INNATE IMMUNITY
INFLAMMATION



People are exposed daily to an environment containing a large variety of toxic substances and potentially infectious and disease-causing microorganisms. Without an efficient system of protection most individuals would succumb to these hazards early in life. That system consists of multiple complementary and interdependent layers. An outer layer of specialized epithelium, including the skin and mucosal surfaces, is relatively resistant to most environmental hazards and resists infection with disease-causing microorganisms.1 If the epithelial barrier is damaged, a highly efficient local and systemic response (inflammation) is mobilized to limit the extent of damage, protect against infection, and initiate repair of the damaged tissue. The natural epithelial barrier and inflammation confer innate resistance and protection, commonly referred to as innate, native, or natural immunity. Inflammation associated with infection usually initiates an adaptive process that results in a long-term and very effective immunity to the infecting microorganism, referred to as adaptive, acquired, or specific immunity. Adaptive immunity is relatively slow to develop but has memory and more rapidly targets and eradicates a second infection with a particular disease-causing microorganism.
The information presented in this chapter introduces the components and processes of innate immunity and sets the stage for Chapter 7, which discusses adaptive immunity. Although inflammation and acquired immunity provide protection, either genetic or acquired aberrations in these processes can lead to disease. Diminution of innate or acquired immunity may lead to critically decreased resistance to infection. Excessive inflammation or acquired immunity may lead to damage to normal tissue or organs. Both may result in severe and potentially fatal disease, examples of which are discussed in Chapter 8. Many microorganisms that cause disease have developed methods of bypassing our protective systems. These are discussed in Chapter 9. Each chapter is designed to render an overview and is not intended to be all-inclusive. Protective mechanisms consist of a very large number of soluble factors and cells and would require many more pages to discuss in adequate detail. Different classes or groups of molecules and cells will be discussed, but only a few examples will be described in detail. Some components directly participate in the protective response, whereas others are designed to limit the extent of the response.
HUMAN DEFENSE MECHANISMS
Innate immunity includes two lines of defense: natural barriers and inflammation (Table 6-1). Natural barriers are physical, mechanical, and biochemical barriers at the body’s surfaces and are in place at birth to prevent damage by substances in the environment and thwart infection by pathogenic microorganisms. If the surface barriers are breached, the second line of defense, the inflammatory response, is activated to protect the body from further injury, prevent infection of the injured tissue, and promote healing. The inflammatory response is a rapid activation of biochemical and cellular processes that is relatively nonspecific, with similar responses being initiated against a wide variety of causes of tissue damage.
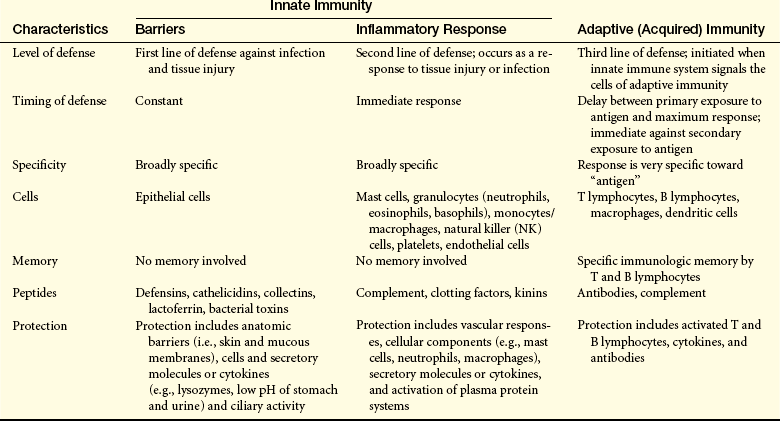
FIRST LINE OF DEFENSE: PHYSICAL, MECHANICAL, AND BIOCHEMICAL BARRIERS
Physical and Mechanical Barriers
The physical barriers that protect against damage and infection are composed of tightly associated epithelial cells including those of the skin and of the membranous sheets lining the gastrointestinal, genitourinary, and respiratory tracts (Figure 6-1).2 The mucosal epithelial cells are highly interconnected junctions that prohibit the passage of microorganisms. The normal turnover of the cells in these sites as well as mechanisms for “washing” the surfaces may mechanically remove many infectious microorganisms and prevent their residence on the epithelial surfaces. For instance, the routine sloughing off and replacement of dead skin cells also removes adherent bacteria. Mechanical cleansing of the surfaces includes vomiting and urination. Goblet cells of the upper respiratory tract produce mucus that coats the epithelial surface and traps microorganisms that are removed by hairlike cilia that mechanically move the mucus upward to be expelled by coughing or sneezing. Additionally, the low temperature on the skin generally inhibits microorganisms, most of which prefer temperatures near 37° C for more efficient growth.
Biochemical Barriers
Epithelial surfaces also provide biochemical barriers by synthesizing and secreting substances meant to trap or destroy microorganisms. Mucus, perspiration (or sweat), saliva, tears, and earwax are all examples of biochemical secretions that can trap potential disease-causing microorganisms and contain substances that will kill the microorganisms. Sebaceous glands in the skin also secrete antibacterial and antifungal fatty acids and lactic acid. Perspiration, tears, and saliva contain an enzyme (lysozyme) that attacks the cell walls of gram-positive bacteria. These glandular secretions result in an acidic skin surface (pH 3 to 5), making it an inhospitable environment for most bacteria.
In addition, the body has a complex array of proteins that function to destroy pathogens before they can colonize a human host. Some of these proteins function on the surface of the same epithelial sheets that provide the physical barrier. Others, however, are meant to defeat microorganisms that have already crossed the physical barrier.
Epithelial-Derived Chemicals
Epithelial cells secrete small-molecular-weight antimicrobial peptides. These are generally positively charged polypeptides of approximately 15 to 95 amino acids and can be divided into two classes—cathelicidins and defensins—based on their three-dimensional structures. Both classes are in very high local concentrations and are toxic to several bacteria, fungi, and viruses.3–5 Cathelicidins have a linear α-helical shape, and only one is currently known to function in humans. In contrast, about 50 different defensins have been identified thus far. All are triple-stranded β-sheet structures. Defensin molecules contain three intrachain disulfide bonds and can be further subdivided into α (at least 6 identified in humans) and β types (at least 10 identified, but perhaps up to 40 different molecules), depending on how the cysteine residues are connected during formation of the disulfide linkages. The α-defensins often require activation by proteolytic enzymes, whereas the β-defensins are synthesized in active forms. Bacteria have cholesterol-free cell membranes, which may allow cathelicidins to insert into and disrupt their membranes. Given the similarity in their chemical charges, defensins may kill bacteria in the same way. These same chemicals also may contribute to other means of protection because they are also produced by monocytes, macrophages, and neutrophils, which are components of the inflammatory response. Cathelicidin is stored in neutrophils, mast cells, and a variety of epithelial cells. The α-defensins are particularly rich in the granules of neutrophils and may contribute to the killing of bacteria by those cells. They are also found in Paneth cells lining the small intestine, where they protect against a variety of disease-causing microorganisms. The β-defensins are found in a variety of epithelial cells lining the respiratory, urinary, and intestinal tracts, as well as in the skin. In addition to antibacterial properties, β-defensins may also help protect epithelial surfaces from human immunodeficiency virus (HIV) infection. Both classes of antimicrobial peptides also can activate cells of innate and acquired immunity.
The lung also produces and secretes a family of glycoproteins, collectins, which includes surfactant proteins A through D and mannose-binding lectin.6 The binding site of each collectin reacts with different affinities to a range of monosaccharides, enabling collectins to recognize a wide array of pathogenic microorganisms. Collectin binding facilitates recognition of the microorganism by macrophages, enhancing macrophage attachment, phagocytosis, and killing. Collectins play a major role in protection against respiratory infections.7
Other epithelial antimicrobials include resistin-like molecule β, bactericidal/permeability-inducing protein, and antimicrobial lectins.2,8 Resistin-like molecule β is found in the intestinal goblet cells, where it appears to protect against helminth infections. Bactericidal/permeability-inducing protein (BPI) is stored in neutrophils and intestinal epithelium. BPI specifically reacts with lipopolysaccharide on the surface of gram-negative bacteria, resulting in bacterial lysis. Antimicrobial lectins are carbohydrates that are found in intestinal epithelium and have activity against gram-positive bacteria.
Bacteria-Derived Chemicals
Many of the body’s surfaces are colonized with a spectrum of bacteria that do not normally cause disease, the normal bacterial flora. These microorganisms are frequently beneficial. For instance, the intestine becomes progressively colonized after birth with hundreds of different species of bacteria, some of which help digest food, releasing nutrients that are taken up by the body. They are responsible for digesting fatty acids, large polysaccharides, and other dietary substances, producing vitamin K, and assisting in the absorption of various ions, such as calcium, iron, and magnesium. The normal flora also contributes to our innate protection by producing several chemicals (e.g., ammonia, phenols, indols) that inhibit colonization by disease-causing microorganisms.9 The normal intestinal flora can be altered by prolonged antibiotic treatment, decreasing its protective activity, and leading to overgrowth of other microorganisms, such as the yeast Candida albicans or the bacterium Clostridium difficile. The bacterium Lactobacillus is a major constituent of the normal vaginal flora in healthy women. This microorganism produces a variety of chemicals (e.g., hydrogen peroxide, lactic acid, bacteriocins) that help prevent infections of the vagina and urinary tract by other bacteria and yeast. Prolonged antibiotic treatment can also diminish colonization with Lactobacillus and increase the risk for urologic or vaginal infections, such as toxic shock syndrome.
SECOND LINE OF DEFENSE: THE INFLAMMATORY RESPONSE
If cells and tissues are damaged the inflammatory response is usually activated. Injury can have a variety of causes including infection, mechanical damage, oxygen deprivation (ischemia), nutrient deprivation, genetic or immune defects, chemical agents, temperature extremes, or ionizing radiation (Figure 6-2). Inflammation (1) depends on the activity of both cellular and chemical components and (2) is nonspecific, meaning that it takes place in approximately the same way regardless of the type of stimulus or whether exposure to the same stimulus has occurred in the past.
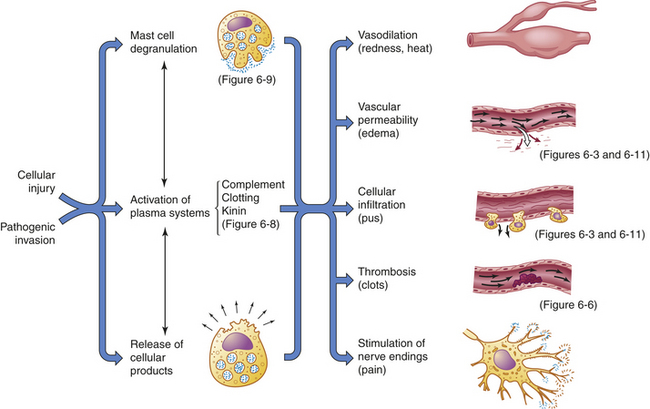
Figure 6-2 Acute inflammatory response. Inflammation is usually initiated by cellular injury, which results in mast cell degranulation, the activation of three plasma systems, and the release of subcellular components from the damaged cells. These systems are interdependent, so that induction of one (e.g., mast cell degranulation) can result in activation of the other two. The result is the development of microscopic changes in the inflamed site, as well as characteristic clinical manifestations. The figure numbers refer to those in which more detailed information may be found on that portion of the response.
Vascular Response
Inflammation occurs in tissue that has a blood supply (vascularized) and results in a group of easily observable characteristics: redness, heat, swelling, and pain. This tetrad represents the “cardinal signs of inflammation” and was identified in the first century by a Roman writer, Celsus. Microscopically, inflammatory changes occur at the vascular level (Figure 6-3). The three characteristic changes in the microcirculation (arterioles, capillaries, and venules) near the site of an injury include the following:
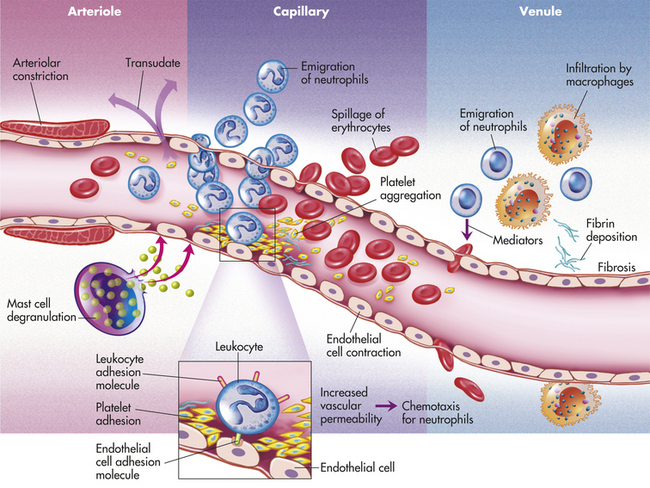
1. Blood vessel dilation (vasodilation)
2. Increased vascular permeability and leakage of fluid out of the vessel
3. White blood cell adherence to the inner walls of vessels and their migration through vessel walls to the site of injury (diapedesis)
The effects of inflammation are visible within seconds. First, arterioles near the site of infection or injury constrict briefly. Vasodilation then causes slower blood velocity and increases local blood flow to the injured site. The increased flow and capillary permeability result in leakage of plasma from the vessels, causing swelling (edema) in the surrounding tissue. As plasma moves outward, blood remaining in the microcirculation flows more slowly and becomes more viscous. The increased blood flow and increasing concentration of red cells at the site of inflammation cause locally increased warmth and redness. Leukocytes adhere to vessel walls. At the same time, biochemical mediators (e.g., histamine, bradykinins, leukotrienes, prostaglandins) stimulate the endothelial cells that line capillaries and venules to retract, creating spaces at junctions between the cells, allowing leukocytes and plasma to enter the surrounding tissue (intercellular junctions are described in Chapter 1).
Each of the characteristic changes associated with inflammation is the direct result of the activities and interactions of a host of chemicals and cellular components found in the blood and tissues. The vascular changes deliver leukocytes, plasma proteins, and other biochemical mediators to the site of injury. Once in the tissues, the cells and chemicals associated with the inflammatory response act in concert to do the following:
1. Prevent infection and further damage by contaminating microorganisms through the influx of fluid to dilute toxins produced by bacteria and released from dying cells, the influx and activation of plasma protein systems that help destroy and contain bacteria (e.g., complement system, clotting system), and the influx of cells (e.g., neutrophils, macrophages) that “eat” and destroy cellular debris and infectious agents.
2. Limit and control the inflammatory process through the influx of plasma protein systems (e.g., clotting system), plasma enzymes, and cells (e.g., eosinophils) that prevent the inflammatory response from spreading to areas of healthy tissue.
3. Interact with components of the adaptive immune system to elicit a more specific response to contaminating pathogen(s) through the influx of macrophages and lymphocytes.10
4. Prepare the area of injury for healing through removal of bacterial products, dead cells, and other products of inflammation (e.g., by way of channels through the epithelium or drainage by lymphatic vessels) and initiation of mechanisms of healing and repair.
Fluid and debris that accumulate at an inflamed site are drained by lymphatic vessels. This process also facilitates the development of acquired immunity because microbial antigens in lymphatic fluid pass through the lymph nodes, where they activate both B and T lymphocytes. (This process is discussed in Chapter 7, and the lymphatic system is described in Chapter 25.)
Inflammation and repair can be divided into several phases (Figure 6-4). The characteristics of the early (i.e., acute) inflammatory response differ from those of the later (i.e., chronic) response, and each phase involves different biochemical mediators and cells that function together. The acute inflammatory response is of short duration, that is, it continues only until the immediate threat to the host is eliminated. This usually takes 8 to 10 days from onset to healing. The acute inflammatory response begins immediately after cellular injury or infection occurs (see Figure 6-4) and involves a vascular response, activation of plasma protein systems, and activation of a variety of cells. (Mechanisms of cellular injury are described in Chapter 2.)
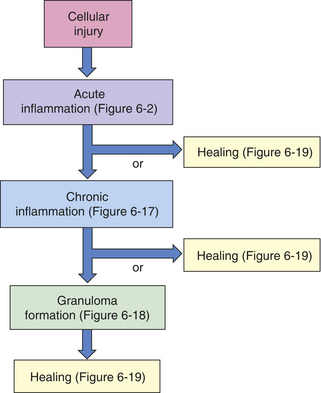
Figure 6-4 Inflammatory phases. Cellular injury leads to acute inflammation and may result in resolution and healing of the injured site or progress into chronic inflammation. Chronic inflammation in turn may result in healing or progress to development of a granuloma. The final step of the inflammatory process is usually healing and reconstruction of the damaged tissue. The figure numbers refer to those in which more detailed information on that portion of the process may be found.
Plasma Protein Systems
Three key plasma protein systems are essential to an effective inflammatory response. These are the complement system, the clotting system, and the kinin system (see Figures 6-5, 6-6, and 6-7). Although each system has a unique role in inflammation, they also have many similarities. Each system consists of multiple proteins in the blood. To prevent activation in unnecessary situations, each protein is normally in an inactive form. Several of the proteins are enzymes that circulate in inactive forms as proenzymes. Each system contains a few proteins that can be activated by products of tissue damage or infection. Activation of the first component of a system results in sequential activation of other components, leading to a biologic function that helps protect the individual. This sequential activation is referred to as a cascade. Thus, we refer to the complement cascade, the clotting cascade, or the kinin cascade. In some cases, activation of a protein may require that it be enzymatically cut into two pieces or fragments of different size. Usually the larger fragment continues the cascade by activating the next component, and the smaller fragment frequently has potent biologic activities to promote inflammation.
Complement System
The complement system consists of several plasma proteins (sometimes called complement components) that together constitute about 10% of the total circulating serum protein.11 The complement system is extremely important because activation of the complement cascade may destroy pathogens directly and can activate or collaborate with virtually every other component of the inflammatory response.12,13 For these reasons, proteins of the complement system are among the body’s most potent defenders against bacterial infection.
Activation of the complement system can be accomplished in three different pathways or cascades, all of which converge at the third component (C3) of the pathway:
1. Classical pathway: activated by proteins of the acquired immune system (antibodies) bound to their specific targets (antigen)
2. Lectin pathway: activated by certain bacterial carbohydrates
3. Alternative pathway: activated by gram-negative bacterial and fungal cell wall polysaccharides
The principal routes by which the complement cascade may be activated are shown in Figures 6-5.
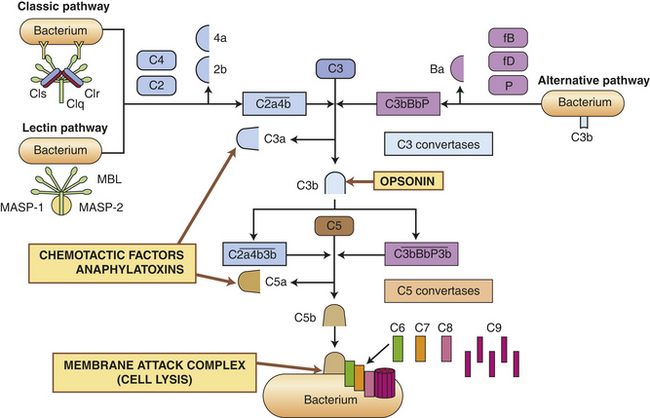
Figure 6-5 Pathways of complement cascade activation. The complement system is activated by three pathways: the classic pathway, the lectin pathway, and the alternative pathway. During activation, many complement components are cleaved into fragments (2b, 4a, Ba, 3a, and 5a). The smaller fragments frequently have potent biologic activities and may serve as chemotactic factors and anaphylatoxins. The larger activated fragments are usually converted into active enzymes (indicated by the bar above the names) and form complexes with additional components in the cascade. The classic pathway is usually activated by antigen-antibody complexes through component C1, which consists of C1q and two C1r and C1s molecules. As indicated, the C1q must simultaneously bind to two antibody molecules (indicated by Y-shaped structures). The lectin pathway is activated by mannose-binding lectin (MBL), which binds to two mannose-rich pathogen-associated molecular patterns on the surface of a bacterium. MBL contains two associated enzymes, MASP-1 and MASP-2, and functions in a manner similar to C1. C1 and MBL each activate complement components C4 and C2. The alternative pathway is activated by many agents, such as bacterial polysaccharides, which bind and stabilize C3b, which is produced by normal breakdown of C3 in the blood. The C3b forms the site of binding of factor B (fB), which is activated by factor D (fD) into Bb and the small fragment Ba. Properdin (P) helps stabilize the complex. Each pathway produces C3 and C5 convertases, which are enzymatically active complexes that activate C3 and C5, respectively. C3b produced by the C3 convertase can function as an opsonin. C5b initiates assemblage of the membrane attack complex (MAC), which results in multiple C9 molecules forming a pore in the bacterial membrane.
Activation of the classical pathway begins with the activation of protein C1 and is preceded by formation of a complex between an antigen and an antibody to form an antigen-antibody complex (immune complex) (discussed in Chapter 7).14 The antigen may be a unique chemical component of the surface of a bacterium or other microorganism. Most pathogens express multiple antigens; therefore, multiple antibodies are usually bound in the complex. The first component of the classical complement cascade, C1, has six sites that can bind to antibodies, and efficient activation of the complement cascade usually requires concurrent binding of C1 to at least two antibody molecules.15 The complex formed by antigen-antibody-complement binding is shown in Figure 6-5. C1 is a macromolecular complex consisting of C1q and two molecules each of C1r and C1s. A conformational change in C1 results in an enzymatically active molecule whose substrates are C4 and C2. The resultant complex formed by the interaction of C1, C4, and C2 uses C3 as a substrate resulting in the production of C3a and C3b. A complex that has C3 as a substrate is generally referred to as a C3 convertase. The complex formed by the activation of C3 then has C5 as a substrate, resulting in the conversion of C5 to C5a and C5b. A complex that has C5 as a substrate is generally called a C5 convertase. Thus activation of C1 initiates the sequential enzymatic activation of all other components of the classic pathway, ultimately resulting in the activation of C5. The classical pathway also can be activated to a lesser degree by biologic molecules other than antibody, including heparin (a charged molecule that prevents clotting), deoxyribonucleic acid (DNA) or ribonucleic acid (RNA), and C-reactive protein, which is increased in the blood during inflammation.
Even under normal conditions small amounts of circulating C3 are spontaneously broken down into C3b and C3a by a number of naturally occurring enzymes in the blood. The rate of C3 spontaneous activation is generally very low, and C3b is usually readily inactivated by complement regulator proteins in the blood (e.g., factor H and factor I). However, materials produced by some infectious microorganisms (e.g., lipopolysaccharides [endotoxin] on the bacterial surface, yeast cell wall carbohydrates [zymosan]) can bind the naturally produced C3b and protect it from inactivation. This will initiate activation of the alternative complement pathway.16 The C3b bound to bacterial products can react with another normally occurring component, factor B. The complex of C3b and factor B is recognized by an enzyme, factor D, which activates factor B, producing factor Bb. The resultant C3b/Bb complex is very unstable unless it binds to properdin (P). The C3b/Bb/P complex is a C3 convertase that produces further C3b, resulting in a C3b/Bb/P/C3b complex that is a C5 convertase, which activates C5.
The lectin pathway is similar to the classical pathway but is antibody independent. It is activated by a plasma protein called mannose-binding lectin (MBL).17 MBL is similar to C1q and binds to bacterial polysaccharides containing the carbohydrate mannose. MBL-associated serine proteases (MASP-1 and MASP-2) substitute for C1r and C1s and activate C4 and C2 to create a C3 convertase.
After activation of C5, the cascade continues through the terminal components C6, C7, C8, and C9. Components C5b through C9 assemble to form complexes (membrane attack complex, or MAC) capable of creating pores in cell membranes and permitting the influx of water and ions and may ultimately result in cell lysis.
The most important result of complement activation is the production of fragments during the activation of C4, C2, C3, and C5. The fragments C4a, C2b, C3a, and C5a are soluble and of low-molecular-weight that contribute in other ways to the inflammatory response. C2b affects smooth muscle, causing vasodilation and increased vascular permeability. C3a, C5a, and to a limited extent C4a, are anaphylatoxins, that is, they induce rapid mast cell degranulation (release of granular contents) and the release of histamine (see Figure 6-9) causing vasodilation and increased capillary permeability.18 C5a is the major chemotactic factor for neutrophils. C3a is approximately 100 times less potent in chemotactic and anaphylatoxic activity. A chemotactic factor is a biochemical substance that attracts leukocytes to the site of inflammation.
The dual functions of a chemotactic factor and an anaphylatoxin are not needed simultaneously or to the same degree. Anaphylatoxic activity is necessary early in inflammation and occurs close to the inflammatory site to induce local mast cell degranulation and to increase the number of soluble mediators available to enhance vascular permeability and vasodilation. Chemotactic activity, on the other hand, is required for a much longer period and occurs distal to the inflammatory site to attract leukocytes from the circulation. Thus it is beneficial to an effective inflammatory response to limit the range of anaphylatoxic activity while allowing widespread chemotactic activity. A plasma enzyme, a carboxypeptidase, removes a terminal arginine on both C3a and C5a peptides, thereby producing “C3a desArg” and “C5a desArg,” which are inactive as anaphylatoxins but retain chemotactic activity. Thus chemotactic activity is retained, while not inducing distal mast cell degranulation that would result in considerable enlargement of the inflammatory response to the detriment of surrounding healthy tissue.
C3b adheres to the surface of a pathogenic microorganism and serves as an efficient opsonin. Opsonins are molecules that “tag” microorganisms for destruction by cells of the inflammatory system (primarily neutrophils and macrophages [see p. 190]). C3b on the cell surface also can be broken down by several enzymes in the blood into inactive fragments (e.g., iC3b), which retain opsonic activity.
In summary, the complement cascade can be activated by at least three different means, and its products have four functions: (1) anaphylatoxic activity resulting in mast cell degranulation, (2) leukocyte chemotaxis, (3) opsonization, and (4) cell lysis.
Clotting System
The clotting (coagulation) system is a group of plasma proteins that form a fibrinous meshwork at an injured or inflamed site.19 This (1) prevents the spread of infection to adjacent tissues, (2) traps microorganisms and foreign bodies at the site of inflammation for removal by infiltrating cells (e.g., neutrophils and macrophages), (3) forms a clot that stops bleeding, and (4) provides a framework for future repair and healing. The main substance in this fibrinous mesh is an insoluble protein called fibrin that is the end product of the coagulation cascade.
Like the complement cascade, the coagulation cascade can be activated through different convergent pathways (see Figure 6-6). The coagulation cascade consists of the extrinsic pathway and the intrinsic pathway that converge at factor X.20 From that point on, a common pathway leads to formation of a fibrin clot. The coagulation cascade is discussed in more detail and illustrated again in Chapter 25.
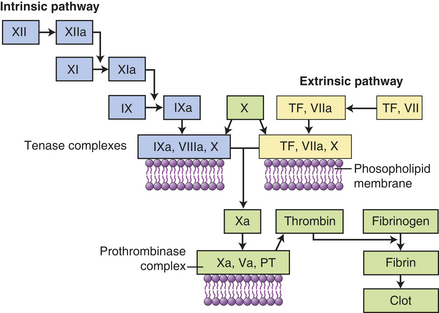
Figure 6-6 Coagulation cascade. Clotting is activated through two pathways: the intrinsic pathway and the extrinsic pathway. The intrinsic pathway is initiated by the activation of Hageman factor (XII) into XIIa (activated factors are enzymes and are indicated by a lowercase a). The sequential activation of other intrinsic pathway components results in formation of a complex of IXa, VIIIa, and X. The extrinsic pathway is activated by exposure of tissue factor (TF) during tissue damage. TF complexes with factor VII, which is activated (VIIa) and forms a complex with factor X (TF, VIIa, X). Both the intrinsic and extrinsic pathway complexes are dependent on calcium, form on phospholipid membranes that are rich in phosphatidylserine, and have “tenase” activity (can activate factor X into Xa). Factor X begins a common pathway in which Xa complexes with Va and prothrombin (PT), with calcium and phospholipid membranes, to form an active prothrombinase (activates prothrombin into thrombin). Thrombin is an enzyme the cuts high-molecular-weight fibrinogen into fibrin molecules. Fibrin polymerizes to form a clot.
The clotting system can be activated by many substances that are released during tissue destruction and infection, including collagen, proteinases, kallikrein, and plasmin, as well as by bacterial products such as endotoxins. As with the complement cascade, activation of the clotting cascade produces fragments that enhance the inflammatory response. Two low-molecular-weight fibrinopeptides, A and B, are released from fibrinogen when fibrin is produced. Both fibrinopeptides (especially fibrinopeptide B) are chemotactic for neutrophils and increase vascular permeability by enhancing the effects of bradykinin (formed from the kinin system).
Kinin System
The third plasma protein system, the kinin system, augments inflammation in several ways.21 The primary kinin produced from the kinin system is bradykinin, which causes dilation of blood vessels, acts with prostaglandins to stimulate nerve endings and induce pain, causes smooth muscle cell contraction, increases vascular permeability, and may increase leukocyte chemotaxis (see Figure 6-2). Bradykinin induces smooth muscle contraction more slowly than histamine and, along with prostaglandins of the E series, is probably responsible for endothelial cell retraction and increased vascular permeability in the later phases of inflammation (endothelial cell retraction is shown in Figures 6-3 and 6-11).
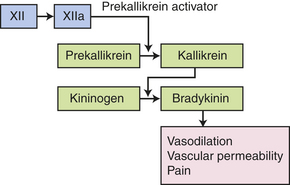
The kinin system is activated by stimulation of the plasma kinin cascade (see Figure 6-7). The conversion of plasma prekallikrein to kallikrein is induced by prekallikrein activator, which is identical to factor XIIa (the product that results from activation of Hageman factor—factor XII) of the clotting cascade. Kallikrein then converts kininogen to bradykinin. Although the plasma kinin cascade is one pathway that leads to the production of bradykinin, tissue kallikreins in saliva, sweat, tears, urine, and feces provide another source for this inflammatory mediator. These tissue kallikreins convert serum kininogens to kallidin, also known as Lys-bradykinin, which may be converted to bradykinin by plasma aminopeptidase. In order to control the extent of inflammation, kinins are rapidly degraded by kininases, enzymes present in plasma and tissues.
Interactions Among the Plasma Protein Systems
The three plasma protein systems are highly interactive so that activation or regulation of one system results in a similar effect on the others (Figure 6-8).22 As an example, plasmin regulates clot formation by degrading fibrin and fibrinogen, and it can activate the complement cascade through components C1, C3, and C5. Plasmin can activate the plasma kinin cascade as well by activating Hageman factor (factor XII) and producing prekallikrein activator. This activation of Hageman factor has four effects that impact all three of the plasma protein systems:
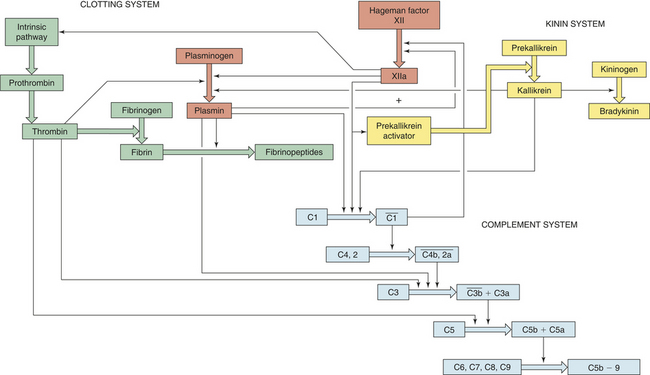
Figure 6-8 Interactions between the complement, clotting, kinin, and fibrinolytic (plasmin) systems. Thick colored arrows denote the activation of factors within a system. Thin arrows denote where a particular factor activates another system.
1. Activation of the clotting cascade through factor XI
2. Control of clotting through conversion of plasminogen proactivator to plasminogen activator, resulting in the generation of plasmin
3. Activation of the kinin system by a Hageman factor fragment, prekallikrein activator
The activity of plasmin itself is also regulated because it is synthesized as a proenzyme, plasminogen. Plasminogen is converted to plasmin by several factors, including plasminogen activator generated from the kallikrein system, thrombin generated from the clotting system, bacterial factors such as streptokinase produced by hemolytic streptococci, plasminogen activators produced by endothelial cells, and several cellular enzymes released during tissue destruction.
Activation of any of the plasma protein systems results in production of a large number of very potent, biologically active substances that further activate the inflammatory response and assist in protection against infection. Very tight control of these processes is essential for two reasons:
1. The inflammatory process is critical for an individual’s survival, thus efficient activation must be guaranteed regardless of the cause of tissue injury.
2. The biochemical mediators generated during these processes are so potent and potentially detrimental to the host itself that their actions must be strictly confined to injured or infected tissues only.
Therefore, multiple mechanisms are available to either activate or inactivate (regulate) these plasma protein systems.
As mentioned, many enzymes from the plasma regulate the activity of these pathways, such as carboxypeptidase inactivating the anaphylatoxic activities of C3a and C5a and kininases degrading kinins. Many other natural inhibitors are present, including enzymes that degrade histamine (histaminase), activated complement components, kallikrein, and plasmin. Another example of a common regulator is C1 esterase inhibitor (C1 inh).23 C1 inh inhibits complement activation through reactivity with C1 (classic pathway), MASP-2 (lectin pathway), and C3b (alternative pathway). It is also a major inhibitor of the clotting and kinin pathways (e.g., kallikrein, activated Hageman factor XIIa). A genetic defect in C1 inh (C1 inh deficiency) results in hereditary angioedema, which is a self-limiting edema of cutaneous and mucosal layers resulting from stress, illness, or relative minor or unapparent trauma. The disease is characterized by hyperactivation of all three plasma protein systems, although excessive production of bradykinin appears to be the principal cause of increased vascular permeability.
Cellular Mediators of Inflammation
The cellular components of the inflammatory response consist primarily of cells of the granulocytic or monocytic lines of leukocytes. Most are generally found in the blood, but several are also found in the tissues. The primary circulating white blood cells are granulocytes, so called because of the many enzyme-containing granules in their cytoplasm. These include neutrophils, eosinophils, and basophils. Other blood components include platelets, agranular monocytes (precursors of macrophages), and various forms of lymphocytes. Circulating cells continually contact endothelial cells lining the blood vessels. Changes in the interactions of endothelium with circulating leukocytes and platelets occur during inflammation and account for several of the characteristics of the inflammatory response. Other cellular members of the inflammatory system are found in various tissues and organs. These include mast cells and cells derived from the monocytes/macrophage lineage. Lymphoid-derived natural killer cells are found in the circulation and tissues and can recognize and destroy cells that have been altered by viral infection or malignancy.
The cells of the inflammatory system secrete and respond to biochemical mediators. Thus most of these cells are recruited and activated by products of the plasma protein systems and by biochemicals released during cell destruction, secreted by other inflammatory cells, or produced by microbes. All of these inflammatory cells and protein systems, along with the substances they produce, preferably act at the site of tissue injury to confine the extent of damage, kill microorganisms, and remove the debris of “battle” in preparation for healing: tissue regeneration or repair (processes known as resolution).
Inappropriate or exaggerated inflammatory processes have deleterious effects on the host. Even appropriate inflammation can be painful and harm healthy tissues. Because inflammation is complex, is nonspecific, and can be triggered and maintained by a great variety of stimuli, it is often difficult to control with drugs.
Cellular Receptors
Cells of both innate and acquired immunity must recognize and respond to their environment, whether to products of damaged cells or to potential pathogenic microorganisms. Each cell has receptors on the cell surface that specifically bind soluble substances produced during tissue damage or infection. Receptor binding results in activation of intracellular signaling pathways and activation of the cell itself. As will be discussed in Chapter 7, B and T lymphocytes of the acquired immune system have evolved surface receptors (i.e., the T-cell receptor, or TCR, and the B-cell receptor, or BCR) that bind a large spectrum of antigens. Cells involved in innate resistance have evolved a different set of receptors that recognize a much more limited array of specific molecules. These are referred to as pattern recognition receptors (PRRs), and they recognize molecular “patterns” on infectious agents or their products (pathogen-associated molecular patterns, or PAMPs), or products of cellular damage (necrosis or apoptosis).24 PRRs are generally found on cells at the interface of the host and environment (i.e., skin, respiratory tract, gastrointestinal tract, genitourinary tract), where they monitor for products of cellular damage and potentially infectious microorganisms. Although most PRRs are on the cell surface, some are secreted. An example of a secreted PRR is mannose-binding lectin of the lectin pathway of complement activation. Cellular PRRs include Toll-like receptors, complement receptors (CRs), scavenger receptors, glucan receptors, and mannose receptors.
In humans, at least 11 different Toll-like receptors (TLRs) have been described, 10 of which are functional.25 They are expressed on the surface of many cells that have direct and early contact with potential pathogenic microorganisms. These include mucosal epithelial cells, mast cells, neutrophils, macrophages, dendritic cells, and some subpopulations of lymphocytes. (Dendritic cells are found in the skin, mucosa, and lymphoid tissues, where they have developed from Langerhans cells and function as highly specialized initiators of the acquired immune response.) TLRs recognize a large variety of PAMPs located on the microorganism’s cell wall or surface (e.g., bacterial lipopolysaccharide [LPS], peptidoglycans, and lipoproteins, yeast zymosan, viral coat proteins), other surface structures (e.g., bacterial flagellin), or microbial nucleic acid (e.g., bacterial DNA, viral double-stranded RNA). Some TLRs recognize host factors that are produced by “stressed” or damaged cells (e.g., breakdown products of extracellular matrix proteins, chromatin). Interactions between PAMPs and TLRs, with the collaboration of other cellular receptors (e.g., CD14), can result in activation of the cell and the release of soluble products (e.g., cytokines) that increase local resistance to the pathogenic microorganism. TLRs are also one of the bridges between innate resistance and the acquired immune response through the induction of cytokines that increase the response of lymphocytes to foreign antigens on the pathogens. Genetic polymorphisms in TLRs may explain some observed differences among individuals’ resistance and susceptibility to infections. Information on each of the TLRs found in humans is shown in Table 6-2.26,27
Table 6-2
Cellular Source and Microbial Target for Each Toll-like Receptor (TLR)
| Receptor | Cellular Expression Pattern | PAMP Recognition |
| TLR1 | Cell surface (ubiquitous): neutrophils, monocytes/macrophages, dendritic cells, T cells, B cells, NK cells | Fungal, bacterial, viral; forms heterodimer with TLR2 (see TLR2 recognition) |
| TLR2 | Cell surface: neutrophils, monocytes/macrophages, dendritic cells | Fungal (yeast zymosan), bacterial (gram-positive bacterial peptidoglycan, lipoproteins), viral (lipoproteins) |
| TLR3 | Intracellular: monocytes/macrophages, dendritic cells, T cells, NK cells, epithelial cells | Double-stranded RNA produced by many viruses |
| TLR4 | Cell surface: granulocytes, monocytes/macrophages, dendritic cells, T cells, B cells, epithelial cells | Bacterial (primarily gram-negative bacterial LPS, lipoteichoic acids), viral (RSV F protein, hepatitis C) |
| TLR5 | Cell surface: granulocytes, monocytes/macrophages, dendritic cells, NK cells, epithelial cells | Bacterial (flagellin); forms heterodimer with TLR4 |
| TLR6 | Cell surface: monocytes/macrophages, dendritic cells, B cells, NK cells | Fungal, bacterial, viral; forms heterodimer with TLR2 (see TLR2 recognition) |
| TLR7 | Intracellular: monocytes/macrophages, dendritic cells, B cells | Natural ligand uncertain; may bind viral single-strand RNA |
| TLR8 | Cell surface: monocytes/macrophages, dendritic cells, NK cells | Natural ligand uncertain; may bind fungal PAMPs or viral single-stranded RNA |
| TLR9 | Intracellular: monocytes/macrophages, dendritic cells, B cells | Bacterial (unmethylated DNA [CpG dinucleotides]) |
| TLR10 | Cell surface: monocytes/macrophages, dendritic cells, B cells | Natural ligand uncertain; may form heterodimers with TLR2 |
| TLR11 | TLR11 gene does not code a full length protein in humans | No known immune response |
DNA, Deoxyribonucleic acid; LPS, lipopolysaccharide; NK, natural killer; PAMPs, pathogen-associated molecular patterns; RNA, ribonucleic acid; RSV, respiratory syncytial virus.
Complement receptors are found on many cells of the innate and acquired immune responses (e.g., granulocytes, monocytes/macrophages, lymphocytes, mast cells, erythrocytes, platelets), as well as some epithelial cells.28 They recognize several fragments produced through activation of the complement system. Under a variety of normal and disease-related conditions, immune complexes of antibody, antigen, and complement form in the blood and are removed by cells expressing surface complement receptor-1 (CR1), which binds to C4b, C3b, and C3b breakdown products (e.g., iC3b). CR2 is found on B lymphocytes, as well as dendritic cells and some epithelial cells, and recognizes C3b breakdown products (particularly iC3b). CR2 appears to facilitate B-cell function and antibody production. Both CR3 and CR4 are integrins that primarily recognize C3b breakdown products (particularly iC3b). CR3 (integrin αMβ2, also called CD11b/CD18) facilitates phagocytosis by neutrophils and monocytes/macrophages. CR4 (αXβ2, also called CD11c/CD18) is found primarily on platelets. (Integrins are cell surface receptors that have a role in cell adhesion and attachment and mediate intracellular signaling within the extracellular matrix [see Figure 1-14]).
Scavenger receptors are primarily expressed on macrophages and facilitate recognition and phagocytosis of bacterial pathogens, as well as damaged cells and altered soluble lipoproteins associated with vascular damage (e.g., HDL, acetylated LDL, oxidized LDL). More than eight receptors have been identified. Some scavenger receptors (e.g., SR-PSOX) recognize the cell membrane phospholipid phosphatidylserine (PS). PS is normally sequestered on the cytoplasmic surface of the cell membrane, but is externalized under a very limited variety of conditions, including erythrocyte senescence and cellular apoptosis. Thus macrophages, through this receptor, can identify and remove old red blood cells and cells undergoing apoptosis. Another important scavenger receptor is CD14, which recognizes the complex of LPS and LPS-binding protein. LPS-binding protein is up-regulated during inflammation by the cytokines interleukin-6 (IL-6) and IL-1 and helps remove bacterial lipopolysaccharide (endotoxin) from the circulation.29–31
Mast Cells
A central cell in inflammation is the mast cell.32 Mast cells, first described by Paul Ehrlich in 1877,33 are cellular bags of granules located in the loose connective tissues close to blood vessels (Figure 6-9). They are found in large numbers in areas directly exposed to the environment including the skin and lining the gastrointestinal and respiratory tracts. A great number of stimuli cause mast cells to become activated, resulting in initiation of the inflammatory response. Typical causes of mast cell activation include (1) physical injury (e.g., heat, mechanical trauma, ultraviolet light, and x-rays), (2) chemical agents (e.g., toxins, snake and bee venoms, proteolytic enzymes, and antimicrobial peptides), (3) immunologic means (e.g., anaphylatoxins released during activation of complement components or particular types of antibody [e.g., immunoglobulin E (IgE)] produced by cells of the acquired immune response [see Chapter 7]), and (4) activation of TLRs by bacteria and viruses.34,35 Soluble and extremely potent chemicals from the mast cell are responsible for its effects on inflammation. These are released in two ways: by release of the contents of their preformed granules (degranulation) and by new synthesis of lipid-derived inflammatory mediators. Mast cells are also involved in initiating many allergic responses (discussed in Chapters 7 and 8).
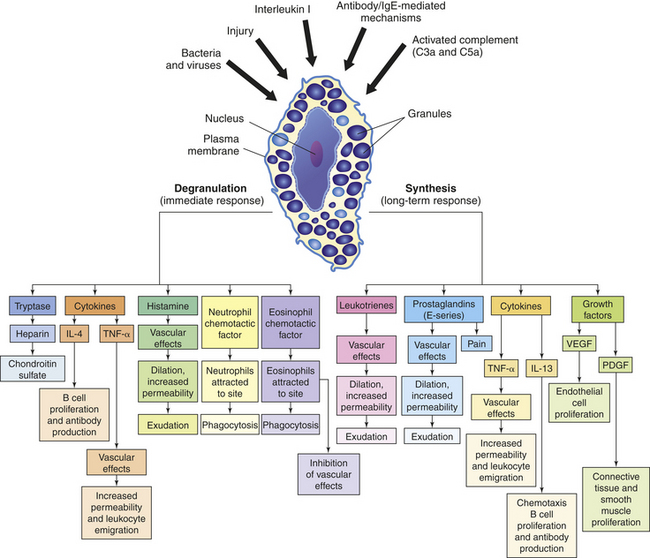
Figure 6-9 Effects of degranulation (left) and synthesis (right) by mast cells. The depiction of a tissue mast cell shows darkly stained granules in the cytoplasm. PDGF, Platelet-derived growth factor; VEGF, vascular endothelial growth factor.
Mast Cell Degranulation: In response to a stimulus, biochemical mediators in the mast cell granules, including histamine, chemotactic factors (e.g., neutrophil chemotactic factor, eosinophil chemotactic factor of anaphylaxis or ECF-A), and cytokines (e.g., tumor necrosis factor-alpha [TNF-α], IL-4) are released within seconds and exert their effects immediately (see Figure 6-9).
Histamine is a vasoactive amine that causes temporary, rapid constriction of the large vessel walls and dilation of the postcapillary venules, both of which result in increased blood flow into the microcirculation. Histamine also causes increased vascular permeability resulting from retraction of endothelial cells lining the capillaries (see Figure 6-3). The pharmacologic effects of histamine are partially determined by histamine receptors on the person’s target cells. Two main histamine receptors are the H1 and H2 receptors (Figure 6-10), and two other receptors, H3 and H4, have been described.36 Binding of histamine to the H1 receptor is essentially proinflammatory, that is, it promotes inflammation. On the other hand, binding to the H2 receptor is generally anti-inflammatory because it results in suppression of leukocyte function. The H1 receptor is present on smooth muscle cells, especially those of the bronchi, and causes bronchial smooth muscle to contract (bronchoconstriction) when stimulated. Both types of receptors are distributed among many different cells and are often present on the same cells and may act in an antagonistic fashion. For instance, neutrophils express both types of receptors, with stimulation of H1 receptors resulting in the augmentation of neutrophil chemotaxis, and H2 stimulation resulting in its inhibition. The H2 receptor is especially abundant on parietal cells of the stomach mucosa and induces the secretion of gastric acid as part of the normal physiology of the stomach. The role of H1 and H2 receptors is discussed further in Chapter 8.
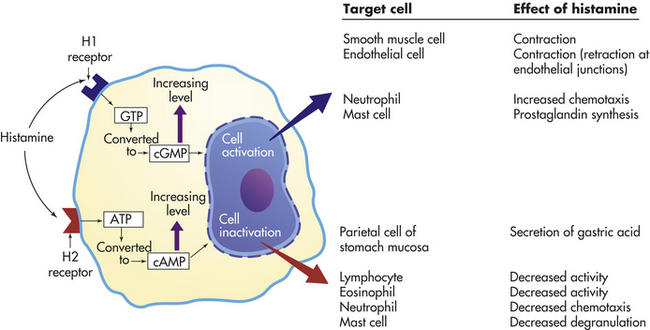
Figure 6-10 Effects of histamine through H1 and H2 receptors. Effects depend on (1) density and affinity of H1 or H2 receptors on the target cell and (2) the identity of the target cell. ATP, Adenosine triphosphate; cAMP, cyclic adenosine monophosphate; cGMP, cyclic guanosine monophosphate; GTP, guanosine triphosphate.
Two chemotactic factors, neutrophil chemotactic factor and ECF-A, are also released during mast cell degranulation. Chemotaxis is directional movement of cells along a chemical gradient formed by a chemotactic factor (Figure 6-11). Neutrophil chemotactic factor attracts neutrophils, and ECF-A attracts eosinophils to the site of inflammation. Neutrophils are the predominant leukocytes at work during the early phases of acute inflammation, and eosinophils have several functions in the inflammatory process; both of these important inflammatory cells are discussed in more detail later in this chapter.
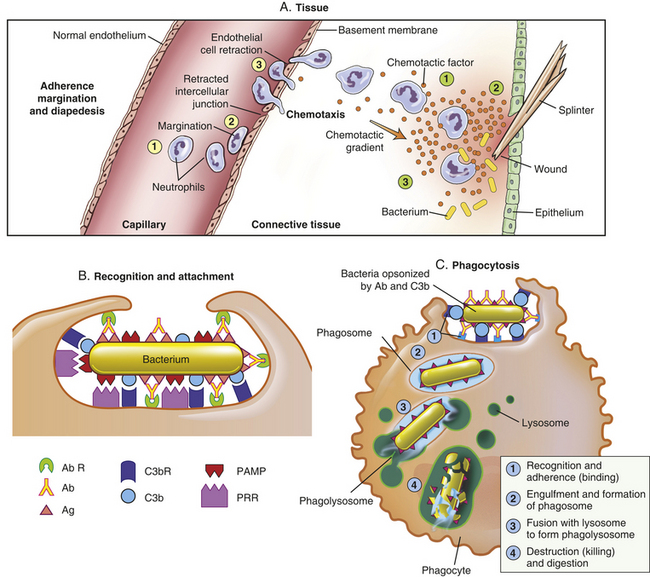
Figure 6-11 Process of phagocytosis. The process that results in phagocytosis is characterized by three interrelated steps: adherence and diapedesis, tissue invasion by chemotaxis, and phagocytosis. A, Adherence, margination, diapedesis, and chemotaxis. The primary phagocyte in the blood is the neutrophil, which usually moves freely within the vessel (1). At sites of inflammation, the neutrophil progressively develops increased adherence to the endothelium, leading to accumulation along the vessel wall (margination or pavementing) (2). At sites of endothelial cell retraction the neutrophil exits the blood by means of diapedesis (3). Chemotaxis: In the tissues, the neutrophil detects chemotactic factor gradients through surface receptors (1) and migrates towards higher concentrations of the factors (2). The high concentration of chemotactic factors at the site of inflammation immobilizes the neutrophil (3). B, Specific receptors for recognition and attachmentC, Phagocytosis. Opsonized microorganisms bind to the surface of a phagocyte through specific receptors (1). The microorganism is engulfed (ingested) into a phagocytic vacuole, or phagosome (2). Lysosomes fuse with the phagosome, resulting in the formation of a phagolysosome (3). During this process the microorganism is exposed to products of the lysosomes, including a variety of enzymes and products of the hexose-monophosphate-shunt (e.g., H2O2, O2−). The microorganism is killed and digested (4). Ab, Antibody; AbR, antibody receptor; C3b, complement component C3b; C3bR, complement C3b receptor; PAMP, pathogen-associated molecular pattern; PRR, pattern recognition receptor.
Mast Cell Synthesis of Mediators: Activated mast cells begin new synthesis of other mediators of inflammation, including those derived from plasma membrane lipids, cytokines (TNF-α, various interleukins), and factors that stimulate cell growth and angiogenesis. Leukotrienes, prostaglandins, and platelet-activating factor are lipid-derived products that are synthesized during mast cell activation (Figure 6-12). Leukotrienes are a product of another lipid, arachidonic acid, which is released from mast cell membranes by an intracellular phospholipase that acts on membrane phospholipids.37 Leukotrienes are acidic, sulfur-containing lipids that produce effects similar to those of histamine, namely, smooth muscle contraction, increased vascular permeability, and perhaps neutrophil and eosinophil chemotaxis. Leukotrienes appear to be important in the later stages of the inflammatory response because they stimulate slower and more prolonged responses than do histamines.
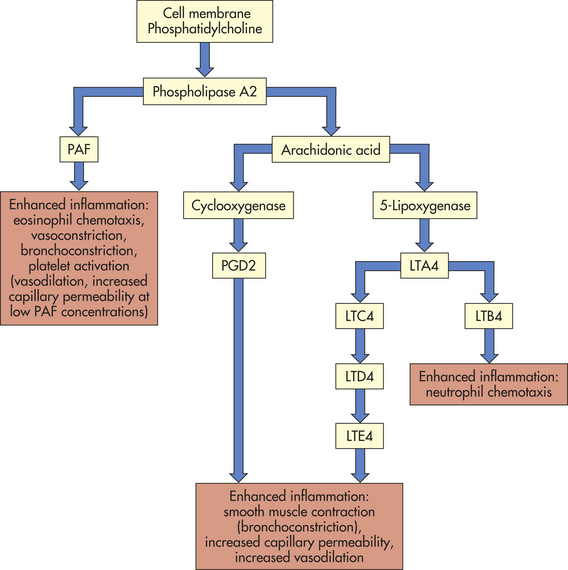
Figure 6-12 Production of lipid vasoactive substances by mast cells. LTA4, LTC4, LTD4, LTE4, LTB4, various leukotriene molecules; PAF, platelet-activating factor; PGD2, prostaglandin D2.
The mast cell also synthesizes prostaglandins, which, like leukotrienes, are a product of arachidonic acid and cause increased vascular permeability and neutrophil chemotaxis. Prostaglandins also induce pain. They are long-chain unsaturated fatty acids produced by the action of the enzyme cyclooxygenase and are classified into groups (E, D, A, F, and B) according to their structure. Prostaglandins E1 and E2 cause increased vascular permeability and smooth muscle contraction, apparently acting directly on postcapillary venules. They can inhibit some aspects of inflammation by suppressing both the release of histamine from mast cells and the release of lysosomal enzymes (enzymes responsible for killing and digesting microorganisms) from neutrophils. Enhancement or suppression of the inflammatory response may be related to the concentration of prostaglandins. Aspirin and some other nonsteroidal anti-inflammatory drugs (NSAIDs) block the synthesis of prostaglandins of the E series and other arachidonic acid derivatives, thereby inhibiting inflammation.
Platelet-activating factor (PAF), another mast cell–derived lipid, is produced by removal of a fatty acid from the plasma membrane phospholipid phosphatidylcholine by phospholipase A2. Although mast cells are a major source of PAF, this molecule also can be produced during inflammation by neutrophils, monocytes, endothelial cells, and platelets. The biologic activity of PAF is virtually identical to that of leukotrienes, namely causing endothelial cell retraction to increase vascular permeability, leukocyte adhesion to endothelial cells, and platelet activation.
Phagocytosis
Phagocytosis is the process by which a cell ingests and disposes of damaged cells and foreign material, including microorganisms (see Figure 6-11). Because most phagocytes are circulating in the blood, they must leave the bloodstream and migrate to the site of inflammation before initiating phagocytosis. Under normal conditions, the circulation in the capillaries and venules is rapidly moving with red blood cells in the main stream and neutrophils and other leukocytes tending to flow more slowly along the vessel’s periphery. Many of the biochemical products produced early at inflammatory sites (e.g., histamine, TNF-α, bradykinin, leukotrienes, prostaglandins) diffuse to the vessels and affect both leukocytes and endothelial cells. Both cell populations respond by producing new adhesion molecules (selectins and integrins)
on their surfaces (Table 6-3).38 (See page 195 for integrins). Selectins are adhesion molecules that bind carbohydrate ligands. The reciprocal change in adhesion molecules on leukocytes, as well as platelets, promotes their interaction with the endothelial cells.39 The initial change of surface molecules increases the adhesion, or stickiness, between leukocytes and endothelial cells, causing the leukocytes to adhere more avidly to the walls of the capillaries and venules in a process called margination, or pavementing.40,41 Adhesion molecules that are expressed later lead to diapedesis, or emigration of the cells through the endothelial junctions that have retracted in response to the same mediators (see Figure 6-11). The leukocytes digest the basement membrane and migrate into the surrounding tissues.
Table 6-3
Examples of Cellular Adhesion Molecules (CAMs) Involved in Leukocyte Interaction with Endothelial Cells
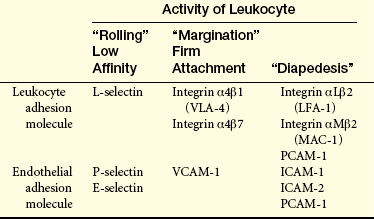
Selectins (lectin-like molecules): L-selectin, leukocyte selectin; P-selectin, platelet selectin; E-selectin, endothelial selectin.
Integrins (noncovalent heterodimers of alpha [α] and beta [β] subunits): VLA-4, very late antigen-4; LFA-1, lymphocyte function antigen-1; MAC-1, macrophage antigen-1.
Immunoglobulin-like molecules: VCAM-1, vascular cell adhesion molecule-1; ICAM-1, ICAM-2, immunoglobulin-like molecules-1 and 2; PCAM-1, platelet-endothelial cell adhesion molecule-1.
Additionally, endothelial cells release nitric oxide (NO), a gas that under normal conditions maintains vascular tone. Inflammation induces additional endothelial nitric oxide synthase, increasing the amount of NO production.42 Effects of NO on inflammation include vasodilation by inducing relaxation of vascular smooth muscle, a response that is local and short-lived, and suppression of mast cell function as well as platelet adhesion and aggregation.
Once inside the connective tissue in the perivascular space, leukocytes migrate to the inflammatory site by means of chemotaxis. They detect chemotactic factors in the environment through chemoreceptors at multiple locations on their plasma membranes and migrate in the direction of highest concentration (see Figure 6-11). The primary chemotactic factors include many bacterial products, complement fragments C3a and C5a, kallikrein, plasminogen activator, products of fibrin degradation, and chemokines. Eosinophils and neutrophils also respond to chemotactic factors released from mast cells. Monocytes are attracted toward a factor (monocyte chemotactic factor) that has been released by neutrophils already at the site of injury. And although histamine is not itself chemotactic, it may facilitate the chemotactic effects of other factors.
Once the phagocytic cell enters the inflammatory site, the process of phagocytosis involves five steps: (1) opsonization, recognition of the target and adherence of the phagocyte to it, (2) engulfment (ingestion or endocytosis) and formation of phagosome, (3) fusion with lysosomal granules within the phagocyte (phagolysosome), and (4) destruction of the target (see Figure 6-11) (lysosomes are described in Chapter 1). Throughout the process, both the target and digestive enzymes are isolated within membrane-bound vesicles. Isolation protects the phagocyte itself from the harmful effects of the target microorganisms, as well as its own enzymes.
Most phagocytes can trap and engulf bacteria using cellular PRRs and PAMPs normally expressed on the bacterial surface (see Figure 6-11). However, that process is slow and inefficient. Opsonization, usually by antibody or complement component C3b, greatly enhances both recognition and adherence. Phagocytosis of an opsonized (antibody and/or complement-protein coated) red blood cell is illustrated in Figure 6-13. Opsonins function as “glue” between the phagocyte and the target cell because receptors on the phagocyte are specific for sites on the opsonin (Fc receptors for antibody, C3b receptors for C3b). This enables the phagocyte to bind an opsonized target very tightly to its surface. Antibody forms a stronger attachment, but C3b facilitates phagocytosis to a greater extent.
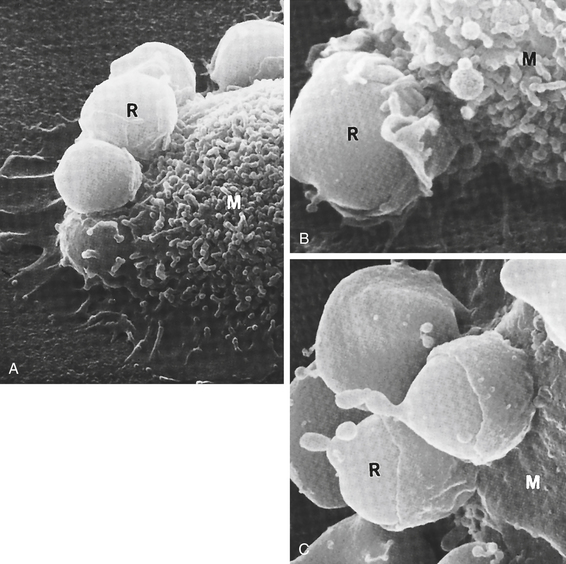
Figure 6-13 Steps in phagocytosis. This scanning electron micrograph shows the progressive steps in phagocytosis. A, Red blood cells (R) attach to the surface of a macrophage (M). B, Part of macrophage (M) membrane starts to enclose the red cell (R). C, The red blood cells are almost totally engulfed by the macrophage. (From King DW, Fenoglio CM, Lefwitch JH: General pathology: principles and dynamics, Philadelphia, 1983, Lea & Febiger.)
Although the inflammatory response is considered to be nonspecific, opsonins and other recognition molecules add a degree of specificity to efficient phagocytosis. Antibodies on the surface of bacteria are directed against antigens that are highly specific to that particular microorganism. If the complement fragment C3b serves as an opsonin, those bacteria with certain polysaccharide coatings are particularly sensitive to activation of the alternative and lectin pathways of complement activation.
Engulfment (endocytosis) is carried out by small pseudopods that extend from the plasma membrane and surround the adherent microorganism (see Figures 6-11 and 6-13) forming an intracellular phagocytic vacuole, or phagosome.43 The membrane that surrounds the phagosome consists of inverted plasma membrane. After the formation of the phagosome, lysosomes converge, fuse with the phagosome, and discharge their contents, creating a phagolysosome. The primary lysosomal granules (azurophilic granules) contain a variety of bactericidal molecules, including myeloperoxidase, lysozyme, defensins, acid hydrolases, elastase, and others. Most phagocytes also contain secondary granules (specific granules) with molecules that are bactericidal and involved in remodeling the surrounding tissue, including lysozyme, collagenase, lactoferrin, and other proteases. Destruction of the bacterium takes place within the phagolysosome and is accomplished by both oxygen-dependent and oxygen-independent mechanisms.44
Phagocytosis is accompanied by a burst of oxygen uptake by the phagocyte, termed the “respiratory burst,” which results from a shift in much of the cell’s glucose metabolism to the hexose-monophosphate shunt. The nicotinamide adenine dinucleotide phosphate (NADPH) that is produced because of this shift is used by a membrane-associated enzyme, NADPH oxidase, to generate superoxide, a reactive oxygen intermediate that is converted to hydrogen peroxide and other reactive oxygen species.45 These steps comprise the oxygen-dependent killing mechanism. Many of the reactive oxygen species are directly toxic to the microorganism. Hydrogen peroxide also can collaborate with the lysosomal enzyme myeloperoxidase and halide anions (Cl– and Br–) to form acids, such as hypochlorous (HClO) and hypobromous (HBrO) acids. These acids probably kill bacteria and fungi by adding Cl– or Br– to the surface of these cells. Oxygen-independent mechanisms of microbial killing are likely the result of (1) the acidic pH (3.5 to 4.0) of the phagolysosome caused by lactic acid production; (2) cationic proteins, such as defensins and cathelicidins, that bind to and damage target cell membranes; (3) enzymatic attack of the mucopeptides in the target cell wall by lysozyme and elastase; and (4) inhibition of bacterial growth by lactoferrin binding of iron.
When a phagocyte dies at an inflammatory site, it frequently lyses (breaks open) and releases its cytoplasmic contents, including the lysosomal enzymes, into the tissue. Enzymes released from lysosomes can digest the connective tissue matrix, causing much of the tissue destruction associated with inflammation. The destructive effects of many enzymes released by dying phagocytes are minimized by natural inhibitors found in the blood, such as α1-antitrypsin, a plasma protein produced by the liver. An inherited deficiency of α1-antitrypsin often results in chronic lung damage and emphysema as a result of inflammation. (The pulmonary effects of α1-antitrypsin deficiency are described in Chapter 33.) Released lysosomal products also may contribute to inflammation by increasing vascular permeability, attracting additional monocytes, and activating the complement and kinin systems.
Neutrophils
The neutrophil, or polymorphonuclear neutrophil (PMN), is a member of the granulocytic series and is named for the characteristic staining pattern of its granules as well as its multilobed nucleus.46 Neutrophils are the predominant phagocytes in the early inflammatory site, arriving within 6 to 12 hours after the initial injury, where they ingest (phagocytose) bacteria, dead cells, and cellular debris. Several inflammatory mediators (e.g., some bacterial proteins, complement fragments C3a and C5a, and mast cell neutrophil chemotactic factor) specifically attract neutrophils from the circulation and activate them. Macrophages and lymphocytes, on the other hand, enter the site later, usually after 24 hours, and gradually replace the neutrophils.
Because the neutrophil is a mature cell incapable of division and sensitive to the acidic environment of inflammatory lesions, it is short lived at the inflammatory site and becomes a component of the purulent exudate, or pus, which is removed from the body through the epithelium or via the lymphatic system. (The lymphatic system is described in Chapter 25) The primary roles of the neutrophil are removal of debris in sterile lesions, such as burns, and phagocytosis of bacteria in nonsterile lesions.
Monocytes and Macrophages
The next phagocytes on the scene are monocytes and macrophages, which perform many of the same functions as neutrophils but for a longer time and in a later stage of the inflammatory response.47,48 Monocytes are the largest normal blood cells (14 to 20 μm in diameter) and have a nucleus that is often indented or horseshoe shaped. Monocytes are produced in the bone marrow, enter the circulation, and migrate to the inflammatory site, where they develop into macrophages. Monocytes also appear to be the precursors of macrophages that are found in tissues (tissue macrophages, discussed in Chapter 7), including Kupffer cells in the liver, alveolar macrophages in the lungs, and microglia in the brain. Macrophages are generally larger (20 to 40 μm) and are more active as phagocytes than their monocytic precursors. Macrophages, particularly those residing in the tissues, are often important cellular initiators of the inflammatory response (Figure 6-14).
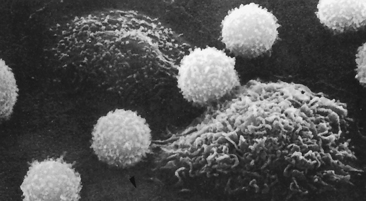
Figure 6-14 Scanning electron micrograph of lymphocytes and macrophages. The lymphocytes are small and spherical; the macrophages are larger and more irregular in shape. (From Raven PH, Johnson GB: Biology, St Louis, 1992, Mosby.)
Monocyte-derived macrophages from the circulation may appear at the inflammatory site as soon as 24 hours after the initial neutrophil infiltration, but usually arrive 3 to 7 days later. They migrate to the site more slowly than neutrophils because they move more sluggishly and also because many of the chemotactic factors that attract them, such as macrophage chemotactic factor, must first be released by neutrophils. Macrophages are better suited than neutrophils to long-term defense against infectious agents because macrophages can survive and divide in the acidic inflammatory site or where there is low oxygen tension.
Neutrophils and monocytes/macrophages differ chiefly in the following ways:
1. Speed: Neutrophils arrive at the injury site first.
2. Active life span: Macrophages survive and divide in the inflammatory site, whereas neutrophils cannot.
3. Chemotactic factors: Neutrophils and macrophages are not attracted by the same factors.
4. Enzymatic content of their lysosomes, or digestive vacuoles
5. Role in the immune response: Macrophages, but not neutrophils, are involved in activation of the adaptive immune system.
6. Role in wound repair: Macrophages are the primary cells that infiltrate tissue in wounds, remove cells and cellular debris, and produce cytokines that suppress further inflammation and initiate healing.
Macrophage Activation: Several bacteria are resistant to killing by granulocytes and can even survive inside macrophages. Microorganisms such as Mycobacterium tuberculosis (tuberculosis), Mycobacterium leprae (leprosy), Salmonella typhi (typhoid fever), Brucella abortus (brucellosis), and Listeria monocytogenes (listeriosis) can remain dormant or even multiply inside the phagolysosomes of macrophages. However, the bactericidal activity of macrophages can be markedly increased with the help of inflammatory cytokines produced by cells of the acquired immune system (subsets of T lymphocytes) or cells activated through Toll-like receptors. (Cytokines are discussed in detail later in this chapter.) Macrophages have cell surface receptors for these cytokines and are further activated to become more effective killers of infectious microorganisms.
Macrophage activation results in increased (1) phagocytic activity, (2) size, (3) plasma membrane area, (4) glucose metabolism, and (5) number of lysosomes.49 Activated macrophages also secrete factors that stimulate the growth, differentiation, and activation of additional inflammatory cells as well as control the initiation of healing processes. These include granulocyte colony-stimulating factor (G-CSF), gamma interferon (IFN-γ), interleukin-1 (IL-1β), angiogenic factor, fibroblast activating factor, and growth factors that promote regrowth of damaged tissues. Macrophages are also the primary cells that infiltrate wounds to remove cellular debris and initiate the regenerative process.50 In some cases, inadequate macrophage activation results from defects in acquired immune responses and deficits in the production of appropriate cytokines. For example, a form of leprosy called lepromatous leprosy is characterized by the survival of phagocytosed M. leprae bacteria in macrophage phagolysosomes. In individuals with lepromatous leprosy, cells of the acquired immune system have failed to secrete the cytokines necessary to transform macrophages into highly efficient killing cells.
Eosinophils
Another population of granulocytes is the eosinophil. Although eosinophils are only mildly phagocytic, they have two specific functions: (1) they serve as the body’s primary defense against parasites and (2) they help regulate vascular mediators released from mast cells.51 Their role in resistance to parasites occurs in collaboration with specific antibodies produced by the acquired immune system and will be discussed in Chapter 7.
The second function, regulation of mast cell-derived inflammatory mediators, is a critical function of eosinophils. As with most defense systems of the body, the acute inflammatory response is usually needed only in a circumscribed area and for a limited time. Therefore, control mechanisms are necessary to prevent biochemical mediators from evoking more inflammation than is needed. Mast cells produce ECF-A, which attracts eosinophils to the site of inflammation. Eosinophil lysosomes contain several enzymes that degrade vasoactive molecules, thereby controlling the vascular effects of inflammation. These enzymes include histaminase, which mediates the degradation of histamine, and arylsulfatase B, which mediates the degradation of some of the lipid-derived mediators produced by mast cells.
Basophils
The basophil is the least prevalent granulocyte in the blood. It is very similar to mast cells in the content of its granules and, in addition, is an important source of the cytokine IL-4, which is a key regulator of the acquired immune response.52 Although often associated with allergies and asthma, its primary role is yet unknown.
Natural Killer Cells
The main function of natural killer (NK) cells is recognition and elimination of cells infected with viruses, although they are also somewhat effective at elimination of other abnormal host cells, specifically cancer cells. NK cells seem to be more efficient in this role when they encounter an infected cell within the circulatory system as opposed to within tissues.53 Along with TLRs, NK cells have additional inhibitory and activating receptors that allow differentiation between infected or tumor cells and normal cells. If the NK cell binds to a target cell through activating receptors, it produces several cytokines and toxic molecules that can kill the target. (Mechanisms of cell to cell killing by NK cells and T cells are discussed further in Chapter 7.)
Platelets
Platelets (thrombocytes) are cellular fragments formed from megakaryocytes. They circulate in the bloodstream until vascular injury occurs. After injury, platelets can be activated by many products of both the innate and adaptive immune responses, including collagen, thrombin, thromboxane, PAF, and antigen-antibody complexes. Activation results in (1) their interaction with components of the coagulation cascade to stop bleeding and (2) degranulation. Platelets contain alpha (α) granules and dense granules. Alpha granules generally contain polypeptides that affect inflammation, including coagulation proteins (e.g., fibrinogen, factor V), soluble adhesion molecules (e.g., von Willebrand factor, vitronectin), growth factors (e.g., platelet-derived growth factor, epidermal growth factor), protease inhibitors (e.g., plasminogen activator inhibitor-1, α2-antiplasmin), and membrane adhesion molecules (e.g., P-selectin, αIIbβ3). Dense granules contain several small molecules, including adenosine diphosphate (ADP), serotonin, calcium, and magnesium. Serotonin is a vasoactive amine with vascular effects similar to those of histamine. (Platelet function is described in detail in Chapter 25.)
Cellular Products
To elicit an effective inflammatory (or acquired immune) response, it is necessary that many different kinds of cells cooperate. Many cells secrete soluble factors that contribute to the regulation of innate or acquired resistance by affecting other neighboring cells (Figure 6-15). These factors are referred to as chemokines or cytokines and are either pro-inflammatory or anti-inflammatory in nature, depending on whether they tend to induce or inhibit the inflammatory response.54 These molecules usually diffuse over short distances, bind to the appropriate target cells, and affect the function of the target cell. Some effects occur over long distances, such as the systemic induction of fever by some cytokines (i.e., endogenous pyrogens) that are produced at an inflammatory site. The binding of chemokines or cytokines to a target cell often induces synthesis of additional cellular products. For example, binding of the cytokine TNF-α to a cell may result in synthesis and release of IL-1. Chemokine and cytokine binding is mediated through specific cell-surface receptors that are themselves sometimes under the regulation of secreted cellular products.
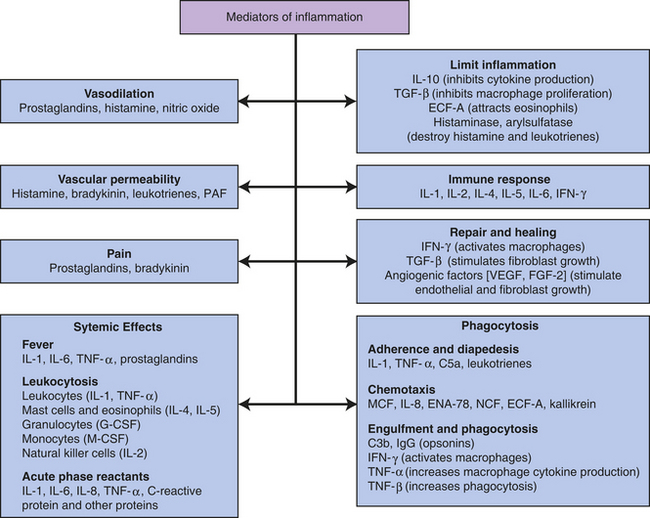
Figure 6-15 Principal mediators of inflammation. C3b, Large fragment produced from complement component C3; C5a, small fragment produced from complement component C5; ECF-A, eosinophil chemotactic factor of anaphylaxis; ENA, epithelial-dermoid neutrophil attractant; FGF, fibroblast growth factor; G-CSF, granulocyte colony-stimulating factor; IFN, interferon; IgG, immunoglobulin G (predominant class of antibody in the blood); IL, interleukin; MCF, monocyte chemotactic factor; M-CSF, monocyte colony-stimulating factor; NCF, neutrophil chemotactic factor; PAF, platelet-activating factor; TGF, T-cell growth factor; TNF, tumor necrosis factor; VEGF, vascular endothelial growth factor.
The actions of chemokines and cytokines are pleiotropic, indicating that the same molecule may have a large variety of different biologic activities depending on the particular target cell to which it binds. In addition, the same molecule may be produced by a large spectrum of cells, many of which are not part of inflammation or the immune system. These molecules may be synergistic, so that their combined activity exceeds the sum of their individual activities, or have antagonistic properties that cause them to inhibit each other. (A partial list of relevant cytokines is provided in Chapter 7, Table 7-5.)
Cytokines
The majority of important cytokines are classified as ILs or IFNs. Other critical cytokines, however, are not classified as either. Many of these same cytokines are produced by cells of the acquired immune system in response to specific antigens and are discussed further in Chapter 7.
Interleukins: The interleukins (ILs) are biochemical messengers produced predominantly by macrophages and lymphocytes in response to their recognition of a microorganism or stimulation by other products of inflammation. One important function of this class of cytokines is enhancement of the acquired immune response against pathogenic microorganisms and other foreign substances. Interleukins, however, are both produced by and have effects on a large variety of cells, often independent of infection.
IL-1 is a proinflammatory cytokine produced mainly by macrophages that have been stimulated by substances associated with infection, including many of the PAMPs discussed earlier in this chapter, as well as by other cytokines. IL-1 is synthesized in two forms, α and β that often elicit the same biologic responses. IL-1 is an endogenous pyrogen (i.e., fever-causing cytokine) that reacts with receptors on cells of the hypothalamus and affects the body’s thermostat. It also activates phagocytes and lymphocytes, thereby enhancing both the innate and acquired immunity, and acts as a growth factor for many cells. It has several effects on neutrophils, including induction of proliferation (resulting in an increase in the number of circulating neutrophils), chemotaxis, increased cellular respiration, and increased lysosomal enzyme activity.
IL-10 is an example of an anti-inflammatory cytokine and is primarily produced by lymphocytes to down-regulate both the inflammatory and acquired immune responses. IL-10 suppresses growth of lymphocytes and production of proinflammatory cytokines by macrophages.
More than 30 human interleukins have been identified, although the functions of several have not yet been defined. Their varied effects include the following:
1. Alteration of adhesion molecule expression on many types of cells
2. Induction of leukocyte chemotaxis
3. Induction of proliferation and maturation of leukocytes in the bone marrow
4. General enhancement or suppression of inflammation (see Table 7-5)
Interferons: Interferons (INFs) are low-molecular-weight proteins that primarily protect against viral infections and modulate the inflammatory response. (Mechanisms of viral infection are described in Chapter 9.) INFs are produced and released by virally infected cells in response to viral double-stranded RNA. Different kinds of INFs are produced by different types of cells—macrophages are the primary producers of both IFN-α and IFN-β, whereas T lymphocytes release IFN-γ. These INFs do not kill viruses directly but instead prevent them from infecting additional healthy cells. Interferons also enhance the efficiency of developing an acquired immune response.
IFN-α and IFN-β induce production of antiviral proteins, thereby conferring protection on uninfected cells. IFN-α or IFN-β is released from virally infected cells, attaches to a receptor on a neighboring cell, and if the neighboring cell is uninfected, stimulates the production of antiviral proteins that will interfere with transcription of viral nucleic acids or with viral replication (Figure 6-16). These interferons have no effect on cells that have already been virally infected. IFN-γ enhances the inflammatory response by increasing the microbicidal activity of macrophages. This cytokine also facilitates development of the acquired immune response against viral antigens on infected cells. Interferons are species specific, meaning that human interferon is effective only in humans; however, these cytokines are not virus specific, meaning that they are effective against almost all viruses.
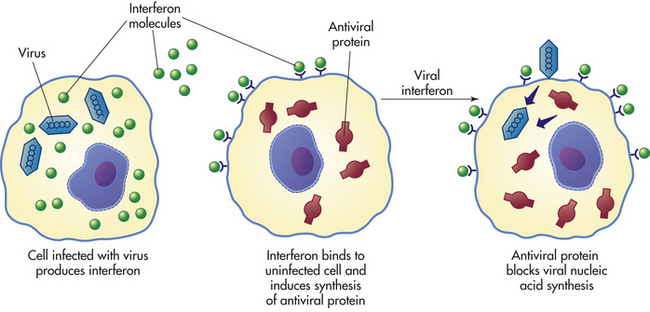
Other Cytokines: Despite the numerous interleukins and interferons, other essential cytokines are needed to mount an efficient inflammatory response. One of the most important of these is tumor necrosis factor-alpha (TNF-α). Macrophages secrete TNF-α in response to recognition of PAMPs by TLRs. Other cells, such as mast cells, are additional and crucial sources of this proinflammatory cytokine.55 TNF-α is initially synthesized as a membrane-spanning protein, which is cleaved into a soluble form by a membrane-associated protease, TNF-converting enzyme (TACE). Soluble TNF-α induces a multitude of proinflammatory effects, including enhancement of endothelial cell adhesion molecule expression and induction of chemokine production by both endothelial cells and macrophages. When secreted in large amounts, TNF-α has systemic effects as well:
Chemokines
Chemokines are members of a family of low-molecular-weight (8 to 10 kDa) peptides that function primarily to induce leukocyte chemotaxis.56 This response can be elicited either by soluble chemokines or by chemokines that are bound to extracellular glycosaminoglycan carbohydrates. Chemokines can be synthesized by multiple cell types, including macrophages, fibroblasts, and endothelial cells, in response to pro-inflammatory cytokines. Macrophages can be stimulated to produce chemokines by recognition of either infectious microorganisms or a β-defensin (both through TLR-4). To date, more than 40 different human chemokines have been described, the vast majority of which are classified as either CC-chemokines (β-chemokines) or CXC-chemokines (α-chemokines), depending on the arrangement of cysteine amino acids in the protein.57 This amino acid arrangement also determines which target cell(s) will respond to a given chemokine. CC-chemokines affect mainly monocytes, lymphocytes, and eosinophils, whereas CXC-chemokines generally affect neutrophils. Examples of CC-chemokines include RANTES (regulated on activation, normal T expressed and secreted), monocyte/macrophage chemotactic proteins (MCP-1, MCP-2, and MCP-3), and macrophage inflammatory proteins (MIP-1α and MIP-1β). CXC-chemokines include IL-8 and epithelial-dermoid neutrophil attractant (ENA-78).
LOCAL MANIFESTATIONS OF INFLAMMATION
The cells and plasma protein systems described previously interact to produce all the characteristics of inflammation, whether local or systemic, as well as determine the duration of inflammation, either acute or chronic. Local inflammation accompanies all types of cellular and tissue injury, whether infected or sterile, from fractures or strains of the musculoskeletal system to burn injuries (see Chapter 2) and is responsible for initiating healing.
All the local manifestations of acute inflammation (i.e., swelling, pain, heat, and redness) result from vascular changes and the subsequent leakage of circulating components into the tissue. Heat and redness are the result of vasodilation and increased blood flow through the injured site. Swelling occurs as exudate (fluid and cells) accumulates. Swelling is usually accompanied by pain caused by pressure exerted by exudate accumulation, as well as the presence of soluble biochemical mediators such as prostaglandins and bradykinin. Loss of function may be associated with these manifestations.
Exudate varies in composition, depending on the stage of the inflammatory response and, to some extent, the injurious stimulus. In early or mild inflammation, the exudate is watery (serous) with very few plasma proteins or leukocytes. An example of serous exudate is the fluid in a blister. In more severe or advanced inflammation, the exudate may be thick and clotted (fibrinous exudate), such as in the lungs of individuals with pneumonia. If a large number of leukocytes accumulate, as in persistent bacterial infections, the exudate consists of pus and is called a purulent (suppurative) exudate. Purulent exudate is characteristic of walled-off lesions (cysts or abscesses). If bleeding occurs, the exudate is filled with erythrocytes and is described as a hemorrhagic exudate.
Although the local manifestations of inflammation can affect all vascularized tissues, lesions vary depending on the organ or tissue involved. The lesion resulting from widespread cellular death (necrosis), for example, differs in myocardial (heart muscle), brain, and hepatic (liver) tissues. Cellular death resulting from myocardial infarction (deprivation of oxygen caused by cessation of blood flow) causes a response that proceeds to replacement of the dead tissue with a fibrinous scar. The same injury to brain tissue is more likely to result in the formation of an abscess filled with necrotic tissue (types of necrosis are described in Chapter 2). Destruction of liver tissue stimulates the regrowth, or regeneration, of liver cells.
SYSTEMIC MANIFESTATIONS OF ACUTE INFLAMMATION
The three primary systemic changes associated with the acute inflammatory response are fever, leukocytosis (a transient increase in circulating leukocytes), and increased levels in circulating plasma proteins.
Fever
An early systemic response is fever, which is partially induced by specific cytokines, for example, IL-1 released from neutrophils and macrophages.58 These fever-causing cytokines are known as endogenous pyrogens to differentiate them from pathogen-produced exogenous pyrogens. Pyrogens act directly on the hypothalamus, the portion of the brain that controls the body’s thermostat. The release of endogenous pyrogens by inflammatory cells occurs after phagocytosis, after exposure to bacterial endotoxin, or after exposure to antigen-antibody complexes. (Mechanisms of temperature regulation are discussed in Chapter 15.)
The generation of a febrile response can be beneficial because the microorganisms that cause some conditions (e.g., syphilis, gonococcal urethritis) are highly sensitive to small increases in body temperature. On the other hand, fever may have some harmful side effects because it may enhance the person’s susceptibility to the effects of endotoxins associated with gram-negative bacterial infections (bacterial toxins are described in Chapter 2).
Leukocytosis
Another systemic change associated with acute inflammation is leukocytosis. During many infections, numbers of circulating leukocytes, primarily neutrophils, increase. This increase is usually accompanied by a “left shift” in the ratio of immature to mature neutrophils, so that the more immature forms of neutrophils, such as band cells, metamyelocytes, and occasionally myelocytes, are present in relatively greater than normal proportions. (Chapter 25 discusses the development and maturation of blood cells.) Production of immature leukocytes increases primarily because proliferation and release of granulocyte and monocyte precursors in the bone marrow are stimulated by several products of inflammation, including complement product C3a and G-CSF.
Plasma Protein Synthesis
The synthesis of many plasma proteins, most of which are products of the liver, is increased during the primary stages of inflammation. These proteins, which can be either pro- or anti-inflammatory in nature, are referred to as acute-phase reactants (Table 6-4). Acute-phase reactants reach maximal circulating levels within 10 to 40 hours of initial infection. In addition to inducing fever, IL-1 also indirectly induces the synthesis of acute-phase reactants. IL-1 up-regulates release of IL-6, which then increases synthesis of acute-phase reactants directly by stimulating liver cells. Administration of IL-1 into animals leads to both fever and elevation of most acute-phase reactants, including fibrinogen, C-reactive protein, haptoglobin, amyloid A, α1-antitrypsin, and ceruloplasmin.
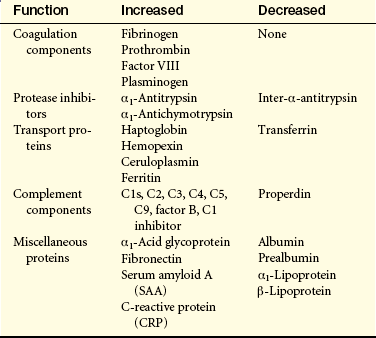
Acute inflammation can be verified by a series of hematologic tests, which are described in detail in Chapter 25. For example, an increase in blood levels of acute-phase reactants, primarily fibrinogen, is usually associated with an increased erythrocyte sedimentation rate. The alteration in plasma proteins probably leads to an enhanced erythrocyte rouleaux formation (stacking of erythrocytes, as in a stack of coins) and thereby an increased rate of sedimentation. Although increased erythrocyte sedimentation is a nonspecific reaction, it is considered a good indicator of an acute inflammatory response. Other symptoms of acute inflammation include somnolence (drowsiness), malaise (generalized feeling of discomfort or illness), anorexia (lack of desire to eat), and muscle aching.
CHRONIC INFLAMMATION
Superficially, the difference between acute and chronic inflammation is purely one of duration, in that chronic inflammation lasts 2 weeks or longer, regardless of cause. Characteristic histologic and mechanistic differences also may be present (Figure 6-17). Chronic inflammation is sometimes preceded by an unsuccessful acute inflammatory response. For example, if bacterial contamination or foreign objects (e.g., dirt, wood splinter, glass) persist in a traumatic wound, an acute response may be prolonged beyond 2 weeks. Pus formation, suppuration (purulent discharge), and incomplete wound healing may characterize this type of chronic inflammation.
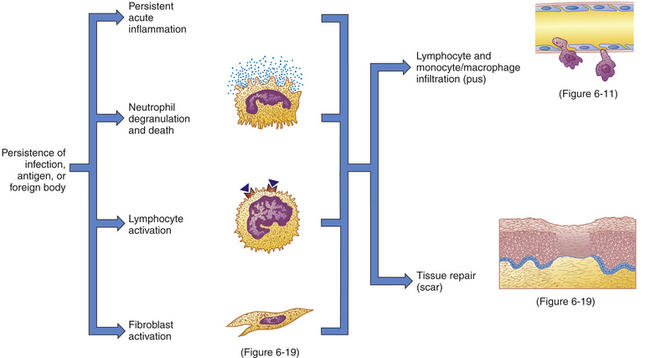
Figure 6-17 The chronic inflammatory response. Inflammation usually becomes chronic because of the persistence of an infection, an antibody, or a foreign body in the wound. Chronic inflammation is characterized by the persistence of many of the processes of acute inflammation. In addition, large amounts of neutrophil degranulation and death, the activation of lymphocytes, and the concurrent activation of fibroblasts result in the release of mediators that induce the infiltration of more lymphocytes and monocytes/macrophages and the beginning of wound healing and tissue repair.
Chronic inflammation can occur also as a distinct process without much previous acute inflammation. Some microorganisms (e.g., mycobacteria that cause tuberculosis) have cell walls with a very high lipid and wax content, making them relatively insensitive to degradation by phagocytes and therefore relatively resistant to clearance in an acute inflammatory response. Other microorganisms, such as those that cause leprosy, syphilis, and brucellosis, can survive within the macrophage and thereby also avoid clearance by the acute inflammatory response. In addition, some microorganisms produce toxins that stimulate tissue-damaging reactions even after they themselves are killed. Persistent inflammation can result from prolonged irritation by these toxins. Finally, chemicals, particulate matter, or physical irritants (e.g., inhaled dusts, wood splinters, and suture material) also can cause an inflammatory response that lasts longer than 2 weeks.
Chronic inflammation is characterized by a dense infiltration of lymphocytes and macrophages. If macrophages are unable to limit the tissue damage or infection, the body attempts to wall off and isolate the infected area, thus forming a granuloma (Figure 6-18). Granulomas may form if neutrophils and macrophages are unable to destroy microorganisms during the acute inflammatory response. For example, infections caused by some bacteria (Listeria sp., Brucella sp.), fungi (histoplasmosis, coccidioidomycosis) and parasites (leishmaniasis, schistosomiasis, toxoplasmosis) can result in granuloma formation. Large antigen-antibody complexes such as those present in rheumatoid arthritis also can result in the formation of these structures. The process of granuloma formation begins when some of the macrophages differentiate into large epithelioid cells, cells that are incapable of phagocytosing large bacteria but are capable of taking up debris and other small particles. Other macrophages fuse into multinucleated giant cells, which are active phagocytes that can engulf very large particles—larger than can be engulfed by a single macrophage. These two types of differentiated macrophages form the center of the granuloma, which is surrounded by a wall of lymphocytes. The granuloma itself is also often encapsulated by fibrous deposits of collagen and may become cartilaginous or possibly calcified by deposits of calcium carbonate and calcium phosphate.
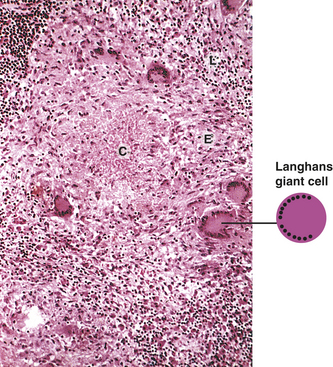
Figure 6-18 Tuberculous granuloma. A central area of amorphous caseous necrosis (C) is surrounded by a zone of lymphocytes (L) and enlarged epithelioid cells (E). Activated macrophages frequently fuse to form multinucleated cells (Langhans giant cells). In tuberculoid granulomas the nuclei of the giant cells move to the cellular margins in a horseshoe-like formation.
The classic granuloma associated with tuberculosis is characterized by a wall of epithelioid cells surrounding a center of dead and decaying tissue (caseous necrosis, see Chapter 2) and mycobacteria. Decay of cells within the granuloma results in the release of acids and the enzymatic contents of lysosomes from dead phagocytes. In this inhospitable environment, the cellular debris is broken down and a clear fluid remains (liquefaction necrosis, see Chapter 2). Eventually this fluid diffuses out and leaves a hollow, thick-walled structure in the tissue that may remain for the life of the individual.
RESOLUTION AND REPAIR
Destruction of tissue is followed by a period of healing that begins during acute inflammation and may not be complete for as long as 2 years.59 The most favorable outcome of healing is tissue regeneration with complete return to normal structure and function. This is an ideal that is often not possible, particularly in adults. However, if damage is minor, no complications occur, and destroyed tissues are capable of regeneration, it is possible to return injured tissues to an approximation of their original structure and physiologic function. This restoration is called resolution. However, if extensive damage is present, if injury occurs in tissues not capable of regeneration, if infection results in abscess or granuloma formation, or if fibrin persists in the lesion, resolution is not possible and repair takes place instead. Repair is the replacement of destroyed tissue with scar tissue. Scar tissue is composed primarily of collagen that fills in the lesion and restores tensile strength but cannot carry out the physiologic functions of destroyed tissue.
Both regeneration and repair actually begin with phagocytosis of the particulate matter found at the site of injury (fibrin from dissolved clots, microorganisms, erythrocytes, and dead tissue cells). This cleanup of the lesion, which also involves dissolution of fibrin clots (or scabs) by fibrinolytic enzymes, is called débridement. After débridement, the remaining debris is drained away and the vascular dilation and permeability associated with inflammation are reversed, thus preparing the lesion for either regeneration or repair.
Healing always involves processes that (1) fill in, (2) seal, and (3) shrink the wound. These common denominators of healing vary in importance and duration among different types of wounds. A clean incision, such as a paper cut or a sutured surgical wound, heals primarily through the process of collagen synthesis. Because sealing of this type of wound has already been facilitated by minimal tissue loss and close apposition of the wound edges, very little sealing (epithelialization) and shrinkage (contraction) are required for healing. Wounds that heal under conditions of minimal tissue loss are said to heal by primary intention (Figure 6-19).
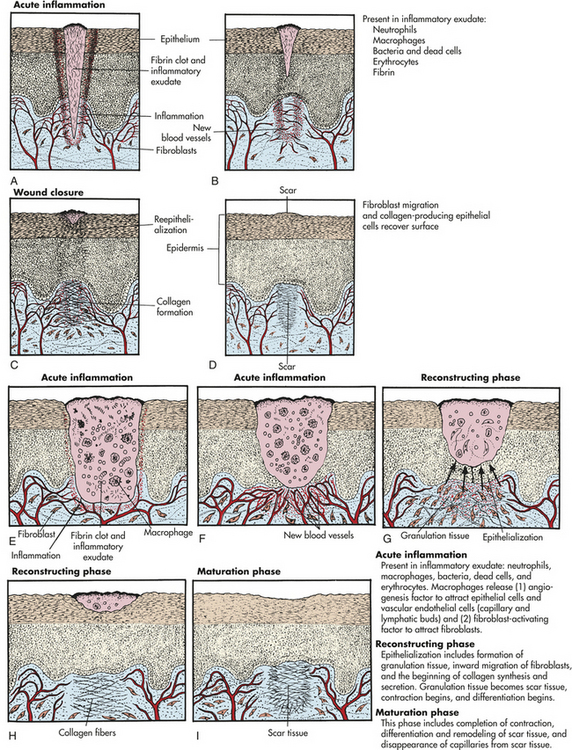
Figure 6-19 Wound repair by primary or secondary intention. A to D, Healing by primary intention. E to I, Healing by secondary intention.
Other wounds do not heal so neatly and easily. Healing of an open wound, such as a stage IV pressure sore (decubitus ulcer), requires a great deal more tissue replacement than healing of a sutured surgical incision. With an open wound, epithelialization, scar formation, and contraction take longer and healing occurs through secondary intention (see Figure 6-19). Healing by either primary or secondary intention may occur at different rates for different types of tissue injury.
Both resolution and repair occur in two overlapping phases. The first phase, called the reconstructive phase, begins 3 to 4 days after the initial injury and continues for as long as 2 weeks. During this phase the lesion is characterized by fibroblast (connective tissue cell) proliferation, followed by collagen synthesis by the fibroblasts, epithelialization, contraction of the wound, and cellular differentiation. The second phase, the maturation phase, begins several weeks after injury and is normally complete within 2 years. During this phase cellular differentiation, scar formation, and scar remodeling continue.
Reconstructive Phase
Because surgical wounds exhibit both the reconstructive and maturation phases, they are useful models of both normal and abnormal (dysfunctional) healing. Such wounds are initially sealed off by a blood clot containing fibrin and trapped cells. The cross-linked mesh of fibrin is created by activation of the coagulation cascade and initially traps platelets to form a platelet plug that further seals damaged vessels (see Chapter 25). Most surgical wounds are completely sealed with platelet plugs within hours after closure. This sealing helps unite the wound edges and acts to create a physical barrier to bacterial invasion, although pathogenic invasion is not always prevented. The fibrin mesh ultimately acts as a scaffold for the collagen or regenerated tissue cells that ultimately fill the wound.
For healing to proceed, the fibrin clot must be dissolved and then replaced by normal tissue (for resolution) or scar tissue (for repair).60 Enzymatic digestion of the clot usually occurs after activation of the plasma fibrinolytic system (plasmin generation, see Chapter 25) or release of lysosomal enzymes from dead neutrophils. Macrophages invade the dissolving clot and, by phagocytosis, clear away debris and dead cells. Débridement by macrophages and remaining neutrophils is followed by regeneration of destroyed cells (resolution) or, if regeneration is not possible, by repair (see Figure 6-19).
The process of healing begins as granulation tissue grows inward from surrounding healthy connective tissue. Granulation tissue is filled with new capillaries (angiogenesis) that give it a red, granular appearance and is surrounded by fibroblasts and macrophages. First, capillary buds sprout from vascular endothelial cells around the wound and extend into the débrided areas. Loops form when the young capillaries join (anastomose). The loops are more fragile and permeable than mature vessels, resulting in leakage of erythrocytes and neutrophils. The erythrocytes are phagocytosed by macrophages and the neutrophils assist in further débridement of the inflammatory lesion. Many of the new capillaries differentiate into larger vessels as repair continues promoting influx of nutrients and removal of metabolic wastes. New lymphatic vessels also grow into the granulation tissue by a similar process.
In addition to their role in débriding, macrophages at the site of injury secrete several biochemical mediators and cytokines that orchestrate and promote healing. Examples of these mediators include:
1. Transforming growth factor-beta (TGF-β) stimulates fibroblasts entering the lesion to synthesize and secrete the collagen precursor procollagen.
2. Angiogenesis factors, such as vascular endothelial growth factor (VEGF) and fibroblast growth factor-2 (FGF-2), stimulate vascular endothelial cells to form capillary buds that grow into the wound.
3. Matrix metalloproteinases (MMPs) may function in the degradation and remodeling of extracellular matrix proteins (e.g., collagen and fibrin) at the site of injury.
As the clot or scab is being dissolved and granulation tissue is being formed, the healing wound must be protected. This is accomplished by epithelialization, the process by which epithelial cells grow into the wound from surrounding healthy tissue. Inching along the exposed collagenous matrix, epithelial cells migrate under the clot or scab using MMPs to unravel collagen as they travel. Unraveling the collagen enables the epithelial cells to move rather than remain immobile (see Figure 6-19); the intact collagen ahead of them provides a pathway on which they can maneuver forward. Eventually the migrating epithelial cells contact similar cells from all sides of the wound and seal it, thereby halting migration and proliferation. The epithelial cells undergo differentiation to give rise to the various epidermal layers (see Chapter 44). Epithelialization of a skin wound can be hastened if the wound is kept moist, preventing the fibrin clot from becoming a scab.
Fibroblasts are the most important cells during the reconstructive phase of wound healing because they synthesize and secrete collagen and other connective tissue proteins. Fibroblasts are stimulated by macrophage-derived TGF-β to proliferate, enter the lesion, and produce these proteins. The collagen and connective tissue proteins produced by fibroblasts are deposited in débrided areas about 6 days after the fibroblasts have entered the lesion. Collagen is the most abundant protein in the body. It contains high concentrations of the amino acids glycine, proline, and lysine, although many of the proline and lysine amino acids are enzymatically modified as the protein is being synthesized. Modification of these amino acids requires several cofactors and is absolutely necessary for proper collagen polymerization and function. The required cofactors include iron, ascorbic acid (vitamin C), and molecular oxygen (O2); absence of any of these results in incomplete or impaired wound healing.
Immature collagen (i.e., procollagen) is secreted by fibroblasts as a complex of three polypeptide chains cross-linked by intermolecular bonds. Procollagen is converted to mature collagen by the proteolytic removal of small polypeptide sequences at both ends of the trimer. As healing progresses, collagen molecules are cross-linked by intramolecular covalent bonds to form collagen fibrils that are further cross-linked to form collagen fibers. The process of complete collagen matrix assembly takes several months because collagen is initially deposited randomly but then is remodeled by repeated dissolution (by MMPs) and reassembly. During this remodeling period, collagen fibers orient along the lines of mechanical stress; further cross-linking adds strength to the final collagen matrix.
Wound contraction is the final process of the reconstructive phase of healing. It is necessary for closure of all wounds, but especially those that heal by secondary intention. Contraction is noticeable 6 to 12 days after injury and may amount to inward movement of the wound edge by approximately 0.5 mm/day in normal healing. The granulation tissue of a healing wound contains myofibroblasts—specialized cells that are likely responsible for wound contraction. As their name implies, myofibroblasts have features of both smooth muscle cells and fibroblasts. They appear microscopically similar to fibroblasts, but differ in that their cytoplasm contains bundles of parallel fibers similar to those found in smooth muscle cells. Wound contraction occurs as extensions from the plasma membrane of myofibroblasts establish connections between neighboring cells, contract their fibers, and exert tension on the neighboring cells while anchoring themselves to the wound bed.
Maturation Phase
Collagen matrix assembly, tissue regeneration, and wound contraction all begin during the reconstructive phase but are not yet completed when the reconstructive phase ends, about 2 weeks after injury. Therefore, these processes continue into the maturation phase—a phase that can persist for years. During the maturation phase scar tissue is remodeled and capillaries disappear, leaving the scar avascular. Within 2 to 3 weeks after maturation has begun, the scar tissue has gained about two thirds of its eventual maximum strength.
Neither epidermal wounds that heal by secondary intention nor unsutured internal lesions are completely restored by healing. At best, repaired tissue regains 80% of its original tensile strength. Only epithelial, hepatic (liver), and bone marrow cells are capable of the complete mitotic regeneration known as compensatory hyperplasia (hyperplasia is described in Chapter 2). In fibrous connective tissue such as joints and ligaments, normal healing results in replacement of the original tissue with new tissue that does not have exactly the same structure or function as that of the original. Some tissues heal without replacement of cells. For example, damage resulting from myocardial infarction heals with a scar composed of fibrous tissue rather than with cardiac muscle cell replacement. Although the composition of various healed tissues may differ, the healing process—reconstruction followed by wound maturation—is essentially the same for all wounds.
Dysfunctional Wound Healing
Dysfunctional wound healing may occur if any of the involved processes occurs abnormally. This can include abnormalities in the inflammatory response itself, insufficient or excessive repair, or if a wound is reinfected. Abnormalities may result from a predisposing disease, such as diabetes mellitus, or from an acquired condition, such as hypoxemia (insufficient oxygen in arterial blood). Numerous drugs and nutrients can affect wound healing as well.
Dysfunction During Inflammatory Response
Healing may be prolonged if bleeding is not stopped during acute inflammation. Hemorrhage in a damaged area delays healing for several reasons. Initially the excess blood cells that accumulate at the site of injury must be cleared—a process that requires additional time. In addition to the cellular accumulation caused by excessive bleeding, formation of a clot increases the amount of space that granulation tissue has to fill and serves as a mechanical barrier to oxygen diffusion. The great amount of fibrin that is released during hemorrhage also must eventually be reabsorbed in order to prevent its organization into fibrous adhesions. Once formed, these adhesions can bind organs together by fibrous bands; with time, shrinkage of these bands can distort or strangulate nearby organs. This is clinically significant, particularly if they form within the pleural, pericardial, or abdominal cavities.
Accumulated blood as a result of hemorrhage also serves as an excellent culture medium for bacteria, promoting continued infection and prolonging inflammation by increasing purulent exudate formation. Prolonged infection can promote excess scar formation or even prevent healing completely. Continued infection of a wound, termed wound sepsis, can be clinically treated in several ways. Most important is the débridement of necrotic tissue and foreign bodies. This removal is accomplished either through surgery or through the use of absorbent dressings. Wound irrigation and antibiotic therapy also may assist in combating continued infection.
Although local hemorrhage during the inflammatory process can pose a huge impediment to healing, many additional factors, both physiologic and pharmacologic, also may adversely affect the healing of an inflamed tissue. Hypovolemia—decreased blood volume—hinders inflammation. The physiologic response to hypovolemia is vessel constriction rather than the dilation required to deliver inflammatory cells to the site of injury. Optimal nutrition is important during all phases of healing because metabolic needs are increased. The most essential nutrients for healing are glucose, oxygen, and amino acids. Because leukocytes need glucose to produce the energy needed for chemotaxis, phagocytosis, and intercellular killing, the wounds of persons with diabetes who receive insufficient insulin heal poorly, mainly due to a prolonging of infection. Persons with diabetes are also at risk for ischemic wounds because they are likely to have both small-vessel diseases that impair the microcirculation and altered (glycosylated) hemoglobin, which has an increased affinity for oxygen and thus does not readily release oxygen in tissues.61 (Hemoglobin’s function as the oxygen-carrying component of blood is described in Chapter 25.) Oxygen delivery is also compromised by hypoxemic states because ischemic tissue is susceptible to infection. Hypoproteinemia prolongs inflammation because the associated decrease in available amino acids is an impediment to fibroblast proliferation. Finally, anti-inflammatory steroids can have an impact upon wound healing. These drugs prevent macrophages from migrating to the site of injury and inhibit their release of collagenase and plasminogen activator. Anti-inflammatory steroids also inhibit fibroblast migration into the wound during the reconstructive phase of healing and impair angiogenesis, wound contraction, and reepithelialization.
Dysfunction During Reconstructive Phase of Healing
Three of the essential processes that occur during the reconstructive phase are assembly and remodeling of the collagen matrix, epithelialization of the wound bed, and contraction of the wound. Dysfunctional wound healing can result from the impairment of any of these processes.
Impaired Collagen Matrix Assembly: A number of factors may interfere with the production of collagen in healing tissues, most being nutritional. Scurvy, for example, is a condition caused by a deficiency in ascorbic acid, one of the cofactors required for the amino acid modification that is necessary for proper collagen matrix assembly. The complication of scurvy is a poorly formed collagen matrix and, therefore, greatly impaired wound healing. Other nutrients, including iron, copper, and calcium, play additional roles in the enzymatic reactions required for collagen modification and assembly. Usually, however, such minute amounts of these substances are required that deficiencies are not clinically significant. Nutritionally, appropriate protein intake is also essential for collagen synthesis. The amino acid methionine that is found in proteins is converted to cysteine, the role of which in collagen synthesis is twofold: (1) it functions as an important cofactor in the enzymatic reactions required for collagen synthesis; and (2) it contains sulfur, which contributes to formation of the strong covalent bonds in cross-linked collagen fibrils.
Dysfunctional healing also may result from excessive production of collagen. Overproduction of collagen causes surface overhealing, which is manifested in the skin by formation of a keloid or a hypertrophic scar (Figure 6-20). A keloid is a raised scar that extends beyond the original boundaries of the wound. It invades surrounding tissue and is likely to recur after surgical removal. A familial tendency toward keloid formation has been observed, with a greater incidence in blacks relative to whites. Similar to a keloid, a hypertrophic scar is also raised but differs in that it remains within the original boundaries of the wound. Hypertrophic scars tend to regress over time, whereas keloids do not. Both keloids and hypertrophic scars are caused by an imbalance between collagen synthesis and collagen degradation in which synthesis is increased relative to degradation. Although the precise mechanism of this imbalance is unknown, recent evidence suggests that keloid fibroblasts have lower rates of apoptosis and an inability to respond to normal suppressive feedback.62
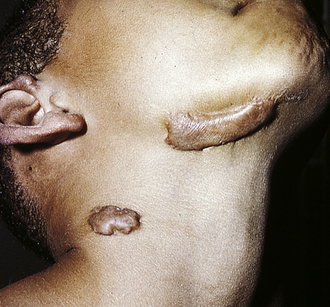
Impaired Epithelialization: The process of epithelialization is suppressed by anti-inflammatory steroids, hypoxemia, and nutritional deficiencies. Anti-inflammatory steroids inhibit phagocyte production of the biochemical mediators required for epithelialization, hypoxemia deprives cells of the energy required for the process, and dietary zinc is necessary for the MMP activity that is crucial to cellular migration.
Wound care techniques also may greatly influence epithelial cell migration. External wounds that are draining or healing by secondary intention often are clinically débrided and protected with dressings. The ideal dressing is one that absorbs some drainage without being incorporated into the clot or granulation tissue. Because epithelial cells must migrate across the wound during healing, dressings that débride healthy epithelial cells along with necrotic tissue prolong epithelialization. Many solutions that traditionally have been used to clean or irrigate wounds are now known to be deleterious to the fragile new cells in the wound bed. Normal saline is the most innocuous solution that can be used to cleanse or irrigate a wound that is healing primarily by epithelialization. Solutions such as povidone-iodine and hydrogen peroxide are desiccating (drying) and, as such, inhibit rather than promote epithelial cell migration.
Impaired Contraction: Excessive wound contraction may result in a deformity or contracture. Burn wounds are especially susceptible to the development of contractures. Internal contractures may occur as well, and are common in cirrhosis of the liver. Internally, scar tissue that becomes contracted constricts blood flow that may contribute to the development of portal hypertension and esophageal varices. Other types of internal contraction deformity include duodenal strictures caused by dysfunctional healing of an ulcer and esophageal strictures caused by chemical burns.
Proper positioning and range-of-motion exercises, as well as surgery, are among the physical means used to overcome the excessive myofibroblast-derived tension that results in contractures. Clinical use of pharmacologic methods for control of wound contracture is still largely experimental, but includes control of myofibroblast contraction by the administration of smooth muscle cell inhibitors such as colchicine and inhibition of proper collagen matrix assembly with drugs that prevent either collagen cross-linking or MMP activity. These latter treatments are based on the knowledge that myofibroblast binding to collagen can “lock” contracted cells into position.
Wound Disruption
Finally, a potential complication in the healing of wounds that are sutured closed is dehiscence, in which the wound pulls apart at the suture line. The greatest incidence of dehiscence occurs 5 to 12 days after suturing, paradoxically at the time when collagen synthesis is at its peak. Approximately 50% of dehiscence occurrences are associated with wound sepsis, although dehiscence also may occur when sutures break as a result of excessive strain. Obesity increases the risk of suture breakage because adipose tissue is difficult to suture. Wound dehiscence usually is heralded by an increase in serous drainage from the wound. In addition, patients may report a feeling that “something gave way.” Prompt surgical ttention is required.
Pediatrics and Mechanisms of Self-Defense
Neonates commonly have transiently depressed inflammatory and immune function. For example, neutrophils and perhaps monocytes may not be capable of efficient chemotaxis. Insufficient response to chemotactic factors appears to be caused by lack of fluidity in the phagocyte’s plasma membrane so that pseudopod formation and migration are impaired. Neonates are prone to infections associated with chemotactic defects, including cutaneous abscesses caused by staphylococci and cutaneous candidiasis. Further, neutrophils in neonates who were stressed by in utero infection or respiratory insufficiency have diminished oxidative and bacterial responses. (Acquired phagocytic defects, which may be induced by a variety of infections, metabolic disorders, nutrition deficiencies, or drugs, are described in Chapter 8.)
Neonates also are partially deficient in complement, especially components of the alternative pathway. They tend to have a relative deficiency of factor B and to develop severe, overwhelming sepsis and meningitis when infected with bacteria against which there is no transferred maternal antibody. Low levels of mannose-binding lectin increase the risk for neonatal hospital-acquired sepsis.63 Neonates also may be deficient in some of the collectins and collectin-like proteins. This is especially true of preterm neonates.64 Some preterm infants with respiratory distress syndrome are deficient in at least one collectin, which provides innate defense against respiratory infections.
Aging and Mechanisms of Self-Defense
The older adult population is also at risk for impaired inflammation and wound healing. In some cases, impaired healing is not directly associated with aging in general but can instead be linked to a chronic illness such as cardiovascular disease or diabetes mellitus. In addition, many older adults require medications such as anti-inflammatory steroids that can interfere with the healing process.
Older adults have increased susceptibility to bacterial infections of the lungs, urinary tract, and skin. Because of impaired sensation or mobility and physiologic changes in the skin, older adults are at increased risk for sustaining various wounds. With aging, subcutaneous fat is lost, diminishing a layer of protection. Collagen fibers become thicker and a certain percentage of elastin is lost, further contributing to loss of protection. The regenerative capability of the skin is maintained with aging, but the epidermis undergoes age-associated changes that include atrophy of the underlying capillaries. The consequent decrease of perfusion makes older adults more susceptible than others to the adverse effects of hypoxia in the wound bed. In addition, aging fibroblasts may have a slower rate of proliferation and therefore wound healing is attenuated.65
Infections of other organ systems in older adults may be due to a diminished natural ability to ward off infection. Several cellular components of innate resistance are deficient in number (e.g., alveolar macrophages) or have diminished activity (e.g., neutrophil chemotaxis, degranulation, and phagocytosis). One explanation for this diminished inflammatory cellular activity is an age-related decrease in expression and function of several, if not all, TLRs.
α1-antitrypsin 201
Abscess 205
Acute-phase reactant 206
Acquired immunity 183
Adaptive immunity 183
Adhesion molecule 198
Adhesions 211
Alternative pathway 188
Anaphylatoxin 189
Angiogenesis factor 210
Antigen-antibody complex (immune complex) 188
Antimicrobial lectin 186
Antimicrobial peptide 185
Bactericidal/permeability-inducing protein 186
Basophil 202
Bradykinin 191
C1 esterase inhibitor (C1 inh) 192
C3 convertase 189
C5 convertase 189
Carboxypeptidase 189
Cathelicidin 185
Cell lysis 189
Chemokine 205
Chemotactic factor 189
Chemotaxis 196
Classical pathway 188
Coagulation cascade 190
Collagen 210
Collectin 186
Complement cascade 188
Complement receptor 194
Contraction 208
Contracture 212
Cyst 205
Cytokine 202
Débridement 208
Defensin 185
Dehiscence 212
Diapedesis 199
Endogenous pyrogen 206
Endothelial cell 199
Eosinophil 202
Eosinophil chemotactic factor of anaphylaxis (ECF-A) 196
Epithelialization 208, 210
Epithelioid cell 207
Extrinsic pathway 190
Exudate 205
Fever 206
Fibrinous exudate 205
Fibroblast 210
Giant cell 208
Granulation tissue 208
Granulocyte 192
Granuloma 207
Hageman factor (factor XII) 192
Heat 205
Hemorrhagic exudate 205
Hereditary angioedema 192
Histamine 196
Hypertrophic scar 211
Inflammation 183
Inflammatory response 184, 186
Innate immunity 183
Integrins 195
Interferon (INF) 204
Interleukin (IL) 204
Intrinsic pathway 190
Keloid 211
Kinin system 191
Kininase 191
Lectin pathway 188
Leukocytosis 206
Leukotriene 197
Macrophage 201
Margination (pavementing) 199
Mast cell 195
Mast cell degranulation 189
Matrix metalloproteinase (MMP) 210
Maturation phase 208, 210
Monocyte 201
Myofibroblast 210
Native immunity 183
Natural barrier 184
Natural immunity 183
Natural killer (NK) cell 202
Neutrophil 201
Neutrophil chemotactic factor 196
Normal bacterial flora 186
Opsonin 190
Pain 205
Pathogen-associated molecular pattern (PAMP) 194
Pattern recognition receptor (PRR) 192
Phagocyte 201
Phagocytosis 198
Phagolysosome 201
Phagosome 200
Plasma kinin cascade 191
Plasma protein system 187
Plasmin 192
Platelet 202
Platelet-activating factor (PAF) 198
Polymorphonuclear neutrophil (PMN) 201
Primary intention 208
Primary lysosomal granule 201
Procollagen 210
Proenzyme 187
Prostaglandin 197
Purulent (suppurative) exudate 205
Reconstructive phase 208
Redness 205
Regeneration 208
Repair 208
Resistin-like molecule β 186
Resolution 208
Scar tissue 208
Scavenger receptor 195
Secondary granule 201
Secondary intention 208
Selectins 199
Serous 205
Swelling 205
Toll-like receptor (TLR) 194
Transforming growth factor-beta (TGF-β) 210
Tumor necrosis factor-alpha (TNF-α) 205
Wound contraction 210
REFERENCES
1. Tosi, M.F. Innate immune responses to infection. J Allergy Clin Immunol. 2005;116(2):241–249.
2. Dann, S.M., Eckmann, L. Innate immune defenses in the intestinal tract. Curr Opin Gastroenterol. 2007;23(2):115–120.
3. Braff, M.H., Gallo, R.L. Antimicrobial peptides: an essential component of the skin defensive barrier. Curr Top Microbiol Immunol. 2006;306:91–110.
4. Yamasaki, K., Gallo, R.L. Antimicrobial peptides in human skin disease. Eur J Dermatol. 2008;18(1):11–21.
5. Ooi, E.H., Wormald, P.J., Tan, L.W. Innate immunity in the paranasal sinuses: a review of nasal host defenses. Am J Rhinol. 2008;22(1):9–13.
6. Holmskov, U., Theil, S., Jensenius, J.C. Collections and ficolins: humoral lectins of the innate immune defense. Ann Rev Immunol. 2004;21:547–578.
7. Kuroki, Y., Takahashi, M., Nishitani, C. Pulmonary collectins in innate immunity in the lung. Cell Microbiol. 2007;9(8):1871–1879.
8. Wershil, B.K., Furuta, G.T. Gastrointestinal mucosal immunity. J Allergy Clin Immunol. 2008;121(2 Suppl):S380–S383.
9. Magalhaes, J.F., Tattoli, I., Girardin, S.E. The intestinal epithelial barrier: how to distinguish between the microbial flora and pathogens. Semin Immunol. 2007;19(2):106–115.
10. Dempsey, P.W., Vaidya, S.A., Cheng, G. The art of war: innate and adaptive immune responses. Cell Mol Life Sci. 2003;60(12):2604–2621.
11. Goldfarb, R.D., Parrillo, J.E. Complement. Crit Care Med. 2005;33(12):S482–S484.
12. Markiewski, M.M., Lambris, J.D. The role of complement in inflammatory diseases from behind the scenes into the spotlight. Am J Pathol. 2007;171(3):715–727.
13. Wills-Karp, M. Complement activation pathways: a bridge between innate and adaptive immune responses in asthma. Proc Am Thorac Soc. 2007;4(3):247–251.
14. Lutz, H.U., Jelezarova, E. Complement amplification revisited. Mol Immunol. 2006;43(1-2):2–12.
15. Gros, P., Milder, F.J., Janssen, B.J.C. Complement driven by conformational changes. Nat Rev Immunol. 2008;8(1):48–58.
16. Thurman, J.M., Holers, V.M. The central role of the alternative complement pathway in human disease. J Immunol. 2006;176(3):1305–1310.
17. Degn, S.E., Thiel, S., Jensenius, J.C. New perspectives on mannan-binding lectin-mediated complement activation. Immunobiol. 2007;212(4-5):301–311.
18. Haas, P.-J., van Strijp, J. Anaphylatoxins: their role in bacterial infection and inflammation. Immunol Res. 2007;37(3):161–175.
19. Aird, W.C. Coagulation. Crit Care Med. 2005;33(12 Suppl):S485–S487.
20. Lwaleed, B.A., et al. Tissue factor: a critical role in inflammation and cancer. Biol Res Nurs. 2007;9(2):97–107.
21. Schmaier, A.H., McCrae, K.R. The plasma kallikrein-kinin system: its evolution from contact activation. J Thromb Haemost. 2007;5(12):2323–2329.
22. Markiewski, M.M., et al. Complement and coagulation: strangers or partners in crime? Trends Immunol. 2007;28(4):184–192.
23. Cicardi, M., Zingale, L.C. The deficiency of C1 inhibitor and its treatment. Immunobiol. 2007;212(4-5):325–331.
24. Kawai, T., Akira, S. Pathogen recognition and Toll-like receptors. Curr Opin Immunol. 2005;17(4):338–344.
25. Kawai, T., Akira, S. TLR signaling. Semin Immunol. 2007;19(1):24–32.
26. Uematsu, S., Akira, S. Toll-Like receptors (TLRs) and their ligands. Handb Exp Pharmacol. 2008;183:1–20.
27. Arancibia, S.A., et al. Toll-like receptors are key participants in innate immune responses. Biol Res. 2007;40(2):97–112.
28. Hawlisch, H., Kohl, J. Complement and Toll-like receptors: key regulators of adaptive immune responses. Mol Immunol. 2006;43(1-2):13–21.
29. Azhar, S., Leers-Sucheta, S., Reaven, E. Cholesterol uptake in adrenal and gonadal tissues: the SR-BI and “selective” pathway connection. Front Biosci. 2003;8:S998–S1029.
30. Horiuchi, S., Sakamoto, Y., Sakai, M. Scavenger receptors for oxidized and glycated proteins. Amino Acids. 2003;25(3-4):283–292.
31. Rhainds, D., Brissette, L. The role of scavenger receptor class B type I (SR-B1) in lipid trafficking: defining the rules for lipid traders. Int J Biochem Cell Biol. 2004;36(1):39–77.
32. Vliagoftis, H., Befus, A.D. Mast cells at mucosal frontiers. Curr Mol Med. 2005;5(6):573–589.
33. Ehrlich, P. Dietbage zur Kenntnis der Anilinsfarb und Ihrer Verwendung nin ungen der Mikroskopichen technik. Arch Mikr Anat. 1877;13:263.
34. Galli, S.J., Tsai, M. Mast cells: versatile regulators of inflammation, tissue remodeling, host defenses and homeostasis. J Dermatol Sci. 2007;49(1):7–19.
35. Hide, M., Yanase, Y., Greaves, M.W. Cutaneous mast cell receptors. Dermatol Clin. 2007;25(4):563–575.
36. Huang, J.F., Thurmond, R.L. The new biology of histamine receptors. Curr Allergy Asthma Rep. 2008;8(1):7–21.
37. Peters-Golden, M., Henderson, W.R., Jr. Leukotrienes. N Engl J Med. 2007;357(18):1841–1854.
38. Garrood, T., Lee, L., Pitzalis, C. Molecular mechanisms of cell recruitment to inflammatory sites: general and tissue-specific pathways. Rheumatol. 2006;45(3):250–260.
39. Pober, J.S., Sessa, W.C. Evolving functions of endothelial cells in inflammation. Nat Rev Immunol. 2007;7(10):803–815.
40. Ley, K., et al. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol. 2007;7(9):678–689.
41. Woodfin, A. Voisin M-B, Nourshargh S: PECAM-1: a multi-functional molecule in inflammation and vascular biology. Arterioscler Thromb Vasc Biol. 2007;27(12):2514–2523.
42. Stephens, C., Fawcett, T.N. Nitric oxide and nursing: a review. J Clin Nurs. 2007;16(1):67–76.
43. Nauseef, W.M. How human neutrophils kill and degrade microbes: an integrated view. Immunol Rev. 2007;219(1):88–102.
44. Segal, A.W. How neutrophils kill microbes. Ann Rev Immunol. 2005;23:197–223.
45. Babior, B.M. NADPH oxidase. Curr Opin Immunol. 2004;16(1):42–47.
46. Marshall, J.S., King, C.A., McCurdy, J.D. Mast cell cytokine and chemokine responses to bacterial and viral infection. Curr Pharm Des. 2003;9(1):11–24.
47. Cavaillon, J.-M., Adib-Conquy, M. Monocytes/macrophages and sepsis. Crit Care Med. 2005;33(12 Suppl):S506–S509.
48. Gordon, S. The macrophage: past, present and future. Eur J Immunol. 2007;37(Suppl1):S9–S17.
49. Ma, J., et al. Regulation of macrophage activation. Cell Mol Life Sci. 2003;60(11):2334–2346.
50. Zhang, X., Mosser, D.M. Macrophage activation by endogenous danger signals. J Pathol. 2008;214(2):161–178.
51. Hogan, S.P., et al. Eosinophils: biological properties and role in health and disease. Clin Exp Allergy. 2008;38(5):709–750.
52. Min, B., Paul, W.E. Basophils and type 2 immunity. Curr Opin Hematol. 2008;15(1):59–63.
53. Vivier, E., et al. Functions of natural killer cells. Nat Immunol. 2008;9(5):503–510.
54. Steinke, J.W., Borish, L. Cytokines and chemokines. J Allergy Clin Immunol. 2006;117(2 Suppl Mini-Primer):S441–S445.
55. Galli, S.J., Nakae, S. Mast cells to the defense. Nat Immunol. 2003;4(12):1160–1162.
56. Allen, S.J., Crown, S.E., Handel, T.M. Chemokine: receptor structure, interactions, and antagonism. Annu Rev Immunol. 2007;25:787–820.
57. Rot, A., von Andrian, U.H. Chemokines in innate and adaptive host defense: basic chemokinese grammar for immune cells. Ann Rev Immunol. 2004;22:891–928.
58. Thompson, H.J. Fever, a concept analysis. J Adv Nurs. 2005;51(5):484–492.
59. Broughton, G., 2nd., Janis, J.E., Attinger, C.E. Wound healing: an overview. Plast Reconstr Surg. 2006;117(7 Suppl):1e-S–32e-S.
60. Broughton, G., 2nd., Janis, J.E., Attinger, C.E. The basic science of wound healing. Plast Reconstr Surg. 2006;117(7 Suppl):12S–34S.
61. Sweitzer, S.M., et al. What is the future of diabetic wound care? Diabetes Educ. 2006;32(2):198–210.
62. Robles, D.T., Berg, D. Abnormal wound healing: keloids. Clin Dermatol. 2007;25(1):26–32.
63. de Benedetti, F., et al. Low serum levels of mannose binding lectin are a risk for neonatal sepsis. Pediatr Res. 2007;61(3):325–328.
64. Awasthi, S., et al. Deficiencies in lung surfactant proteins A and D are associated with lung infection in very premature neonatal baboons. Am J Respir Crit Care Med. 2001;163(2):389–397.
65. Renshaw, M., et al. Cutting edge: impaired Toll-like receptor expression and function in aging. J Immunol. 2002;169(9):4697–4701.