STRESS AND DISEASE


Modern society is full of stress. As a culture, Westerners are champions of the work ethic, a Protestant philosophy originating in the sixteenth century that views idleness as taboo. In Poor Richard’s Almanac, Ben Franklin counseled people not to waste time. Driven by this perspective, Westerners have devoted time and energy to inventing time-saving devices such as cell phones and portable internet access. These inventions have fueled the drive of the “workaholic” American and, despite our prosperity, have not resulted in more leisure hours. The pressure to remain in contact despite illness, travel, vacation, and other events that used to provide socially acceptable absences is now customary in American, and indeed global, culture. This prevalent assumption of constant availability is a newly identified stressor contributing to the more global and better studied stressors of work related and relationship stressors. Tensions may be created due to the need to balance work and leisure time. When added to other well identified stressors such as financial problems, the result may be potential suffering from the so-called stress-related disorders.
It is often reported that use of the term stress in a biologic sense began with Hans Selye in 1946. Selye defined physiologic stress as a chemical or physical disturbance in the cells or tissue fluid produced by a change, either in the external environment or within the body itself, that requires a response.1 In 1914, however, Walter B. Cannon used the term in both a physiologic and a psychologic sense in a paper reporting psychoendocrine studies. In his report Cannon used such phrases as “great emotional stress” and “times of stress.”1 In 1935 Cannon published another paper called “Stresses and Strains of Homeostasis.” In it he applied the engineering concept of stress and strain in a physiologic context.2 Cannon thought also that stress involved psychologic factors; his paper stated that physical as well as emotional stimuli can cause stress. The popularization of the term, however, began with Selye’s work.
In the past decade it was been demonstrated that the interactions among social, psychologic, biologic, and behavioral factors are inherent in the causes and courses of many diseases. Molecular biologists, immunologists, neurologists, clinicians, and behavioral scientists began exploring the role of the neglected half of the mind-body (dualistic) model—that is, the mind. What is now emerging is a more holistic and complex model of health and disease states. This model involves the biochemical relationships of the central and autonomic nervous systems, the endocrine system, and the immune system and their relationships to stress-elicited coping behaviors, such as smoking and poor diet, that can also modify the integrity of the immune system. Discoveries of these complex links have led to the creation of the field of Psychoneuroimmunology.
CONCEPTS OF STRESS
Psychologic stress may cause or exacerbate (worsen) several disease states, including many of the diseases implicated as the leading causes of death in the United States, such as cardiovascular disease and infectious diseases.3,4 (Table 10-1). Important effects of acute emotional stress, such as the death of a loved one, on the heart include three areas: (1) left ventricular contractile dysfunction, (2) myocardial ischemia, and (3) disturbances of heart rhythm (see What’s New? Acute Emotional Stress and Adverse Heart Effects). Evidence implicates stress as a precipitating factor for some diseases and conditions and worsens symptoms and outcomes in a number of others, including irritable bowel syndrome, ulcers, asthma, autoimmune disorders, delayed wound healing, reproductive dysfunction, diabetes, and depression.5,6 Effects of stress on inflammatory and immune processes influence coronary artery disease, depression, autoimmune disorders (e.g., rheumatoid arthritis, lupus, multiple sclerosis) and some cancers (e.g., virally mediated).7 Evidence published since 2000 has generally supported a role for stress on human immunodeficiency virus (HIV) progression.8 Moreover, stress has been linked to the recurrence of genital herpes virus in women with HIV.8 Further, stress-induced, chronic inflammation is suggested as being important in the functional decline that leads to frailty, disability, and untimely death.9,10 As evidence has mounted concerning the important role that stress plays in certain disease processes, research has focused on the mechanisms responsible for these mind-body interactions. Along with a greater understanding of the relationship between the human stress response and disease, new strategies for treatment of stress-related disorders are emerging. This chapter describes definitions of stress, the history of stress research, and recent findings on the role of stress in disease.
Table 10-1
Examples of Stress-Related Diseases and Conditions
| Target Organ or System | Disease or Condition |
| Cardiovascular system | Coronary artery disease |
| Hypertension | |
| Stroke | |
| Disturbances of heart rhythm | |
| Muscle | Tension headaches |
| Muscle contraction (backache) | |
| Connective tissues | Rheumatoid arthritis (autoimmune disease) |
| Related inflammatory diseases of connective tissue | |
| Immune system | Asthma (hypersensitivity reaction) |
| Hay fever (hypersensitivity reaction) | |
| Immunosuppression or deficiency | |
| Autoimmune diseases | |
| Gastrointestinal system | Ulcer |
| Irritable bowel syndrome | |
| Diarrhea | |
| Nausea and vomiting | |
| Ulcerative colitis | |
| Genitourinary system | Diuresis |
| Impotence | |
| Frigidity | |
| Skin | Eczema |
| Neurodermatitis | |
| Acne | |
| Endocrine system | Type 2 diabetes mellitus |
| Amenorrhea | |
| Central nervous system | Fatigue and lethargy |
| Type A behavior | |
| Overeating | |
| Depression | |
| Insomnia | |
| Impaired learning and memory |
The term stress has been used persistently and widely in specialties such as biology, health sciences, and social sciences despite numerous disagreements over its definition. Nevertheless, in recent years stress has been more usefully defined as a transactional or interactional concept. Transactionally, stress is viewed as the state of affairs arising when a person relates to (i.e., interacts or transacts with) situations in certain ways. People are not disturbed by situations per se but by the ways they appraise and react to situations. In general, a person experiences stress when a demand exceeds a person’s coping abilities, resulting in reactions such as disturbances of cognition, emotion, and behavior that can adversely affect well-being.
General Adaptation Syndrome
Selye11 originally sought to discover a new sex hormone when he discovered the biologic syndrome of stress. In his attempts to discover the new hormone, Selye injected crude ovarian extracts into rats. Repeatedly, he found that the following triad of structural changes occurred: (1) enlargement of the cortex of the adrenal gland, (2) atrophy of the thymus gland and other lymphoid structures, and (3) development of bleeding ulcers of the stomach and duodenal lining. Selye soon discovered that this triad of manifestations was not specific for his ovarian extracts but also occurred after he exposed the rats to other noxious stimuli, such as cold, surgical injury, and restraint. He called these stimuli stressors. Selye concluded that this triad or syndrome of manifestations represented a nonspecific response to noxious stimuli. Because many diverse agents caused the same syndrome, Selye suggested that it be called the general adaptation syndrome (GAS).
Selye later defined three successive stages in development of the GAS: (1) the alarm stage, in which the central nervous system (CNS) is aroused and the body’s defenses are mobilized (i.e., flight or fight); (2) the stage of resistance or adaptation, during which mobilization contributes to flight or fight; and (3) the stage of exhaustion, in which continuous stress causes the progressive breakdown of compensatory mechanisms (acquired adaptations) and homeostasis. The stage of exhaustion, Selye believed, marked the onset of certain diseases he called diseases of adaptation.
The nonspecific physiologic response identified by Selye consists of interaction among the sympathetic branch of the autonomic nervous system (ANS) (see Chapter 14) and other neural signals that activate the endocrine system, known as the hypothalamic-pituitary-adrenal (HPA) axis, or interactions among the hypothalamus, pituitary gland, and the adrenal gland. The alarm phase of the GAS begins when a stressor triggers the actions of the hypothalamus and the sympathetic nervous system (SNS) (Figure 10-1). The resistance or adaptation phase begins with the actions of the adrenal hormones cortisol, norepinephrine, and epinephrine. Exhaustion occurs if stress continues and adaptation is not successful, ultimately causing impairment of the immune response, heart failure, and kidney failure, leading to death.
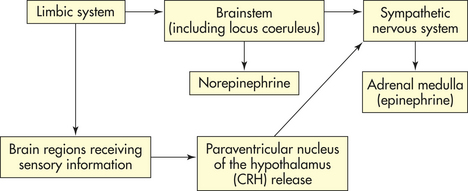
Selye defined physiologic stress as a chemical or physical disturbance in the cells or tissue fluid produced by a change, either in the external environment or within the body itself, that requires a response (i.e., begins the GAS) to counteract the disturbance. Selye identified three components of physiologic stress: (1) the exogenous or endogenous stressor initiating the disturbance, (2) the chemical or physical disturbance produced by the stressor, and (3) the body’s counteracting (adaptational) response to the disturbance.11
Psychologic Mediators and Specificity
Although Selye’s identification of the GAS is regarded as tremendously important and the cornerstone of stress research, the idea that stress is a purely physiologic response is oversimplified. In the mid-1950s, studies showed that activation of the adrenal cortex occurred in humans in response to psychologic stressors12; in monkeys with conditioned emotional responses13; and in humans subjected to a stressful interview technique.14 In the early 1960s, researchers found that plasma cortisol levels in groups of subjects increased while they watched war movies and decreased while they viewed Disney nature films.15,16 Later, Mason17 demonstrated in a series of experiments that occurrence of the GAS depended on psychologic factors surrounding the stressors. Mason demonstrated that several factors, including degrees of discomfort, unpleasantness, or suddenness of the stress, could account for the presence or absence of the physiologic stress response.
Selye believed that stressors cause a general or nonspecific response. However, research in the past 30 years has shown the remarkable sensitivity of the central nervous system and endocrine system to psychologic influences (emotion is included in psychologic and social stress and acts through psychologic mechanisms).
As with a physically mediated stress response, psychologic stressors can elicit a reactive stress response. The reactive response is a physiologic response derived from psychologic stressors. For example, the stress of taking an examination may produce an increased heart rate and dry mouth in the unprepared student. Although no physical stressor is involved, the psychologic effect of taking an examination elicits a reactive physiologic response in the body.18
Another type of psychologic-mediated stress response is the anticipatory response. Rather than reacting to an obvious stressor, the body mounts a physiologic stress response in anticipation of disruption of the optimal steady-state, also known as homeostasis. These anticipatory responses can be generated either by species-specific innate programs, such as reacting to the presence of predators and unfamiliar situations, or by experience-dependent memory programs created by conditioning.18 In a conditional response the organism learns that specific stimuli (i.e, objects or situational context) are associated with danger, and anticipation of subsequent encounters with the stimulus produces a physiologic stress response. For example, a child that is abused by a parent may experience a physiologic stress response in anticipation of further abuse when the parent enters the room. Under some circumstances these memory programs may become so strong that psychologic disorders, such as phobias, develop. In a similar fashion, some persons develop post-traumatic stress disorder (PTSD) in response to the memory as opposed to the anticipation of traumatic events, characterized by flashback memories, sleep disturbances, depression, and other symptoms. These symptoms often render a person incapable of employment and frequently disrupt personal relationships.
Anticipatory responses are learned responses under fine control by brain regions located in the limbic system. These regions are those most frequently associated with learning and memory and include the hippocampus, amygdala, and prefrontal cortex. In order for these regions to elicit a stress response, the paraventricular nucleus (PVN) of the hypothalamus must be stimulated (see Chapter 20). The limbic structures rarely interact directly with the PVN and are believed to influence the stress response through intermediary neurons, some of which are primarily used for the reactive response.
Psychoneuroimmunologic Mediators of Stress
Psychoneuroimmunology (PNI) is the study of the interaction of consciousness (psycho), brain and spinal cord (neuro), and the body’s defense against external infection and abnormal cell division (immunology). Psychoneuroimmunology assumes that all immune-related disease is multifactorial, or the result of interrelationships among psychosocial, emotional, genetic, neurologic, endocrine, and immune systems and behavioral factors.19,20 The immune system is integrated with other physiologic processes and is sensitive to changes in CNS and endocrine functioning, such as those that accompany psychologic states. Stressors can elicit the stress response or stress system through the action of the nervous and endocrine systems. Stressors include, but are not limited to, infection, noise, decreased oxygen supply, pain, malnutrition, heat, cold, trauma, prolonged exertion, radiation, responses to life events (including anxiety, depression, anger, fear, and excitement), obesity, old age, drugs, disease, surgery, and medical treatment. The volume of psychoneuroimmunologic research is growing rapidly and represents a substantial presence in the psychosomatic literature. Sufficient data now exist to conclude that immune modulation by psychosocial stressors or interventions leads directly to health outcomes. The strongest support for the link between psychosocial stressors and health outcomes is found in studies of infectious disease and wound healing.21–26
STRESS RESPONSE
The stress response is initiated by the central nervous system and endocrine system (see Figure 10-1). Specifically, corticotropin-releasing hormone (CRH) is released from the hypothalamus, the sympathetic nervous system, the pituitary gland, and the adrenal gland (Figure 10-2). CRH is also released peripherally at inflammatory sites called peripheral, or immune, CRH. The activation of these systems redirects adaptive energy to the CNS and stressed body sites.
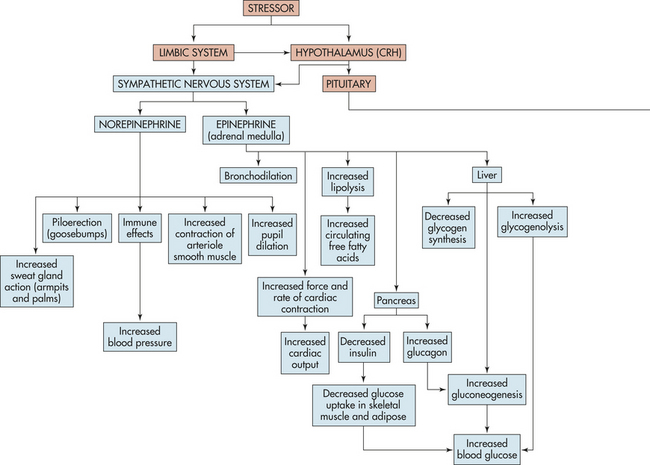
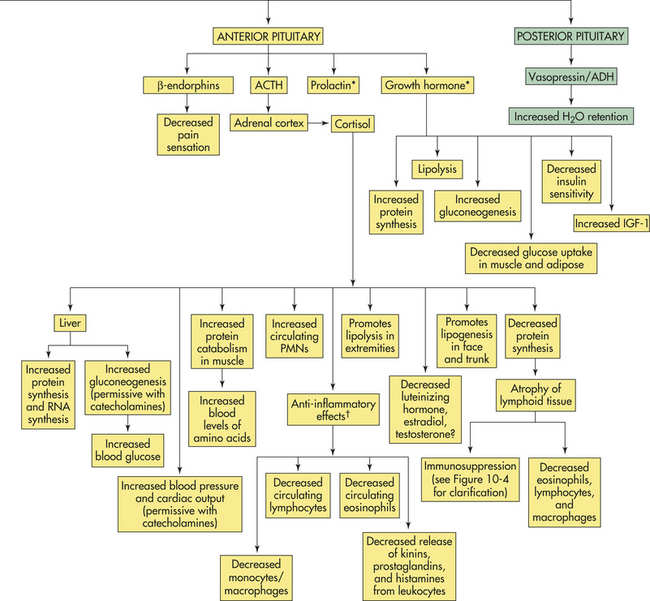
Figure 10-2 The stress response. ACTH, Adrenocorticotropic hormone; CRH, corticotropin-releasing hormone; ADH, antidiuretic hormone; IGF-1, insulin-like growth factor-1; PMNs, polymorphonuclear leukocytes; RNA, ribonucleic acid. See text for explanation of hormone functions. (∗See p. 344 for immune effects. †Explained in text.)
Where the stress response begins depends on whether the stressor is perceived or real. Perceived stressors elicit an anticipatory response that usually begins in the limbic system of the brain, the area responsible for emotions and cognition. The limbic system indirectly elicits an endocrine stress response by stimulating neural pathways responsible for receiving sensory information and a central response by directly stimulating the locus coeruleus (LC) to release norepinephrine (Figure 10-1). Norepinephrine release promotes arousal, increased vigilance, increased anxiety, and other protective emotional responses. Real stressors elicit a reactive response that can begin either in the limbic system or in regions of the brain receiving specific sensory information (see Figure 10-1). This information is then relayed to the PVN. The PVN stimulates the LC and both central and endocrine stress responses.
Central Stress Response
The sympathetic nervous system (SNS) is aroused during the stress response and causes the medulla of the adrenal gland to release catecholamines (80% epinephrine and 20% norepinephrine) into the bloodstream. The adrenal medulla is actually an extension of the SNS because preganglionic fibers from the splanchnic nerve terminate in the medulla, where they innervate the chromaffin cells that produce the catecholamine hormones. Simultaneously, hypothalamic CRH stimulates the pituitary gland to release a variety of hormones, including antidiuretic hormone and oxytocin from the posterior pituitary gland, and prolactin, endorphins, growth hormone (GH), and adrenocorticotropic hormone (ACTH) from the anterior pituitary gland. ACTH stimulates the cortex of the adrenal gland to release cortisol.
Catecholamines
In response to stress, the chromaffin cells of the adrenal medulla produce large quantities of epinephrine and small amounts of norepinephrine. Once released, catecholamines circulate bound to the plasma protein albumin. Epinephrine is rapidly transported to and acts on several organs, but it is metabolized quickly making it short acting. Further, very little adrenal norepinephrine reaches distal tissue; thus, the effects caused by norepinephrine during the stress response are primarily elicited from the SNS.27,28
The catecholamines stimulate two major classes of receptors: α-adrenergic receptors and β-adrenergic receptors. These two classes are divided further into two subclasses: (1) α1 and α2 and (2) β1 and β2. Table 10-2 summarizes the actions of the two subclasses of adrenergic receptors. (A thorough discussion of receptors can be found in Chapters 1, 20, and 29.) Epinephrine binds to and activates both α- and β-adrenergic receptors. Norepinephrine at physiologic concentrations binds primarily to α-adrenergic receptors.29
Table 10-2
Physiologic Actions of α- and β-Adrenergic Receptors
| Receptor | Physiologic Actions |
| α1 | Increased glycogenolysis; smooth muscle contraction (blood vessels, genitourinary tract) |
| α2 | Smooth muscle relaxation (gastrointestinal tract); smooth muscle contraction (some vascular beds); inhibition of lipolysis, renin release, platelet aggregation, and insulin secretion |
| β1 | Stimulation of lipolysis; myocardial contraction (increased rate, increased force of contraction) |
| β2 | Increased hepatic gluconeogenesis; increased hepatic glycogenolysis; increased muscle glycogenolysis; increased release of insulin, glucagon, and renin; smooth muscle relaxation (bronchi, blood vessels, genitourinary tract, gastrointestinal tract) |
The circulating catecholamines essentially mimic direct sympathetic stimulation. (Sympathetic function is described in Chapter 14.) Norepinephrine regulates blood pressure by constricting smooth muscle in all blood vessels. During stress, norepinephrine raises blood pressure by constricting peripheral vessels; it dilates the pupils of the eye, causes piloerection, and increases sweat gland action in the armpits and palms (see Figure 10-2).
Epinephrine has a greater influence on cardiac action and is the principal catecholamine involved in metabolic regulation. Epinephrine enhances myocardial contractility (inotropic effect), increases the heart rate (chronotropic effect), and increases venous return to the heart, all of which increase cardiac output and blood pressure. Epinephrine dilates blood vessels of skeletal muscle, allowing for greater oxygenation. Metabolically, epinephrine causes transient hyperglycemia (high blood sugar) by activating enzymes whose actions promote glucose formation (gluconeogenesis) and glycogen breakdown (glycogenolysis) in the liver while inhibiting glycogen formation. Epinephrine decreases glucose uptake in the muscles and other organs and decreases insulin release from the pancreas. The decrease in insulin release prevents glucose from being taken up by peripheral tissue and thus preserves it for the CNS. Epinephrine also mobilizes free fatty acids and cholesterol by stimulating lipolysis, freeing triglycerides and fatty acids from fat stores, and inhibiting the degradation of circulating cholesterol to bile acids. The metabolic actions of epinephrine aid the metabolic actions of cortisol, which are similar. Table 10-3 summarizes other well-known effects of adrenal catecholamines. All of these effects prepare the body to take physical action: to fight or flee. Stressors commonly associated with catecholamine release by the adrenal medulla include exercise, thermal changes, and acute emotional states.
Table 10-3
Physiologic Effects of Catecholamines∗
| Organ/Tissue | Process or Result |
| Brain | Increased blood flow; increased glucose metabolism |
| Cardiovascular system | Increased rate and force of contraction |
| Peripheral vasoconstriction | |
| Pulmonary system | Bronchodilation |
| Skeletal muscle | Increased glycogenolysis |
| Increased contraction | |
| Increased dilation of muscle vasculature | |
| Decreased glucose uptake and utilization (decreases insulin release) | |
| Liver | Increased glucose production |
| Increased glycogenolysis | |
| Adipose tissue | Increased lipolysis |
| Decreased glucose uptake | |
| Skin | Decreased blood flow |
| Gastrointestinal and genitourinary tracts | Decreased protein synthesis |
| Decreased smooth muscle contraction | |
| Increased renin release | |
| Increased gastrointestinal sphincter tone | |
| Lymphoid tissue | Acute and chronic stress inhibits several components of cellular immunity, particularly decreasing natural killer cells |
| Macrophages | Inhibit and stimulate macrophage activity |
| Depends on availability of type 1/proinflammatory cytokines, the presence or absence of antigenic stressors, and peripheral corticotropin-releasing hormone (CRH) |
∗Some of these responses require glucocorticoids (e.g., cortisol) for maximal activity (see text for explanation).
Data from Elenkov IJ, Chrousos GP: Ann N Y Acad Sci 966:290-303, 2002; Granner DK: Hormones of the adrenal medulla. In Murray RK et al, editors: Harper’s biochemistry, ed 25, New York, 2000.
Catecholamines can modify the numbers of cells of the immune system circulating in the blood.1 Injection of epinephrine into healthy human subjects is associated with a transient increase of the number of lymphocytes (e.g., T cells and natural killer [NK] cells) in the peripheral blood. Specifically, T cytotoxic and NK cells increase, whereas little change occurs in B lymphocytes. The main change involves the NK cells.1 Qualitatively, lymphocyte responsiveness of T and B lymphocytes is reduced. Similar quantitative and qualitative changes are found 5 to 6 minutes after exposure to a psychologic or physical stressor.30 The effects of acute elevation of catecholamines on the alteration of lymphocyte function are short lived, lasting only about 2 hours.2 However, a study of stress duration and susceptibility to infection22 found that chronic elevation of catecholamines is immunosuppressive, but it is unclear whether the increase in lymphocytes comes from the bone marrow or the peripheral tissues.
Cortisol
The adrenal cortex is activated during stress by ACTH (see Figure 10-1), which increases adrenocortical secretion of glucocorticoid hormones, primarily cortisol (a synthetically produced but chemically identical version of cortisol known also as hydrocortisone). Cortisol circulates in the plasma, both protein bound and free. The main plasma-binding protein is called transcortin or corticosteroid-binding globulin. The unbound, or free, fraction is approximately 8% of the total plasma cortisol and is biologically active.29 Cortisol mobilizes substances needed for cellular metabolism. One of the primary effects of cortisol is the stimulation of gluconeogenesis, or the formation of glucose from noncarbohydrate sources, such as amino or free fatty acids in the liver. In addition, cortisol enhances the elevation of blood glucose promoted by other hormones, such as epinephrine, glucagon, and growth hormone. This action by cortisol is said to be permissive for the actions of other hormones. Cortisol also inhibits the uptake and oxidation of glucose by many body cells. The overall action of cortisol increases blood glucose, thereby enabling the body to combat the stressor. The physiologic effects of cortisol are summarized in Table 10-4.
Table 10-4
Physiologic Effects of Cortisol
| Functions Affected | Physiologic Effects |
| Carbohydrate and lipid metabolism | Diminishes peripheral uptake and use of glucose; promotes gluconeogenesis in liver cells; enhances the gluconeogenic response to other hormones; promotes lipolysis in adipose tissue |
| Protein metabolism | Increases protein synthesis in the liver and decreased protein synthesis (including immunoglobulin synthesis) in muscle, lymphoid tissue, adipose tissue, skin, and bone; increases plasma level of amino acids; stimulates deamination in the liver |
| Anti-inflammatory effects (systemic effects) | High levels of cortisol used in drug therapy suppress the inflammatory response; inhibits proinflammatory activity of many growth factors and cytokines; however, over time some patients may develop tolerance to glucocorticoids, causing an increased susceptibility to inflammatory and autoimmune diseases |
| Proinflammatory effects (possible local effects) | Cortisol levels released during the stress response may increase proinflammatory effects (this very complex physiology is reviewed on p. 344). |
| Lipid metabolism | Lipolysis in the extremities and lipogenesis in the face and trunk |
| Immune effects | Immunosuppression of T-cell or cellular immunity at therapeutic levels; nontherapeutic levels, such as those seen with stress, can suppress cellular (Th1) and increase humoral (Th2) immunity—the so-called Th2 shift |
| Digestive function | Promotes gastric secretion |
| Urinary function | Enhances excretion of calcium |
| Connective tissue function | Decreases proliferation of fibroblasts in connective tissue (thus delaying healing) |
| Muscle function | Maintains normal contractility and maximal work output for skeletal and cardiac muscle |
| Bone function | Decreases bone formation |
| Vascular system and myocardial function | Maintains normal blood pressure; permits increased responsiveness of arterioles to the constrictive action of adrenergic stimulation; optimizes myocardial performance |
| Central nervous system function | Modulates perceptual and emotional functioning, essential for normal arousal and initiation of daytime activity |
| Possible synergism with estrogen in pregnancy? | Suppresses maternal immune system to prevent rejection of fetus? |
Cortisol also affects protein metabolism. It has an anabolic effect, that is, it increases the rate of synthesis of proteins and ribonucleic acid (RNA) in liver. The anabolic effect of cortisol, however, is countered by its catabolic effect on protein stores in other tissues. Protein catabolism acts to increase circulating amino acids, and chronic exposure to excess cortisol can severely deplete protein stores in muscle, bone, connective tissue, and skin. Further, cortisol acts to reduce protein synthesis in nonhepatic tissues, a loss for which dietary protein cannot compensate. Some evidence suggests that cortisol depresses transport of amino acids into muscle cells while enhancing their uptake into the liver.
Cortisol also has a powerful effect that reverses the insulin-induced suppression of hepatic gluconeogenesis, and basal levels of cortisol stimulate the activity of hepatic enzymes responsible for glycogen and glucose production. The increased amino acid uptake into liver and glucose-producing enzymes favors the production of glucose. Although diseases of excess cortisol secretion, such as Cushing disease, produce characteristics of type 2 (non-insulin dependent) diabetes mellitus, recent studies found that chronic stress also may facilitate the development of type 2 diabetes31,32 (see What’s New? Glucocorticoids, Insulin, Inflammation, and Obesity). The mechanism for this action is under investigation, but it is believed that the development of diabetes is secondary to cortisol-induced obesity. Cortisol promotes lipogenesis in certain regions of the body and to a lesser extent promotes lipolysis in other regions by increasing the actions of lipolytic hormones, such as catecholamines and growth hormone. Chronic cortisol excess induces lipogenesis in the abdomen, trunk, and face resulting in central obesity.
WHAT’S NEW?
Glucocorticoids, Insulin, Inflammation, and Obesity
The signs and symptoms of Cushing syndrome (e.g., excess glucocorticoids [GCs]) include truncal obesity, relatively thin extremities, a “moon face,” and a “buffalo [neck] hump.” In such individuals the possibility of associated hypertension is high as well as increased risk of infection and metabolic syndrome or frank type 2 diabetes. In addition, the likelihood of an elevated ratio of intra-abdominal subcutaneous fat mass to nonabdominal fat mass is high because the glucocorticoids mediate the redistribution of stored calories into the abdominal region. The specific increase in abdominal fat stores is a consequence of elevated glucocorticoids combined with increased insulin action. However, the increased glucocorticoids need not be present in the circulation, but can be generated locally in fat through conversion of inactive cortisone to active cortisol through the action of the isoenzyme 11-β-hydroxysteroid dehydrogenase (11-β-HSD) type-1. This conversion is referred to as “pre-receptor” metabolism of cortisol. The active steroid is secreted directly to the liver through the portal vein. In vitro insulin synthesis and secretion from the pancreas are inhibited by the glucocorticoids. However, increasing glucocorticoids in vivo are associated with increasing insulin secretion possibly because of an anti-insulin effect on the liver, which appears to be vulnerable to the negative effects of glucocorticoids on insulin action. Hepatic insulin resistance is strongly associated with abdominal obesity.
Recent data reveal that the plasma concentration of inflammatory mediators, such as tumor necrosis factor-α (TNF-α) and interleukin-6 (IL-6), is increased in the insulin-resistant states of obesity and type 2 diabetes. Two mechanisms might be involved in the pathogenesis of inflammation: (1) glucose and macronutrient intake (i.e., which can be mediated through chronic stress) causes oxidative stress; and (2) the increased concentrations of TNF-α and IL-6 associated with obesity and type 2 diabetes might interfere with insulin signal transduction. This interference might promote inflammation. Chronic overnutrition (obesity) might thus be a proinflammatory state with oxidative stress.
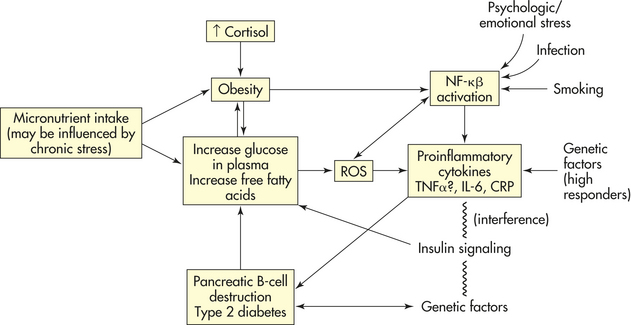
Stress, inflammation, obesity, and type 2 diabetes. The induction of reactive oxygen species (ROS) generation and inflammation through the proinflammatory transcription factor, NF-kβ, activates most proinflammatory genes. Macronutrient intake, obesity, free fatty acids, infection, smoking, psychologic stress, and genetic factors increase the production of ROS. Interference with insulin signaling (insulin resistance) leads to hyperglycemia and proinflammatory changes. Proinflammatory changes increase TNF-α, increase IL-6, and also lead to the inhibition of insulin signaling and insulin resistance. Inflammation in pancreatic B cells leads to B-cell dysfunction, which in combination with insulin resistance leads to type 2 diabetes. CRP, C-reactive protein.
Data from Dallman MF et al: Endocrinology 145(6):2633-2638, 2004; Dandona P, Aljada A, Brandyopadhyay A: Trends Immunol 25(1):4-7, 2004; Kim SP et al: Diabetes 52:2453-2460, 2003; Masuzaki H et al: Science 94:2166-2170, 2001; Padgett DA, Glaser R: Trends Immunol 24(8):444-448, 2003; Strack AM et al: Am J Physiol 268:R142-R149, 1995; Thakore JH et al: Biol Psychiatry 47:1140-1142, 1998; Wagen Knecht LE et al: Diabetes 52:2490-2496, 2003.
In the gastrointestinal tract, cortisol promotes gastric secretion. This effect is opposite that of norepinephrine, which reduces gastric secretion. Excessive cortisol may stimulate gastric secretion enough to cause ulceration of the gastric mucosa. This could account for the gastrointestinal ulceration observed by Selye.
Cortisol and the Immune System
Stress hormones, especially glucocorticoids (cortisol), have been used therapeutically as powerful anti-inflammatory/immunosuppressive agents. Data suggest, however, that glucocorticoids and catecholamines (epinephrine and norepinephrine) at concentration levels reached during stress may paradoxically result in decreased cellular immunity and increased autoimmune (humoral) responses. These data may help explain the seemingly contradictory response to stress of immunosuppression and increased risk of infection (decreased cellular immunity) and a heightened antibody response and autoimmune disease (increased humoral immunity).
As discussed in Chapters 6 and 7, immune responses are regulated by cells of innate immunity called antigen-presenting cells (APCs), such as monocytes/macrophages, dendritic cells, and other phagocytic cells, and by cells of adaptive immunity, such as lymphocyte subclasses T-helper 1 (Th1) and T-helper 2 (Th2) cells. These cells regulate the immune system by the secretion of chemicals called cytokines. Cytokines are a group of chemicals such as interferons, interleukins, and tumor necrosis factors that can stimulate or inhibit various components of the immune system. APCs release cytokines that induce T cells to differentiate into Th1 cells. Th1 cells and APC cytokines work together to stimulate the immune activity of cytotoxic T cells, NK cells, and activated macrophages—the major components of cellular immunity. These cytokines also stimulate the synthesis of nitric oxide and other inflammatory mediators that increase chronic delayed-type inflammatory responses. Because of this effect, these cytokines are considered to be the major proinflammatory cytokines.32–35 The cytokines secreted by the Th2 cells act to inhibit Th1 cells and can promote humoral immunity by stimulating growth and activation of mast cells and eosinophils, as well as the differentiation of B-cell immunoglobulins. Thus these cytokines are considered to be the major anti-inflammatory cytokines33 (Figure 10-3).
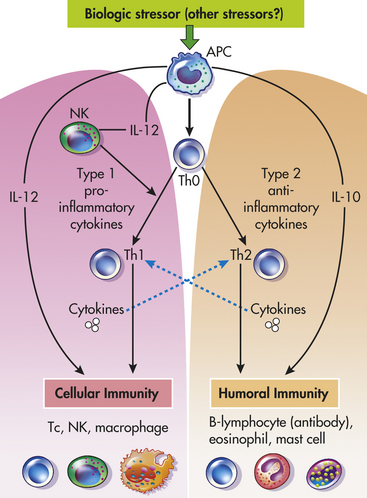
Figure 10-3 Role of Th1 and Th2 cells in the regulation of cellular and humoral immunity. Humoral immunity provides protection against multicellular parasites, extracellular bacteria, some viruses, soluble toxins, and allergens. Cellular immunity provides protection against intracellular bacteria, fungi, protozoa, and several viruses. Type 1 cytokines or proinflammatory cytokines include IL-12, interferon-gamma (IFN-γ), and tumor necrosis factor-alpha (TNF-α). Type 2 cytokines or anti-inflammatory cytokines include IL-10 and IL-4. Solid lines (black) represent stimulation, whereas dashed lines (blue) represent inhibition (i.e., Th1 and Th2 are mutually inhibitory, IL-12 and IFN-γ inhibit Th2, and vice versa; IL-4 and IL-10 inhibit Th1 responses). APC, Antigen-presenting cell; B, B cell; IL, interleukin; NK, natural killer cell; Th, T-helper cell; Tc, cytotoxic T cell. (Redrawn from: Elenkov IJ, Chrousos GP: Trends Endocrinol Metab 10[9]:359-368, 1999.)
Stress influences immunity by stimulating cortisol and epinephrine secretion from the adrenal glands and norepinephrine from the sympathetic nervous system. Cortisol acts to suppress the activity of Th1 cells, which leads to a decrease in cellular immunity and to the proinflammatory response. Cortisol also stimulates the activity of the Th2 cells, which leads to an increase in adaptive humoral immunity and the anti-inflammatory response. Epinephrine and norepinephrine have a similar effect: the decrease in Th1 activity and the increase in Th2 activity. This decrease in Th1 activity and increase in Th2 activity is sometimes called a Th1 to Th2 shift. Individuals experiencing a Th1 to Th2 shift are more likely to experience allergic responses, infections and temporary worsening of autoimmune conditions such as arthritis.
The above description of the effect of stress hormones on the Th1-Th2 balance may not be accurate for certain local responses.33 It has been documented that the release of catecholamines (epinephrine and norepinephrine) can cause certain epithelial cells of the lung to release cytokines that promote recruitment of leukocytes potentially enhancing inflammation and worsening lung function. This paradoxical stress-induced potentiation of inflammation in the lungs may explain why “acute respiratory distress syndrome” often develops in individuals with major infections associated with profound activity of the stress response.36
CRH influences the immune system indirectly by the activation of cortisol (glucocorticoids) and catecholamines. CRH is secreted by the hypothalamus and also peripherally at inflammatory sites.33,37,38 Peripheral (immune) CRH is proinflammatory, causing an increase in vasodilation and vascular permeability.39 Therefore, it appears that mast cells are the target of peripheral CRH. Mast cells release histamine, which is a well-known mediator of acute inflammation and allergic reactions (Figure 10-4). Recent evidence has indicated that immune cells may have histamine receptors and that histamine may have an effect similar to catecholamines. This finding suggests that histamine induces acute inflammation and allergic reactions while suppressing Th1 activity (decreasing cellular immunity) and promoting Th2 activity (increasing humoral immunity).39–42
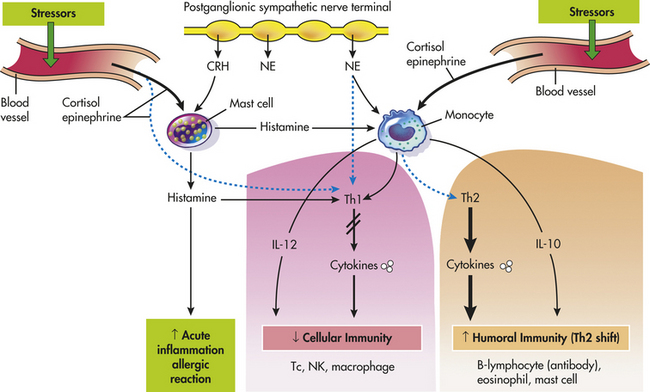
Figure 10-4 Effect of corticotropin-releasing hormone—mast cell—histamine axis, cortisol, and catecholamines on the Th1/Th2 balance—cellular and humoral immunity. Stress and CRH modulate inflammatory/immune and allergic responses by stimulating cortisol (glucocorticoid), catecholamines, and peripheral (immune) CRH secretion and by changing the production of regulatory cytokines and histamines. Solid lines (black) represent stimulation, and dashed lines (blue) represent inhibition. B, B cell; CRH (peripheral, immune), corticotropin-releasing hormone; NE, norepinephrine; Th, T-helper cell; IL, interleukin; Tc, cytotoxic T cell; NK, natural killer cell; ↓ decreased (inhibited); ↑ increased (stimulation). (Redrawn from Elenkov IJ, Chrousos GP: Stress hormones, Th1/Th2 patterns, pro-/anti-inflammatory cytokines and susceptibility to disease, Trends Endocrinol Metab 10[9]:359-368, 1999.)
In summary, stress can activate an excessive immune response and, through cortisol and catecholamines, suppress the Th1 response and cause a Th2 shift. Locally, stress can exert proinflammatory or anti-inflammatory effects depending on what chemicals are released in the local environment and how the cells of the local environment respond to those chemicals. Finally, recent evidence indicates that responses to stressful events can be considered variable and may even be specific.25 Moreover, different types of stressors may have variable effects on the immune response. Thus systemic responses to stress may cause a decrease in cellular immunity and enhance humoral immunity, whereas local responses to stress, under certain conditions, can induce proinflammatory activities that may influence the onset and cause of infection, autoimmune/inflammatory, allergic, and neoplastic disease.
Therapeutic levels of glucocorticoids inhibit the accumulation of leukocytes at the site of inflammation and inhibit the release of substances involved in the inflammatory response (i.e., kinins, plasminogen-activating factor, prostaglandins, and histamine) from the leukocytes. Glucocorticoids inhibit fibroblast proliferation and function at the site of an inflammatory response. This inhibition accounts for the poor wound healing, increased susceptibility to infection, and decreased inflammatory response that often are noted in individuals with chronic glucocorticoid excess.
It is not entirely clear why cortisol secretion during stress is beneficial. It has been suggested that gluconeogenesis promoted by cortisol ensures an adequate source of glucose (energy) for body tissues, and nerve cells in particular. The pooling of amino acids from catabolized proteins may ensure amino acid availability for protein synthesis in certain cells. The redistribution of protein to sites where replacement is critical, such as muscle or cells of damaged tissue, would be beneficial. Short-term, cortisol-induced alterations in immune cell distribution (e.g., traffic) patterns may be adaptive, with a decrease in peripheral blood cell numbers as effector cells locate to sites of injury or inflammation. In addition, with high concentrations of cortisol, decreased immune-cell activity (both T cell and B cell) prevents immune-mediated tissue damage by prolonged cell exposure to high levels of certain cytokines.43 Whether cortisol-induced effects are adaptive or destructive may depend on the intensity, type, and duration of the stressor, and the subsequent concentration and length of cortisol exposure that target cells of the individual experience.
Stress and the Immune System
Many immune-related conditions and diseases are associated with stress. Several conditions with variable pathophysiologic characteristics appear to have a common origin44,45 relating to chronic inflammatory processes. These conditions include
cardiovascular disease, osteoporosis, arthritis, type 2 diabetes mellitus, chronic obstructive pulmonary disease (COPD), other diseases associated with aging, and some cancers; all are characterized by the prolonged presence of proinflammatory cytokines.44,46 (Inflammation is discussed in Chapter 6.) Stress and negative emotions are associated directly with the production of increased levels of proinflammatory cytokines, providing a possible link between stress, immune function, and disease (see What’s New? Psychosocial Stress and Progression to Coronary Heart Disease). The specific stress-induced mechanisms causing these illnesses are not clearly defined. Research is focused on the regulatory interactions between the immune system and the nervous and endocrine systems, which may represent mechanistic pathways for stress-associated immune-mediated diseases (see Table 10-1).
The immune, nervous, and endocrine systems communicate through similar pathways involving hormones, neurotransmitters, neuropeptides, and immune cell products. Immune system responses are potentially affected by all known neuroendocrine-produced factors involved in the stress reaction. Conversely, immune cell–derived cytokines and other products have effects on neurocrine and endocrine cells47,48 (see Table 7-4). Several pathways regulate communication among these systems with both direct and indirect patterned effects (Figure 10-5).
Figure 10-5 Nervous system—endocrine system—immune system interactions. Interconnections of pathways of communication among the immune, nervous, and endocrine systems.
The stress response directly influences the immune system through hypothalamic and pituitary peptides and through products of the sympathetic branch of the ANS. These factors include CRH, ACTH, endorphins, substance P, epinephrine, norepinephrine, dopamine, serotonin, histamine, GH, vasoactive intestinal polypeptide (VIP), β-endorphin, methionine-enkephalin, leucine-enkephalin, and somatostatin49–52 (Table 10-5). Direct suppressive effects of CRH have been reported also on two immune cell types possessing CRH receptors—the monocyte/macrophage and CD4 (T helper) lymphocyte.53 Release of endogenous opiates occurs during stress, and these peptides have been shown to have concentration-dependent, enhancing or suppressive effects on various immune cells.49 Immune cells have been shown to have surface receptors for epinephrine, serotonin, ACTH, CRH, endorphins, GH, and prolactin, as well as intracellular steroid receptors.
Table 10-5
Other Hormones that Probably Influence the Stress Response
| Hormone | Source | Comments |
| Melatonin | Produced by pineal gland | Increases during the stress response; release is suppressed by light and increased in the dark; receptors have been identified on lymphoid cells, possibly higher density of receptors on T cells than B cells; suppression of lymphocyte function by trauma was reversed by melatonin (Maestroni, 1999) |
| Somatostatin (SOM) | Produced by sensory nerve terminals found in and released from lymphoid cells and hypothalamus | Natural killer (NK) cell function and immunoglobulin synthesis is decreased by SOM; growth hormone secretion decreased by SOM |
| Vasoactive intestinal peptide (VIP) | Found in neurons of the central nervous system (CNS) and in peripheral nerves | VIP increases during stress; VIP-containing nerves are located in primary and secondary lymphoid tissues, around blood vessels, and the gastrointestinal tract; VIP receptors are on T and B cells; VIP may influence lymphocyte maturation; cytokine production by T cells is modified by VIP; B-cell and antibody production is influenced by VIP |
| Calcitonin gene–related peptide (CGRP) | Found in spinal cord motor neurons and in sensory neurons near dendritic cells of the skin and in primary and secondary lymphoid tissues | CGRP receptors are present on T and B lymphocytes, thus it is likely that CGRP can modulate immune function; CGRP may enhance the acute inflammatory response because it is a vasodilator; maturation of immune B lymphocytes is inhibited by CGRP; IL-1 is inhibited by CGRP, which is important for the activation of T cells; it has been shown to interfere with lymphocyte activation |
| Neuropeptide Y (NPY) | Present in the neurons of the CNS and in neurons throughout the body; co-localized in nerve terminals in lymphatic tissues with norepinephrine | Lymphocytes have receptors for NPY and thus may modulate their function (Pettito et al, 1994); several lines of evidence suggest that NPY is a neurotransmitter and neurohormone involved in the stress response; increased levels of NPY occur in plasma in response to severe or prolonged stress; it may be responsible for stress-induced regional vasoconstriction (splanchnic, coronary, and cerebral); it may also increase platelet aggregation (Rabin, 1999) |
| Substance P (SP) | Produced by a neuropeptide classified as a tachykinin (increases heart rate subsequent to lowering blood pressure) found in the brain, as well as nerves innervating secondary lymphoid tissues | SP increases in response to stress; receptors for SP are found on the membrane of T and B cells, mononuclear phagocytic cells, and mast cells; proinflammatory activity induces the release of histamine from mast cells during the stress response; causes smooth muscle contraction; causes macrophages and T cells to release cytokines, and increases antibody production |
Data from Maestroni GJ: Adv Exp Med Biol 460:396, 1999; Pettito JM et al: J Neuro Immunol 54:81, 1994; Rabin BS: The nervous system—immune system connection. In Rabin BS: Stress, immune function, and health: the connection, Wiley-Liss, 1999, New York.
Products of the sympathetic branch of the ANS also influence immune cell behavior. There is direct innervation of the thymus, spleen, lymph nodes, and bone marrow,52 and histochemical studies have verified the presence of cholinergic and adrenergic nerve terminals in the lymphoid organs and tissues. There is evidence for interaction of norepinephrine released from nerve endings with lymphocytes and macrophages in the spleen. This finding implicates the presence of a route of communication between the ANS and immune system through direct delivery of chemical mediators that alter immune cell behavior in a paracrine (cell to adjacent cell) fashion in the microenvironment of the lymphoid organ.
The pineal gland regulates the immune response and mediates the apparent effects of circadian rhythm on immunity. Blockage of production of melatonin (by continuous light or by pharmacologic means) results in suppression of immune response, whereas administration of melatonin reverses these effects.54 This immunomodulation pathway may affect immune changes found with dysregulation of circadian rhythm, a common occurrence among older adults, acutely ill, and stressed individuals.55 Melatonin also modulates seasonal changes in immune function and affects tumor development.56
The HPA axis may produce indirect effects on the CNS that modulate immune responses. This is the most extensively studied pathway, with original interest stemming from early studies showing profound effects of prolonged severe stress on immunologic structures.57 It was noted that the adrenal gland enlarged with simultaneous involution of the thymus and lymph nodes. Increased levels of GCSs may be an important mechanism in stress-related immune structure alterations and in suppression of immune responses.57 The GCS level increases are attributable to pituitary ACTH production—a result of increased hypothalamic CRH. A number of stress factors initiate CRH production, including high levels of interleukin-1 (IL-1) and IL-6. Increased CRH secretion results in an increase in cortisol secretion. Cortisol feeds back to inhibit further cytokine release by macrophages and monocytes.
The observation that IL-1 can elicit changes in the nervous and endocrine systems by stimulating CRH production in the hypothalamus is part of a growing body of evidence demonstrating immune-induced regulation of the CNS. The release of immune inflammatory mediators IL-6, tumor necrosis factor-beta (TNF-α), and interferon is triggered by bacterial or viral infections, cancer, and tissue injury that in turn initiate a stress response through the HPA pathway described previously. Enhanced systemic production of these cytokines also induces other CNS and behavior changes seen frequently during the acute phase of an infectious episode, acting either directly in a distant systemic “endocrine” way or through the mediation of neuropeptides.47,58–60 These effects include pyrogenesis (fever), induction of slow wave sleep, and anorexia, all of which are adaptive responses to infection and possibly cancer. Slow wave sleep is associated with enhanced release of GH and a reduction in levels of cortisol, which is beneficial for tissue repair and enhanced immune response.61 Normal and predictive changes in sleep occur in response to infections, and these changes appear to be of important recuperative value to the individual.62
Lymphocytes also are known to produce ACTH and endorphins in small amounts, which probably influence immune response in an autocrine or a paracrine manner in the local microenvironment of an ongoing immune response.63 The T-cell growth factor (IL-2) can up-regulate pituitary ACTH. Immune-derived cytokines have significant influence on neuroendocrine function, with evidence for direct and indirect cytokine effects on nervous and adrenal cell functions. Thus the immune system has an adaptive role as a “signal” organ to alert other systems of inner threatening stimuli (e.g., infection, tissue damage, tumor cells) that may upset the dynamic steady state.
Neuropeptides and hormones have a significant effect on the immune response. Whether this effect on immune function is suppressive or potentiating depends on the type of factor secreted, with some factors enhancing, some suppressing activities, and some doing both, depending on the concentration and length of exposure, the target cell, and the specific immune function studied.48 Neuropeptides and neuroendocrine hormones may directly control biochemical events affecting cell proliferation, differentiation, and function or may indirectly control immune cell behavior by affecting the production or activity of cytokines.48
In summary, a significant body of evidence supports a link among the nervous, endocrine, and immune systems. The bidirectional communication among these systems involves common use of signal molecules and their receptors, which in turn regulates the behavior of cells in each system. Thus the most recent findings are that (1) there are direct effects of CNS neuropeptides on immune cells; (2) stress-induced endocrine products influence immune cell and neurologic cell function; and (3) immune cell products (cytokines) affect nervous and endocrine cell function through direct and indirect pathways.
Stress-Induced Hormonal Alterations
Elevation of glucocorticoid concentration associated with psychologic stress or physical exercise is not as high as that achieved by pharmacologic means.1 Stress produces changes in levels of hormones other than glucocorticoids; the interaction of these various hormones in modifying the function of other physiologic systems still needs investigation.
Female Reproductive System: Cortisol exerts inhibiting effects by suppressing levels of luteinizing hormone (LH), estradiol, progesterone, and possibly testosterone.64,65 The HPA axis exerts powerful, multilevel effects on the female reproductive system. Stress generally inhibits the female reproductive system (Figure 10-6), primarily through the HPA axis by (1) suppression of hypothalamic gonadotropin-releasing hormone (GnRH) secretion by CRH and CRH stimulation of β-endorphin release; (2) inhibition of GnRH, pituitary LH, and ovarian estradiol (E2) secretion by cortisol; and (3) cortisol-induced target tissue resistance by estradiol.65,66 The locus coeruleus-norepinephrine (LC/NE) system (see Figure 10-6) provides positive input to the reproductive system, which is frequently altered by the stress-activated HPA axis. Sexual stimulation and GnRH neuron activation, however, may cause the gonadal axis to be resistant to suppression by the HPA axis. Through estradiol, the reproductive system provides positive input to both components of the stress system by stimulating CRH secretion and inhibiting reuptake and catabolism of catecholamines. Table 10-6 presents potential pathologic effects of central and peripheral CRH in women.
Table 10-6
Potential Pathologic Effects of Central and Peripheral Corticotropin-Releasing Hormone (CRH) in Women
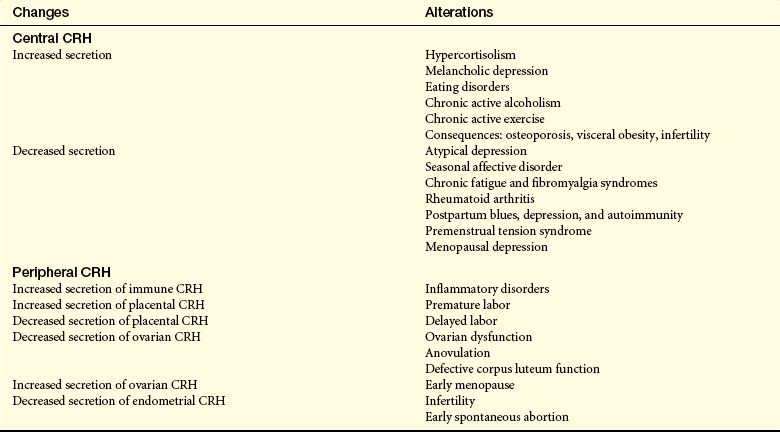
Data from Chrousos GP et al: Ann Intern Med 129(3):229-240; Kalantaridou SN et al: J Reprod Immunol 62(1-2):61-68, 2004.
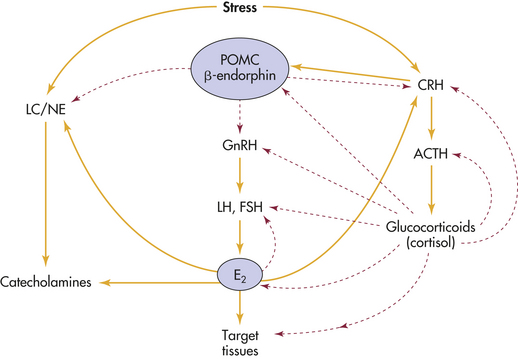
Figure 10-6 Stress and the female reproductive system. Interactions of the reproductive system with the hypothalamic-pituitary-adrenal (HPA) axis and locus coeruleus–norepinephrine system (LC/NE). Corticotrophic cells of the pituitary gland express proopiomelanocortin (POMC) peptides. Stress generally inhibits the female reproductive system primarily through the HPA by (1) suppressing hypothalamic gonadotropin-releasing hormone (GnRH) secretion by corticotropin-releasing hormone (CRH) and CRH-induced β-endorphins; (2) inhibiting GnRH, pituitary luteinizing hormone (LH), and ovarian estradiol (E2) secretion by cortisol; and (3) cortisol-induced target tissue resistance to estradiol. The LC/NE system provides positive input to the reproductive system, which can be overridden by the stress-activated HPA. Estradiol can cause the reproductive system to stimulate the stress system by stimulating CRH secretion and inhibiting reuptake and catabolism of catecholamines. ACTH, Adrenocorticotropic hormone; FSH, follicle-stimulating hormone. Dashed lines refer to inhibitory pathways. Solid lines refer to direct stimulatory pathways. (Adapted from Chrousos GP et al: Interactions between the hypothalamic-pituitary-adrenal axis and the female reproductive system, Ann Intern Med 129[3]:229-240, 1998.)
Estrogen stimulates the HPA axis. Compared with controls, pregnant women and women receiving high-dose estrogen therapy had elevated levels of cortisol in morning and evening plasma samples.67 In addition, it appears that the HPA axis responsiveness is greater in women than men.68 Estrogen directly stimulates the CRH gene promoter and the central noradrenergic (norepinephrine) system, which may help explain adult women’s slight hypercortisolism, increases in affective anxiety and eating disorders, mood cycles, and vulnerability to autoimmune and inflammatory disease, all of which follow estradiol fluctuations. Estradiol down-regulates glucocorticoid receptor binding in the anterior pituitary, hypothalamus, and hippocampus—this tends to increase HPA activity by interfering with glucocorticoid-negative feedback, whereas progesterone opposes these effects.69 Thus alterations in estradiol during normal menses, perimenopause (including increases as well as decreases), and menopause alter the regulatory feedback loop, and adaptations over time develop as a new equilibrium is established in the relation as shown in Figure 10-6. Over time, these changes increase the incidence of mood alterations, eating disorders, anxiety, depression, weight alterations, and inflammatory and immune disorders.
The adipocyte-derived peptide hormone leptin interacts directly and indirectly with the adrenal and gonadal axes; leptin levels are higher in women than in men. Leptin regulates appetite (satiety) and energy balance. It also inhibits the HPA axis at both hyopothalamic and adrenocortical levels. In addition, leptin positively influences the female reproductive axis by inhibition of the HPA axis and arcuate proopiomelanocortin (POMC) neuronal system and through activation of the LC/NE system. By promoting satiety and sympathetic system outflow, leptin is thought to provide the peripheral signal to a central mechanism regulating the size of body fat stores.70 Thus leptin may be significant in control of the onset of puberty because of its relationship to the amount of fat mass, in the adaptive activation of the HPA axis, and in inhibition of gonadal function that takes place in cases of starvation and anorexia nervosa.71–73
Endorphins and Enkephalins: Endorphins and enkephalins (endogenous opiates) are released into the blood as part of the response to stressful stimuli. They are proteins found in the brain that have pain-relieving capabilities. Stressful stimuli include traumatic injury and an acute, intense stress situation, such as first-time parachute jumping. In inflamed tissue, immune cell–derived endorphins activate endorphin receptors on peripheral sensory nerves, leading to pain relief or analgesia.74 Hemorrhage increases β-endorphin levels that appear to inhibit blood pressure increase or delay compensatory changes that would increase blood pressure.75 Thus endogenous opiates modulate blood pressure instability and neuroendocrine and cytokine responses to blood losses.76,77
The secretion of ACTH and β-endorphin is stimulated by CRH; β-endorphins are released from the pituitary gland.78 Enkephalin is released from the adrenal medulla. Evidence is accumulating that β-endorphins can modulate ACTH secretion and, with ACTH, inhibit hypothalamic CRH secretion, a possible down-regulation pathway of the stress response.79
In a number of conditions or activities in which endogenous opiate activity is increased, subjects not only experience insensitivity to pain but also report increased feelings of excitement, positive well-being, or euphoria. In addition, cells of the immune system synthesize and release opioids when the lymphoid cells are activated.80 T and B lymphocytes and mononuclear phagocytic cells have receptors for opioids.1 Endorphins may play a role in the excitement and exhilaration produced by dancing, contact sports, and combat. There is little direct evidence, however, documenting the endorphin system in most of these activities.
Growth Hormone (Somatotropin): GH is synthesized from the anterior pituitary gland and is produced by lymphocytes and mononuclear phagocytic cells.81 GH affects protein, lipid, and carbohydrate metabolism and counters the effects of insulin. It is involved in tissue repair and may participate in the growth and function of the immune system.1 Receptors for GH are present on lymphoid cells.82 This finding suggests a role for GH in regulating phagocytic function and possibly antigen presentation.1 GH appears to have enhancing effects on immune function.1 GH levels increase in the blood after a variety of acutely stressful stimuli, such as cardiac catheterization, electroshock therapy, gastroscopy, surgery, fever, and physical exercise. Psychologic stimuli associated with increased levels of GH include taking examinations, viewing of violent or sexually arousing films, anticipation of exhausting exercise, and certain psychologic performance tests. However, prolonged activation of the stress response (chronic stress) leads to suppression of GH and other growth factor effects on target tissues.83 (This is not to be confused with a congenital GH deficiency which leads to short stature, decreased muscular strength and bone density problems.) When under stress, chronic glucocorticoid stimulation decreases the release of GH from the pituitary gland, resulting in potentially decreased skeletal muscle, bone catabolism, reduced oxygen uptake, increased fat production, worsening sleep patterns, decreased force of cardiac contractions, decreased thyroid function, and poorer mood state. In other words, glucocorticoids antagonize the beneficial actions of GH.84
Prolactin: Prolactin is released from the anterior pituitary gland as well as numerous extrapituitary tissue sites.85 It is necessary for lactation and breast development.86 Prolactin receptors are present in many different tissues, including the liver, kidney, intestine, and adrenals. Prolactin is also produced by lymphoid cells.1,87 Prolactin levels in plasma increase as a result of a variety of stressful stimuli, including gastroscopy, proctoscopy, pelvic examination, and surgery.88 The level of prolactin also rises during parachute jumping, during motion sickness, after taking examinations, and after receiving various sexual stimuli, for example, stimulation of the nipple or areola in women. Unlike GH, prolactin levels show little change after exercise. Like GH, however, a prolactin increase appears to require more intense stimuli than those leading to increases in catecholamine or cortisol levels. Immune cells also are influenced by prolactin. Prolactin acts as a second messenger for IL-2 and is known to have a positive influence on B-cell activation and differentiation.89 Several classes of lymphocytes have receptors for prolactin, suggesting a direct effect of prolactin on immune function.
Oxytocin: Oxytocin is well known as a hormone produced in high levels by the hypothalamus during childbirth and lactation. It is also produced during orgasm in both sexes and has been shown to promote bonding and social attachment.90 Oxytocin also has antistress properties, as has been shown in animal experiments in which elevations in endogenous oxytocin were associated with reduced HPA activation levels and reduced anxiety.91 Oxytocin in some tissues works in concert with estrogen; these two hormones have a calming effect during stressful situations.92 In contrast, another hormone closely resembling oxytocin, vasopressin, acts in concert with testosterone to increase blood pressure and heart rate, thus enhancing the “fight or flight” stress response. A recent proposal is that the oxytocin-mediated stress response may promote the “tend and befriend” response, more commonly experienced by women because estrogen is a co-mediator.93 Studies in animals have identified a wide group of affiliative behaviors involving social encounters, pair bonding, and attachment as being increased by oxytocin.94 Thus different effects of stress on males and females may be explained, in part, by gender-related hormonal profiles that dictate to some extent the characteristics, quality, and outcomes of the stress response.
Testosterone: Testosterone, a hormone secreted by Leydig cells, regulates male secondary sex characteristics and libido. Testosterone levels decrease after stressful stimuli. The decrease in testosterone occurs after stimuli such as ether or anesthesia, surgery, marathon running, and mountain climbing.95 The mechanism causing decreased levels of testosterone is thought to be exerted by cortisol and β-endorphin.
Psychologic stimuli also lead to a decrease in testosterone levels. Men engaged in rigorous combat training and those engaged in the first several weeks of officer candidate school experience significant drops in testosterone levels.96,97 However, recent data indicate that the psychologic stress associated with some types of competition (e.g., pistol shooting) increases both testosterone and cortisol, especially in athletes older than 45 years.98 Individuals with acute illness, such as respiratory failure, burns, and congestive heart failure, show a marked reduction in plasma testosterone.99
The direct immunologic effects of sex hormones contribute to the sexual dimorphism seen in the incidence of autoimmune disease100 and the greater susceptibility to sepsis and mortality in males following injury.101 Estrogens generally are associated with a depression of T-cell–dependent immune function and enhancement of B-cell functions, and androgens suppress both T- and B-cell responses.99 In injury, however, males produce greater amounts of proinflammatory cytokines, a profile that is associated with poor outcome.102 Additionally, androgens appear to induce a greater degree of immune cell apoptosis following injury, a mechanism that may elicit a greater immunosuppression in injured males versus females.103 (See Table 10-5 for other hormones, including melatonin, substance P, neuropeptide Y, calcitonin gene–related peptide, somatostatin, and vasoactive intestinal peptide.)
STRESS, PERSONALITY, COPING, AND ILLNESS
It is not entirely clear why cortisol secretion during stress is beneficial. It has been suggested that gluconeogenesis prompted by cortisol ensures an adequate source of glucose (energy) for body tissues, and nerve cells in particular. The pooling of amino acids from catabolized proteins may ensure amino acid availability for protein synthesis in certain cells. The redistribution of protein to sites where replacement is critical, such as muscle or cells of damaged tissue, would be beneficial. Short-term, cortisol-induced alterations in immune cell distribution (e.g., traffic) patterns may be adaptive, with a decrease in peripheral blood cell numbers as effector cells locate to sites of injury or inflammation. In addition, decreased immune cell activity by cortisol may be beneficial in some situations because it prevents immune-mediated tissue damage by prolonged cell exposure to high levels of certain cytokines. Whether cortisol-induced effects are adaptive or destructive may depend on the intensity, type, and duration of the stressor, and the subsequent concentration and length of cortisol exposure that target cells of the individual experience.
Extreme physiologic stressors, such as severe burn injury, represent a predictable stimulus for the stress responses described previously. A less severe and defined event or situation, however, can be a stressor for one person and not for another. Many stressors, such as fasting or temperature changes, do not necessarily cause a physiologic stress response if psychologic factors are minimized. Stress itself is not an independent entity but a system of interdependent processes that are moderated by the nature, intensity, and duration of the stressor and the perception, appraisal, and coping efficacy of the affected individual, all of which in turn mediate the psychologic and physiologic response to stress. Further, adjustment to repetitive stressors is known to be individualized, based on a person’s appraisal of a situation.104 Illustrating the influence of an individualized stress appraisal on physiologic processes, a meta-analysis of the relationships between stressors and immunity found that a higher perception of stress was associated with reduced T-cytotoxic (Tc)-cell cytotoxicity although not with levels of circulating Th or Tc lymphocytes.105
Psychosocial distress may be predictive of psychologic and physical health outcomes. In psychologic distress the individual feels a general state of unpleasant arousal after life events that manifests as physiologic, emotional, cognitive, and behavioral changes.106 Periods of depression and emotional upheaval often are associated with adverse life events and place the affected individual at risk for immunologic deficits, increasing the risk of ill health.107 A meta-analysis of studies shows a relationship between depression and reduction in lymphocyte proliferation and NK cell activity.108 Multiple moderating factors may be important in immune modulation in depressed individuals, including comorbidities such as alcoholism. Examples of triggering mechanisms include bereavement, academic and job-related pressures, life events (positive and negative changes),107 and aging. Adverse life events having the most negative effect on immunity are characterized as uncontrollable, undesirable, and overtaxing the individual’s ability to cope.109,110
Studies have strengthened the association of stress with potential for illness in humans. One study examined medical students who were immunized with hepatitis B vaccine on the third day of a stressful examination period; the time to seroconversion and level of antibody titer to the vaccine were measured later. The students with the most rapid seroconversion and the highest titers also reported being less stressed and had a good social support system (which may reduce stress).111 Even more convincing is a study in which the psychologic stress status was determined in healthy individuals after experimentally controlled exposure to respiratory virus by nasal inoculation. Individuals reporting more stress had an increased incidence of clinical cold and respiratory symptoms compared with subjects reporting less stress, and other infections, including HIV, were shown to be potentially influenced by psychosocial factors.112–115
Evidence also suggests adverse changes in immune function following intense exercise, with increased cortisol levels, changes in lymphocyte counts, and alterations in cytokine production.116 Some of these immune changes could be reduced by administration of carbohydrate beverages to endurance athletes, implying that dehydration and decreased tissue perfusion were catalysts for the exercise stress–induced immune changes.
Animal studies have found that stress contributes to the initiation, growth, and metastasis of certain tumors.7,117 Studies of mechanisms in humans reveal stress affects important processes in cancer including antiviral responses, deoxyribonucleic acid (DNA) repair, and aspects of cellular aging.117 But the overall evidence is mixed from prospective studies linking stress with cancer incidence. Stress may be more likely to influence progression and recurrence of cancer; however, the critical prospective studies have been mostly unsupportive.7 Studies examining impairments in antiviral immunity and chronic activation of hormonal responses (e.g., HIV-related tumors, hepatocellular carcinoma, and cervical cancer) may be more successful designs for defining the stress-related mechanisms.117
Evidence is showing a relationship between immune stimulation and heart disease.118 The relationship between stress and cardiovascular health may be mediated by stress-induced changes in immune function, which may potentiate proinflammatory processes and permit alterations that lead to heart disease119 (see What’s New? Acute Emotional Stress and Adverse Heart Effects, p. 337, and What’s New? Psychosocial Stress and Progression to Coronary Heart Disease, p. 347).
In the past decade a significant amount of evidence has accumulated linking severe psychosocial stress resulting from negative life events to a chronic syndrome with mental and physical consequences. Posttraumatic stress disorder (PTSD) has been described in many populations.120–122 A cascade model has been proposed to describe the pathogenesis and clinical course illustrating the clinical, epidemiologic, neurobiologic, and psychosocial components of PTSD.123 The study of PTSD has contributed to the knowledge concerning mechanisms involved in the chronic stress and disease relationship. Recently an appreciation of the association of chronic stress with high levels of cortisol production and paradoxical biounavailability (i.e., bound to plasma protein and therefore not bioavailable) of cortisol has been gained.124
The interaction with healthcare providers in a clinical setting, the diagnosis of a major illness, and various clinical procedures (e.g., blood draws, injections, examinations, surgical procedures) also may represent significant negative life events to many individuals (Figure 10-7). For example, mammography and the activation of the HPA axis.
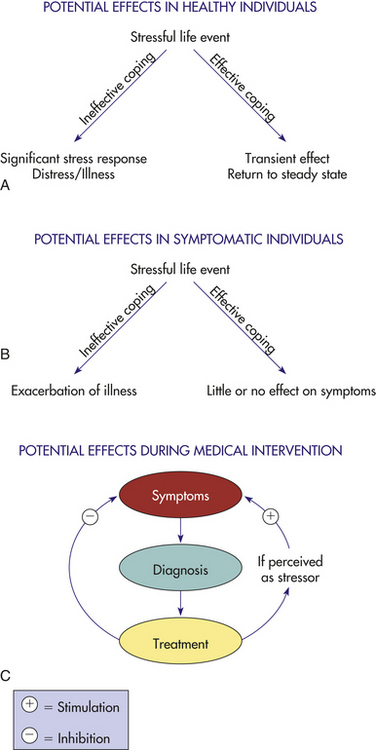
Figure 10-7 Health outcome determination in stressful life situations is moderated by numerous factors. Whether a life-challenged individual experiences distress or illness depends on the subject’s appraisal of the event and the coping strategies used during the stressful period. A and B reflect possible outcomes in stressed healthy and symptomatic individuals. C illustrates the dynamic clinical setting in which the diagnosis of a serious illness and subsequent medical interventions may be perceived as stressful challenges and have potentially detrimental influences on physical outcome.
The influence of repetitive but episodic stress on cancer survivors demonstrates a connection between events such as mammography and activation of the HPA axis. Early research with breast cancer survivors by Cordova and colleagues125 demonstrated a link between sympathetic activity and HPA axis activation, noting that some women reported symptoms of PTSD (heart palpitations, panic, shakiness, nausea) during thoughts of recurrence triggered by events such as finding themselves near the hospital where they received initial treatment.125 HPA axis activation also influences organs and tissues that enable bidirectional communication processes (feedback loops) between neuroendocrine and immune processes.126 For example, among breast cancer survivors 3 to 5 years post diagnosis, elevated baseline cortisol levels and blunted cortisol reactivity were reported in response to the anticipation of a real, regularly scheduled mammogram, and alterations in cortisol and heart rate variability were reported in women simulating the threat of cancer with a controlled laboratory stressor.127,128 Further, Ma and colleagues127 reported that the threat of cancer recurrence (using a simulated mammography event as a stressor to elicit thoughts of cancer recurrence) elicited greater alterations in heart rate variability when compared with another simulated controlled stressor. These studies suggest activation of the autonomic nervous system to events, such as mammography, that occur repeatedly throughout breast cancer survivorship, although the timing of onset of these autonomic activation responses to a stressor is unclear. Similar ANS activation may occur in association with events particular to the management of many other types of chronic illnesses as well as interactions with the healthcare system.
These additional stresses may affect the course of illness as well as interfere with the efficacy of the medical intervention. Identifying and reducing stress in the clinical setting have particular applicability in both disease prevention and illness management. In addition to medical procedures, patient-provider communication also provides an important area for future research. Recent studies of cancer communication and patient-provider interaction have demonstrated a link between communication events and emotional outcomes, such as uncertainty and mood state in breast cancer survivors.129,130 Although a logical extension, it remains to be seen if these emotional outcomes affect physiologically-based health outcomes caused by activation of the HPA axis and subsequent immune processes.
Personality characteristics are associated with individual differences in appraisal and response to stressors.131 The coping response of individuals may exaggerate or moderate physical consequences of the stress response. Coping is defined as the process of managing stressful demands and challenges that are appraised as taxing or exceeding the resources of the person.132 One response may be a negative change in behavior resulting in potentially adverse health effects (e.g., increased smoking, change in eating habits). Serious disturbances of the sleep-wake cycle are observed in many people under stress and in many clinical settings. Further, sleep disturbances may exacerbate the pathophysiologic status of certain patient populations.133–135 Investigators have reported that sleep deprivation and circadian disruption, even in young, otherwise healthy individuals, have detrimental influences on respiratory and immune system function. Even partial sleep deprivation was associated with reduced NK cell activity in healthy subjects, and only recently have seriously ill patients been assessed for adequacy and structure of sleep during recovery.133
Adaptive coping strategies, especially those that are problem focused and those that encourage seeking social support, are beneficial during stressful experiences. The extent to which an individual responds to distress, using effective positive coping strategies, determines the degree of successful moderation of the stress challenge. Conversely, ineffective negative coping attempts may exacerbate the effects of distress on health, thus augmenting the potential for illness. Mediating factors that may influence stress susceptibility or resilience include age, socioeconomic status, gender, social support status, personality and lifestyle, self-esteem, genetics, life events, past experiences, and current health status.136 Evidence suggests that effective intervention may result in greater stress resilience and improved psychologic and physiologic outcomes. In a study of nursing home residents randomly assigned to control or social support intervention groups, improved psychologic measures and immune function (NK cell activity) were observed in the experimental group at 6 weeks.137 In another study, women with recurrent metastatic breast cancer were given either routine follow-up (routine care) or weekly support group sessions. Survival in the support treatment group was an average of 19 months longer than in the routine care group, suggesting a mediating influence of additional support for these women.32,138
The importance of social support for seriously ill individuals has focused attention on the health and well-being of family members who function as caregivers. Significant stress manifested as depression, anxiety, and fatigue has been noted in family caregivers of those with cancer, Alzheimer disease, and burn trauma. Individuals and caretakers exhibited suppression of various measures of immune function, with improved function associated with better perceived social support.139–141 Gender-based coping differences may be attributed, in part, to the hormonal milieu of the individual, with females more likely to offer social support, a behavior with an oxytocin/estrogen association.92
Interventions to prevent or manage stress-related psychologic or physical problems include both short- and long-term coping strategies. Stress management consists of educational components specific to the individual’s problems and relaxation techniques, which may include meditation, imagery, massage, and biofeedback. These approaches may be used on an individual or a support group basis. Incorporation of these approaches into clinical training facilitates their use in the clinical arena. Research should focus on the efficacy of such approaches with various populations.
Aging and Stress: Stress-Age Syndrome
With aging, sometimes a set of neurohormonal and immune alterations, as well as tissue and cellular changes, develops. These changes, which recently have been defined as stress-age syndrome, include the following136,142:
• Irregular excitability of structures of the limbic system influencing the relationship with the hypothalamus
• Rise of the blood concentration of catecholamines, antidiuretic hormone (ADH), ACTH, and cortisol
• Decrease of testosterone, thyroxine, and other hormones
• Alterations in opioid peptide concentration
Some of the alterations are adaptational, whereas others are potentially damaging. These stress-related alterations of aging can influence the course of developing stress reactions and lower adaptive reserve and coping.
Alarm stage 338
Anticipatory response 339
Conditional response 339
Coping 354
Corticosteroid-binding globulin 343
Corticotropin-releasing hormone (CRH) 339
Disease of adaptation 338
General adaptation syndrome (GAS) 338
Homeostasis 339
Hypothalamic-pituitary-adrenal (HPA) axis 338
Peripheral (immune) CRH 339
Physiologic stress 338
Post-traumatic stress disorder (PTSD) 339
Psychologic distress 352
Psychoneuroimmunology (PNI) 339
Reactive response 338
Stage of exhaustion 338
Stage of resistance or adaptation 338
Stress 337
Stress response 339
Stressor 338
Th2 shift 345
Transcortin 343
REFERENCES
1. Rabin, B.S. The nervous system—immune system connection. In: Rabin B.S., ed. Stress, immune function, and health: the connection. New York: Wiley-Liss, 1999.
2. Kapcala, L.P., et al. The protective role of the hypothalamic-pituitary-adrenal axis against lethality produced by immune, infections, and inflammatory stress. Ann N Y Acad Sci. 1995;771:419–437.
3. Black, P.H., Garbutt, L.D. Stress, inflammation and cardiovascular disease. J Psychosom Res. 2002;52(1):1–23.
4. Kiecolt-Glaser, J.K., et al. Psychoneuroimmunology: psychological influences on immune function and health. J Consult Clin Psychol. 2002;70(3):537–547.
5. Liu, L.Y., et al. School examinations enhance airway inflammation to antigen challenge. Am J Respir Crit Care Med. 2002;165(8):1062–1067.
6. Cohen, S., Janicki-Deverts, D., Miller, G.E. Psychological stress and disease. JAMA. 2007;298(14):1685–1687.
7. Vedhara K., Irwin M., eds. Human pyschoimmunology. Oxford, England: Oxford University Press, 2005.
8. Pereira, D.B., et al. Stress as a predictor of symptomatic genital herpes virus recurrence in women with human immunodeficiency virus. J Psychosom Res. 2003;54(3):237–244.
9. Hamerman, D. Toward an understanding of frailty. Ann Intern Med. 1999;130(11):945–950.
10. Taaffe, D.R., et al. Cross-sectional and prospective relationships of interleukin-6 and C-reactive protein with physical performance in elderly persons: MacArthur studies of successful aging. J Gerontol A Biol Sci Med Sci. 2000;55(12):M709–M715.
11. Selye, H. The general adaptation syndrome and the diseases of adaptation. J Clin Endocrinol Metab. 1946;6:117.
12. Hill, S.R., et al. Studies on adrenocortical and psychological responses to stress in man. Arch Intern Med. 1956;97:269.
13. Mason, J.W., Brady, J.V. Plasma 17-hydroxycortico-steroid changes related to reserpine effects on emotional behaviors. Science. 1956;124:983.
14. Hetzel, B.S., et al. Changes in urinary 17-hydroxycorticosteroid excretion during stressful life experiences in man. J Clin Endocrinol Metab. 1955;15(9):1057–1068.
15. Handlon, J.H., et al. Psychological factors lowering plasma 17-hydroxycorticosteroid concentration. Psychosom Med. 1962;24:535–541.
16. Wadeson, R.W., et al. Plasma and urinary 17-OHCS responses to motion pictures. Arch Gen Psychiatry. 1963;14:146–156.
17. Mason, J.W. Organization of psychoendocrine mechanisms: a review and reconsideration of research. In: Greenfield N.S., Steinbach R.A., eds. Handbook of psychophysiology. New York: Holt, Rinehart, & Winston, 1972.
18. Herman, J.P., et al. Central mechanisms of stress integration: hierarchical circuitry controlling hypothalmo-pituitary-adrenocortical responsiveness. Front Neuroendocrinol. 2003;24(3):151–158.
19. Bauer-Wu, S.M. Psychoneuroimmunology. Part I: Physiology. Clin J Oncol Nurs. 2002;6(3):167–170.
20. Bauer-Wu, S.M. Psychoneuroimmunology. Part II: Mind-body interventions. Clin J Oncol Nurs. 2002;6(4):243–246.
21. Cohen, S., et al. Types of stressors that increase susceptibility to the common cold in healthy adults. Health Psychol. 1998;17(3):214–223.
22. McCain, N.L., et al. A randomized clinical trial of alternative stress management interventions in persons with HIV infection. J Consult Clin Psychol. 2008;76(3):431–441.
23. Glaser, R., et al. Chronic stress modulates the immune response to a pneumococcal pneumonia vaccine. Psychosom Med. 2000;62(6):804–807.
24. Marucha, P.T., Kiecolt-Glaser, J.K., Favagehi, M. Mucosal wound healing is impaired by examination stress. Psychosom Med. 1998;60(3):362–365.
25. Pacak, K., et al. Heterogeneous neurochemical responses to different stressors: a test of Selye’s doctrine of nonspecificity. Am J Physiol. 1998;275(4Pt2):R1247–R1255.
26. Repka-Ramirez, M.S., Baraniuk, J.N. Histamine in health and disease. Clin Allergy Immunol. 2002;17:1–25.
27. Dimsdale, J.E., Ziegler, M.G. What do plasma and urinary measures of catecholamines tell us about human response to stressors? Circulation. 1991;83(4 Suppl):II36–II42.
28. Herd, J.A. Cardiovascular response to stress. Physiol Rev. 1991;71(1):305–330.
29. Sapolsky, R.M., Romero, L.M., Munck, A.U. How do glucocorticoids influence stress responses? Integrating permissive, suppressive, stimulatory, and preparative actions. Endocr Rev. 2000;21(1):55–89.
30. Moyna, N.M., et al. The effects of incremental submaximal exercise on circulating leukocytes in physically active and sedentary males and females. Eur J Appl Physiol Occup Physiol. 1996;74(3):211–218.
31. Drapeau, V., et al. Is visceral obesity a physiological adaptation to stress? Panminerva Medica. 2003;45(3):189–195.
32. Rosmond, R. Stress induced disturbances of the HPA axis: a pathway to type 2 diabetes? Med Sci Monit. 2003;9(2):RA35–RA39.
33. Elenkov, I.J., Chrousos, G.P. Stress hormones, proinflammatory and antiinflammatory cytokines, and autoimmunity. Ann N Y Acad Sci. 2002;966:290–303.
34. Fearon, D.T., Locksley, R.M. The instructive role of innate immunity in the acquired immune response. Science. 1996;272(5258):50–53. [review].
35. Mosmann, T.R., Sad, S. The expanding universe of T-cell subsets: Th1, Th2, and more. Immunol Today. 1996;17(3):138–146. [review].
36. Meduri, G.U., Chrousos, G.P. Duration of glucocorticoid treatment and outcome in sepsis: is the right drug used the wrong way? Chest. 1998;114(2):355–360.
37. Chrousos, G.P. The hypothalamic-pituitary-adrenal axis and immune-mediated inflammation. N Engl J Med. 1995;332(20):1351–1362.
38. Karalis, K., et al. Autocrine or paracrine inflammatory actions of corticotropin-releasing hormone in vivo. Science. 1991;254(5030):421–423.
39. Chrousos, G.P., Elenkov, I.J. Interactions of the endocrine and immune systems. In DeGroot L.G., Jameson J.L., eds.: Endocrinology, ed 4, Philadelphia: Saunders, 2001.
40. Elenkov, I.J. Glucocorticoids and the Th1/Th2 balance. Ann N Y Acad Sci. 2004;1024:138–146. [review].
41. Lagier, B., et al. Different modulation by histamine of IL-4 and interferon-gamma (IFN-gamma) release according to the phenotype of human Th0, Th1, and Th2 clones. Clin Exp Immunol. 1997;108(3):545–551.
42. Rocklin R.E., ed. Histamine and H2 antagonists in inflammation and immunodeficiency. New York: Marcel Dekker, 1990.
43. Kiecolt-Glaser, J.K., et al. Chronic stress and age-related increases in the proinflammatory cytokine IL-6. Proc Natl Acad Sci U S A. 2003;100(15):9090–9095.
44. Rohleder, N., et al. Age and sex steroid-related changes in glucocorticoid sensitivity of proinflammatory cytokine production after psychosocial stress. J Neuroimmunol. 2002;126(1-2):69–77.
45. Bone, R.C. Toward an epidemiology and natural history of SIRS (systemic inflammatory response syndrome). JAMA. 1992;268(24):3452–3455.
46. Poynter, M.E., Daynes, R.A. Peroxisome proliferator–activated receptor alpha activation modulates cellular redox status, represses nuclear factor-kappaB signaling, and reduces inflammatory cytokine production in aging. J Biol Chem. 1998;273(49):32833–32841.
47. Aoki, N., Ohno, Y., Imamura, M. Physiological interactions between the immune and endocrine systems: are cytokines hormones? Med Sci Res. 1990;18:195–201.
48. Khansari, D.N., Murgo, A.J., Faith, R.E. Effects of stress on the immune system. Immunol Today. 1990;11(5):170–175.
49. Dunn, A.J. Psychoneuroimmunology for the psychoneuroendocrinologist: a review of animal studies of nervous system-immune system interactions. Psychoneuroendocrinology. 1989;14(4):251–274.
50. Friedman, E.M., Irwin, M.R. A role for CRH and the sympathetic nervous system in stress-induced immunosuppression. Ann N Y Acad Sci. 1995;771:396–418.
51. Jankovic, B.D. Neuroimmunomodulation: facts and dilemmas. Immunol Lett. 1989;21(2):101–118.
52. Rabin, B.S., et al. Bidirectional interaction between the central nervous system and the immune system. Clin Rev Immunol. 1989;9(4):279–312.
53. Jain, R., et al. Corticotropin-releasing factor modulating the immune response to stress in the rat. Endocrinology. 1991;128(3):1329–1336.
54. Maestroni, G.J., Conti, A. Anti-stress role of the immuno-opioid network: evidence for a physiological mechanism involving T cell–derived, immunoreactive beta-endorphin and MET-enkephalin binding to thymic opioid receptors. Int J Neurosci. 1991;61(3-4):289–298.
55. Schwab, R.J. Disturbances of sleep in the intensive care unit. Crit Care Clin. 1994;10(4):681–694.
56. Nelson, R.J., Drazen, D.L. Melatonin mediates seasonal adjustment in immune function. Reprod Nutr Dev. 1999;39(3):383–398.
57. Ader, R., Felten, D., Cohen, N. Interactions between the brain and the immune system. Annu Rev Pharmacol Toxicol. 1990;30:561–602.
58. Busbridge, N.J., Grossman, A.B. Stress and the single cytokine: interleukin modulation of the pituitary-adrenal axis. Mol Cell Endocrinol. 1991;82(2-3):C209–214.
59. Hori, T., et al. Immune cytokines and regulation of body temperature, food intake, and cellular immunity. Brain Res Bull. 1991;27(3-4):309–313.
60. Navarra, P., et al. Interleukins-1 and -6 stimulate the release of corticotropin-releasing hormone-41 from rat hypothalamus in vitro via the eicosanoid cyclooxygenase pathway. Endocrinology. 1991;128(1):37–44.
61. Hall, N.R.S., O’Grady, M.P. Regulation of pituitary peptides by the immune system. BioEssays. 1989;11(5):141–144.
62. Krueger, J.M., et al. Sleep, microbes, and cytokines. Neuroimmunomodulation. 1994;1(2):100–109.
63. Weigent, D.A., Carr, D.J., Blalock, J.E. Bidirectional communication between the neuroendocrine and immune systems: common hormones and hormone receptors. Ann N Y Acad Sci. 1990;579:17–27.
64. Chrousos, G.P., Gold, P.W. The concepts of stress and stress system disorders: overview of physical and behavioral homeostasis. JAMA. 1992;267(9):1244–1252.
65. Chrousos, G.P., et al. Interactions between the hypothalamic-pituitary-adrenal axis and the female reproductive system. Ann Intern Med. 1998;129(3):229–240.
66. Kalantaridou, S.N., et al. Stress and the female reproductive system. Review. J Reprod Immunol. 2004;62(1-2):61–68.
67. Lindholm, J., Schultz-Moller, N. Plasma and urinary cortisol in pregnancy and during estrogen-gestagen treatment. Scand J Clin Lab Invest. 1973;31(1):119–122.
68. Gallucci, W.T., et al. Sex differences in sensitivity of the hypothalamic-pituitary-adrenal axis. Health Psychol. 1993;12(5):420–425.
69. Peiffer, A., Lapointe, B., Barden, H. Hormonal regulation of type II glucocorticoid receptor messenger ribonucleic acid in rat brain. Endocrinology. 1991;129(4):2166–2174.
70. Caro, J.F., et al. Leptin: the tale of an obesity gene. Diabetes. 1996;45(11):1455–1462.
71. Ahima, R.S., et al. Role of leptin in the neuroendocrine response to fasting. Nature. 1996;382(6588):250–252.
72. Boden, G., et al. Effect of fasting on serum leptin in normal human subjects. J Clin Endocrinol Metab. 1996;81(9):3419–3423.
73. Grinspoon, S., et al. Serum leptin levels in women with anorexia nervosa. J Clin Endocrinol Metab. 1996;81(11):3861–3863.
74. Machelska, H., et al. Opioid control of inflammatory pain regulated by intercellular adhesion molecule-1. J Neurosci. 2002;22(13):5588–5596.
75. Molina, P.E. Stress-specific opioid modulation of haemodynamic counter-regulation. Clin Exp Pharmacol Physiol. 2002;29(3):248–253.
76. Jochem, J., Josko, J., Gwozdz, B. Endogenous opioid peptides system in haemorrhagic shock—central cardiovascular regulation. Med Sci Monit. 2001;7(3):545–549.
77. Molina, P.E. Opiate modulation of hemodynamic, hormonal, and cytokine responses to hemorrhage. Shock. 2001;15(6):471–478.
78. Curtis, A.L., et al. Previous stress alters corticotropin-releasing factor neurotransmission in the locus coeruleus. Neuroscience. 1995;65(2):541–550.
79. Calogero, A.E., et al. Multiple feedback regulatory loops in hypothalamic corticotropin-releasing hormone secretion: potential clinical implications. J Clin Invest. 1988;82(3):767–774.
80. Cabot, P.J., et al. Immune cell-derived beta-endorphin: Production, release, and control of inflammatory pain in rats. J Clin Invest. 1997;100(1):142–148.
81. Weigent, D.A., et al. Characterization of the promoter-directing expression of growth hormone in a monocyte cell line. Neuroimmunomodulation. 2000;7(3):126–134.
82. Kelly, P.A., et al. The prolactin/growth hormone receptor family. Endocr Rev. 1991;12(3):235–251.
83. Burguera, B., et al. Dual and selective actions of glucocorticoid upon basal and stimulated growth hormone release in man. Neuroendocrinology. 1990;51(1):51–58.
84. Tsigos, C., Chrousos, G.P. Hypothalamic-pituitary-adrenal axis, neuroendocrine factors and stress. J Psychosom Res. 2002;53(4):865–871.
85. Ben-Jonathan, N., et al. Extrapituitary prolactin: distribution, regulation, functions, and clinical aspects. Endocr Rev. 1996;17(6):639–669. [Review].
86. Shiu, R.P., Friesen, H.G. Mechanisms of action of prolactin in the control of mammary gland function. Annu Rev Physiol. 1980;42:83–96.
87. Van De Weerdt, C., et al. Far upstream sequences regulate the human prolactin promoter transcription. Neuroendocrinology. 2000;71(2):124–137.
88. Noel, G.L., et al. Human prolactin and growth hormone release during surgery and other conditions of stress. J Clin Endocrinol Metab. 1972;35(6):840–851.
89. Prystowsky, M.B., Clevenger, C.V. Prolactin as a second messenger for interleukin 2. Immunomethods. 1994;5(1):49–55.
90. Anderson-Hunt, M., Dennerstein, L. Oxytocin and female sexuality. Gynecol Obstet Invest. 1995;40(4):217–221.
91. Neumann, I.D. Alterations in behavioral and neuroendocrine stress coping strategies in pregnant, parturient, and lactating rats. Prog Brain Res. 2001;133:143–152.
92. Liu, Y., et al. Differential expression of vasopressin, oxytocin, and corticotrophin-releasing hormone messenger RNA in the paraventricular nucleus of the prairie vole brain following stress. J Neuroendocrinol. 2001;13(12):1059–1065.
93. Taylor, S.E., et al. Biobehavioral responses to stress in females: tend-and-befriend, not fight-or-flight. Psychol Rev. 2000;107(3):411–429.
94. Insel, T.R. A neurobiological basis of social attachment. Am J Psychiatry. 1997;154(6):726–736.
95. Matsumoto, K., et al. Plasma testosterone levels following surgical stress in male patients. Acta Endocrinol. 1970;65(1):11–17.
96. Aakvaag, A., et al. Testosterone and testosterone-binding globulin (TeBG) in young men during prolonged stress. Int J Androl. 1978;1:22.
97. Kreuz, L.E., Rose, R.M., Jennings, J.R. Suppression of plasma testosterone levels and psychological stress: a longitudinal study of young men in Officer Candidate School. Arch Gen Psychiatry. 1972;26(5):479–482.
98. Guezennec, C.Y., et al. Effect of competition stress on tests used to assess testosterone administration in athletes. Int J Sports Med. 1995;16(6):368–372.
99. Rose, R.M. Psychoendocrinology. In Wilson J.D., Foster D.W., eds.: Williams textbook of endocrinology, ed 7, Philadelphia: Saunders, 1985.
100. Da Silva, J.A. Sex hormones and glucocorticoids: interactions with the immune system. Ann N Y Acad Sci. 1999;876:102–117.
101. Offner, P.J., Moore, E.E., Biffl, W.L. Male gender is a risk factor for major infections after surgery. Arch Surg. 1999;134(9):935–938.
102. Angele, M.K., et al. Sex steroids regulate pro- and anti-inflammatory cytokine release by macrophages after trauma-hemorrhage. Am J Physiol. 1999;277(1Pt1):C35–42.
103. Angele, M.K., et al. Gender dimorphism in trauma-hemorrhage–induced thymocyte apoptosis. Shock. 1999;12(4):316–322.
104. McEwen, B.S. Protective and damaging effects of stress mediators. N Engl J Med. 1998;338(3):171–179.
105. Segerstrom, S.C., Miller, G.E. Psychological stress and the human immune system: a meta-analytic study of 30 years of inquiry. Psychol Bull. 2004;130(4):601–630.
106. Thoits, P.A. Dimensions of life events that influence psychological distress: an evaluation and synthesis of the literature. In: Kaplan H.B., ed. Psychosocial stress: trends in theory and research. Orlando, FL: Academic Press, 1983.
107. Rozlog, L.A., et al. Stress and immunity: implications for viral disease and wound healing. J Periodontol. 1999;70(7):786–792.
108. Irwin, M. Immune correlates of depression. Adv Exp Med Biol. 1999;461:1–24.
109. Irwin, M., et al. Impaired natural killer activity during bereavement. Brain Behav Immun. 1988;1:98.
110. Kiecolt-Glaser, J., et al. Modulation of cellular immunity in medical students. J Behav Med. 1986;9(1):5–21.
111. Glaser, R., et al. Stress-induced modulation of the immune response to recombinant hepatitis B vaccine. Psychosom Med. 1992;54(1):22–29.
112. Cohen, S. Psychosocial stress and susceptibility to upper respiratory infections. Am J Respir Crit Care Med. 1995;152(4Pt2):S53–58.
113. Evans, D.L., et al. Stress-associated reductions of cytotoxic T lymphocytes and natural killer cells in asymptomatic HIV infection. Am J Psychiatry. 1995;152(4):543–550.
114. Nott, K.H., Vedhara, K., Spickett, G.P. Psychology, immunology and HIV. Psychoneuroendocrinology. 1995;20(5):451–474.
115. Sheridan, J.F., et al. Psychoneuroimmunology: stress effects on pathogenesis and immunity during infection. Clin Microbiol Rev. 1994;7(2):200–212.
116. Nieman, D.C. Nutrition, exercise, and immune system function. Clin Sports Med. 1999;18(3):537–548.
117. Antoni, M.H., et al. The influence of bio-behavioral factors on tumor biology: pathways and mechanisms. Nat Rev Cancer. 2006;6(3):240–248.
118. Sharma, R., Coats, A.J., Anker, S.D. The role of inflammatory mediators in chronic heart failure: cytokines, nitric oxide, and endothelin-1. Int J Cardiol. 2000;72(2):175–186.
119. Sher, L. Effects of psychological factors on the development of cardiovascular pathology: role of the immune system and infection. Med Hypotheses. 1999;53(2):112–113.
120. Bremner, J.D., et al. Neural correlates of memories of childhood sexual abuse in women with and without posttraumatic stress disorder. Am J Psychiatry. 1999;156(11):1787–1795.
121. Clohessy, S., Ehlers, A. PTSD symptoms, response to intrusive memories and coping in ambulance service workers. Br J Clin Psychol. 1999;38(pt 3):251–265.
122. Donnelly, C.L., Amaya-Jackson, L., March, J.S. Psychopharmacology of pediatric posttraumatic stress disorder. J Child Adolesc Psychopharmacol. 1999;9(3):203–220.
123. Heim, C., Ehlert, U., Hellhammer, D.H. The potential role of hypocortisolism in the pathophysiology of stress-related bodily disorders. Psychoneuroendocrinology. 2000;25(1):1–35.
124. Alarcon, R.D., Glover, S.G., Deering, C.G. The cascade model: an alternative to comorbidity in the pathogenesis of posttraumatic stress disorder. Psychiatry. 1999;62(2):114–124.
125. Cordova, M.J., et al. Frequency and correlates of posttraumatic-stress-disorder-like symptoms after treatment for breast cancer. J Consult Clin Psychol. 1995;63(6):981–986.
126. Reiche, E.M., Nunes, S.O., Morimoto, H.K. Stress, depression, the immune system, and cancer. Lancet Oncol. 2004;5(10):617–625.
127. Ma, Z., Faber, A., Dube, L. Exploring women’s psychoneuroendocrine responses to cancer threat: insights from a computer-based guided imagery task. Can J Nurs Res. 2007;39(1):98–115.
128. Porter, L.S., et al. Cortisol levels and responses to mammography screening in breast cancer survivors: a pilot study. Psychosom Med. 2003;65(5):842–848.
129. Clayton, M.F., Dudley, W.N., Musters, A. Communication with breast cancer survivors. Health Commun. 2008;23(3):207–221.
130. Clayton, M.F., Mishel, M.H., Belyea, M. Testing a model of symptoms, communication, uncertainty, and well-being, in older breast cancer survivors. Res Nurs Health. 2006;29(1):18–39.
131. Kiecolt-Glaser, J.K., et al. Psychoneuroimmunology and psychosomatic medicine: back to the future. Psychosom Med. 2002;64(1):15–28. [Review].
132. Folkman, S., Lazarus, R.S. The relationship between coping and emotion: implications for theory and research. Soc Sci Med. 1988;26(3):309–317.
133. Irwin, M., et al. Partial sleep deprivation reduces natural killer cell activity in humans. Psychosom Med. 1994;56(6):493–498.
134. Pollmacher, T., et al. Influence of host defense activation on sleep in humans. Adv Neuroimmunol. 1995;5(2):155–169.
135. White, D., et al. Sleep deprivation and the control of ventilation. Am Rev Respir Dis. 1983;128(6):984–986.
136. Frolkis, V.V. Stress-age syndrome. Mech Aging Dev. 1993;69(1-2):93–107.
137. Kiecolt-Glaser, J., et al. Psychosocial enhancement of immunocompetence in a geriatric population. Health Psychol. 1985;4(1):25–41.
138. Spiegel, D. Psychosocial intervention in cancer. J Natl Cancer Inst. 1993;85(15):1198–1205.
139. Baron, R.S., et al. Social support and immune function among spouses of cancer patients. J Pers Soc Psychol. 1990;59(2):344–352.
140. Kiecolt-Glaser, J., et al. Chronic stress and immune function in family caregivers of Alzheimer’s disease victims. Psychosom Med. 1987;45(5):523.
141. Shelby, J., et al. Severe burn injury: effects on psychologic and immunologic function in noninjured close relatives. J Burn Care Rehabil. 1992;13(1):58–63.
142. Mazzeo, R.S. Aging, immune function, and exercise: hormonal regulation. Int J Sports Med. 2000;21(Suppl 1):S10–S13. [Review].