PAIN, TEMPERATURE REGULATION, SLEEP, AND SENSORY FUNCTION


Alterations in sensory function may involve dysfunctions of the general or the special senses. Dysfunctions of the general senses include chronic pain, abnormal temperature regulation, tactile dysfunction, and proprioceptive dysfunction. Dysfunctions of the special senses include visual, auditory, vestibular, olfactory, and gustatory (taste).
Pain is a unique sensory experience that although universally described as unpleasant, is nonetheless essential to our survival. Pain provides protection by signaling the presence of disease or injury. Unlike pain, which need not be a part of everyday life, temperature is carefully monitored and regulated within clearly defined normal limits. Like pain, however, variations in temperature can signal disease. Fever is a common manifestation of dysfunction and is often the first symptom observed in an infectious or inflammatory condition. If the body’s temperature regulatory mechanism is out of balance, the result may be death.
Sleep is a normal, cyclic process that restores the body’s energy and maintains normal functioning. Sleep is so essential to physiologic and psychologic function that sleep deprivation causes a wide range of clinical manifestations. Prolonged deprivation or disruption of sleep ultimately leads to serious dysfunction.
The special senses of vision, hearing, touch, smell, and taste are the means by which individuals perceive stimuli that are essential for interacting with the environment. Special sensory receptors are connected to specific areas of the brain through the afferent pathways of the peripheral and central nervous system (CNS). Each of the special senses thus involves a connected system of organs and tissues that receives stimuli and sends sensory messages to areas of the CNS, where they are processed and guide behavior.
PAIN
Pain is one of the body’s most important adaptive and protective mechanisms and all definitions suggest it is a complex phenomenon and cannot be characterized as only a response to injury. A widely accepted definition of pain is that drafted by the International Association for the Study of Pain (IASP) and accepted by the American Pain Society and the World Health Organization: “Pain is an unpleasant sensory and emotional experience associated with actual or potential tissue damage or described in terms of such damage.”1 McCaffery maintains that “pain is whatever the experiencing person says it is, existing whenever he says it does.”2 Waddell defines pain as “a symptom, not a clinical sign, diagnosis or disease.…”3 A clear understanding of the complexities of the pain experience—specifically one that encompasses an individual’s emotions, cognition, motivation, prior history, and even issues of secondary gain—is needed to manage pain and to further understanding the pain processes.
Theories of Pain
In the seventeenth century, the French philosopher and mathematician René Descartes proposed that the body works like a machine that can be studied by scientific methods and that injury activates specific pain receptors and fibers that project to the brain. He further postulated that the intensity of pain is directly related to the amount of associated tissue injury. For instance, pricking one’s finger with a needle would cause minimal pain, whereas cutting one’s hand with a knife would produce more pain. This theory—the specificity theory—is generally accurate when applied to certain types of injuries and the acute pain associated with them. But the specificity theory did not allow for psychologic contributions, such as attention to pain, prior experience, and the emotions involved in the “meaning” of the situation.4 Nevertheless, it was this theory of pain, with some modification, that was still operational entering the twentieth century.
The gate control theory, proposed in 1965 by Melzack and Wall, provided the first cohesive explanation for the emerging complexities of pain phenomena, particularly chronic pain, and this has had a powerful effect on pain research and therapy.5 According to this theory, pain transmission is modulated by a balance of impulses transmitted to the spinal cord by large A-delta (Aδ) and small C fibers. These fibers terminate on inhibitory interneurons in the substantia gelatinosa (laminae in the dorsal horn of the spinal cord). Cells in the substantia gelatinosa function as a gate, regulating transmission of impulses to the CNS. Stimulation of non-nociceptive larger A fibers such as touch, vibration or thermal stimuli, cause the cells in the substantia gelatinosa to “close the pain gate” which diminishes pain perception. Small fiber input inhibits cells in the substantia gelatinosa and “opens the pain gate”, enhancing pain perception. The CNS, through efferent pathways, may also close, partially close, or open the gate. The gate control theory is inadequate to explain some chronic pain problems such as phantom limb pain, and the neuromatrix theory was proposed to explain such pain (see p. 494). Over the past 20 years exceptional progress has been made in strengthening the gate control theory of pain by elucidating the neuroanatomy and neuropharmacology of pain pathways in the peripheral and central nervous systems.
Neuroanatomy of Pain
The perception of pain is called nociception and depends on specifically dedicated receptors and afferent pathways that detect and transmit noxious or damaging or potentially damaging stimuli. The gate control theory describes these pathways in great detail, and explains the experience of pain by emphasizing the activation of non-nociceptive afferent input coming into the dorsal horn of the spinal cord to inhibit pain signals as well as the dynamic role of the brain in modulating pain processes.
Nociceptors
Nociceptive impulses arising from skin, muscle, joints, arteries, and the viscera are transmitted from unspecialized, bare sensory nerve endings called nociceptors that respond to chemical, mechanical, and thermal stimuli (Table 15-1). The variable nature and distribution of nociceptors affects relative sensitivity to pain in different areas of the body. For example, fingertips have more nociceptors than the skin of the back, and all skin has many more nociceptors than the internal organs. Unlike sensory neurons of the special senses of vision, gustation, and olfaction (discussed later), which are required to detect only one type of sensory stimulus (e.g., light for the sense of vision), primary nociceptive afferents have the remarkable ability to detect a wide range of stimuli. To do this nociceptors are equipped with an array of transduction channels that can sense different forms of noxious stimulation and at different intensities. In addition to the previously well studied voltage-gated potassium, sodium, and calcium channels, research has delineated the presence of up to seven other types of transmembrane receptors (called transient receptor potential [TRP] channels), which reside on “naked nerve endings” and respond to a variety of physical, chemical, and thermal stimuli.6
Table 15-1
Stimuli That Activate Nociceptors (Pain Receptors)
| Location of Receptor | Provoking Stimuli |
| Skin | Pricking, cutting, crushing, burning, freezing |
| Gastrointestinal tract | Engorged or inflamed mucosa, distention or spasm of smooth muscle, traction on mesenteric attachment |
| Skeletal muscle | Ischemia, injuries of connective tissue sheaths, necrosis, hemorrhage, prolonged contraction, injection of irritating solutions |
| Bone | Periosteal injury, inflammation, fractures, tumors |
| Joints | Synovial membrane inflammation |
| Arteries | Piercing, inflammation |
| Head | Traction, inflammation, or displacement of arteries, meningeal structures, and sinuses; prolonged muscle contraction |
| Heart | Ischemia and inflammation |
Nociceptors are categorized according to the stimulus to which they respond and by the properties of the axons associated with them. Severe mechanical deformation excites mechanonociceptors, whereas mechanothermal nociceptors are stimulated by mechanical deformation and/or extremes of temperature. These two receptors are associated with lightly myelinated, medium-sized Aδ fibers. Other types of mechanical, thermal, and chemical nociception are transmitted by excitation of polymodal nociceptors and are carried on small, unmyelinated C fibers.
The nerve action potentials generated by excitation of any of these nociceptors travel along these two fiber types to reach the spinal cord. Nociceptive transmission through the larger Aδ fibers occurs more quickly than it does through C fibers. Aδ fibers carry well-localized, sharp pain sensations and are important in initiating rapid reactions to stimuli (fast pain). The small unmyelinated C polymodal nociceptors are responsible for the transmission of the diffuse burning or aching sensations that follow (slow pain) (Figure 15-1).
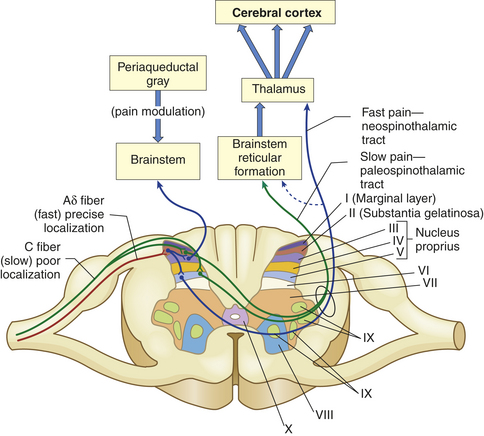
Figure 15-1 Pain fibers that terminate primarily in laminated II and V of the dorsal horn. The myelinated Aδ fibers (fast localized pain) synapse on a second set of neurons that carry the signal to the thalamus via the neospinothalamic tracts. The C fibers (slow pain) synapse on laminae II and V interneurons that connect with neurons in laminae II, IV, and V and carry the pain signal to the reticular formation and midbrain via the paleospinothalamic tract. The axons of the spinothalamic tracts cross over the spinal cord to ascend in the anterior and lateral spinal cord white matter.
Pathways of Nociception
The cell bodies of primary-order neurons or pain-transmitting neurons reside in the dorsal root ganglia just lateral to the spine along the sensory pathways that penetrate the posterior part of the cord. Once the axons of the primary afferents (Aδ and C fibers) enter the cord (Figure 15-2), they synapse with second-order neurons that may branch into ascending or descending collaterals for one or two cord segments in neuronal projections called the dorsolateral tract of Lissauer (named after the German neurologist who first described it in the late nineteenth century). Eventually all of the primary afferents terminate in the gray matter of the dorsal horn in distinctive layers or laminae originally described by Rexed.7 The gray matter of the cord has been divided into 10 of these laminae based on the size, shape, and plane of the projection of the neurons found there. Many of the primary afferents carrying nociceptive information were originally found to terminate in lamina II, which was given the name substantia gelatinosa. Lamina I is called the marginal layer, and laminae III to V are known as the nucleus proprius (see Figure 15-1).
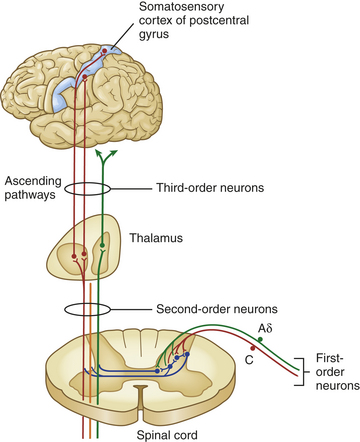
Figure 15-2 Nociception pathways. Aδ and C fibers comprise the primary, first-order sensory afferents coming into the gate at the posterior part of the spinal cord. Here we see second-order neurons crossing the cord (“decussating”) and ascending to the thalamus as part of the spinothalamic tract. Third-order afferents project to higher brain centers of the limbic system, the frontal cortex, and the primary sensory cortex of the postcentral gyrus of the parietal lobe.
Three classes of second-order neurons are found in the dorsal horn: (1) projection cells, which relay information to higher brain areas (cephalad); (2) excitatory interneurons, which relay nociceptive transmissions to projection cells, other interneurons, or to motor cells concerned with local reflexes such as the pain withdrawal reflex; and (3) inhibitory interneurons, which modulate nociceptive transmission. The synaptic connections between cells of primary and second-order neurons located in the substantia gelatinosa and other Rexed laminae function as a “pain gate,” providing one of the major tenets advanced by the gate control theory. This “gate” in the spinal cord regulates the transmission of pain impulses that ascend to the brain for further processing and interpretation (see p. 482).
From the gate in the dorsal horn, nociception continues on the axons of the second-order neurons as they cross the midline of the cord and ascend to various areas of the brain. These ascending fibers are organized into tracts or funiculi that are found in the white matter of the spinal cord, and are named according to their location in the cord and to where they project—either to the higher cord and brainstem or to the diencephalon (thalamus and hypothalamus) and limbic structures. Most nociceptive information travels by means of ascending columns in the lateral spinothalamic tract (also called the anterolateral funiculus). Other bits of nociceptive signals travel in the posterior columns of the cord to the dorsal column nuclei of the medulla, and from there ascend in the medial lemniscus to the lateral thalamus. Several other spinal cord projection systems convey nociceptive information directly or indirectly to the reticular formation of the brainstem and the periaqueductal gray (PAG) matter of the midbrain. These include the postsynaptic dorsal column, and the spinocervical, spinoreticular, spinomesencephalic, spinoparabrachial, spinohypothalamic, and other spinolimbic pathways8 (Figure 15-3).
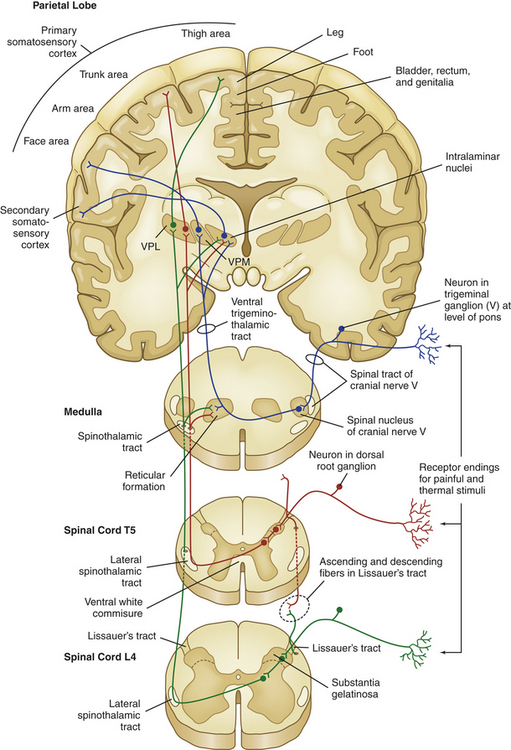
Figure 15-3 Central nervous system pathways that mediate the sensations of pain and temperature. VPM, Ventral posterior medial thalamic nuclei; VPL, ventral posterior lateral thalamic nuclei.
Although the organization of all of the ascending tracts is complex, the principal target for nociceptive afferents is the thalamus (the major relay station of sensory information in general). The thalamus is divided into medial and lateral groups by a band of fibers called the internal medullary lamina. The ventral posterior lateral (VPL) and ventral posterior medial (VPM) nuclei of the thalamus facilitate the localization of pain and integrate these perceptions into a neuroendocrine response (e.g., the response to fright or to surgical stress).9,10
From the thalamus, brainstem, and midbrain, third-order neurons project to portions of the CNS involved in the processing and interpretation of pain, the chief areas being the reticular and limbic systems and cerebral cortex.
Pain Processing in the Brain
Cerebral Cortex: The third-order neurons of the ventral posterior nuclear complex of the thalamus project in a highly organized manner to the primary and secondary somatosensory cortex11 (Brodmann areas 3, 1, 2; see Figure 14-7, B). On the postcentral gyrus of the parietal lobe there is a topographically organized representation of the body that mirrors the concentration of peripheral sensory receptors known as the sensory homunculus (see Figure 14-8, p. 453). This area of the brain is thought to be involved in the discriminative and cognitive aspects of pain; that is, what we think about the pain.12
The frontal lobe of the cerebral cortex receives diffuse projections from the medial thalamic nuclei, which are thought to subserve the affective expression of pain (how your expression of pain appears to an observer) through their frontal-limbic connections. Frontal lobectomies were once used in an effort to treat some cases of intractable pain. Postsurgically individuals continued to report pain if questioned, but they seldom asked for medications and no longer seemed to care about their pain. This response is also observed in bilateral thalamic lesions.13
Subcortical Systems: The limbic and reticular tracts are involved in alerting the body to danger, initiating arousal of the organism, and emotionally processing the perceived afferent signals not just as stimuli, but as pain. The limbic system consists of a ring of cortex on the medial aspect of each cerebral hemisphere, the subcortical nuclei, parts of the thalamus and midbrain, and the hypothalamus. The reticular activating system (see Figure 14-6) is composed of a number of vaguely defined nuclei situated in the core of the brainstem extending throughout its rostrocaudal extent. Many reticular neurons respond to noxious stimulation by initiating escape behaviors. The limbic and reticular systems are phylogenetically very old. Together, they regulate the complex emotional responses to pain; that is, what we feel about the pain. Pain signals to these areas serve to arouse the whole organism to ongoing tissue damage, thereby activating protective neuroendocrine and autonomic reflexes such as the “fight or flight” response, the release of stress hormones, and beneficial cardiovascular changes. The gate control theory has been very successful in integrating the functions of these very “old” systems into our understanding of the emotional and motivational aspects of pain.
Neuromodulation of Pain
The extraordinary advances of gate control theory moved the focus of pain research away from the periphery and into the spinal cord and brain. The theory helps to better explain the psychologic component of pain—that the pain experience itself need not be proportional to the actual peripheral injury or disease. The pain pathways, no longer viewed as merely labeled lines of electric wires, were finally understood to function holistically as a single peripheral CNS complex, promoting further investigation into spinal sensitization and CNS plasticity.
By the mid-1970s and into the 1980s, two other developments played key roles in extending the theory to explain how, under certain stressful conditions, significant traumatic injuries can be experienced as completely painless in awake, neurologically intact individuals. The first was the discovery of specific descending pathways from the brain to the spinal cord that could produce significant and selective analgesia in those experiencing pain14 (Figure 15-4). The second event was the identification of ubiquitous opioid receptors found throughout the body, and shortly thereafter, the isolation, purification, and sequencing of endogenous opioid peptides.15 What followed was a virtual explosion of scientific research aimed at elucidating the pathways and chemicals that modify, or neuromodulate, the pain experience.
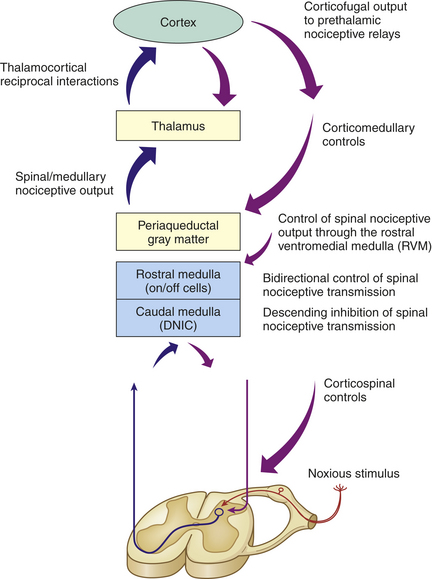
Figure 15-4 Diagram representing the central mechanisms of pain modulation. A noxious peripheral stimulus activates both segmental and bulbospinal heterosegmental modulatory mechanisms, which either accentuate or inhibit afferent pain transmission to the brain. The most important and widespread source of top-down (corticofugal) modulation arises from the cortex. Both thalamic and prethalamic nociceptive relays are under the influence of this corticofugal control. The dorsal horn of the spine is also under the influence of the caudal medulla through diffuse noxious inhibitory control (DNIC). (Modified from Villanueva L, Fields HL: The pain system in normal and pathological states: a primer for clinicians, Seattle, 2004, IASP Press.)
Pathways of Neuromodulation
How does central processing, including memory and interpretation of pain in the brain, lead to changes in how much algogenic (pain related) information passes through the spinal cord gate? When Melzack and Wall originally proposed the gate control theory, they described the possibility of segmental inhibition of pain, elicited by activity in large-diameter cutaneous afferent fibers at the dermatome level, which can be activated naturally by innocuous mechanical stimuli.4 The reality of segmental inhibition was quickly verified when research into peripheral nerve anatomy demonstrated that stimulation of a group of large, fast, heavily myelinated A-beta (Aβ) fibers (which synapse in the dorsal horn along with their nociceptive Aδ and C fiber counterparts) can close the pain gates through an inhibitory interneuron. These afferent Aβ fibers carry non-noxious low-threshold mechanical information gained by touch, vibration, and pressure. This is intuitive to anyone who has hit his or her thumb with a hammer and knows that holding the thumb or putting it into the mouth conveys distraction input that lessens the pain.
The vast body of work completed since the inauguration of the gate control theory has focused on the complexity of inhibitory modulation beyond the segmental level (i.e., heterosegmental modulation). This newer work emphasizes a functional basis for pain control outside the dorsal horn, which was the focus of the original gate control hypothesis.
Over the past 3 decades a wealth of information has added to our understanding of heterosegmental, supraspinal mechanisms elicited by noxious and non-noxious stimuli.16 Powerful heterosegmental control of nociception probably originates from the cortex because almost all nociceptive relays within the CNS are under so-called top-down (corticofugal) modulation that often occurs even in the absence of painful stimuli.17 Farther down, the caudal medulla participates in widespread inhibitory phenomena called diffuse noxious inhibitory controls (DNICs) whereby an unpleasant (noxious) peripheral stimulation remote from the pain site relieves pain—the basis for pain relief with acupuncture or deep massage.18 Several ascending and descending bulbospinal pathways (between the medulla and spinal cord) respond simultaneously to the noxious stimulus and participate in DNIC. The net effect of these supraspinal structures is to precisely encode the intensity of the noxious stimulus and transmit descending feedback, mainly to the deep dorsal horn neurons.19 Figure 15-4 illustrates how higher brain regions participate in corticofugal and bulbospinal pain modulation as intricately incorporated descending pathways complete the circle back to the Rexed laminae of the spinal cord. Of interest is the recent demonstration that expectancy-related cortical activation also can exert control over analgesic systems of the brainstem to attenuate pain.20,21 In other words, cognitive expectations (also known as the placebo effect or nocebo [adverse] effect) can cause real, measurable, often powerful physiologic effects that share some of the same descending corticofugal pathways as our pain modulation system.22
The entire complex of pathways now can be visualized as an integration of peripheral sensory axon terminals, spinal interneurons, and top-down control pathways that converge on the spinal dorsal horns. The result is to modify, dampen, or augment nociceptive transmission, depending on the many factors existing both within and without the organism.
Neurotransmitters of Neuromodulation
Many neurotransmitters mediate the transmission of pain in the periphery, the spinal cord, and the brain. In the periphery, local injury and inflammation can result in direct or indirect excitation of nociceptors. Pain neurotransmitters can be classified as inflammatory, pain excitatory, or pain inhibitory (Box 15-1).

Direct excitation occurs when nociceptors respond with a threshold depolarization initiated by the application of heat, radiation, toxic chemicals, or tissue trauma. Indirect excitation occurs through the release of inflammatory mediators after the tissue is injured. The tissue injury results in inflammation and the release of prostaglandins such as PGE2 and PGI2, tumor necrosis factor-alpha (TNF-α), nitric oxide, bradykinins, and histamine (see Chapter 6). For example, it has been shown that lymphokines released in chronic lymphocytic inflammatory lesions contribute to some types of chronic pain. In addition, activity within the nociceptors causes them to release peptides and neurotransmitters such as substance P, neurokinin A, calcitonin gene-related peptide (CGRP), and adenosine triphosphate (ATP), which promote the spread of pain locally and further contribute to vasodilation, increased vascular permeability, and degranulation of even more mast cell cytokines. The resultant “inflammatory soup” serves to lower the threshold for nociceptive depolarization resulting in peripheral sensitization and pain augmentation—hyperalgesia. This is readily recognized by anyone who has suffered a bad sunburn and then notices the resulting extrasensitivity of the skin to stimuli (such as mild heat or touch) that normally would be considered non-noxious. Normally peripheral sensitization phenomena extinguish themselves as the tissue heals and inflammation subsides. However, when primary afferent function is altered in an enduring way by injury or disease of the nervous system, hyperalgesia may persist and be highly resistant to treatment.
In the spinal cord and brain, a wide variety of biogenic amines and other neurotransmitters act to modulate control over the transmission of pain impulses. Serotonin, norepinephrine, glutamate, aspartate, glycine, gamma-aminobutyric acid, and an array of endogenous opioids have been found to stimulate or inhibit interneurons in the CNS. This in turn may serve to stimulate or inhibit the pain gate and the primary nociceptive tracts.
Excitatory Neurotransmitters: Glutamate and aspartate, amino acid precursors, are the most common excitatory neurotransmitters in the brain and spinal cord. Glutamate activates two different kinds of receptors: AMPA/kinate (alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionate) receptors, which are very fast, and N-methyl-D-aspartate (NMDA) receptors, which are implicated in memory and long-term potentiation of synapses. Both of these receptors lead to excitation of the membrane and depolarization of the cell, be it an inhibitory or excitatory neuron. Glutamate receptors mediate many spinal and central responses to painful stimulation.23 High levels of glutamate and aspartate have been found in the PAG as well as at the synapses of first-order nociceptors with ascending spinothalamic tract neurons. High levels of activity in the nociceptive afferents may result in an activity-dependent increase in the excitability of neurons in the dorsal horn of the cord. Glutamate, which accumulates in the dorsal horn, results in the displacement of a magnesium ion that serves to inhibit the NMDA receptor. With the loss of the blocking magnesium ion, receptor “wind-up” or sensitization in the CNS to further nociception becomes evident. As a result of this central sensitization (increased excitability of neurons), activity levels that were subthreshold before the sensitizing event become sufficient to open the more sensitive pain gate.
Although central sensitization is triggered in dorsal horn neurons by activity in nociceptors, innocuous activation of fibers carrying low-threshold mechanoreception (such as lightly brushing the surface of the skin with a cotton swab) will activate second-order nociceptive neurons, giving rise to a sensation (often quite distressing) of pain. The induction of pain by what is normally considered an innocuous or nonpainful stimulus (i.e., light touch) is referred to as allodynia and can be a major feature of neuropathic pain syndromes.
Inhibitory Neurotransmitters: Gamma-aminobutyric acid (GABA) and glycine have major inhibitory effects in the spinal cord and brain. For example, dorsal horn laminae interneurons are rich in GABA (GABA-A, GABA-B, etc.) and function to inhibit release of pain neurotransmitters. Norepinephrine and 5-hydroxytryptamine (serotonin) contribute to pain modulation (inhibition) in the medulla and pons.
Endogenous opioids are a family of morphine-like neuropeptides that inhibit transmission of pain impulses in the spinal cord, brain, and periphery.24 There are four types of opioid neuropeptides: (1) enkephalins, (2) endorphins, (3) dynorphins, and (4) endomorphins. These substances are neurohormones that act as neurotransmitters by binding to one or more G-protein–coupled opioid receptors. Three distinct types of opioid receptors are found in the body: mu (μ) (with subtypes μ-1 and μ-2), kappa (κ), and delta (δ). Each receptor type binds differently with the various types of opioids.
Agonist activity at the opioid receptors by endogenous opioids inhibits the release of excitatory neurotransmitters such as substance P in the dorsal horn (blocking the transmission of the painful stimulus) or in other areas of the brain such as the PAG or the rostral ventromedial nuclei in the brainstem25 (Figure 15-5). Opioids from the midbrain release adrenergic and serotonergic descending pathways from GABAergic inhibition and decrease pain. Leukocytes release opioids and participate in peripheral pain control26 (see What’s New? Opioid-Producing Leukocytes and Pain Control).
WHAT’S NEW?
Opioid-Producing Leukocytes and Pain Control
At sites of peripheral inflammation, leukocytes release inflammatory mediators that cause pain. However, infiltrating leukocytes also produce anti-inflammatory cytokines and opioid peptides that produce analgesia. Inflammation in peripheral tissue results in up-regulation of opioid receptors at peripheral sensory nerve terminals with enhanced peripheral efficacy of opioids. Opioid-containing immune cells migrate to inflamed tissues using chemokines and adhesion molecules. Under stressful conditions or in response to releasing agents such as corticotropin-releasing factor, cytokines, chemokines, and catecholamines, leukocytes secrete opioids. Once released opioid peptides activate peripheral opioid receptors and produce analgesia by inhibiting the excitability of sensory nerves and/or the release of excitatory neuropeptides. Of clinical significance, these effects occur without central untoward side effects such as respiratory depression, somnolence, clouding of consciousness, or addiction. Future pain management strategies include the selective targeting of opioid-containing leukocytes and peripheral endogenous opioid systems to achieve analgesia with few side effects.
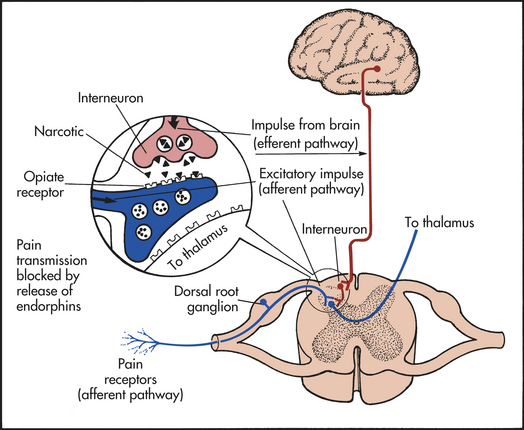
Figure 15-5 Descending pathway and endorphin response. The biologic receptors of the enkephalins and endorphins are located close to known pain receptors in the periphery and ascending and descending pain pathways.
Data from: Smith HS: Pain Physician 11(2 Suppl):S121-S132, 2008; Machelska H: Neuropeptides 41(6):355-363, 2007; Rittner HL et al:Br J Anaesth 101(1):40-44, 2008.
Perhaps the best known and the most prevalent of these natural opioids are the enkephalins. There are two types of enkephalins, methionine enkephalin (met-enkephalin) and leucine enkephalin (leu-enkephalin), and their ratio is 4:1, respectively. They were the first endogenous opioids extracted in research. Enkephalins, which like the other endogenous opioids can be identified immunohistochemically, are found concentrated in the hypothalamus, the PAG matter, the nucleus raphe magnus of the medulla, and the dorsal horns of the spine.
Endorphins were first discovered in the human PAG in 1979, β-endorphin being the best studied of the group. The synthesis and activity of β-endorphin is concentrated in the hypothalamus and the pituitary gland; β-endorphin is purported to produce a greater sense of exhilaration, or “high,” than all of the other endorphin types. It is a strong μ-receptor agonist and is generally believed to provide substantial natural pain relief.
Dynorphins (the most potent of these endogenous neurohormones) are found in the hypothalamus, the brainstem PAG rostral ventromedial medulla (PAG-RVM) system, and the spine. Dynorphins, which bind strongly to κ receptors located in the dorsal horn of the spinal cord, generally serve to impede pain signals but can, in certain areas, incite pain by activation of bradykinin receptors.27,28
Endomorphins have potent analgesic and gastrointestinal (GI) effects. Endomorphin-1 (Tyr-Pro-Trp-Phe-NH2, EM-1) and endomorphin-2 (Tyr-Pro-Phe-Phe-NH2, EM-2) are peptides isolated from the brain and the spinal cord and show the highest affinity and selectivity for the μ (morphine) opiate receptor.29 Chemical (capsaicin) and surgical (rhizotomy) disruption of nociceptive primary afferent neurons deplete levels of EM-2, implicating the peripheral nervous system as being the principal site of action. Thus EM-2 is well-positioned to serve as an antinociceptive modulator of pain in its earliest stages (i.e., in the peripheral transmission). In contrast to EM-2, which is more prevalent in the spinal cord and lower brainstem, EM-1 is more widely and densely distributed throughout the brain. The distribution is consistent with the role peptides play in the modulation of diverse functions, including adaptation to pain and stress and enhancement of reward perceptions.
In addition to its analgesic effects, endogenous opioids are involved in a variety of other functions throughout the body—one being maintenance of feeding behavior. This finding is supported by research indicating enhanced feeding responses being elicited after heavy exertion or injection of β-endorphin. Administration of naloxone, a μ-receptor antagonist, was found to negate this effect. Endogenous opioids also have been linked to moderation of drinking behavior and cough suppression. Stress, excessive physical exertion, acupuncture, intercourse, and other factors increase the levels of circulating endogenous opioids—serotonin, norepinephrine, and other neurotransmitters—and consequently raise the pain threshold.29 Endogenous opioids of one type or another are found to bind to almost all tissues in the body and may be responsible for general sensations of well-being or lack thereof.24
Pain Threshold and Pain Tolerance
The pain threshold is the point at which a stimulus is perceived as pain. The threshold does not vary significantly among people or in the same person over time. Intense pain at one location, however, may cause an increase in the threshold in another location. For example, a person with severe pain in one knee is less likely to experience chronic back pain that is less intense. This phenomenon is called perceptual dominance. Because of perceptual dominance, an individual with many painful sites may report only the most painful one. After the dominant pain is diminished, the individual may then identify other painful areas.4
Pain tolerance is the duration of time or the intensity of pain that an individual will endure before initiating overt pain responses. It is the amount of pain the person will tolerate before outwardly responding to it. Pain tolerance is influenced by the person’s cultural perceptions, expectations, role behaviors, and physical and mental health. Pain tolerance generally is decreased with repeated exposure to pain. Tolerance is decreased also by fatigue, anger, boredom, apprehension, and sleep deprivation. Tolerance may be increased by alcohol consumption, persistent use of pain medication, hypnosis, warmth, distracting activities, and strong beliefs or faith.
Pain tolerance varies greatly among people and in the same person over time because of the body’s ability to respond differently to noxious stimuli. No direct relationship exists between the objectively measured intensity of painful stimuli and an individual’s perception of pain or response to pain.
Clinical Description of Pain
The most widely used clinical classifications for pain are based on the inferred neurophysiologic mechanisms, temporal aspects, etiology, and region affected. The mechanistic approach to categorizing pain is common, but from a clinical viewpoint, this approach has shortcomings. Usually pain conditions are described clinically by mechanism as either nociceptive or non-nociceptive and by duration as either acute or chronic.
Nociceptive pain, such as the pain of a crushed finger or a heart attack, is pain with a cause resulting from normal tissue injury. Nociceptive pain is either somatic (derived from the Greek word soma for “body” or “body wall”—meaning the whole axial portion of the body including trunk, head, and neck) or visceral (derived from the internal organs).
Non-nociceptive pain, for the purposes of this text, is defined as neuropathic pain (discussion of psychogenic pain is not covered in this chapter). Neuropathic pain is subdivided into peripheral and central categories (see page 493).
Additionally, somatic, visceral, and neuropathic pains can all occur as acute or chronic presentations. These broad categories have been summarized in Box 15-2. Some of the most common clinical pain presentations are detailed below.
Acute Pain
Acute pain is a protective mechanism that alerts the individual to a condition or experience that is immediately harmful to the body. The onset of acute pain is sudden, and usually dissipates after the stimulus is removed and the tissues have healed. A direct one-to-one relationship between physical signs of disease and accompanying symptoms is almost always present. Peripheral and central sensitization, that is, wind-up (see p. 486), is not evident. Anxiety is common in acute pain states and is usually apparent in the alterations of vital signs. Tachycardia, hypertension, fever, diaphoresis, dilated pupils, and outward pain behavior such as moaning, touching, or rocking motions are discernible to an observer. Other physical manifestations include elevation of blood sugar levels, decreases in gastric acid secretion and intestinal motility, and a general decrease in blood flow to the viscera and skin. Nausea occasionally occurs.
The signs, symptoms, and behaviors of acute pain are a predictable response to the threat inherent in the painful experience, including issues surrounding the cause of pain, its treatment, and prognosis (Table 15-2). Individuals often psychologically respond to acute pain with fear (e.g., fear of diagnosis, fear of continued pain), anxiety, and a general sense of unpleasantness or unease. The stress of fear itself may in turn contribute to the physiologic signs of pain. Some individuals are reluctant to discuss or report their pain,30 although hope of recovery is usually present.
Table 15-2
Comparison of Acute and Chronic Pain
| Characteristic | Acute Pain | Chronic Pain |
| Experience | An event | A situation; state of existence |
| Source | External agent or internal disease usually known | Unknown; if known, treatment is prolonged or ineffective |
| Onset | Usually sudden | May be sudden or develop insidiously |
| Duration | Transient (up to 6 months) | Prolonged and persistent (months to years) |
| Pain identification | Painful and nonpainful areas generally well identified | Painful and nonpainful areas less easily differentiated: change in sensations becomes more difficult to evaluate |
| Clinical signs | Typical response pattern with more visible signs | Response patterns vary; fewer overt signs (adaptation) |
| Significance | Significant (informs person something is wrong) | Person looks for significance |
| Pattern | Self-limiting or readily corrected | Continuous or intermittent; intensity may vary or remain constant |
| Course | Suffering usually decreases over time | Suffering usually increases over time |
| Actions | Leads to actions to relieve pain | Leads to actions to modify pain experience |
| Prognosis | Likelihood of eventual complete relief | Complete relief usually not possible |
Data from Black RG: Surg Clin North Am 55(4):999, 1975.
Acute pain arises from cutaneous and deep somatic tissue, or from visceral organs and can be classified as (1) acute somatic, (2) acute visceral, and (3) referred.
Somatic pain arises from connective tissue, muscle or bone, and skin and is either sharp and well localized (especially fast pain carried by Aδ fibers) or dull, aching, and poorly localized pain as seen in polymodal C fiber transmissions.
Visceral pain refers to pain in internal organs and the abdomen and is transmitted by sympathetic afferents. It is poorly localized because of the lesser number of nociceptors in the visceral structures, which often can be cauterized or cut without any sensation of pain being felt. However, any stretching or distention of internal organs or the abdomen will elicit a violent response.
Visceral pain is associated with nausea and vomiting, hypotension, restlessness, and, in some cases, shock. It often radiates (spreads away from the actual site of the pain) or is referred. Visceral pain that is primarily nociceptive and not referred is carried by second-order neurons that travel in the dorsal column pathway to the gracilis nucleus of the medulla and then in contralateral pathways of the medial lemniscus to finally synapse in the thalamus.31
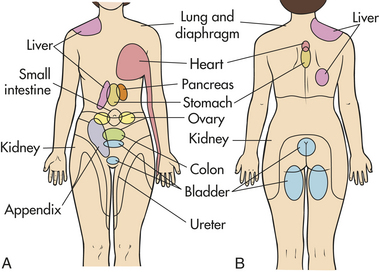
Referred pain is pain that is present in an area removed or distant from its point of origin. Referred pain usually, but not always, originates from the viscera and so is discussed here. The most familiar examples are pain in the shoulder from myocardial infarction, pain in the back from pancreatic or renal disease, and pain in the right shoulder from an inflamed gallbladder. Referred visceral pain is carried by second-order neurons that travel in the contralateral spinothalamic tracts. The area of referred pain is supplied by the same spinal segment as the actual site of pain because impulses from many cutaneous and visceral neurons converge. The presumed mechanism is one of afferent fibers from this conjoint area in the spinal cord, relaying the mistaken perception that the pain arises from the referral site. Because the skin has more receptors, the painful sensation is more often experienced there instead of at the site of origin.32 (Common areas of referred pain and their associated sites of origin appear in Figure 15-6.)
Chronic Pain
Chronic pain also can be nociceptive, but it is prolonged—usually defined as lasting at least 3 months. Perhaps even more important than the duration of the pain are the attendant physical and emotional issues that often appear to be way out of proportion to any observable tissue injury. Thus the cause of chronic pain often is unknown, and even if the cause is known or suspected, the pain does not respond to usual therapy. Chronic pain may be persistent as in chronic low back pain or occur intermittently as seen in migraine or muscle tension–migraine variant headache syndromes. The onset may be sudden, but chronic pain often develops insidiously so that the individual generally experiences more suffering over time. Individual behavior is adaptive and directed toward modifying the pain. Chronic pain is often associated with a sense of hopelessness and helplessness as relief becomes more elusive and the timeframe more protracted. The pain is perceived as meaningless, and depression is often a concomitant finding, as either a result of the chronic pain state or as a contributor to its development. It is significantly more difficult to manage than acute pain, and complete relief usually is never obtained (see Table 15-2).
Physiologic responses to chronic pain depend on whether it is persistent or intermittent. Intermittent pain produces a physiologic response similar to that of acute pain (i.e., tachycardia, diaphoresis, elevated blood pressure, etc). Persistent, chronic pain, however, allows for physiologic adaptation so that a person with chronic pain may have normal heart and respiratory rates and normal blood pressure. The absence of acute physiologic responses has led many to mistakenly assume that people in chronic pain are malingering because they do not appear to be in pain.
Chronic pain produces significant behavioral and psychologic changes.33 Individuals with chronic pain often are depressed, have difficulty sleeping and eating, and may become preoccupied with the pain.34 Living with chronic pain requires constant attention to the earliest signs of pain so that the pain-provoking stimuli can be identified and avoided. People with chronic pain generally attempt to keep pain-related behavior to a minimum so that they appear as normal as possible. A desire for the validation of the pain and the need to hide the pain are usually conflicting drives for those with chronic pain. They tend not to report pain for fear of being labeled complainers. They often deny pain and sometimes engage in activities that provoke pain in an effort to keep up with others. Even in learning to pace themselves throughout the day’s activities, they may inadvertently aggravate the pain.35
Chronic pain states are thought to arise from a misinterpretation of nociceptive input—an imbalance of neuromodulation controls such as might occur with a decreased level of endorphins or a predominance of C-neuron stimulation36,37 (Table 15-3). The following mechanisms have been implicated in the initiation and entrenchment of chronic pain states38,39:
Table 15-3
Common Chronic Pain Conditions
| Condition | Description |
| Persistent low back pain | Most common chronic pain condition |
| Results from poor muscle tone, inactivity, muscle strain, or sudden, vigorous exercise | |
| Myofascial pain syndromes | Second most common chronic pain condition |
| Pain results from muscle spasm, tenderness, and stiffness | |
| Examples include myositis, fibrositis, myofibrositis, myalgia, and muscle strain—conditions that involve injury to the muscle and fascia | |
| As disorder progresses, pain becomes increasingly generalized | |
| Chronic postoperative pain | Chronic pain that can occur with disruption or cutting of sensory nerves |
| Cancer pain | Can be pain attributed to advance of disease, associated with treatment, or attributed to coexisting disease entities |
| Deafferentation pain | Painful condition resulting from damage to a peripheral nerve |
| Common types include severe burning pain triggered by various stimuli, such as cold, light touch, or sound, and reflex sympathetic dystrophies (occur after peripheral nerve injury and are characterized by continuous, severe, burning pain associated with vasomotor changes and muscle wasting) | |
| Hyperesthesias | Increased sensitivity and decreased pain threshold to tactile and painful stimuli |
| Pain is diffuse, modified by fatigue and emotion and mixed with other sensations | |
| May result from chronic irritations of central nervous system areas | |
| Hemiagnosia | Loss of ability to identify source of pain on one side of the body |
| Painful stimuli on that side produce discomfort, anxiety, moaning, agitation, and distress but no attempt to withdraw from the stimulus | |
| Associated with stroke | |
| Phantom limb pain | Pain experienced in amputated limb after stump has completely healed; may be immediate or occur months later |
| Influenced by emotions or sympathetic stimulation | |
| Trigger points—small hypersensitive regions in muscle or connective tissues that, when stimulated, produce pain in a specific area |
• Changes in sensitivity of neurons—lower threshold with peripheral and central sensitization
• Spontaneous impulses from regenerating peripheral nerves
• Alterations in the dorsal root ganglion in response to peripheral nerve injury and neurotransmitters—reorganization of nociceptive neurons (deafferentation pain)
The most common chronic, disabling pain condition in the United States is persistent low back pain, which became a medical disaster in the twentieth century. Over the past few decades much has been learned about back pain—where back pain fits into the pain spectrum and how people react to and are affected by it. Predictions that chronic low back pain (and the disability arising from it) would be reducing are unfounded; instead both the report of low back pain and the accompanying disability are on the rise. Recent figures estimate that 7% to 14% of adults in the United States have some disability related to back pain, and 1% to 2% of the population are totally disabled by back pain at any given time.40,41
Myofascial pain syndrome (MPS) is associated with injury to muscle, fascia and tendons and include myositis, fibrositis, myofibrositis, myalgia, and muscle strain. These conditions involve myofascial trigger points within a taut band of skeletal muscle.42 The pain may be the result of low-threshold mechanosensitive afferents projecting to sensitized dorsal horn neurons.43 Compression of the trigger point causes referred pain, motor dysfunction, and autonomic responses. During the early stages of the disorder the pain is localized, but as the disorder progresses it becomes deep, aching, and more generalized. These, like many other chronic conditions, begin as a result of poor muscle tone, inactivity, muscle or tendon strain, or sudden vigorous exercise and can evolve into a chronic pain state.
Chronic postoperative pain occurs in some individuals and includes complex regional pain syndrome, phantom limb pain, chronic donor site pain, post-thoracotomy pain syndrome, postmastectomy pain syndrome, and joint arthroplasty pain. Plastic changes in the peripheral nervous system (PNS) and CNS contribute to pain development, and multimodal approaches to analgesia are needed for pain management.44,45
Cancer pain is often chronic and associated with neuropathies. Studies done at Memorial Sloan-Kettering Cancer Center indicate three major categories of pain syndromes that result in chronic pain in the individual with cancer.46 The categories are (1) pain attributed to the advance of the disease, (2) pain associated with treatment of the disease, and (3) pain attributed to coexisting entities (e.g., osteoarthritis) that are unrelated to the disease. Cancer pain is by far the most common cause of chronic pain attributed to the advance of an identifiable disease.46 Pain can be caused by infection and inflammation, increasing pressure of a growing tumor on nerve endings, tissue destruction, stretching of visceral surfaces, or obstruction of ducts and intestine. Therapeutic approaches to the management of cancer pain have advanced significantly in recent years, particularly in palliative care and hospice programs.47 Frequent pain assessment, management of breakthrough pain and implementation of therapeutic strategies including pharmacotherapy, anesthetic, neurosurgical, psychologic, and rehabilitation techniques and frequent evaluations are essential to optimal cancer pain management.48–50
Neuropathic Pain
Neuropathic pain is the result of trauma or disease of nerves and leads to long term plastic changes along somatosensory pathways from the periphery to cortex and abnormal processing of sensory information by the PNS and CNS.51 Most neuropathic pain seen commonly in clinical practice is chronic. Chronic neuropathic pain syndromes can be divided into two groups based on a central or peripheral location of the nervous system lesion.52 Examples of peripheral causes of neuropathic pain include trauma, diabetic or alcohol abuse–induced neuropathy, carcinoma, and human immunodeficiency virus (HIV). Examples of central causes of neuropathic pain include brain or spinal cord trauma, tumors, vascular lesions, multiple sclerosis, postherpetic neuralgia (PHN), phantom limb pain, and reflex sympathetic dystrophy. Diabetic neuropathy (PDN) and post herpetic neuralgia are both significant causes of neuropathic pain.53 Neuropathic pain is often paroxysmal with paresthesias (tingling sensations of pins and needles), burning, shooting, or stabbing sensations. Neuropathic pain is often described as “gnawing” and miserable. Pain sensation can occur in the absence of a stimulus, be evoked by movement (incident pain), and hypersensitivity and/or allodynia may be present in the involved body part. No single diagnostic test for neuropathic pain (or for pain in general) is available, and differentiating some neuropathies from other chronic somatic pain syndromes can be difficult.54 When injured nerves become hyperexcitable, they generate ectopic discharges, resulting in spontaneous firing of some neurons with low thresholds for mechanical, chemical, or thermal stimuli. 55,56 Injury resulting in a permanent loss of sensory input from a part of the body is called deafferentation, and differentiates peripheral neuropathic pain syndromes from central forms of the disease.
Deafferentation pain results from trauma or chemical injury to the peripheral nervous system; tumor infiltration of nerve tissue; or damage from radiation, chemotherapy, or surgical sectioning of a nerve with loss of sensory input to the CNS. It is associated with hyperactivity of the somatosensory thalamus and cortex.57 Deafferentation pain, which is poorly controlled by many analgesics, is usually described as a constant, dull, viselike ache, accompanied by paroxysms of burning or electric shock-like sensations.58,59
Central pain is neuropathic and is caused by a lesion or dysfunction in the CNS (brain or spinal cord) and may be related to alterations in thalamocortical transmission.60 The lesions of central pain can include infarction, hemorrhage, abscess, degeneration, tumors, or traumatic injury and can develop in diseases such as Parkinson disease and multiple sclerosis. The pain may manifest over a large and diffuse area or be well defined and circumscribed. It is usually irritating and constant, can be difficult to treat, and can cause considerable suffering. Hemiagnosia pain is one form of central pain associated with stroke that produces paralysis and a hypersensitivity/allodynia on one half of the body. In this painful condition, often a concomitant loss of ability to identify the source of pain through normal sensory pathways occurs. The result is a confusing picture in which even mild stimulation of the affected side of the body produces discomfort, anxiety, moaning, agitation, and distress, with a diminished ability to withdraw from the offending stimulus. Thalamic pain is another form of central pain that involves lesions in the thalamus.
Phantom limb pain is pain that an individual feels in an amputated limb after the stump has completely healed. Nonpainful phantom limb sensations occur in almost all amputees, but the sensations usually fade with time. This is distinguished from the syndrome of phantom limb pain, a chronic pain occurring in 80% to 100% of amputees.61,62 It is more likely to appear in individuals who experienced pain in the limb before amputation. Theories about the cause of phantom limb pain include regeneration or activity of injured or cut peripheral nerves, scar tissue or neuroma formation in the cut peripheral nerves, spinal cord deafferentation, and alterations in the thalamus and cortex. It has been proposed that CNS integration, including reorganization and plastic changes of the somatosensory cortex, results in the perception of pain from receptors associated with the amputated limb even though the limb itself is no longer present63,64 (see What’S New? The Concept of a Neuromatrix and Pain). The cause is likely multifactorial, contributing to the difficulty of prevention or effective treatment.
Sympathetically maintained pain (SMP) is another type of neuropathic pain that occurs after peripheral nerve or extremity injury and is characterized as continuous and severe with a burning quality. Sympathetically maintained pain is often associated with vasospasm and vasomotor changes in the affected limb. The diseases formerly called reflex sympathetic dystrophy and causalgia, both thought to arise from sympathetic nervous system imbalance, are now called complex regional pain syndromes (CRPSs), types I and II. CRPS develops 1 to 2 weeks after an extremity injury (i.e., a fracture without identifiable nerve injury) (type I—reflex sympathetic dystrophy) or injury to the brachial plexus, the median, sciatic, or other peripheral nerves (type II—causalgia). The exact pathophysiology is unclear, but a combination of injury and the presence of inflammatory cytokines and neuropeptides may lead to peripheral nociceptive sensitization and physiologic change in pain transmission and autonomic and motor systems.65 The severe, diffuse, and persistent pain occurs in the extremity supplied by the injured nerve. Discoloration and changes in the texture of the skin may appear in the affected area. Vasomotor changes usually begin with vasodilation and are followed by vasoconstriction and cool, cyanotic, and edematous extremities. Excessive nail growth may be noted, and swelling and stiffness of proximate joints may occur. Hair loss is usually noted, and allodynia is often prominent, disabling, and difficult to treat.66–68
In the evaluation and management of neuropathic pain, as well as all other chronic pain states, psychologic components (e.g., depression or anxiety), sleep disturbances,
work-related issues of impairment and disability, treatment expectations, the availability of social support from family and friends (or lack thereof) and legal compensation should not be overlooked.69
Pediatrics and Perception of Pain
Children and infants have the anatomic and functional ability to perceive pain. Pain pathways and cortical and subcortical centers for pain perception, as well as neurochemicals associated with pain transmission and modulation, are functional in preterm and newborn infants.70 The nociceptor system is functional in fetuses by 20 to 24 weeks of gestation although the cortical experience may be minimal.71 Repetitive, painful experiences and prolonged exposure to analgesic drugs in infants during the neonatal period may permanently alter synaptic and neuronal organization,72 and fetal pain may have an enduring effect on behavior and pain perception.73
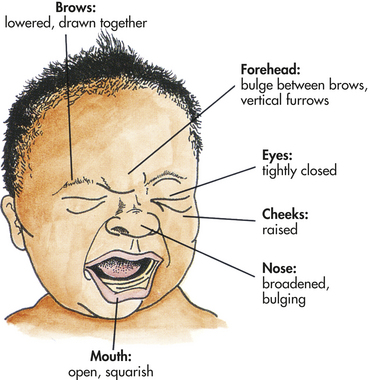
Facial expression, crying, body movements, and lack of consolability are the most consistent expressions of pain in infants. The painful facial expression includes lowered brows drawn together; presence of a vertical bulge and furrows in the forehead between the brows; broadened nasal root; tightly closed, scourged eye fissures; and angular, squarish mouth and chin quiver (Figure 15-7). Physiologic responses include increases in heart rate, blood pressure, and respiratory rate. There may be flushing or pallor, sweating, and decreased oxygen saturation.74 Toddlers also express pain with crying, facial expression, and body language (tensed body, guarding, and hands holding body). Older children, between ages 5 and 18 years, tend to have a lower pain threshold than do adults. Children, like adults, have highly individual responses to pain. Any pain must be carefully and accurately assessed 75 and adequately treated for children of all ages.76
Aging and Perception of Pain
Studies on pain perception in the older adult population have yielded conflicting evidence. Some studies show an increase in pain threshold with aging; others show no change.77,78 The varied results are probably a function of independent variation in the sensory-discriminative, motivational-affective, and cognitive-evaluative components of the pain experience. In general, studies confirm that an increase in the pain threshold occurs in some older adults. This change may be caused by peripheral neuropathies and changes in the thickness of the skin.79 (Neuropathies are discussed in Chapter 17.) A decrease in pain tolerance is also evident in some older adults, and women appear to be more sensitive to pain than are men80 (see What’S New? Pain and Gender). Pain in the older adult is also influenced by liver and renal function, including alterations in metabolism of drugs and metabolites and age-associated brain changes. Pain must be accurately assessed in relation to its effect on cognitive function, coexisting disease, drug interactions, other reactions to treatment, and an individual’s ability to express pain.81 Poorly managed pain can result in depression, inactivity, and failure to maintain activities of daily living.82–84
TEMPERATURE REGULATION
In all homeothermic animals, temperature regulation is achieved through precise balancing of heat production, heat conservation, and heat loss. In humans, body temperature is maintained around 37° C (98.6° F) and rarely exceeds 41° C. The normal range is 36.2° to 37.7° C (97.2° to 99.9° F), but all parts of the body do not have the same temperature. The extremities, for example, are generally cooler than the trunk. The temperature at the core of the body (as measured by rectal temperature) is generally 0.5° C higher than at the surface (as measured by oral temperature) and has minimal fluctuations. The internal temperature varies normally in response to activity, environmental temperature, and daily fluctuation of circadian rhythm (the pattern of each 24-hour day). Oral temperatures generally fluctuate within 0.2° to 0.5° C over a 24-hour period. Women tend to have wider fluctuations that follow the menstrual cycle, with a sharp rise in temperature just before ovulation. In both sexes the daily fluctuating temperature peaks around 6 PM and is at its lowest during sleep.85 Maintenance of body temperature within the normal range is necessary for life.
Hypothalamic Control of Temperature
Temperature regulation is mediated by thermoregulatory centers in the hypothalamus.86 Peripheral thermoreceptors in the skin and central thermoreceptors in the hypothalamus, spinal cord, abdominal organs, and other central locations provide the hypothalamus with information about skin and core temperatures. If these temperatures are low or high, the hypothalamus responds by triggering heat production, heat conservation, or heat loss mechanisms.
Increased heat production is initiated by a series of hormonal mechanisms involving the hypothalamus and its connections with the endocrine system (see Chapter 20). The heat-producing mechanism begins with a hypothalamic hormone, thyroid-stimulating hormone-releasing hormone (TSH-RH). In turn, TSH-RH stimulates the anterior pituitary to release TSH, which acts on the thyroid gland, stimulating release of thyroxine (T4), one of the thyroid hormones. This hormone then acts on the adrenal medulla, causing the release of epinephrine (a catecholamine and vasopressive hormone) into the bloodstream (see Chapter 20). Epinephrine causes vasoconstriction, stimulates glycolysis, and increases metabolic rates, thus increasing heat production.
The hypothalamus also triggers heat conservation. The mechanisms of heat conservation involve stimulating the sympathetic nervous system, which is responsible for stimulating the adrenal cortex, increasing skeletal muscle tone, initiating the shivering response, and producing vasoconstriction. The hypothalamus also functions in raising body temperatures by relaying information to the cerebral cortex. Awareness of cold provokes voluntary responses such as increased body movement and adding protective clothing.
The hypothalamus responds to warmer core and peripheral temperatures by reversing the same mechanisms. The TSH-RH pathway is shut down. The sympathetic pathway is prompted to produce cutaneous vasodilation, decreased muscle tone, and increased sweat production. Hypothalamic stimulation of the cerebral cortex provokes voluntary measures to reduce heat production and promote heat loss.
Mechanisms of Heat Production
Body heat is produced by the chemical reactions of metabolism, skeletal muscle tone and contraction, and chemical thermogenesis. Heat is distributed by the circulatory system.
Chemical Reactions of Metabolism: The chemical reactions that occur during the ingestion and metabolism of food and those required to maintain the body at rest (basal metabolism) require energy and give off heat. These processes occur in the body core (primarily the liver) and are in part responsible for the maintenance of core temperature.
Skeletal Muscle Contraction: Skeletal muscles produce heat through two mechanisms: (1) gradual increase in muscle tone and (2) rapid muscle oscillations (shivering—which does not occur in neonates). Both increasing muscle tone and shivering are controlled by the posterior hypothalamus and occur in response to cold. As peripheral temperature drops, muscle tone increases and shivering begins. Shivering is a fairly effective method for increasing heat production because no work is performed and all the energy produced is retained as heat.87
Chemical Thermogenesis: Chemical thermogenesis, also called nonshivering thermogenesis or adrenergic thermogenesis, results from the release of epinephrine and norepinephrine. Epinephrine and norepinephrine produce a rapid, transient increase in heat production by raising the body’s basal metabolic rate. Chemical thermogenesis seems to be different from hormone-triggered increases in the basal metabolic rate. Chemical thermogenesis produces a quick, brief rise in basal metabolic rate, whereas the hormone thyroxine triggers a slow, prolonged rise.88 Chemical thermogenesis occurs in brown adipose tissue present mainly in newborn infants that have high surface:volume ratios. Brown adipose tissue is rich with mitochondria and blood vessels and is essential for nonshivering thermogenesis. Infants lose more heat through conduction and convection than they are capable of generating through normal metabolic mechanisms, and brown adipose tissue therefore plays an important role in maintaining infant body temperature. As with most mammals reared in a temperature-controlled environment, humans gradually lose the capacity for chemical thermogenesis as brown adipose cells dedifferentiate. This can occur as early as 4 weeks after birth.89,90 Because of the decrease in brown adipose tissue in the adult, the role of this mechanism of heat production is less significant than in infants, but may be important for thermal adaptation in cold environments.91,92
Mechanisms of Heat Loss
Heat loss is achieved through many mechanisms: (1) radiation, (2) conduction, (3) convection, (4) vasodilation, (5) decreased muscle tone, (6) evaporation, (7) increased respiration, (8) voluntary measures, and (9) adaptation to warmer climates.
Radiation: Radiation refers to heat loss through electromagnetic waves. These waves emanate from surfaces with temperatures higher than the surrounding air. Thus if the temperature of the skin is higher than that of the air, the skin and therefore the body lose heat to the air.
Conduction: Conduction refers to heat loss by direct molecule-to-molecule transfer from one surface to another. Through conduction, the warmer surface loses heat to the cooler surface. Thus the skin loses heat through direct contact with cooler air, water, or another surface. In the same manner, the core of the body loses heat to the cooler body surface.
Convection: Convection is the transfer of heat through currents of gases or liquids. It greatly aids heat loss through conduction by exchanging warmer air at the surface of the body with cooler air in the surrounding space. Convection occurs passively as warmer air at the surface of the body rises away from the body and is replaced by cooler air, but the process may be aided by fans or wind. (The combined effect of conduction and convection by wind is conventionally measured as the windchill factor.)
Vasodilation: Peripheral vasodilation increases heat loss by diverting core-warmed blood to the surface of the body. As the core-warmed blood passes through the periphery, heat is transferred by conduction to the skin surface and from the skin to the surrounding environment. Because heat loss through conduction depends on the surrounding temperature, it is minimal to nonexistent if the surrounding air or water is warmer than the body surface.
Vasodilation occurs in response to autonomic stimulation under the control of the hypothalamus. It is useful in instances of moderate temperature elevation. As core temperature increases, vasodilation increases until maximal dilation is achieved. At that point the body must use additional heat loss mechanisms.
Decreased Muscle Tone: To decrease heat production, muscle tone may be moderately reduced and voluntary muscle activity curtailed. These mechanisms explain in part the “washed-out” feeling associated with high temperatures and warm weather. Decreased muscle tone and reduced activity have a limited effect on decreasing heat production; however, because muscle tone and heat production cannot be reduced below basal body requirements.
Evaporation: Evaporation of body water from the surface of the skin and the linings of the mucous membranes is a major source of heat reduction. Insensible water loss (in the absence of perceptible sweating) accounts for a loss of about 600 ml of water per day. Heat is lost as surface fluid is converted to gas, so that heat loss by evaporation is increased if more fluids are available at the body surface. To speed this process, fluids are actively secreted through the sweat glands. As much as 2.2 L of fluid per hour may be lost by sweating. Electrolytes are lost with the water. Therefore, loss of large volumes through sweating may result in decreased plasma volume, decreased blood pressure, weakness, and fainting. (Alterations in fluid balance are discussed in Chapter 3.)
Like other heat reduction mechanisms, stimulation of sweating occurs in response to sympathetic neural activity and depends on a favorable temperature difference between the body and the environment. In addition, heat loss through evaporation is affected by the relative humidity of the air. If the humidity is low, sweat evaporates quickly, but if the humidity is high, sweat does not evaporate and instead remains on the skin or drips off.
Increased Pulmonary Ventilation: Exchanging air with the environment through the normal pulmonary ventilation provides some heat loss, although it is minimal in humans. As air is inhaled, the air draws heat from the upper respiratory tract. The air is further warmed in the alveoli by blood in the microcirculation. This warmed air then is exhaled into the environment. This normal process occurs faster at higher body temperatures through an increase in ventilatory rates. Thus hyperventilation is associated with hyperthermia. (Normal pulmonary function is discussed in Chapter 32.)
Voluntary Mechanisms: In response to high body temperatures, people physically “stretch out,” thereby increasing the body surface area available for heat loss. They also “slow down” or “take it easy,” thereby decreasing skeletal muscle work, and they “dress for warm weather” with light-colored, loose-fitting garments to reflect heat and promote convection, conduction, and evaporation.
Adaptation to Warmer Climates: The body of an individual who moves from a cooler to a much warmer climate undergoes a period of adjustment, a process that takes several days to weeks. At first the individual experiences feelings of lassitude, weakness, and faintness with even moderate activity. Body temperatures rise with any work. Within several days, however, the individual experiences an earlier onset of sweating, the volume of sweat is increased, and the sodium content is lowered. Heart rate is decreased and stroke volume increased so that cardiac output remains unchanged. Extracellular fluid volume increases, as does plasma volume. These physiologic adaptations result in improved warm weather functioning and decreased symptoms of heat intolerance. People’s work output, endurance, and coordination increase, and their subjective feelings of discomfort decrease.93
Mechanisms of Heat Conservation
The body conserves heat and protects core temperature through two important mechanisms: (1) involuntary vasoconstriction mediated by the sympathetic nervous system and (2) voluntary mechanisms.94 To preserve core temperature, the skin and periphery are used as an insulating cover.93
Vasoconstriction: By constricting peripheral blood vessels, centrally warmed blood is shunted away from the periphery (where radiation, conduction, and convection would allow heat loss) to the core of the body, where heat can be retained. This mechanism takes advantage of the insulating layers of the skin and subcutaneous fat to protect core temperature.
Voluntary Mechanisms: In response to lower body temperatures, individuals typically “bundle up,” “keep moving,” or “curl up in a ball.” Bundling up involves dressing with several layers of clothes that allow air to be trapped between the skin and the clothing, thus providing an additional layer of insulation. Keeping moving, stamping feet, clapping hands, jogging, and other types of physical activity increase skeletal muscle activity and thus promote heat production. Curling up in a ball decreases the amount of skin surface available for heat loss through radiation, convection, and conduction.
Pediatrics and Changes in Temperature Regulation
Infants and older adults require special attention to maintenance of body temperature. Infants produce sufficient body heat but are unable to efficiently conserve it. This poor heat conservation is caused by the infant’s small body size and greater ratio of body surface to body weight, which gives the infant more surface area for heat loss. Infants also have a very thin layer of subcutaneous fat and thus are not as well insulated as adults.95
Aging and Changes in Temperature Regulation
Older adults have poor responses to environmental temperature extremes as a result of slowed blood circulation, structural and functional changes in the skin, and an overall decrease in heat-producing activities. Other factors affecting thermal regulation in the older adult population include decreased shivering response (delayed onset and decreased effectiveness), slowed metabolic rate, sedentary lifestyle, decreased vasoconstrictor and vasodilator responses, diminished or absent sweating, desynchronization of circadian rhythm, undernutrition, and decreased perception of heat and cold.96,97
Pathogenesis of Fever
Fever is part of the acute phase response to infection or inflammation (see Chapter 9). Fever is a complex, integrated cascade of behavioral, neurologic, and endocrine responses to an immune challenge initiated by endogenous pyrogens or disorders of the hypothalamus. It is a normal adaptive response to cytokines and prostaglandin E2 (PGE2).98 The thermoregulatory mechanisms of the hypothalamus and brainstem adjust heat production, conservation, and loss to maintain body core temperature at a normal level. During fever this level is raised so that the thermoregulatory centers adjust heat production, conservation, and loss to maintain the core temperature at the new, higher temperature, which functions as a new balance point or set point.86,98
The pathophysiology of fever begins with the introduction of exogenous pyrogens (i.e., endotoxins from gram-negative bacteria) (Figure 15-8). The most frequently encountered exogenous pyrogens are the lipopolysaccharide complex in the cell wall of gram-positive bacteria and viruses.99 Endogenous pyrogens, including PGE2, interleukin-1 (IL-1), IL-6, TNF-α), and interferon-γ, are produced by phagocytic cells as they destroy microorganisms within the host.100 The endogenous pyrogens act on the preoptic nucleus of the hypothalamus to produce the late phases of fever. An integrated behavioral, endocrine, and autonomic nervous system response is then initiated. Centers in the hypothalamus and brainstem signal an increase in heat production and heat conservation to raise body temperature to the new set point. Peripheral vasoconstriction occurs with shunting of blood from the skin to the body core. Epinephrine release increases metabolic rate, and muscle tone increases. Decreased release of vasopressin reduces the volume of body fluid to be heated. Shivering also may occur. The individual dresses more warmly, decreases body surface area by curling up, and may go to bed in an effort to get warm. Body temperature is maintained at the new level until the fever “breaks.”
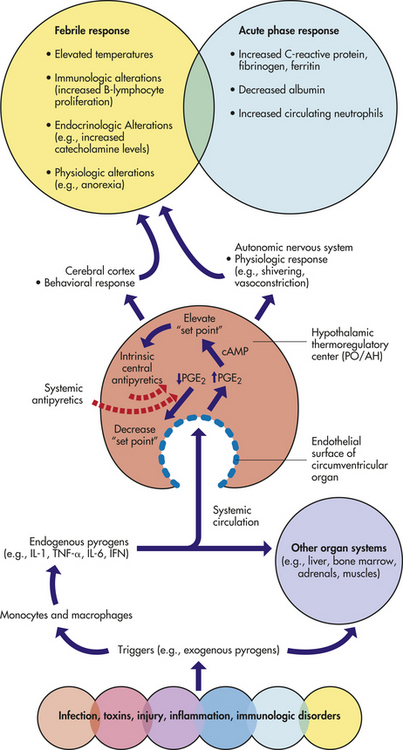
Figure 15-8 Pathogenesis of fever and acute-phase response. Certain disease states, through the elaboration of exogenous pyrogens, stimulate monocytes and macrophages to produce endogenous pyrogens such as IL-1, IL-6, TNF-α, and interferon-γ. These pyrogenic cytokines act at the endothelial surface of the circumventricular organ of the preoptic area of the anterior hypothalamus (PO/AH) to induce the production of PGE2, which elevates the body’s thermal set point. Physiologic and behavioral responses may be invoked to raise body temperature to a new set point. This febrile response must be considered in the context of an overlapping acute-phase response as a global nonspecific response to the original insult. Intrinsic central antipyretics and systemic antipyretics exert their effects by decreasing levels of PGE2, decreasing the “set point” and lowering body temperature. The red dotted line represents a negative feedback response. cAMP, Cyclic adenosine monophosphate; IFN, interferon; IL-1, IL-6, interleukin-1, interleukin-6; PGE2, prostaglandin E2, TNF-α, tumor necrosis factor-alpha. (Modified from Armstrong D, Cohen J: Infectious diseases, 2nd ed, St Louis, 2004, Mosby.)
During fever, arginine vasopressin (AVP), α-melanocyte-stimulating hormone (α-MSH), and corticotropin-releasing factor are released from the brain, and systemic anti-inflammatory cytokines (i.e., IL-1 receptor agonist and IL-10) can act as endogenous cryogens or antipyretics to help diminish the febrile response.101 This antipyretic effect constitutes a negative-feedback loop (see Figure 15-8). The antipyretic effect may help explain fluctuations in the febrile response. When the fever breaks, the set point is returned to normal. The hypothalamus responds by signaling a decrease in heat production and an increase in heat-reduction mechanisms. The result is decreased muscle tone, peripheral vasodilation, flushed skin, and sweating. The individual feels very warm, replaces warm clothing with cooler clothes, throws off the covers, and stretches out. Once the body has returned to a normal temperature, the individual feels more comfortable and the hypothalamus adjusts thermoregulatory mechanisms to maintain the new temperature.
Benefits of Fever
Fever production aids responses to infectious processes through several mechanisms.102 A raised body temperature kills many microorganisms and has adverse effects on the growth and replication of others. Higher body temperatures decrease serum levels of iron, zinc, and copper, all of which are needed for bacterial replication. The body switches from burning glucose to a metabolism based on lipolysis and proteolysis, thereby depriving bacteria of a food source. Anorexia and somnolence reduce the demand for muscle glucose.103 Increased temperature also causes lysosomal breakdown and autodestruction of cells, thus preventing viral replication in infected cells. Acute-phase proteins produced by the liver during inflammation bind cations necessary for bacterial reproduction. Heat increases lymphocytic transformation and motility of polymorphonuclear neutrophils, thus facilitating the immune response. Phagocytosis is enhanced, and production of antiviral interferon may be augmented.104,105
Because fever is a beneficial response to infection, suppressing fever with antipyrogenic medications should be reviewed carefully.106 Such treatment should be used only if the fever is high enough to produce serious side effects such as nerve damage or convulsion.
Infection and fever responses in older adults and in children may vary from those in the normal adult. Older individuals may have decreased or no fever response to infection. The absence of fever responses to infection and therefore the beneficial aspects of fever production may explain the increase in morbidity and mortality rates seen in very old individuals.107 In contrast, children develop higher temperatures than do adults for relatively minor infections. Febrile seizures may occur with temperatures above 39° C (102.2° F), although most children do not develop febrile seizures until temperatures are much higher. Febrile seizures are more predominant in boys before age 5 years, and genetic factors contribute to susceptibility.108 Febrile seizures are generally brief and self-limiting, lasting less than 5 minutes in 40% of children and less than 20 minutes in 75% of children. Although in most instances there appear to be no long-term effects on the child, a small percentage of children (1% to 2%) may develop epilepsy.109 Prolonged febrile seizures are associated with the development of temporal lobe epilepsy in children and are probably associated with functional changes in neurons and neural networks.110
Disorders of Temperature Regulation
Hyperthermia (marked warming of core temperature) can produce nerve damage, coagulation of cell proteins, and death. At 41° C (105.8° F), nerve damage produces convulsions in the adult. At 43° C (109.4° F), death results. Hyperthermia is not mediated by pyrogens, and there is no resetting of the hypothalamic set point. Hyperthermia may be accidental or therapeutic. Therapeutic hyperthermia is a form of local or general body-induced hyperthermia. Its purpose is to destroy pathologic microorganisms or tumor cells by facilitating the host’s natural immune process through elevated body temperature.111 As a form of treatment, it is generally controversial. The four forms of accidental hyperthermia are (1) heat cramps, (2) heat exhaustion, (3) heat stroke, and (4) malignant hyperthermia.112
Heat Cramps: Heat cramps are severe spasmodic cramps in the abdomen and extremities that follow prolonged sweating and associated sodium loss. Heat cramps usually appear in individuals who are not accustomed to heat or in those who are performing strenuous work in very warm climates. Fever, rapid pulse, and increased blood pressure often accompany the cramps. Treatment involves administration of dilute salt solutions through oral or parenteral routes.
Heat Exhaustion: Heat exhaustion, or collapse, is a result of prolonged high core or environmental temperatures. These high temperatures cause the appropriate hypothalamic response of profound vasodilation and profuse sweating. Over a prolonged period the hypothalamic responses produce dehydration, decreased plasma volumes, hypotension, decreased cardiac output, and tachycardia. The individual feels weak, dizzy, nauseated, and faint. The symptoms of heat exhaustion cause the individual to stop work, lie down, and rest. Ceasing activity decreases muscle work, causing decreased heat production. Lying down redistributes vascular volume. The individual should be encouraged to drink warm fluids to replace fluid lost through sweating.
Heat Stroke: Heat stroke is a potentially lethal result of a breakdown in control of an overstressed thermoregulatory center. The brain cannot tolerate temperatures over 40.5° C (104.9° F). When core temperature reaches or exceeds 40.5° C (104.9° F), the brain may be preferentially cooled by maximal blood flow through the veins of the head and face, specifically the forehead. Sweat production on the face is maintained even during dehydration. Evaporation of the sweat cools the blood in the veins of the face and forehead; the blood then is returned to the endocranial venous network and sinus cavernosus, cooling the blood in the cerebral arterial vessels that lie in proximity. Fanning the face enhances this mechanism. In this way the brain can be maintained temporarily at 40° C (104° F), even when core temperatures are higher.93,113 In instances of very high core temperatures (40° to 43° C [104° to 109.4° F]), the cardiovascular and thermoregulatory centers may cease to function appropriately. Sweating ceases, and the skin becomes dry and flushed. The individual may be irritable, confused, stuporous, or comatose. Visual disturbances may occur.
As heat loss through the evaporation of sweat ceases, core temperatures increase rapidly. High core temperatures and vascular collapse produce cerebral edema, degeneration of the CNS, swollen dendrites, and renal tubular necrosis. Death results unless immediate, effective treatment is initiated.114
Treatment includes removing the person from the warm environment, if possible, and using a cooling blanket or cool water bath. Care must be taken to prevent too rapid cooling of the surface, which causes peripheral vasoconstriction and prevents core cooling. Individuals who recover from heat stroke may have permanent damage to the thermoregulatory center and thus may have difficulty tolerating environmental temperature changes.115
Children are more susceptible to heat stroke than adults because (1) they produce more metabolic heat when exercising, (2) they have a greater surface area:mass ratio, and (3) their sweating capacity is less than that of adults.116
Malignant Hyperthermia: Malignant hyperthermia is a potentially lethal complication of a rare inherited muscle disorder. The condition is precipitated by the administration of volatile anesthetics and neuromuscular-blocking agents. About 1 in 200 individuals may be at risk for the muscle disorder. Malignant hyperthermia is caused by either increased calcium release or decreased calcium uptake with muscle contraction. This allows intracellular calcium levels to rise, producing sustained, uncoordinated muscle contractions, which in turn increase muscle work, oxygen consumption, and lactic acid production. As a result of these contractions, acidosis develops and temperature rises (body temperature may rise 1° C [1.8° F] every 5 minutes); approximately 5% of those who develop malignant hyperthermia do not survive.117 Malignant hyperthermia occurs most often in children and young adults immediately after the induction of anesthesia. Sympathetic responses and acidosis produce tachycardia and cardiac dysrhythmias, followed by hypotension, decreased cardiac output, and eventually, cardiac arrest. Increasing temperature, acidosis, hyperkalemia, and hypoxia produce coma-like symptoms in the CNS (including unconsciousness, absent reflexes, fixed pupils, apnea, and sometimes a flat electroencephalogram [EEG]). Oliguria and anuria are common, probably resulting from shock, ischemia, and low cardiac output.118
Treatment includes withdrawal of the provoking agents and administration of dantrolene sodium (a skeletal relaxant that inhibits calcium release during muscle contraction). Procainamide (Pronestyl) is used to treat cardiac dysrhythmias. Sodium bicarbonate also may be used. Body temperature can be decreased through use of ice bags, a cooling blanket, and iced saline lavage.119
Hypothermia
Hypothermia (core body temperature less than 35° C [95° F]) is caused by prolonged exposure to cold—from 1999 to 2002 there were 4607 deaths reported in the United States.120 Hypothermia produces vasoconstriction, shivering, alterations in microcirculation, coagulation, and ischemic tissue damage. In a controlled situation, such as a surgical procedure, most tissues can tolerate temperatures as low as 33° C (91.4° F). In severe hypothermia (less than 28° C [82.4° F]), ice crystals forming on the inside of the cell cause cells to rupture and die. Tissue hypothermia slows the rate of chemical reactions (tissue metabolism), increases the viscosity of the blood, slows blood flow through the microcirculation, facilitates blood coagulation, and stimulates profound vasoconstriction. Hypothermia may be accidental or therapeutic. In accidental hypothermia, high energy phosphates (e.g., ATP) are depleted and in therapeutic hypothermia, ATP storage is preserved.121
Accidental Hypothermia: Accidental hypothermia (temperature below 35° C [95° F]) is generally the result of sudden immersion in cold water or prolonged exposure to cold environments.122 At particular risk for accidental hypothermia are young persons and older adults, because thermoregulatory mechanisms are altered in these two groups.123 Also at risk are individuals with conditions that diminish the ability to generate heat. Such conditions include hypothyroidism, hypopituitarism, decreased liver function, malnutrition, Parkinson disease, and rheumatoid arthritis. Other risk factors include chronic increased vasodilation and decreased thermoregulatory control caused by cerebral injuries, ketoacidosis, uremia, and drug overdoses.124,125 In acute hypothermia, peripheral vasoconstriction shunts blood away from the cooler skin to the core in an effort to decrease heat loss, which produces peripheral tissue ischemia. Intermittent reperfusion of the extremities (the Lewis phenomenon) helps preserve peripheral oxygenation. Intermittent peripheral perfusion continues until core temperatures drop dramatically.
The hypothalamic center stimulates shivering in an effort to increase heat production. Severe shivering occurs at core temperatures of 35° C (95° F) and continues until core temperature drops to about 30° to 32° C (86° to 89.6° F). Prolonged shivering can lead to exhaustion of liver glycogen stores. Thinking becomes sluggish and coordination is decreased at 34° C (93.2° F). As hypothermia deepens, paradoxical undressing may occur as hypothalamic control of vasoconstriction is lost and vasodilation occurs with loss of core heat to the periphery. The hypothermic individual therefore feels suddenly warm and begins to remove clothing.126
At 30° C (86° F), the individual becomes stuporous; heart rate and respiratory rate decline; and cardiac output is diminished. Cerebral blood flow is decreased. Metabolic rate declines, further decreasing core temperature. Sinus node depression occurs with slowing of conduction through the atrioventricular node. In severe hypothermia (core temperature of 26° to 28° C [78.8° to 82.4° F]), pulse and respirations may be undetectable. Acidosis is moderate to severe. Ventricular fibrillation and asystole are common.124 Surface cooling may cause frostbite and fat necrosis.
If hypothermia is mild, passive rewarming may be sufficient. Passive rewarming includes provision of warm, dry clothes and warm drinks and performance of isometric exercises to increase heat production and minimize heat loss. Core temperature should be checked as soon as possible.127,128
If core temperature is greater than 30° C (86° F), active rewarming also may be required. Active rewarming uses warm-water baths, warm blankets, heating pads, and warm oral fluids when the individual is fully alert. Active core rewarming is performed when core temperatures have dropped below 30° C (86° F) or when severe cardiovascular abnormalities appear. Core rewarming may be accomplished through administration of warm intravenous (IV) solutions, warm gastric lavage, warm peritoneal lavage, inhalation of warmed gases, and, in extreme cases, exchange transfusions, warming blood in a pump oxygenator circuit, and mediastinal lavage.129,130
Rewarming generally should proceed no faster than a few degrees per hour. (Short-term complications of rewarming are listed in Table 15-4.) Long-term complications include congestive heart failure, hepatic and renal failure, abnormal erythropoiesis, myocardial infarction, pancreatitis, and neurologic dysfunctions.
Table 15-4
Accidental Hypothermia: Complications of Rewarming
| Complication | Mechanism |
| Acidosis | Rewarming stimulates peripheral vasodilation; peripheral blood, returning to the core from the ischemic peripheral tissues, causes a reduction in the pH of core blood |
| Rewarming shock | As rewarming and vasodilation progress, the body is unable to maintain blood pressure because of reduced fluid volume (from “cold diuresis”), catecholamine depletion (prolonged shivering), and myocardial injury |
| Deep-ended hypothermia | As colder surface blood is returned to the core, core temperature may drop; this is also referred to as “after fall” or “after drop” |
| Dysrhythmia | Rewarming places an additional stress on an already severely stressed myocardium |
Therapeutic Hypothermia: Therapeutic hypothermia is used to slow metabolism and preserve ischemic tissue after brain trauma or during brain surgery, after cardiac arrest, and in neonatal hypoxic encephalopathy. Hypothermia protects the brain by reduction in metabolic rate, ATP consumption and oxidative stress, reduction of the critical threshold for oxygen delivery, modulation of excitotoxic neurotransmitters, calcium antagonism, preservation of protein synthesis, preservation of the blood-brain barrier and decreased edema formation, and modulation of the inflammatory response.131 Survival from accidental hypothermia has been reported in individuals with core temperatures at 16° C (60.8° F) and from therapeutic hypothermia with temperatures at 9° C (48.2° F).132–135
Trauma
Major body trauma has varying effects on temperature regulation, depending on the body systems involved. Five types of traumatic injury that usually affect temperature regulation are (1) CNS trauma (discussed in Chapter 17), (2) accidental injury, (3) hemorrhagic shock, (4) major surgery, and (5) thermal burns.
Central Nervous System Trauma: CNS trauma that causes CNS damage, inflammation, increased intracranial pressures, or intracranial bleeding typically produces a fever greater than 39° C (102.2° F). This temperature, often referred to as neurogenic fever, appears with or without relative bradycardia and is not caused by infection. The temperature is sustained, does not induce sweating, and is highly resistant to antipyretic therapy.136
Accidental Injuries: Mild accidental injuries may produce a slight elevation in core temperature. Moderate to severe injuries result in peripheral vasoconstriction with decreased surface and core temperatures. Core temperature is thought to be inversely related to the severity of the injury and may be a result of decreased oxygen transport to the tissues. In severe injuries, shivering is absent and some alteration in thermoregulation is evident.137
Hemorrhagic Shock: Loss of blood volume in hemorrhage triggers peripheral vasoconstriction and hypoxia contributing to hypothermia.138 Risk for subsequent decreases in core temperature occurs when hemorrhagic shock is treated with unwarmed, volume-expanding solutions and surgery. Volume expansion with warmed solutions is recommended to prevent the deleterious effects of hypothermia on cardiac output, cardiac rhythm, and the immune system.
Major Surgery: Major surgery often induces significant hypothermia through exposure of body cavities to the relatively cool operating room environment, irrigation of body cavities with room temperature solutions, infusion of room temperature intravenous solutions, use of drugs that impair thermoregulatory mechanisms, and inhalation of unwarmed anesthetic agents. Anesthesia induces hypothermia, reduces platelet function, and impairs the coagulation cascade contributing to transfusion requirements and postoperative complications.139 Use of warmed irrigating and intravenous solutions and perioperative forced air and other warming procedures reduces intraoperative hypothermia and postoperative complications.140,141
Thermal Burns: Large burn injuries produce significant hypothermia because of the loss of the skin barrier to fluid evaporation and the loss of control of the microcirculation in the skin. Severe burns also compromise the normal insulation of the skin and subcutaneous tissues. (Burns are discussed in Chapter 46.)
SLEEP
Sleep is an active, multiphase, complex brain process that provides restorative functions and promotes memory consolidation. Several areas of the brain are associated with sleep and sleep-wake cycles. A small group of hypothalamic nerve cells, the suprachiasmatic nucleus (SCN), controls the timing of the sleep-wake cycle and coordinates this cycle with circadian rhythms (24-hour rhythm cycles) in other areas of the brain and other tissues.142
Normal sleep has two phases that can be documented by EEG: rapid eye movement (REM) sleep and non-REM (NREM), or slow-wave, sleep. REM and NREM sleep succeed each other in 90- to 110-minute intervals in a predictable pattern.143 Four to six cycles occur during a normal sleep period. NREM sleep is divided into four stages based on changes in the EEG pattern (Figure 15-9):
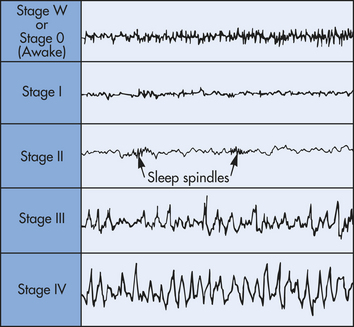
Figure 15-9 Electroencephalogram (EEG) stages of wakefulness and NREM sleep. Stage W or stage 0 awake, low-voltage fast activity; stage I, falling asleep; stage II, light sleep with sleep spindles; stage III, moderately deep sleep; stage IV, deep sleep with slow delta waves. Rapid eye movement (REM) sleep looks similar to awake and stage I.
Stage W or stage 0—Wakefulness with eyes closed and predominated by alpha waves (8 to 25 Hz).
Stage I—Light sleep, with alpha waves (6 to 8 Hz) interspersed with low-frequency theta waves; slow eye movements (3% to 8% of sleep time)
Stage II—Further slowing of the EEG (4 to 7 Hz) with the presence of sleep spindles and slow eye movements (45% to 55% of sleep time)
Stage III—Low-frequency (1 to 3 Hz) high-amplitude delta waves with occasional sleep spindles—also known as slow-wave sleep; no slow eye movements (15% to 20% of sleep time)
Stage IV—Delta waves, slow-wave sleep (15% to 20% of sleep time)
Non–Rapid Eye Movement (NREM) Sleep
NREM (slow-wave) sleep accounts for 75% to 80% of sleep time and is initiated by the withdrawal of neurotransmitters from the reticular formation and by the inhibition of arousal mechanisms in the cerebral cortex. During NREM sleep, respiration is controlled by metabolic processes.144 The basal metabolic rate is decreased by 10% to 15%. Temperature is decreased 0.5° to 1° C (0.9° to 1.8° F). Heart rate decreases by 10 to 30 beats per minute. Respiration, blood pressure, and muscle tone all decrease. Knee-jerk reflexes are absent. Pupils are constricted. During stages I and II, cerebral blood flow to the brainstem and cerebellum is decreased. During stages III and IV, cerebral blood flow to the cortex is decreased.145,146 Growth hormone is released during stage IV, and levels of corticosteroids and catecholamines are depressed.
Rapid Eye Movement Sleep
Rapid eye movement (REM) sleep accounts for 20% to 25% of sleep time and is characterized by desynchronized, low-voltage, fast activity that occurs for 5 to 60 minutes about every 90 minutes beginning after 1 to 2 hours of NREM sleep. The brain is quite active in REM sleep with vivid dreaming. REM sleep is also known as paradoxic sleep because the EEG pattern is similar to the normal awake pattern. Alternating periods of REM and NREM sleep occur throughout the night, with lengthening intervals of REM sleep and fewer intervals of deeper stages of NREM sleep toward morning. REM sleep is characterized by bursts of conjugate rapid eye movement; atonia of antigravity muscles; suppressed temperature regulation; alteration in heart rate, blood pressure,147 and respiration; penile erection in men and clitoral engorgement in women; and a high rate of memorable dreams. Steroids are released in short bursts. During REM sleep, respiratory control is thought to be largely independent of metabolic requirements and oxygen variation. Loss of normal voluntary muscle control in the tongue and upper pharynx may produce some respiratory obstruction. Cerebral blood flow to both hemispheres is increased. REM sleep is controlled by the pontine reticular formation.
An individual progresses through REM and NREM sleep in a predictable cycle while asleep. The first cycle of the night begins with stage I. The individual then progresses through stages II, III, IV, and REM sleep (Stage V). A new cycle, beginning with stage II, follows each REM sleep. With each successive cycle, the amount of time spent in stage IV sleep decreases and the amount of time spent in REM sleep (Stage V) increases (Figure 15-10). The individual who is awakened begins the next cycle with stage I.
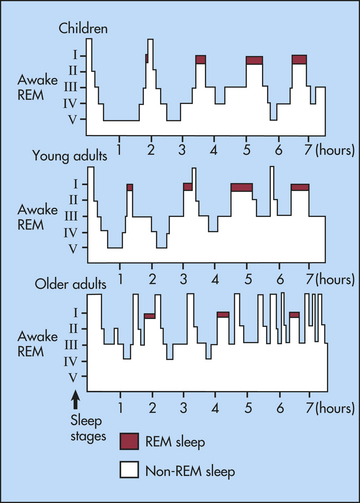
Figure 15-10 Normal sleep cycles. Rapid eye movement (REM) sleep (Stage V) occurs cyclically throughout the night at intervals of approximately 90 minutes in all age groups. REM sleep shows little variation in the different age groups, whereas stage IV sleep decreases with age. In addition, older adults awaken frequently and show a marked increase in total time awake.
The hypothalamus as a major sleep center secretes hypocretins (orexins), neuropeptides that promote wakefulness and REM sleep as well as appetite, energy consumption, and pleasure or reward. In addition to the hypocretins, acetylcholine and glutamate are important waking factors. The preoptic area of the hypothalamus promotes sleep through inhibitory modulation of multiple arousal systems with involvement of the inhibitory neurotransmitter GABA.148 The reticular formation is primarily responsible for generating REM sleep and projections from the reticular formation and other areas of the mesencephalon and brainstem produce NREM sleep.149
Many neurotransmitters are associated with excitatory and inhibitory sleep mechanisms. Sleep-promoting neurotransmitters include prostaglandin D2, L-tryptophan, serotonin, adenosine, melatonin, GABA, and growth hormones.150–152 Awake-promoting neurotransmitters include hypocretin (orexin), acetylcholine, and glutamate.153 Their mechanism of action is complex and not clearly understood. Growth hormone is associated with initiation of sleep, and cortisol rises in the morning just before waking.154 Acetylcholine and somatostatin play a role in stages of sleep transition. Forced awakenings in the middle of the night may result in increased difficulty returning to sleep or may alter the normal progression of sleep, or both.155,156 The purpose of sleep is unknown, although restorative processes, particularly neuronal repair and generation of new neural pathways, have been proposed. Loss of REM sleep impairs learning and memory. Sleep is important enough that people spend about one third of their life sleeping.
Pediatrics and Sleep Patterns
The sleep patterns of the newborn and young child vary from those of the adult in total sleep time, cycle length, and percentage of time spent in each sleep cycle.157 Newborns sleep about 16 to 17 hours per day. About 53% of that time is spent in active sleep (REM sleep), 23% in quiet sleep (NREM sleep), and the remainder in an indeterminate phase. The infant sleep cycle is approximately 50 to 60 minutes long, with 20 minutes of NREM sleep and 10 to 45 minutes of REM sleep, in contrast to the adult sleep cycle. Newborns enter REM sleep immediately on falling asleep.158,159 At about 1 year of age, an infant spends approximately 45% of total sleep time in quiet sleep and 41% in REM sleep. Total sleep time decreases slightly from birth to 1 year. Bottle-fed infants that do not share sleeping space with the mother increase maximum sleep time from 4 to 5 hours to 8 to 10 hours by 4 months of age. They begin to “sleep through the night.” Infants that are breast-fed for up to 2 years and share sleeping space with the mother continue to sleep in short bouts and wake frequently to nurse.160 In the young child, the sleep cycle length is 45 to 60 minutes, in contrast to 90 to 100 minutes in the adult. The child assumes the adult sleep pattern at some point during the first 2 to 5 years of life.161 Sleep for infants and children is important for growth and neurocognitive development. Sleep disorders are common in children and include insomnia and obstructive sleep apnea syndrome (OSAS). OSAS is related to adenotonsillar inflammation, enlargement of the adenoids and tonsils, or to obesity.162 Sudden infant death syndrome (SIDS) is presented in Chapter 34.
Aging and Sleep Patterns
The sleep pattern of the older adult differs from that of the younger adult or child. Total sleep time is decreased, and the older individual takes longer to fall asleep. Older adults tend to go to sleep earlier in the evening and awaken more frequently during the night and earlier in the morning. REM and slow-wave sleep decreases. On EEG, the spindle indicating stage II sleep is less well formed.163,164 Changes in the older adult’s sleep pattern may be associated with changes in lifestyle, chronic disease, lack of daily routine, desynchronization of circadian rhythm, and use of sedatives. Growth hormone and cortisol are diminished in the older adult and may affect sleep patterns.165 The alteration in sleep pattern typically appears about 10 years later in women than in men. Older adults are less able than younger individuals to tolerate sleep deprivation.163
Sleep Disorders
The classification of sleep disorders is complex and a system has been produced by the American Academy of Sleep Medicine166 that includes four general classifications: (1) dyssomnias (intrinsic and extrinsic sleep disorders and circadian rhythm sleep disorders); (2) parasomnias (arousal and sleep-wake transition disorders and REM sleep disorders); (3) sleep disorders associated with mental, neurologic, or other medical disorder; and (4) proposed sleep disorders. The most common dyssomnias and parasomnias are presented here.
Common Dyssomnias
The dyssomnias are sleep disorders related to difficulty in initiating or maintaining sleep or excessive sleepiness. The most common dyssomnias include insomnia, obstructive sleep apnea syndrome, restless leg syndrome, circadian rhythm disorder, and hypersomnia.
Insomnia is the inability to fall or stay asleep and is more common in women.167 Primary insomnia may be transient, lasting a few days, and related to travel across time zones, disrupted sleep schedules, or acute stress. Secondary insomnia is associated with drug or alcohol abuse, chronic pain disorders, or chronic depression, obesity, and aging.168 Drugs known to produce insomnia include amphetamines, steroids, central adrenergic blockers, bronchodilating agents, and caffeine.169
Obstructive sleep apnea syndrome (OSAS) is a disorder of breathing during sleep related to upper airway obstruction that is associated with reduced blood oxygen saturation and hypercapnia. Risk factors include obesity, male gender, and age.170 Premenstrual women may be protected from sleep-disordered breathing because the female hormone progesterone is a respiratory stimulant.171,172 The longer pharyngeal airway in males also may contribute to increased risk for OSAS. Obese individuals often have a short, thick neck; impaired respiratory mechanics; and depressed respiratory control, particularly during sleep. Obesity hypoventilation syndrome may be related to leptin resistance because leptin is also a respiratory stimulant.
OSAS is characterized by repetitive increases in resistance to airflow within the upper airway with loud snoring, gasping, intervals of apnea lasting from 10 to 30 seconds, fragmented sleep, and chronic daytime sleepiness. The obstruction is caused by the soft palate or base of the tongue, or both, collapsing against the pharyngeal walls because of decreased muscle tone especially during REM sleep. In the supine position, the tongue may fall farther back, obstructing the airway. The level of negative intrathoracic pressure is the most likely stimulus for arousal, possibly mediated by mechanoreceptors in the upper airway. Potentially fatal systemic illnesses often associated with OSAS include hypertension, pulmonary hypertension, heart failure, nocturnal cardiac dysrhythmias, myocardial infarction, and ischemic stroke.173 The combination of intermittent hypoxia and hypercapnia, arousals, increased sympathetic tone, altered baroreflex control during sleep, and cardiovascular changes induced by increased negative intrathoracic pressure contribute to these complications. Many individuals are not aware of their heavy snoring and nocturnal arousals and may remain undiagnosed. Fatigue, decline in cognitive function, car accidents, and poor work performance are common.
Diagnosis of OSAS is confirmed with polysomnography. Treatments include continuous positive airway pressure, dental devices that modify the position of the tongue or jaw, upper airway surgical reconstruction, and weight reduction.174
Restless leg syndrome (RLS) is a common sensorimotor disorder associated with unpleasant sensations (prickling, tingling, crawling) that occurs at rest and is worse in the evening or at night. There is a compelling urge to move the legs for relief, with a significant effect on sleep and quality of life. The disorder is more common in women, older adults, and individuals with iron deficiency. RLS has a familial tendency and is associated with a circadian fluctuation of dopamine in the substantia nigra. Iron is a cofactor in dopamine production, and some individuals respond to iron administration as well as dopamine agonists.175–177
Hypersomnia is excessive daytime sleepiness and is commonly associated with voluntary sleep deprivation. There may also be an underlying sleep disorder such as OSAS or narcolepsy. Individuals may fall asleep while driving, working, or even conversing with significant concerns for safety. Treatment is symptomatic with reinforcement of good sleeping habits.
Narcolepsy is characterized by hypersomnia, cataplexy (brief spells of muscle weakness), hallucinations, and sleep paralysis. The disorder is associated with hypothalamic hypocretin (orexin) deficiency and may be related to immune-mediated destruction of hypocretin-secreting cells. There is a genetic component to the disorder in animals.178
Circadian rhythm sleep disorders are common disorders of the sleep-wake schedule and include rapid time-zone change (jet-lag syndrome), changing sleep schedule with an advance or a delay of 3 hours or more in sleep time, or a change in total sleep time from day to day. These changes in the timing of established sleep schedules have been shown to desynchronize circadian rhythm. Degree of vigilance, performance of psychomotor tasks, and subjective reports of levels of arousal are markedly depressed after alterations in the sleep-wake schedule. Individuals may experience short sleep episodes called microsleeps without being aware of decreased vigilance.179,180
It is well established that industrial shift workers exhibit a decrease in accuracy and increased accident proneness.181 For similar reasons, people suffering from jet lag require several days to adapt to a new time zone. Travel across time zones requires 2 days to adjust the sleep-wake schedule, 5 days to adjust the body temperature cycle, and 8 days to adjust cortisol secretion. Transmeridian travel requires up to 10 days to adjust the body clock when traveling from east to west. Czeisler’s experiments with timed bright-light stimulation have had some success in retiming or resetting the body clock before or after time-zone shifts.182
Common Parasomnias
Parasomnias are complex behaviors related to awakening from REM sleep or partial arousal from NREM sleep and disorders of sleep stage transitions. Three types of parasomnias include arousal disorders such as confusional arousals, sleepwalking (somnambulism), night terrors (dream anxiety attacks), rearranging furniture, eating food, violent behavior, bruxism (teeth grinding), and sleep enuresis; sleep-wake transition disorders such as rhythmic movements (head banging), sleep talking, and nocturnal leg cramps; and disorders associated with REM sleep such as sleep paralysis and nightmares, sleep apnea, and SIDS. SIDS affects children primarily in the first 2 years of life and may be related to central sleep apnea (a disorder of thalamic breathing control) episodes (see Chapter 34). Parasomnias are more common in children and may be familial.183
Sleep Disorders Associated with Mental, Neurologic, or Medical Disorders
Many mental and physical disorders are associated with alterations in sleep (secondary sleep disorders). Many disorders produce alterations in the quantity and quality of sleep or affect sleep stages including mental disorders (i.e., depression and anxiety), disorders of neurocognitive function (i.e., dementia or Parkinson disease), and physical disorders (i.e., alterations in thyroid hormone, pain, and many acute and chronic diseases). In other instances, sleep stages may contribute to disease (sleep-provoked disorders) such as chronic obstructive pulmonary disease (COPD), asthma, cardiac ischemia diabetes mellitus, and gastroesophageal reflux.
REM sleep behavior disorder is loss of normal skeletal muscle atonia during REM sleep and may cause injury from acting out of dreams. It is more common in older adult men and is associated with Parkinson disease and other neurodegenerative diseases.184
Individuals who are depressed have difficulty falling asleep and exhibit less slow-wave sleep, less time spent in REM sleep, early awakening, and less total sleep time. In addition, depressed individuals move through the sleep stages more quickly than do individuals who are not depressed. The same neurotransmitters that may be disturbed in depression also regulate sleep.185
Coronary artery disease is most affected during REM sleep. During REM, dreams may provoke nocturnal angina, increased heart rate, and electrocardiogram (ECG) changes. In adults, attacks of bronchial asthma may occur at any time during the night. The attacks cause the individual to spend more of the sleep period awake and thus cause a decrease in stage IV sleep. In children, bronchial asthma attacks are uncommon during the first one third of the night, when stage IV sleep predominates, and occur more frequently during the final two thirds of the night. Stage IV sleep is decreased overall in the child with bronchial asthma. In addition to these changes, asthmatic individuals may experience bronchial spasm during REM sleep.186
People with COPD experience significantly lowered oxygen tension and increases in carbon dioxide retention during sleep. The lowered oxygen tension is most significant in the tonic phase of REM sleep when voluntary neuromuscular control, including intercostal muscle function, is depressed. Pulmonary spasm and transient pulmonary hypertension result. These changes are particularly evident in the “blue bloater” individual and may contribute to early pulmonary hypertension and cor pulmonale in these people.187
Because blood glucose levels vary during sleep, individuals with uncontrolled diabetes may need to pay careful attention to blood sugar levels. Studies show that people with duodenal ulcers secrete 3 to 20 times more gastric acid during REM sleep than do people without duodenal ulcers. This increased gastric acid secretion often produces nocturnal epigastric pain.