NEUROBIOLOGY OF SCHIZOPHRENIA, MOOD DISORDERS, AND ANXIETY DISORDERS


Mental illnesses are common and have a long history of afflicting humanity. They appear in different cultures and across the socioeconomic spectrum. When mental disorders are left untreated, the consequences can be devastating. This chapter provides an introduction to the neurobiology of schizophrenia, mood disorders, and some anxiety disorders. The etiology and pathophysiology of these major mental illnesses are diverse and complex. Diagnostic criteria are constantly being updated to more precisely diagnose and effectively treat the disorders. Every mental disorder manifests a range of symptoms that vary in intensity for each individual. Symptom variations likely reflect individual differences in neural pathologic brain structures or functions, or both, which affect the options for treatment. To further complicate diagnosis and treatment, comorbid disorders such as depression and anxiety are often present.
Insight into the pathophysiologic basis of mental disorders has been greatly aided by the development of structural and functional neuroimaging techniques that provide a visual and quantitative evaluation of diseased brain regions. In schizophrenia, neuroanatomic, functional, and neurochemical alterations associated with this debilitating disorder have been uncovered along with a host of candidate genes that confer risk. Similarly, in mood and anxiety disorders, brain scans provide new information on structural and functional abnormalities. Use and overuse, however, of brain scans is a continuing focus of concern and study. Many of the altered brain regions found in those with schizophrenia, mood, and anxiety disorders involve structures implicated in normal cognitive and emotional processes, suggesting that their exaggerated or diminished functions underlie the illness.
Knowledge of the pathophysiology associated with a specific mental illness has guided the development of pharmacologic medications. Although more effective second- and third-generation drugs with fewer side effects are available, many individuals continue to suffer. Future identification and characterization of diseased genes and how they contribute to psychopathology may eventually lead to medications that produce effective relief of symptoms and stop or reverse the neural alterations that produce the disorder.
SCHIZOPHRENIA
Schizophrenia is a serious psychiatric illness that strikes 1% of the world’s population. The illness is equally prevalent in males and females and emerges in young adults during the late teens and early 20s, with a slightly earlier onset in males than in females. Schizophrenia is the term coined originally by Eugene Bleuler in 1911 to describe a collection of illnesses characterized by thought disorders. According to Bleuler, thought disorders reflected a break in reality or splitting of the cognitive from the emotional side of one’s personality. A schizophrenic individual may exhibit a feeling of happiness when recollecting a terrible event or emotional indifference when describing a joyful occasion. Today, disorganized thought in schizophrenia is characterized by symptoms including hallucinations, delusions, and cognitive deficits. The impairment of cognitive functions, such as attention, planning, and social skills results in devastating effects on the individual and families. Although treatment of schizophrenia has been improving, the illness remains difficult to treat because of heterogeneity of the schizophrenia phenotype and the inability to identify the specific underlying neuropathology. However, advances in genetics, neurobiology, and neuroimaging provide unprecedented insights that hold promise for the effective treatment of schizophrenia.
Etiology and Pathophysiology
Genetic epidemiologic studies demonstrate that schizophrenia is a heritable disorder. In monozygotic twins, the concordance rate varies from 30% to 50%. This variability may stem from different diagnostic criteria and methodologic or sampling differences across studies. In dizygotic twins and siblings the concordance rate decreases to 12%, which is still considerably higher than the 1% figure found in the general population.
Nonetheless, schizophrenia is not a simple genetic disorder in which inherited disease alleles generally lead to illness. Schizophrenia likely involves several genes located on different chromosomes and differs from mendelian disorders, in which genes are fully penetrant. That is, in mendelian disorders, individuals with genes for a disease (e.g., Huntington disease) will usually develop the disorder. In contrast, as indicated by the 50% concordance rate in monozygotic twins, the genes for schizophrenia show reduced penetrance, resulting in individuals who may carry the disease genes without manifesting the illness. Further complicating the search for the genes that confer risk of schizophrenia is the variability in biologic and phenotypic traits among individuals who manifest the illness (see What’s New? Novel Deletions and Duplications of Genes Controlling Neural Development Contribute to Schizophrenia).
Prenatal and Perinatal Factors: A leading hypothesis for the etiology of schizophrenia suggests that the illness results from neurodevelopmental defects that occur in fetal life. According to this hypothesis, environmental factors interfere with genetically programmed
neural development, leading to alterations in brain structure and function.1 An early brain defect may remain silent and not affect the individual until subsequent development requires extensive use of that brain structure.2 Several early environmental factors have been suggested to increase the risk of developing schizophrenia including viral infection during pregnancy, prenatal nutritional deficiencies, and perinatal complications, such as birth defects and neonatal hypoxia.
Neuroanatomic and Functional Abnormalities
Neuroanatomic Alterations: The use of advanced neuroimaging techniques has provided strong evidence for structural brain abnormalities of individuals with schzophrenia.3,4 A consistent finding is the enlargement of the lateral and third ventricles and widening of frontocortical fissures and sulci (Figure 18-1). Schizophrenic individuals with cerebral ventricular enlargement often exhibit cognitive impairments and negative symptoms, and respond poorly to treatment. Other imaging studies reveal reductions in the thalamus and temporal lobe (which includes the amygdala, hippocampus, and parahippocampal gyrus) brain areas important in emotional regulation and memory functions.5 A reduction in thalamus size may disrupt communication between broad regions of the cortex and primary sensory and motor areas. Temporal lobe alterations may be responsible for the production of positive schizophrenic symptoms, such as hallucinations, delusions, thought disorder, and bizarre behavior.
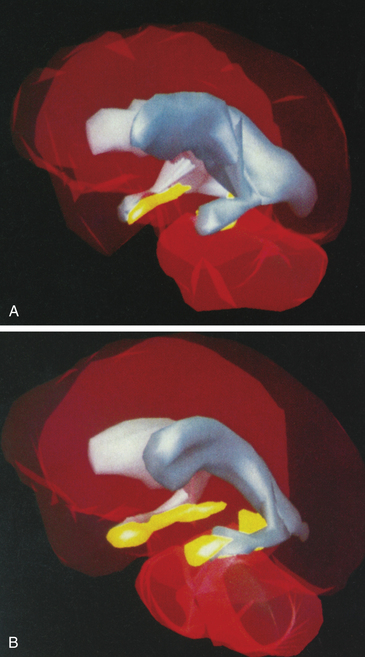
Figure 18-1 Magnetic resonance imaging (MRI) comparison of normal brain and brain with schizophrenia. Three-dimensional MRI reconstructions showing A, the cerebroventricles (gray regions) and hippocampus (yellow regions) of a schizophrenic patient, and B, a normal individual. Note enlarged cerebroventricles and reduced hippocampal volume of the brain of the schizophrenic individual. (From Gershon ES, Rieder RO: Sci Am 267:128, 1992. Original illustrations by Nancy C. Andreason, University of Iowa.)
Brain imaging work further reveals progressive loss of cortical gray matter in adolescent individuals with early onset schizophrenia (Figure 18-2). Loss of gray matter was found in temporal lobes, somatosensory and motor cortices, and the dorsolateral prefrontal cortex. Of particular concern is the loss of cortical tissue, which is evident by the time the individual seeks treatment and progressively worsens throughout the course of the illness despite the use of antipsychotic medication.6 In particular, the frontal lobe undergoes a progressive loss in volume that is accompanied by increased severity of negative symptoms and reduced cognitive functioning. These results highlight the progressive nature of the structural brain abnormalities and the ineffectiveness of current treatments in attenuating or reversing the frontal brain alterations present in schizophrenia.
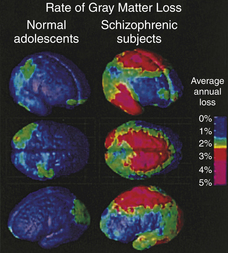
Figure 18-2 Accelerated gray matter loss in brains of early onset schizophrenic adolescents. Annual gray matter loss ranging from 2% to 5% was found in 13- to 18-year-old schizophrenics—compared with age-matched healthy—adolescents. (From Thompson PM et al: Proc Natl Acad Sci U S A 98:11650, 2001.)
The abnormalities documented in schizophrenic brains are believed to originate during the prenatal period of cell proliferation and migration. Reelin, an extracellular matrix protein involved in neuronal migration during development and synaptic function in the adult, is reduced in the prefrontal cortex and hippocampus of schizophrenic individuals.7,8 Reelin is concentrated in interneurons that contain gamma-aminobutyric acid (GABA), the most widespread inhibitory neurotransmitter in the brain. Furthermore, in the dorsal prefrontal cortex of schizophrenic brains, glutamic acid decarboxylase, the major enzyme in GABA biosynthesis, is diminished, which likely impairs synaptic performance and cognitive and behavioral functions associated with this brain region.
Data suggest that the pathophysiologic processes in the dorsal prefrontal cortex are responsible for the negative symptoms of schizophrenia (Figure 18-3). In particular, the dorsolateral prefrontal cortex (DLPFC) is intricately involved in the initiation and maintenance of goal-directed activities and is actively involved in solving cognitive problems related to working memory. Working memory involves the brief storage and use of information that may be important in simple number calculations as well as complex tasks such as winning a chess match. Working memory is also essential in verbal comprehension and reasoning. During cognitive problem solving, blood flow and metabolism normally increase in the DLPFC. However, individuals with schizophrenia often perform poorly on these tests and fail to show an increase in cortical blood flow and metabolism. These studies suggest that the dorsal prefrontal cortex is hypoactive in schizophrenia.
Neurotransmitter Alterations: A long-standing neurotransmitter hypothesis of schizophrenia involves dopamine. The dopamine hypothesis initially suggested that abnormal elevation in dopaminergic transmission contributes to the onset of schizophrenia. This hypothesis was based on pharmacologic studies showing that antipsychotic drugs are potent blockers of brain dopamine receptors. A strong positive correlation is found between the clinical potencies of traditional antipsychotic drugs (e.g., chlorpromazine, fluphenazine, and haloperidol) and their affinity for the dopamine D2 receptor. In addition, drugs that increase dopaminergic transmission—such as levodopa (L-dopa), cocaine, and amphetamine—produce schizophrenic-like psychosis at high doses. Dopamine blockers reverse these drug-induced psychotic states.
A more current view of the dopamine hypothesis of schizophrenia is that brain dopamine pathways are altered in different ways (Figure 18-4). For example, the mesocortical dopamine pathway plays an essential role in dorsal prefrontal cortical functions. Depletion of prefrontal cortical dopamine in monkeys produces deficits in cognition and affect. Some investigators suggest that negative symptoms of schizophrenia result from decreased dopaminergic neurotransmission in frontocortical regions.9 This hypodopaminergic transmission in the dorsal prefrontal cortex contrasts with the hypothesized hyperdopaminergic secretion in mesolimbic pathways. The mesolimbic dopamine pathway innervates temporal lobe structures including the hippocampal formation and amygdala, as well as the nucleus accumbens and anterior cingulate cortex. Hypersecretion of mesolimbic dopamine may contribute to the manifestation of positive symptoms.
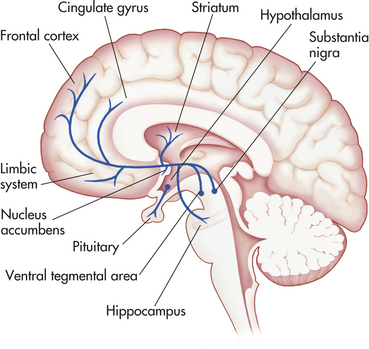
Figure 18-4 The dopamine system. Dopamine cell bodies are located in the substantia nigra, where they project to the stratum (nigrostriatal pathway); and in the ventral tegmental area where they project to the frontal and cingulate cortex (mesocortical pathway), the striatum, the hippocampus, and other limbic structures (mesolimbic pathway). Dopamine nuclei are also located in the hypothalamus and project to the pituitary.
Of additional relevance to brain dopamine systems and schizophrenia is the potential involvement of a genetic loci at chromosome 22q11.10 Microdeletion of 22q11 causes velocardiofacial syndrome characterized by cardiac defects, learning impairments, and deformities of the palate and face. In addition, 25% to 30% of individuals with velocardiofacial syndrome exhibit psychotic symptoms. Deletion of 22q11 is also found in 2% of schizophrenic patients, a rate much higher than the 0.025% estimate occurring in the general population. Interestingly, 22q11 contains a gene encoding catechol-O-methyltransferase (COMT), an enzyme that catabolizes dopamine. COMT is normally active in dorsal prefrontal brain regions linked to schizophrenia. Thus deletion of a gene encoding an enzyme involved in dopamine regulation increases the risk of schizophrenia.
Another neurotransmitter system that may be involved in schizophrenia is the excitatory neurotransmitter glutamate and its actions on the N-methyl-D-aspartate (NMDA) receptor subtype.11 This receptor is implicated in learning and memory, and NMDA antagonists, such as the cyclohexylamine anesthetics phencyclidine and ketamine, cause psychosis in healthy subjects and exacerbate symptoms in schizophrenic individuals. In schizophrenia, glutamate concentrations in the cerebrospinal fluid (CSF) are reduced along with a reduction in cortical glutamate synthesis. These data suggest that potential reduction in NMDA function may contribute to cognitive disorders in schizophrenia.
Clinical Manifestations
The characteristic symptoms of schizophrenia are manifested in the dysfunctions of basic human processes of perception, emotion, language, memory, and judgment. The symptoms are traditionally divided into positive and negative groups (Box 18-1). Positive symptoms frequently occur during a psychotic episode, when an individual loses touch with reality and experiences something that should be absent (e.g., hallucinations). Negative symptoms are characterized by the absence of something that should be occurring. According to the Diagnostic and Statistical Manual of Mental Disorders (DSM-IV-TR),11 schizophrenia is diagnosed when an individual exhibits delusions, hallucinations, negative symptoms, or social/occupational dysfunctions for at least 6 months.
Psychotic Dimension
Psychotic dimension refers to hallucinations and delusions and reflects a person’s confusion or loss of touch with the external world. Hallucinations and delusions are classified as positive symptoms and are the most common in schizophrenia.
Hallucinations: A hallucination is a perception experienced without external stimulation of the sense organs. Sensory hallucinations can be auditory, tactile, visual, gustatory, and olfactory. For example the schizophrenic individual may hear voices, experience touch or electrical sensations, report images of animate and inanimate objects, or complain of unpleasant tastes and odors. These hallucinations may occur alone or together.
Delusions: A delusion is a persistent belief contrary to the educational and cultural background of the individual. Delusions may involve grandiose, nihilistic, persecutory, somatic, sexual, and religious themes. A common delusion revolves around paranoid beliefs that may involve spying, conspiracy, persecution, and ridicule. Delusions also may be referential in that certain stimuli or events become highly personalized. For example, schizophrenic individuals may believe that a television talk show host is directing information specifically at them.
Disorganized Behavior
Disorganized behavior includes disorganized speech and disorganized or bizarre behavior. Incongruity of affect is another dimension of disorganized behavior.
Disorganized Speech: A common form of disorganized speech is formal thought disorder, which involves fluent speech that is difficult to comprehend. The speech is often incoherent as the individual moves from one topic to another unexpectedly (loose associations). Answers to questions are illogical or unrelated, and the person becomes easily distracted when talking.
Another form of disorganized speech is called poverty of content. In this case, the use of vocabularies to convey information is severely retarded despite a fair amount of spoken words. For instance, the same phrases are used repeatedly throughout a conversation.
Disorganized Behavior: Disorganized (or bizarre) behavior is the conceptual equivalent of disorganized speech. The individual has difficulty engaging in goal-directed activities. Behavior may be repetitive (e.g., stereotyped rocking) or aimless, and personal hygiene is poorly maintained. Another aspect is the incongruity of affect or the manifestation of inappropriate situational affect as exemplified by hostility without provocation or childlike silliness in sober situations.
Negative Dimensions
Negative dimensions reflect a deficit in normal functioning. These symptoms are disabling and include affective flattening, anhedonia, alogia (poverty of speech), and avolition. Affective flattening is the near absence of emotional or facial expression. A fixed expression is maintained throughout a conversation or in different situations. In anhedonia, individuals are unable to experience emotions such as pleasure or pain and report a sense of detachment from the environment. Alogia is the absence of spontaneous speech production for the purpose of answering questions or expressing oneself. Avolition is a deficit in spontaneous or goal-directed behavior in which an individual may sit for prolonged periods and must be prodded into completing simple daily tasks.
Treatment
Prior to the early 1950s, there was a steady increase in the number of schizophrenic patients requiring extensive hospitalization in mental institutions. The use of chlorpromazine dramatically changed the treatment of schizophrenia. The drug was especially effective in reducing positive symptoms such as hallucinations and delusions as well as thought disorders and hyperactivity. The widespread use of chlorpromazine and similar drugs such as haloperidol led to a marked reduction in the number of patients requiring hospitalization. These antipsychotic drugs are also known as neuroleptics from the Greek term “to clasp the neuron.” The beneficial effects of these first-generation neuroleptics on positive symptoms were due to their ability to block the dopamine D2-receptor subtype, especially in limbic and prefrontal brain regions associated with emotionality. The mesolimbic dopamine pathways are hypothesized to be overly active in schizophrenia, which contributes to the expression of positive symptoms. However, blockade of the D2 receptor in brain regions such as the striatum also produced a notable neurologic side effect resembling Parkinson disease, a disorder associated with degeneration of dopamine cell bodies in the substantia nigra that projects to the striatum. Other side effects may include sedation, hypotension, akathisia (motor restlessness), constipation, weight gain, amenorrhea, and less frequently, hepatotoxicity and electrocardiographic changes. Although the pharmacologic blockade of D2 receptors occurs rapidly, clinical efficacy does not appear until after 1 or 2 weeks of treatment. Thus the antipsychotic effects are not related directly to D2-receptor blockade or acute suppression of dopamine hypersecretion.
Although the majority of schizophrenic individuals obtained some positive symptom relief from the first-generation or conventional antipsychotic drugs, approximately 20% failed to respond to D2-blocking drugs (Box 18-2). Some of these treatment-resistant individuals responded to a second generation of drugs that became known as atypical antipsychotic drugs.12 Some studies suggest that atypical antipsychotic drugs have superior efficacy in reducing not only the positive but also the negative symptoms in comparison with conventional neuroleptics. For example, clozapine may improve some cognitive functions, such as verbal fluency, verbal learning, and memory, and physical functions, such as psychomotor speed. In addition, the notable neurologic side effects that accompany the use of the conventional neuroleptics were diminished.
Although the mechanism of action of these drugs is not known, atypical medications clearly differ from conventional antipsychotic drugs in blocking a range of neurotransmitter receptors. For example, clozapine blocks a variety of receptors that include D2 receptors, as well as D1, D3, D4, and D5 receptors and serotonin (5-hydroxytryptamine, i.e., HT2, 5-HT6, 5-HT7); norepinephrine; cholinergic, and histamine receptors. Risperidone and ziprasidone have higher affinity for blocking 5-HT2 than D2 receptors. The higher 5-HT2:D2-receptor–binding ratio of atypical antipsychotics in comparison with conventional neuroleptics may reflect a normalization of serotonin-dopamine interactions leading to clinical efficacy not observed with D2-receptor blockade alone.
Atypical antipsychotics are not without adverse effects, most notably metabolic abnormalities including glucose regulation, lipids, and weight gain. Weight gain is a risk factor for diabetes and cardiovascular disease in schizophrenics who are maintained particularly on long-term clozapine or olanzapine treatment.
In conjunction with antipsychotic medication, psychosocial therapy can facilitate the management of schizophrenia. Psychosocial relationships may assist the individual in developing coping strategies and in identifying stressors and relapse symptoms. The addition of cognitive-behavioral therapy (CBT) may alleviate some of the schizophrenic symptoms resistant to medication.13 An important benefit of psychosocial and family support is the encouragement of compliance with antipsychotic medication that requires a period before the emergence of clinical efficacy.
MOOD DISORDERS: DEPRESSION AND BIPOLAR DISORDER
Mood refers to a sustained emotional state as opposed to brief emotional feelings, which are termed affective states. Healthy individuals are normally capable of experiencing a variety of affective states including euphoria, joy, surprise, fear, sadness, anxiety, and depression. When emotional states, such as sadness, become predominant and uncontrollable, individuals may be diagnosed with a mood disorder called depression. The two major classifications of mood disorder are (1) unipolar or major depressive disorder, also known as major depression or clinical depression, which consists of episodes of depression; and (2) bipolar disorder, also known as manic-depressive illness, which is further classified into bipolar I and bipolar II disorders. Bipolar I disorder features manic episodes and at least one major depressive episode and bipolar II disorder is characterized by recurrent major depressive episodes with one or more hypomanic (milder than manic) episodes. Box 18-3 presents the major criteria of depression and bipolar disorder according to the American Psychiatric Association’s DSM-IV-TR.11
Major (unipolar) depression is the most common mood disorder and the leading cause of disability in the United States and throughout the world. Unipolar depression appears in all age groups including young children. In the United States, the lifetime prevalence rate of depression is 16.2% of the population with a twofold greater risk in women than men after adolescence. In children and adolescents, 2% to 6% suffer from depression. The prevalence of bipolar disorder ranges from 3% to 5% in the general population. Bipolar I disorder occurs equally in men and women in comparison with bipolar II disorder, which afflicts more women than men. When left untreated, a number of depressed and bipolar individuals are at risk of developing a host of medical illnesses, including cardiovascular disease, obesity, diabetes, and thyroid disease.
Etiology and Pathophysiology
Genetic Predisposition and Environmental Influences
Results from family and twin studies indicate a strong basis for mood disorders. A study using the Danish twin registry for bipolar disorder yielded concordance rates of 79% and 24% for monozygotic and dizygotic twins, respectively. For unipolar disorder, concordance rates of 54% and 19% have been reported in monozygotic and dizygotic twins, respectively. Even among adoptees with a biologic family history of mood disorders, the incidence of developing major depression or manic-depressive illness is higher than among control adoptees. The strong tendency for mood disorders to run in families has encouraged a search for the abnormal gene or genes. Interestingly, loci on chromosomes 18 and 22 have been linked to both bipolar disorder and schizophrenia. Bipolar individuals, who may exhibit psychotic behavior, have deficits in reelin expression linked to genetic loci, located on chromosome 22, which confers susceptibility to schizophrenia (see section on schizophrenia). However, the large variation in clinical symptoms suggests that developmental and environmental factors are as important as genetic factors in contributing to the etiology of mood disorders.
A current view of mood disorders is that the illness stems from a complex interplay between susceptible genes and environmental influences. For example, the interplay between life stressors and a potentially dysfunctional serotonin (5-HT) system appears to elevate the risk of depression.14 In particular, researchers have identified a polymorphic variant of the serotonin transporter (5-HT-T) that exists either as a short (s) allele or long (l) allele. The serotonin transporter serves in the reuptake of serotonin at the synapse and may moderate the serotonergic response to stress. However, individuals with one or two copies of the s allele were more likely to develop major depression and have suicidal thoughts in response to stressful events than individuals homozygous for the l allele. In addition, among individuals who carry two s alleles, the risk of a major depressive episode increased twofold after experiencing four or more stressful events. This work14 has implications for a gene-by-environment interaction underlying major depression by showing that adverse life events and a genetic alteration in 5-HT function increase the risk of a major depressive episode.
Neurochemical Dysregulation
Modern theories of the pathophysiology of mood disorders began with the important observation that imipramine reduced the reuptake of norepinephrine into presynaptic neurons, and the resultant increase in norepinephrine within the synapse was then responsible for antidepressant effects. This key finding, along with other studies work showing that drugs (e.g., reserpine) that deplete monoamines produce depression, led to the dominant monoamine hypothesis of depression. Accordingly, depression occurs following a deficit in brain norepinephrine, dopamine, and/or serotonin, whereas mania results from elevated concentrations of monoamines. The three major classes of antidepressant medications include monoamine oxidase inhibitors (MAOIs), tricyclic antidepressants (TCAs), and selective serotonin reuptake inhibitors (SSRIs). All available antidepressants share the common property, albeit through different mechanisms, that increasing monoamine neurotransmitter levels within the synapse is the basis for their antidepressant effects (Figure 18-5).
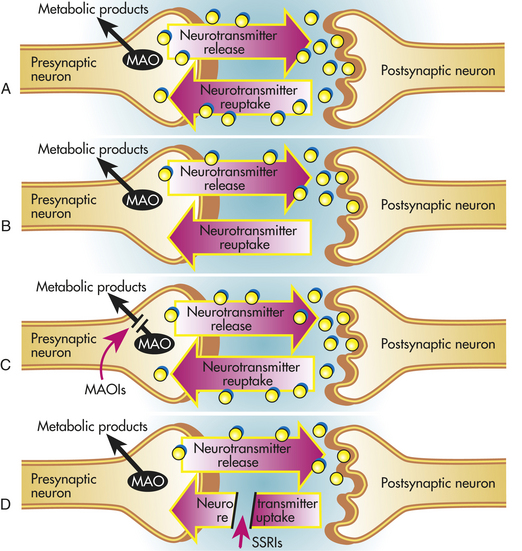
Figure 18-5 Schematic diagrams showing the sites of actions of antidepressants and their effects on neurotransmitter levels. A, In normal individuals an action potential generated in the presynaptic neuron results in neurotransmitter release into the synapse. Some neurotransmitters bind to receptors on the postsynaptic neuron that leads to activation of second messenger systems (not shown). Neurotransmitters are also removed from the synapse by reuptake into the presynaptic neuron and deaminated by monoamine oxidase (MAO). B, In depressed individuals, neurotransmitter levels are hypothesized to be reduced. The mechanisms responsible for this reduction are not understood. C, MAO inhibitors act by preventing the degradation of neurotransmitters, such as norepinephrine and serotonin. As a result, neurotransmitter levels are elevated. D, The tricyclic antidepressants (TCAs) and selective serotonin reuptake inhibitors (SSRIs) act by reducing the uptake of neurotransmitters from the synapse, leading to increased neurotransmitter levels. TCAs, such as nortriptyline and desipramine, tend to block norepinephrine reuptake, whereas amitriptyline and imipramine also have effects on serotonin reuptake. SSRIs are highly effective in blocking serotonin reuptake.
Further support for the monoamine hypothesis of depression came from examination of the major metabolites of norepinephrine (3-methoxy-4-hydroxyphenylglycol [MHPG]), dopamine (homovanillic acid [HVA]), and serotonin (5-hydroxyindoleacetic acid [5-HIAA]), in depressed individuals. The major metabolites of these monoamines were reduced in the CSF of depressed individuals. Other work demonstrated that dietary depletion of tryptophan, the precursor of serotonin synthesis, or alpha-methylparatyrosine (AMPT), a drug that inhibits dopamine and norepinephrine synthesis, produced a rapid return to depression in individuals who had been treated successfully with antidepressants.15
Although TCAs and SSRIs rapidly block synaptic reuptake of monoamines, resulting in elevations in synaptic monoamine concentrations, the onset of clinical efficacy requires several weeks or months of treatment. Antidepressant treatment appears to produce the delayed clinical effects through alterations in receptor-mediated functions. For example, the cyclic adenosine monophosphate (cAMP) signal transduction pathway is regulated by direct coupling of monoamine receptors to adenylyl cyclase or indirectly through other second messenger pathways. Antidepressants increase adenylyl cyclase leading to subsequent augmentation in cAMP signal transduction, phosphorylation of intracellular proteins and transcription factors, and genomic effects that may underlie the therapeutic actions of antidepressant drugs. Similarly, the mode of action of mood stabilizers for bipolar disorders appears to be mediated by altering postsynaptic signal transduction pathways. For example, the clinical effects of the mood stabilizer lithium result from an inhibition of the overactive phosphatidylinositol pathway (see Chapter 1).
Neuroendocrine Dysregulation
Hypothalamic-Pituitary-Adrenal System Dysregulation: The hypothalamic-pituitary-adrenal (HPA) system plays an essential role in an individual’s ability to cope with stress (see Chapter 10). However, excessive activation of the HPA system resulting in elevated glucocorticoid secretion is found in a large percentage (30% to 70%) of people with major depression suggesting that mechanisms responsible for HPA hormone alterations contribute to the pathophysiology of depression.16 Research has focused on inflammation, proinflammatory cytokines, and depression (see What’s New? Does Inflammation Contribute to the Development of Depression?). Altered HPA function can be demonstrated in the dexamethasone suppression test.
Administration of dexamethasone, a potent synthetic glucocorticoid, normally suppresses adrenal cortisol secretion because of its negative-feedback effects on the HPA system. Individuals with depression, however, fail to suppress cortisol secretion after dexamethasone administration. In addition, whereas healthy individuals typically exhibit a diurnal rise in cortisol secretion at the onset of wakefulness followed by a trough 12 hours later, depressed people continue to exhibit elevated plasma cortisol levels throughout the evening and early morning hours. Notably, antidepressant drugs effective in normalizing the HPA system are associated with a good clinical response, whereas persistent HPA system dysregulation, as evidenced in the dexamethasone suppression test, is related to continued depression or relapse.
Hypothalamic-Pituitary-Thyroid System Dysregulation: Thyroid hormones are involved in the modulation of mood and behavior. Approximately 20% to 30% of persons with unipolar depression have an altered hypothalamic-pituitary-thyroid (HPT) system. These individuals exhibit increased CSF levels of thyrotropin-releasing hormone (TRH), blunted thyrotropin-stimulating hormone (TSH) response to TRH challenge, and decreased nocturnal rise in TSH that normally occurs between midnight and the early morning hours.17 Persistent blunting of the TSH response to TRH is associated with an increased probability of relapse. Abnormalities in thyroid function, such as increased basal TSH concentrations with normal thyroxine levels, also are observed in bipolar disorder, especially among individuals with rapid cycling.
Neuroanatomic and Functional Abnormalities
The dorsal and median raphe nuclei, located in the central gray of the caudal mesencephalon and rostral pons, contain a large group of serotonin-synthesizing neurons that project extensively to all regions of the cortex, basal ganglia, limbic system, hypothalamus, cerebellum, and brainstem (Figure 18-6). Postmortem and/or brain imaging studies of depressed individuals revealed a widespread decrease in serotonin 5-HT1A receptor subtype binding in frontal, temporal, and limbic cortex as well as serotonin transporter binding in cerebral cortex and hippocampus. Mood disorders may reflect a dysfunctional raphe-serotonin system, which normally modulates homeostasis, emotionality, and tolerance to aversive experiences.
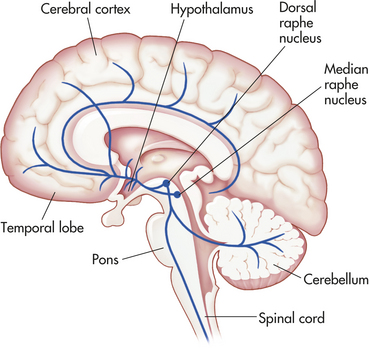
Figure 18-6 The serotonin system. Serotonin neurons are located in the brain stem raphe nuclei. They project diffusely to all regions of the cortex, temporolimbic regions, hypothalamus, basal ganglia, cerebellum, the brainstem, and spinal cord.
A group of norepinephrine-containing cells located in the locus ceruleus of the rostral pons project to vast areas of the forebrain, brainstem, and spinal cord (Figure 18-7). The locus ceruleus-norepinephrine system is implicated in global psychologic processes including attention, vigilance, and orientation to novel, aversive, or threatening stimuli. Activation of the locus ceruleus-norepinephrine system is also capable of inhibiting the raphe-serotonin system, suggesting an indirect role in modulating serotonin functions. Norepinephrine receptor alterations (e.g., α- and β-adrenergic receptor subtypes) are found in the frontal cortex of some suicide victims with major depression. Alterations in norepinephrine systems may be linked to attention or concentration difficulties as well as sleep and arousal disturbances in depression.
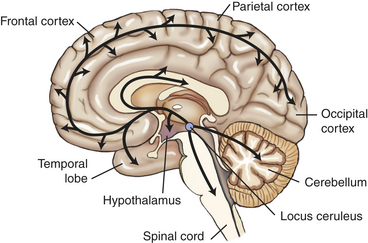
Figure 18-7 The norepinephrine system. The norepinephrine cell bodies originate in the locus ceruleus and project throughout the brain, including the hypothalamus, the temporal lobe, the entire cortex, the cerebellum, and spinal cord.
Postmortem and brain imaging studies further reveal structural and functional abnormalities associated with mood disorders, especially in frontal and limbic regions such as the amygdala.18,19 Postmortem studies report a reduction in glial cell numbers in people with unipolar and bipolar disorders. There are also reports of reduced frontal lobe volume in depressed individuals and decreased or asymmetric temporal lobe volume in bipolar illness and depression. One study reported an increased volume of the amygdala in bipolar illness. In some cases, specific brain abnormalities are associated with a subtype of depression. For instance, enlarged lateral ventricles are found more often in late onset or depressed older adults than in midlife depressed individuals, depressed older adults with an early age of illness, or bipolar individuals.
Functional neuroimaging studies indicate decreased cerebral blood flow and glucose metabolism in the dorsolateral and dorsomedial prefrontal cortex of major and bipolar individuals. Dorsolateral abnormalities in depression may be responsible for the retardation in cognitive processing and speech deficits similar to those found in schizophrenia. Dorsomedial frontal dysfunction may be associated with mnemonic and attentional impairments that accompany mood disorders. Other frontocortical regions, including the ventrolateral, ventromedial, and orbital areas, exhibit increased blood flow and metabolism in unipolar depression (Figure 18-8). These frontal brain areas have extensive interconnections with the amygdala, and increased blood flow and metabolism, especially in the right amygdala, is positively related to negative affect in depressed individuals. These functional changes in brain activity begin to normalize with successful antidepressant treatments, suggesting they are state rather than trait related.
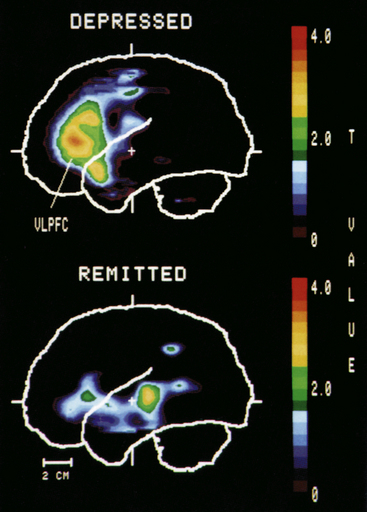
Figure 18-8 Positron-emission tomography (PET) comparison of brain activity in depression and in remittance. PET scan showing increased activity in the left prefrontal cortex in a depressed person but not in the remitted person. VLPFC, Ventrolateral prefrontal cortex. (From Drevets WC et al: J Neurosci 12:3628, 1992. Copyright ©1992 by the Society for Neuroscience.)
Clinical Manifestations
Major depression is characterized by unremitting feelings of sadness and despair (see Box 18-3). The dysphoric mood or intensely painful mood is accompanied frequently by insomnia, loss of appetite and body weight, and reduced interest in pleasurable activities and interpersonal relationships. Sleep disturbances may include difficulty in initially falling asleep, awakening in the middle of the night and lying awake for several hours, and early morning wakefulness with an inability to subsequently fall asleep. Individuals may have reduced motor activity and suffer marked fatigue. Others complain of restlessness and agitation. Feelings of worthlessness and guilt are common, and pessimistic or negative outcomes are often perceived even in routine situations. The ability to function (e.g., work) and concentrate is greatly diminished. Suicidal risk increases in depression. Factors such as living alone or divorced, prior history of drug abuse or suicide attempt, or having depression at midlife or older ages than younger ages contribute to suicide in 10% to 15% of depressed individuals.
The symptoms of depression may occur suddenly or gradually for a period ranging from a few weeks to months or years. Depressive episodes recur in a substantial number of individuals and 20% may exhibit a chronic form of depression.
Bipolar Disorder: Mania
Manic individuals experience elevated levels of euphoria. Self-esteem and feelings of grandiosity are abnormally elevated and may result in psychoses such as delusions and hallucinations. Energy levels are greatly enhanced even after only a few hours of sleep each night. The increased energy, however, does not lead to organized plans and thoughts. The individual may show poor judgment in spending money, may become hypersexual, or may make poor business commitments. Other hallmarks of mania are excessive, rapid, loud, and pressured speech. The manic person frequently skips from one topic of conversation to another and is easily distracted both when speaking and when performing tasks. Approximately 50% of manic individuals develop psychotic symptoms, such as delusions or hallucinations, which require hospitalization. The onset and termination of manic symptoms (see Box 18-3) are usually abrupt and may last for a few days or months followed by depression. The risk of recurrence of bipolar disorder is high, especially without immediate treatment.
Treatment
Unipolar depression is one of the more treatable psychiatric disorders. Approximately 80% of depressed persons will respond to antidepressant drugs, psychotherapy, or a combination of both. MAOIs, TCAs, and SSRIs are used in the treatment of depression (Box 18-4). In addition, atypical antidepressants, such as nefazodone, trazodone, and mirtazapine, presumably produce their clinical effects by blocking specific receptors (e.g., 5-HT2A). A new generation of antidepressants that selectively block serotonin and norepinephrine reuptake is available in the United States (i.e., venlafaxine) or Europe (i.e., milnacipran, reboxetine). Although SSRIs have become the standard first-line treatment for major depression, initial selection of an antidepressant often includes an assessment of the person’s symptoms, age, side effects, safety, cost, and convenience. For example, medications that produce sedation may be helpful for the treatment of sleep disturbances. Approximately 50% of depressed individuals may not show a favorable response during initial treatment to an antidepressant drug, and 10% to 20% may continue to exhibit symptoms after 2 years. Individuals who are nonresponsive to a specific antidepressant during a 2-month period may be given another antidepressant medication. At present, there are no criteria that indicate whether selection of the next antidepressant drug will be efficacious. Among children and adolescents, only fluoxetine, which is approved for use in children by the U.S. Food and Drug Administration (FDA), appears to have a favorable risk-benefit profile (see What’s New? Advisory Warnings of SSRI Use in Pediatric Depression).20
In bipolar depression, antidepressant medications may lead to cycle acceleration or induction of mania. However, SSRIs and bupropion, which have effects on norepinephrine (NE) and dopaminergic function, may be less likely to induce these effects than MAOIs or TCAs.
A number of side effects are reported with MAOIs, TCAs, and SSRIs. Commonly reported side effects of MAOIs include sedation or agitation, insomnia, dry mouth, impotence, and weight gain. MAOIs also may induce acute and heightened elevations in blood pressure (e.g., hypertensive crisis) after intake of tyramine-rich foods, such as aged cheeses, sour cream, pods of broad beans, pickled herring, liver, canned figs, raisins, and avocados. In addition, MAOI interactions with TCAs,
SSRIs, stimulants, and over-the-counter flu medications are dangerous and should be avoided. Because of these adverse side effect issues, MAOIs are used less often than other antidepressant medications.
TCAs may produce sedation, insomnia, orthostatic hypotension, seizures, and weight gain. Some TCAs have moderate anticholinergic side effects, including constipation, urinary hesitancy or retention, dry mouth, blurred vision, and memory impairment. These side effects may be an issue when considering TCA treatment of older adults, in which case, the TCAs desipramine and nortriptyline may be preferred because of their reduced anticholinergic, cardiovascular, and sedating effects.
Common side effects of SSRIs include sleep disturbances (e.g., insomnia) and nausea. However, agitation, allergic skin reactions, dry mouth, anxiety, altered appetite, and sexual dysfunction have been reported. Unlike MAOIs and TCAs, SSRIs do not have pronounced effects on the cardiovascular or cholinergic systems. SSRIs are potent inhibitors of cytochrome P-450 isoenzymes, which are involved in drug metabolism. Therefore, SSRIs may lead to dangerous elevations in blood concentrations of other psychiatric medications when taken together. SSRIs should not be taken with MAOIs or immediately after discontinuing MAOI treatment. A serotonin syndrome characterized by excitement or autonomic hyperactivity, abdominal pain, rigidity, and hyperthermia may develop, leading to coma or death.
Side effects of atypical antidepressants may include sedation, dry mouth, weight gain, and constipation. Nefazodone and trazodone have been associated with hepatic toxicity. Venlafaxine and reboxetine lack many of the serious side effects associated with TCAs; however, sweating, dry mouth, and some sedation may occur.
Electroconvulsive therapy (ECT) is used when individuals fail to respond to antidepressants or when they are severely depressed, pregnant, suicidal, or psychotic. ECT is effective in alleviating depressive symptoms in about 50% to 80% of people who may then begin to respond to antidepressant medications. Although the mechanism of action of ECT is not clear, the procedure is known to produce alterations in monoamine systems.
Bipolar Disorder
A number of FDA-approved treatments are available for bipolar disorders.21 Individuals with bipolar I disorder are usually treated with lithium, the first choice of treatment that also reduces the risk of suicide. In some cases, lithium in combination with SSRIs is used to treat bipolar disorder. In addition to lithium, a number of medications for bipolar disorders, colloquially referred to as mood stabilizers, are available, including anticonvulsants (e.g., carbamazepine, divalproex, gabapentin, lamotrigine, topiramate) or atypical antipsychotics (e.g., clozapine, olanzapine). Some individuals also may benefit from thyroid augmentation (levothyroxine). As in depression, ECT is administered when manic individuals fail to respond to medication, are pregnant, or have cardiovascular disease.
Frequently reported side effects of lithium treatment include increased thirst, tremors, diarrhea, and weight gain, which diminish over time. A potentially serious side effect is lithium toxicity. Lithium is normally removed from the kidneys; however, when the body is sodium depleted, the kidneys will reabsorb sodium along with lithium. Individuals receiving lithium treatment are advised to avoid physically demanding activities that may dehydrate the body and to seek medical attention during fever or other conditions that may increase sweating. Anticonvulsant treatment may produce unsteadiness, dizziness, tremors, nausea, and blurry vision. Adverse effects of atypical antipsychotics may include movement problems (e.g., akathisia) and metabolic disturbances that lead to weight gain.
In addition to pharmacotherapy, psychotherapy can be beneficial for those who have difficulty with psychosocial stressors, such as self-esteem, legal problems, fear of recurrence, and interpersonal conflicts. Treatment involves a combination of making the individual aware of the bipolar disorder, coping with psychosocial stressors, facilitating drug compliance, and monitoring symptom recurrences.
Unlike the use of mood stabilizers to primarily treat the mania and rapid cycling in bipolar I disorder, a major focus in the treatment of bipolar II disorder is on the recurrent depressive symptoms. Although mood stabilizers are used, antidepressants alone (e.g., escitalopram, fluoxetine, venlafaxine) are reported to be effective in treating bipolar II disorder.
ANXIETY DISORDERS
Fear and anxiety are normal feelings expressed in threatening or harmful situations. The symptoms may include arousal, tenseness, and increased autonomic activity such as heart rate, blood pressure, and respiration. In addition, individuals often engage in protective behavioral responses such as flight or avoidance. These physiologic and behavioral responses reflect the individuals’ evolutionary heritage. Their expression allowed humans to adapt and cope under a variety of situational challenges. However, when fear and anxiety become too intense and undermine the ability to function on a daily basis, the individual may develop an anxiety disorder. Anxiety disorders are the most prevalent psychiatric disorder, occurring in approximately 10% to 30% of the general population. Notably, many individuals with anxiety disorders develop major depression, and those with major depression often suffer from anxiety disorders. Comorbidity of anxiety disorders and depression suggest a common neural pathophysiologic basis linking these two mental illnesses.
Freud used the term anxiety neurosis to denote feelings of fearfulness, panic, terror, and doom. Anxiety neurosis is classified into a number of distinct anxiety disorders based on refined clinical observations. The DMS-IV-TR lists eight recognized anxiety disorders: panic disorder, agoraphobia, generalized anxiety disorder, social phobia, specific phobia, obsessive-compulsive disorder, posttraumatic stress disorder, and acute stress disorder. This section presents an overview of panic disorder, generalized anxiety disorder, posttraumatic stress disorder, and obsessive-compulsive disorder.
Panic Disorder
Panic disorder is a well-studied psychiatric condition that consists of multiple disabling panic attacks. Approximately 2% to 3% of women and 0.5% to 1.5% of men have panic disorder. Between panic attacks the individual spends an excessive amount of time worrying about future panic attacks. Panic attacks are characterized by intense autonomic arousal involving a wide variety of symptoms, including lightheadedness, a racing heart, difficulty breathing, chest discomfort, generalized sweating, general weakness, trembling, abdominal distress, and chills or hot flashes. In addition, the individual experiences the fear of losing control and dying. Symptoms originally occur spontaneously and vary in length from several minutes to an hour. If the symptoms are prolonged, they can be disabling.
A notable complication of panic disorder is the development of agoraphobia or phobic avoidance of places or situations where escape or help is not readily available. The agoraphobic individual will avoid being away from home, standing in line or in a crowd, or riding a train, plane, or automobile. Severe agoraphobia leads to individuals being housebound.
Etiology and Pathophysiology
Genetic factors appear to play a major role in panic disorder. The risk is nearly 20% among first-degree relatives of panic disorder individuals. The prevalence of panic disorder is about 1.5% in men and up to 3.0% in women with no family history of the illness. Some studies suggest that the cholecystokinin (CCK) receptor gene on chromosome 11p may be linked to panic disorder.22
Although the etiology of panic attacks is not known, some factors may be acting on vulnerable brainstem regions to provoke symptoms. For example, physiologic information from the peripheral cardiovascular and respiratory systems is regulated closely by cells in the brainstem, which may activate central autonomic pathways. Fearful perceptions and thoughts emanating from the cerebral cortex may further contribute by activating neural circuits in the temporal lobe and brainstem, which may then facilitate the production of panic symptoms.
Brain regions likely to be involved in panic attacks include the locus ceruleus–NE system and temporal lobe structures, including the amygdala and hippocampus23 (see Figures 18-7 and 18-9). Both the hippocampus and locus ceruleus monitor internal and external signals and increase their activity when aroused. The locus ceruleus is sensitive to respiratory and cardiovascular changes in the periphery, such as hyperventilation. Abnormal firing of locus ceruleus neurons in response to peripheral autonomic signals may contribute to the onset of panic attacks.
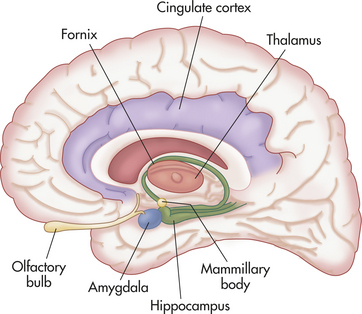
Figure 18-9 The limbic system. Structures of the limbic system play important roles in emotion, learning, and memory. Pathophysiology in limbic structures is frequently found in mental disorders.
Panic disorder also may involve the GABA-benzodiazepine (BZ) receptor system. BZ receptors and GABAA receptors are functionally coupled. BZ increases the GABAA ion channel response to GABA, thereby elevating chloride ion influx and producing a neuronal inhibitory effect. Brain imaging work reveals a reduction in BZ receptor binding in brain regions including the hippocampus, insular, and prefrontal cortex.24 Thus an alteration in inhibitory neuromodulation may contribute to panic disorder.
Individuals with panic disorder respond to some pharmacologic challenges differently than do healthy controls. Physical symptoms of panic attacks are reproduced to a certain degree in panic-prone persons by inhalation of carbon dioxide, caffeine ingestion, sodium lactate, and NE receptor compounds. Many of these agents appear to directly or indirectly modulate the locus ceruleus–NE system, thereby contributing to the production of panic. For example, panic-prone individuals are sensitive to the anxiety-provoking effects of yohimbine, an α2-adrenergic receptor antagonist, and exhibit elevated levels of plasma MHPG (a norepinephrine metabolite), cortisol, and cardiovascular responses. Clonidine, the α2-adrenergic receptor agonist, produces less sedation and less plasma MHPG, but greater hypotension when given to individuals with panic disorder. Other drugs that block the benzodiazepine receptor or stimulate the cholecystokinin receptor are reported to increase panic attacks and feelings of anxiety in people with panic disorder. As indicated, these receptor systems may be involved in the pathophysiology of panic disorder.
Treatment
Panic disorder is highly treatable. Up to 80% of panic disorder individuals respond to CBT and antidepressant medication, either separately or in combination. In CBT, the individual learns that the physical symptoms are not fatal and attempts to exert control over the anxiety and panic. For example, breathing exercises to control hyperventilation serve to lessen the intense physiologic symptoms of panic, such as elevated heart and respiration rates. Another benefit of CBT is awareness of the benefits associated with drug medications and compliance. For those individuals who have mild agoraphobia, CBT alone may be effective.
Antidepressants such as SSRIs are considered first-line medications for panic disorder. Among the SSRIs, paroxetine and sertraline have received FDA approval specifically for panic disorder. Venlafaxine, a serotonin-norepinephrine reuptake blocker, also is effective.
BZs such as alprazolam and clonazepam are another class of medications used in the treatment of panic disorder; their efficacy may be related in part to their inhibitory effects on locus ceruleus neuronal activity. These drugs also are used as an adjunct or augmentation therapy for individuals who do not fully respond to SSRIs. Short-term effects of BZ treatment include sedation, ataxia, and cognitive impairments. A potential complication of long-term BZ treatment is physiologic and psychologic dependence. Abrupt BZ withdrawal may produce a withdrawal syndrome that includes a heightened reemergence or rebound of anxiety, insomnia, photophobia, and diarrhea. These symptoms may be lessened with gradual tapering off of BZ medication. Individuals taking BZs may benefit from CBT, which could reduce their reliance on these drugs.
Generalized Anxiety Disorder
Excessive and persistent worries are the hallmarks of generalized anxiety disorder (GAD). The individual worries about life events such as marital relationships, job performance, health, money, or social status. The lifetime prevalence rates of GAD range from 4.1% to 6.6%, with somewhat higher rates in women than in men. GAD usually emerges in the early 20s, but can occur in childhood. Six major symptoms of GAD have been identified including restlessness, muscle tension, irritability, being easily fatigued, difficulty concentrating, and difficulty sleeping. The individual startles easily and frequently suffers from depression and panic attacks. The severity of symptoms fluctuates over time and may be linked to the changing nature of environmental stress. Although GAD tends to be a chronic disorder, the symptoms may become less severe with age. A frequent complication of GAD is the development of depression or substance abuse. The latter may occur as a result of self-medication with alcohol or drugs to relieve the symptoms.
Etiology and Pathophysiology
The etiology and pathophysiology of GAD are poorly understood. Female twin studies suggest a concordance rate of 30%, but disease genes linked to specific chromosomes have yet to be identified. Abnormalities in the norepinephrine and serotonin systems were reported in GAD.25 For example, there is a reduction in α2-adrenergic receptor binding, a decrease in serotonin levels in CSF, and reduced platelet binding of paroxetine, an SSRI.
A prominent alteration of GAD involves the GABA-BZ receptors. In GAD there is a reduction in peripheral BZ receptors, which increase after treatment with BZ drugs. Brain imaging work indicates GAD subjects have greater homogeneity of the cerebrum, BZ receptor distribution, and a significant reduction in the left temporal hemisphere.26 The therapeutic effects of BZs may lie in part in their ability to normalize these BZ receptor alterations.
Treatment
GAD is diagnosed when an individual spends at least 6 months worrying excessively and exhibits at least three of the six symptoms.11 5-HT/NE reuptake inhibitors, such as venlafaxine or the SSRIs paroxetine and escitalopram, have become first-line therapeutics for managing GAD. These medications may produce relief of GAD symptoms within 1 week and are effective in treating comorbid symptoms of depression. Buspirone, which has affinity for serotonin receptors (5-HT1A) is another treatment option, although the onset of clinical efficacy may take 2 weeks. The primary side effects of buspirone, which lessen over time, include dizziness, headaches, nausea, and mild nervousness. GAD nonresponders to 5-HT/NE reuptake inhibitors or buspirone may be placed on BZs. However, because GAD tends to be chronic, and comorbid with depression or other anxiety disorders,27 BZs are usually limited to uncomplicated cases of GAD. SSRIs also have been used in the treatment of the young with GAD. In addition to drug therapy, treatment of GAD may involve behavioral therapy, during which the individual learns relaxation techniques to control anxiety.
Posttraumatic Stress Disorder
Exposure to a terrifying or life-threatening trauma may produce posttraumatic stress disorder (PTSD).28,29 Although the disorder was initially described in combat situations and called “shell shock,” “war neurosis,” or “traumatic neurosis,” PTSD does not arise solely from exposure to the battlefield. Exposure to any major trauma, including serious accidents, natural disasters (such as earthquakes), child abuse, kidnapping, and violent attacks (such as rape or muggings), that involves intense fear, threat of death, or helplessness may induce PTSD. The disorder may develop within hours of the traumatic experience or after several months or years. In PTSD, the individual reexperiences the traumatic event as intrusive recollections or flashbacks during the day and during persistent nightmares. During a flashback, images, odors, sounds, and negative emotions are recalled and lead to marked distress. The duration of the flashback varies from seconds to hours or, in rare cases, several days. Nightmares replicate the traumatic experiences and often prevent further sleep. Exposure to cues associated with the life-threatening event also may trigger psychologic distress, intense autonomic arousal, and avoidance behavior. The individual shows emotional numbing or detachment from others and avoids activities that may lead to a recollection of thoughts, feelings, places, or people involved in the trauma. Persistent symptoms of PTSD include sleeping difficulties, irritability, lack of concentration, hypervigilance, and exaggerated startle response.
The lifetime prevalence rate of PTSD is 7% to 8%. In men, PTSD is usually found among combat veterans, whereas PTSD in women is often related to rape or assault. Abused children also may develop PTSD. Certain individuals appear to be vulnerable to PTSD. Those with a history of psychiatric illness (major depression, panic disorder) or those lacking strong social support are at increased risk and may be more sensitive to the effects of traumatic stress.
Etiology and Pathophysiology
The primary etiology of PTSD is exposure to a terrifying life-threatening event and may involve several neural structures and neurotransmitter systems. Structural brain imaging studies reported that combat-related PTSD victims have a smaller hippocampus, a brain structure susceptible to the damaging effects of the stress hormone cortisol and excitatory amino acids. Pediatric PTSD studies reveal a more generalized effect of trauma on reducing total brain volume. In functional imaging studies, PTSD individuals exposed to trauma-related stimuli generally exhibit increased activation in the amygdala and diminished activity in some prefrontal cortical areas. Reduced activation of the anterior cingulate cortex also has been reported in some but not all studies.30
As in panic disorder, BZ binding is altered in those with PTSD. Individuals with PTSD have reduced distribution of BZ receptor binding in the prefrontal cortex compared with healthy controls.31 This reduction in BZ receptor distribution was not found in other brain regions.
These structural and functional data involving the hippocampus, amygdala, and prefrontal cortex are highly relevant to the pathophysiology of PTSD because these brain regions normally play important roles in how fearful memories are stored, retrieved, and forgotten. The pathogenesis of PTSD is hypothesized to stem from an altered fear-learning process that may involve, for example, amygdala hyperresponsiveness, lack of prefrontal cortical inhibition, hippocampal dysfunction, and/or susceptibility to the adverse effects of stress.32 Support of this hypothesis is based on PTSD studies showing reduced hippocampal volume and increased metabolic activity in the amygdala. Continued dysfunction and deterioration of this fear-based memory system may underlie chronic PTSD.
Treatment
PTSD is diagnosed according to the duration and timing of symptoms (DSM-IV-TR). When the duration of symptoms is less than 3 months, PTSD is diagnosed as acute. PTSD is diagnosed as chronic when symptoms persist for 3 months or longer or when the delay of onset is longer than 6 months after the trauma. As in GAD, the severity of symptoms fluctuates over time. Chronic PTSD lasting for years may occur in 30% of diagnosed individuals. Treatment methods involve drug medications and CBT. The individual learns to control the anxiety symptoms and memories of the event during therapy. Among war veterans, group or family therapy is supported by the Veterans Administration.
Paroxetine and sertraline are considered first-line SSRI medications for chronic PTSD because of their tendency to lessen the recurrent nightmares and flashbacks. For chronic PTSD, long-term SSRI therapy is required for steady improvement of symptoms and quality of life. Other antidepressants such as the TCAs (amitriptyline and imipramine) have moderate effects and are second-line drugs. Some data suggest that nefazodone and bupropion may provide benefits. The tendency to use antidepressants is because of the high prevalence of comorbid depression and substance abuse. BZs may be used during the aftermath of a traumatic event to control hyperarousal symptoms such as irritability, insomnia, and muscle tension. However, there is no clear evidence that BZs have clinical efficacy or provide prophylaxis against the development of chronic PTSD—they should be carefully monitored among individuals with a history of drug abuse.
Obsessive-Compulsive Disorder
Repetitive, intrusive thoughts and/or compulsions are the hallmarks of obsessive-compulsive disorder (OCD). These thoughts and acts are irrational, impair normal functioning, and may cause marked distress. Obsessions may involve a preoccupation with contamination, doubting, religious or sexual themes, or the belief that a negative outcome will occur if a specific act is not performed. Compulsions are physical and mental ritualized acts such as washing, cleaning, checking, counting, organizing, hoarding, and repeating specific thoughts or prayers. The lifetime prevalence rates of OCD range from 1.2% to 3.3% with an age of onset between 20 to 25 years. OCD occurs equally in adult men and women; however, many begin to experience symptoms during childhood or adolescence.
OCD individuals are often diagnosed when obsessions or compulsions cause severe distress, are time consuming, or interfere with normal daily activities.11 In many cases the OCD individual may present with other psychiatric illnesses.33 In adulthood, comorbidity with major depression, other anxiety disorders (especially panic disorders and GAD), and Tourette syndrome are common. Among children, tic disorders, attention deficit/hyperactivity disorder, and depression coexist with OCD.
Etiology and Pathophysiology
Family studies indicate a risk of about 10% to 12% among first-degree relatives. These first-degree relatives are also at increased risk (4.6%) of Tourette disorder and tics in comparison with control relatives (1%). Thus OCD and Tourette syndrome may share common genes and pathophysiology.
Abnormalities of the basal ganglia–frontocortical circuitry are found in OCD.34 Some studies have shown increased orbitofrontal and thalamic volumes in OCD but not in the caudate nucleus of the basal ganglia (Figure 18-10). More consistent data are obtained from functional imaging studies, which report an increase in orbitofrontal and anterior cingulate cortical activity. Orbitofrontal and anterior cortical hyperactivity may be responsible for intrusive thoughts, obsessions, and anxiety, which drives the basal ganglia to engage in compulsive ritualized acts as a means to alleviate the anxious obsessions. Studies further suggest that when provoked, OCD individuals consistently show increased activity in the orbitofrontal, anterior cingulate, and caudate nucleus areas of the brain. Of particular relevance to prognosis, OCD individuals with very high frontocortical activity are likely to respond more poorly to treatment than those with lower pretreatment activity in the orbitofrontal cortex.
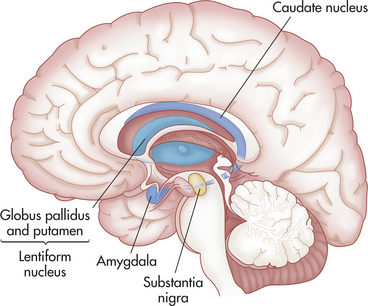
Figure 18-10 Basal ganglion. Structures of the basal ganglion, which include the caudate nucleus, putamen, globus pallidus, and substantia nigra, are important in movement.
Abnormalities in serotonin and dopamine functions may contribute to the pathophysiology of OCD. Serotonin agonists exacerbate the symptoms of OCD, and serotonin synthesis is decreased in the prefrontal cortex and caudate nucleus. Stimulation of the dopamine system increases repetitive acts, which may be blocked by dopamine antagonists. However, the dopamine stimulant-induced compulsions are not accompanied by anxiety, which has led some investigators to suggest that lack of serotonin control over the dopamine system may be a primary defect in OCD.
Treatment
Because of the chronic nature of OCD, long-term treatment consisting of a combination of pharmacotherapy and CBT is often required. SSRIs, including citalopram, fluvoxamine, paroxetine, and sertraline, are the first drugs of choice for OCD.35 Approximately 70% to 80% of OCD individuals will show a partial response that may be further improved by other medications. For example, clonazepam, a BZ, is found to improve the effects of fluoxetine and clomipramine therapy. Antipsychotic drugs, haloperidol and risperidone, in combination with SSRIs, are also effective especially in comorbid OCD and tic disorders. Normalization of dysfunctional serotonin and dopamine systems in OCD may be the basis for the therapeutic effects of SSRIs and dopamine receptor–related drugs.
CBT involves daily exposure to cues that elicit distress followed by preventing the individual from engaging in compulsive rituals for at least an hour or until the anxiety subsides. This exposure and response prevention therapy can produce long-term symptom remission and is effective in adults and children who are able to tolerate the exposure-induced anxiety component.
For individuals with severe treatment-resistant OCD, neurosurgery is performed to disconnect the basal ganglia from the frontal cortex.36 This lesioning procedure results in significant relief of obsessions and compulsions in nearly 50% of individuals and provides further evidence of a pathophysiology in the basal ganglia–frontocortical circuitry in OCD.
Affective flattening 651
Agoraphobia 659
Alogia 651
Anhedonia 651
Anxiety disorder 658
Avolition 651
Bipolar disorder 652
Delusion 650
Depression 655
Dopamine hypothesis 648
Dorsolateral prefrontal cortex (DLPFC) 648
Dysphoric mood 655
Formal thought disorder 651
Gamma-aminobutyric acid (GABA) 648
Generalized anxiety disorder (GAD) 660
Hallucination 650
Major (unipolar) depression 652
Manic 656
Monoamine hypothesis of depression 653
Mood 652
Neuroleptics 651
Obsessive-compulsive disorder 661
Panic disorder 659
Posttraumatic stress disorder (PTSD) 660
Poverty of content 651
Psychotic episode 650
Schizophrenia 647
Thought disorder 647
Working memory 648
REFERENCES
1. Lewis, D.A., Levitt, P. Schizophrenia as a disorder of neurodevelopment. Annu Rev Neurosci. 2002;25:409–432.
2. Marenco, S., Weinberger, D.R. The neurodevelopmental hypothesis of schizophrenia: following a trail of evidence from cradle to grave. Dev Psychopathol. 2000;12(3):501–527.
3. Berman, K.F., Meyer-Lindenberg, A. Functional brain imaging studies in schizophrenia. In Charney D.S., Nestler E.J., eds.: Neurobiology of mental illness, ed 2, New York: Oxford, 2004.
4. Shenton, L.D., et al. A review of MRI findings in schizophrenia. Schizophr Res. 2001;49(1-2):1–52.
5. Ross, C.A., et al. Neurobiology of schizophrenia. Neuron. 2006;52(1):139–153.
6. Ho, B.C., et al. Progressive structural brain abnormalities and their relationship to clinical outcome. Arch Gen Psychiatry. 2003;60:585–594.
7. Costa, E., et al. Dendritic spine hypoplasticity and downregulation of reelin and GABAergic tone in schizophrenia vulnerability. Neurobiol Dis. 2001;8(5):723–742.
8. Fatemi, S.H., Earle, J.A., McMenomy, T. Reduction in reelin immunoreactivity in hippocampus of subjects with schizophrenia, bipolar disorder and major depression. Mol Psychiatry. 2000;5(6):654–663.
9. Duncan, G.E., Sheitman, B.B., Lieberman, J.A. An integrated view of pathophysiological models of schizophrenia. Brain Res Brain Res Rev. 1999;29(2-3):250–264.
10. Kennedy, J.L., et al. The genetics of adult-onset neuropsychiatric disease: complexities and conundra? Science. 2003;302(5646):822–826.
11. American Psychiatric Association. Diagnostic and statistical manual of mental disorders, ed 4, Text Revision (DSM-IV-TR). Washington, DC: American Psychiatric Association; 2000.
12. Tamminga, C.A. Principles of the pharmacotherapy of schizophrenia. In Charney D.S., Nestler E.J., eds.: Neurobiology of mental illness, ed 2, New York: Oxford, 2004.
13. Rathod, S., et al. Cognitive-behavioral therapy for medication-resistant schizophrenia: a review. J Psychiat Pract. 2008;14(1):22–33.
14. Caspi, A., et al. Influence of life stress on depression: moderation by a polymorphism in the 5-HTT gene. Science. 2003;301(5631):386.
15. Heninger, G.R., et al. The revised monoamine theory of depression: a modulatory role for monoamines, based on new findings from monoamine depletion experiments in humans. Pharmacopsychiatry. 1996;29(1):2–11.
16. Holsboer, F. The corticosteroid receptor hypothesis of depression. Neuropsychopharmacology. 2000;23:477.
17. Bartalena, L., et al. Nocturnal serum thyrotropin (TSH) surge and the TSH response to TSH-releasing hormone: disassociative behavior in untreated depression. J Clin Endocrinol Metab. 1990;71(3):650–655.
18. Drevets, W.C. Prefrontal cortical-amygdalar metabolism in major depression. Ann N Y Acad Sci. 1999;877:614–637.
19. Rajkowska, G. Depression: what we can learn from postmortem studies. Neuroscientist. 2003;9(4):273–284.
20. Whittington, C.J., et al. Selective serotonin reuptake inhibitors in childhood depression: systematic review of published versus unpublished data. Lancet. 2004;363(9418):1341–1345.
21. Goldberg, J.F. What psychotherapists should know about pharmacotherapies for bipolar disorder. J Clin Psychol. 2007;63(5):475–490.
22. Kennedy, J.L., et al. Investigation of cholecystokinin system genes in panic disorder. Mol Psychiatry. 1999;4(3):284.
23. Goddard, A.W., Charney, D.S. Toward an integrated neurobiology of panic disorder. J Clin Psychiatry. 1997;58(Suppl 2):4–11.
24. Malizia, A.L., et al. Decreased brain GABAA-benzodiazepine receptor binding in panic disorder. Arch Gen Psychiatry. 1998;55(8):715–720.
25. Jetty, P.V., et al. Neurobiology of generalized anxiety disorder. Psychiatr Clin North Am. 2001;24(1):75.
26. Tiihonen, J., et al. Cerebral benzodiazepine receptor binding and distribution in generalized anxiety disorder. Mol Psychiatry. 1997;2(6):463–471.
27. Bruce, S.E. Infrequency of “pure” GAD: impact of psychiatric comorbidity on clinical course. Depress Anxiety. 2001;14(4):219–225.
28. Charney, D.S., et al. Psychobiologic mechanisms of posttraumatic stress disorder. Arch Gen Psychiatry. 1993;50(4):294–305.
29. Southwick, S.M., et al. Neurotransmitter alterations in PTSD: catecholamines and serotonin. Semin Clin Neuropsychiatry. 1999;4(4):242–248.
30. Pissota, A., et al. Neurofunctional correlates of posttraumatic stress disorder: a PET symptom provocation study. Eur Arch Psychiatry Clin Neurosci. 2002;252(2):68–75.
31. Bremner, J.D., et al. Decreased benzodiazepine receptor binding in prefrontal cortex in combat-related posttraumatic stress disorder. Am J Psychiatry. 2000;157(7):1120–1126.
32. Bremner, J.D., et al. Structural and functional plasticity of the human brain in posttraumatic stress disorder. Prog Brain Res. 2008;167:171–186.
33. Fireman, B., et al. The prevalence of clinically recognized obsessive-compulsive disorder in a large health maintenance organization. Am J Psychiatry. 2001;158(11):1904–1910.
34. Saxena, S., Rauch, S.L. Functional neuroimaging and the neuroanatomy of obsessive-compulsive disorder. Psychiatr Clin North Am. 2000;23(3):563–586.
35. Swedo, S.E., Snider, L.A. The neurobiology and treatment of obsessive-compulsive disorder. In Charney D.S., Nestler E.J., eds.: Neurobiology of mental illness, ed 2, New York: Oxford, 2004.
36. Greenberg, B.D., Murphy, D.L., Rasmussen, S.A. Neuroanatomically based approaches to obsessive-compulsive disorder. Psychiatr Clin North Am. 2000;23(3):671–686.