SPECIAL SENSES
The eyes are complex sense organs responsible for vision. Within a protective casing, each eye has receptors, a lens system for focusing light on the receptors, and a system of nerves for conducting impulses from the receptors to the brain. Visual dysfunction may be caused by abnormal ocular movements or alterations in visual acuity, refraction, color vision, or accommodation. Visual dysfunction also may be the secondary effect of another neurologic disorder.
External Eye Structures
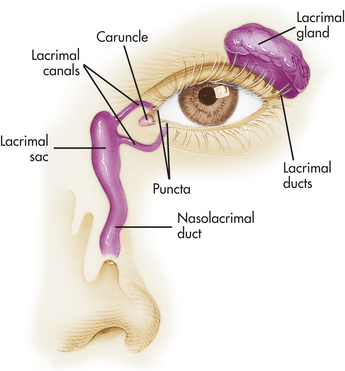
The external structures protecting the eye include the eyelids (palpebrae), conjunctivae, and lacrimal apparatus (Figure 15-11). Infection and inflammatory responses are the most common conditions affecting the supporting structures of the eyes. Blepharitis is an inflammation of the eyelids caused by Staphylococcus or seborrheic dermatitis. Redness, edema, and itching are common symptoms. A hordeolum (stye) is an infection of the sebaceous glands of the eyelids, and a chalazion is an infection of the meibomian (oil-secreting) gland. These conditions are treated symptomatically.188
Conjunctivitis
Conjunctivitis is an inflammation of the conjunctiva (mucous membrane covering the front part of the eyeball). Conjunctivitis may be caused by bacteria, viruses, allergies, or chemical irritations. The inflammatory response produces redness, edema, pain, and lacrimation. Treatment is related to cause.189
Acute bacterial conjunctivitis (pinkeye) is highly contagious and often is caused by gram-positive organisms (Staphylococcus, Haemophilus, Proteus), although other bacteria may be involved. The onset is acute, characterized by mucopurulent drainage from one or both eyes. Preventing spread of the organism with meticulous handwashing and use of separate towels is important. The disease often is self-limiting and resolves spontaneously in 10 to 14 days. Antibiotic eyedrops usually are effective.
Viral conjunctivitis is caused by an adenovirus. Symptoms vary from mild to severe. Some strains of virus cause conjunctivitis and pharyngitis (pharyngoconjunctival fever), and others cause keratoconjunctivitis. Both diseases are contagious, with watering, redness, and photophobia. Treatment is symptomatic.
Allergic conjunctivitis is associated with a variety of antigens, including pollens. Ocular itching is associated with photophobia, burning, and gritty sensations in the eye. Treatment is symptomatic and may include antihistamines, steroids, and vasoconstrictors.
Chronic conjunctivitis is the result of any persistent conjunctivitis. The cause requires identification for effective treatment.
Trachoma (chlamydial conjunctivitis) is caused by Chlamydia trachomatis. It often is associated with poor hygiene and is the leading cause of preventable blindness in the world. The severity of the disease varies, but it can involve inflammation with scarring of the conjunctiva and eyelids causing distorted lashes to abrade the cornea leading to corneal scarring and blindness. Chlamydial organisms are sensitive to local or systemic antibiotics. The World Health Organization aims to eliminate trachoma as a public health problem by 2020 using the SAFE Strategy: Surgery for inturned lashes, Antibiotics, Facial cleanliness, and Environmental improvement.190
Keratitis: Keratitis is an infection of the cornea that can be caused by bacteria or viruses. Bacterial infections often cause corneal ulceration and require intensive antibiotic treatment. Type I herpes simplex virus can involve the cornea and conjunctiva. Common symptoms include photophobia, pain, and lacrimation. Severe ulcerations with residual scarring require corneal transplantation.
The Eye
The wall of the eye is formed of three layers: sclera, choroid, and retina (Figure 15-12). The sclera is the thick, white, outermost layer. It becomes transparent at the cornea, the portion of the sclera in the central anterior region that allows light to enter the eye. The choroid is the deeply pigmented middle layer that prevents light from scattering inside the eye. The iris, part of the choroid, has a round opening, the pupil, through which light passes. Smooth muscle fibers control the size of the pupil so that in close vision and bright light the pupil constricts and in distant vision and dim light the pupil dilates.
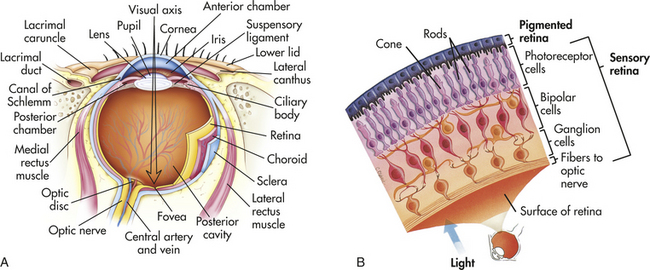
Figure 15-12 Structure of the eyeball and cell layers of the retina. A, Horizontal section through the left eyeball. The eye is viewed from above. B, Pigmented and sensory layers of the retina. (From Patton KT, Thibodeau GA: Anatomy & physiology, ed 7, St Louis, 2010, Mosby.)
The innermost layer of the eye, the retina, contains millions of rods and cones, special photoreceptors that convert light energy into nerve impulses. In the retina, rods mediate peripheral and dim light vision and are densest at the periphery. Cones, densest in the center of the retina, are color and detail receptors. The photoreceptive rods and cones are distributed over the entire retina, except where the optic nerve leaves the eyeball. Lack of rods and cones in this area results in the optic disc, or blind spot. Lateral to each optic disc is the fovea centralis, a tiny area that contains only cones and provides the greatest visual acuity (see Figure 15-12).
As shown in Figure 15-13, nerve impulses pass through the optic nerves after leaving the retinas. At the optic chiasm the fibers from the inner (nasal) halves of the retinas cross to the opposite side, where they join fibers from the outer (temporal) halves of the retinas to form the optic tracts. The fibers of the optic tracts synapse in the dorsal lateral geniculate nucleus, and from there the geniculocalcarine fibers pass by way of the optic radiation (or geniculocalcarine tract) to the primary visual cortex in the occipital lobe of the brain.
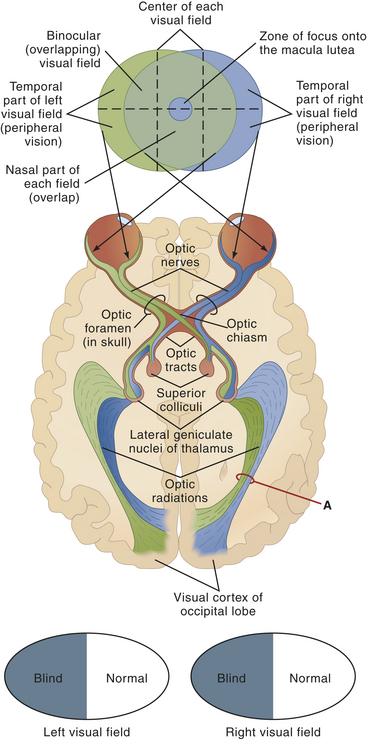
Figure 15-13 Visual fields and neuronal pathways. Note the structures that make up each pathway: optic nerve, optic chiasm, lateral geniculate body of thalamus, optic radiations, and visual cortex of occipital lobe. Fibers from the nasal portion of each retina cross over to the opposite side at the optic chiasm and terminate in the lateral geniculate nuclei. Location of a lesion in the visual pathway determines the resulting visual defect. Damage at point A, for example, would cause blindness in the right nasal and left temporal visual fields (as the black and white ovals indicate; trace the visual pathway from point A back to the visual field map to see why this is so). What would be the effect of pressure on the optic chiasm, by a pituitary tumor, for instance? (Answer: It would produce blindness in both temporal visual fields. Why? Because it destroys fibers from the nasal side of both retinas.) (From Patton KT, Thibodeau GA: Anatomy & physiology, ed 7, St Louis, 2010, Mosby.)
Light entering the eye is focused on the retina by the lens—a flexible, biconvex, crystal-like structure. In youth the lens is transparent and has the consistency of hardened jelly. With age the lens becomes increasingly hard and opaque. The lens divides the anterior chamber into (1) the aqueous chamber and (2) the vitreous chamber. Aqueous humor, which fills the aqueous chamber, helps maintain the pressure inside the eye and provides nutrients to the lens and cornea. Aqueous humor is free-flowing fluid, secreted by the ciliary processes and reabsorbed into the canal of Schlemm. If drainage is blocked, pressure within the eye increases (as it does with glaucoma). The vitreous chamber is filled with a gel-like substance called vitreous humor. Vitreous humor helps prevent the eyeball from collapsing inward.
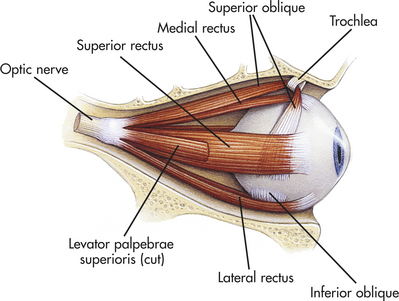
The central retinal artery provides blood to the inner retinal surface. Nutrients are supplied to the outer surface of the retina by the choroid. Six extrinsic eye muscles, attached to the outer surface of each eye, allow gross eye movements and permit the eyes to follow a moving object (Figure 15-14).
Aging and Vision
Changes in the structural components of the eye caused by aging begin at an early age, particularly in the lens of the eye. Changes caused by aging are summarized in Table 15-5. Structural changes combined with chronic diseases including diabetes mellitus result in a decline in visual acuity.191
Table 15-5
Changes in the Eye Caused by Aging
| Structure | Change | Consequence |
| Cornea | Thicker and less curved | Increase in astigmatism |
| Formation of a gray ring at the edge of cornea (arcus senilis) | Not detrimental to vision | |
| Anterior chamber | Decrease in size and volume caused by thickening of lens | Occasionally exerts pressure on Schlemm canal and may lead to increased intraocular pressure and glaucoma |
| Lens | Increase in opacity | Decrease in refraction with increased light scattering and decreased color vision (green and blue); can lead to cataracts |
| Ciliary muscles | Reduction in pupil diameter, atrophy of radial dilation muscles | Persistent constriction (senile miosis); decrease in critical flicker frequency∗ |
| Retina | Reduction in number of rods at periphery, loss of rods and associated nerve cells | Increase in the minimum amount of light necessary to see an object |
∗The rate at which consecutive visual stimuli can be presented and still be perceived as separate.
Visual Dysfunction
Alterations in Ocular Movements: Abnormal ocular movements occur as a result of oculomotor, trochlear, or abducens cranial nerve dysfunction (see Table 14-6). The three types of eye movement disorders are (1) strabismus, (2) nystagmus, and (3) paralysis of individual extraocular muscles.
Strabismus is the deviation of one eye from the other when a person is looking at an object resulting in failure of the two eyes to simultaneously focus on the same image with loss of binocular vision. It is caused by weak or hypertonic muscle in one of the eyes. The deviation may be upward, downward, inward, or outward. Strabismus in children requires early intervention to prevent the development of amblyopia (reduced vision in the affected eye without ocular pathology and with full optical correction). Treatment of amblyopia is patching. The primary symptom of strabismus is diplopia (double vision). Strabismus may be caused by a neuromuscular disorder of the eye muscle, diseases involving the cerebral hemispheres, or thyroid disease.192,193
Nystagmus is an involuntary unilateral or bilateral rhythmic movement of the eyes and can occur in infants (congenital) or adults (acquired). It may be present at rest, or it may occur with eye movement. The two major forms of nystagmus are pendular nystagmus and jerk nystagmus. Pendular nystagmus is characterized by a regular to-and-fro movement of the eyes in which both phases of the movement are equal in length. In jerk nystagmus one phase of the eye movement is faster than the other. Nystagmus may be caused by an imbalance in the normally coordinated reflex activity of the inner ear, vestibular nuclei (connecting the vestibular nerve with vestibulospinal tracts), cerebellum, medial longitudinal fascicle (connecting the mesencephalon with the upper portion of the spinal cord), or nuclei of the oculomotor, trochlear, and abducens cranial nerves (see Table 14-6). Drugs, retinal disease, and diseases involving the cervical cord also may produce nystagmus. Acquired untreated nystagmus can lead to loss of visual acuity.194
Paralysis of specific extraocular muscles may cause a variety of abnormalities, including limited abduction, abnormal closure of the eyelid, ptosis (drooping of the eyelid), and diplopia. The abnormalities occur as a result of unopposed muscle activity. Trauma or pressure in the area of the cranial nerves may cause paralysis of specific extraocular muscles. Diseases such as diabetes mellitus and myasthenia gravis also may affect specific extraocular muscles.
Alterations in Visual Acuity: Visual acuity is the ability to see objects in sharp detail. With advancing age the eye’s lens becomes less flexible and less adjustable. In addition, the sclera changes shape, causing light to fall on the macula (an opaque portion of the cornea). Thus visual acuity declines with age. Visual acuity also may change or diminish for many other reasons. Specific causes of visual acuity changes include (1) amblyopia, (2) scotoma, (3) cataracts, (4) papilledema, (5) dark adaptation, (6) glaucoma, (7) retinal detachment, and (8) macular degeneration.
Amblyopia is a reduction or dimness of vision for unknown reasons. It does not result from a change in refraction (i.e., deviation of light rays) or from any visible changes in the eye. Amblyopia is associated with strabismus and anisometropia (refractive error in one eye differs from the other eye) and diseases such as diabetes mellitus, renal failure, and malaria and with toxic substances such as alcohol and tobacco. Amblyopia is the most common cause of vision loss in children and is usually treated by patching the unaffected eye for extended times to ensure a period of use of the affected eye or with atropine drops.195 Refractive errors are treated with corrective lenses.
A scotoma is a circumscribed defect of the central field of vision. It is most often a sequel to demyelinating optic neuritis, an inflammatory lesion of the optic nerve frequently associated with multiple sclerosis (see Chapter 17). Less common causes include the compression of one optic nerve by a retroorbital tumor, neuromyelitis optica (autoantibody-related inflammation of the optic nerve and spinal cord), pernicious anemia, and toxic or metabolic causes such as methyl alcohol poisoning and use of tobacco. The precise mechanisms for these conditions causing a scotoma are uncertain, but the result is always a serious impairment in visual acuity.196
A cataract is a cloudy or opaque area in the ocular lens. The incidence of cataracts increases with age as the lens enlarges. Cataracts develop because of alterations of metabolism and transport of nutrients within the lens. Although the most common form of cataract is degenerative, cataracts also may occur congenitally or as a result of infection, radiation, trauma, drugs, or diabetes mellitus. Cataracts cause decreased visual acuity, blurred vision, glare, and decreased color perception. Cataracts are treated by removal of the entire lens and replacement with an intraocular artificial lens.197
Papilledema is edema and inflammation of the optic nerve at its point of entrance into the eyeball. Generally, papilledema is caused by some obstruction to the venous return from the retina. An early sign is distention of the retinal vein. Obliteration of the physiologic cup (a bright area normally located in the center of the optic disc) follows. Later the optic disc becomes raised above the level of the surrounding retina, and the margins become blurred and indistinct. With severe swelling, hemorrhage and patches of white exudate develop around the disc margins. The three principal causes of papilledema are (1) increased intracranial pressure, (2) retrobulbar neuritis, and (3) changes in the retinal blood vessels. Retinal blood vessel changes are especially prevalent in individuals with diabetes mellitus or hypertension. Such changes account for a large percentage of individuals newly affected with blindness each year. Typically the blood vessels narrow, and hemorrhages and white exudate appear. Ultimately papilledema occurs.
Dark adaptation also affects visual acuity. Low illumination causes impaired visual acuity, particularly in older adults. The average 80-year-old needs more than twice as much light as a 20-year-old to see equally well. Changes in the quantity and quality of rhodopsin, a substance found in the rods and responsible for low-light vision, are thought to be responsible for reduced dark adaptation in older adults.198 Vitamin A deficiencies can cause the same phenomenon in individuals of any age.
Glaucoma is a leading cause of visual impairment and blindness. It is characterized by intraocular pressures above the normal pressures of 12 to 20 mmHg maintained by the aqueous fluid. Family history is a risk factor, and glaucoma can be inherited.199 The types of glaucoma are summarized in Table 15-6 and Figure 15-15. Chronic increased intraocular pressure causes death of retinal ganglions and optic nerve degeneration with loss of peripheral vision, followed by central vision impairment and blindness.200 Extremely high pressures can cause blindness within days or hours. Loss of visual acuity results from pressure on the optic nerve, which is believed to block the flow of cytoplasm from neuronal bodies in the retina to peripheral optic nerve fibers entering the brain. Lack of nutrients, ischemia, cytotoxic factors, and altered immune mechanisms may lead to death of the involved neurons.201 Acute pain may result. Early detection and treatment prevent optic neuropathy and visual impairment. Glaucoma often is treated with pharmaceutical eyedrops to reduce secretion or increase absorption of aqueous humor. Surgery may be needed to open the spaces of the trabeculae and reduce intraocular pressure. Neuroprotective therapies are being evaluated.202
Table 15-6
| Type | Mechanism of Increased Pressure |
| Open-angle | Obstruction of outflow of aqueous humor at trabecular meshwork or Schlemm canal; myopia may be a risk factor |
| Normal or low tension | A form of open-angle glaucoma with symptomless damage to the optic nerve and gradual vision loss when intraocular pressure is within normal range (12-20 mmHg) |
| Narrow-angle (angle closure) | Forward displacement of iris toward cornea with narrowing of iridocorneal angle and obstruction to outflow of aqueous humor from anterior chamber |
| Acute-angle closure | Acute closure of iridocorneal angle with a sudden rise in intraocular pressure, producing pain, redness, and visual disturbances |
| Chronic-angle closure | Progressive, permanent closure of anterior chamber angle |
| Secondary | Open or closed angle obstruction caused by, for example, uveitis, hemorrhage, rupture of lens or tumors |
| Congenital glaucoma | Malformation of trabecular meshwork and excess extracellular matrix in outer meshwork. |
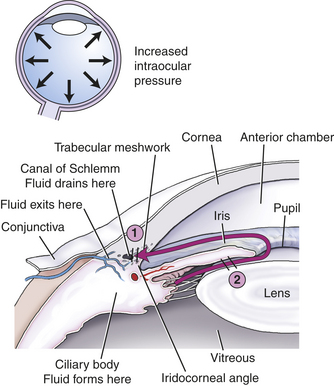
Figure 15-15 Glaucoma. 1, Open-angle glaucoma. The obstruction to aqueous flow lies in the trabecular meshwork. 2, Closed-angle glaucoma. The trabecular meshwork is covered by the root of the iris.
Retinal detachment is a common cause of visual impairment and blindness. Risk factors include retinal holes and vitreoretinal traction. Fluid (exudate, hemorrhage, or liquid vitreous) separates the photoreceptors from the retinal pigment epithelium. The separation deprives the outer retina of oxygen and nutrients because the diffusion distance is increased. Communication is also disrupted between the pigment epithelium and photoreceptors. Rhegmatogenous retinal detachment (retinal breaks caused by vitreoretinal traction) is the most common form of retinal detachment. Causes include intracapsular cataract extraction, severe myopia, lattice degeneration, vitreoretinal traction, and trauma. Contraction of fibrous membranes can cause tractional separation of the retinal layers as occurs in proliferative diabetic retinopathy. Treatment involves surgical retinal reattachment.203
Age-related macular degeneration (AMD), loss of central vision, is the major cause of vision loss in individuals older than 60 years. Hypertension, cigarette smoking, diabetes mellitus, and genetic predisposition are risk factors that contribute to oxidative stress and capillary injury.204 The atropic or dry form of AMD is most prevalent and involves loss of retinal pigment epithelium and photoreceptors with overall atrophy of cells. The wet exudative form (neovascular AMD) is the more severe form and involves proliferation of abnormal choroidal vessels, which leak and bleed, causing retinal detachment.205 Symptoms include blurred vision, loss of central vision, difficulty reading, and poor night vision. Progress is being made in antiangiogenic treatments and in understanding genetic factors contributing to AMD.206
Alterations in Accommodation: Accommodation is the process whereby the thickness of the lens changes. Accommodation is needed for clear vision and is mediated through the oculomotor nerve. Pressure, inflammation, and disease of the oculomotor nerve may alter accommodation. Symptoms include diplopia, blurred vision, and headache. Accommodation is affected also by the decreased flexibility of the lens that occurs with aging. By 60 years of age the lens has become so inelastic that accommodation is not possible.
Loss of accommodation in older adults is termed presbyopia, a condition in which the ocular lens becomes larger, firmer, and less elastic. The major symptom is reduced near vision, causing the individual to hold reading material at arm’s length. Correction is accomplished through reading glasses or bifocal lenses, accommodative intraocular lenses, or surgical treatment.207,208
Alterations in Refraction: Alterations in refraction are the most common visual problem. Errors in refraction are caused by irregularities of the corneal curvature, the focusing power of the lens, and the length of the eye. The major symptoms of refraction alterations are blurred vision and headache. Three types of refraction alterations are myopia, hyperopia, and astigmatism (Figure 15-16).
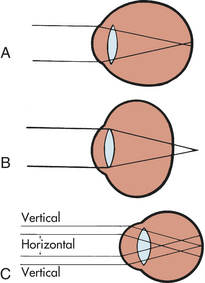
Figure 15-16 Alterations in refraction. A, Myopic eye. Parallel rays of light are brought to a focus in front of the retina. B, Hyperopic eye. Parallel rays of light come to a focus behind the retina in the unaccommodative eye. C, Simple myopic astigmatism. The vertical bundle of rays is focused on the retina; the horizontal rays are focused in front of the retina. (From Stein HA, Slatt BJ, Stein RM: The ophthalmic assistant: fundamentals in clinical practice, St Louis, 1988, Mosby.)
In myopia (nearsightedness), light rays are focused in front of the retina when a person is looking at a distant object, resulting in burred vision. A concave lens is needed for correction. Myopia requires frequent changes of eyeglasses while the eyeball is lengthening in childhood. Myopia is a risk factor for retinal detachment.
In hyperopia (farsightedness), light rays are focused behind the retina when a person is looking at a near object. Hyperopia is corrected with a convex lens. Astigmatism is caused by an unequal curvature of the cornea. In astigmatism, light rays are bent unevenly and do not come to a single focus on the retina. Astigmatism may coexist with myopia, hyperopia, or presbyopia. Correction is accomplished with a cylinder lens.
Alterations in Color Vision: Normal sensitivity to color diminishes with age because of the progressive yellowing of the lens that occurs with aging. All colors become less intense, although color discrimination for blue and green is most greatly affected. Color vision deteriorates more rapidly for individuals with diabetes mellitus than for the general population. The deterioration is thought to be an accelerated version of senile color vision deterioration.
Abnormal color vision also may be caused by color blindness, an inherited trait. Color blindness is generally an X-linked recessive characteristic affecting 8% of the male population and 0.5% of the female population. Although many forms of color blindness exist, most commonly the affected individual cannot distinguish red from green.209,210
Neurologic Disorders Causing Visual Dysfunction: Various neurologic disorders may cause visual dysfunction. Vision may be disrupted at many points along the visual pathway, causing a variety of defects in fields of vision. Visual changes do not always cause defects or blindness in the entire visual field; hemianopia is the term that describes defective vision in half of a visual field. (Figure 15-17 illustrates the many areas along the visual pathway that may be damaged and the associated visual changes.)
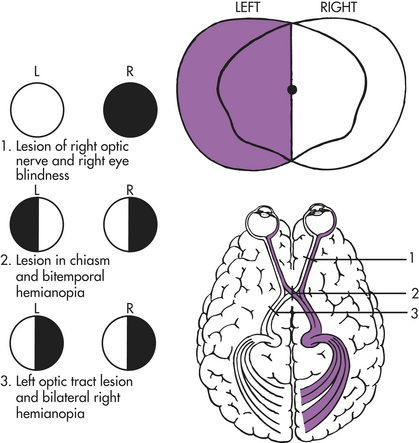
Figure 15-17 Visual pathway defects. (From Thompson JM et al: Mosby’s clinical nursing, ed 5, St Louis, 2002, Mosby.)
Because of the anatomy of the optic nerves, injury to the optic nerve causes ipsilateral (same side) blindness but a normal contralateral (opposite side) visual field. Injury to the optic chiasm (the X-shaped crossing of the optic nerves), often caused by atherosclerotic ischemia or external compression from trauma or aneurysm, can cause a variety of defects, depending on the location of injury. These defects vary because at the optic chiasm, nerve fibers from the medial half of each retina separate from the lateral half and enter the opposite optic tract.
Because of the normal structure of the visual pathways, destruction of one optic tract causes homonymous hemianopsia (complete loss of vision in the inner half of one eye and the outer half of the other). Thus, if an injury to the left optic tract occurs, the individual is blind in the right eye’s lateral (outer) field and the left eye’s medial (inner) field. If the compression of the optic tract is asymmetric, an incongruous (or uneven) homonymous defect results. Injury to one optic radiation (an ocular pathway in the internal capsule, temporal lobe, or occipital lobe) also causes a homonymous (same field) defect. A major injury in the optic radiation causes homonymous hemianopsia. A lesser injury may cause an upper quadrant homonymous defect. Generally the defects are the same size in both eyes. When the homonymous hemianopsia is caused by an occipital lobe lesion, the area of hemianopsia is split. Although visual acuity may remain unimpaired, reading is difficult because of the inability to group words.
Hearing
The external auditory canal is surrounded by the bones of the cranium. Its opening (meatus) is just above the mastoid process (Figure 15-18), which contains air-filled sinuses called mastoid air cells. These promote conductivity between the external and the middle ear.
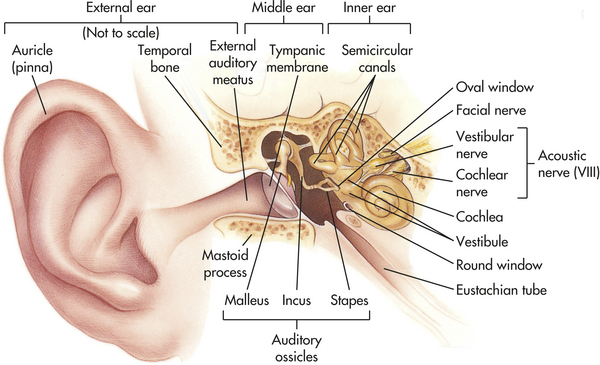
Figure 15-18 The ear. External, middle, and inner ear structures. (Anatomic structures are not drawn to scale. Middle and inner ears enlarged for better visualization here.) (From Patton KT, Thibodeau GA: Anatomy & physiology, ed 7, St Louis, 2010, Mosby.)
The Normal Ear
The ear is divided into three areas: (1) the external ear, involved only with hearing; (2) the middle ear, involved only with hearing; and (3) the inner ear, involved with both hearing and equilibrium.
The external ear is composed of the pinna (auricle), which is the visible portion of the ear, and the external auditory canal, a tube that leads to the middle ear (see Figure 15-18). Sound waves entering the external auditory canal hit the tympanic membrane (eardrum) and cause it to vibrate. The tympanic membrane separates the external ear from the middle ear.
The middle ear is composed of the tympanic cavity, a small chamber in the temporal bone. Three ossicles (small bones) transmit the vibration of the tympanic membrane to the inner ear. The three ossicles are termed the malleus (hammer), incus (anvil), and stapes (stirrup). When the tympanic membrane moves, the malleus moves with it and transfers the vibration to the incus, which passes it on to the stapes. The stapes presses against the oval window, a small membrane of the inner ear. The movement of the oval window sets the fluids of the inner ear in motion (Figure 15-19).
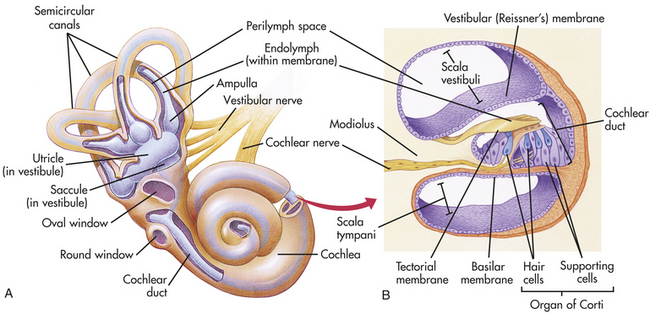
Figure 15-19 The inner ear. A, The bony labyrinth (orange) is the hard outer wall of the entire inner ear and includes semicircular canals, vestibule, and cochlea. Within the bony labyrinth is the membranous labyrinth (purple), which is surrounded by perilymph and filled with endolymph. Each ampulla in the vestibule contains a crista ampullaris that detects changes in head position and sends sensory impulses through the vestibular nerve to the brain. B, The inset shows a section of the membranous cochlea. Hair cells in the organ of Corti detect sound and send the information through the cochlear nerve. The vestibular and cochlear nerves join to form the eighth cranial nerve. (From Patton KT, Thibodeau GA: Anatomy & physiology, ed 7, St Louis, 2010, Mosby.)
The eustachian (pharyngotympanic) tube connects the middle ear with the thorax. Normally flat and closed, the eustachian tube opens briefly when a person swallows or yawns, and it equalizes the pressure in the middle ear with atmospheric pressure. Equalized pressure permits the tympanic membrane to vibrate freely. Through the eustachian tube the mucosa of the middle ear is contiguous with the mucosal lining of the throat.
The inner ear is a system of osseous labyrinths (bony, mazelike chambers) filled with a fluid called perilymph. The bony labyrinth is divided into the cochlea, the vestibule, and the semicircular canals (see Figure 15-18). Suspended in the perilymph is a membranous labyrinth that basically follows the shape of the bony labyrinth. The membranous labyrinth is filled with a thicker fluid called endolymph.
Within the cochlea is the organ of Corti, which contains hair cells (hearing receptors). Sound waves that reach the cochlea through vibrations of the tympanic membrane, ossicles, and oval window set the cochlear fluids into motion. Receptor cells on the basilar membrane are stimulated when their hairs are bent or pulled by the movement. Once stimulated, hair cells transmit impulses along the cochlear nerve (a division of the vestibulocochlear nerve) to the auditory cortex of the temporal lobe in the brain (see Figure 15-19), where interpretation of the sound occurs. Directional hearing is controlled by the angle of the sound source to both ears and axonal delay in conduction in groups of neurons.211
The semicircular canals and vestibule of the inner ear contain equilibrium receptors. In the semicircular canals the dynamic equilibrium receptors respond to changes in direction of movement. Within each semicircular canal is the crista ampullaris, a receptor region composed of a tuft of hair cells covered by a gelatinous cupula. When the head is rotated, the endolymph in the canal lags behind and moves in the direction opposite to the head’s movement. The hair cells are stimulated, and impulses are transmitted through the vestibular nerve (a division of the vestibulocochlear nerve) to the cerebellum.
The vestibule in the inner ear contains maculae, receptors essential to the body’s sense of static equilibrium. As the head moves, otoliths (small pieces of calcium salts) move in a gel-like material in response to changes in the pull of gravity. The otoliths pull on the gel, which in turn pulls on the hair cells in the maculae. Nerve impulses in the hair cells are triggered and transmitted to the brain (see Figure 15-19). Thus the ear not only permits the hearing of a large range of sounds but also assists with maintaining balance through the sensitive equilibrium receptors.
Aging and Hearing
Auditory changes caused by aging are common and incremental. Changes in hearing with aging are summarized in Table 15-7. Approximately one third of people older than 65 years have hearing loss caused by genetic and environmental factors.212 Changes may occur in the structural and functional components of the peripheral or central auditory system. Loss of hearing for sounds in the high-frequency range (presbycusis) is most common and interferes with understanding speech, particularly high-frequency consonant sounds (e.g., s, sh, f). Hearing may be lost in both ears but not at the same time. Older adults from rural areas have less hearing loss than those in noisy cities. The ability to discriminate localization of sound varies with high and low frequencies and diminishes with age.213 In the low-frequency range, sound localization is a function of the timing of sound arrival between the two ears; localization of high-frequency sounds is a function of sound intensity. Because older adults tend to lose high-frequency hearing first, they may have difficulty localizing high-frequency sounds.
Table 15-7
Changes in Hearing Caused by Aging
| Changes in Structure | Changes in Function |
| Cochlear hair cell degeneration | Inability to hear high-frequency sounds (presbycusis, sensorineural loss); interferes with understanding speech; hearing may be lost in both ears at different times |
| Loss of auditory neurons in spiral ganglia of organ of Corti | Inability to hear high-frequency sounds (presbycusis, sensorineural loss); interferes with understanding speech; hearing may be lost in both ears at different times |
| Degeneration of basilar (cochlear) conductive membrane of cochlea | Inability to hear at all frequencies, but more pronounced at higher frequencies (cochlear conductive loss) |
| Decreased vascularity of cochlea | Equal loss of hearing at all frequencies (strial loss); inability to disseminate localization of sound |
| Loss of cortical auditory neurons | Equal loss of hearing at all frequencies (strial loss); inability to disseminate localization of sound |
Ear Infections
Otitis Externa: Otitis externa is the most common infection of the outer ear usually caused by bacteria and less commonly a fungus.214 The most frequently found microorganisms are Pseudomonas, Escherichia coli, and Staphylococcus aureus. Infection usually follows prolonged exposure to moisture (swimmer’s ear). The earliest symptoms are inflammation with swelling and clear drainage progressing to purulent drainage with obstruction of the canal. Tenderness and pain with earlobe retraction accompany inflammation.
Otitis Media: Otitis media is an infection of the middle ear and is the most common infection of infants and children. Most children have one episode by 3 years of age. The most common pathogens are Streptococcus pneumoniae, Haemophilus influenzae, and Moraxella catarrhalis. Respiratory viruses also may have an etiologic role.215 Predisposing factors include allergy, sinusitis, submucous cleft palate, adenoidal hypertrophy, and immune deficiency. Breast-feeding is a protective factor.
Acute otitis media (AOM) is associated with ear pain, fever, irritability, inflamed tympanic membrane, and fluid in the middle ear. The tympanic membrane progresses from erythema to opaqueness with bulging as fluid accumulates. An increasing prevalence of AOM is caused by methicillin-resistant microorganisms. Otitis media with effusion (OME) is the presence of fluid in the middle ear without symptoms of acute infection.Treatment includes antimicrobial therapy for AOM, particularly in infants 2 years and younger.
Chronic otitis media is persistent or recurring infection of the middle ear. Placement of tympanostomy tubes is considered when bilateral effusion persists for 3 months and for significant hearing loss.216 Mastoidectomy combined with tympanostomy tubes may be required when there is cholesteatoma (skin growth into the middle ear associated with perforation of the eardrum).217 Complications include mastoiditis, brain abscess, meningitis, and chronic otitis media with hearing loss. Speech, language, and cognitive disabilities may be affected by persistent middle ear effusions.218
Auditory Dysfunction
Between 5% and 10% of the general population have a hearing impairment. The major categories of auditory dysfunction are conductive hearing loss, sensorineural hearing loss, mixed hearing loss, and functional hearing loss.
Conductive Hearing Loss: Conductive hearing loss occurs when a change in the outer or middle ear impairs sound from being conducted from the outer to the inner ear. Conductive hearing loss occurs when there is interference in air conduction. Conditions that commonly cause a conductive hearing loss include impacted cerumen, foreign bodies lodged in the ear canal, benign tumors of the middle ear, carcinoma of the external auditory canal or middle ear, eustachian tube dysfunction, otitis media, acute viral otitis media, chronic suppurative otitis media, cholesteatoma, and otosclerosis.
Symptoms of conductive hearing loss include diminished hearing and soft speaking voice. The voice is soft because often the individual hears his or her voice, conducted by bone, as loud. In addition, although the cause is unknown, the individual often hears better in a noisy environment than in a quiet one (a condition called paracusia willisiana). Treatment of the underlying cause generally improves hearing.219 A hearing aid is used if the hearing loss is between 40 and 50 decibels.
Sensorineural Hearing Loss: A sensorineural hearing loss is caused by impairment of the organ of Corti or its central connections. The hearing loss may be gradual or sudden. Conditions that commonly cause sensorineural hearing loss include congenital and hereditary factors, noise exposure, aging, Ménière disease, ototoxicity, and systemic disease (syphilis, Paget disease, collagen diseases, diabetes mellitus). Congenital and neonatal sensorineural hearing loss may be caused by maternal rubella, ototoxic drugs, prematurity, traumatic delivery, erythroblastosis fetalis, and congenital hereditary malfunction. Diagnosis often is made when delayed speech development is noted.
Presbycusis is age-related hearing loss usually in the high frequencies. It is the most common form of sensorineural hearing loss and is especially common in older adutls.220 Presbycusis may occur because of atrophy of the basal end of the organ of Corti, a loss in the number of auditory receptors, vascular changes, or stiffening of the basilar membranes. Because of the slow progression of hearing loss, onset of symptoms is gradual. In addition, drug ototoxicities (drugs that cause destruction of auditory function) have been observed after exposure to a variety of chemicals, for example, antibiotics such as streptomycin, neomycin, gentamicin, and vancomycin; diuretics such as ethacrynic acid and furosemide; and chemicals such as salicylate, quinine, carbon monoxide, nitrogen mustard, arsenic, mercury, gold, tobacco, and alcohol. Because of increased concentrations of antibiotics in the endolymph, these drugs generally cause damage to the cells of the cristae and maculae (located in the inner ear) or the cells of the organ of Corti. The increased concentration of drugs in the endolymph is preferentially toxic to the cells.
Diuretics affect hearing primarily by altering the sodium-potassium balance, causing extracellular fluid accumulation and changes in the microstructure of secretory cells. Quinine, mercury, and lead affect the neural pathways of hearing, including the spinal ganglia, the eighth cranial nerve, and the cochlear nucleus. The site of action for the other chemicals, including alcohol and tobacco, has not yet been determined. In most instances the drugs and chemicals listed previously initially cause tinnitus (ringing in the ear), followed by a progressive high-tone sensorineural hearing loss. Care is aimed at prevention of further hearing loss because the loss is usually permanent.
Olfaction and Taste
Olfaction is a function of cranial nerve I and part of cranial nerve V. Taste is a function of multiple nerves in the tongue, soft palate, uvula, pharynx, and upper esophagus, including cranial nerves VII and IX. Olfaction (smell) dysfunction and taste (gustation) dysfunction may occur separately or jointly. The strong relationship between smell and taste creates the sensation of flavor. If either sensation is impaired, the perception of flavor is altered. (Olfactory structures are illustrated in Figure 15-20.)
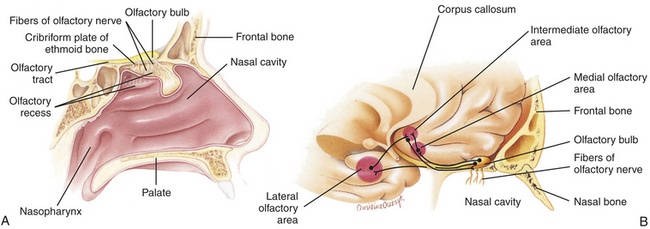
Figure 15-20 Olfaction. Location of olfactory epithelium, olfactory bulb, and neuronal pathways involved in olfaction. A, Midsagittal section of the nasal area shows the locations of major olfactory sensory structures. B, Major olfactory integration centers of the brain. (Modified from Patton KT, Thibodeau GA: Anatomy & physiology, ed 7, St Louis, 2010, Mosby.)
Olfactory cells, which are located in the olfactory epithelium, are the receptor cells for smell. Seven primary classes of olfactory stimulants have been identified: (1) camphoraceous, (2) musky, (3) floral, (4) peppermint, (5) ethereal, (6) pungent, and (7) putrid. The primary sensations of taste are sour, salty, sweet, and bitter. Taste buds sensitive to each of the primary sensations are located in specific areas of the tongue.221
Aging and Olfaction and Taste
Sensitivity to odors declines steadily with aging.222 A study of odor identification indicates an increasing ability from childhood to adolescence and then a decline after 60 years of age. The most significant impairments develop after 80 years.223 Women generally have better olfactory abilities than men, but the patterns of decline are similar.224
The sense of smell begins to degenerate with loss of olfactory sensory neurons and loss of cells from the olfactory bulbs.225,226 Loss of olfactory sensitivity and odor identification may diminish appetite and food selection and thus may lead to malnutrition. Safety also may be compromised by an inability to smell toxic or hazardous gases.
Taste
The decline in taste sensation is more gradual than that of smell and may be associated with loss of olfaction. Higher concentrations of flavors are required, and older adults have difficulty differentiating combinations of flavors.227 The best-known change with aging is the decline in the number of fungiform papillae on the tongue, which decrease by 50% by about 50 years of age.227 Taste also may be affected by decreased salivary gland secretion. Amylase, contained in saliva, facilitates perception of sweet sensations and also is reduced with aging.
Olfactory Dysfunction
Olfactory dysfunctions include hyposmia, anosmia, hallucinations, and parosmia. Hyposmia is the impaired sense of smell, and anosmia is the complete loss of smell. Both conditions are associated with aging, neurodegenerative and nasal/sinus disorders and head trauma.228 When hyposmia or anosmia occurs bilaterally, it is usually the result of rhinitis (inflammation of nasal mucosa), sinusitis, nasal polyps, or excessive smoking.229 Unilateral hyposmia or anosmia may indicate compression of one olfactory bulb (a bulblike portion of the olfactory nerves) or nerve tract (olfactory nerve pathway), possibly by tumor or head trauma. Olfactory hallucinations arise from hyperactivity in cortical neurons and involve smelling odors that are not really present. They are associated with temporal lobe seizures and rarely with schizophrenia. Parosmia, an abnormal or perverted sense of smell, may occur with severe depression.230
Taste Dysfunction
The sense of taste can be impaired by injury, medications, oral infections, or aging. An alteration in taste also may be attributable to impairment of smell associated with injury near the hippocampus.
Hypogeusia is decrease in taste sensation, and ageusia is the absence of taste. Ageusia affecting the entire tongue may follow head injury. Damage to the glossopharyngeal nerve (cranial nerve IX, which innervates the posterior one third of the tongue) causes the loss of the ability to detect bitterness. This loss occurs because the receptors for bitter are located on the base of the tongue. Damage to the facial nerve (cranial nerve VII, which innervates the anterior two thirds of the tongue) causes loss of the ability to detect sour, sweet, and salty tastes. Only bitter tastes can be detected. These losses occur because sour, sweet, and salt receptors are located on the anterior portion of the tongue. Parageusia is a perversion of taste in which substances possess an unpleasant flavor. Parageusia occasionally develops for no apparent reason in older adults and is common in individuals receiving chemotherapy for cancer. In both cases, parageusia often leads to anorexia and malnutrition.
SOMATOSENSORY FUNCTION
Touch is not a uniform sensory experience. The sensation of touch involves the fusion of several qualities, including modality, intensity, location, and duration of the sensory stimulus. Receptors sensitive to touch are present in the skin. Meissner and pacinian corpuscles are rapidly adapting receptors, whereas Merkel disks and Ruffini endings are slowly adapting touch receptors. Touch receptors are most numerous in the skin of the fingers and lips and are more scarce in the skin of the trunk. Specific sensory input is carried to the higher levels of the CNS by the dorsal column of the spinal cord and the anterior spinothalamic tract.
Much of the development of the cutaneous senses takes place before birth, but structural growth of the cutaneous senses continues into early adulthood at a reduced rate. Then a gradual decline occurs.231 Studies have documented loss in tactile sensitivity with advancing age.232,233 This occurs simultaneously with an increase in the size of pacinian corpuscles and a decrease in the number of corpuscles.
Abnormal tactile perception may be caused by alterations at any level of the nervous system, from the receptor to the cerebral cortex. Any factor that interrupts or impairs reception, transmission, perception, or interpretation of touch also alters tactile sensation. Trauma, tumor, infection, metabolic changes, vascular changes, and degenerative diseases thus may cause tactile dysfunction, which may involve heightened or diminished tactile perceptions.
In addition, most tactile sensations evoke affective responses that determine whether the sensation is unpleasant, pleasant, or neutral. Cerebral and hypothalamic centers influence this response. Sedative drugs and prefrontal injury, which interrupt connections between the prefrontal cortex and subcortical centers, diminish the interpretation of tactile sensations.
Proprioception
Perception and awareness of the position of the body and its parts depend on impulses from the inner ear and from receptors in joints and ligaments. The role of muscle, tendon, and cutaneous receptors is indefinite. Sensory data are transmitted to higher centers, primarily through the dorsal columns and the spinocerebellar tracts, with some data passing through the medial lemnisci and thalamic radiations to the cortex. These stimuli are necessary for the coordination of movements, the grading of muscular contraction, and the maintenance of equilibrium.
A progressive loss of proprioception has been reported in older adults.233 Proprioceptive dysfunction may be caused by alterations at any level of the nervous system. As with tactile dysfunction, any factor that interrupts or impairs the reception, transmission, perception, or interpretation of proprioceptive stimuli also alters proprioception. Two common causes of proprioceptive dysfunction are vestibular dysfunction and neuropathy.
Specific vestibular dysfunctions are vestibular nystagmus and vertigo. Vestibular nystagmus is the constant, involuntary movement of the eyeball caused by ear disturbances. This condition occurs when the semicircular canal system is overstimulated. Vertigo is the sensation of spinning that occurs with inflammation of the semicircular canals in the ear. The individual may feel either that he or she is moving in space or that the world is revolving. Vertigo often causes loss of balance. Vertigo and nystagmus may occur in a variety of conditions, including labyrinthitis, vestibular neuritis, acute toxic labyrinthitis, benign paroxysmal positional vertigo, migrainous vertigo, and Ménière disease.234
Ménière disease is an idiopathic vestibular disorder that can cause proprioceptive dysfunction. The individual with Ménière disease also can experience vertigo, hearing loss, or tinnitus, and standing or walking may be impossible.
Peripheral neuropathies also can cause proprioceptive dysfunctions. Neuropathies may be caused by a variety of conditions and commonly are associated with renal disease and diabetes mellitus. Although the exact sequence of events is unknown, neuropathies are thought to be caused by a metabolic disturbance of the neuron itself. The result is a diminished or absent sense of body position or position of body parts. Gait changes often occur. (Neuropathies are further discussed in Chapter 17.)
Acute bacterial conjunctivitis (pinkeye) 506
Aδ fiber 483
Acute otitis media (AOM) 515
Acute pain 490
Age-related macular degeneration (AMD) 510
Ageusia 516
Allergic conjunctivitis 507
Allodynia 488
Amblyopia 509
Anosmia 516
Antipyretic 498
Aqueous humor 508
Astigmatism 511
Blepharitis 506
Cancer pain 493
Cataract 509
Central pain 494
Central sensitization 488
Chalazion 506
Choroid 507
Chronic conjunctivitis 507
Chronic otitis media 515
Chronic pain 492
Chronic postoperative pain 492
Circadian rhythm 496
Circadian rhythm sleep disorder 505
Cochlea 514
Color blindness 511
Complex regional pain syndrome (CRPS) 494
Conduction 497
Conductive hearing loss 515
Cone 507
Convection 497
Cornea 507
Crista ampullaris 514
Dark adaptation 510
Deafferentation pain 494
Diffuse noxious inhibitory controls (DNICs) 486
Diplopia 509
Dynorphin 489
Dyssomnia 504
Endogenous cryogen 498
Endogenous opioid 488
Endogenous pyrogen 498
Endolymph 514
Endomorphin 490
Endorphin 489
Enkephalin 489
Equilibrium receptor 514
Eustachian (pharyngotympanic) tube 513
Exogenous pyrogen 498
External auditory canal 512
Fovea centralis 507
Functional hearing loss 515
Gamma-aminobutyric acid (GABA) 488
Gate control theory 482
Glaucoma 510
Glutamate 488
Glycine 488
Hair cells 514
Heat cramps 500
Heat exhaustion 500
Heat production 496
Heat stroke 500
Hemianopia 512
Homonymous hemianopsia 512
Hordeolum (stye) 506
Hyperalgesia 487
Hyperopia 511
Hypersomnia 505
Hyperthermia 501
Hypogeusia 516
Hyposmia 516
Hypothermia 501
Incus (anvil) 512
Insomnia 504
Iris 507
Jerk nystagmus 509
Lens 508
Limbic system 486
Macula 509
Malignant hyperthermia 500
Malleus (hammer) 512
Mastoid air cell 512
Mastoid process 512
Ménière disease 517
Mixed hearing loss 515
Myofascial pain syndrome (MPS) 492
Myopia 511
Narcolepsy 505
Neuropathic pain 493
Nociception 482
Nociceptive pain 490
Nociceptor 482
Non-nociceptive pain 490
NREM (slow-wave) sleep 503
Nystagmus 509
Obesity hypoventilation syndrom 505
Obstructive sleep apnea syndrome (OSAS) 505
Olfactory hallucination 516
Opioid receptor 488
Optic chiasm 510
Optic disc 507
Organ of Corti 514
Otitis externa 514
Otitis media 514
Otoliths 514
Oval window 513
Pain threshold 490
Pain tolerance 490
Papilledema 510
Parageusia 516
Parasomnia 505
Parosmia 516
Pendular nystagmus 509
Perceptual dominance 490
Perilymph 514
Peripheral sensitization 487
Phantom limb pain 494
Pinna 512
Presbycusis 515
Presbyopia 511
Pupil 507
Radiation 497
Rapid eye movement (REM) sleep 503
Referred pain 491
Restless leg syndrome (RLS) 505
Reticular activating system 486
Retina 507
Retinal detachment 510
Rhegmatogenous retinal detachment 510
Rods 507
Sclera 507
Scotoma 509
Segmental inhibition 486
Semicircular canal 514
Sensorineural hearing loss 515
Shivering 501
Sleep 502
Somatic pain 491
Specificity theory 482
Stapes (stirrup) 512
Strabismus 509
Suprachiasmatic nucleus (SCN) 502
Sympathetically maintained pain (SMP) 494
Tinnitus 515
Trachoma 507
Tympanic cavity 512
Tympanic membrane 512
Unmyelinated C polymodal nociceptor 483
Vertigo 517
Vestibular nystagmus 517
Vestibule 514
Viral conjunctivitis 507
Visceral pain 491
Vitreous humor 508
REFERENCES
1. American Pain Society: www.ampainsoc.org/ce/npc/I/b_definitions.htm. Accessed July 24, 2004.
2. McCaffery, M. Understanding your patient’s pain. Nursing. 1980;10(9):26–31.
3. Waddell, G. The back pain revolution, ed 2. London: Churchill Livingstone; 2004.
4. Melzack, R., Wall, P.D. Pain mechanisms: a new theory. Science. 1965;150:971.
5. Melzack, R., Wall, P.D. Pain mechanisms: a new theory. Science. 1965;150:971–979.
6. Tominaga, M., Nociception and TRP channels. Handb Exp Pharmacol. 2007(179):489–505.
7. Rexed, B. A cytoarchitectural atlas of the spinal cord in the cat. J Comp Neurol. 1954;100:297.
8. Willis, W.D., Coggeshall, R.E. Sensory mechanisms of the spinal cord, ed 3. New York: Kluwer; 2004.
9. Romanelli, P., Esposito, V. The functional anatomy of neuropathic pain. Neurosurg Clin North Am. 2004;15(3):257–268.
10. Herrero, M.T., Barcia, C., Navarro, J.M. Functional anatomy of thalamus and basal ganglia. Childs Nerv Syst. 2002;18(8):386–404.
11. Partridge, L.D. Nervous system actions and interactions: concepts in neurophysiology. Boston: Kluwer; 2003.
12. Villemure, C., Bushnell, M.C. Cognitive modulation of pain: how do attention and emotion influence pain processing? Pain. 2002;95(3):195–199.
13. Mayer, D.J., Price, D.D. Central nervous system mechanisms of analgesia. Pain. 1976;2(4):379–404.
14. Ruda, M.A., Bennett, G.J., Dubner, R. Neurochemistry and neurocircuitry in the dorsal horn. Prog Brain Res. 1986;66:219–268.
15. Hughes, J., et al. Identification of two related pentapeptides from the brain with potent opiate agonist activity. Nature. 1975;258(5536):577–579.
16. Calvino, B., Grilo, R.M. Central pain control. Joint Bone Spine. 2006;73(1):10–16.
17. Rainville, P. Brain mechanisms of pain affect and pain modulation. Curr Opin Neurobiol. 2002;12(2):195–204.
18. Villenueva, L., Le Bars, D. The activation of bulbospinal controls by peripheral nociceptive inputs: diffuse noxious inhibitory controls (DNIC). Biol Res. 1995;28:113–125.
19. Fields, H.L., Basbuam, A.I., Heinricher, M.M. Central nervous system mechanisms of pain modulation. In McMahon S., Koltzenburg M., eds.: Wall and Melzack’s textbook of pain, ed 5, Edinburgh, Scotland: Churchill Livingstone, 2005.
20. Ploghaus, A., et al. Neural circuitry underlying pain modulation: expectation, hypnosis, placebo. Trends Cogn Sci. 2003;7(5):197–200.
21. Petrovic, P., et al. Placebo and opioid analgesia—imaging a shared neuronal network. Science. 2002;295:1737–1740.
22. Scott, D.J., et al. Placebo and nocebo effects are defined by opposite opioid and dopaminergic responses. Arch Gen Psychiatry. 2008;65(2):220–231.
23. Bleakman, D., Alt, A., Nisenbaus, E.S. Glutamate receptors and pain. Semin Cell Dev Biol. 2006;17(5):592–604.
24. Bodnar, R.J. Endogenous opiates and behavior, 2006. Peptides. 2007;28(12):2435–2513.
25. von Zastrow, M. Opioid receptor regulation. Neuromolec Med. 2004;5(1):51–58.
26. Rittner, H.L., Brack, A., Stein, C. Pain and the immune system. Br J Anaesth. 2008;101(1):40–44.
27. Wollemann, M., Benyhe, S. Non-opioid actions of opioid peptides. Life Sci. 2004;75(3):257–270.
28. Lai, J., et al. Pronociceptive actions of dynorphin via bradykinin receptors. Neurosci Lett. 2008;437(3):175–179.
29. Fichna, J., et al. The endomorphin system and its evolving neurophysiological role. Pharmacol Rev. 2007;59(1):88–123.
30. Coward, D.D., Wilkie, D.J. Metastatic bone pain: meanings associated with self-report and self-management decision making. Cancer Nurs. 2000;23(2):101–108.
31. Purves, D., et al. Neuroscience, ed 3. Sunderland, MA: Sinauer; 2004.
32. Thibodeau, G.A., Patton, K.T. Anatomy & physiology, ed 5. St Louis: Mosby; 2003.
33. Turk, D.C. The role of psychological factors in pain management. Acta Anaesthesiol Scand. 1999;43(9):885–888.
34. Turk, D.C., Okifuji, A. Psychological factors in chronic pain: evolution and revolution. J Consult Clin Psychol. 2002;70(3):678–690.
35. Miles, A., et al. Managing constraint: the experience of people with chronic pain. Soc Sci Med. 2005;61(2):431–441.
36. Almay, B.G., et al. Relationships between CSF levels of endorphins and monoamine metabolites in chronic pain patients. Psychopharmacology. 1980;67(2):139–142.
37. Schaible, H.G., Richter, F. Pathophysiology of pain. Langenbecks Arch Surg. 2004;389(4):237–243.
38. Whitehead, W., III., Kuhn, W.F. Miller, T.W., eds. Chronic pain, vol 1. Madison, WI: International Universities, 1990.
39. White, F.A., Jung, H., Miller, R.J. Chemokines and the pathophysiology of neuropathic pain. Proc Natl Acad Sci U S A. 2007;104(51):20151–20158.
40. Jones, G.T., Macfarlane, G.J. Epidemiology of low back pain in children and adolescents. Arch Dis Child. 2005;90(3):312–316.
41. Ehrlich, G.E. Back pain. J Rheumatol. 2003;67(Suppl):26–31.
42. Lavelle, E.D., Lavelle, W., Smith, H.S. Myofascial trigger points. Med Clin North Am. 2007;91(2):229–239.
43. Cummings, M., Baldry, P. Regional myofascial pain: diagnosis and management. Best Pract Res Clin Rheumatol. 2007;21(2):367–387.
44. Kehlet, H., Jensen, T.S., Woolf, C.J. Persistent postsurgical pain: risk factors and prevention. Lancet. 2006;367(9522):1618–1625.
45. Macrae, W.A. Chronic post-surgical pain: 10 years on. Br J Anaesth. 2008;101(1):77–86.
46. Goudas, L.C., et al. The epidemiology of cancer pain. Cancer Invest. 2005;23(2):182–190.
47. Seymour, J., Clark, D., Winslow, M. Pain and palliative care: the emergence of new specialties. J Pain Symptom Manage. 2005;29(1):2–13.
48. Cherny, N.I. The pharmacologic management of cancer pain. Oncology (Huntingt). 2004;18(12):1499–1515.
49. De Leon-Casasola, O.A. Current developments in opioid therapy for management of cancer pain. Clin J Pain. 2008;24(Suppl 10):S3–S7.
50. Dy, S.M., et al. Evidence-based standards for cancer pain management. J Clin Oncol. 2008;26(23):3879–3885.
51. Zhuo, M. Neuronal mechanism for neuropathic pain. Mol Pain. 2007;3:14.
52. Schaible, H.G., Peripheral and central mechanisms of pain generation. Handb Exp Pharmacol. 2007(177):3–28.
53. Edwards, J.L., et al. Diabetic neuropathy, mechanisms, to management. Pharmacol Ther. 2008;120(1):1–34.
54. Horowitz, S.H. The diagnostic workup of patients with neuropathic pain. Anesthesiol Clin. 2007;25(4):699–708.
55. Aurillo, C., et al. Ionic channels and neuropathic pain: physiopathology and applications. J Cell Physiol. 2008;215(1):8–14.
56. Ji, R.R., Strichartz, G., Cell signaling and the genesis of neuropathic pain. Sci STKE. 2004(252):reE14.
57. De Ridder, D., et al. Somatosensory cortex stimulation for deafferentation pain. Acta Neurochir Suppl. 2007;297(Pt 2):67–74.
58. Backonja, M.M., Serra, J. Pharmacologic management part 2: lesser-studied neuropathic pain diseases. Pain Med. 2004;5(Suppl 1):S48–S59.
59. Payne, R. Anatomy, physiology, and neuropharmacology of cancer pain. Med Clin North Am. 1987;71(2):153–167.
60. Canavero, S., Bonicalzi, V. Central pain syndrome: elucidation of genesis and treatment. Expert Rev Neurother. 2007;7(11):1485–1497.
61. Chahine, L., Kanazi, G. Phantom limb syndrome: a review. Middle East J Anesthesiol. 2007;19(2):345–355.
62. Woodhouse, A. Phantom limb sensation. Clin Exp Pharmacol Physiol. 2005;32(1-2):132–134.
63. Flor, H., Cortical reorganization and chronic pain: implications for rehabilitation. J Rehabil Med. 2003(41 Suppl):66–72.
64. Guimmarra, M.J., et al. Central mechanisms in phantom limb perception: the past, present and future. Brain Res Rev. 2007;54(1):219–232.
65. Schwartzman, R.J., Alexander, G.M., Grothusen, J. Pathophysiology of complex regional pain syndrome. Expert Rev Neurother. 2006;6(5):669–681.
66. Baron, R. Mechanistic and clinical aspects of complex regional pain syndrome (CRPS). Novartis Found Symp. 2004;261:220–233.
67. Littlejohn, G. Regional pain syndrome: clinical characteristics, mechanisms and management. Nat Clin Pract Rheumatol. 2007;3(9):504–511.
68. Akbazaz, R., Wong, Y.T., Homer-Vanniasinkam, S. Complex regional pain syndrome: a review. Ann Vasc Surg. 2008;22(2):297–306.
69. Tunks, E.R., Crook, J., Weir, R. Epidemiology of chronic pain with psychological comorbidity: prevalence, risk, course, and prognosis. Can J Psychiatry. 2008;53(4):224–234.
70. Gupta, A., Giordano, J. On the nature, assessment, and treatment of fetal pain: neurobiological bases, pragmatic issues, and ethical concerns. Pain Physician. 2007;10(4):525–532.
71. Lowery, C.L., et al. Neurodevelopmental changes of fetal pain. Semin Perinatol. 2007;31(5):275–282.
72. Grunau, R.E., Holsti, L., Peters, J.W. Long-term consequences of pain in humans and neonates. Semin Fetal Neonatal Med. 2006;11(4):268–275.
73. Van de Velde, M., et al. Fetal pain perception and pain management. Semin Fetal Neonatal Med. 2006;11(4):232–236.
74. Hummel, P., van Dijk, M. Pain assessment: current status and challenges. Semin Fetal Neonatal Med. 2006;11(4):237–245.
75. Duhn, J.L., Medves, J.M. A systematic integrative review of infant pain assessment tools. Adv Neonatal Care. 2004;4(3):126–140.
76. Golianu, B., et al. Pediatric acute pain management. Pediatr Clin North Am. 2000;47(3):559–587.
77. Gibson, S.J., Farrell, M. A review of age differences in the neurophysiology of nociception and the perceptual experience of pain. Clin J Pain. 2004;20(4):227–239.
78. Zheng, Z., et al. Age-related differences in the time course of capsaicin-induced hyperalgesia. Pain. 2000;85(1-2):51–58.
79. Gibson, S.J., Farrell, M. A review of age differences in the neurophysiology of nociception and the perceptual experience of pain. Clin J Pain. 2004;20(4):227–239.
80. Woodrow, K.M., et al. Pain tolerance: differences according to age, sex, and race. Psychosom Med. 1972;34(6):548–556.
81. Karp, J.F., et al. Advances in understanding the mechanisms and management of persistent pain in older adults. Br J Anaesth. 2008;101(1):111–120.
82. McCleane, G. Pharmacological pain management in the elderly patient. Clin Interv Aging. 2007;2(4):637–643.
83. McCleane, G. Pain perception in the elderly patient. Clin Geriatr Med. 2008;24(2):203–211. [V].
84. Weiner, D.K. Office management of chronic pain in the elderly. Am J Med. 2007;120(4):306–315.
85. Dinarello, C.A., Wolff, S.M. Pathogenesis of fever in man. N Engl J Med. 1978;298(11):607–612.
86. Boulant, J.A. Role of the preoptic-anterior hypothalamus in thermoregulation and fever. Clin Infect Dis. 2000;31(Suppl 5):S157–S161.
87. Mahmood, M.A., Zweifler, R.M. Progress in shivering control. J Neurol Sci 15. 2007;261(1-2):218–227.
88. Himms-Hagen, J. Current status of nonshivering thermogenesis. In: Ross Laboratories, ed. Assessment of energy metabolism in health and disease: a report of the first Ross conference in medical research. Columbus, OH: Ross Laboratories, 1980.
89. Sell, H., Deshaies, Y., Richard, D. The brown adipocyte: update on its metabolic role. Int J Biochem Cell Biol. 2004;36(11):2098–2104.
90. Asakura, H. Fetal and neonatal thermoregulation. J Nippon Med Sch. 2004;71(6):360–370.
91. Nedergaard, J., Bengtsson, T., Cannon, B. Unexpected evidence for active brown adipose tissue in adult humans. Am J Physiol Endocrinol Metab. 2007;293(2):E444–E452.
92. Steegmann, A.T., Jr. Human cold adaptation: an unfinished agenda. Am J Hum Biol. 2007;19(2):218–227.
93. Yousef, M.K. Effects of climatic stresses on thermoregulatory processes in man. Experientia. 1987;43(1):14–19.
94. Rintamaki, H. Human responses to cold. Alaska Med. 2007;49(2 Suppl):29–31.
95. Sherman, T.I., et al. Optimizing the neonatal thermal environment. Neonatal Netw. 2006;25(4):251–260.
96. Holowatz, L.A., et al. Altered mechanisms of vasodilation in aged human skin. Exerc Sport Sci Rev. 2007;35(3):119–125.
97. Degroot, D.W., Kenney, W.L. Impaired defense of core temperature in aged humans during mild cold stress. Am J Physiol Regul Integr Comp Physiol. 2007;292(1):R99–R102.
98. Roth, J., et al. Molecular aspects of fever and hyperthermia. Neurol Clin. 2006;24(3):421–439.
99. Steiner, A.A., Branco, L.G. Fever and anapyrexia in systemic inflammation: intracellular signaling by cyclic nucleotides. Front Biosci. 2003;8:s1398–s1408.
100. Blatteis, C.M. The onset of fever: new insights into its mechanism. Prog Brain Res. 2007;162:3–14.
101. Roth, J. Endogenous antipyretics. Clin Chim Acta. 2006;371(1-2):13–24.
102. Gregson, A.L., Mackowiak, P.A., Pathogenesis of fever. Cohen, J., Powderly, W.G., eds. Infectious diseases, vol 1. St Louis: Mosby, 2004.
103. Luheshi, G., Rothwell, N. Cytokines and fever. Int Arch Allergy Immunol. 1996;109(4):301–307.
104. Dinarello, C.A., Wolff, S.M. Molecular basis of fever in humans. Am J Med. 1982;72(5):799–819.
105. Kluger, M.S. The adaptive value of fever. In: Mackowiak P.A., ed. Fever: basic mechanisms and management. New York: Raven, 1991.
106. Griesman, L.A., Mackowiak, P.A. Fever: beneficial and detrimental effects of antipyretics. Curr Opin Infect Dis. 2002;15(3):241–245.
107. Yoshikawa, T.T., Norman, D.C. Approach to fever and infection in the nursing home. J Am Geriatr Soc. 1996;44(1):74–82.
108. Nakayama, J., Arinami, T. Molecular genetics of febrile seizures. Epilepsy Res. 2006;70(Suppl 1):S190–S198.
109. Fruthaler, G.J. Fever in children: phobia vs. facts. Hosp Pract. 1985;20(11A):49–53.
110. Dubé, C.M., et al. Fever, febrile seizures and epilepsy. Trends Neurosci. 2007;30(10):490–496.
111. Roti Roti, J.L. Cellular responses to hyperthermia (40-46 degrees C): cell killing and molecular events. Int J Hyperthermia. 2008;24(1):3–15.
112. Cleary, M. Predisposing risk factors on susceptibility to exertional heat illness: clinical decision-making considerations. J Sports Rehabil. 2007;16(3):204–214.
113. Cabanac, M., Brinnel, H. The pathology of human temperature regulation: thermiatrics. Experientia. 1987;43(1):19–27.
114. Bouchama, A., Dehbi, M., Chaves-Carballe, E. Cooling and hemodynamic management in heatstroke: practical recommendations. Crit Care. 2007;11(3):R54.
115. Glazer, J.L. Management of heatstroke and heat exhaustion. Am Fam Physician. 2005;71(11):2133–2140.
116. Bytomski, J.R., Squire, D.L. Heat illness in children. Curr Sports Med Rep. 2003;2(6):320–324.
117. Rosenberg, H., et al. Malignant hyperthermia. Orphanet J Rare Dis. 2007;2:21.
118. Wappler, F. Malignant hyperthermia. Eur J Anaesthesiol. 2001;18(10):632–652.
119. Ali, S.Z., Taguchi, A., Rosenberg, H. Malignant hyperthermia. Best Pract Res Clin Anaesthesiol. 2003;17(4):519–533.
120. Centers for Disease Control and Prevention: Hypothermia-related deaths—United States 2003-2004. MMWR Morb Mortal Wkly Rep. 2004;54(7):173–175.
121. Hildebrand, F., et al. Pathophysiologic changes and effects of hypothermia on outcome in elective surgery and trauma patients. Am J Surg. 2004;187(3):363–371.
122. Antretter, H., Dapunto, O.E., Bonatti, J. Management of profound hypothermia. Br J Hosp Med. 1995;54(5):215–220.
123. Reuler, J.B. Hypothermia: pathophysiology, clinical settings, and management. Ann Intern Med. 1978;89(4):519–527.
124. Aslam, A.F., et al. Hypothermia: evaluation, electrocardiographic manifestations, and management. Am J Med. 2006;119(4):297–301.
125. Long, W.B., 3rd., et al. Cold injuries. J Long Term Eff Med Implants. 2005;15(1):7–78.
126. DeLapp, T.D. Accidental hypothermia. Am J Nurs. 1983;83(1):62–67.
127. Wittmers, L.E., Jr. Pathophysiology of cold exposure. Minn Med. 2001;84(11):30–36.
128. Lee-Chiong, T.L., Jr., Stitt, J.T. Accidental hypothermia: when thermoregulation is overwhelmed. Postgrad Med. 1996;99(1):87–88. [7780, 83-84].
129. Jurkovich, G.J. Environmental cold-induced injury. Surg Clin North Am. 2007;87(1):247–267. [viii].
130. McCullough, L., Arora, S. Diagnosis and treatment of hypothermia. Am Fam Physician. 2004;70(12):2325–2332.
131. Sahuquillo, J., Vilalta, A. Cooling the injured brain: how does moderate hypothermia influence the pathophysiology of traumatic brain injury. Curr Pharm Des. 2007;13(22):2310–2322.
132. Axelrod, Y.K., Diringer, M.N. Temperature management in acute neurologic disorders. Crit Care Clin. 2006;22(4):767–785.
133. Azzopardi, D., Edwards, A.D. Hypothermia. Semin Fetal Neonatal Med. 2007;12(4):303–310.
134. Cushman, L., Warren, M.L., Livesay, S. Bringing research to the bedside: the role of induced hypothermia in cardiac arrest. Crit Care Nurs Q. 2007;30(2):143–153.
135. Kabon, B., Bacher, A., Spiss, C.K. Therapeutic hypothermia. Best Pract Res Clin Anaesthesiol. 2003;17(4):551–568.
136. Agrawal, A., Timothy, J., Thapa, A. Neurogenic fever. Singapore Med J. 2007;48(6):492–494.
137. Little, R.A., Stoner, H.B. Body temperature after accidental injury. Br J Surg. 1981;68(4):221–224.
138. Saski, H., et al. Is there a self-preserving hypothermic mechanism in shock? Shock. 2007;27(4):354–357.
139. Rajagopalan, S., et al. The effects of mild perioperative hypothermia on blood loss and transfusion requirement. Anesthesiology. 2008;108(1):71–77.
140. Okeke, L.I. Effect of warm intravenous and irrigating fluids on body temperature during transurethral resection of the prostate gland. BMC Urol. 2007;7:15.
141. Scott, E.M., Buckland, R. A systematic review of intraoperative warming to prevent postoperative complications. AORN J. 2006;83(5):1090–1104. [1107–1113].
142. Moore, R.Y. Suprachiasmatic nucleus in sleep-wake regulation. Sleep Med. 2007;8(Suppl 3):27–33.
143. Voss, U. Functions of sleep architecture and the concept of protective fields. Rev Neurosci. 2004;15(1):33–46.
144. Phillipson, E.A. State-of-the-art control of breathing during sleep. Am Rev Respir Dis. 1978;118:909.
145. Sakai, F., et al. Normal human sleep: regional cerebral hemodynamics. Ann Neurol. 1980;7(5):471.
146. Meadows, G.E., et al. Cerebral blood flow response to isocapnic hypoxia during slow-wave sleep and wakefulness. J Appl Physiol. 2004;97(4):1343–1348.
147. Silvania, A. Physiological sleep-dependent changes in arterial blood pressure: central autonomic commands and baroreflex control. Clin Exp Pharmacol Physiol. 2008;35(9):987–994.
148. Ganjavi, H., Shapiro, C.M. Hypocretin/Orexin: a molecular link between sleep, energy regulation, and pleasure. J Neuropsychiatry Clin Neurosci. 2007;19(4):413–419.
149. Siegel, J.M. Mechanisms of sleep control. J Clin Neurophysiol. 1990;7(1):49–65.
150. Dugovic, C. Role of serotonin in sleep mechanisms. Rev Neurol (Paris). 2001;157(11 Pt 2):S16–S19.
151. Siegel, J.M. Hypocretin (orexin): role in normal behavior and neuropathology. Annu Rev Psychol. 2004;55:125–148.
152. Sutcliffe, J.G., de Lecea, L. The hypocretins: setting the arousal threshold. Nat Rev Neurosci. 2002;3(5):339–349.
153. Siegel, J.M. The neurotransmitters of sleep. J Clin Psychiatry. 2004;65(Suppl 16):4–7.
154. Steiger, A. Neurochemical regulation of sleep. J Psychiatr Res. 2007;41(7):537–552.
155. Campbell, S.S. Evolution of sleep structure following brief intervals of wakefulness. Electroencephalogr Clin Neurophysiol. 1987;66(2):175–184.
156. Kedas, A., Lux, W., Amodeo, S. A critical review of aging and sleep research. West J Nurs Res. 1989;11(2):196–206.
157. Meltzer, L.J., Mindell, J.A. Sleep and sleep disorders in children and adolescents. Psychiatr Clin North Am. 2006;29(4):1059–1076.
158. Anders, T.F., Keener, M. Developmental course of nighttime sleep-wake patterns in full-term and premature infants during the first year of life. Sleep. 1985;8(3):173.
159. Keefe, M.R. Comparison of neonatal nighttime sleep-wake patterns in nursing versus rooming-in environments. Nurs Res. 1987;36(3):140.
160. Elias, M.F., et al. Sleep/wake patterns of breast-fed infants in the first 2 years of life. Pediatrics. 1986;77(3):322.
161. Leo, G. Parasomnias. WMJ. 2003;102(1):32–35.
162. Ivanhoe, J.R., Lefebvre, C.A., Stockstill, J.W. Sleep disordered breathing in infants and children: a review of the literature. Pediatr Dent. 2007;29(3):193–200.
163. Espiritu, J.R. Aging-related sleep changes. Clin Geriatr Med. 2008;24(1):1–14. [v].
164. Harrington, J., Lee-Chiong, T., Jr. Sleep and older patients. Clin Chest Med. 2007;28(4):673–684.
165. Kern, W., et al. Changes in cortisol and growth hormone secretion during nocturnal sleep in the course of aging. J Gerontol A Biol Sci Med Sci. 1996;51(1):M3–M9.
166. American Academy of Sleep Medicine. Diagnostic Classification Steering Committee: International classification of sleep disorders, revised: diagnostic and coding manual. Westchester, IL: American Academy of Sleep Medicine; 2005.
167. Voderh olzer, U., et al. Are gender differences in objective and subjective sleep measures? A study of insomniacs and healthy controls. Depress Anxiety. 2003;17(3):162–172.
168. Ancoli-Israel, S. Insomnia in the elderly: a review for the primary care practitioner. Sleep. 2000;23(Suppl 1):S23–S30. [discussion S36-S38].
169. Rigndahl, E.N., Pereira, S.L., Delzell, J.E., Jr. Treatment of primary insomnia. J Am Board Fam Pract. 2004;17(3):212–219.
170. Patil, S.P., et al. Adult obstructive sleep apnea: pathophysiology and diagnosis. Chest. 2007;132(1):325–337.
171. Netzer, N.C., Eliasson, A.H., Strohl, K.P. Women with sleep apnea have lower levels of sex hormones. Sleep Breath. 2003;7(1):25–29.
172. Saaresranta, T., et al. Effect of medroxyprogesterone on inspiratory flow shapes during sleep in postmenopausal women. Respir Physiol Neurobiol. 2003;134(2):131–143.
173. Bradley, T.E., Floras, J.S. Obstructive sleep apnoea and its cardiovascular consequences. Lancet. 2008;373(9657):82–93.
174. Ballard, R.D. Management of patients with obstructive sleep apnea. J Fam Pract. 2008;57(8 Suppl):S24–S30.
175. Lohmann-Hedrich, K., et al. Evidence for linkage of restless legs syndrome to chromosome 9p: are there two distinct loci? Neurology. 2007;70(9):686–694. [epub].
176. Patrick, L.R. Restless legs syndrome: pathophysiology and the role of iron and folate. Altern Med Rev. 2007;12(2):101–112.
177. Paulus, W., et al. Pathophysiological concepts of restless legs syndrome. Mov Disord. 2007;22(10):1451–1456.
178. Nishino, S. Clinical and neurobiological aspects of narcolepsy. Sleep Med. 2007;8(4):373–399.
179. Monk, T.H. Shift work. In Kryger M.H., Roth T., Dement W.C., eds.: Principles and practice of sleep medicine, ed 4, Philadelphia: Saunders, 2005.
180. Sack, R.L., et al. Circadian rhythm sleep disorders: part I, basic principles, shift work and jet lag disorders, An American Academy of Sleep Medicine review. Sleep. 2007;30(11):1460–1483. [review].
181. Taub, J.M., Berger, R.J. The effects of changing the phase and duration of sleep. J Exp Psychol Human Percept Perform. 1976;2(1):30.
182. Eastman, C.I., et al. Advancing circadian rhythms before eastward flight: a strategy to prevent or reduce jet lag. Sleep. 2005;28(1):33–44.
183. Sheldon, S.H. Parasomnias in childhood. Pediatr Clin North Am. 2004;51(1):69–88. [vi].
184. Gugger, J.J., Wagner, M.L. Rapid eye movement sleep behavior disorder. Ann Pharmacother. 2007;41(11):1833–1841.
185. Modell, S., Lauer, C.J. Rapid eye movement (REM) sleep: an endophenotype for depression. Curr Psychiatry Rep. 2007;9(6):480–485.
186. Lewis, D.A. Sleep in patients with respiratory disease. Respir Care Clin North Am. 1999;5(3):447–460. [ix].
187. McNicholas, W.T. Impact of sleep on COPD. Chest. 2000;117(2 Suppl):48S–53S.
188. Papier, A., Tuttle, D.J., Mahar, T.J. Differential diagnosis of the swollen red eyelid. Am Fam Physician. 2007;76(12):1815–1824.
189. Rao, S.K., et al. The itching, burning eye: diagnostic algorithm and management options. Compr Ophthalmol Update. 2006;7(4):157–167.
190. Wright, H.R., Turner, A., Taylor, H.R. Trachoma. Lancet. 2008;371(9628):1945–1954.
191. Rosenberg, E.A., Sperazza, L.C. The visually impaired patient. Am Fam Physician. 2008;77(10):1431–1436.
192. Doshi, N.R., Rodriquez, M.L. Amblyopia. Am Fam Physician. 2007;74(3):361–367.
193. Ticho, B.H. Strabismus. Pediatr Clin North Am. 2003;50(1):173–188.
194. Rucker, J.C. An update on acquired nystagmus. Semin Ophthalmol. 2008;23(2):91–97.
195. Shotton, K., Elliott, S. Interventions for strabismic amblyopia. Cochrane Database Syst Rev. (2):2008. [CD006461].
196. Brass, S.D., Zivadinov, R., Bakshi, R. Acute demyelinating optic neuritis: a review. Front Biosci. 2008;13:2376–2390.
197. West, S. Epidemiology of cataract: accomplishments over 25 years and future directions. Ophthalmic Epidemiol. 2007;14(4):173–178.
198. Jackson, R.G., Owsley, C., McGwin, G., Jr. Aging and dark adaptation. Vision Res. 1999;39(23):3975–3982.
199. Wiggs, J.L. Genetic etiologies of glaucoma. Arch Ophthalmol. 2007;125(1):30–37.
200. Legrun-Julien, F., Di Polo, A. Molecular and cell-based approaches for neuroprotection in glaucoma. Optom Vis Sci. 2008;85(6):417–424.
201. Terzel, G., Yang, J., Wax, M.B. Heat shock proteins, immunity and glaucoma. Brain Res Bull. 2004;62(6):473–480.
202. Chidlow, G., Wood, J.P., Casson, R.J. Pharmacological neuroprotection for glaucoma. Drugs. 2007;67(5):725–759.
203. Sodhi, A., et al. Recent trends in the management of rhegmatogenous retinal detachment. Surv Ophthalmol. 2008;53(1):50–67.
204. Montezuma, S.R., Sobrin, L., Seddon, J.M. Review of genetics in age related macular degeneration. Semin Ophthalmol. 2007;22(4):229–240.
205. Zayit-Soudry, S., Moroz, I., Loewenstein, A. Retinal pigment epithelial detachment. Surv Ophthalmol. 2007;52(3):227–243.
206. Gohel, P.S., et al. Age-related macular degeneration: an update on treatment. Am J Med. 2008;121(4):279–281.
207. Beiko, G. Status of accommodative intraocular lenses. Curr Opin Ophthalmol. 2007;18(1):74–79.
208. Callina, T., Reynolds, T.P. Traditional methods for the treatment of presbyopia: spectacles, contact lenses, bifocal contact lenses. Ophthalmol Clin North Am. 2006;19(1):25–33.
209. Deeb, S.S., Kohl, S. Genetics of color vision deficiencies. Dev Ophthalmol. 2003;37:170–187.
210. Swanson, W.H., Cohen, J.M. Color vision. Ophthalmol Clin North Am. 2003;16(2):179–203.
211. Javer, A.R., Schwartz, D.W. Plasticity in human directional hearing. J Otolaryngol. 1995;24(2):111–117.
212. Timiras, P.S. Physiological basis of aging and geriatrics, ed 3. Boca Raton, FL: CRC Press; 2003.
213. Liu, X.Z., Yan, D. Ageing and hearing loss. J Pathol. 2007;211(2):188–197.
214. Osguthorpe, J.D., Nielsen, D.R. Otitis externa: review and clinical update. Am Fam Physician. 2006;74(9):1510–1516.
215. Cripps, A.W., Otczyk, D.C. Prospects for a vaccine against otitis media. Expert Rev Vaccines. 2006;5(4):517–534.
216. Smith, J.A., Danner, C.J. Complications of chronic otitis media and cholesteatoma. Otolaryngol Clin North Am. 2006;39(6):1237–1255.
217. Yoon, T.H., et al, Tympanoplasty, with or without mastoidectomy, is highly effective for treatment of chronic otitis media in children. Acta Otolaryngol Suppl. 2007(558):44–48.
218. Paradise, J.L., et al. Tympanostomy tubes and developmental outcomes at 9 to 11 years of age. N Engl J Med. 2007;356(3):248–261.
219. De la Cruz, A., Angeli, S., Slattery, W.H. Stapedectomy in children. Otolaryngol Head Neck Surg. 1999;120(4):487–492.
220. Liu, X.Z., Yan, D. Ageing and hearing loss. J Pathol. 2007;211(2):188–197.
221. Temple, E.C., et al. Taste development: differential growth rates of tongue regions in humans. Brain Res Dev Brain Res. 2002;135(1-2):65–70.
222. Boyce, J.M., Shone, G.R. Effects of ageing on smell and taste. Postgrad Med J. 2006;82(966):239–241.
223. Doty, R.L. Influence of age and age-related diseases on olfactory function. Ann N Y Acad Sci. 1989;561:76–86.
224. Cain, W.S., Reid, F., Stevens, J.C. Missing ingredients: aging and the discrimination of flavor. J Nutr Elderly. 1990;9(3):3.
225. Seiberling, K.A., Conley, D.B. Aging and olfactory and taste function. Otolaryngol Clin North Am. 2004;37(6):1209–1228. [vii].
226. Kovacs, T. Mechanisms of olfactory dysfunction in aging and neurodegenerative disorders. Ageing Res Rev. 2004;3(2):215–232.
227. Winkler, S., et al. Depressed taste and smell in geriatric patients. J Am Dent Assoc. 1999;130(12):1759–1765.
228. Nordin, S., Bramerson, A. Complaints of olfactory disorders: epidemiology, assessment and clinical implications. Curr Opin Allergy Clin Immunol. 2008;8(1):10–15.
229. Bromley, S.M. Smell and taste disorders: a primary care approach. Am Fam Physician. 2000;61(2):427–438.
230. Strous, R.D., Shoenfeld, Y. To smell the immune system: olfaction, autoimmunity and brain development. Autoimmun Rev. 2006;6(1):54–60.
231. Besne, I., Descombes, C., Breton, L. Effect of age and anatomical site on density of sensory innervation in human epidermis. Arch Dermatol. 2002;138(11):1445–1450.
232. Stevens, J.C. Age and spatial acuity of touch. J Gerontol. 1992;47(1):P35–P40.
233. Shaffer, S.W., Harrison, A.L. Aging of the somatosensory system: a translational perspective. Phys Ther. 2007;87(2):193–207.
234. Neuhauser, H.K. Epidemiology of vertigo. Curr Opin Neurol. 2007;20(1):40–46.