaesthesia; however, care must be taken when placing matrix bands.
• Use rubber dam to protect the soft tissues.
• Endodontic treatment can be safely carried out without factor cover.
• Periodontal treatment with deep scaling and subgingival curettage requires factor replacement.
• Multiple extractions require hospital admission and haematological work-up in conjunction with the haematology team.
Medical management
Haemophilia A
• All children and most adult patients are treated with recombinant factor VIII. A small number of adults continue to receive either recombinant or purified plasma derived factor VIII.
• Severe haemorrhage is treated to 100% replacement, although minor bleeds can be controlled with partial replacement between 30% and 50%.
• Minor trauma may also be life-threatening, especially with intracerebral bleeds.
• Some patients will form antibodies (inhibitors) to factor VIII, severely complicating medical management.
• A single unit of factor VIII concentrate per kg will raise blood levels by approximately 2% and has a half-life of 10–12 h.
von Willebrand’s disease
• Type I may be treated with 1-deamino (8-D-arginine) vasopressin (DDAVP).
• Types II and III require treatment with purified plasma derived factor VIII concentrate (which contains both factor VIII and von Willebrand’s factor).
• Avoid platelet transfusions, if possible, due to the development of antiplatelet antibodies and the risk of transmission of viral diseases such as hepatitis B and C.
1-Deamino (8-D-arginine) vasopressin (DDAVP)
• Can be used for people with mild haemophilia and those with von Willebrand’s disease.
• Can result in an up to two-fold release of factor VIII from endothelial cells – this is adequate if levels of factor VIII are >10% and the patient is responsive to DDAVP.
Post-surgical administration of antifibrinolytic agents such as tranexamic acid (Cyklokapron) 25 mg/kg loading dose and 15–20 mg/kg three times daily for 5–7 days is helpful in preventing clot lysis. During the time that antifibrinolytics are given, the parent and child should be instructed not to use straws, metal utensils, pacifiers or baby bottle teats.
Characteristically, haemophilia bleeds are delayed 12–24 h, as primary haemostasis is not impaired, and local pressure has little effect. It is worth noting that mild haemophilia can go undiagnosed. The APTT is not sensitive to detect mild deficiencies of FVIIIc and levels of FVIIIc 25–30 IU/dL can be associated with a normal APTT. In addition, FVIIIc values in mild haemophilia are temporarily increased (as occurs in unaffected persons) by stress, exercise and bleeding. If there is a convincing history of a bleeding tendency always do a specific factor assay even if the initial screening tests are normal.
The normal regimen for DDAVP is 0.3 µg/kg intravenous infusion over 1 h before surgery followed by tranexamic acid 15–20 mg/kg orally every 8 h for 7 days. After 9–12 h, if the FVIIIc levels are still low (50–60%), then the original dose of DDAVP may be repeated. If repeated doses are planned or required it is important to fluid restrict the patient and monitor electrolytes. Repeated doses of DDAVP may cause fluid retention and hyponatraemia. This regimen is useful in von Willebrand’s disease and children on renal dialysis.
Clinical Hints
Questions commonly asked by parents are:
• Will my child’s teeth erupt normally? Usually yes, but there is often more bleeding from a traumatized operculum that may require active intervention.
• Will my child’s teeth fall out normally? Usually yes, unless continually traumatized, there is normally no abnormal bleeding associated with exfoliating primary teeth. However, if there is prolonged mobility and oozing occurs, then extraction may be necessary under appropriate factor cover to reduce the risk of persistent bleeding (Figure 12.3).
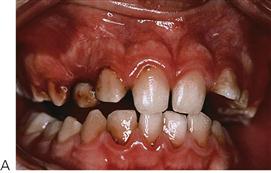
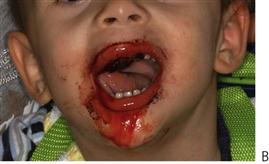
Figure 12.3 (A) Gingival haemorrhage around an exfoliating maxillary right primary canine in a child with Christmas disease (factor IX deficiency). Normally, exfoliation of primary teeth is not of major concern and bleeding is locally controllable. (B) A boy with haemophilia presenting following minor trauma to the labial frenum. Note the poorly formed clot in the mouth and continued oozing after several days.
• Can a child with a bleeding disorder have orthodontic treatment? Yes, provided extractions are performed after appropriate consultation with the haematologist and there is vigilant maintenance of the appliances.
Anticoagulant therapy
Management of children on anticoagulant therapy needs special consideration. Anticoagulants are usually prescribed for children with valvular heart disease and prosthetic valves to reduce the risk of remobilization. If extractions or surgery are required, it is necessary to decrease the clotting times to facilitate adequate coagulation but not to such an extent so as to cause emboli or clotting around the valves. The dental management of these children is also complicated by their congenital cardiac defect and antibiotics are required for prophylaxis against infective endocarditis.
Therapeutic drugs used
• Oral warfarin sodium (Coumadin):
• Vitamin K antagonist depleting factors II, VII, IX and X.
• Usually 3–4 days are required for full anticoagulation onset and its efficacy is assessed by PT level (factor VII levels).
• Shorter acting and has an immediate onset (inhibits factors IX, X and XII).
• Can be administered either subcutaneously using a low-molecular-weight derivative or intravenously under the supervision of a paediatric haematologist.
• Enoxaparin sodium (Clexane):
• Low-molecular-weight heparin which inhibits factor Xa and thrombin.
Children on anticoagulant therapy should stop taking warfarin 3–5 days prior to the surgery date. In those in whom there is a significant risk for thrombosis with sub-therapeutic warfarin level, parenteral anticoagulation may be necessary. This is generally achieved with enoxaparin sodium (Clexane) 1.5 mg/kg subcutaneously once daily (mane) via Insuflon. This drug is omitted on the morning of surgery. With the use of this regimen, the child may be admitted to hospital on the day of dental surgery. Warfarin is recommenced in normal dose on the evening of surgery. If further enoxaparin sodium prophylaxis is required, it should be given the morning after surgery and continued until the PT and international normalized ratio (INR) are therapeutic. Monitoring of enoxaparin sodium is rarely required. In emergency situations with prolonged bleeding from oral wounds post-surgery, following recommencement of warfarin, FFP (fresh frozen plasma) may also be of benefit.
Local haemostatic measures
• Application of topical thrombin (Avitene).
• Packing of the socket with microfibrillar collagen haemostat (MCH or CollaTape), oxidized regenerated cellulose (Gelfoam or Surgicel).
• Suturing of attached gingivae to maintain pressure.
• Splints or stomo-adhesive bandages may also be of benefit.
• There have been recent reports of the efficacy of ‘fibrin glue’ in the management of coagulopathies, but its use on moist oral mucosa is limited.
Management of oral haemorrhage
Unexpected bleeding from the oral cavity can occur at any time. There may have been a slow ooze for several days or, in the other extreme, there may be a significant sudden oral bleed. Such bleeding can occur without warning and may not be associated with any prior investigative or operative work. As well, haemorrhage from the mouth can occur following such routine procedures as biopsy, restorative work or tooth extraction.
The initial management of such cases involves identifying the exact site of haemorrhage, controlling the bleeding and then preventing a recurrence. In the cases of haemorrhage from the mouth that has not been associated with any dental procedure, clinicians should take an accurate history of the bleeding, the duration, lost volume and any causative factors. Abnormal bleeding may occur around an erupting tooth, from an exfoliating tooth site or may be associated with physical and sexual child abuse or congenital vascular anomalies such as arteriovenous malformations. The possibility of a childhood malignancy should also be considered.
In cases of oral haemorrhage following dental procedures, the following steps should be taken (it is important to prevent or minimize bleeding in the first instance):
• A sensible limitation of surgical trauma.
• Digital compression of the alveolus after tooth extraction.
• Packing of the socket with a resorbable gel.
• Adequate suturing of extraction sites to help reduce postoperative complications.
• Pressure application to the surgical site with gauze packs.
• Construction of a removable splint is recommended following more extensive surgery.
• Written postoperative instructions to ensure adequate rest, avoidance of hard foods and early mouth rinsing.
• Prescription of non-aspirin medication are necessary to avoid any parent misunderstanding.
In cases of severe uncontrollable haemorrhage following tooth extraction that can occur due to arteriovenous malformations, remember that the best method of controlling the bleeding is to replant the extracted tooth back into the socket and suture it well.
Red cell disorders
Anaemia
Anaemia is considered to be present if the haemoglobin level falls below 100 g/L. The cause of anaemia in children may be due to blood loss, iron, folate and vitamin B12 deficiency, bone marrow failure, haemolysis of red blood cells or anaemia of chronic disorders. It is usually an incidental finding in the routine dental management of children. A full blood count (FBC) is usually ordered when children present with pallor, lethargy, fever, bruising, undiagnosed systemic or oral pathology after major trauma associated with excessive blood loss or on work-up before surgery for other medical conditions. When unexpected anaemia is discovered, follow-up by the paediatrician is required.
Haemolytic anaemia
Acute haemolytic disease of the newborn or erythroblastosis fetalis is caused by ABO incompatibility and Rhesus (Rh) iso-immunization. There will be discolouration of those primary teeth that are calcifying at the time of birth. The cusp tips of the first permanent molars may also be affected. A yellow-green staining is most commonly seen as a result of high levels of unconjugated bilirubin.
Glucose 6-phosphate dehydrogenase (G6PD)
G6PD deficiency also results in acute haemolytic anaemia when the child is exposed to certain drugs (sulphonamides, chloramphenicol, aspirin, antimalarials) or infection (hepatitis).
Aplastic anaemia
Aplastic anaemia is defined as a decrease or absence of haemopoiesis in the bone marrow that is not due to marrow involvement or recognized disease process. FBC and bone marrow aspirate confirms the diagnosis. Bone marrow transplantation is the treatment of choice for moderate to severe aplastic anaemia.
Haemoglobinopathies
Thalassaemia
The haemoglobinopathies are a group of genetic disorders involving the globin chains of the haemoglobin (Hb) complex. These diseases comprise two main groups: the structural haemoglobinopathies, resulting in abnormal globins (HbE, HbS) and the thalassaemias. The thalassaemias represent a group of autosomal recessive disorders, common in patients from the Mediterranean, North Africa, the Middle East, India and Central Asia, expressing mutations of genes responsible for the production of any of the haemoglobin chains.
Haemoglobin is a tetrameric protein comprising four globin protein subunits. Adult blood contains haemoglobin A (HbA), comprised of two α-chains and two β-chains and a small amount of haemoglobin A2 (HbA2) comprised of two α-chains and two δ-chains. Children also produce fetal haemoglobin (HbF – two α-chains and two γ-chains), which has a much higher oxygen affinity. Fetal haemoglobin levels decrease after 6 months of age from around 70% at birth to trace amounts in adulthood.
α-Thalassaemia – is caused by deletions or mutations of the four alpha globin genes on chromosome 16. One to four genes may be affected, resulting in a relative overproduction of β-chains. Homozygous α-zero thalassaemia (four alpha globin genes deleted) is incompatible with life, while carriers (1–2 gene deletions) have no clinical symptoms. Children with HbH disease (three alpha genes deleted/abnormal) may have mild anaemia or a transfusion dependent anaemia.
β-Thalassaemia – Of more clinical significance is homozygous β-thalassaemia major (Cooley’s anaemia). Due to the absence of the β-chain, there is a compensatory increased production of HbA2 and HbF. As erythropoiesis is inadequate, the bone marrow is reactive and there is compensatory intermedullary haemopoiesis in the maxilla and diploe of the skull. There may be severe haemolytic anaemia with marked hepatosplenomegaly and failure to thrive. Those children with sickle/β-thalassaemia show evidence of vascular thrombosis with ischaemia to organs, especially bones.
Due to maxillary and zygomatic overgrowth there is often a severe Class II Division 1 malocclusion with separation of teeth and widening of the periodontal ligament space. Lateral skull radiographs demonstrate a ‘hair on end’ appearance. Children are given regular packed red cell hypertransfusions until the haemoglobin rises to 140–150 g/L and desferrioxamine, an iron-chelating agent, to increase iron excretion. When excessive haemosiderosis in the spleen adds significantly to the haemolysis rate, elective splenectomy is performed.
Sickle cell disease
This is different from the other haemoglobinopathies in that the red blood cells are more susceptible to haemolysis and have difficulty passing through small blood vessels causing infarcts and ischaemia of organs and bone. These patients are usually asymptomatic unless subjected to low oxygen concentrations and this may be an issue when a general anaesthetic is required. Blood transfusions, analgesics, antimicrobials, adequate hydration and other life-supportive measures are necessary.
Dental management
Consultation with the child’s haematologist prior to treatment is essential to arrange haematological preparation and transfusion. It is important to schedule dental treatment shortly after blood transfusions and provide antibiotic prophylaxis, especially if the child has had splenectomy. Avoid elective treatment if haemoglobin level is <100 g/L. Minimize stress that might compromise the child’s ability to oxygenate the tissue adequately. Respiratory depressants should be avoided and additional oxygenation during conscious sedation or general anaesthesia is desirable along with the use of pulse oximetry. Local anaesthesia is not contraindicated but the use of prilocaine (Citanest) is not advised due to the formation of methaemoglobin. Vasoconstrictors in the standard dose are not contraindicated. Orthodontic treatment may be undertaken but teeth will move quickly through the bone and relapse will most likely occur.
Immunodeficiency
Immunodeficiency may be caused by quantitative or qualitative defects in neutrophils, primary immunodeficiencies, involving T cells, B cells, complement or combined defects and secondary immunodeficiency or acquired disorders.
Qualitative neutrophil disorders
Quantitative neutrophil disorders
Neutropenia
This is defined as <1.8 × 109 cells/L. Life-threatening sepsis is associated with a level of neutrophils <0.5 × 109 cells/L. Neutropenia can occur in the following situations:
• Infiltration of bone marrow by neoplastic cells.
• After administration of cytotoxic drugs used for treatment of childhood malignancy.
• Cyclic neutropenia (21–28 day cycling).
• Nutritional: protein-calorie malnutrition, vitamin B12 deficiency, copper deficiency.
• Pseudo neutropenia: usually mild and spontaneously resolves.
Primary immunodeficiencies
Secondary or acquired immunodeficiencies
Secondary or acquired immunodeficiencies include those conditions acquired during childhood, such as:
• Human immunodeficiency virus (HIV) infection.
• Drug-induced immunodeficiency (cytotoxics, corticosteroids, cyclosporin A, tacrolimus).
These can also occur in children who have undergone bone marrow transplantation and radiotherapy (radiotherapy-induced immunodeficiency).
Combined immunodeficiencies
Dental implications
Both neutrophil and T-cell-mediated immunodeficiencies predispose the child to infection by compromising the host defence system. Opportunistic organisms that do not usually cause disease in a healthy child can proliferate in the oral cavity of the immunodeficient host. Common oral manifestations seen are:
• Acute pseudomembranous candidiasis (Figure 12.5B).
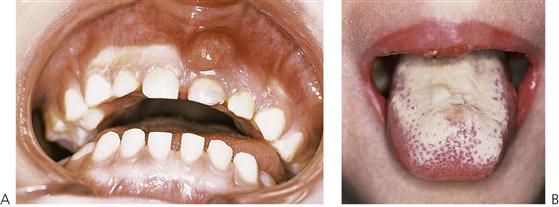
Figure 12.5 Two manifestations of immunodeficiency. (A) Abscess formation above the maxillary right primary lateral incisor tooth after administration of high-dose steroids for asthma, in an area that was previously quiescent. (B) Candidal infection of the tongue in an immunocompromised child.
• Generalized prepubertal periodontitis (Figure 12.4).
• Recurrent aphthous ulceration.
Generally, B-cell deficiencies exhibit fewer oral complications but are often associated with chronic bacterial infections such as pneumonia, otitis media and skin lesions.
Dental management
Regular review of the developing dentition, gingivae and mucosa and the institution of a preventive programme are essential for maintenance of healthy hard and soft tissues. Elimination of any potential oral focus of infection during the course of medical treatment is the primary objective.
The underlying deficiency must be fully assessed and the likelihood of oral complications endangering the child’s medical status should be evaluated. An individual risk–benefit assessment of any oral lesion must be considered with regard to the overall management plan.
A decision whether to extract or maintain carious teeth and exfoliating primary teeth must be based on the worst case scenario during the immunodeficiency period. If a carious lesion cannot be stabilized with an adequate interim restoration, then extraction is the preferred treatment. In a case being prepared for bone marrow transplantation, all mobile primary teeth should be removed at least 2 weeks prior to the conditioning phase.
Thorough dental scaling and prophylaxis and the provision of custom trays for delivery of medication (antiseptic or fluoride gels) prior to commencement of head and neck radiotherapy is also recommended to prevent oral sepsis and radiation-induced dental caries.
Prophylactic antimicrobials specific for commensal oral organisms determined from culture and sensitivity tests are indicated during the course of medical treatment. Biopsy specimens may assist the diagnostic process. The antimicrobial protocol may include appropriate antibiotics (amoxicillin trihydrate and ampicillin, vancomycin), acyclovir sodium if HSV-positive, ganciclovir if cytomegalovirus-positive, antifungals (topical nystatin and amphotericin B) and twice daily 0.2% chlorhexidine gluconate (Curasept) mouthwashes during the active therapy phase.
Acquired immunodeficiency syndrome (AIDS)/HIV
HIV infection has been identified in increasing numbers of children with otherwise unexplained immune deficiency and opportunistic infections of the type found in adults with acquired immune deficiency syndrome (AIDS). For the limited purposes of epidemiological surveillance, the Centers for Disease Control (CDC) characterizes a case of paediatric HIV infection as a reliably diagnosed disease in children that is at least moderately indicative of underlying cellular immunodeficiency, and with which no known cause of underlying cellular immunodeficiency or any other reduced resistance is reported to be associated.
Transmission
The main transmission media are body fluid, such as blood and semen. Saliva contains low and inconsistent levels of the HIV virus and is unlikely to provide a significant mode of transmission. Consequently, the two major routes of transmission in children are vertical (from an infected mother) and from blood products, with children with haemophilia being most at risk. Vertical transmission rates are up to 39% and occur before, during or after birth. Infection from breast-feeding may be up to 29%.
Risk factors
• The risk factors for paediatric HIV infection vary depending on the age group.
• Most children with AIDS are under 5 years of age.
• The primary risk factors are perinatal.
• Infants born to women who are intravenous drug users or who have bisexual partners comprise the largest group.
• About one-third of the infants weigh less than 2500 g at birth and are small for gestational age. Of these babies, 25–30% of children develop AIDS in the first year of life.
• The presenting pattern of encephalopathy varies with age and significant growth failure occurs in early infancy.
Serodiagnosis and immune function
The screening ELISA test for HIV antibodies is liable to give false negatives and any apparently positive results must be confirmed by the western blot assay. Antigen assays are far more reliable, but a failure to detect virus or antigen in a young antibody-positive child does not exclude infection. A positive virus or antigen test is likely to indicate infection. Due to the long incubation period and the limitations of medical history and serodiagnosis, it must be assumed that all blood derivatives may be infectious.
The human immunodeficiency virus attaches to the CD4 variant of the T4 helper lymphocyte and remains within infected cells throughout their life, being transmitted to other cells mainly by cell-to-cell contact. Other cells that may be affected include macrophages, and possibly endothelial, neuroglial, epithelial and dendritic cells. The principal effect of HIV infection on the immune system is depletion of CD4 lymphocytes (helper cells), which results in a drop in the absolute CD4 count and a reversal of the CD4/CD8 ratio. These are immune indicators of disease progression.
Oral manifestations (Figure 12.6)
Oral lesions are often early warning signs of HIV infection. Common disorders may manifest in different ways in the presence of HIV. In children, the most common lesions are:
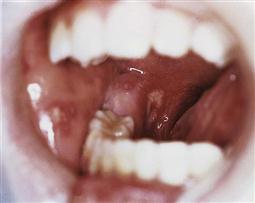
Figure 12.6 Severe oral ulceration in a child in the terminal stages of HIV/AIDS. The ulceration was most likely due to disseminated HSV infection, to which the child succumbed 7 days after this photograph was taken.
Candidosis
The most common oral lesion in HIV infection is acute pseudomembranous candidiasis. It is an early lesion and suggests the presence of other opportunistic infections. The severity of the candida infection may be related to the T4/T8 ratio and occurs when CD4 counts are <300/mL. Oesophageal candidiasis occurs when CD4 counts drop below 100/mL. Fungal infections can be related to reduced salivary flow and S-IgA. It responds well to treatment with systemic antifungals and an improvement in oral hygiene.
Ulceration
Recurrent herpes simplex infections are frequent and are typically intra-oral and circum-oral. Other parts of the body may also be affected. Aphthous-type ulcers are persistent and very common in children. Treatment is palliative with adequate hydration, analgesia and the use of Diflamm-C mouth rinses.
Atypical gingivitis
HIV-related gingivitis manifests as red erythematous gingival tissues and can extend to the free gingival margin. There is often spontaneous gingival haemorrhage and petechiae within the gingival margin, either localized or generalized. Consideration must be given to a fungal component. Treatment involves improved tooth brushing and flossing and the use of daily 0.2% chlorhexidine gluconate (Curasept) mouthwashes and gels.
Salivary gland enlargement
Parotitis or HIV associated parotid gland disease (HIV-PGD) occurs more frequently in paediatric than in adult patients and is similar to the presentation of mumps. It may be unilateral or bilateral and results in xerostomia and pain. Reduced salivary flow may lead to pseudomembranous candidiasis and dental caries. There have been mixed results with the use of antibiotics and glucocorticosteroids in treating this condition. Artificial saliva substitutes or oral lubricants can alleviate the xerostomia.
Hairy leukoplakia
This is uncommon in children, with only a few reported child cases. It occurs predominantly on the lateral border of the tongue and occasionally on the buccal mucosa and the soft palate.
HIV-related periodontitis
HIV-related periodontitis presents with deep pain and spontaneous bleeding, inter-proximal necrosis and cratering, and intense erythema more severe than acute necrotizing ulcerative gingivitis (ANUG). HIV periodontitis appears more frequently in HIV-infected patients who have reduced T4/T8 ratios and symptomatic opportunistic infection. Organisms such as black-pigmented bacteroides and Gram-positive bacilli, which are similar to those found in adult periodontitis, have been identified in HIV periodontitis.
Kaposi’s sarcoma
Uncommon in children and adolescents. The lesion mainly affects the palate, and also the gingivae and the tongue. Treatment is by chemotherapy, radiotherapy or laser excision.
Outcomes
Primary colonization by commensal organisms rather than reactivation of opportunistic infections usually occurs (cytomegalovirus, retinitis and toxoplasmosis are rare). Bacterial infections are also rare, although Streptococcus pneumoniae and Haemophilus influenzae are common respiratory complications. Kaposi’s sarcoma is seen infrequently but lymphomas (especially with central nervous system (CNS) involvement) can occur. The progression of disease process can vary and in many instances, oral and physical symptoms do not often present for years after infection with the immunodeficiency virus. Lymphocytic interstitial pneumonitis is frequently the cause of death for children with AIDS, but is often asymptomatic. There have been major advances in the management of HIV/AIDS with antiretroviral medications and consequently, many children may lead a normal and effective life.
Oncology
Childhood cancer accounts for about 1% of all cancer cases in the population. In Australia, the annual incidence of malignant tumours in children under 15 years is approximately 11 per 100 000 children. Approximately 600–700 children between birth and 15 years of age develop cancer each year. Whereas most adult cancers are carcinomas with strong aetiological associations, childhood cancers are a wide range of different histological types of tumour with less aetiological connection.
The incidence, either in childhood cancer as a whole or in individual types of cancer, varies little from one country to the next and no racial group is exempt. Among more than 50 types of childhood cancers, the most common forms include leukaemias, lymphomas, CNS tumours, primary sarcomas of bone (Figure 12.7A) and soft tissues, Ewing sarcoma, Wilms’ tumours, neuroblastomas and retinoblastomas. Acute leukaemias and tumours of the CNS account for approximately one-half of all childhood malignancies. Multimodal therapy (chemotherapy, radiotherapy and surgery) has resulted in an overall 5-year survival rate for childhood cancer of approximately 70%.
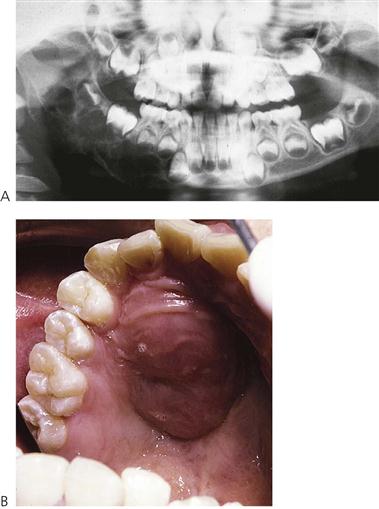
Figure 12.7 Neoplasms may arise as primary lesions within the jaws, invade from local tumours or may seed as metastases from distant primaries. (A) The panoramic radiograph shows an extensive primary neoplasm of the right mandible involving the infratemporal fossa. Histologically, this lesion was a desmoplastic fibroma and required a hemimandibulectomy. (B) Presentation of a lymphoma in the palate of a 15-year-old girl. The lesion was asymptomatic and the child had merely presented for a routine check-up.
Leukaemia
Leukaemia is a heterogeneous group of haematological malignancies caused by clonal proliferation of primitive white blood cells.
Acute lymphoblastic leukaemia (ALL)
• Accounts for 80–85% of acute childhood leukaemias.
• Defined by the presence of more than 25% lymphoblasts in the bone marrow.
• Therapy is tailored to the risk of relapse dependent on cytogenetic markers and includes a combination of induction chemotherapy, CNS prophylaxis and maintenance chemotherapy for approximately 2 years’ duration.
• Intrathecal therapy (commonly methotrexate) has been used to replace cranial irradiation.
• Cure rates for standard risk ALL are now over 90–92% on current protocols. If relapse occurs 40–50% can be cured with chemotherapy and/or haematopoietic stem cell transplantation.
• Prognosis depends on age of onset, initial white cell count, cytogenetic abnormalities and other features.
• Bone marrow transplantation is reserved for very high risk or patients with relapse.
Acute myeloid leukaemia (AML)
• Accounts for 15–20% of acute childhood leukaemias.
• In this disease, the bone marrow is infiltrated with primitive myeloid cells, classified by their morphological appearance (FAB subtypes M1–M7). The clinical features of AML are similar to other leukaemias.
• AML with monocytic morphology (M4/M5) can manifest gingival infiltration and promyelocytic morphology (M3) is associated with disseminated intravascular coagulation.
• Induction chemotherapy therapy is often followed by early allogenic bone marrow transplantation for high-risk patients.
Chronic myeloid leukaemia (CML)
• Rare in childhood and accounts for less than 5% of leukaemic cases.
• Two types: one identical to adult CML and characterized by the presence of the Philadelphia chromosome (Ph) in malignant cells; the second or juvenile form (JCML) occurs earlier in infancy with a rapid course, infection, haemorrhage and poor survival rate.
• Bone marrow aspirate reveals granulocytic proliferation without an excess of lymphoblasts.
• The chronic phase of this disease is now effectively treated with specific Bcr-Abl tyrosine kinase inhibitors (imatinib or dasatinib), which can lead to remission lasting for years.
• Allogeneic bone marrow transplantation remains the only definitive curative therapy but is now generally reserved for patients relapsing on tyrosine kinase inhibitors or for children who have an HLA-matched sibling.
Problems in medical management
The main problems in medical management are bone marrow suppression initially at diagnosis due to malignant infiltration and later due to chemotherapy, subsequent anaemia, infection and mucosal ulceration and bleeding. Infection in the immunocompromised child is a life-threatening condition and may be due to bacteria, viruses, fungi or parasites. Broad-spectrum triple antibiotic treatment is usually required. Disease relapse may occur in the marrow, CNS or other organs (e.g. testes).
Solid tumours in childhood
Brain tumours
• These are most frequent solid tumours of childhood.
• Approximately 70% are gliomas, mostly low-grade astrocytomas or medulloblastoma.
• More than 50% of paediatric intracranial tumours occur in the posterior cranial fossa region. Surgical excision combined where possible with chemotherapy and radiotherapy is the standard approach to treatment.
• Chemotherapy can be used to delay or avoid cranial radiotherapy in infants.
• The overall survival rate is approximately 60% at 10 years.
Non-Hodgkin’s lymphoma (Figure 12.7B)
Wilms’ tumour
• Occurs in the kidney around 3–4 years of age.
• Usually presents as an asymptomatic abdominal mass.
• Often associated with aniridia and other congenital anomalies.
• The tumour responds well to combined therapy: chemotherapy with or without radiotherapy to reduce the tumour mass and surgical removal depending on disease stage. Commonly lung, hepatic and skeletal metastases occur.
Neuroblastoma
• Arises from neural crest cells anywhere along the sympathetic chain.
• Most common site is abdominal, either in the adrenal gland or paraspinal ganglia. Other sites include thorax, neck or pelvis.
• Tumour spread to lymph nodes, bone marrow, liver or subcutaneous tissues.
• Diagnosis is confirmed by raised levels of urinary catecholamines and tissue biopsy.
• Prognosis depends on patient age at diagnosis, tumour stage and biological features of the tumour, especially presence of amplification of the n-myc gene. Children with high-risk disease (approximately 50% of cases) have 25% survival rates even with aggressive chemotherapy, surgery, radiation and autologous bone marrow transplantation.
Rhabdomyosarcoma
• Arises from embryonal mesenchymal tissue with potential for differentiation to skeletal (striated) muscle.
• Children often present with a painless, usually rapidly enlarging subcutaneous lump, almost anywhere in the body.
• Common sites include head and neck, genitourinary tract and extremities.
• Large lesions in the head and neck invade bone and jaw lesions are quite common in advanced cases.
• Treatment involves surgery with adjuvant chemotherapy and radiotherapy.
• Prognosis is influenced by site, subtype of rhabdomyosarcoma, and stage at diagnosis.
Hodgkin’s disease
• Lymphoid malignancy characterized by presence of Reed–Sternberg cells in the tumour.
• Usually affects teenagers and young adults.
• Presents most commonly as a painless enlargement of the lower cervical or mediastinal lymph nodes accompanied by unexplained fever and weight loss.
• Excellent response (cure rate approaches 90% for low stage disease) to chemotherapy.
Retinoblastoma
• Tumour of the retinoblasts in children under 5 years of age.
• Strong hereditary component.
• Diagnosis is usually a white or yellow pupillary reflex (normally red reflex).
• Treatment often requires enucleation of the globe and post-surgical radiotherapy. Occasionally adjunct chemotherapy is also required.
Ewing’s sarcoma
• Malignant tumour of bone in teenagers, commonly involving the midshaft of long bones, although any bone may be involved.
• Occurs most commonly in the proximal femur or pelvis and is characterized by densely packed small round cells.
• Treatment involves surgery, chemotherapy and local irradiation.
• The prognosis worsens with pelvic primary or metastatic disease.
Langerhans’ cell histiocytosis (see Chapter 8)
• A rare neoplasm similar to ALL, often presenting with eczematous, purpuric rash on the hands, scalp and trunk.
• Osteolytic lesions of the skull and mandible can occur and premature exfoliation of primary teeth has been reported.
• Prognosis depends on the extent of disease at diagnosis and the progression of lesions.
Dental management
Close collaboration between the child’s oncologist and the paediatric dentist is essential when planning appropriate dental care. At the time of diagnosis and during the initial stages of chemotherapy, dental care should be provided by the paediatric dentist at the hospital. Once the child has achieved remission, or has successfully completed chemotherapy, routine dental care can often be provided by the child’s own dentist.
Where dental treatment is needed prior to or during chemotherapy, planning with the oncology team is essential. If extractions are required, a FBC including differential white cells and platelets is necessary. If the platelet count is <30 × 109/L, then platelet infusion is indicated and antifibrinolytic agents (doses similar to management of haemostatic disorders) may be helpful. As with immunocompromised children, if the neutrophil count is <1.8 × 109/L, specific antimicrobial prophylaxis should be administered. As many children have been receiving systemic corticosteroids, the possibility of adrenocortical suppression should be considered and additional steroid cover provided as appropriate.
Elective dental treatment should be delayed until the child is in remission or on maintenance chemotherapy. Children in full remission can be treated as normal for most routine dental treatment, although an FBC is prudent if an invasive procedure is planned. Pulpal therapy of primary teeth during the induction and consolidation phase of chemotherapy is contraindicated. When pulpal therapy of permanent teeth is needed, the risk of bacteraemia and potential septicaemia must be weighed against the potential benefits.
Oral hygiene and prevention
It is important to maintain meticulous oral hygiene by using a soft toothbrush during chemotherapy. Four times daily 0.2% chlorhexidine gluconate mouthwashes or gel application to the mucosa helps reduce the symptoms of mucositis and topical and systemic antifungal agents (nystatin or fluconazole) help prevent candidiasis during immunosuppression. Topical lidocaine hydrochloride (Xylocaine Viscous 2%) is helpful during acute episodes of mucositis prior to eating (if possible) or drinking. Prophylactic parenteral antibiotics and antiviral medications, if indicated, are always given during febrile episodes and periods of severe neutropenia to prevent further medical complications.
Immediate oro-dental effects of childhood neoplasia and treatment
Dramatic advances in the treatment of childhood cancer in the past three decades have led to the long-term cure of 70% of the children diagnosed today. Since about 1 in 600 children develop cancer before the age of 15 years, almost 1 young adult in every 1000 will be a long-term survivor of childhood cancer.
As the number of survivors of a variety of paediatric cancers increases, the oro-dental sequelae of effective medical treatment in these patients are emerging. These effects are unique because of the impairment of active growth and development during the cancer therapy. Other late effects include short stature, growth hormone deficiency, cognitive defects, secondary malignancy. Adverse sequelae caused by the cancer treatment can be grouped into postsurgical, post-radiotherapy, post-chemotherapy and combined effects.
Post-surgery
Surgical removal of a solid tumour in the oral cavity can cause:
• Disfigurement (temporary or permanent) (see Figure 12.8A).
Figure 12.8 (A) A child in remission from acute lymphoblastic leukaemia with typical alopecia resulting from chemotherapy. (B) Late effects of surgery and radiotherapy for a rhabdomyosarcoma of the right mandible involving the parotid, neck and infratemporal fossa. This child underwent a hemimandibulectomy and radical neck dissection, followed by reconstruction with a free vascular rib graft. Note the limited oral opening and the facial deformity. Access for restorative work on the carious molars was extremely difficult. Caries resulted from reduced salivary flow after removal of the parotid gland.
Post-radiotherapy (Figure 12.8B)
Radiotherapy produces an initial mucosal inflammation that is often followed by surface sloughing and ulceration (mucositis). The extent of inflammation depends on the location and dosage of radiotherapy and whether fractionated versus whole-dose radiation is used. The most common symptoms following cranial irradiation are oral pain and difficulty in eating and drinking, which are most severe 10–14 days following commencement of radiotherapy. The mucositis usually resolves in another 2–3 weeks after radiotherapy.
When radiotherapy involves the major salivary glands, xerostomia frequently occurs within a few days producing a viscous, acidic saliva. Loss or alteration of taste (hypo- or dysgeusia) may also occur prompting the patient to change to a softer, more cariogenic diet to alleviate soreness and dryness of the oral cavity. This is probably the major factor in the aetiology of rapid dental caries that has been reported in these patients if they are not given adequate preventive therapy. Radiation-induced dental caries has a distinctive generalized cervical pattern and sometimes the complete dentition can be destroyed in a relatively short period.
Progressive endarteritis is a complication that can occur in irradiated bone and can lead to osteoradionecrosis. The mandible is particularly prone to this complication and if such an area of dead bone should become infected following dental extraction, a refractory osteomyelitis may ensue. Endarteritis may also cause fibrosis in the masticatory muscles and subsequent trismus.
Chemotherapy
The cytotoxic drugs used during chemotherapy can cause damage to several organs:
Direct stomatotoxicity is caused by the cytotoxic action of the chemotherapeutic agents on oral mucosal cells leading to inflammation, thinning and ulceration of the mucosa (mucositis). Saliva function may also be diminished although this response has not been reported as common in children. These problems are commonly encountered in the induction and consolidation phases of chemotherapy when relative high doses of multi-agent therapy are employed. Recent case reports suggest that the incidence and severity of stomatotoxicity is reduced with the concomitant administration of granulocyte colony-stimulating factor (G-CSF) during chemotherapy.
The effects of chemotherapy and radiotherapy appear to be synergistic. Since craniofacial and dental development have not been completed until the adolescent period, it is not surprising that dental late effects are commonly found in survivors of childhood cancer. Chronic problems involving target tissues lead to impairment of growth and development of hard and soft tissues, which may result in orofacial asymmetry, xerostomia, dental caries, trismus and a variety of dental abnormalities. Generally, the nature and degree of these complications vary widely and depend on several factors including the type and location of malignancy, the age of the patient, total dosage and timing of chemotherapeutic agents, and the initial oral health status and the level of dental care before, during and after therapy.
Late oro-dental effects of childhood neoplasia and treatment (Figure 12.9)
The majority of those children for whom oncology treatment results in a stable remission can expect to follow a healthy life. Recurrence of the original malignancy may occur although the likelihood of this becomes increasingly remote as time passes. Consequently, successfully treated paediatric oncology patients are never ‘discharged’, their health being regularly monitored throughout their life.
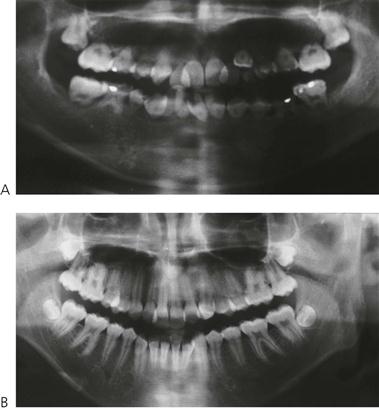
Figure 12.9 Effects of radiation to the head and neck. A set of identical twins, the first of which (A) had acute lymphoblastic leukaemia diagnosed at 18 months. She relapsed during the first remission and received a bone marrow transplant and total body irradiation (TBI). The comparison with her sister (B) at 15 years of age is dramatic. There is agenesis of some permanent teeth, arrested root development of the incisors and first permanent molars, and microdontia.
With the exception of those children treated with radiotherapy to the orofacial region, the majority of children are no more prone to dental and periodontal disease than the well child, and often exhibit excellent oral health. However, long-term oro-dental effects of radiotherapy can influence dental management.
Growth disturbances
Following head and neck radiotherapy, facial growth can be impaired and alterations to developing teeth can occur. Children younger than 5 years of age are affected more severely than older patients. An altered craniofacial growth pattern with diminished mandibular growth, is often associated with a field of irradiation that includes a portion of ascending ramus and the entire condyle of the mandible. Dental effects may include:
• Arrested or altered root development, and premature closure of the root apices can complicate permanent tooth development.
• Microdontia and agenesis of teeth are also common (Figure 12.9A).
The exact nature and extent of damage depends on the stage of dental development and the timing and dosage of irradiation. The lack of specificity of cytotoxic agents in terms of differentiating neoplastic cells from metabolically active normal cells, such as ameloblasts and odontoblasts, can result in abnormalities of dental development. Microdontia, enlarged pulp chambers, shortening, thinning and blunting of the root apex and delayed tooth eruption have been frequently reported in children receiving chemotherapy. Enamel opacities, hypocalcification and a high rate of dental caries have also been reported in several studies, however, it remains unclear whether these findings are due to direct alteration of enamel formation or maturation or to alterations in the oral environment (saliva and flora), diet and home care often observed among young patients on chemotherapy.
Xerostomia
Cranial irradiation can also irreversibly damage the acini cells of the major salivary glands and xerostomia can occur in children. This condition is often transient due to the lower dosage of radiation used and greater regenerative capacity of the exocrine cells in children. Generally, there is a lower incidence of radiation-induced dental caries in children compared with adults. Regular nightly fluoride mouth rinsing is required to prevent enamel demineralization during this critical period.
Other less common effects
Epidermal and mucosal changes include skin hyperpigmentation, cutaneous telangiectasia, subcutaneous tissue atrophy and permanent thinning or loss of hair. Disturbances of intellectual, endocrine and germ cell development have also been reported following cranial irradiation. However, the mean age of dental maturation in children following cranial irradiation is within the normal range.
Since most craniofacial tumours are treated by combined chemotherapy and head and neck irradiation, it is difficult to know the exact effect of each treatment. In general, late oro-dental effects are more severe in patients who receive a higher-dosage treatment either with chemotherapy or radiotherapy. Dental aberrations are more severe and extensive in patients younger than 6 years of age due to the immature development of the permanent teeth. Total body irradiation in bone marrow transplantation appears to increase the risk of disturbance to dental development.
Complications associated with bone marrow transplantation
Almost all children undergoing bone marrow transplantation develop the typical oral mucosal changes of ulceration, keratinization and erythema that develops in 4–14 days post-transplantation. Mucosal atrophy is also frequently associated with ulceration between 1 and 3 weeks after bone marrow transplantation. During this period, oral pain is often severe with many patients requiring narcotic analgesia. The use of keratinocyte growth factor (palifermin) has been demonstrated to reduce this complication in adults undergoing autologous transplantation and paediatric studies of this promising treatment are in progress.
As mentioned previously, oral infection with Candida albicans, herpes simplex, cytomegalovirus and varicella zoster are the major infective agents seen in children undergoing bone marrow transplantation, if inadequate prophylaxis is given. These conditions can be life-threatening if not treated aggressively at diagnosis.
Oral manifestations of defective haemostasis are common but seldom serious and include mucosal bleeding or crusting of the lips and gingival oozing.
Graft-versus-host disease (GVHD)
This condition occurs when transplanted T cells recognize the host tissues as foreign. GVHD is a major problem following bone marrow transplantation with clinical manifestations in up to half of patients. The acute form of GVHD tends to occur within weeks of bone marrow transplantation, with signs of fever, rash, diarrhoea and abnormal liver function leading to jaundice. Chronic GVHD may follow some months later and is characterized by lichenoid or scleroderma-like changes of the skin, keratogingivitis, abnormal liver function, pulmonary insufficiency and intestinal problems. Oral manifestations of GVHD vary with the severity of the conditions but often include:
• Painful desquamative gingivitis.
• Lichenoid patches of the buccal mucosa.
• Striae on the buccal mucosa with subsequent restriction of mouth opening.
Management of chronic GVHD necessitates a multidisciplinary approach. The goal of treatment entails effective care as well as minimizing toxicity and relapse. Long-term systemic immunosuppression with prednisone and other agents is often needed. Recently, extracorporeal photopheresis has been known to produce disease remission. Topical treatments can be effective such as dexamethasone mouthwashes, Biotene moisturizing drops, Difflam-C, and sodium bicarbonate diluted in water helps with disturbances in taste (dysgeusia). Careful attention to oral health with close communication with the treating medical team is needed to give the best outcomes.
Bisphosphonate-related osteonecrosis of the jaw (BRONJ)
Osteonecrosis of the jaw is a well-described complication of bisphosphonate therapy in adults and has not yet manifested as a disorder in children. While there have been no reported cases of bisphosphonate-related osteonecrosis (BRONJ) in children, there has been a significant increase in the use of these drugs in the management of children with connective tissue disorders and decreased bone density including:
• Congenital osteoporosis – osteogenesis imperfecta.
• Secondary osteoporosis – immobility, steroid induced.
• Focal orthopaedic conditions – avascular necrosis (AVN), Perthes’ disease; fracture non-union; Ilizarov limb lengthening; bone cysts.
• Other bone disorders – fibrous dysplasia, idiopathic juvenile osteoporosis.
There is an increased association of BRONJ with any invasive dental procedure such as extractions. While the risks for children are unknown, clinicians should be aware of this potentially destructive condition. There is no concurrence regarding the optimal bisphosphonate drug in children, dosage or duration of treatment. The more serious side-effects linked to bisphosphonates in adults such as uveitis, thrombocytopenia or oesophageal or oral ulcerations are rare in children. Potent bisphosphonates have established therapeutic half-lives of over 30 years, so there may be long term, a low to very low, residual risk of BRONJ in these patients.
Adolescents and young adults requiring bisphosphonate therapy, particularly in the setting of malignancy may also be at increased risk of BRONJ and caution needs to be applied in the undertaking of extractions and oral surgery in this select patient group.
Current management of established BRONJ
| Stage 1 conservative: | Chlorhexidine 0.12% t.d.s. |
| Stage 2 conservative + symptomatic: | Chlorhexidine 0.12% t.d.s.; antibiotics |
| Stage 3 conservative + surgery: | As above + sequestrectomy/resection |
Prevention of BRONJ in children requiring invasive dental procedures
Pre-bisphosphonate therapy: dental evaluation with radiographic screening, within 3 months of starting treatment.
Management for children requiring dental interventions:
• If possible stop bisphosphonate 3 months prior and for 3–6 months after procedure.
• Prophylactic antibiotics 10 days prior to and after procedure.
• Gentle handling of dental tissue and consider primary closure of wounds.
• Chlorhexidine mouthwash 10 days prior to and after procedure.
Dental follow-up for children receiving bisphosphonates: 6-monthly dental reviews with 1–2-yearly radiographic survey as required.
Nephrology
Renal disorders
Renal diseases are classified as acute, chronic, acquired or congenital conditions.
Chronic renal disease
The most common chronic conditions affecting the kidneys are:
End-stage renal failure (Figure 12.10)
• Leads to a progressive drop in glomerular filtration rate that results in hypertension, fluid retention and build-up of metabolites that are not excreted normally.
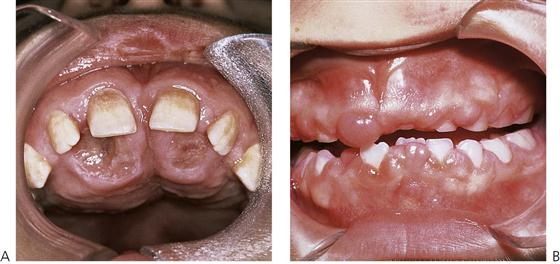
Figure 12.10 (A) Severe renal osteodystrophy in a child in end-stage renal failure. There has been gross expansion of the maxilla in an attempt to produce red blood cells because of failure of erythropoiesis. This is similar to events in β-thalassaemia. (B) Gingival overgrowth due to cyclosporin A treatment after kidney transplantation. The teeth are also hypoplastic and small because of renal disease in infancy.
• Medical management is directed toward prevention of fluid and electrolyte imbalance, restriction of proteinuria, correction of hypoalbuminaemia, hypocalcaemia, hyperphosphataemia and control of anaemia and hypertension.
• In children with severe renal failure, drug treatment is often inadequate and artificial filtration by either peritoneal dialysis or haemodialysis becomes necessary.
Acquired conditions
• Urinary tract infections, usually from coliform bacteria from the intestinal tract and cystitis (bladder infection).
• Acute glomerulonephritis usually accompanies β-haemolytic streptococcal infections and often resolves with antibiotic therapy in most children. However, 3–4% may develop post-infection chronic renal failure and subsequently need dialysis.
Medical complications can be overcome with successful renal transplantation, which is now the preferred treatment of choice for children with end-stage renal failure. Despite the restricted availability of donor organs, renal transplantation has a high success rate. Complications associated with immunosuppression due to cyclosporin (Sandimmune) and prednisone therapy to prevent organ rejection need to be considered in dental management. The most common oral manifestations following renal transplant are gingival hyperplasia (Figure 12.10B) and opportunistic infection from commensal oral flora.
Dental implications
Impaired renal function can result in several oral manifestations including:
• Uraemic stomatitis: as a result of high levels of urea due to breakdown of ammonia.
• Pallor or petechiae and ecchymosis of oral mucosa.
• Intrinsic and extrinsic tooth staining.
• Excessive supragingival calculus due to increased salivary urea and phosphate levels.
Uraemic stomatitis may develop when the serum urea level is >300 mg/mL. It occurs as ulcerated or non-ulcerative forms involving the tongue and buccal mucosa predominantly. Both forms have a tendency to bleed and are susceptible to secondary infection by oral commensal organisms.
Renal osteodystrophy (Figure 12.9A)
Lytic lesions of the mandible or maxilla, known as Brown’s tumours, can also occur in severe renal failure due to secondary hyperparathyroidism. Histologically, these lesions are similar to giant cell tumours and usually resolve following correction of hypocalcaemia and hyperphosphataemia with vitamin D metabolites. Hypocalcaemia occurs due to increased phosphate retention and decreased calcium absorption. Active calcium absorption from the gut depends on the presence of the active metabolite, 25-hydroxy-cholecalciferol (vitamin D3). However, vitamin D metabolism is impaired due to failure of the hydroxylation of 25-hydroxy-cholecalciferol to 1,25-dihydroxy-cholecalciferol in the diseased kidney. In an attempt to raise serum calcium there is a secondary hyperparathyroidism and calcium is removed from bone stores giving rise to the characteristic radiographic appearance of renal osteodystrophy. Other dental manifestations include demineralization, decreased trabeculation, loss of lamina dura, macrognathia, tooth mobility, malocclusion, enamel hypoplasia and pulp stones.
Dental management
The observed dental changes depend on the time of onset of renal disease. Those teeth calcifying during renal failure will exhibit chronological hypoplasia or hypomineralization of the enamel and dentine. Developing teeth are often stained green or brown due to the incorporation of blood products such as unconjugated bilirubin or haemosiderin, respectively. Caries is often minimal in these children, possibly due to urea metabolites in the saliva, but supragingival calculus formation is markedly increased, even when oral hygiene is adequate.
• Consultation with a renal physician or nephrologist is often required before these children can receive routine dental treatment.
• Children with acute renal conditions should have elective dental treatment postponed until restoration of their renal function.
• Emergency or palliative care is only indicated following pre-treatment screening for elevated bleeding time or APTT.
• Extraction of pulpally involved primary teeth is the preferred treatment option due to the risk of chronic bacteraemia following pulpotomy or pulpectomy.
• Symptomatic patients with proteinuria or on long-term steroid therapy are best managed in the hospital environment where blood pressure and fluid balance can be monitored before treatment. Fluids and electrolytes in such children can be adjusted by the nephrologist and steroid supplementation can be given prior to general anaesthesia or a major dental procedure.
Dialysis
Children receiving dialysis often exhibit somatic growth retardation, and are pale and anaemic on presentation. They also have a bleeding tendency due to increased capillary fragility and thrombocytopenia. In addition, children on haemodialysis receive anticoagulation with intravenous heparin, and can experience other complications such as infection of the port site and increased risk of hepatitis. In children receiving peritoneal dialysis, complications can occur with catheter placement including peritonitis and exit-site infections. However, peritoneal dialysis is easier to manage in children, requiring less time for the fluid exchange, less restriction of food and fluid intake, and fewer haemodynamic problems compared with haemodialysis.
• Children on haemodialysis and anticoagulant therapy can be successfully managed with pretreatment DDAVP and antibiotic prophylaxis to prevent infection of the access device.
• Any dental treatment, especially extractions, should be performed the day after dialysis when the heparin is no longer active (heparin half-life is 4 h but residual effects can occur for 24 h).
• Sockets should be packed with a haemostatic agent and sutured well. Platelet transfusions are to be avoided if possible.
• Children on continuous ambulatory peritoneal dialysis can be managed more conservatively.
Drug interactions
Drug interactions can occur in children with end-stage renal failure who are managed with long-term antihypertensives and steroids. Medications that are metabolized in the kidney or nephrotoxic should be avoided in children with renal insufficiency. These include:
Dentists should also be aware that renal excretion of drugs is also impaired and their half-life may be extended. However, if children are adequately haemodialysed, it may be necessary to increase dosage of drugs to obtain the necessary pharmacological effect. Adjustment of a drug dosage as well as timing of intake should be made in consultation with the child’s nephrologist.
Renal transplantation
A pre-transplant dental assessment is essential to reduce the risk of oral complications following organ transplant and immunosuppression. The presence of dental caries and oral infections will necessitate delay in transplantation until all potential foci of infection are eliminated. Comprehensive dental treatment and institution of a rigorous preventive programme are recommended prior to transplant to reduce the risk of subsequent oral diseases. Antibiotic prophylaxis as per the protocol for prevention of infective endocarditis is essential prior to invasive surgical procedures.
Gastroenterology
Hepatic and biliary disorders
Biliary atresia
In this condition, there is congenital obliteration or hypoplasia of the bile ducts, resulting in biliary cirrhosis and portal hypertension. In severe cases, transplantation is necessary (Figure 12.11).
Dental implications
• Coagulation problems are of major concern due to the reduction in production of vitamin-K-dependent clotting factors (II, VII, IX, X).
• Patients are immunocompromised. Children are usually mildly anaemic due to destruction of red blood cells. High levels of circulating red-blood-cell degradation products may become incorporated in developing enamel. High levels of unconjugated bilirubin will cause green developmental staining of enamel.
Dental management
Hepatitis A (infectious hepatitis)
Transmission period is very short (3 weeks). The important point to note is that there is no carrier state. If a patient is hepatitis-A positive treatment should be delayed for at least 4 weeks.
Hepatitis B (serum hepatitis)
Chronic active carrier (HBsAg +ve, e +ve)
• The chronic active carrier has active viral replication and is very infective. These patients have active liver disease, and their liver function may be abnormal.
• It is important to liaise directly with the patient’s doctor in the first instance when planning dental treatment and to do so again at regular intervals while the patient is under the care of the paediatric dental unit.
Liver function tests
These are useful in chronic active carriers. Liver function tests should be considered in chronic healthy carriers (HBsAg +ve, e −ve), if recently diagnosed or just recently become negative for HBsAg, and in patients who have just recovered from hepatitis A.
Hepatitis C (non-A, non-B hepatitis)
The hepatitis C virus (HCV) is the cause of what was previously termed non-A, non-B hepatitis. Patients with antibodies to HCV are chronic carriers and are potentially infectious. They are usually asymptomatic although their liver enzymes may be intermittently abnormal. Transmission of HCV is primarily by blood or blood products; however, the virus can be detected, by polymerase chain reaction (PCR), in the saliva of chronic carriers.
Liver transplantation
Children with end-stage liver disease and doubtful prognosis are candidates for liver transplantation. Unfortunately, there is an acute shortage of organs for transplantation and many children succumb to their illness before a donor organ is found. Complete oral and radiographic evaluation is essential prior to transplantation. Any potential foci of oral infection, including exfoliating primary teeth should be eliminated before transplantation can proceed. Teeth with large carious lesions, even if not pulpally involved, should be extracted.
Blood transfusion with packed red cells or partial exchange transfusion with coagulation factors is often required. Prophylactic antibiotic therapy before surgical procedures should be instituted. The main management problem with children undergoing liver transplantation is that of immunodeficiency.
Immunosuppressive therapy includes various corticosteroids and cyclosporin. Cyclosporin is both nephrotoxic and hepatotoxic and can cause hypertension. However, most children are more easily treated post-transplantation due to the cure of their original disease. This is especially true of children with end-stage hepatic failure.
Oesophageal disorders
Neuromuscular control of the lower oesophageal sphincter is inadequate in some children resulting in chronic gastro-oesophageal reflux disease (GORD). The exact pathophysiology of GORD is unknown, although gastric motor abnormalities characterized by delayed gastric emptying have been observed in some children. Regurgitation is not always clinically obvious or symptomatic and 24-hour oesophageal pH monitoring may be necessary to demonstrate significant GORD. Symptomatic children are given oral metoclopramide hydrochloride (Maxolon) to prevent reflux, whereas in children with concurrent oesophagitis, a histamine H2 antagonist such as cimetidine is helpful. Surgical treatment is required if medical therapy fails.
Increasingly, paediatric dentists may be the first professionals to observe the chronic effects of undiagnosed GORD on the primary dentition, namely severe dental erosion of the canine and molar teeth. If a diagnosis of GORD is suspected, prompt referral to a gastroenterologist is recommended for endoscopy and a pH monitor. Fluoride mouth rinses and varnishes help in remineralization. Comprehensive dental treatment with stainless steel crowns is usually required to restore the lost vertical dimension.
Inflammatory bowel disease (see Chapter 8)
Commonly, ‘chronic inflammatory bowel disease’ entails ulcerative colitis and Crohn disease that may represent ends of a spectrum of tissue reaction to a common agent. Establishing an aetiological agent is difficult. Both conditions present as a chronic inflammatory process of the gastrointestinal tract with acute exacerbations. Of particular interest to the dentist, is the association with orofacial granulomatosis that sometimes precedes the onset of ulcerative colitis by 1–2 years.
Oral changes reported in some children include linear ulceration or fissuring of the buccal and labial mucosa, diffuse swelling of the lips, angular cheilitis and diffusely swollen erythematous gingivae, chronic granulomatous lesions. These lesions may improve with sulfasalazine (Salazopyrin) therapy but usually reappear on the buccal mucosa periodically.
Endocrinology
Diabetes mellitus
Type 1 or insulin-dependent diabetes mellitus (TIDM) is the most common form of diabetes in children. Approximately 1 in 1200 children between the ages of 5 and 18 years have the disease. Type 1 diabetes results from autoimmune destruction of the insulin-producing β cells in the pancreas, which is thought to be triggered by viral or toxic insults to the pancreas in the child genetically predisposed to developing the disorder. The goal of treatment is to maintain blood glucose levels in a range that prevents the development of short-term and long-term complications. The target blood glucose ranges are 5–12 mmol/L in children less than 5 years of age, 4–10 mmol/L for middle childhood and 4–8 mmol/L thereafter.
Management protocols
While there are individual variations, contemporary management generally involves administration of a long-acting insulin once or twice per day supplemented with short-acting insulin at meals. For those children with insulin pumps, a subcutaneous continuous infusion of short acting insulin maintains a (variable, but programmed) basal level throughout the day. Additional bolus doses are given with meals (Figure 12.12).
Figure 12.12 An insulin pump showing the position of the subcutaneous canula, usually sited in the abdomen. The pump can be programmed to deliver a basal level of insulin and bolus doses as required with meals mimicking the function of the pancreas.
Pancreatic islet cell transplantation and stem cell research raises the possibility for cure for juvenile diabetes and is currently under critical investigation.
Relatives of patients with diabetes are 2.5 times more likely to develop the disease than the population at large. Presenting symptoms include:
A diagnosis of diabetes is made when fasting blood glucose level above 7.7 mmol/L is recorded or a random blood glucose level >11.0 mmol/L is recorded. Occasionally, children will have diabetes diagnosed following an abnormal oral glucose tolerance test.
Dental implications
• Periodontal disease is the most consistent oral finding in children with diabetes and is more common if control of blood glucose levels is suboptimal. These children exhibit increased alveolar bone resorption and inflammatory gingival changes. This may mimic the clinical manifestation of chronic generalized juvenile periodontitis.
• Xerostomia and recurrent intraoral abscesses may also be present in severe cases.
• Enamel hypocalcification and hypoplasia along with reduced salivary flow can predispose these patients to an increased risk of early childhood caries.
• Altered oral flora changes can occur with an increase in Candida albicans, haemolytic streptococci and staphylococci.
Dental management
• Children with well-controlled diabetes can receive dental treatment in the usual way, except when a general anaesthetic is required. The best measure of control is the level of glycosylated haemoglobin (HbA1c). The target for most children is a value of <6.5–7.0% (≤53 mmol/mol). For routine dental appointments, the child should eat a normal meal prior to the dental procedure although a glucose source should always be available to treat hypoglycaemia.
• Fasting before a general anaesthetic requires careful blood glucose monitoring and adjustment of insulin doses during the fasting period. This is to prevent extremes of hyper- and hypoglycaemia. Insulin doses and the possibility of intravenous fluid therapy should be discussed with the child’s treating paediatrician/endocrinologist.
• Post-surgical healing can be delayed, particularly if blood glucose control is suboptimal and oral sepsis can be an additional risk.
• It is often possible to manage many T1DM children requiring general anaesthesia in day-stay facilities, provided that children can begin taking fluids shortly after their procedure. Other children may require admission to a paediatric hospital, with a dextrose and insulin infusion set-up to maintain BGL levels and avoid complications during the fasting and postoperative periods. It is obviously essential to liaise with the treating endocrinologist.
• Prophylactic antibiotic therapy is recommended prior to surgical procedures.
Pituitary disorders
Hypopituitarism
Hypopituitarism (complete or partial) may be either congenital or secondary to pituitary or hypothalamic disease (tumours, infections, trauma or after exposure to ionizing radiation). These children need to be managed in conjunction with a paediatric endocrinologist to prevent potentially serious complications of pituitary hormone deficiency.
Isolated growth hormone (GH) deficiency is the most common pituitary hormone deficiency, but evolution of other deficiencies may take place over time. Treatment consists of supplemental GH that stimulates the differentiation of epiphyseal growth plate precursor cells and induces clonal expansion of cartilage cells. Timely detection and treatment of GH deficiency is essential to minimize the growth disturbance.
Dental implications
• It is important to ensure that any child with ACTH deficiency having general anaesthesia, be given stress doses of hydrocortisone (100 mg/m2) as an IV injection at the onset of anaesthesia. Until the child has recovered from the surgery, stress doses of hydrocortisone will be required. The exact dose and route of administration will vary depending on the situation. An endocrinologist should always be consulted prior to any procedure requiring a GA in these children to guide the hydrocortisone therapy.
• Hypopituitarism can decrease linear facial measurements (particularly in the posterior facia height) and linear cranial base measurements.
• Children often present with an open bite accompanied by the typical immature hypopituitary facies.
• Somatic skeletal development is consistently more retarded than craniofacial development, although tooth eruption and root formation can be delayed or incomplete.
• Early orthodontic assessment is required to monitor orofacial growth and development.
Pituitary hormone excess
Gigantism results from primary hypersecretion of growth hormone during childhood. Gigantism is extremely rare in children and is associated with pituitary tumours and McCune–Albright syndrome.
Dental implications
• Precocious and accelerated development of the craniofacial skeleton.
• Accelerated dental development and eruption.
• Enlarged crenated tongue, and coarse facial features.
• Radiographically, there is a marked thickening of the cranium and cortical bone of the mandible.
• Osseous structures exhibit overdevelopment with poor maturation and bone quality (osteoporosis) and hypercementosis of tooth roots is common.
Dental management
Dental management of patients with pituitary disorders focuses mainly on the management of the associated craniofacial malformations. Treatment needs to be planned carefully and coordinated in a multidisciplinary setting. No contraindications exist for comprehensive dental healthcare but the treating endocrinologist must be consulted prior to any invasive treatment or general anaesthesia.
Thyroid disorders
Hypothyroidism
Thyroid hormone deficiency (hypothyroidism) is most often due to a primary disorder of the thyroid gland and less commonly secondary to hypothalamic and/or pituitary insufficiency. Primary hypothyroidism may be either congenital or acquired (thyroiditis, after radiation exposure or surgical excision). Untreated congenital hypothyroidism leading to severe developmental delay (Cretinism) is rare in those countries where neonatal testing is performed (TSH with or without fT4) and treatment with synthetic thyroxine started in the first 1–2 weeks of life. Congenital hypothyroidism can be attributed to aplasia, hypoplasia or maldescent of the thyroid gland, abnormal thyroid hormone production (dyshormonogenesis) or prenatal iodide deficiency. Juvenile hypothyroidism can stem from autoimmune thyroiditis, thyroidectomy, thyroid irradiation, infection or medication.
Although hypothyroid changes can include: growth failure, diminished physical activity, decreased circulation, poor muscle tone, speech disorders, delayed mental development and craniofacial manifestations, these changes are obviously dependent on the age of onset of the disease, the degree of hypothyroidism and the timing of diagnosis and treatment. Most hypothyroid children have very few clinical features, despite a quite severe deficiency state.
Dental implications of untreated or suboptimally treated hypothyroidism
• Decreased vertical facial growth.
• Decreased cranial base length and flexure, maxillary protrusion, and open bite with typical immature facial patterns.
• Delayed eruption of teeth and increased spacing between both primary and permanent teeth.
• Developmental anomalies such as enamel hypoplasia have been reported.
Hyperthyroidism
Hyperthyroidism can occur due to autoimmune hyperstimulation of the gland (Grave’s disease or Hashimoto’s thyroiditis), inflammatory disorders (subacute thyroiditis), iodine excess or rarely neoplasm. In children, the most common causes are Graves’ disease, and thyroiditis. Thyrotoxicosis is more common in females and is most likely to appear between 8 and 16 years of age and is usually associated with a goiter.
Changes associated with hypothyroidism are largely related to hyperactivity of the sympathetic nervous system. Typical clinical manifestations include nervousness, poor school concentration with deteriorating performance, muscle weakness, heat intolerance, loss of weight despite increased appetite, insomnia, tachycardia, palpitations, marked perspiration and gastrointestinal disturbances. In addition, ocular abnormalities such as eyelid lag, exophthalmos and widening of the palpebral fissures may be seen in some cases.
Dental implications
• Accelerated growth and development of the craniofacial complex and skeleton.
• Precocious eruption of teeth.
• Increased susceptibility to caries and periodontal disease, burning mouth syndrome.
• Osteopenia or poor bone mass accrual.
• Typical orofacial changes are increased vertical facial height with anterior open bite and mild mandibular prognathism.
Dental management
The principal concern in children with thyroid disorders is the increased risk associated with general anaesthesia. The hypothyroid patient is at risk of extreme delay to wake normally after general anaesthetic.
If a child with inadequately controlled hyperthyroidism is anaesthetized, there is a risk of precipitating thyroid storm, a very dangerous condition of tachycardia, acute onset of fever, with possible cardiac failure and death. The untreated patient is also at risk from oral infection or surgical procedures as thyroid crisis may be precipitated.
Rarely, intercurrent infections may precipitate significant worsening of hyperthyroidism. As such, oral infections should be treated aggressively and thyroid function (TSH and fT4) monitored. Rare side-effects of antithyroid drugs include parotitis and agranulocytosis, which predispose the patient to bleeding episodes, ulceronecrotic lesions and chronic oral infections.
In general, a child with well-controlled thyroid disease can undergo dental management similar to any other child. Where possible, thyroid function should be normalized prior to dental intervention. In the unusual situation where this is not possible, close collaboration with a paediatric endocrinologist is recommended.
Parathyroid disorders
Hypoparathyroidism
Hypoparathyroidism results from structural or functional deficiencies in the parathyroid glands. In children, it most commonly presents as hypocalcaemia in the neonatal period in association with complex disorders, e.g. Di George syndrome and CATCH22 disorders. During childhood it may result from autoimmune destruction of the parathyroid gland, after parathyroidectomy for gland over-activity in association with multiple endocrine neoplasia or after inadvertent parathyroidectomy at the time of thyroidectomy.
Treatment focuses on maintaining near normal serum calcium by calcium and calcitriol supplementation. Trials are underway to supplement with recombinant PTH. If untreated, hypocalcaemia can lead to neuromuscular excitability, tetany, muscle weakness, lethargy and fatigue. Associated syndromes have a variety of clinical features including cardiovascular dysfunction, epilepsy, ectodermal defects, immune dysfunction and craniofacial manifestations.
Dental implications of untreated hypoparathyroidism
• Circumoral paraesthesia and spasm of the facial muscles has been reported in severe hypocalcaemia.
• Hypoplasias of enamel, wide pulp chambers, pulp calcifications.
• Hypodontia and root anomalies are common clinical findings.
• Tooth eruption can be markedly delayed or arrested.
• Increased risk of acute and chronic oral candidiasis has also been reported due to associated immune dysfunction.
Pseudohypoparathyroidism (PHP)
This is a heterogeneous disorder that has at its core, resistance to PTH action. Inheritance of PHP is classically an X-linked dominant disease in which there is resistance to the effect of PTH with resultant increase in PTH but low serum calcium. It is characterized by low serum calcium, high phosphate levels and high PTH levels. If inherited from the mother, the child will have typical physical features of short stature, round face and short hands (Albright’s hereditary osteodystrophy), but if inherited from the father, biochemical changes will only be present.
Hyperparathyroidism
Excessive production of parathyroid hormone may result from a primary defect in the gland (adenoma, hyperplasia, hypertrophy) or secondarily as a compensatory phenomenon, usually correcting hypocalcaemic states due to rickets or from chronic renal disease. Tertiary hyperparathyroidism (autonomous hyperparathyroidism) can occur following prolonged and uncontrolled secondary hyperparathyroidism. Primary and tertiary disease results in hypercalcaemia and hypercalcuria, muscle weakness, abdominal pain and constipation, polyuria, polydipsia, kidney stones and bone loss. In infants and young children, there may be failure to thrive, poor feeding and muscular hypotonia.
The bony lesions are rarely seen in children but include brown tumours, so-called because they contain areas of haemorrhage, an abundance of multinucleated giant cells, fibroblasts and haemosiderin. Hypophosphataemia can lead to rickets in children and osteomalacia in adults. Generalized osteoporosis with cortical resorption is the most common bone lesion in adults and radiographic signs can also include multiple rarefactions, loss of typical trabeculation, ground-glass appearance and metastatic calcifications.
Dental implications
• Severe malocclusion and drifting of teeth with no apparent pathological periodontal pocketing have been reported.
• Radiographic changes with metastatic soft-tissue calcifications, periapical radiolucencies, root resorption, loss of lamina dura, and generalized loss of radiodensity.
• Dental abnormalities associated with underlying disorder if secondary or tertiary hyperparathyroidism.
Dental management
Generally, routine dental treatment involves no treatment modifications provided there are no associated medical complications present. Pitting enamel hypoplasia and failure of tooth eruption may occur. Addison’s disease (adrenal insufficiency), on the basis of autoimmunity, may accompany hypoparathyroidism and thus the child may be at risk from stressful procedures such as oral surgery and general anaesthesia.
Hypo- and hypercalcaemia may be associated with cardiac arrhythmias that increase the risk of cardiac arrest during general anaesthesia. Risk of pathological fracture in advanced cases of hyperparathyroidism should be a consideration during oral surgical procedures. Splinting of mobile teeth is a useful adjunct to prevent further drifting following stabilization of the dentition.
Adrenal gland disorders
Adrenal glands have two endocrine functions, located within the cortical and medullary areas. The adrenal cortex produces three major classes of steroid hormones: glucocorticoids, mineralocorticoids, and sex hormones. Glucocorticoids (cortisol) have an important role in carbohydrate, fat and protein metabolism, assist in the maintenance of normal blood pressure and protect the body against stresses of various types. Mineralocorticoids (aldosterone) help maintain salt and water balance through their action on the kidney. Adrenal sex hormones help complement the actions of the gonadal steroids in the development of sexual characteristics and reproductive capability.
Adrenal insufficiency
The major problems associated with adrenal hypofunction are the result of glucocorticoid and mineralocorticoid deficiency. Primary adrenal insufficiency (Addison’s disease) is a chronic condition characterized by:
There are low blood cortisol levels, and an acute adrenal crisis (hypotension and collapse, electrolyte disturbance and hypoglycaemia) can be precipitated by a relatively minor stress. This is a medical emergency, and management is with immediate steroid supplementation. Secondary adrenal insufficiency is caused either by prolonged administration of steroids resulting in the suppression of endogenous cortisol or by a central lack of adrenocortical stimulating hormone (ACTH). Children do not usually present with obvious clinical signs unless stressed and an adrenal crisis can occur without warning.
Congenital adrenal hyperplasia
Congenital adrenal hyperplasia (CAH) is the most common congenital cause of adrenal insufficiency. CAH arises when there is an enzymatic block in the complex biosynthetic pathways from cholesterol to cortisol and mineralocorticoid. Females usually present in the neonatal period with ambiguous genitalia. Untreated, CAH is fatal with babies becoming desperately unwell – vomiting, electrolyte disturbance, hypoglycaemia and shock. Management includes immediate and long-term replacement of cortisol, together with restoring electrolyte balance with mineralocorticoid replacement therapy.
Adrenocortical hyperfunction
Cushing syndrome refers to a clinical condition characterized by manifestations of adrenocortical hyperfunction, with physical features of growth failure together with weight gain, ‘moon facies’, truncal obesity, formation of striae, hirsutism and poor wound healing. It is caused by a tumour in the pituitary gland secreting ACTH (Cushing’s disease) or in the adrenal, in which case treatment requires removal of the lesion surgically. It may also be iatrogenic or secondary to treatment of inflammatory or immune diseases with high-dose glucocorticoids.
Dental management of adrenal insufficiency
It is important to confirm the diagnosis and medical management with the endocrinologist before commencement of any dental treatment. Any child with adrenal insufficiency must have a plan for stress glucocorticoid therapy prior to undertaking dental treatment. Failure to do so may result in serious medial complications for the child. An assessment of the potential for adrenal suppression can be considered from the duration and dosage of previous corticosteroid treatment but if a child has used steroids for more than 3–4 weeks, there is a risk for adrenal suppression and advice should be sought prior to treatment as to the necessity of giving steroid cover during any procedure. Supplemental steroids should be given following medical consultation and proportional to the degree of adrenal suppression and the perceived stress of the dental procedure. It is beyond the scope of this chapter to outline the various treatment strategies that best suit an individual patient. As such, individual specialist paediatric endocrine consultation is required.
Neurology
Febrile convulsion
This is a term used to describe a child under 5 years of age who has a seizure in response to a febrile illness. It usually occurs with body temperatures over 38°C and when no other cause can be determined. Recurrent, non-febrile seizures are termed as epilepsy.
Epilepsy
Epilepsy is the most common disorder in paediatric neurology and the predominant aetiologies are birth injury and congenital abnormalities.
Convulsions can be classified as:
Lennox–Gastaut syndrome is an intractable form of severe epilepsy with clinical features of head-dropping attacks, atypical petit mal and brief tonic–clonic seizure usually at night.
Modern treatment is usually restricted to the use of one anticonvulsant medication and slowly increasing the dose to achieve therapeutic blood levels and minimizing side-effects. About 70% of children do very well, with only minimal problems, even if treatment is long term. However, drowsiness, ataxia, excessive salivation, hyperactivity and aggressive behaviour are common complications of anticonvulsant therapy. Gradual reduction of multiple medications has a beneficial effect in terms of increased alertness, and a reduced number of seizures. Phenytoin sodium (Dilantin) is still probably the most effective drug for grand mal seizures but the cosmetic side-effects such as hirsutism and gingival hypertrophy have reduced its use in favour of carbamazepine (Tegretol).
Dental implications
The major oro-dental concerns with epileptic children are gingival enlargement (Figure 12.11) and the precipitation of a seizure in the dental surgery.
Dental management
Management of gingival hypertrophy (Figure 12.9B) is dependent on oral hygiene and dental development at diagnosis. In the permanent dentition, full mouth gingivectomy may be required, but gingival overgrowth will recur if oral hygiene is not optimal. Maintenance of adequate oral hygiene may be especially difficult in children with additional intellectual disability and is highly dependent on the motivation and skill of the parents and caregivers. Battery-operated plaque removers with small circular heads are beneficial in the maintenance of good oral hygiene in the more intractable cases. The use of daily chlorhexidine-containing gels is effective in reducing the inflammatory component of the gingival overgrowth. It is important to always keep the interests of the child in mind, particularly with regard to aggressive surgical treatment that may not benefit the child in the long term.
The general goal of dental management is the avoidance of a seizure. It is important to know the type of epilepsy and any precipitating factors, medications and dosage, compliance and degree of seizure control before commencing treatment. In addition, drug interactions with anticonvulsants are common and their half-life and blood levels can be increased substantially. Consultation with the child’s neurologist is essential before commencement of treatment.
The following management protocol is recommended for prevention and control of seizures in the dental surgery:
• Reduce stress to the child by behavioural management and conscious sedation techniques.
• Reduce direct overhead lighting, particularly for the photosensitive form of epilepsy.
• Avoid seizure-promoting medications, such as CNS stimulants and local anaesthetics containing adrenaline (epinephrine).
• Emergency drugs such as oxygen, intravenous or rectal diazepam (Valium) and intravenous phenobarbital sodium should be readily available.
• Pre-arranged transfer to a paediatric hospital, in case required.
General anaesthesia is preferable in children with poor seizure control as the abnormal neural activity is completely ablated during the procedure. Dental trauma is an obvious consequence in the child with frequent, poorly controlled seizures. Removable appliances are contraindicated in an epileptic child due to potential airway obstruction.
Respiratory disease
Asthma
Australia has one of the highest rates of childhood asthma in the world, with 1 in 5 children and 1 in 7 adolescents affected. It is a respiratory condition characterized by increased responsiveness of the airways to a wide variety of stimuli, leading to widespread narrowing of the airways resulting in symptoms of dyspnoea, wheezing and coughing. Precipitating factors include emotional stress, exercise, cold air, viral respiratory infections, air pollution and aspirin. The condition is reversible, either spontaneously or as a result of bronchodilator therapy. Currently, there is an emphasis on prophylactic medications to prevent episodes rather than simply treating acute attacks.
Bronchodilators include β2-adrenergic drugs such as salbutamol sulphate (Ventolin) and theophylline (Nuelin). Preventive agents include disodium cromoglycate (Intal) and oral corticosteroids such as prednisone and inhaled corticosteroids such as beclomethasone dipropionate (Beclovent) and salmeterol xinafoate (Seretide).
Dental implications
• The major concern to the paediatric dentist is the exacerbation of an acute attack in the dental surgery.
• Avoid any known precipitating factors prior to dental treatment and ensure that the child has the appropriate medication (inhaler) for emergency use if an asthmatic attack occurs during dental treatment.
• Some bronchodilator and corticosteroid medications may cause extrinsic staining of the teeth due to changes in oral flora and may also predispose to oral candidiasis.
• Children on high-dose corticosteroids (>1600 µg/day) may be immunocompromised and may also require additional supplemental corticosteroids on the day of dental treatment due to adrenal suppression.
• Children are often mouth breathers causing gingivitis and swelling of anterior gingival tissues.
• Disturbances in taste, xerostomia and gastro-oesophageal reflux due to β-2-agonist that may also contribute to tooth surface loss, are other concerns.
Dental management
• Regular dental prophylaxis may be necessary if extrinsic staining is present.
• The use of inhalers with a spacer device minimizes medication deposits in the oral cavity and oropharynx, thus allowing more of the particles to be inhaled into the lung.
• The use of saliva substitutes can improve xerostomic effects. Xylitol-containing products reduce the acidogenic potential of plaque.
• Advise parents to brush their child’s teeth following administration of medication or use an aqueous mouth rinse if oral hygiene is not possible, particularly during the night to reduce the risk of dental erosion and counteract the acidic pH of the medication.
• Children who have been admitted to hospital with frequent episodic asthma within the past 12 months or those managed with high-dose oral corticosteroids are not suitable for day surgery and should be admitted preoperatively to be adequately prepared by the paediatric respiratory team.
• Avoidance of adrenaline-containing local anaesthetic solutions is recommended due to their potential adrenergic effects if injected intravenously.
• There are no known contraindications to the use of nitrous oxide or sedative doses of benzodiazepines or antihistamines for asthmatic children.
Cystic fibrosis
This multisystem disorder is an autosomal recessive condition affecting all mucus-secreting exocrine glands of the body, especially the respiratory and gastrointestinal systems. The incidence is 1 in 2500 and the heterozygote frequency is estimated to be 1 in 25 in the Caucasian population. Cystic fibrosis is characterized by chronic respiratory obstruction and infection, intestinal malabsorption and growth retardation. Sexual development is often delayed. Although the disease is currently incurable, aggressive symptomatic treatment has improved survival and quality of life in recent years. Long-term complications include sinusitis, pneumothorax, osteopenia, diabetes, liver disease with problems in bleeding and drug metabolism. The major cause of death is respiratory failure from recurrent chest infections.
Children were previously treated with broad-spectrum tetracyclines and often exhibited classic intrinsic staining and enamel hypoplasia of the permanent dentition. However, in Australia, this practice stopped in the early 1970s and the incidence of enamel anomalies has decreased dramatically in recent years. Increased and altered rates of salivary flow seem not to predispose to dental caries, possibly due to long-term use of antibiotics.
Dental management
• Dental management should be based on optimal prevention, including strict oral hygiene and diet control. Routine dental treatment can be undertaken provided pulmonary involvement is stable and well controlled with medication.
• Local anaesthetics with vasoconstrictors are not contraindicated.
• Long appointments should be avoided. However, the use of conscious sedation or general anaesthesia must be carefully discussed with the respiratory paediatrician and only used when routine pain management is not possible.
• Physiotherapy and prophylactic antibiotic treatment is required both preoperatively and postoperatively in cases of severe pulmonary involvement.
• Although, life expectancy has increased with current medical therapies, there should be a rational and pragmatic approach to treatment planning and expectations.
Tuberculosis
Tuberculosis is an infectious disease caused by Mycobacterium tuberculosis that involves granulomatous inflammation and caseating necrosis in the lung tissues. The most common presentation in children is malaise, anorexia and weight loss, fever and cough with signs of cervical lymphadenitis. Children are managed with long-term antibiotic treatment specific to the infectious organism. Chemoprophylaxis is given to other family members and contacts, to prevent spread of the disease. The bacille Calmette–Guérin (BCG) vaccine is used routinely in communities with a high prevalence of tuberculosis.
Dental treatment should not be performed until the diagnosis and causative agent have been identified and only then be performed following consultation with the respiratory physician.
Genetics and dysmorphology
Diagnosis
Although individually uncommon, many children with genetic disorders will present to the paediatric dentist with specific dental anomalies associated with their condition or medical problems that complicate their dental management. Never assume that all conditions will have been diagnosed before they present, as many children are often diagnosed as having a significant genetic disorder quite late in childhood, either because the condition has late manifestations or because features have simply been missed. When taking a history, it is always useful to draw a simple three-generation family pedigree (Figure 12.13).
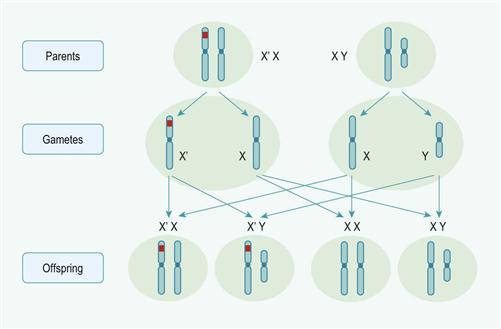
Figure 12.13 Calculation of risk in an X-linked recessive disorder. For a carrier mother (illustrated), there is a 50% chance that sons will be affected and a 50% chance that daughters will be carriers; for an affected father, no sons will be affected but all daughters will be carriers.
Many disorders do not follow Mendelian inheritance patterns, but are clearly of ‘familial’ or hereditary nature. Many important and common conditions fall into this group; they are often not single entities but causally heterogeneous and are seen as the end result of multiple gene effects against a variable environmental background (multifactorial or polygenic), e.g. cleft lip and palate.
One in 170 live-born births has a major chromosomal abnormality. Chromosomal abnormalities, particularly those involving imbalance of the autosomes, usually result in developmental delay and dysmorphic features and often include multi-system anomalies. These include numerical abnormalities (trisomy 21 or Down syndrome) and structural abnormalities such as segmental deletions (cri-du-chat syndrome), duplications and unbalanced translocations. Numerical abnormalities of the sex chromosomes are better tolerated than those of the autosomes and include conditions such as Turner syndrome (45, X) in females and Klinefelter syndrome (47, XXY) in males. Standard cytogenetic karyotyping is now being replaced by DNA-based chromosome micro-arrays (CMA) that can detect many more significant submicroscopic deletions and duplications (pathological copy number variations). Many other genetic disorders are caused by mutations within genes, which cannot be detected by karyotype or CMA analysis, but may be able to be confirmed by targeted gene testing after a syndrome is recognized (Table 12.1).
Table 12.1
Classification of genetic abnormalities
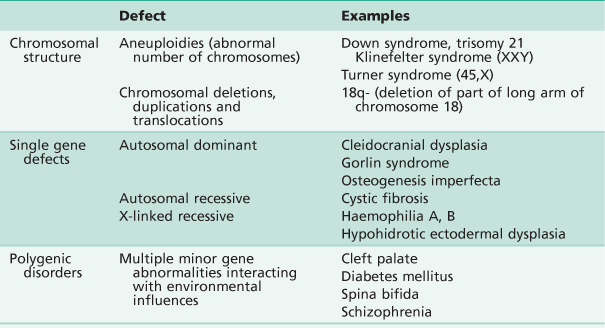
Adapted from Jones, K.L., 1997. Smith’s recognizable patterns of human malformation, fifth ed. WB Saunders, Philadelphia.
The first step in making a diagnosis is the recognition that the child is dysmorphic or unusual looking. It is the pattern of dysmorphism and associated malformations rather than a single feature that aids diagnosis. Accurate diagnosis is the key to prognosis, management and sometimes the underlying genetic cause of the disorder. It also helps the parents, as it removes the anxiety of uncertain aetiology and prognosis.
The diagnostic procedure involves history, clinical examination and laboratory investigations. Based on the above information, children can be divided into three basic groups:
Referral to a genetic clinic is recommended for all children with multiple anomalies or dysmorphic features of unknown cause. Syndrome diagnosis aids (available by paid subscription) such as POSSUM (www.possum.net.au) or the London Dysmorphology Databases (www.lmdatabases.com/) can assist. Laboratory investigations include radiographic survey, chromosomal or microarray analysis, biochemical screening of urine or blood or cultured fibroblasts for specific enzymatic or protein deficiencies if a metabolic disorder is suspected. Referral to a genetics clinic is usually recommended to further elaborate a diagnosis and to assist with specialized genetic testing and genetic counselling.
It is not the intention of this section to detail those anomalies with a major craniofacial or orofacial manifestation, of which dental clinicians should be aware. The reader is directed to texts and online sources that more comprehensively cover these conditions (see References and further reading, below).
Terms used in morphogenesis
Sequence
A pattern of multiple anomalies arising from a single structural defect or event; previously termed anomalad. They usually originate early in development with single problems that create secondary anomalies and manifest with multiple defects at birth or later. These sequences may be divided into three basic groups:
Malformation sequence
• May be single or multiple but are primary structural abnormalities.
Malformations may present as:
• Accessory tissue (e.g. polydactyly, preauricular skin tags).
• Hamartomas (e.g. haemangioma/lymphangioma).
• Agenesis (e.g. salivary gland agenesis).
• Hypoplasia (e.g. enamel hypoplasia in amelogenesis imperfecta).
• Incomplete septation (e.g. ventral septal defect, VSD).
• Incomplete migration of neuroectoderm cells (e.g. Di George association).
• Failure of cell death (apoptosis) (e.g. spina bifida, syndactyly).
Disruption sequence
• Destruction or breakdown of normal tissue, which may result from vascular, infective or physical causes.
• Low risk of recurrence, poor prognosis for normal growth in affected areas.
• Deafness from congenital rubella.
• Facial clefting from amniotic bands.
• Hemifacial microsomia (from stapedial artery haemorrhage).
Risk of recurrence in genetic disorders
Autosomal dominant
There is a 50% risk to each offspring of a single affected parent. Many dominant conditions have variable expression and so the manifestations may be increased or reduced compared with the parent. New dominant genetic conditions can also occur.
Autosomal recessive
Carriers do not express the trait. If both parents are carriers, the risk of the child being affected (homozygous; the trait will be expressed) is 25%; the risk of being a carrier is 50% and the chance of neither being a carrier nor affected is 25%. If one parent is a carrier, there is a 50% chance of the child being a carrier.
X-linked (sex-linked)
• 50% risk of transmission from female carriers to sons (who would then be affected) or daughters (who would then be a carrier).
• No male-to-male transmission from affected fathers to sons, but all daughters will be carriers.
The terms ‘X-linked recessive’ and ‘X-linked dominant’ are used to describe sex-linked conditions with an altered frequency of phenotypic expression. Males usually have only one X chromosome; males with an X-linked abnormality are described as hemizygous for the trait, and will be affected. Females usually have two X-chromosomes; female carriers of an X-linked trait are heterozygous. In X-linked recessive disorders, female carriers can be unaffected or affected (manifesting carrier) but the latter are usually much less severely affected than males. Rarely, females are homozygous for an X-linked trait, so will be affected as severely as a hemizygous male. ‘X-linked dominant’ traits manifest in females and males, but males are often more severely affected.
The degree of phenotypic expression in heterozygous females is determined by the pattern of X inactivation (Lyonization, see Chapter 11) in each tissue. For example, some female carriers of haemophilia A will show a measurable (but subclinical) reduction in factor VIII; and those that carry X-linked hypohidrotic ectodermal dysplasia may also show some phenotypic variation in the dentition such as microdontia and oligodontia, but not to the same extent as in hemizygous males. The markedly increased frequency of females seen with some ‘X-linked dominant’ conditions, such as incontinentia pigmenti or focal dermal hypoplasia (Goltz–Gorlin syndrome) is explained by male lethality for these mutations in most hemizygous males.
Prenatal tests for genetic disorders
Ultrasound
Ultrasound has become a routine investigation for most pregnancies. It is non-invasive to the mother and the fetus and there are many anomalies that may be diagnosed by this technique. Common ultrasound techniques include the first trimester nuchal translucency measurement (a screening examination for Down syndrome risk) and the mid-trimester fetal morphology scan.
Amniocentesis
This is the sampling of cells from amniotic fluid at around 15–18 weeks. A number of tests can be performed including:
Chorionic villus sampling
This test is performed earlier than amniocentesis, at around 11–13 weeks. Similar tests are performed to those done with amniocentesis, although it has the advantage of earlier diagnosis.
Preimplantation genetic diagnosis (PGD)
This is DNA testing of cells biopsied from IVF embryos. This is an option increasingly chosen by couples where genetic risk is high, abortion is not an acceptable option and targeted testing is possible (prior work-up is required).
Postnatal tests
Chromosome analysis (cytogenetic) or chromosomal microarrays (DNA-based)
For any baby with dysmorphic features or multiple abnormalities.
DNA analysis
For specific disorders; direct mutation detection in selected gene(s).
Neonatal screening tests
A range of newborn screening tests is undertaken in each country, but the tests offered vary significantly between centres. Commonly performed tests include those for:
Genetic counselling
Genetic counselling is the process of providing diagnostic assessment, information and support to families or individuals who have, or are at increased risk for, birth defects, chromosomal abnormalities or a variety of inherited conditions. Genetic counselling specialists include clinical geneticists and genetic counsellors. Clinical geneticists provide medical diagnostic assessment and investigation, interpretation of information about the disorder, analysis of inheritance patterns and risks of recurrence and review of available management options with the family. Genetic counsellors take a major role in providing information and supportive counselling to individuals and families, including facilitation of access to resources such as genetic support groups or other community or state support services.
Dental management
The aim of dental management should be a part of a team approach in the care of the child. The overall aim is to reduce the handicapping consequences of the condition. Restorative and surgical management of the specific dental disability (e.g. enamel hypoplasia or hypodontia) should be addressed with a long-term comprehensive management plan. This must involve several dental disciplines such as orthodontics, periodontics, oral surgery and restorative dentistry, working together to coordinate a treatment plan. Overall success in management is measured by the total rehabilitation of the child and family.
Further reading
Endocarditis
See Appendix E.
Haematology
1. Johnson WT, Leary JM. Management of dental patients with bleeding disorders: review and update. Oral Surgery, Oral Medicine, and Oral Pathology. 1988;66:297–303.
2. Morimoto Y, Yoshioka A, Sugimoto M, et al. Haemostatic management of intraoral bleeding in patients with von Willebrand disease. Oral Diseases. 2005;11:243–248.
3. Piot B, Sigaud-Fiks M, Huet P, et al. Management of dental extractions in patients with bleeding disorders. Oral Surgery, Oral Medicine, Oral Pathology, Oral Radiology, and Endodontics. 2002;93:247–250.
4. Warrier AI, Lusher JM. DDAVP: a useful alternative to blood components in moderate hemophilia A and von Willebrand’s disease. Journal of Paediatrics. 1983;102:228.
Immunodeficiency
1. Flaitz CM, Hicks MJ. Oral candidiasis in children with immune suppression: clinical appearance and therapeutic considerations. ASDC Journal of Dentistry for Children. 1999;66:161–166.
2. Malech HL, Nauseef WM. Primary inherited defects in neutrophil function: etiology and treatment. Seminars in Hematology. 1997;34:279–290.
3. Moyer IN, Kobayashi RH, Cannon ML. Dental treatment of children with severe combine immunodeficiency. Pediatric Dentistry. 1983;5:79.
4. Porter SR, Scully C. Orofacial manifestations in the primary immunodeficiency disorders. Oral Surgery, Oral Medicine, and Oral Pathology. 1994;78:4–13.
5. Woroniecka M, Ballow M. Office evaluation of children with recurrent infection. Pediatric Clinics of North America. 2000;47:1211–1224.
Acquired immunodeficiency syndrome
1. Eldridge K, Gallagher JE. Dental caries prevalence and dental health behaviour in HIV infected children. International Journal of Paediatric Dentistry. 2000;10:19–26.
2. Frezzini C, Leao JC, Porter S. Current trends of HIV disease of the mouth. Journal of Oral Pathology and Medicine. 2005;34:513–531.
3. Hicks MJ, Flaitz CM, Carter AB. Dental caries in HIV-infected children: a longitudinal study. Pediatric Dentistry. 2000;22:359–364.
4. Ramos-Gomez FJ, Flaitz C, Catapano P. Classification, diagnostic criteria, and treatment recommendations for orofacial manifestations in HIV-infected pediatric patients Collaborative Workgroup on Oral Manifestations of Pediatric HIV Infection. Journal of Clinical Pediatric Dentistry. 1999;23:85–96.
Oncology
1. American Academy of Pediatric Dentistry Clinical Affairs Committee. American Academy of Pediatric Dentistry Council on Clinical Affairs 2005–2006 Guideline on dental management of pediatric patients receiving chemotherapy, hematopoietic cell transplantation, and/or radiation. Pediatric Dentistry Reference Manual Volume. 2008;34(6):280–286.
2. da Fonseca MA. Dental care of the pediatric cancer patient. Pediatric Dentistry. 2004;26:53–57.
3. Dahllöf G, Barr M, Bolme P, et al. Disturbances in dental development after total body irradiation in bone marrow transplant recipients. Oral Surgery, Oral Medicine, and Oral Pathology. 1988;65:41–44.
4. Dignan FL, Scarisbrick JJ, Cornish J, et al. Organ-specific management and supportive care in chronic graft-versus-host disease. British Journal of Haematology. 2012;158:62–78.
5. Ferretti GA. Chlorhexidine prophylaxis for chemotherapy and radiotherapy induced stomatitis: a randomized double-blind trial. Oral Surgery, Oral Medicine, and Oral Pathology. 1990;69:331–338.
6. Reid H, Zietz H, Jaffe N. Late effects of cancer treatment in children. Pediatric Dentistry. 1995;17:273–284.
Bisphosphonate-associated osteonecrosis
1. Bachrach LK, Ward LM. Clinical review 1: Bisphosphonate use in childhood osteoporosis. Journal of Clinical Endocrinology and Metabolism. 2009;94:400–409.
2. Brown JJ, Ramalingam L, Zacharin MR. Bisphosphonate-associated osteonecrosis of the jaw: does it occur in children? Clinical Endocrinology. 2008;68:863–867.
3. Malmgren B, Astrom E, Soderhall S. No osteonecrosis in jaws of young patients with osteogenesis imperfecta treated with bisphosphonates. Journal of Oral Pathology and Medicine. 2008;37:196–200.
4. Milano M, Wright T, Loechner KJ. Dental implications of osteogenesis imperfecta: treatment with IV bisphosphonate: report of a case. Pediatric Dentistry. 2011;33:342–352.
5. Sambrook P, Olver I, Goss A. Bisphosphonates and osteonecrosis of the jaw. Australian Family Physician. 2006;35:801–803.
6. Schwartz S, Joseph C, Iera D, et al. Bisphosphonates, osteonecrosis, osteogenesis imperfecta and dental extractions: a case series. Journal of the Canadian Dental Association. 2007;74:537–542.
Nephrology
1. Lucas VS, Roberts GJ. Oro-dental health in children with chronic renal failure and after renal transplantation: a clinical review. Pediatric Nephrology. 2005;20:1388–1394.
Gastroenterology
1. Dodds AP, King D. Gastroesophageal reflux and dental erosion: case report. Pediatric Dentistry. 1997;19:409–412.
2. Little JW, Rhodus NL. Dental treatment of the liver transplant patient. Oral Surgery. 1992;73:419–426.
3. Seow WK, Shepherd RW, Thong YH. Oral changes associated with end-stage liver disease and liver transplantation: implications for dental management. Journal of Dentistry for Children. 1991;58:474.
4. Wondimu B, Németh A, Modéer T. Oral health in liver transplant children administered cyclosporin A or tacrolimus. International Journal of Paediatric Dentistry. 2001;11:424–429.
Endocrinology
1. Dahms WT. An update in diabetes mellitus. Pediatric Dentistry. 1991;13:79.
2. Fabue LC, Soriano YJ, Pérez MG. Dental management of patients with endocrine disorders. Journal of Clinical and Experimental Dentistry. 2010;2:e196–e203.
3. Lalla E, Cheng B, Lal S. Diabetes-related parameters and periodontal conditions in children. Journal of Periodontal Research. 2007;42:345–3490.
4. Milenkovic A, Markovic D, Zdravkovic D, et al. Adrenal crisis provoked by dental infection: case report and review of the literature. Oral Surgery, Oral Medicine, Oral Pathology, Oral Radiology and Endodontics. 2010;110:325–329.
5. Twetman S, Petersson GH, Bratthall D. Caries risk assessment as a predictor of metabolic control in young type 1 diabetics. Diabetic Medicine. 2005;22:312–315.
6. Whitlock RI. The jaw lesions associated with hyperparathyroidism. Transactions of the International Conference on Oral Surgery. 1970;1970:322–329.
7. Wray L. The diabetic patient and dental treatment: An update. British Dental Journal. 2011;211:209–215.
Neurology
1. Nelson LP, Ureles SD, Holmes G. An update in pediatric seizure disorders. Pediatric Dentistry. 1991;13:128–135.
Respiratory disease
1. Stensson M, Wendt LK, Koch G, et al. Oral health in pre-school children with asthma – followed from 3 to 6 years. International Journal of Paediatric Dentistry. 2010;20:165–172.
2. Zhu JF, Hidalgo HA, Holmgreen WC, et al. Dental management of children with asthma. Pediatric Dentistry. 1996;18:363–370.
Genetics and dysmorphology
1. Hennekam RCM, Krantz ID, Allanson JE. Gorlin’s Syndromes of the Head and Neck. Fifth edn New York: OUP; 2010.
2. Jones KL. Smith’s Recognizable Patterns of Human Malformation. Sixth ed Philadelphia: Elsevier Saunders; 2006.
3. Online Mendelian Inheritance in Man. In: www.ncbi.nlm.nih.gov/omim;.