DISORDERS OF THE CENTRAL AND PERIPHERAL NERVOUS SYSTEMS AND THE NEUROMUSCULAR JUNCTION



Alterations in central nervous system (CNS) function are caused by traumatic injury, vascular disorders, tumor growth, infectious and inflammatory processes, metabolic derangements (including those arising from nutritional deficiencies and drugs/chemicals), and degenerative processes. Alterations in peripheral nervous system function involve the nerve roots (radiculopathies), a nerve plexus, or the nerves themselves (neuropathies). Disorders of the neuromuscular junction also occur.
CENTRAL NERVOUS SYSTEM DISORDERS
Brain Trauma
Traumatic brain injury (TBI) is defined by the Brain Injury Association of America as a traumatic insult to the brain capable of producing physical, intellectual, emotional, social, and vocational changes. Of the 1.4 million traumatic brain injuries in the United States each year, 1.1 million are treated and released from emergency departments and 235,000 require hospitalization1: 80% have mild TBIs, 10% moderate TBIs, and 10% severe TBIs.2 The Glasgow Coma Scale (GCS) is used to describe injury severity by the international and United States National Traumatic Coma Data Banks. The hallmark of a severe TBI is loss of consciousness for 6 hours or more. TBI classifications using the GCS are (1) mild TBI with GCS of 13 to 15, associated with mild concussion; (2) moderate TBI with GCS of 9 to 12, associated with structural injury such as hemorrhage or contusion; and (3) severe TBI with GCS of 3 to 8, associated with cognitive and/or physical disability or death. Age and admission GCS are important diagnostic factors in TBI.2
At highest risk for TBI are young persons 15 to 35 years of age, infants 6 months to 2 years, young school-age children, and adults older than 70 years of age. Males are 1.5 times as likely to sustain a TBI.1 TBI is highest among blacks in lower-median income families. Persons living in high-crime areas are at greater risk and blacks have the highest mortality rates.1
TBI is broadly categorized into blunt (closed, nonmissile) trauma and open (penetrating, missile) trauma. Blunt trauma, the more common injury, involves the head striking a hard surface or a rapidly moving object striking the head. The dura mater remains intact, and brain tissues are not exposed to the environment. Blunt trauma may result in both focal brain injuries and diffuse axonal injuries (Table 17-1). When a break in (penetration of) the dura mater results in exposure of the cranial contents to the environment, open trauma has occurred, which results in focal brain injuries.
Table 17-1
Severity of Trauma Related to Injury, Onset, and Persistence
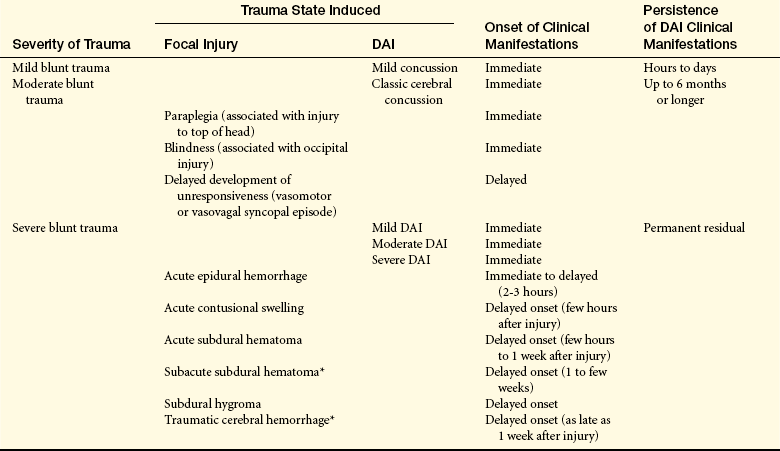
∗May be seen after moderate head injury, especially in older adults.
The most common types of brain injury are mild concussion and classic cerebral concussion (see page 590). Of all head injuries, 75% to 90% are not severe. Focal brain injury and diffuse axonal injury (DAI) each account for half of all injuries. Focal brain injury accounts for more than two thirds of head injury deaths; DAI, for less than one third. However, DAI accounts for the greatest number of severely disabled survivors, including persons who persist in an unresponsive state or reduced level of consciousness.
In recent years the surviving TBI population has changed, mostly because of focus on reducing severity of injury (e.g., passive seat restraints, air bags), reduced transport time, and improved on-the-scene medical management. Management of secondary and tertiary injury has improved; acute care professionals are focusing more on morbidity than mortality. As a result, individuals with more severe TBIs are admitted to rehabilitation programs.
Causes of Brain Trauma: Most TBIs are caused by falls (28%), motor vehicle crashes (20%), being struck by moving objects or moving against stationary objects (19%), and assaults (11%).1 Sports-related events also account for a portion of TBIs. Blasts are the leading cause of TBIs for active duty personnel.3
Compound fractures are caused by objects striking the head with great force or by the head striking an object forcefully. The comments regarding contusion (see page 585) hold true for compound fractures. Temporal blows, related to basilar skull fractures, may produce a fracture involving the middle fossa. An occipital blow may result in a basilar fracture down the occipital bone and across the petrous pyramid. The cervical vertebrae upwardly impacting the base of the skull can produce a posterior fossa basilar skull fracture.
Causes of penetrating injuries are missiles (most commonly bullets fired from rifles and handguns) and sharp projectiles (e.g., knives, ice picks, axes, screwdrivers). Most through-and-through (enter the head on one side and exit on the other) injuries are from high-velocity bullets.
Brain damage originates from primary and secondary brain injury. Primary injury is caused by the direct impact and involves the initial tear, neural injury and hemorrhage. In primary glial injury, oligodendroglia are affected by axon injury and by direct mechanical disruption caused by debris and leakage. Secondary injury includes intracranial and extracranial causes of brain damage. Intracranial brain damage is complex and occurs as a result of impairment of cerebral blood flow autoregulation, alterations in the blood-brain barrier, cerebral edema, increased intracranial pressure (ICP), brain herniation, a decrease in cerebral perfusion pressure and inflammation. Significant to secondary injury is tissue hypoxia arising from cerebral ischemia (inadequate perfusion and tissue hypoxia). Consequences of ischemia are as follows: (1) ischemic neurons release substances that produce glial permeability to sodium (cytotoxic edema); (2) with energy failure, influxes of calcium through incompetent channels produce axonal injury, mitochondrial swelling, and cell death; and (3) lactic acidosis. Tertiary causes of brain injury occur from compromised systemic circulation with hypotension and shock or inadequate pulmonary ventilation, or both. Traumatic brain injury can be focal or diffuse. Focal injury occurs in a specific area of the brain and includes contusions and hematomas. Diffuse injury involves more than one area and includes diffuse axonal injury and concussion.
Focal Brain Injury: Focal brain injury is specific and involves grossly observable brain lesions. The force of impact (translational acceleration) typically produces brain contusions (bruises from blood seeping from microhemorrhages into brain tissue) and intracranial bleeding that displaces brain tissue (i.e., extradural, subdural, and intracerebral hematomas). Contusion and bleeding occur because of small tears in blood vessels resulting from these forces. The focal injury may be coup (directly below the point of impact) or contrecoup (on the pole opposite the site of impact) (Figure 17-1). Objects (e.g., baseball bat, weapon) striking the front of the head usually produce only coup injuries (contusions and fractures) because the inner skull in the occipital area is smooth. Objects striking the back of the head usually result in both coup and contrecoup injuries because of the irregularity of the inner surface of the frontal bones (see Figure 17-1). Objects striking the side of the head may produce coup or contrecoup injuries. The same is true when the head strikes an immovable object with little velocity (e.g., a short fall). Brain edema forms around and in damaged neural tissues, contributing to the increasing ICP. Within the contused areas are infarction and necrosis, multiple hemorrhages, and edema. The tissue has a pulpy quality. The maximum effects of injury related to contusion, bleeding, and edema peak 18 to 36 hours after severe head injury.
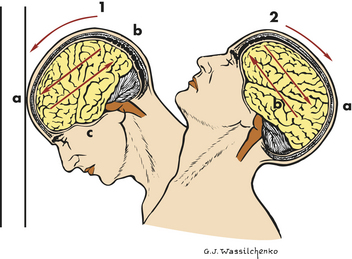
Figure 17-1 Coup and contrecoup brain injury following blunt trauma. 1, Coup injury: impact against object; a, site of impact and direct trauma to brain; b, shearing of subdural veins; c, trauma to base of brain. 2, Contrecoup injury: impact within skull, a, site of impact from brain hitting opposite side of skull; b, shearing forces through brain. These injuries occur in one continuous motion—the head strikes the wall (coup) and then rebounds (contrecoup). (Modified from Rudy EB: Advanced neurological and neurosurgical nursing, St Louis, 1984, Mosby.)
Contusions (Figure 17-2) are found most commonly in the frontal lobes, particularly at the poles and along the inferior orbital surfaces; in the temporal lobes, especially in the anterior poles and along the inferior surface; and at the frontotemporal junction. The severity of contusion is associated with the amount of energy transmitted by the skull to underlying brain tissue. In addition, the smaller the area of impact, the greater the severity of injury because the force is concentrated into a smaller area. Contusions result in changes in attention, memory, executive attentional function (motivation, goal selection or formation, planning, self-monitoring, and use of feedback), affect, emotion, and behavior. Less commonly, contusions occur in the parietal and occipital lobes. Focal cerebral contusions are superficial, involving just the gyri. Hemorrhagic contusions may coalesce into a large, confluent intracranial hematoma.
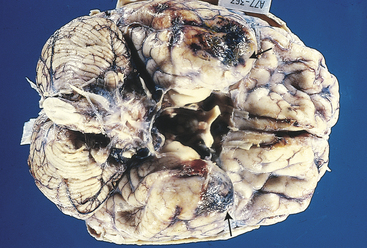
Figure 17-2 Cerebral contusions. The temporal poles are discolored by areas of hemorrhage (arrows). Such lesions represent “bruises” on the surface of the brain caused by violent contact between the delicate brain parenchyma and the hard inner surface of the skull. (From Kumar V, Cotran RS, Robbins SL: Robbins basic pathology, ed 7, Philadelphia, 2003, Saunders.)
The clinical manifestations of a contusion may include immediate loss of consciousness (generally accepted to last no longer than 5 minutes); loss of reflexes, which results in the individual falling to the ground; transient cessation of respiration; brief period of bradycardia; and decrease in blood pressure (lasting 30 seconds to a few minutes). A momentary increase in cerebrospinal fluid (CSF) pressure and changes on electrocardiogram (ECG) and electroencephalogram (EEG) have been demonstrated to occur on impact. Vital signs may stabilize to normal values in a few seconds. Reflexes return next and the person begins to regain consciousness. Returning to being fully awake and alert can vary from minutes to days. Regaining a full level of consciousness may be extremely slow and residual deficits may persist. In some persons, full level of consciousness never returns. Evaluation should include a complete history and physical examination. Skull and spinal radiographs are taken frequently and a computed tomography (CT) scan or magnetic resonance imaging (MRI) may be done. Large contusions and lacerations with hemorrhage may be excised surgically. Otherwise, treatment is directed at controlling ICP and managing symptoms.
Extradural hematomas (epidural hematomas or epidural hemorrhages) represent 1% to 2% of major head injuries and occur in all age groups, but usually in people 20 to 40 years of age. Extradural hematomas are caused most commonly by motor vehicle accidents (MVAs), occasionally by minor falls and sporting accidents. A temporal fracture causes 90% of temporal lobe extradural hematomas. Direct frontal lobe trauma is associated with frontal extradural hematomas. Posterior extradural hematomas are associated with a fracture across the transverse sinus from an occipital blow.
An artery is the source of bleeding in 85% of extradural hematomas (Figure 17-3); 15% result from injury to the meningeal vein or dural sinus. Ninety percent of individuals also have a skull fracture. The temporal fossa is the most common site of extradural hematoma caused by injury to the middle meningeal artery or vein. The resulting shift of the temporal lobe medially precipitates uncal and hippocampal gyrus herniation through the tentorial notch. Extradural hemorrhages are found occasionally in the subfrontal area (especially in the young and older adult populations), caused by injury to the anterior meningeal artery or a venous sinus, and in the occipital-suboccipital area, which results in herniation of the posterior fossa contents through the foramen magnum. CT and MRI show a lens-shaped mass over the surface of the cortex.
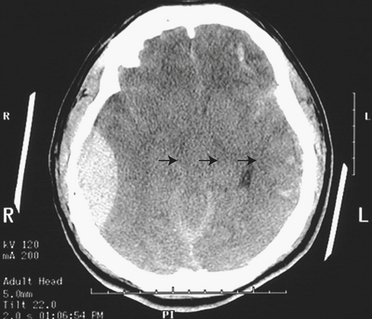
Figure 17-3 Epidural hematoma, CT image. Note the large right epidural hematoma with a lens-shaped outline as the smooth dura becomes indented against the underlying cortex on the right lateral aspect of the cerebrum. The epidural hematoma is confined within an area bounded by cranial sutures where the dura is firmly adherent to the skull. Note the mass effect with effacement of the lateral ventricles and the shift of midline to the left (arrows). In this case the individual fell from a height and struck the right side of his head, severing the middle meningeal artery. This epidural hematoma collected within hours. CT, Computed tomography. (From Klatt EC: Robbins and Cotran atlas of pathology, Philadelphia, 2006, Saunders.)
Individuals with classic temporal extradural hematomas (i.e., over the temporal lobe) experience loss of consciousness at the time of injury, followed by a lucid period that lasts from a few hours to a few days in one third of individuals (if bleeding from a vein). As the hematoma accumulates, a headache of increasing severity, vomiting, drowsiness, confusion, seizure, and hemiparesis may develop. Level of consciousness may dwindle rapidly as temporal lobe herniation begins. Clinical manifestations of temporal lobe herniation also include ipsilateral pupillary dilation and contralateral hemiparesis.
The diagnosis of an extradural hematoma is usually made by CT or MRI. In some instances, diagnosis is made by history and clinical findings, because time for a CT or MRI is not available. The prognosis is usually good if intervention is initiated before bilateral dilation of the pupils. Surgical therapy is evacuation of the hematoma through burr holes, followed by ligation of the bleeding vessel or vessels. Extradural hematomas are almost always medical emergencies.
Subdural hematomas arise in 10% to 20% of TBIs. MVAs are the most common cause of subdural hematomas; 50% of subdural hematomas are associated with skull fractures. Falls, especially in older adults or in those with long-term alcohol abuse, are associated with chronic subdural hematomas.
Acute subdural hematomas rapidly develop (within 48 hours) and usually are located at the top of the skull (the cerebral convexities). On CT they appear as a high-density mass. Bilateral hematomas occur in 15% to 20% of persons. Subacute subdural hematomas develop more slowly, often over 48 hours to 2 weeks. On CT they appear as a mixed-density mass. Chronic subdural hematomas (commonly found in older adults and those who abuse alcohol who have some degree of brain atrophy with a subsequent increase in the extradural space) develop over weeks to months. Tearing of the bridging veins is the major cause of rapidly developing and subacutely developing subdural hematomas, although torn cortical veins or venous sinuses and contused tissue may be the source. These subdural hematomas act as expanding masses, giving rise to increased ICP that eventually compresses the bleeding vessels (Figures 17-4 and 17-5). The displacement of brain tissue can result in a herniation syndrome.
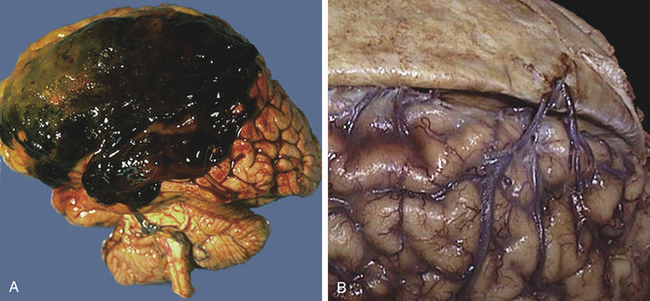
Figure 17-4 Subdural hematoma, gross, and bridging veins, gross. A large subdural hematoma (A) is seen in the frontoparietal region. A subdural hematoma forms after head trauma that severs the bridging veins from dura to brain, shown in the right panel (B) where the dura has been reflected to reveal the normal appearance of the bridging veins that extend across to the superior aspect of the cerebral hemispheres. Older adultss and the very young are at greater risk because their cerebral veins are more vulnerable to injury. Because the bleeding is venous, blood collects over hours to weeks, with variable onset of symptoms. Because the blood collects beneath the dura, a subdural hematoma can be seen to cross the region of cranial sutures. (From Klatt EC: Robbins and Cotran atlas of pathology, Philadelphia, 2006, Saunders.)
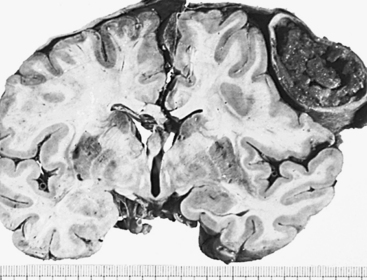
Figure 17-5 Chronic subdural hematoma. Compression of underlying brain and lateral ventricle. Note bone formation in falx and uncal herniation on the side of hematoma. (From Kissane JM, editor: Anderson’s pathology, ed 9, St Louis, 1993, Mosby.)
In acute, rapidly developing subdural hematomas the expanding clots directly compress the brain, giving rise to the clinical manifestations. As the ICP rises the bleeding veins are compressed and thus bleeding is self-limiting, although cerebral compression and displacement of brain tissue can cause temporal lobe herniation.
An acute subdural hematoma classically begins with headache, drowsiness, restlessness or agitation, slowed cognition, and confusion. These symptoms worsen over time and progress to loss of consciousness, respiratory pattern changes, and pupillary dilation (the symptoms of temporal lobe herniation). These manifestations are more pronounced than focal manifestations such as dysphasia, dyspraxia, or hemiparesis. Other clinical manifestations may include homonymous hemianopia (defective vision in either the right or the left field), disconjugate gaze, and gaze palsies.
The pathogenesis of a chronic subdural hematoma is different. The existing subdural space gradually fills with blood. A vascular membrane forms around the hematoma in approximately 2 weeks. Further enlargement takes place in some persons, but the mechanism of this enlargement is unclear.
Presenting manifestations of chronic subdural hematomas vary. Of those affected, 80% have chronic headaches and tenderness over the hematoma on percussion. Most appear to have a progressive dementia accompanied by generalized rigidity (paratonia).
Whereas most acute and subacute subdural hematomas are treated with clot evacuation through a burr hole, chronic subdural hematomas (and some that are subacute) require a craniotomy to evacuate the gelatinous blood. The membrane around a chronic subdural hematoma is then dissected away from the dura mater and arachnoid membranes. A technique for percutaneous drainage for chronic subdural hematomas has proved successful.
Intracerebral hematomas (intraparenchymal hemorrhages)4 occur in 2% to 3% of head injuries usually associated with MVAs and falls from some distance. Intracerebral hematomas may be single or multiple, and they are associated with contusions. Although most commonly located in the frontal and temporal lobes, intracerebral hematomas may occur in the hemispheric deep white matter. Small blood vessels are traumatized by penetrating injury or shearing forces. The intracerebral hematoma then acts as an expanding mass, resulting in increased ICP and compression of brain tissues with resultant edema (Figure 17-6). Delayed intracerebral hematomas may appear 3 to 10 days after the head injury.
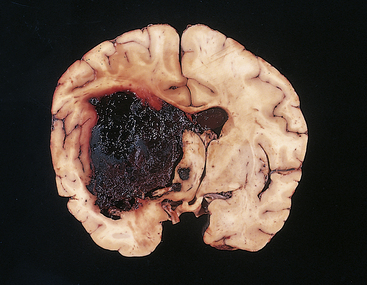
Figure 17-6 Acute intracerebral hemorrhage. A fresh hematoma has disrupted and expanded the left cerebral hemisphere, causing the midline structures to shift to the right. Uncontrolled hypertension is an important cause of this catastrophic lesion. (From Kumar V, Cotran RS, Robbins SL: Robbins basic pathology, ed 7, Philadelphia, 2003, Saunders.)
A decreasing level of consciousness is associated with an intracerebral hematoma. Coma or a confusional state from other injuries, however, can make the cause of this increasing unresponsiveness difficult to detect. Contralateral hemiplegia also may occur. As the ICP rises, clinical manifestations of temporal lobe herniation may appear. In delayed intracerebral hematoma, the presentation is similar to that of hypertensive brain hemorrhage: sudden, rapidly progressive decreased level of consciousness with pupillary dilation; breathing pattern changes; hemiplegia; and bilateral positive Babinski reflexes.
Evacuation of a singular intracerebral hematoma has only occasionally been helpful, mostly for subcortical white matter hematomas. Otherwise, treatment is directed at reducing the ICP and allowing the hematoma to reabsorb slowly.
Open trauma produces discrete (focal) injuries and includes compound fractures and missile injuries. A compound fracture opens a communication between the cranial contents and the environment and should be investigated whenever there are lacerations of the scalp, tympanic membrane, a sinus, an eye, or mucous membranes. Such fractures may involve the cranial vault or the base of the skull (basilar skull fracture). The injury incurred from bone fragments is mainly a tangential injury (injury caused by direct contact) and occasionally a penetrating injury. Bone fragments may lacerate or contuse brain tissues or blood vessels. In addition, cranial nerves may be damaged with a basilar skull fracture.
Missiles include bullets, rocks, shell fragments, knives, and blunt instruments. The mechanisms of injury are crush injury and stretch injury. Crush injury is the laceration and crushing of whatever tissue the missile touches, with the amount of crush related to the degree of fragmentation, deformity, size, and shape. A tangential injury is injury to the coverings of the brain (scalp lacerations), skull fractures, laceration of the meninges, and cerebral lacerations. Projectiles and debris from scalp and skull injury, when driven into the brain substance, produce a penetrating brain injury. Occasionally projectiles are so forceful that they exit the cranial vault in addition to entering it, producing a through-and-through injury. Primary damage is localized along the path of the penetrating object, and direct tissue disruption along the projectile tract results. A high-velocity bullet produces contusions at the site of entry, caused by bone striking the brain tissue on impact. Bone fragments are driven inward.
Stretch injury involves blood vessels and nerves that are damaged without direct contact due to the amount of tissue stretched secondary to shape, deformation, and striking velocity. Air compressed in front of a bullet exerts an explosive effect on entry, producing extreme distant tissue damage and an immediate primary increase in ICP; a cavity many times greater than the size of the bullet is produced because the brain tissue is propelled away from the tract. The cavity and pressure produce contrecoup injuries. The intracranial volume is increased directly by the projectile and the debris. The temporary cavity collapses back onto itself, leaving a smaller, permanent cavity. Intracranial bleeding occurs into the permanent cavity and may cause the cavity to expand. Edema in and around the injured brain tissue rapidly develops; edema and bleeding contribute markedly to ICP. This second rise in ICP to 60 to 100 mmHg may last 2 to 5 minutes. Because of acute ischemic damage to the tract, necrosis of tissue begins. Within hours after bullet-induced injury, tissue within 1 cm adjacent to the tract disintegrates. Demyelination of white matter affected by hemorrhage and edema occurs by the second day. Unconsciousness, flaccidity, or decerebrate posture (see Chapter 16) are associated with a 94% mortality.
With open-head injury, most victims lose consciousness. The depth of the coma and the length of the unresponsive state are related to the location of injury, extent of damage, and amount of bleeding. Open-head injury often requires surgery to débride the traumatized tissues to prevent infection and to remove blood clots to help reduce the ICP. ICP also is managed with steroids, dehydrating agents, osmotic diuretics, or a combination of these drugs. Broad-spectrum antibiotics are administered.
The diagnosis of a compound fracture is made through physical examination, skull radiographs, or both. The diagnosis of a basilar skull fracture is made on the basis of clinical findings. Skull radiographs often do not demonstrate the fracture, although intracranial air or air in the sinuses on radiograph, CT, or MRI is indirect evidence of a basilar skull fracture.
A compound linear fracture is débrided nonsurgically in cooperative adults and surgically in children and uncooperative adults. Cranioplasty with insertion of bone or an artificial graft may be necessary but often is delayed until antibiotics have been given. Antibiotics are administered after surgery.
Bed rest and close observation for meningitis and other complications are prescribed for a basilar skull fracture. Use of prophylactic antibiotics is controversial because studies have failed to demonstrate that they reduce the rate of infection.
Diffuse Brain Injury: Diffuse brain injury (diffuse axonal injury [DAI]) results from a shaking effect (inertial effects of mechanical input to the head associated with high levels of acceleration and deceleration, effects of head motion). Rotational acceleration (twisting movement) is the primary mechanism of injury, producing strains and distortions within the brain (see Figure 17-1). The brain tissues experience shearing stresses set up by the rotational forces that operate when a freely moving head is struck because of the skull’s motion from its attachment to the neck. Shearing, tearing, or stretching of nerve fibers with subsequent axonal damage results. Forces applied axially as a result of centrifugal acceleration of the head establish a gradient of injury severity from the hemispheres to the brain stem. The most severe axonal injuries are located more peripheral to the brainstem, thus accounting for the tremendous cognitive and affective impairments seen in survivors of traumatic brain injury from MVAs. The frontal and temporal axonal tracts are particularly vulnerable. Damage reduces the speed of informational processing and responding and disrupts attention.
The common pathologic substrate in diffuse brain injury is axonal damage (disruption). Pathophysiologically, at the time of injury, the damage can be seen only with an electron microscope and involves either numerous axons alone or axonal injury in conjunction with actual tissue tears (Figure 17-7). Areas where axons and small blood vessels are torn appear as small hemorrhages, located particularly in the corpus callosum and dorsolateral quadrant of the rostral brain stem at the superior cerebellar peduncle.
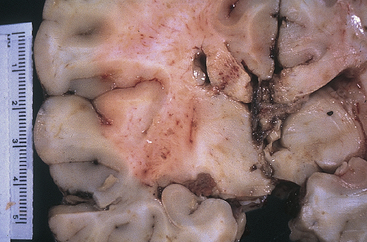
Figure 17-7 Diffuse axonal injury. Gross photograph demonstrating characteristic hemorrhage lesions within the corpus callosum. Courtesy of Walter Kemp, MD, Department of Pathology, University of Texas Southwestern Medical School, Dallas.) (From Kumar V, Cotran RS, Robbins SL: Robbins basic pathology, ed 7, Philadelphia, 2003, Saunders.)
Oxygen-free radicals contribute to secondary injury. Free radicals damage proteins and the phospholipid components of cells and organelle membranes. Membrane depolarization caused by the trauma permits nonselective opening of voltage-sensitive calcium channels, resulting in abnormal calcium accumulation in neurons and glial cells. These calcium shifts are associated with activation of lipolytic and proteolytic enzymes, protein kinases, protein phosphatases, dissolution of microtubules, and altered gene expression. Abnormal calcium influx occurs through activation of excitatory amino acid receptors. Widespread exotoxicity occurs after trauma, resulting in cell swelling, vacuolization, and death.4
Progressively increasing numbers of damaged axons are visible 12 hours to several days after the injury. Chromatolysis of the neurons involving eccentric relocation of the nucleus, swelling of the axon hillock, and redistribution of the rough endoplasmic reticulum in the cell body is evident. During this time the torn axons, which resemble dilated sausage links, also regress into round balls called retraction balls. These retraction balls are visible with light microscopy.
The number of retraction balls increases during the first week or two but begins to diminish in 2 to 3 weeks. Clusters of microglia appear in their place. Lastly, astrocytosis (gliosis, equivalent to scarring) occurs at the sites of axonal damage. Demyelination is seen particularly in the long axon tracts of the upper brainstem.
Severity of the diffuse injury correlates with the direction and velocity of rotation, that is, how much shearing force was applied to the brainstem. Figure 17-8 illustrates the spectrum of the diffuse injury as the magnitude increases. DAI is not associated with intracranial hypertension soon after injury, but acute brain swelling (increased intravascular blood within the brain, vasodilation, and increased cerebral blood volume) is seen often.
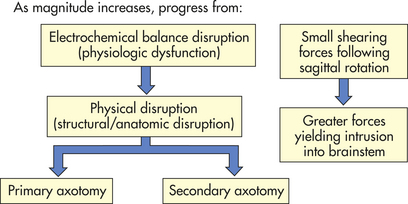
Several categories of diffuse brain injury exist: mild concussion, classic concussion, mild DAI, moderate DAI, and severe DAI. An organic component is present within each category, in contrast to the previous conceptualization that concussion had no structural injury component.
Mild concussion involves temporary axonal disturbances. Cerebrocortical dysfunction related to attentional and memory systems results, but consciousness is not lost. Three forms have been described:
Grade I: Confusion and disorientation accompanied by amnesia (momentary)
Grade II: Momentary confusion and retrograde amnesia that develops after 5 to 10 minutes (memory loss involves only events occurring several minutes before injury)
Grade III: Confusion and retrograde amnesia present from impact (also anterograde amnesia) (persists for several minutes)
Recommended guidelines for return to play after sports-related concussive injuries are contained in What’s New? Sports-Related Concussion.
Mild concussion is characterized by an immediate onset of clinical manifestations at the time of injury and the transitory nature of clinical manifestations. A momentary rise in CSF pressure and changes in ECG and EEG have been demonstrated to occur on impact in the laboratory. No loss of consciousness is experienced. The initial confusional state exists for a moment to several minutes. Amnesia for events preceding the trauma (retrograde amnesia) may be experienced. Anterograde amnesia may exist transiently. Persons may experience head pain and complain of nervousness and “not being oneself” for up to a few days.
Classic cerebral concussion (grade IV) involves diffuse cerebral disconnection from the brainstem reticular activating system and is a phenomenon of physiologic, neurologic dysfunction without substantial anatomic disruption. Evidence of this disconnection is the immediate loss of consciousness, which lasts less than 6 hours. Retrograde and anterograde (posttraumatic) amnesia is present. This type of diffuse injury frequently is associated with focal pathologic findings, especially cerebral contusions that yield focal signs, not loss
of consciousness. There are two forms of classic cerebral contusion: uncomplicated classic cerebral concussion (without focal injury) and complicated classic cerebral concussion (accompanied by focal injury).
In classic cerebral concussion loss of consciousness lasts as long as 6 hours and reflexes are lost, causing falls. Reflexes are regained as responsiveness returns. Transient cessation of respiration, brief periods of bradycardia, and a decrease in blood pressure lasting 30 seconds or less occur. Vital signs stabilize within a few seconds to within normal limits. Retrograde and anterograde amnesia exist. A confusional state persists for hours to days. The individual experiences head pain, nausea, and fatigue. Attentional and memory system impairments may persist for weeks to months and may include inability to concentrate and forgetfulness. Mood and affect changes may persist for weeks to months and may include nervousness, anxiety reactions, depression, irritability, fatigability, and insomnia.
Some of the effects of a concussion may persist for weeks or months, depending on the severity of the injury. Fifty percent of persons have a postconcussive syndrome that includes headache, cognitive impairments, psychologic and somatic complaints, and cranial nerve signs and symptoms.4 Treatment entails reassurance and symptomatic relief. Close observation for 24 hours by a reliable individual is indicated so that immediate intervention can be obtained if delayed effects become severe.
DAI produces prolonged traumatic coma lasting more than 6 hours because of axonal disruption. Three forms of DAI exist: mild, moderate, and severe. In mild diffuse axonal injury, posttraumatic coma lasts 6 to 24 hours. Death is uncommon but residual cognitive, psychologic, and sensorimotor deficits may persist. Mild DAI is a relatively uncommon lesion, occurring in 8% of all severe head injuries and 19% of all cases of DAI. In mild DAI 30% of persons display decerebrate or decorticate posturing; they may experience prolonged periods of stupor or restlessness (see Figure 16-5).
In moderate diffuse axonal injury, widespread physiologic impairment exists throughout the cerebral cortex and diencephalon. Actual tearing of some axons in both hemispheres occurs. Basal skull fracture, a focal injury, is commonly associated with moderate DAI. Prolonged coma lasting more than 24 hours is present but prominent brainstem signs do not exist with moderate DAI. Recovery often is incomplete in 93% of those individuals who survive. Moderate DAI is the most common type of DAI and is found in 20% of severe head injuries and 45% of all cases of DAI.
In moderate DAI, the GCS score is 4 to 8 initially and 6 to 8 by 24 hours. Thirty-five percent of victims have transitory decerebration or decortication. The person often remains unconscious for days or weeks and on awakening is confused. He or she experiences a long period of posttraumatic anterograde and retrograde amnesia and often has permanent deficits in memory, selective attention, vigilance, detection, working memory, data processing, vision or perception, and language, as well as mood and affect changes ranging from mild to severe.
Severe diffuse axonal injury, formerly called primary brainstem injury or brainstem contusion, involves severe mechanical disruption of many axons in both cerebral hemispheres and those extending to the diencephalon and brainstem. Severe DAI represents 16% of all severe head injuries and 36% of all cases of DAI. With an initial GCS score of 3, the mortality rate is 78%2; with an initial score between 3 and 8, the mortality rate is 36%; and 16% of persons have either a moderate or severe disability and 5% survive in a coma (unresponsive) state.2
Severe DAI is associated with brainstem signs that disappear in a few weeks. The person experiences immediate autonomic dysfunction that resolves in a few weeks. Increased ICP appears 4 to 6 days after injury. Pulmonary complications occur frequently, with profound sensorimotor and cognitive system deficits. Severely compromised coordinated movements and verbal and written communication, inability to learn and reason, and inability to modulate behavior also are found.
CT scan and MRI are the diagnostic tests of choice for TBI.2,5 There is no strong research evidence that any treatment reduces the complications of moderate to severe TBI, although various treatment protocols are instituted. Specifically, the evidence is inconclusive about the effectiveness of hyperventilation, mild hypothermia, and use of mannitol.2 Barbiturates have not been shown to be effective in reducing intracranial pressure or preventing adverse outcomes after TBI.2 The Corticosteroid Randomisation After Significant Head Injury (CRASH) trial showed corticosteroids increase mortality with acute TBI, so these drugs are no longer used. Carbamazepine and phenytoin may reduce the occurrence of early seizures but have not been shown to reduce the onset of late seizures, neurologic disability, or death.2 Prophylactic antibiotics also have not been shown to reduce the risk of meningitis or death with a skull fracture.2 Extensive research is under way to discover effective therapeutic interventions. The role of fluid and nutrition management has emerged as critically important in the care of individuals with severe brain injuries.6
Genetics of Head Injury: Certain genes are up-regulated and others are down-regulated after head trauma. Researchers’ attention has been focused predominantly on the apolipoprotein E (apoE) gene and its various alleles. Certain alleles have been correlated with increased susceptibility to and severity of head injury. Other alleles have been associated with improved or diminished recovery after head injury. The clinical significance of these findings is not clearly known.4,7
Spinal Cord Trauma
The number of spinal cord injuries (SCIs) in the United States is approximately 235,000, with 11,000 annually; of these, 77.8% are men, mostly young adults.8 The average age is 38 years; however, the percent of SCI in individuals older than 60 years of age has increased to 11.5%, up 4.7% since 2000.8 At discharge, since 2000, the extent of injury has been 34.1% incomplete quadriplegia, 18.3% complete quadriplegia, 18.5% incomplete paraplegia, and 23.0% complete paraplegia.8 MVAs have accounted for 46.9% of reported SCI cases since 2000. Falls are now the next most common cause followed by acts of violence, primarily gunshot wounds, then recreational sporting activities.8 Older adults, because of preexisting degenerative vertebral disorders, are particularly at risk for minor trauma resulting in serious spinal cord injury, especially from falls.
PATHOPHYSIOLOGY Spinal cord injuries most commonly occur because of vertebral injuries, as a result of acceleration, deceleration, or deformation forces most frequently applied at a distance. These forces injure the vertebral or neural tissues by compressing the tissues, pulling or exerting a traction (tension) on the tissues, or shearing tissues so that they slide into one another. These forces may be exerted on the vertebral and neural tissues by hyperextension, hyperflexion, vertical compression, or rotation of the spine (Figures 17-9 to 17-12). The bones, ligaments, and joints of the vertebral column may be damaged. The vertebral column may incur fracture and often compression of one or more elements, dislocation of its elements, or both fracture and dislocation (see Figure 14-16 for the structure of the vertebral column). Vertebral injuries can be classified as (1) simple fracture, a single break usually affecting transverse or spinous processes; (2) compressed (wedged) vertebral fracture, in which a vertebral body is compressed anteriorly; (3) comminuted (burst) fracture, in which a vertebral body is shattered into several fragments; and (4) dislocation.
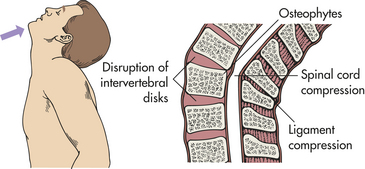
Figure 17-9 Hyperextension injuries of the spine. Hyperextension can result in fracture or nonfracture injuries with spinal cord damage.
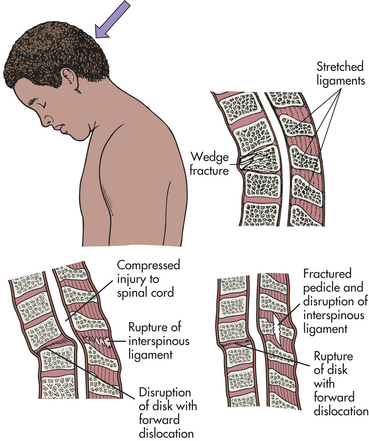
Figure 17-10 Hyperflexion injury of the spine. Hyperflexion produces translation (subluxation) of vertebrae, which compromises the central canal and compresses spinal cord parenchyma or vascular structures.
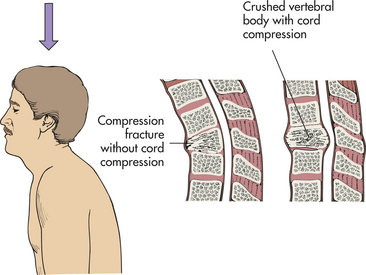
Figure 17-11 Axial compression injuries of the spine. In axial compression the spinal cord is contused directly by retropulsion of bone or disk material into the spinal canal.
Vertebrae fracture readily with direct and indirect trauma. When the supporting ligaments are torn, the vertebrae move out of alignment, and dislocations occur. A horizontal force moves the vertebrae straight forward; if the individual is in a flexed position at the time of injury, the vertebrae are then in an angulated position. Flexion and extension injuries may result in dislocations. (Mechanisms of vertebral injury are presented in Table 17-2.) Vertebral injuries occur mostly at vertebrae C1-C2 (cervical), C4-C7, and T1-L2 (thoracic-lumbar) (see Figure 14-10). These are the most mobile portions of the vertebral column. The cord occupies most of the vertebral canal in the cervical and lumbar regions. The size makes the cord in these areas more easily injured. The primary spinal cord injuries are summarized in Table 17-3. Noncontiguous vertebral injuries are not uncommon. Further, primary injury occurs if an injured spine is not adequately immobilized. A comparison of adult with child spine and spinal cord injuries is contained in Table 17-4.
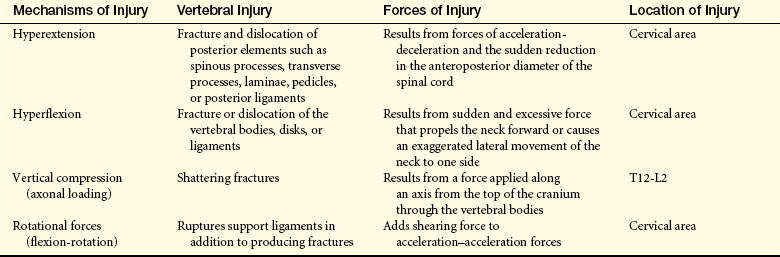
Table 17-3
| Injury | Description |
| Cord concussion | Results in a temporary disruption of cord-mediated functions |
| Cord contusion | Bruising of the neural tissue causing swelling and temporary loss of cord-mediated functions |
| Cord compression | Pressure on the cord causing ischemia to tissues; must be relieved (decompressed) to prevent permanent damage to the spinal cord |
| Laceration | Tearing of the neural tissues of the spinal cord; may be reversible if only slight damage is sustained by the neural tissues; may result in permanent loss of cord-mediated functions if spinal tracts are disrupted |
| Transection | Severing of the spinal cord, causing permanent loss of function |
| Complete | All tracts in the spinal cord completely disrupted; all cord-mediated functions below the transection are completely and permanently lost |
| Incomplete | Some tracts in the spinal cord remain intact, together with functions mediated by these tracts; has the potential for recovery although function is temporarily lost |
| Preserved sensation only | Some demonstrable sensation below the level of injury |
| Preserved motor nonfunctional | Preserved motor function without useful purpose; sensory function may or may not be preserved |
| Preserved motor functional | Preserved voluntary motor function that is functionally useful |
| Hemorrhage | Bleeding into the neural tissue because of blood vessel damage; usually no major loss of function |
| Damage or obstruction of spinal blood supply | Causes local ischemia |
Table 17-4
Comparison of Spine and Spinal Cord Injuries in Adults and Children
| Characteristics | Adult | Pediatric |
| Most common mechanism of injury | Motor vehicle accidents | Falls |
| Level of Injury | ||
| C1-C3 | 1% to 2% | 60% |
| C3-C7 | 85% | 30% to 40% |
| Thoracolumbar | 10% to 15% | 5% |
| Type of Injury | ||
| Fracture-dislocation | >70% | 25% |
| Subluxation | <20% | 50% |
| SCIWORA | Rare | Up to 50% |
| Delayed neurological deficits | Rare | Up to 50% |
SCIWORA, Spinal cord injury without radiologic abnormalities.
From Evans RW, Wilberger JE: Traumatic disorders. In Goetz CG, editor, Textbook of neurology, Philadelphia, 2003, Saunders.
The pathophysiologic cascade of secondary spinal cord injury begins within a few minutes after injury. Microscopic hemorrhages appear in the central gray matter and pia arachnoid that increase in size within 2 hours. Edema in the white matter occurs, impairing the microcirculation of the cord. Within 4 hours, numerous swollen axis cylinders develop. Localized hemorrhaging and edema therefore are followed by loss of autoregulation, vasospasm, impaired venous drainage, and reduced vascular perfusion with development of ischemic areas. Oxygen tension in the tissue at the injury site is decreased. The microscopic hemorrhages and edema are maximal at the level of injury and for two cord segments above and below it.
Cellular and subcellular alterations and tissue necrosis occur. By 5 minutes after injury, venules of the gray matter are congested and distended by erythrocytes. In 15 to 30 minutes, small hemorrhages occur with extravasation of erythrocytes into perivascular spaces of postcapillary and muscular venules. Within 4 hours, disruption of myelin, axonal degeneration, and ischemic endothelial injury occur.
Chemical and metabolic changes in spinal cord tissues include release of toxic excitatory amino acids, accumulation of endogenous opiates, lipid hydrolysis with production of active metabolites, and local free radical release. These changes may produce further ischemia, vascular damage, and necrosis of tissues (autodestruction). Necrosis consumes 40% of cross-sectional cord within 4 hours of trauma and 70% within 24 hours. Cord swelling increases an individual’s degree of dysfunction so that distinguishing the functions to be lost permanently from those that are impaired just temporarily becomes difficult. In the cervical region, cord swelling may be life threatening because of the possibility of resulting impairment of the diaphragm function (phrenic nerves exit C3-C5) and vegetative functions mediated by the medulla oblongata. Within the first few days of injury, progressive axonal changes occur and necrotic zones develop. Progressive cavitation and coagulation necrosis at the site of injury are termed posttraumatic infarction.
Circulation in the white matter tracts of the spinal cord returns to normal in about 24 hours, but gray matter circulation remains altered. Phagocytes appear 36 to 48 hours after injury. There is proliferation of microglia and changes in astrocytes. Red cells then begin to disintegrate, and resorption of hemorrhages begins. Degenerating axons are engulfed by macrophages in the first 10 days after injury. A cyst with fluid forms. The traumatized cord is replaced by acellular collagenous tissue (a scar), usually in 3 to 4 weeks. Meninges thicken as part of the scarring process.
CLINICAL MANIFESTATIONS Normal activity of the spinal cord cells at and below the level of injury ceases because of loss of the continuous tonic discharge from the brain or brainstem and inhibition of suprasegmental impulses immediately after cord injury, thus causing spinal shock. Spinal shock is characterized by a complete loss of reflex function in all segments below the level of the lesion. This condition involves all skeletal muscles, bladder, bowel, sexual function, and autonomic control. Severe impairment below the level of the lesion is obvious; it includes paralysis and flaccidity in muscles, absence of sensation, loss of bladder and rectal control, transient drop in blood pressure, and poor venous circulation. The condition also results in disturbed thermal control because the sympathetic nervous system is damaged. This damage causes faulty control of sweating and radiation through capillary dilation. The hypothalamus cannot regulate body heat through vasoconstriction and increased metabolism; therefore, the individual assumes the temperature of the air.
Spinal shock may last for 7 to 20 days after onset; it may persist for as short a time as a few days or as long as 3 months. Indications that spinal shock is terminating include the reappearance of reflex activity, hyperreflexia, spasticity, and reflex emptying of the bladder. With cervical or upper thoracic cord injury, a form of distributive shock, called neurogenic shock, may be seen in addition to spinal shock, as a result of the loss of sympathetic outflow, causing vasodilation, hypotension, bradycardia, and hypothermia.
Loss of motor and sensory function depends on the level of injury. All motor, sensory, reflex, and autonomic functions cease below any transected area and may cease below concussive, contused, compressed, or ischemic areas (Table 17-5). Paralysis of the lower half of the body with both legs involved is termed paraplegia. Paralysis involving all four extremities is termed quadriplegia (tetraplegia). In complete quadriplegia the level of injury is above C6, and all upper extremity function is lost. In incomplete quadriplegia, function at or above C6 is preserved, leaving the shoulder, upper arm, and some forearm muscle control intact. With acceleration injuries the greatest stress point is C4-C5. With a deceleration force the greatest stress point is at C5-C6.
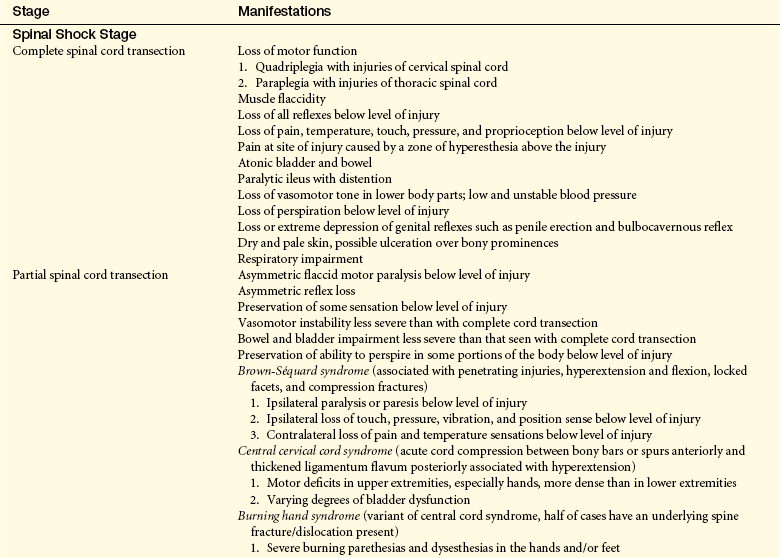

Return of spinal neuron excitability occurs slowly. Depending on the degree of damage, either of the following can occur: (1) motor, sensory, reflex, and autonomic functions return to normal; or (2) autonomic neural activity in the isolated segment develops. The sequence of hyperactivity phases, which vary in length, may include (1) minimal reflex activity, (2) flexor spasms, (3) alternation between flexor and extensor spasms, and (4) predominant extensor spasms.
The initial clinical manifestations associated with acute spinal cord injury are rapid loss of (1) voluntary movement in body parts below the level of injury, (2) sensations in the lower extremities and possibly lower trunk (depending on the level of injury), and (3) spinal and autonomic reflexes below the level of injury. The duration of this areflexic state is highly variable. In most persons, reflex activity returns in 1 to 2 weeks.
Gradually reflexes return and become increasingly easier to elicit. A pattern of flexion reflexes emerges, first involving the toes and later the feet and legs. Reflex voiding and bowel elimination appear. Flexor spasms accompanied by profuse sweating, piloerection, and automatic bladder emptying (together called a mass reflex) may develop. The ability to sweat when overheated may be disrupted, and extensor spasms may develop, usually after full development of flexor spasms. Sometimes after several months, episodes of autonomic hyperreflexia are elicited.
Autonomic hyperreflexia (dysreflexia) is a syndrome of a sudden and dangerous increase in blood pressure that may occur at any time after spinal shock resolves. The syndrome is associated with a massive, uncompensated cardiovascular response to stimulation of the sympathetic nervous system (Figure 17-13). The condition is life threatening and requires immediate treatment. Individuals most likely to be affected have lesions at the T6 level or above. Autonomic hyperreflexia is characterized by paroxysmal hypertension (up to 300 mmHg systolic), a pounding headache, blurred vision, sweating above the level of the lesion with flushing of the skin, nasal congestion, nausea, piloerection caused by pilomotor spasm, and bradycardia (30 to 40 beats/minute).9 The symptoms may develop singly or in combination (syndrome) and often are associated with a distended bladder or rectum.
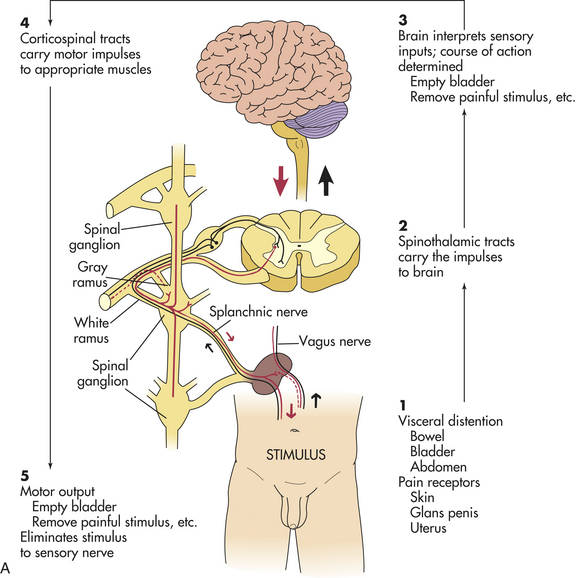
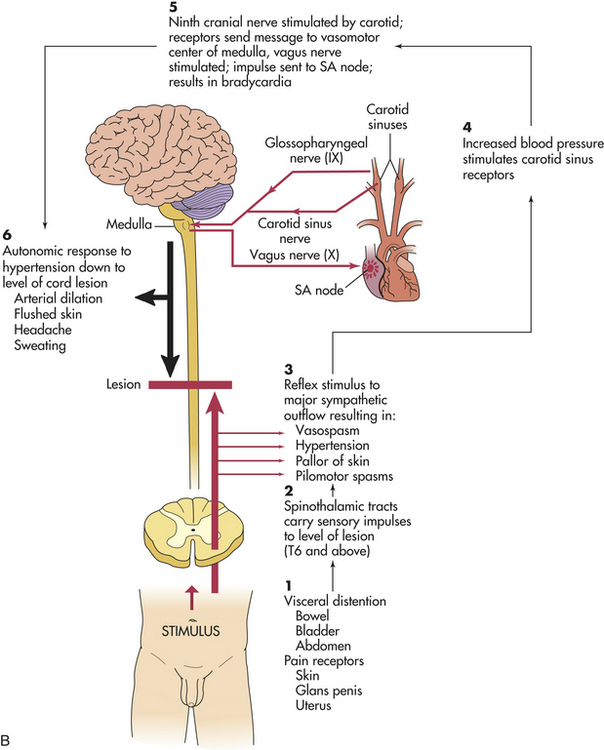
Figure 17-13 Autonomic hyperreflexia. A, Normal response pathway. B, Autonomic dysreflexia pathway. SA, Sinoatrial. (Modified from Rudy EB: Advanced neurological and neurosurgical nursing, St Louis, 1984, Mosby.)
Pathophysiology of hyperreflexia involves the stimulation of sensory receptors below the level of the cord lesion. The intact autonomic nervous system reflexively responds with an arteriolar spasm that increases blood pressure. Baroreceptors in the cerebral vessels, the carotid sinus, and the aorta sense the hypertension and stimulate the parasympathetic system. The heart rate decreases, but the visceral and peripheral vessels do not dilate because efferent impulses cannot pass through the cord.
The most common precipitating cause is a distended bladder or rectum, but any sensory stimulation can elicit autonomic hyperreflexia. Stimulation of the skin or stimulation of the pain receptors may cause autonomic hyperreflexia. Emptying of the bladder or bowel usually relieves the syndrome, and this may be facilitated by drugs, such as phenoxybenzamine.
EVALUATION AND TREATMENT Diagnosis of spinal cord injury is made on the basis of physical, radiologic, and myelographic examination; CT scan; and MRI. For a suspected or confirmed vertebral fracture or dislocation, regardless of the presence or absence of spinal cord injury, the immediate intervention is immobilization of the spine to prevent further injury. Decompression and surgical fixation may be necessary. Corticosteroids are given at the time of injury to decrease secondary cord injury and continued for 24 to 48 hours, depending on time of initiation following injury. The only other agent continuing to show promise is GM-1 ganglioside.4 Nutrition, lung function, skin integrity, and bladder and bowel management must be addressed. Plans for rehabilitation require early consideration.10
In cases of autonomic hyperreflexia, intervention must be prompt because cerebrovascular accident (CVA) is possible. The head of the bed should be elevated, and the stimulus should be found and removed. Medications may be used if these measures do not effectively reduce blood pressure.
Degenerative Disorders of the Spine
Degenerative changes occur in the vertebral disks. Degenerative disk disease (DDD) is a common finding in individuals 30 years of age and older. Only a small percentage of those persons have any functional incapacity because of pain. The causes of DDD include biochemical and biomechanical alterations of the tissue of the intervertebral disk. Fibrocartilage replaces the gelatinous mucoid material of the nucleus pulposus as the disk changes with age. There may be splits in the annulus fibrosis, permitting herniation of elements of nucleus pulposus. There may be shrinkage of the nucleus pulposus that produces prolapse or folding of the annulus with secondary osteophyte formation at the margins of the adjacent vertebral body. The pathologic findings in DDD include disk protrusion, spondylolysis, and/or subluxation and degeneration of vertebrae (spondylolisthesis) and spinal stenosis.11
Symptoms result from either (1) disk or annulus protrusion or (2) narrowing of the spinal canal or intervertebral foramen by osteophytes. A congenital narrow canal or congenitally short pedicles may be present.
Posterior disk protrusion in the cervical and thoracic regions lead to cord compression, and cauda equina compression results in the lumbar area. Both situations are called myelopathy. Posterolateral disk protrusions, with or without a contribution from the vertebral body or apophyseal joint osteophytes, lead to nerve root compression (called radiculopathy).
Cervical spondylolysis is a DDD in the cervical spine predominantly at C5-C6 and C6-C7. It may present as a cervical radiculopathy or a cervical myelopathy. Clinical manifestations of cervical radiculopathy include neck pain as well as pain in the medial aspects of the scapula, the shoulder, or arm. Sensory symptoms, such as tingling or numbness, follow a dermatomal pattern; weakness follows the pattern of innervation of the affected nerve root and occipital or suboccipital headache (some authorities refute this). Clinical manifestations of cervical myelopathy include difficulty walking, altered sensation in the feet, and sphincter disturbances (occurs late).
Thoracic disk disease is rarely symptomatic, but prolapse is found in one seventh of scans. Lumbosacral disk disease (lumbar spondylosis) involves the lower two lumbar disks in 90% of persons. There may be (1) lateral disk protrusion (10% of cases) manifesting as pain referred to the anterior thigh and leg; (2) posterolateral disk protrusion; or (3) central disk protrusion manifesting with pain, lower extremity weakness, impaired sphincter function, and saddle anesthesia. Clinical manifestations of posterolateral protrusions (Figure 17-14) include pain in the back, the sacroiliac joint, and the medial aspect of the buttock and upper thigh; radicular pain exacerbated by movement and straining (medial calf suggests L5, lateral calf suggests S1 root compression); sensory symptoms that are common and segmental in distribution; focal tenderness on palpation of the back; limited range of motion in back and scoliosis secondary to paravertebral spasms; restricted straight-leg raising (root at or below L5); positive femoral stretch test (roots of L2, L3, or L4); and focal signs that are determined by root affected.
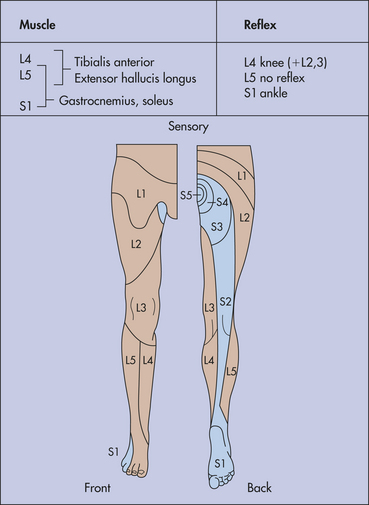
Figure 17-14 Motor, sensory, and reflex changes in lumbosacral root disorders. (From Perkin DG: Mosby’s color atlas and text of neurology, London, 1998, Mosby-Wolfe.)
Spondylolysis: Spondylolysis is a degenerative process of the vertebral column and associated soft tissue. It is characterized by a structural defect of the spine involving the lamina or neural arch of the vertebra. The most common site affected is the lumbar spine. This defect occurs in the portion of the lamina between the superior and inferior articular facets called the pars interarticularis. Mechanical pressure may cause a forward displacement of the deficient vertebra called spondylolisthesis.
Heredity plays a significant role, and spondylolysis is associated with an increased incidence of other congenital spinal defects. As a result of torsional and rotational stress, “microfractures” occur at the affected site and eventually cause dissolution of the pars interarticularis.
Spondylolisthesis: Spondylolisthesis is a stress factor allowing a vertebra to slide forward in relation to the vertebra below, commonly occurring at L5-S1. Spondylolisthesis is graded from 1 to 4 on the basis of the percentage of slip that has occurred. Individuals with grade 3 or 4 are considered for operative decompression or stabilization or both. Grades 1 and 2 usually are managed symptomatically and with nonsurgical methods.
Spinal Stenosis: In spinal stenosis the spinal canal may be congenitally narrowed or narrowed by a bulging annulus, a facet hypertrophy, or a thick/ossified posterior longitudinal ligament entrapping a single nerve involving many roots. It is classified as acquired (more common) or developmental (such as occurs in achondroplastic dwarfism). Surgical decompression is recommended for those with long-term symptoms and those who remain unresponsive to medical management.
Low Back Pain
Low back pain affects the area between the lower rib cage and gluteal muscles and often radiates into the thighs. About 1% of individuals with acute low back pain have sciatica or pain in the distribution of the sciatic nerve or lumbar and sacral nerve roots. Sciatica often is accompanied by neurosensory and motor deficits, such as weakness.
The incidence of, or percentage of population affected with, low back pain at some point in life is 60% to 80%, and the annual incidence is 5%. Men and women are affected equally; however, women report low back symptoms more often after the age of 60 years.
PATHOGENESIS Most cases of low back pain are idiopathic, and clinicians are unable to provide a precise diagnosis for most individuals with this disorder. The local processes involved in low back pain range from tension caused by tumors or disk prolapse, bursitis, synovitis, rising venous and tissue pressure (found in degenerative joint disease), abnormal bone pressures, problems with spinal mobility, inflammation caused by infection (as in osteomyelitis), bony fractures, or ligamentous sprains to pain referred from viscera or the posterior peritoneum. General processes resulting in low back pain include bone diseases, such as osteoporosis or osteomalacia, and hyperparathyroidism.
Several risk factors have been identified in the pathogenesis of low back pain. They include involvement caused by occupations that require repetitious lifting in the forward bent-and-twisted position, exposure to vibrations caused by vehicles or industrial machinery, and perhaps cigarette smoking. Osteoporosis increases the risk of spinal compression fractures and may be the reason older adult women report more symptoms than men. Genetic predispositions for low back pain include isthmic spondylolisthesis (vertebra slides forward or slips in relation to a vertebra below), spinal osteochondrosis, and spinal stenosis associated with achondroplasia. Variations in posture, such as lordosis and scoliosis of less than 60 degrees, do not appear to increase the risk of low back pain or sciatica. Differences in weight, height, and leg length are controversial as risk factors.
Anatomically, low back pain must come from innervated structures, but deep pain is widely referred and varies from person to person. The nucleus pulposus has no intrinsic innervation; however, when extruded or herniated through a prolapsed disk, it irritates the dural membranes and is responsible for pain referred to the segmental area (see Figure 17-14). The interspinous bursae can be a source of low back pain between L3, L4, L5, and S1, but also may affect L1, L2, and L3 spinous processes, depending on the closeness of the adjacent pair of spines. The anterior and posterior longitudinal ligaments of the spine and the interspinous and supraspinous ligaments are abundantly supplied with pain receptors, as is the ligamentum flavum. All of these ligaments are vulnerable to traumatic tears (sprains) and fracture. The role of muscle injury in the production of low back pain remains uncertain, even though sprains and strains are the most common diagnoses. The muscle spasms that often are produced during sieges of low back pain are thought to be produced by as yet unknown sensory or motor-reflex pathways. The most commonly encountered causes of low back pain include lumbar disk herniation, degenerative disk disease, spondylolysis, spondylolisthesis, and spinal stenosis. (For a discussion of disk herniation and rupture, see following text.)
EVALUATION AND TREATMENT Diagnosis of low back injury is made by physical examination, electromyography (EMG), epidurography, diskography, and MRI; CT with or without myelography; and nerve conduction studies. Most individuals with acute low back pain benefit from a nonspecific short-term treatment regimen including bed rest, analgesic medications, exercises, physical therapy, and education. Surgical treatments may be indicated if individuals do not respond to medical management. Surgical treatments include diskectomy and spinal fusions. Individuals with chronic low back pain can be treated with anti-inflammatory and muscle relaxant medications, exercise programs, massage, topical heat, spinal manipulation, cognitive-behavioral therapy, and interdisciplinary care.12
Herniated Intervertebral Disk
Herniation of an intervertebral disk is a displacement of the disk material (nucleus pulposus or the annulus fibrosis) beyond the intervertebral disk space2 (Figure 17-15). Men are more affected than women with a 2:1 ratio. The highest incidence is among those 30 to 50 years of age.2 Between ages 25 and 55, 95% of herniated disks are in the lower lumbar spine (L4-L5); over age 55, herniation level is higher. Disk herniation occasionally occurs in the cervical area, usually at C5-C6 and C6-C7. Herniations at the thoracic level are extremely rare. Risk factors for herniation include smoking, weightbearing sports like weightlifting, and certain work activities such as repeated lifting.2 Rupture of an intervertebral disk usually is caused by trauma or degenerative disk disease or both. Lifting with the trunk flexed and sudden straining when the back is in an unstable position are the most common causes. The injury may have an immediate onset or an onset within a few hours, or the manifestations of injury may take months to years to develop.
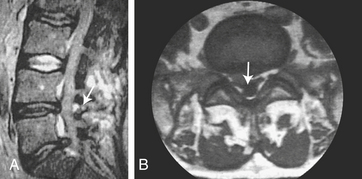
Figure 17-15 Posterolateral disk protrusion. Magnetic resonance imaging (MRI) scan, (A) sagittal (see arrow) and (B) axial (see arrow) sections. (From Perkin DG: Mosby’s color atlas and text of neurology, London, 1998, Mosby-Wolfe.)
PATHOPHYSIOLOGY In a herniated disk the ligament and posterior capsule of the disk usually are torn, allowing the gelatinous material (the nucleus pulposus) to extrude. This extrusion compresses the nerve root. Occasionally the injury tears the entire disk loose, and it protrudes onto the nerve root or compresses the spinal cord. One or more nerve roots may be compressed. This multiple nerve root compression is found especially at the L5-S1 level, where the cauda equina may be compressed. Large amounts of extruded nucleus pulposus or complete disk herniation (i.e., of both the capsule and the nucleus pulposus) may compress the spinal cord.
CLINICAL MANIFESTATIONS The location and size of the herniation into the spinal canal, together with the amount of space that exists inside the spinal canal, determine the clinical manifestations associated with the injury (see Figure 17-15). A herniated disk in the lumbosacral area is associated with pain that radiates along the sciatic nerve course over the buttock and into the calf or ankle. The pain occurs with straining, including coughing and sneezing, and usually on straight-leg raising. Other clinical manifestations include limited range of motion of the lumbar spine; tenderness on palpation in the sciatic notch and along the sciatic nerve; impaired pain, temperature, and touch sensation in the L5-S1 or L4-L5 dermatomes of the leg and foot; decreased or absent ankle jerk; and mild weakness of the foot.
With the herniation of a lower cervical disk, paresthesias and pain are present in the upper arm, forearm, and hand in the affected nerve root distribution. Neck and nerve root pain may be increased by neck motion and straining, including coughing and sneezing. Neck range of motion is diminished. Slight weakness and atrophy of biceps or triceps may occur; the biceps or triceps reflex may decrease. Occasionally signs of corticospinal and sensory tract impairments appear. These include motor weakness of the lower extremities, sensory disturbances in the lower extremities, and presence of a Babinski reflex.
EVALUATION AND TREATMENT Diagnosis of a herniated intervertebral disk is made on the basis of the history and physical examination, EMG, CT, MRI, myelography, and diskography; spinal radiography; and nerve conduction studies. Radiologic evidence of disk herniation does not reliably correlate with symptoms or predict low back pain. Many individuals with disk herniation on imaging have no symptoms.13 Clinical improvement occurs in most people. Only about 10% have sufficient pain after 6 weeks to consider surgery. The herniated disk portion on serial imaging tends to regress over time.2 There is little evidence to support drug treatments including the use of analgesics, antidepressants, or muscle relaxants.14 Nonsteroidal anti-inflammatory drugs (NSAIDs), bed rest, or traction did not improve sciatica caused by herniation. Insufficient evidence exists to judge the effectiveness of epidural injections of nonsteroidals, activity, acupuncture, massage, exercise, heat, or ice.2 However, standard diskectomy and microdiskectomy had self-reported improvement. A surgical approach is indicated if there is evidence of severe compression (weakness, decreased deep tendon reflexes and bladder/bowel reflexes).
Cerebrovascular Disorders
Cerebrovascular disease is the most frequently occurring neurologic disorder. More than 50% of persons admitted to general hospitals with neurologic problems have cerebrovascular disease. Any abnormality of the brain caused by a pathologic process in the blood vessels is referred to as a cerebrovascular disease. Included in this category are lesions of the vessel wall; occlusion of the vessel lumen by thrombus or embolus; rupture of the vessel; and alteration in vessel permeability, such as increased blood viscosity.
The brain abnormalities induced by cerebrovascular disease are of two types: (1) ischemia with or without infarction (death of brain tissues) accounting for 80% of CVAs and (2) hemorrhage. The common clinical manifestation of cerebrovascular disease is a cerebrovascular accident (CVA, stroke), which is a sudden, nonconvulsive focal neurologic deficit. Box 17-1 highlights the differences for strokes in children.
Cerebrovascular Accidents (Stroke Syndromes)
The incidence of new and recurrent stroke is 795,000 approximately 185,000 of these were recurrent.15 CVAs are the third leading cause of death in the United States, resulting in 143,600 deaths (2005) or about one in seventeen deaths per year in the US.15 Globally, 4.5 million people die from CVAs per year.2 About 10% of persons with acute ischemic CVAs die within 30 days of onset. CVAs are the leading cause of disability in the United States—50% of individuals experience some level of disability after 6 months.2 Five percent to 14% of stroke survivors have a second stroke within 1 year of the first CVA. By 5 years, 24% of females and 42% of males have a second stroke.
Fifty percent of CVAs occur in persons over 70 years of age.2 Strokes, however, do occur in a 3:10 ratio (28%) in individuals younger than 65 years of age. Stroke tends to run in families. The incidence of stroke is 2.5 times higher in blacks than whites. Stroke prevalence in 2005 for black men was 2.3 million compared to 3.4 million in black women.16 The risk of first ever stroke in blacks is almost twice that of whites.16 Death rates in blacks was 74.9 in males and 65.5 in females compared to an overall death rate of 50 in whites. Mexican Americans have an increased incidence of stroke compared with non-Hispanic whites.16 Blacks suffer greater physical impairments and are nearly twice as likely to die from their strokes. Intracranial atherosclerosis is more common in black and Asian populations, whereas extracranial disease is more common in the white population.
The mildest outcome of a CVA is so minimal as to be almost unnoticed. The most severe outcomes are hemiplegia, coma, and death. CVAs (stroke syndromes) are classified according to pathophysiology and thus are ischemic (thrombotic or embolic), global hypoperfusion (as in shock), or hemorrhagic. Risk factors for stroke include the following:
1. Arterial hypertension as well as elevated systolic and diastolic blood pressures are independent risk factors.
2. Smoking doubles the risk of stroke.
3. Diabetes is an independent risk factor and increases the risk of ischemic stroke between 2.5 and 3.5 times.17
4. Insulin resistance is an independent risk factor for ischemic stroke.
5. Polycythemia and thrombocythemia increase the risk for ischemic stroke.
6. Presence of elevated lipoprotein-a is an independent risk factor for ischemic stroke.
7. Impaired cardiac function increases the risk for ischemic stroke.
8. Hyperhomocysteinemia is a strong and independent risk factor for ischemic stroke.
9. Nonrheumatic atrial fibrillation is associated with a fivefold increase in the incidence of ischemic stroke.18
10. Chlamydia pneumoniae can increase the risk of stroke by infecting and injuring the endothelium.
Thrombotic Stroke: Thrombotic strokes (cerebral thrombosis) arise from arterial occlusions caused by thrombi formed in the arteries supplying the brain or in the intracranial vessels. The development of a cerebral thrombosis most frequently is attributed to atherosclerosis and inflammatory disease processes (arteritis) that damage arterial walls. Increased coagulation can lead to thrombus formation. Conditions causing inadequate cerebral perfusion (e.g., dehydration, hypotension, prolonged vasoconstriction from malignant hypertension) increase the risk of thrombosis. Over 20 to 30 years atheromatous plaques (stenotic lesions) tend to form at branchings and curves in the cerebral circulation. The smooth stenotic area can degenerate, forming an ulcerated area of vessel wall. Platelets and fibrin adhere to the damaged wall, and clots form, gradually occluding the artery. The thrombus may enlarge both distally and proximally in the vessel. Portions of the clot break off and travel up the vessel to distant sites where occlusion occurs, producing a stroke syndrome.
The distinction between transient ischemic attacks and thrombotic stroke is losing importance. With increasing use of brain imaging, many persons with symptoms lasting less than 24 hours are found to have had a brain infarction. The new definition for transient ischemic attack (TIA) is a brief episode of neurologic dysfunction caused by a focal disturbance of brain or retinal ischemia with clinical symptoms typically lasting less than 1 hour and without evidence of infarction.17 TIAs probably represent thrombotic particles causing an intermittent blockage of circulation or spasm. Recurrence of symptoms is 10.7% at 90 days, 60% at 6 months, and 80% at 1 year without definitive treatment.17
Embolic Stroke: Cardioembolism accounts for 30% of all CVAs, 25% to 30% of strokes in persons less than 45 years of age.19 An embolic stroke involves fragments that break from a thrombus formed outside the brain or in the heart, aorta, common carotid, or thorax. Emboli infrequently arise from the ascending aorta or common carotid artery. The embolus usually involves small vessels and obstructs at a bifurcation or other point of narrowing, thus causing ischemia. An embolus may plug the lumen entirely and remain in place or break into fragments and move up the vessel. High-risk sources for the onset of embolic stroke are atrial fibrillation (15% to 25% of strokes), left ventricular aneurysm or thrombus, left atrial thrombus, recent myocardial infarction, rheumatic valvular disease, mechanical prosthetic valve, nonbacterial thrombotic endocarditis, bacterial endocarditis, patent foramen ovale, and primary intracardiac tumors.19,20 In persons who experience an embolic stroke, a second stroke usually follows at some point because the source of emboli continues to exist. The 7-day recurrence risk is 6.5%, in-hospital mortality is 27.3%, and 5-year prognosis is as high as 80%.19 Embolization is usually in the distribution of the middle cerebral artery.
Hemorrhagic Stroke: Hemorrhagic stroke (intracranial hemorrhage [ICH]) is the third most common cause of CVA (10% of strokes) and accounts for 10% to 15% of CVAs in whites but 30% in blacks and Asians.18 There are 40,000 ICHs in the United States per year.21 The most common causes of spontaneous primary hemorrhagic strokes are hypertension (56% to 81%), ruptured aneurysms, arteriovenous malformation and fistula, amyloid angiopathy, and cavernous angioma.21 ICH can occur secondary to TBI, bleeding into the ischemic brain infarction or tumor, or a bleeding disorder or anticoagulation. Risk factors for hemorrhagic stroke include hypertension, previous cerebral infarct, coronary artery disease, and diabetes mellitus.
A hypertensive hemorrhage is associated with a significant increase in systolic and diastolic pressure over several years and usually occurs within the brain tissue. A mass of blood is formed, and its volume increases. Adjacent brain tissue is displaced and compressed and rupture or seepage into the ventricular system occurs in many cases. Hemorrhages are described as massive, small, slit, or petechial. A massive hemorrhage is several centimeters in diameter; a small hemorrhage is 1 to 2 cm in diameter; a slit hemorrhage lies in the subcortical area; and a petechial hemorrhage is the size of a pinhead bleed. The most common sites for hypertensive hemorrhages are in the putamen of the basal ganglia (a portion of the lentiform nucleus) (40%), the thalamus (15%), the cortex and subcortex (22%), the pons (8%) (Figure 17-16), caudate (7%), and cerebellar hemispheres (8%).
Lacunar Stroke: A lacunar stroke (lacunar infarct) is a microinfarct smaller than 1 cm in diameter and involves the small perforating arteries, predominantly in the basal ganglia, internal capsules, and pons. Lacunar infarcts are caused by lipohyalinosis, subintimal lipid-loading foam cells, and fibrinoid materials that thicken the arterial walls and are associated with smoking,18 hypertension, and diabetes mellitus. Because of the subcortical location and small area of infarction, these strokes may have pure motor and sensory deficits.
Cerebral Infarction: Cerebral infarction results when an area of the brain loses blood supply because of vascular occlusion. The pathologic manifestation is either (1) a global process that affects neurons most susceptible to ischemia (pyramidal and striatal neurons), Purkinje cells of the cerebral hemispheres, and the border zones at the very end of the arteries’ circulation; or (2) a focal process with a central zone of cell loss surrounded by a zone of injured cells, the ischemic penumbra, that if perfused in 1 hour will survive. Proposed pathogenesis may include (1) abrupt vascular occlusion (e.g., embolus), (2) gradual vessel occlusion (e.g., atheroma), and (3) vessels that are stenosed but not completely occluded. Cerebral thrombi and cerebral emboli are the most common causes of occlusion, but atherosclerosis and hypotension are the dominant underlying processes.
Cerebral infarctions are ischemic or hemorrhagic. In ischemic infarcts (pale infarcts, “white stroke”), cytotoxic ischemic events and interaction between blood elements and blood vessels combine to produce brain injury. The affected area becomes slightly discolored and softens about 6 to 12 hours after the occlusion. Necrosis, swelling around the insult, and mushy disintegration have appeared by 48 to 72 hours after infarction. At a microscopic level, neuronal cell bodies change, myelin sheaths and axis cylinders are interrupted and disintegrate, and there is loss of oligodendrites and astrocytes.
Cellular and biochemical events involve the loss of glucose and oxygen delivery resulting in depletion of high-energy phosphate compounds (failure of mitochondrial energy production) allowing cell membrane depolarization. With membrane depolarization, neurotransmitters, including glutamate, are released and cannot be reuptaken. Glutamate promotes excess entry of extracellular calcium. Unregulated, elevated calcium concentrations activate degrading enzymes.22 Production and accumulation of lactic acid also result in an associated focal vasodilation. Infarcted areas may lose autoregulation of blood flow.
A syndrome of luxury perfusion in areas adjacent to the infarct develops first from the loss of autoregulation. The vascular bed in this area dilates. Later, capillary sprouting (neovascularization) supports this luxury perfusion syndrome.
In hemorrhagic infarcts (“red strokes”), bleeding occurs into the infarcted area as a result of restoration of blood flow. Reperfusion occurs when the embolus fragments, or lysis or compressive forces lessen, allowing blood flow to be reestablished into the infarcted area. Most hemorrhagic infarcts are located in the cerebral cortex. Unfortunately, reperfusion has been shown to compromise recovery by accelerating the sequence of metabolically damaging events including oxidative stress (reperfusion injury).
Cerebral Hemorrhage: The primary cause of cerebral hemorrhage is hypertension. (Aneurysms and arteriovenous malformations are discussed on pp. 606-608.) The pathogenesis of hypertensive cerebral hemorrhage is not fully understood. Hypertension involves primarily smaller arteries and arterioles, resulting in thickening of the vessel walls, and increased cellularity of the vessels and hyalinization. Necrosis may be present. Microaneurysms in these smaller vessels or arteriolar necrosis precipitates the bleeding.
A mass of blood is formed as bleeding continues into the brain tissue. In massive ICH (volume greater than 150 ml), cerebral perfusion falls to zero and cerebral blood flow stops, resulting in death.21 Adjacent brain tissue is deformed, compressed, and displaced. Necrosis around the hematoma is present within 6 hours. Edema forms and the blood-brain barrier is disrupted. An inflammatory reaction in surrounding brain tissue appears rapidly and peaks in several days.21 Figure 17-17 illustrates additional detail. Rupture or seepage of blood into the ventricular system occurs in many cases.
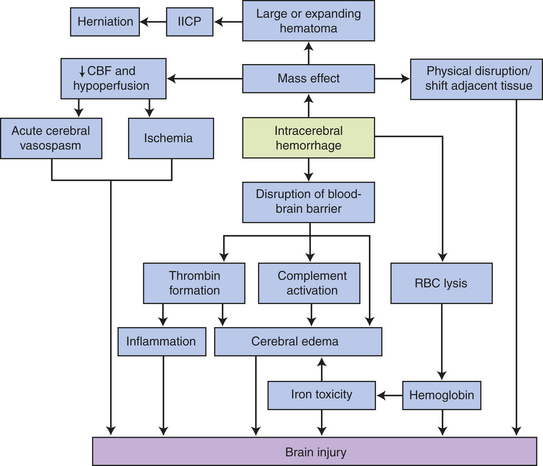
Figure 17-17 Injury mechanisms promoted by intracerebral hemorrhage. Hemorrhages can induce neuronal injury through mass effect, particularly in large hematomas that can cause increased intracranial pressure and herniation. Hemorrhages may also cause tissue damage through cerebral edema and “neurotoxic” mechanisms caused by activation of the coagulation cascade and inflammation. Not all potential interactions are shown (e.g., thrombin may potentiate iron-induced injury; the complement and inflammatory systems overlap and several factors contribute to cerebral edema). CBF, cerebral blood flow; IICP, increased intracranial pressure; RBC, red blood cell. (Data from Mocco J et al: Neurosurg Focus 15:22[5]:E7, 2007; Schubert GA, Thome C: Front Biosci 13:1594-603, 2008; Xi G, Keep RF, Hoff JT: Lancet Neurol 5[1]:53-63, 2006.)
The cerebral hemorrhage resolves through reabsorption. Macrophages and astrocytes appear to clear away the blood. A cavity forms, surrounded by a dense gliosis after removal of the blood.
CLINICAL MANIFESTATIONS Because neurons surrounding the ischemic or infarcted areas undergo changes that disrupt plasma membranes, cellular edema results, causing further compression of capillaries. Most persons survive an initial hemispheric ischemic stroke unless massive cerebral edema develops. However, massive brain stem infarcts, caused by basilar thrombosis or embolism, are almost always fatal.
Clinical manifestations of thrombotic stroke vary, depending on the artery obstructed. Different sites of obstruction create different occlusion syndromes (Table 17-6).
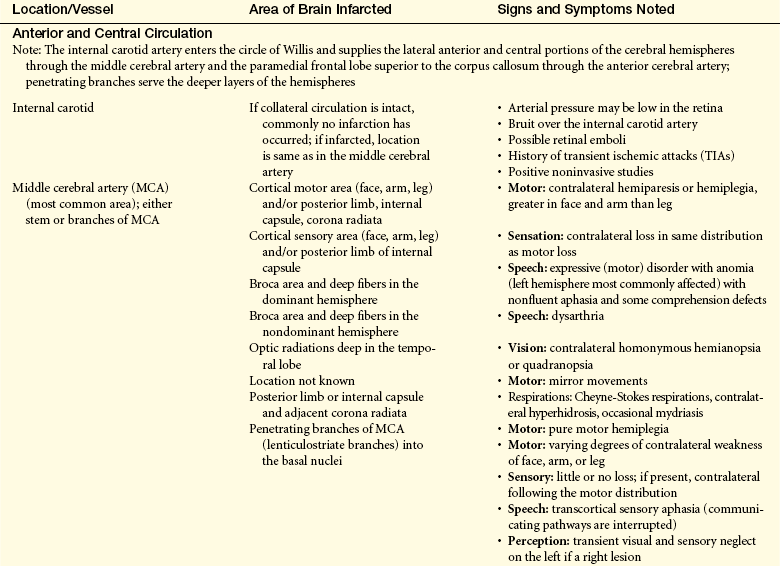



With hemorrhagic stroke, clinical manifestations vary according to the location and size of the bleed. Focal neurologic deficits are found in 80% of individuals experiencing hemorrhagic strokes; altered consciousness occurs in 50%. Once a deep unresponsive state occurs the immediate prognosis is grave, and the individual rarely survives. If the person survives, however, recovery of function frequently is possible.
Individuals experiencing intracranial hemorrhage from a ruptured or leaking aneurysm have one of three sets of symptoms: (1) onset of an excruciating generalized headache with an almost immediate lapse into an unresponsive state; (2) headache, but with consciousness maintained; and (3) sudden lapse into unconsciousness. If the hemorrhage is confined to the subarachnoid space, there may be no local signs. If bleeding spreads into the brain tissue, hemiparesis/paralysis, dysphasia, or homonymous hemianopia may be present. Warning signs of an impending aneurysm rupture may include headache, transient unilateral weakness, transient numbness and tingling, and transient speech disturbance. Warning signs, however, often are not present.
EVALUATION AND TREATMENT No identifiable cause can be established by conventional diagnostic tests in up to 40% of CVAs and are classified as having “undetermined” or “cryptogenic” mechanisms.20 The principle of acute stroke treatment is “time is brain.” Time to treatment is often too great considering the time limits for reversibility of brain ischemia. Treatment needs to be initiated within 6 hours of symptom onset. Research evidence related to acute stroke management is as follows:
1. Specialized stroke rehabilitation appears to be more effective than conventional care at reducing death and dependency and length of hospital stay.23
2. Aspirin has been shown to effectively reduce death and dependency at 6 months when given within 48 hours of ischemic stroke as are systemic anticoagulants (unfractionated heparin, low-molecular-weight heparin, heparinoids, oral anticoagulants, or specific thrombin inhibitors) but with a lower risk of intracranial and extracranial hemorrhage.2,24
3. Thrombolysis (i.e., tissue plaminogen activator [tPA]) given within 3 hours of onset of symptoms reduces dependency at 6 months when the diagnosis of ischemic stroke has been confirmed. Streptokinase has been found to increase the risk of intracranial hemorrhage and is not used to treat acute ischemic stroke.25
4. Acute blood pressure lowering in acute ischemic stroke may actually lead to increased cerebral ischemia.
5. Disappointingly, neuroprotective agents (calcium channel antagonists, citicoline, gamma-aminobutyric acid (GABA) agonists, glycine antagonists, magnesium, N-methyl-D-aspartate antagonists, tirilazid) have not been shown to significantly reduce the risk of poor outcome including death or improve outcomes with ischemic stroke.26
6. Surgical evacuation does not appear to be an effective treatment for supratentorial ICHs but may be indicated in a few specific situations.27
Research evidence preventing a recurrence is as follows:
1. Antiplatelet therapy effectively reduces the risk of recurrence. No evidence shows that alternate antiplatelet regimens to low-dose aspirin are any more or less effective than aspirin alone.2
2. Lowering blood pressure reduces the risk of serious vascular events regardless of stroke etiology.2
3. Anticoagulation does not appear to be of benefit in preventing recurrence in persons with a normal sinus rhythm and the risk for intracranial and extracranial hemorrhage is increased. Oral anticoagulants reduce the risk of initial or recurrent stroke in atrial fibrillation.28 Only 11% of persons with cardioembolic stroke had been receiving anticoagulation therapy29 as reported in one study, and women are less likely to have anticoagulation therapy for atrial fibrillation.30
4. Carotid endarterectomy effectively reduces the risk of recurrence in cases with greater than 50% carotid stenosis, is not effective with 30% to 49% carotid stenosis, and increases the risk of stroke with less than 30% stenosis. No benefit was found for persons with near occlusion.2
5. Cholesterol reduction using statins appears to reduce primary or recurrence risk regardless of baseline cholesterol level or coronary artery disease status. Other methods of cholesterol reduction do not lower recurrence risk.31
6. Sufficient evidence to judge the efficacy of carotid and vertebral percutaneous transluminal angioplasty to prevent recurrence is not yet available.
Rehabilitation is indicated in thrombotic and embolic stroke. Treatment of an intracranial bleed, regardless of cause, is focused on stopping or reducing the bleeding, controlling the increased ICP, preventing a rebleed, and preventing vasospasm. Occasionally an attempt is made to evacuate or aspirate the blood.
Intracranial Aneurysm: Intracranial aneurysms may result from arteriosclerosis, congenital abnormality, trauma, inflammation, or infection. Cocaine use has been linked to aneurysm formation. The size of the aneurysm may vary from 2 mm to 3 cm. Most aneurysms are located at bifurcations in or near the circle of Willis, in the vertebrobasilar arteries, or within the carotid system (see Figure 14-19)—85% to 95% are in the anterior portion of the circle of Willis. Aneurysms may be single, but in 20% to 25% of cases, more than one is present. In these instances the aneurysms may be unilateral or bilateral. The incidence of rupture is 11 in 100,000 per year. Peak incidence of rupture is from 50 to 60 years of age. Women have a slightly greater incidence of aneurysms.
PATHOPHYSIOLOGY No single pathologic mechanism exists. A combination of genetic, congenital, and acquired factors is present.32,33 Abnormalities in multiple layers of the blood vessel are found. The endothelial layer is thin, the internal elastic lamina is not present or fragmented, and the muscularis layer of the media ends at the aneurysm. Atherosclerotic changes are found. Aneurysm development is attributed to hemodynamic stress and is believed to be exacerbated by hypertension and certain connective tissue disorders in which there are abnormalities in the extracellular matrix.33 The aneurysm wall is composed of fibrous tissue. Aneurysms may be classified on the basis of shape and form (Figure 17-18).
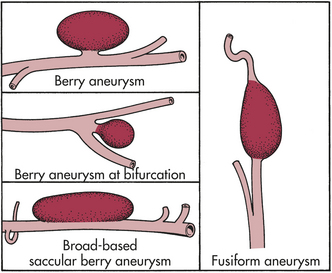
Saccular aneurysms (berry aneurysms) occur frequently (in approximately 2% of the population) and are the result of a combination of a congenital abnormality in the media of the arterial wall and degenerative changes.18 The sac grows over time. A saccular aneurysm may be (1) round with a narrow stalk connecting it to the parent artery (Figure 17-19), (2) broad based without a stalk, or (3) cylindric. Saccular aneurysms are rare in childhood; their highest incidence of rupturing or bleeding is among people 20 to 50 years of age.
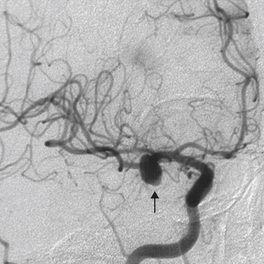
Figure 17-19 Berry aneurysm, angiogram. In this lateral view with contrast filling a portion of the cerebral arterial circulation can be seen a berry aneurysm (arrow) involving the middle cerebral artery of the circle of Willis at the base of the brain. (From Klatt EC: Robbins and Cotran atlas of pathology, Philadelphia, 2006, Saunders.)
Fusiform aneurysms (giant aneurysms), by definition greater than 25 mm in diameter, make up 5% of all intracranial aneurysms. They occur as a result of diffuse arteriosclerotic changes and are found most commonly in the basilar arteries or terminal portions of the internal carotid arteries. They act as space-occupying lesions. Mycotic aneurysms result from arteritis caused by bacterial emboli; these aneurysms are uncommon. Traumatic (dissecting) aneurysms are caused by a weakening of the arterial wall by a fracture line, by a penetrating missile, or after neurosurgical or imaging (e.g., angiographic) procedures.
What causes an aneurysm to rupture is not known33 but rupture causes hemorrhage into the subarachnoid space with rapid spread, producing localized changes in the cerebral cortex and focal irritation of nerves and arteries (see Laplace law, Chapter 29). Because of compression, bleeding ceases with the formation of a fibrin-platelet plug at the point of rupture. Blood undergoes reabsorption through arachnoid villi within 3 weeks.
CLINICAL MANIFESTATIONS Aneurysms are frequently asymptomatic. In routine autopsy, 5% of persons are found to have one or more intracranial aneurysms. Clinical manifestations may arise from cranial nerve compression, but the signs vary, depending on the location and size of the aneurysm. Most often, cranial nerves III, IV, V, and VI are affected (see Table 14-6). Unfortunately the most common first indication of an aneurysm is an acute subarachnoid hemorrhage, intracerebral hemorrhage, or combined subarachnoid-intracerebral hemorrhage (see pages 601 and 607).
EVALUATION AND TREATMENT Diagnosis before a bleeding episode is made using arteriographic examination. After a subarachnoid or an intracerebral hemorrhage, a tentative diagnosis of an aneurysm that has bled is based on clinical manifestations, history, CT, and MRI. The treatment of choice for an aneurysm is surgical management. The location and size of the aneurysm and the person’s clinical status determine whether invasive therapy is feasible.34
Vascular Malformations
Vascular formations are one tenth as common as aneurysms.18 Four types of vascular malformation exist: arteriovenous malformation, cavernous angioma, capillary telangiectasis, and venous angioma. Most are sporadic, although multiple lesions are observed in families.35 Cavernous angiomas (malformations) are sinusoidal collections of blood vessels without interspersed normal brain tissue. They rarely hemorrhage and comprise 8% to 15% of all vascular lesions. A capillary telangiectasis is dilated capillaries with interspersed normal brain tissue found deep in the brain, particularly in the brainstem; hemorrhage is rare. These vascular malformations are associated with Rendu-Osler-Weber disease. Venous angioma, the most common vascular malformation found at autopsy (3% of cases), is considered a subset of developmental venous anomalies that occur secondary to arrested development. The result is primitive embryologic veins in a radial pattern feeding a central vein. These rarely hemorrhage.36
In an arteriovenous malformation (AVM), arteries feed directly into veins through a vascular tangle of malformed vessels (Figure 17-20). AVMs hemorrhage at a rate of 40% a year, occur in any part of the brain, and are usually cone shaped. Their size is highly variable, from malformations of a few millimeters to large ones that extend from the cortex to the ventricle. The large AVMs also may involve the dura mater, including the falx cerebri and the tentorium cerebelli. AVMs occur as frequently in males as in females, and occasionally are found in families. Although usually present at birth, AVMs exhibit a delayed age of onset and symptoms most commonly occur before 30 years of age. They usually rupture in the second and third decades of life.
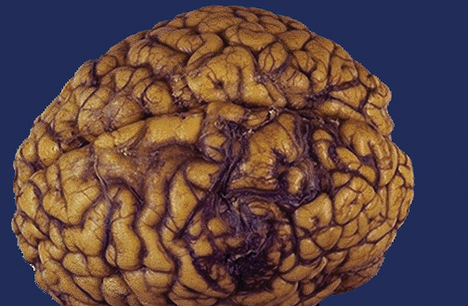
Figure 17-20 Vascular malformation, gross. A vascular malformation represented by a mass of irregular tortuous vessels over the left posterior parietal region of the brain. (From Klatt EC: Robbins and Cotran atlas of pathology, Philadelphia, 2006, Saunders.)
PATHOPHYSIOLOGY AVMs, which are developmental abnormalities that represent persistence of embryonic patterns of blood vessels, do not have a normal blood vessel structure and are abnormally thin. The involved vessels are thought by some to enlarge over time. The AVM may be fed by one or several arteries. These feeder vessels become tortuous over time and often are dilated. With moderate to large AVMs, sufficient blood is shunted into the malformation to deprive surrounding tissue of adequate blood perfusion.
CLINICAL MANIFESTATIONS Clinical manifestations vary: 20% of persons with an AVM have a characteristic chronic nondescript headache, although some experience migraine; 50% experience seizure disorders caused by compression. Initially, the seizures tend to be focal or jacksonian; generalization often occurs over time. (Seizures are discussed in Chapter 16.) The other 50% suffer an intracerebral, a subarachnoid, or a subdural hemorrhage. Bleeding from an AVM into the subarachnoid space causes clinical manifestations identical to those associated with a ruptured aneurysm. If bleeding is into the brain tissue, focal signs that develop resemble a stroke-in-evolution. Ten percent of persons experience hemiparesis or other focal signs. Hemiparesis usually is caused by compression or rupture. At times, noncommunicating hydrocephalus (see Chapter 16) develops with a large AVM that extends into the ventricle lining. AVMs account for up to 1% of all sudden deaths.37
EVALUATION AND TREATMENT A systolic bruit over the carotid in the neck, the mastoid process, or (in a young person) the eyeball is almost diagnostic of an AVM. CT, magnetic resonance angiography (MRA), transcranial Doppler (TCD), and MRI are used in initial diagnosis, followed by an arteriogram to identify feeding vessels. Treatment options are direct surgical approach, embolization, or radiotherapy. The risk of bleeding is 6% in the first year and 2% to 4% each year thereafter with no intervention.
Subarachnoid Hemorrhage
With a subarachnoid hemorrhage (SAH), blood escapes from a defective or injured vasculature into the subarachnoid space (Figure 17-21). First-degree relatives of persons with SAH have seven times the risk of developing an SAH.33 Heavy alcohol use, hypertension, smoking, anticoagulation, and oral contraceptive use are associated with SAH. Individuals at risk for a SAH are those with a saccular intracranial aneurysm (80% of cases), intracranial AVM, or hypertension and those who have sustained head injuries. There is a 50% overall mortality rate, and one third of survivors are dependent. SAHs often recur, especially from a ruptured intracranial aneurysm.
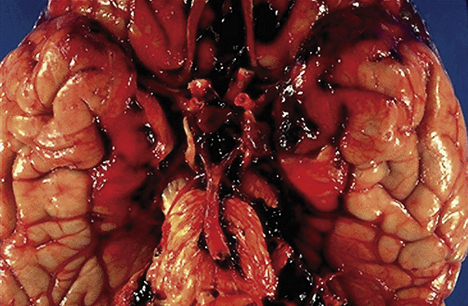
Figure 17-21 Subarachnoid hemorrhage, gross. Subarachnoid hemorrhage resulting from rupture of a berry aneurysm. (From Klatt EC: Robbins and Cotran atlas of pathology, Philadelphia, 2006, Saunders.)
PATHOPHYSIOLOGY When a vessel is leaking, blood oozes into the subarachnoid space. When a vessel tears, blood under pressure is pumped into the subarachnoid space. The blood is extremely irritating to the meningeal and other neural tissues and so produces an inflammatory reaction in these tissues. Additionally, the blood coats nerve roots, clogs arachnoid granulations (impairing CSF reabsorption), and clogs foramina within the ventricular system (impairing CSF circulation). ICP immediately increases to almost diastolic levels. ICP returns to near baseline in about 10 minutes. Cerebral blood flow and cerebral perfusion pressure (CPP) decrease. The expanding hematoma acts like a space-occupying lesion, compressing and displacing brain tissue. Granulation tissue is formed, and scarring of the meninges with resulting impairment of CSF reabsorption and secondary hydrocephalus often results.
Subarachnoid hemorrhage has a unique pathophysiologic cascade triggered by the sudden appearance of blood in the subarachnoid space. Eighty percent of persons with SAH have infarction on MRI, which is the major cause of death and disability and 33% of persons have asymptomatic vasospasm in the first 2 weeks following an SAH.33 The cause for cerebral vasospasm (CVS) and delayed cerebral ischemia (DCI) is unclear. DCI is related to but not explained by the CVS.
Development of CVS requires the presence of blood and its breakdown products as a consequence of the SAH. It also appears that oxyhemoglobin is a powerful precipitator of CVS. It scavenges nitric oxide (a vasodilator), and its breakdown triggers a free radical cascade that disrupts multiple blood vessel layers and initiates release of inflammatory factors.38 It appears that both CVS and disruption of cerebral autoregulation are necessary for DCI to occur. Autoregulation may be impaired in SAH and the combination of narrowed blood vessles, because of spasm and disrupted autoregulation, impairs perfusion, resulting in ischemia.33
CLINICAL MANIFESTATIONS Early manifestations associated with leaking vessels are episodic headache, transient changes in mental status or level of consciousness, nausea or vomiting, focal neurologic defects including visual or speech disturbances, cranial nerve palsies, or stiff neck. A ruptured vessel often is accompanied by a sudden throbbing, “explosive” headache that is associated with nausea and vomiting, visual disturbances, motor deficits, and loss of consciousness. These signs can be related to a dramatic rise in ICP. Meningeal irritation and inflammation often occur, causing neck stiffness (nuchal rigidity), photophobia, blurred vision, irritability, restlessness, and low-grade fever. A positive Kernig sign (in which straightening the knee with the hip and knee in a flexed position produces pain in the back and neck regions) and Brudzinski sign (in which passive flexion of the neck produces neck pain and increased rigidity) may appear. No localizing signs are present if the bleed is confined completely to the subarachnoid space.
The Hunt and Hess SAH grading system is based on description of the clinical manifestations (Table 17-7). Rebleeding is a significant risk with a high mortality (up to 70%). The period of greatest risk is the first month, with the peak incidence of rebleeding during the first 2 weeks after the initial bleed. Rebleeding is manifested by a sudden increase in blood pressure and ICP, along with a deteriorating neurologic status.
Table 17-7
Subarachnoid Hemorrhage Classification Scale
| Category | Description |
| Grade I | Neurologic status intact; mild headache, slight nuchal rigidity |
| Grade II | Neurologic deficit evidenced by cranial nerve involvement; moderate to severe headache with more pronounced meningeal signs (e.g., photophobia, nuchal rigidity) |
| Grade III | Drowsiness and confusion with or without focal neurologic deficits; pronounced meningeal signs |
| Grade IV | Stuporous with pronounced neurologic deficits (e.g., hemiparesis, dysphasia); nuchal rigidity |
| Grade V | Deep coma state with decerebrate posturing and other brainstem dysfunction |
From Cook HA: Aneurysmal subarachnoid hemorrhage: neurosurgical frontiers and nursing challenges. In Winkleman C, editor: AACN clinical issues in critical care nursing, Philadelphia, 1991, Lippincott.
The peak time of CVS onset is 3 to 5 days, with maximal narrowing at 5 to 14 days after the initial bleed, but vasospasm may persist for 2 to 4 weeks. Seizures occur in 25% of SAHs. The incidence of hydrocephalus after a bleed is 20%. Hypothalamic dysfunction, manifested by salt wasting, hyponatremia, and ECG changes, is common.
EVALUATION AND TREATMENT The diagnosis of a subarachnoid hemorrhage is based on the clinical presentation, a noncontrast CT scan, and a lumbar puncture.39 Arteriographic examination is the definitive diagnostic measure for defining and localizing an aneurysm or AVM. Treatment is directed at control of intracranial pressure, prevention of ischemia and hypoxia of neural tissues, and prevention of rebleeding episodes. Antifibrinolytic drugs may be used to stop rebleeding in selected cases. Blood pressure is allowed to remain in the high normal range or is elevated to that level. Platinum coils and balloon embolization to occlude the aneurysm are used, but microsurgical repair remains the treatment of choice. Calcium channel blockers, such as nimodipine, are used to prevent or reverse vasospasm. Volume expansion or hemodilution through continuous or bolus administration of hetastarch and plasma protein factors to maintain a hematocrit of 33% is used to expand blood volume and augment cerebral perfusion. Cerebral angioplasty can be tried for vasospasm. The primary problem must be diagnosed and corrected as well.
Headache
Headache is a common neurologic disorder and is usually a benign symptom. However, it can be associated with serious disease, such as brain tumor, meningitis, and giant cell arteritis. The headache syndromes discussed here are the chronic, recurring type not associated with structural abnormalities or systemic disease and include migraine, cluster, paroxysmal hemicrania, and tension headaches. Characteristics of the major types of headache syndromes are summarized in Table 17-8.
Migraine
Migraine is now viewed as a familial, episodic disorder whose marker is headache40 and is defined as repeated, episodic headache lasting 4 to 72 hours. It is diagnosed when any two of the following features occur: unilateral head pain, worsening with movement, accompanied by photophobia or phonophobia; and presence of any one of the following: throbbing quality, moderate to severe nausea, or vomiting.41 At least 12% of the general population experience an average of 18 migraine attacks per year,41 two thirds of whom are female. Migraine is more common in those 25 to 55 years of age, and can occur in young children. The prevalence in women is highest at 20 to 40 years of age but remains higher than in men into older age. Onset after 50 years of age is rare. Hormonal factors account for most of the gender differences. A positive family history is common as is a genetic predisposition. Migraine is a multifactorial disorder caused by a combination of multiple genetic and environmental factors. Migraine sufferers have an increased risk for epilepsy, depression, anxiety disorders, and stroke.40 Triggers believed to precipitate migraine attacks include altered sleep patterns (becoming tired or too much sleep), skipping meals, overexertion, weather change, stress or relaxation from stress, hormonal changes (such as menstrual periods), excess afferent stimulation (bright lights, strong smells), and chemicals (alcohol or nitrates).41
The International Headache Society has broadly classified migraine with aura (previously called classic migraine) and migraine without aura (previously called common migraine).41 In migraine with aura, at least some of the attacks are temporarily associated with distinct aura symptoms suggestive of focal brain dysfunction (flashing lights, visual loss). There are no associated focal neurologic symptoms in migraine without aura. Two thirds of those with migraines have migraine without aura.
The pathophysiologic basis for migraine is complex and includes neurologic, vascular, hormonal, and neurotransmitter components. The end point is hypothesized to be a disturbance of subcortical sensory modulation systems. The clinical phase of a migraine attack and associated pathophysiology follow:
1. Premonitory phase: up to one third have premonitory symptoms at least some of the time for several hours before aura or headache onset; the pathogenesis is unknown but evidence points to dopaminergic/hypothalamic involvement42
2. Migraine aura: up to one third have aura symptoms at least some of the time that may last 1 hour or sometimes much longer; migraine aura is defined as a spreading, focal, neurologic disturbance manifested as visual, sensory, or motor symptoms; pathophysiology appears to be a cortical spreading depression (CSD), reduction in electrical activity, and decrease in blood flow that slowly spreads across the cerebral cortex from the occipital region43
3. Headache phase: includes associated symptoms and may last from 4 to 72 hours (usually about a day); most research-supported pain mechanism is compensatory overactivity in the trigeminovascular system of the brain; activation of trigeminal sensory nerves produces release of vasoactive peptides that cause a sterile, inflammatory response around vessels in the meninges are present44
Migraine without aura is often located on one side. The pain is throbbing, of moderate to severe intensity, and aggravated by physical activity. In migraine with aura, the most common prodromal symptoms are visual (scotomas with luminous angles and scintillating edges, and hemianopsia). Sensory deficits and aphasia also may be present. The aura develops within 5 to 20 minutes and remits within 60 minutes, followed by headache and other symptoms, including nausea, vomiting, photophobia, scalp tenderness; 10% experience diarrhea.
In susceptible women, migraine occurs most frequently before and during menstruation and is decreased during pregnancy and menopause. The cyclic withdrawal of estrogens may trigger attacks of migraine.45 Cyclic changes in estrogen are absent in pregnancy and after menopause, which could explain the less frequent attacks in some women. Estrogens may act directly on vascular smooth muscle, modulate activity of vasoactive substances at the neurovascular junction, and activate vasoregulatory responses in the hypothalamus. However, no direct evidence has been found to link circulating female sex hormones with the frequency and severity of migraine.
The diagnosis of migraine is made from medical history and physical examination. Clinicians must be skilled in their understanding of different types of headaches, risk factors, family history, and clinical features. Differential diagnosis is confirmed with CT, MRI, and EEG. A significant number of individuals with migraine have depression as a comorbidity.
The management of migraine includes education that migraine is a chronic physiologic, not psychosomatic, disorder. Avoidance of triggers, adequate sleep, regular eating habits, and daily relaxation and meditation can create a headache-protective environment. With the onset of acute migraine, a dark room, ice, and sleep can provide relief. The pharmacologic management of migraine varies with each individual and is related to the severity of the attack. Drug considerations should include antiemetics, NSAIDs, ergotamine and dihydroergotamine, and serotonin receptor agonists (e.g., sumatriptan). Triptans, transcutaneous estrogen, and magnesium administration may help some women with menstrual migraine. Gastric absorption may be decreased during an attack, and routes of administration other than oral (e.g., nasal sprays, intravenous, and rectal) may be used.
The prophylaxis of migraine is considered when attacks cannot be treated effectively. Several drugs may be considered and should not be used in combination. Examples include beta-blockers, a calcium antagonist (flunarizine), serotonin antagonists (lisuride, methysergide), NSAIDs, dihydroergotamine (DHE), valproic acid, and amitriptyline.
Cluster Headache
Cluster headaches are one of a group of disorders referred to as trigeminal autonomic cephalagia44 and occur primarily in men between 20 and 50 years of age. Cluster headache has been known also as histamine cephalalgia, Horton syndrome, and erythromelalgia. These headaches are known as cluster headaches because several attacks can occur during the day for a period of days followed by a long period of spontaneous remission. Cluster headache has an episodic and a chronic form.
The headache attack usually begins without warning and is characterized by severe, unilateral tearing, burning, periorbital, and retrobulbar or temporal pain lasting 30 minutes to 2 hours. One or several attacks may occur in a day, usually at the same time of day or night. The same side is affected in subsequent episodes, and the attack activates the trigeminal-autonomic reflex. Associated symptoms include lacrimation, reddening of the eye, nasal stuffiness, eyelid ptosis, and nausea. Pain often is referred to the midface and teeth. If the cluster of attacks occurs more frequently without sustained spontaneous remission, they are classified as chronic cluster headaches (20% of cases). Alcohol can stimulate an attack during a cluster headache in about 50% to 70% of cases, but it is not a triggering factor during remission.
The cause of trigeminal activation is unclear. The pathogenic mechanism for pain like migraine is probably release of vasoactive peptides and the formation of neurogenic inflammation. Autonomic dysfunction is characterized by sympathetic underactivity and parasympathetic activation.44 The rhythmicity of attacks is associated with changes in the inferior posterior hypothalamus. There may be altered serotonergic nerve transmission but at different loci than in migraine headache.
Prophylactic drugs are used to treat cluster headache. The most effective are prednisone, lithium, methysergide, calcium channel antagonists, and valproate. Acute attacks are managed with oxygen inhalation, sumatriptan, and inhaled ergotamine.
Chronic Paroxysmal Hemicrania: Chronic paroxysmal hemicrania (CPH) is a cluster-type headache that occurs with more daily frequency (4 to 12 times per day) but with shorter duration (20 to 120 minutes). The remission phases are often shorter. The attacks are more common in women, usually after pregnancy. The symptoms are similar to cluster headache. As with cluster headache, there is an episodic and a chronic form. The pathophysiology involves a disorder of sympathetic hyperactivity, but the mechanism is different from cluster headache because there is effective relief of symptoms with indomethacin.
Tension-Type Headache: Tension-type headache is the most common type of headache, occurring in 69% of men and 88% of women. The average age of onset is during the second decade of life. Female/male ratio is 1:1. It is a mild to moderate bilateral headache with a sensation of a tight band or pressure around the head. The onset of pain is usually gradual. The headache occurs in episodes and may last for several hours or several days. It is not aggravated by physical activity. Chronic tension-type headache (CTTH) evolves from episodic tension-type headache and represents headache that occurs at least 15 days per month for at least 3 months.41 Many individuals have both tension-type and migraine headaches.
Both a central mechanism and a peripheral mechanism operate in causing tension headache. The central mechanism probably involves hypersensitivity of pain fibers from the trigeminal nerve. The peripheral mechanism is probably related to contraction of jaw and neck muscles, but the exact mechanisms are unknown. Headache sufferers have more localized pain and tenderness of pericranial muscles.
Mild headaches are treated with ice, and more severe forms are treated with aspirin or NSAIDs. Chronic tension-type headaches are best managed with a tricyclic antidepressant, such as amitriptyline. Amitriptyline and mirtazapine are similarly effective for CTTH in decreasing the duration and frequency of the headache, however, amitriptyline has been found to have fewer adverse effects.2 Cognitive behavioral therapy reduces symptoms of CTTH.2 Naproxen is a second drug of choice. Long-term use of analgesics or other drugs, such as muscle relaxants, antihistamines, tranquilizers, caffeine, and ergot alkaloids, should be avoided.
Tumors of the Central Nervous System
The incidence of primary brain tumor has risen approximately 25% in the past two decades—a rise that may be attributable to better detection.46 No proven causative agents have been established for tumors of the central nervous system (CNS). Carcinogenesis is discussed in Chapter 11.
Cranial Tumors
Tumors within the cranium can be either primary or metastatic. Primary tumors are classified as primary intracerebral tumors or primary extracerebral tumors. Primary intracerebral tumors originate from brain substance, neuroglia, neurons, cells of the blood vessels, and connective tissue (Table 17-9). Primary extracerebral tumors originate outside the substance of the brain and include meningiomas, acoustic nerve tumors, and tumors of the pituitary and pineal glands. Metastatic tumors, or secondary tumors, can be found inside or outside the brain substance. Sites of intracranial tumors are illustrated in Figure 17-22.


CNS tumors include brain and spinal cord tumors. The incidence is 10 per 100,000, which seems to increase up to 70 years of age and then decreases. These tumors represent the second most common group of tumors in children. Approximately 70% of all intracranial tumors in children are located infratentorially, and in adults 70% to 75% are located supratentorially. Peripheral nerve tumors are rare in children and common in adults.
Cranial tumors cause local and generalized clinical manifestations. The local effects are caused by the destructive action of the tumor itself on a particular site in the brain and compression causing decreased cerebral blood flow. The effects are varied and include seizures, visual disturbances, unstable gait, and cranial nerve dysfunction. The generalized effects result from increased ICP (Figure 17-23). Increased ICP may occur because of obstruction of the ventricular system, hemorrhages occurring in and around the tumor, or cerebral edema caused by tumors.
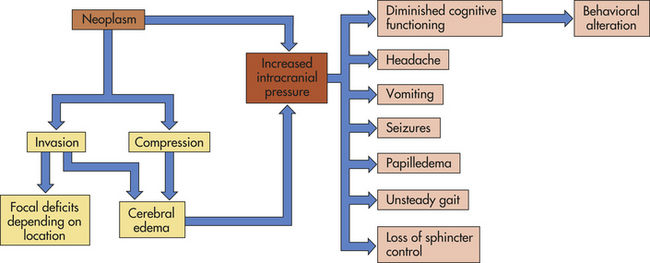
Intracranial brain tumors do not metastasize as readily as tumors in other organs because there are no lymphatic channels within the brain substance. If metastasis does occur, it is usually through seeding of cerebral blood, through CSF, during cranial surgery, or through artificial shunts.
Primary Brain (Intracerebral) Tumors: Primary brain (intracerebral) tumors, also called gliomas, comprise 50% to 60% of all adult brain tumors and include astrocytomas, oligodendrogliomas, mixed oligoastrocytomas, and ependymomas based on histologic and immunohistologic characteristics (Figure 17-24).47 The World Health Organization (WHO) divides gliomas into four grades based on histopathologic features of anaplasia-nuclear atypia, mitotic activity, microvascular proliferation, and/or necrosis.47 Pilocytic astrocytomas (WHO grade I), diffuse astrocytomas (WHO grade II), and oligodendrogliomas (WHO grade II) are classified as low-grade gliomas. Anaplastic astrocytomas (WHO grade III), anaplastic oligodendrogliomas (WHO grade III), and anaplastic oligoastrocytomas (WHO grade III), along with glioblastoma multiforme (WHO grade IV), are classified as high-grade gliomas.47 Glioblastomas are further subdivided into primary glioblastomas that start their development as grade IV gliomas, and secondary glioblastomas that arise from a low-grade precursor glioma through a sequence of genetic changes.47 Low-grade gliomas are found throughout the CNS. Although high-grade gliomas also can be found anywhere in the CNS, they have a cerebral hemispheric predominance.
Figure 17-24 Common glial neoplasms of the central nervous system (hematoxylin & eosin stain). A, Juvenile pilocytic astrocytoma. The left panel (MRI) shows a low-grade slow-growing astrocytic tumor often occurring in children below the tentorium (arrow). The right panel shows microcystic change and pilocytic cells with long, thin processes (arrow). B, Diffuse astrocytoma. The left panel shows a discrete mass that enhances brightly because of vascularity. The right panel shows a low-grade tumor with pleomorphic cells at the top center. C, Oligodendroglioma. The left panel (MRI) shows a well-circumscribed left temporal oligodendroglioma. The right panel shows neoplastic cells with round, relatively uniform nuclei and a fine capillary network. MRI, Magnetic resonance imaging. (From Klatt EC: Robbins and Cotran atlas of pathology, Philadelphia, 2006, Saunders.)
Primary brain tumors make up about 2% of all cancers in the United States. Pilocytic astrocytomas are more common in children and most often present with a median age of 12 years.48 All other gliomas generally present after age 30 with secondary glioblastomas occurring more frequently in younger adults and primary glioblastomas presenting in older adults.47
Etiology for primary brain tumors is unclear. Intracranial irradiation for therapeutic purposes increases the risk. Genetic alterations in three proliferation signaling pathways have been identified: the p53 apoptotic and cell cycle, the retinoblastoma cell cycle, and cell death signaling pathway. Alterations also occur in growth factor signaling.49
It had been thought that gliomas originate from loss of differentiation of a mature brain cell in response to genetic alterations. The thinking now is that gliomas may arise from transformation of resident brain tumor stem cells.47,50
The principal treatment for cerebral tumors is surgical or radiosurgical excision or surgical decompression if total excision is not possible. Chemotherapy, radiation therapy, and hyperthermia also may be used. Supportive treatment is directed at reducing edema. (Cancer treatment is discussed in Chapter 12.)
Astrocytoma: Astrocytomas are the most common primary CNS tumors (50% of all brain and spinal cord tumors). Astrocytomas develop from astrocytes and grow by expansion and infiltration into the normal surrounding brain tissues. These tumor cells are believed to have lost normal growth restraint, and thus they proliferate uncontrollably.
One third of astrocytomas are classified at diagnosis as grade I or grade II. These slow-growing but infiltrative gliomas tend to form cavities (pseudocysts); however, some are firm, noncavitating, avascular, gray-white masses that are difficult to distinguish from normal white matter of the brain. Although these tumors may occur anywhere in the brain or spinal cord, they are located most commonly in the cerebrum, hypothalamus, or pons. Low-grade astrocytomas in adults tend to have a lateral or supratentorial location, and they tend to be midline or near midline in position in children, often in the posterior fossa.
Headache and subtle neurobehavioral changes may be an early symptom. Approximately half of persons with low-grade astrocytomas experience a focal or generalized seizure. Onset of a focal seizure disorder between the second and sixth decades of life is suggestive of an astrocytoma. Other general or focal neurologic manifestations develop gradually. Increased ICP is usually a late clinical manifestation.
Grade I astrocytomas are treated with surgery and follow-up CT scans. Grade II astrocytomas are treated surgically if they are accessible or by conventional external radiation, local radiation, or stereotactic radiosurgery. Following surgery alone, the 5-year survival rate is 25%; with surgery followed by radiotherapy, the 5-year survival rate is 50%.
Grades III and IV astrocytomas are found predominantly in the frontal lobes and cerebral hemispheres (Figures 17-25 and 17-26). These tumors also may be located in the brainstem (Figure 17-27), cerebellum, and spinal cord. They are found twice as frequently in men as in women. Grades III and IV astrocytomas are the third most common cancer in the 15- to 34-year-old age group and the fourth most common in the 35- to 54-year-old age group.46 Grades III and IV astrocytomas are often large and well circumscribed with a variegated pattern. The peripheral rim is pinkish gray and solid with a soft, yellow necrotic center and points of hemorrhage. Microscopically, there is increased cellularity, vascular proliferation, cellular pleomorphism, and necrosis. Necrosis is the main histologic difference between an anaplastic grade III tumor and a grade IV glioblastoma multiforme.
Figure 17-25 Well-differentiated infiltrating astrocytoma. The right temporal lobe contains an infiltrative, homogeneous lesion that has expanded the lobe and obscured the normal boundaries between gray and white matter (compare to left temporal lobe). Because of the ill-defined borders, surgical resection seldomly removes all of the tumor in such cases. (From Kumar V, Cotran RS, Robbins SL: Robbins basic pathology, ed 7, Philadelphia, 2003, Saunders.
Figure 17-26 Glioblastoma multiforme. In contrast to the well-differentiated infiltrating astrocytoma in Figure 17-25, this glioblastoma contains irregular areas of discoloration and cystic change, reflecting the presence of necrosis and hemorrhage. These lesions are widely infiltrative and associated with considerable mass effect. Note the shift of midline structures to the right. (From Kumar V, Cotran RS, Robbins SL: Robbins basic pathology, ed 7, Philadelphia, 2003, Saunders.)
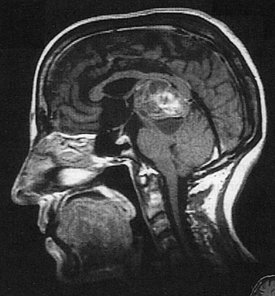
Figure 17-27 Magnetic resonance imaging (MRI) with gadolinium showing a high brainstem glioma. (From Perkin DG: Mosby’s color atlas and text of neurology, London, 1998, Mosby-Wolfe.)
Grade IV glioblastoma multiformes are highly vascular and extensively infiltrative. They may become large enough to extend from the meningeal surface through the ventricular wall. Fifty percent of glioblastomas are bilateral or at least occupy more than one lobe at the time of death. There are reports of grade IV astrocytomas found outside the central nervous system.51
The typical clinical presentation for a glioblastoma multiforme is that of diffuse, nonspecific clinical manifestations, such as headache, irritability, and personality changes, that progress to more clear-cut manifestations of increased ICP, such as headache on position change; papilledema; or vomiting. Of those affected, 30% to 40% experience seizure activity. Symptoms may progress to definite focal signs, such as hemiparesis, dysphasia, dyspraxia, cranial nerve palsies, and visual field deficits, in addition to the generalized signs from increased ICP.
Diagnosis of high-grade astrocytomas most commonly takes 3 to 6 months from onset of the first clinical manifestations because the person does not recognize the need to consult a healthcare provider.
Grade III astrocytomas are treated with surgery if they are accessible; radiotherapy; and chemotherapy possibly before, during, and after other therapies. Chemotherapy is given in cycles. With treatment, 1-year survival for grade III astrocytomas is 55% to 60%, 30% to 35% survive 2 years, and 10% survive longer than 5 years. Grade IV gliomas are also treated with surgery if accessible, radiotherapy and chemotherapy, or placement of wafers. The median survival rate is 3 years.46
Oligodendroglioma: A far less commonly occurring glioma is oligodendroglioma, comprising 2% of all brain tumors and 10% to 15% of all gliomas. Oligodendrogliomas are typically slow-growing well-differentiated tumors, often with cysts and calcification present. Most are macroscopically indistinguishable from other gliomas. They occur most often from 30 to 50 years of age and are more common in males than females. Their etiology is unknown. Most oligodendrogliomas are in the frontal and temporal lobes, often in deep white matter; 20% are in both hemispheres. They may be found also in other parts of the cerebrum, third ventricle, brainstem, cerebellum, and spinal cord. A high incidence of this tumor occurs in young adults with a history of temporal lobe epilepsy. Approximately half of these tumors generally classified as oligodendrogliomas (a grade II tumor) are actually oligoastrocytomas (a grade III tumor).52 Malignant degeneration occurs in approximately one third of persons with oligodendrogliomas. If there is extension to the pia mater or ependymal wall (see Figure 14-15), oligodendrogliomas may metastasize to distant CNS sites through the ventriculoarachnoid spaces.
More than 50% of individuals experience a focal or generalized seizure as the first clinical manifestation; approximately half have experienced increased ICP at the time of diagnosis and surgery, and only one third develop any focal manifestations. The time from first clinical manifestation to surgical intervention often ranges from 2 to 6 years. Treatment options are surgery; radiotherapy (conventional external beam or stereotactic gamma knife and converged beam); and chemotherapy before, during, and after radiation. Median survival, when surgery and radiotherapy are both used, is 5 to 10 years.46
Ependymoma: Ependymomas are gliomas that arise from ependymal cells that form the walls of the ventricles and grow either into the ventricle or into adjacent brain tissue; they are not encapsulated (Figure 17-28 and see Table 17-9). They comprise 6% of all primary brain tumors in adults and 10% in children and adolescents. Among children and adolescents, 50% of those affected are younger than 5 years of age. Seventy percent of ependymomas occur in the fourth ventricle (i.e., in the posterior fossa) and manifest as difficulty with balance, unsteady gait, uncoordinated muscle movement, and difficulty with fine motor skills. Other common sites for ependymomas are the third ventricle, lateral ventricles, and caudal portion of the spinal cord. The clinical presentation of a lateral and third ventricle ependymoma that involves the cerebral hemispheres is seizures, visual changes, and contralateral weakness of a body part on one side of the body. Approximately 40% of infratentorial ependymomas occur in children younger than 10 years. Occurrence of cerebral (supratentorial) ependymomas is distributed among all ages but more common in adults. Etiology for these tumors is unknown.
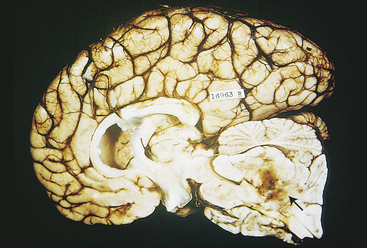
Figure 17-28 Ependymoma. These tumors may arise in both the intracranial compartment and the spine. Intracranial tumors typically originate from a ventricular surface, as in the case of this large lesion arising in the fourth ventricle (arrow). (From Kumar V, Cotran RS, Robbins SL: Robbins basic pathology, ed 7, Philadelphia, 2003, Saunders)
Blockage of the CSF pathway by the tumor clinically results in the presence of headache, nausea, and vomiting related to the hydrocephalus produced. Brainstem or upper spinal cord ependymomas may cause neck pain as well.
Clinical manifestations and progression of dysfunction associated with ependymomas may follow a short or long course. The interval between first manifestations and surgery may be as short as 4 weeks with some ependymoblastomas to as long as 7 to 8 years with others.
Ependymomas are treated surgically and with radiotherapy of the tumor region and operative site (possibly of the entire brain and spine); stereotactic radiosurgery focused on eradication; and chemotherapy. The 5-year survival rate is between 20% and 50%. Some persons benefit from a shunting procedure when the ependymoma has caused a noncommunicating hydrocephalus (see Chapter 16).
Meningioma: Meningioma constitutes about 20% of all intracranial tumors. The annual incidence is 6 cases per 100,000, with a peak incidence in the sixth and seventh decades, and is more common in women.53 Predisposing factors to developing a meningioma are having neurofibromatosis (NF) type 2 (NF2, see p. 616) and ionizing radiation after a several-decade latency period.54 Genetically, formation of benign meningiomas has been linked to NF2 gene mutation and chromosome 22q loss along with DAL-1 loss on chromosome 18. Atypical and anaplastic meningiomas have been linked to additional gene alterations involving multiple other chromosomes.55
A meningioma is a sharply circumscribed mass that derives its shape from the space it occupies; the cause is unknown. These slow-growing, often encapsulated tumors arise from arachnoidal (meningeal) cap cells in the dural coverings of the brain.56 Rarely do meningiomas arise from arachnoid cells of the choroid plexus of the ventricles. Most are attached to the dura mater and arise within the intracranial cavity, the spinal cavity, or, rarely, the orbit (Figure 17-29).56 A meningioma may extend to the dural surface and erode the cranial bones or produce an osteoblastic reaction. Small meningiomas (less than 2 cm in diameter) are often found on postmortem examination in middle-aged and older adults who had experienced no clinical manifestations and died of totally unrelated causes. A few meningiomas exhibit malignant, invasive qualities.
Figure 17-29 Meningioma, gross. Meningioma beneath the dura with compression of underlying cerebral hemisphere. (From Klatt EC: Robbins and Cotran atlas of pathology, Philadelphia, 2006, Saunders.)
Only when meningiomas reach a certain size—at which time they begin to indent the brain parenchyma—do they begin to produce clinical manifestations. Focal seizures are frequently the first manifestation. Other clinical manifestations depend on the tumor’s location. Clinical features based on site of origin are as follows:
1. Sphenoidal wing: ophthalmoplegia, mild proptosis, and involvement of the ophthalmic division of the trigeminal nerve
2. Olfactory groove: anosomia, personality change, and visual failure
3. Parasagittal: focal seizures of a focal motor or sensory deficit
4. Parasellar: evidence of chiasmatic compression; urinary incontinence; dementia; gradual paraparesis, hormonal failure; optic atrophy; bitemporal hemianopia
5. Lateral convexity: variable depending on structures compressed, including slow hemiparesis, speech abnormalities46
Because of the extremely slow-growing nature of most meningiomas, increased ICP is less common than with gliomas.
The most common symptom is seizures (40%). Diagnosis is made using contrast-enhanced CT, MRI, or both. The primary treatment is surgical resection. Sterotactic radiotherapy is used with incomplete resection or recurrence (20% rate). Conventional radiotherapy also is used. Hydroxyurea has been used in recurrence.57
Nerve Sheath Tumors: Nerve sheath tumors are either neurofibroma or schwannoma (neuroma, neurolemma). NF is an inherited autosomal dominant disorder accounting for 5% of all neuromas and are divided into two types: NF1 and NF2, which are clinically and genetically distinct disorders. The gene products are neurofibromin and merlin (schwannomin), both of which are thought to be tumor suppressors.58 Alterations in chromosomes 17 (17q11.2 for NF1) and 22 (NF2) are associated with both neurofibromas and schwannomas.59 NF1 is associated with cutaneous manifestations, iris hamartomas, and tumors primarily involving the peripheral nervous system and, occasionally, the CNS.60 NF2 is associated with cataracts, hearing loss and tumors primarily in the CNS, most commonly vestibular schwannoma.60 Criteria for the diagnosis of neurofibromatosis types 1 and 2 are presented in Box 17-2. The remainder of neuromas is benign tumors that arise from the sheath of Schwann cells surrounding the axons of the cranial nerves. The tumors most commonly affect people older than 50 years, women more often than men. The vestibular division of cranial nerve VIII is most commonly affected, although neuroma of the acoustic division of cranial nerves VIII, V, VII, and IX are found (Figure 17-30).
Figure 17-30 Acoustic neuroma (schwannoma), gross. A mass lesion arising in the right vestibular branch of the eighth cranial nerve at the cerebellopontine angle (arrow). (From Klatt EC: Robbins and Cotran atlas of pathology, Philadelphia, 2006, Saunders.)
The tumor originates most commonly just distal to the junction between the nerve root and the brainstem. As the tumor grows, it extends into the posterior fossa to occupy the cerebropontine angle and compress adjacent nerves. Eventually the brainstem is displaced, and the CSF flow is obstructed.
Initial clinical manifestations may include headache, tinnitus, hearing loss, impaired balance, unsteady gait, facial pain, and loss of facial sensations. Later, vertigo with nausea and vomiting, a sense of pressure in the ear, and moderate to severe unsteadiness with rapid position changes may appear. CT or MRI can establish the diagnosis. Posterior fossa dye studies may be required. Treatment is by surgical excision and radiotherapy of the neuroma. Pituitary tumors are discussed in Chapter 21, and cerebral tumors in children are discussed in Chapter 19.
Brain Metastases: Brain metastases are approximately 10 times more common than primary brain tumors. The incidence may be as high as 200,000 cases per year. At autopsy, 20% to 40% of persons with metastases have brain metastases. Lung and breast are the most common tumors to have brain metastases within 1 to 3 years, but renal cell carcinoma, and malignant melanomas have an even higher brain metastasis incidence.61 Carcinoma of the gallbladder, liver, thyroid, testes, uterus, ovary, and pancreas also may metastasize to the brain. Other tumors, besides carcinomas, that metastasize only occasionally are rhabdomyosarcomas, Ewing tumors, chorioepithelioma, and lymphoma.
Metastasis of a cancer to the brain parenchyma or the meninges is a late occurrence in the disease process. Metastasis to the brain parenchymna is believed to be hematogenous in origin.62 Two thirds of metastatic tumors are located within the brain and one third are located in extradural spaces. The cerebral hemispheres are the site of 75% of metastases, most predominantly in the frontal lobes followed by the parietal, occipital, and temporal lobes in order of frequency of location. Tumors of the pelvis or retroperitoneal space have a predilection to metastasize to the cerebellum, pons, or their coverings.63 In more than three fourths of persons with metastasis, the metastases are multiple and found in both the cerebrum and cerebellum in a scattered distribution. The metastatic tumors often are located in the meninges and near the brain surface in the gray matter and subcortical white matter. These tumors produce little glial cell reaction in the brain tissue but do cause vasogenic, peritumoral edema in the surrounding brain tissue due to blood-brain barrier incompetence.62
The brain metastatic process requires a series of sequential events, called the “metastatic cascade,” as follows:
1. Invasion of primary tumor border
2. Extravasation of the circulatory system
3. Survival and persistence/quiescence in the circulation
4. Extravasation at the distant CNS site
5. Formation of micrometastasis
6. Progressive colonization and growth61
Hematogenesis metastasis to the CNS is an inherently inefficient process depending on an interaction between tumor cells with host defenses and the microenvironment. To produce brain metastasis, tumor cells must reach the brain vasculature by attaching themselves to the endothelial cells of the brain microvessels in the blood-brain barrier, extravasate into the brain parenchyma, induce blood vessel development (angiogenesis), and proliferate in response to growth factors. Local brain invasion, in itself, is a process requiring mechanisms for cell motility, cell adhesion, and enzymatic remodeling of the extracellular components. Cytokines, chemokines, and growth factors have been demonstrated to participate in the process.64
The clinical manifestations of parenchymal brain metastasis are headache or alteration in cognition, mental status, and behavior,62 although several unusual syndromes do exist. Carcinomatous encephalopathy causes headache, nervousness, depressed mood, trembling, confusion, and forgetfulness. In carcinomatosis of the cerebellum, headache, dizziness, and ataxia are found. Carcinomatosis of the craniospinal meninges (carcinomatous meningitis) manifests with headache, confusion, and manifestations of cranial or spinal nerve root dysfunction.
Contrasted enhancing imaging is the most sensitive imaging procedure for metastatic brain tumors. Prognosis is poor. If one to three tumors are found, surgical excision is indicated. Radiotherapy is commonly used to treat solitary as well as multiple tumors. Whole brain radiation prior to stereotactic radiosurgery improves regional control.65 Chemotherapy is increasingly becoming part of the treatment plan.65
Spinal Cord Tumors
Spinal cord tumors are relatively rare. The most common primary spine tumors are listed in Box 17-3 and shown in Figure 17-31. Spinal cord tumors are named to reflect their cell type, growth rate, and structure of origin. They are classified as intramedullary tumors (originating within the neural tissues) or extramedullary tumors (originating from tissues outside the spinal cord). Extramedullary tumors arise from the meninges or roots (forming intradural tumors) or from epidural tissue or vertebral structure (forming extradural tumors). About 5% of spinal cord tumors seen in general hospital settings are intramedullary, 40% are intradural-extramedullary, and 55% are extradural.
Figure 17-31 Distribution of some spinal tumors. (From Perkin DG: Mosby’s color atlas and text of neurology, London, 1998, Mosby-Wolfe.)
The axial skeleton is the third most common site for metastasis behind lung and liver metastasis. Metastatic spinal cord tumors are three to four times more common than primary spinal cord tumors. They are usually carcinomas from breast, lung, and prostate; lymphomas; or myelomas; 25% to 70% involve the vertebral body and are asymptomatic. Metastatic spinal cord tumors are extradural in location. Of extradural tumors, 50% are metastatic and have spread to the spine through direct extension from tumors of the vertebral structures or from extraspinal sources extending through the interventricular foramen or through the bloodstream.
The most common primary extramedullary spinal cord tumors are neurofibromas and meningiomas. These tumors are intradural more often than extradural. Neurofibromas are found most commonly in the thoracic and lumbar regions. Meningiomas are more evenly distributed throughout the spine. Other extramedullary tumors in order of frequency of occurrence are sarcomas, vascular tumors, chordomas, and epidermoid and similar tumors. Of intradural-extramedullary tumors, 70% are meningiomas, neurofibromas, or sarcomas.
Intramedullary tumors have the same cellular origins as brain tumors. Ependymomas account for 40% of intramedullary spinal cord tumors. Astrocytomas, glioblastomas, oligodendrogliomas, ganglioneuromas, medulloblastomas, hemangiomas, and hemangioblastomas are more or less equally distributed in frequency of occurrence.
PATHOPHYSIOLOGY Extramedullary spinal cord tumors produce dysfunction by compression of adjacent tissue, not by direct invasion. The spinal cord is compressed by the tumor from without, and destruction of the white matter tracts occurs. The spinal canal around the cord becomes filled by tumor.
Intramedullary spinal cord tumors produce dysfunction by invasion and compression. The cord enlarges as a result of the tumor that is enlarging inside the cord. In addition, distortion of adjacent white matter tracts occurs. Metastases from spinal cord tumors occur from seeding through the CSF; medulloblastomas and ependymomas establish distant implants in this manner.
CLINICAL MANIFESTATIONS The acute onset of clinical manifestations suggests a vascular insult caused by thrombosis of vessels supplying the spinal cord. Clinical manifestations that are gradual and progressive suggest compression. The clinical manifestations associated with spinal cord tumors fall into three major categories: (1) a compressive syndrome (sensorimotor syndrome), (2) an irritative syndrome (radicular syndrome), and rarely (3) a syringomyelic syndrome.
The compressive syndrome (sensorimotor syndrome) is associated with compression and is caused less frequently by invasion and destruction of the spinal cord tracts. Symptoms are usually gradual and progressive, and initial manifestations may be asymmetric. With tumors located in the cervical area, the motor dysfunction usually has the following pattern: ipsilateral arm involvement, followed by ipsilateral and contralateral leg involvement, and finally involvement of the opposite arm. With thoracic tumors the pattern of motor involvement is paresis and spasticity of one leg, followed by involvement of the opposite leg. The sensory clinical manifestations of tingling paresthesias have a pattern similar to that of the motor signs. Pain and temperature dysfunctions are found more commonly than touch, vibration, and proprioceptive changes, although posterior column signs also are found frequently. Pain is less well localized than with an irritative syndrome caused by root involvement. Initially the pain and temperature changes are contralateral to the motor deficit (Brown-Séquard syndrome, see Table 17-5). Bladder and bowel deficits usually appear when paresis develops in the legs.
The irritative syndrome (radicular syndrome) combines the clinical manifestations of a cord compression with radicular pain, which is pain in the sensory root distribution and indicates root irritation. The segmental manifestations associated with root irritation include segmental sensory changes that include paresthesias and impaired pain and touch perception; motor disturbances, including cramps, atrophy, fasciculations, and decreased or absent deep tendon reflexes; and ache in the spine. Tenderness of the spinous processes over the tumor is present in about half of extramedullary tumors. The segmental changes may appear months and sometimes years before the clinical manifestations of compression in benign tumors. The compressive clinical manifestations include an asymmetric spastic paresis of the lower extremities with tumors in the thoracic or lumbar region, paresis of the arms and legs with tumors in the cervical area, decreased or absent pain and temperature perception below the tumor site, posterior column signs, and spastic bladder.
Because they involve the central gray matter of the cord, intramedullary spinal cord tumors (notably ependymomas) may produce a syringomyelic syndrome, or inflammation of the spinal cord. Inflammation results in the development of tubular (syrinx) cavities in the spinal cord. Occasionally an extramedullary tumor may produce the same effect, although the mechanisms are unknown.
EVALUATION AND TREATMENT The diagnosis of a spinal cord tumor is made through bone scan, needle biopsy guided by CT and positron-emission tomography (PET), or open biopsy. Benign or malignant spinal tumor staging may be done (Box 17-4). Involvement of specific cord segments is established.
Treatment varies, depending on the nature of the tumor and the person’s clinical status. Indications for surgery include establishing a tissue diagnosis, neurologic palliation, spinal stabilization, pain relief, and cancer therapy. Surgical resection may involve curettage (piecemeal removal of the tumor) or may be performed en bloc (removal of tumor in one piece). Surgical approaches to the spine include posterior approach (decompression laminectomy), lateral approach, anterior approach (most favored), and combined approaches. Posterior and anterior reconstructive surgery may be necessary. Oncologic surgical procedures are classified as intralesional, marginal, wide excision, or radical excision. Indications for external radiation versus surgery are a radiosensitive tumor (e.g., lymphoma), soft tissue compression without instability, a person who is a poor surgical candidate, paraplegia or advanced paraparesis of greater than 24 hours’ duration, and an expected survival of less than 3 to 4 months. Chemotherapy, hormonal therapy, and pain management protocols may be appropriate.