ALTERATIONS OF CARDIOVASCULAR FUNCTION IN CHILDREN


Cardiovascular disease in children can be classified as congenital or acquired heart disease. Congenital heart disease is the most common. The diagnosis and management of congenital heart defects continue to improve with the use of fetal echocardiography, early interventional catheterization, and refined surgical repair. Acquired heart defects in children continue to present challenges to the practitioner; although guidelines for diagnosing acquired defects are available, work is needed in developing standards of treatment and long-term follow-up.
DEVELOPMENT OF THE CARDIOVASCULAR SYSTEM
Embryology
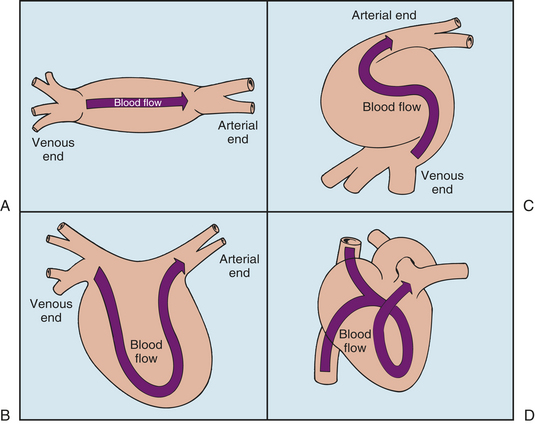
Cardiogenesis begins at approximately 3 weeks of gestation; however, most cardiovascular development occurs between the fourth and seventh weeks.1 The heart arises from the mesenchyme and begins development as an enlarged blood vessel with a large lumen and a muscular wall (Figure 31-1, A). Initially, two lateral endocardial heart tubes fuse to form a single structure (Figure 31-1, B). During the fifth week of gestation, the midsection of this tube begins to grow faster than its ends. This single heart tube elongates and rotates to the right (D-loop formation), creating a bulboventricular loop by approximately the twenty-eighth day1 (Figure 31-1, C). Also at this time the first fetal heart contractions occur. At this stage the primitive heart structures include a common atrium; common ventricle; the sinus venosus, which eventually evolves into the superior and inferior venae cavae; the bulbus cordis, which eventually evolves into the ventricular outflow tracts; and the truncus arteriosus, which eventually yields the main pulmonary artery (PA) and aorta (Figure 31-1, D). By the fourth week of gestation, cardiovascular septation, ventricular development, aortic arch evolution, and circulation begin.
Cardiac Septation
Separation first begins when collections of mesenchymal cells cause the endocardial lining of the heart to bulge into the internal lumen. These changes, known as endocardial cushions, are instrumental in closing the lower portion of the atrial septum, dividing the atrioventricular (AV) canals into the right and left AV orifices, and forming the upper portion of the interventricular septum. Altered formation of the endocardial cushions can result in ostium primum atrial septal defects, inlet ventricular septal defects (VSDs), malformation of the AV valves, or a complete AV defect (also known as atrioventricular septal defect).2
Atrial septation begins when two thin membrane-like structures, known as the septum primum and the septum secundum, grow toward the area of the endocardial cushions (Figure 31-2). The septum primum forms along the posterior wall of the common atrium and grows downward toward the center portion of the heart. The gap between the two structures, known as the ostium primum, normally closes by extensions from the endocardial cushions. At the time of closure, fenestrations or openings develop in the superior portion of the septum primum, creating the ostium secundum. Failure of the septum primum to fuse with the endocardial cushions results in an ostium primum defect in the atrial septum near the AV valve area.
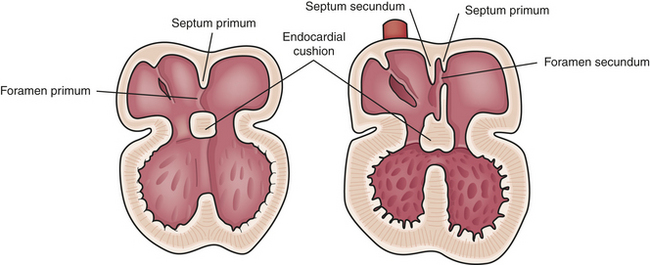
The septum secundum is also a fenestrated, membrane-like structure located anteriorly that grows toward the endocardial cushions. During fetal development this structure does not completely fuse with the endocardial cushions to achieve complete atrial septal closure. The nonfused septum secundum and ostium secundum result in the formation of a flapped orifice known as the foramen ovale, which allows the right-to-left shunting necessary for fetal circulation. Altered development in any of these structures can lead to an atrial septal defect.
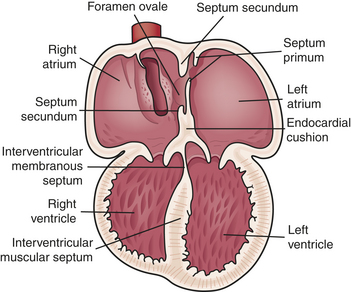
Ventricular septation develops when the muscular ridge located at the apex, the endocardial tissue, and the bulbar ridges in the bulbus cordis fuse (Figure 31-3). Closure of the interventricular septum ensures communication between the right ventricle (RV) and the PA and between the left ventricle (LV) and the aorta. Further evolution of the endocardial tissue gives rise to the membranous ventricular septum and the AV valves. The conal portion of the ventricular septum that separates the aorta from the PA forms from the bulbus cordis.
When the single primitive heart tube begins to form the D-loop, the venous and arterial poles of the heart are fixed, resulting in torsion within the anterosuperior region of the loop, known as the truncus arteriosus. This torsion creates a spiral ridgelike structure or septum within the truncus arteriosus that divides it into the PA and the aorta. The semilunar valves evolve from tubercles after this division is complete.
Before this division occurs, however, two large arteries form at the distal end of the truncus arteriosus. Over time they give rise to a series of arterial vessels, collectively called the six aortic arches. By the fifth week of gestation, the first two pairs disappear and the third eventually evolves into the common carotid artery, the external carotid artery, and part of the internal carotid artery. The fourth pair of aortic arches will form part of the true aortic arch and the proximal segment of the right subclavian artery. The fifth pair disappears; however, the sixth pair yields the proximal and branch pulmonary arteries within the lung parenchyma and the ductus arteriosus.
Swellings in the conal region at the base of the main trunk separate the right ventricular outflow (pulmonary outflow) tract from the left ventricular outflow (aortic outflow) tract. The conus also contributes to complete closure of the interventricular septum, and normal reabsorption of the subaortic conal region ensures rotation of the great arteries so that the aorta is posterior and to the right of the PA and the PA is anterior and to the left of the aorta. Despite division of the truncus arteriosus and separation of the right and left outflow tracts, a communication exists between the aorta and the PA known as the ductus arteriosus.
In order to deliver maximally oxygenated blood to the developing brain, fetal circulation differs from the adult pattern by the presence of alternate pathways known as fetal shunts (Figure 31-4). Fetal oxygenation occurs in the placenta instead of the fetal lungs. The fetal lungs are not aerated, although the fetus does make breathing motions.
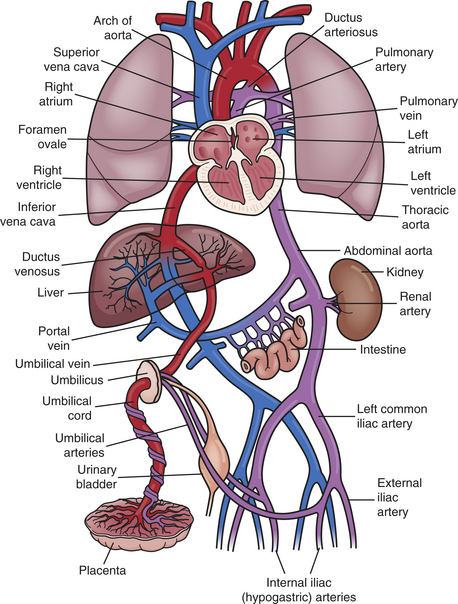
Figure 31-4 Fetal circulation. Circulation of the fetus reflects the fact that oxygenation of fetal blood does not take place in the lungs, but rather in the placenta. Therefore, the pulmonary circulatory system is essentially “bypassed.” Instead of traveling from the right heart to the lungs, as occurs after birth, most blood entering the right heart passes through the ductus arteriosus and into the systemic circulation.
In utero the fetus receives blood carrying oxygen and nutrients from the placenta through the umbilical vein. Fetal arterial oxygen tension is much lower than that found in the postnatal period—approximately 20 to 30 torr (mmHg pressure). Yet, despite this hypoxemic state, tissue hypoxia does not occur because of high fetal cardiac output and fetal hemoglobin. The blood travels to the liver, where a portion enters the portal and hepatic circulation; approximately half the flow is diverted away from the liver through the ductus venosus and into the inferior vena cava. Because the blood received from the inferior vena cava yields a higher pressure, blood entering the right atrium (RA) from the inferior vena cava is shunted through the foramen ovale and into the left atrium (LA) and is then pumped through the LV and into the aorta. Approximately two thirds of the blood flows to the head and upper extremities. Because this blood is mainly from the placenta, the brain and coronary arteries receive the blood with the highest oxygen concentration. The remaining blood flows into the descending aorta.
Less-saturated blood, with an oxygen tension of 15 to 19 torr, returns from the upper body, head, neck, and arms and travels from the superior vena cava into the RA. A small portion of this blood flows into the RV and out the PA and enters the nonfunctioning lungs. Most of the blood, however, bypasses the lungs by flowing through the ductus arteriosus and into the descending aorta. Blood from the descending aorta returns to the placenta through two umbilical arteries.1
The nonaerated lungs and low oxygen tension induce vasoconstriction, creating high pulmonary vascular resistance. This is transmitted to the right side of the heart and the PAs. Conversely, fetal systemic resistance is low because of the large-volume placenta and ductus arteriosus. Therefore, because blood flow follows the path of least resistance, high pulmonary resistance diverts most of the blood flow into the PA, through the ductus arteriosus, and into the aorta. From there it travels into the low-resistance placenta.
Transitional Circulation
At birth a series of circulatory changes occur that affect blood flow, vascular resistance, and oxygen tension. The most important change that takes place in the circulation is the shift of gas exchange from the placenta to the lungs. In addition, alterations in pressure and volume of blood flowing through the heart chambers functionally close the ductus arteriosus, ductus venosus, and foramen ovale. A decrease of pulmonary vascular resistance and an increase of systemic vascular resistance lead to changes in the size and shape of the heart chambers.
Clamping of the umbilical cord and expansion of the lungs at birth shift gas exchange from the placenta to the lungs. Removal of the low-resistance placenta from circulation also causes an immediate increase in systemic vascular resistance to about twice that before birth. Conversely, pulmonary vascular resistance decreases because of expansion of the lungs that results from the infant’s respirations and exposure to more oxygen-rich blood.
Closure of Fetal Shunts
Once the umbilical cord is clamped, the umbilical arteries and vein, which comprise the cord, vasoconstrict and undergo fibrous changes. Therefore, blood flow through the ductus venosus falls instantly; absence of fetal shunting through this vessel usually occurs within the first 7 days of life. Once the ductus venosus closes, its remnants form the ligamentum venosum, or round ligament of the liver.
Increased pulmonary venous return and decreased inferior vena cava return cause functional closure of the foramen ovale within the first month of life. In the fetus the foramen ovale is held open by the blood flow from the high-pressure right side, reflecting pulmonary vascular resistance, to the lower-pressure area on the left side of the heart, reflecting systemic vascular resistance. At birth the pressure gradients reverse (left atrial pressure exceeds right atrial pressure by a small degree), causing the valve flaps of the foramen ovale to close. Functional closure occurs by the adherence of these flaps to the atrial septum. Anatomic closure occurs within the first month of life after deposition of fibrin tissue and cell products permanently seals the flaps closed. Until this occurs, any condition that stimulates an increase in the right-sided pressures or causes dilation of the RA can reopen the foramen ovale. Conditions in which a patent foramen ovale may continue past the first month of life include pulmonary hypertension, RV failure, and tricuspid atresia.
The ductus arteriosus closes more gradually. Increased oxygen saturation in the systemic arterial blood is thought to be the major stimulus causing vasoconstriction of the ductus arteriosus. In addition, a decrease in the amount of endogenous prostaglandins promoting dilation and the release of vasoactive substances stimulate further ductal closure. Vasoconstriction of the ductal medial smooth muscle shortens and thickens the intima of the ductal wall within 15 to 18 hours after birth. Permanent closure is complete 10 to 21 days after birth. Fibrous tissue adheres to the remaining structure, and the ductus arteriosus eventually evolves into the ligamentum arteriosum. Conditions that involve low arterial oxygen saturations, such as cyanotic heart disease, decreased medial muscle layer within the ductus, or increased levels of circulating vasodilating substances in the blood, may delay or prevent ductal closure.3
Postnatal Development
The infant’s cardiopulmonary system is proportionally larger in relation to body surface area than the adult’s. The infant’s heart points at a transverse angle, but as the lungs and heart mature, the heart shifts lower in the chest and is rotated at a more oblique angle. Unlike the adult heart, the newborn heart has RV dominance with a thickened RV wall. This is because of the high pulmonary vascular resistance in the fetal circulation that subjects the RV to high afterload, which in turn causes the right ventricular myocardium to become as thick and strong as the left.
After birth the right ventricular myocardium becomes less dominant as pulmonary vascular resistance drops. As systemic vascular resistance increases, the left ventricular myocardium becomes thicker. By 1 month of age, the newborn’s ventricles are approximately equal in weight. As the child grows, the heart size increases accordingly. The weight of the heart doubles during the first year of life and increases six times that by 9 years of age.3
Postnatal changes involve a rise in arterial oxygen tension and an increase in alveolar oxygenation that stimulates vasodilation, resulting in a decrease in pulmonary vascular resistance. During the first 2 to 9 weeks of life, the inner medial linings of the small pulmonary arterioles thin out in response to decreased pulmonary arterial pressure. This increased diameter of the pulmonary vessels, along with further development of the pulmonary bed in response to lung growth, results in a decrease in pulmonary vascular resistance. By 2 months of age, pulmonary resistance may approximate adult levels. During the neonatal period, however, care must be taken to maintain homeostasis because of hyperactivity of the pulmonary bed. Adverse conditions, such as alveolar hypoxia, acidosis, and hypothermia, may trigger pulmonary vasoconstriction and lead to pulmonary hypertension.
Postnatal Hemodynamics
As stated, systemic vascular resistance begins to rise once the placenta is removed from the circulation. Normal levels in the infant range from approximately 10 to 15 Wood units × body surface area (in square meters) and gradually increase to 15 to 30 Wood units × body surface area (in square meters) by childhood.4 Likewise, the systolic pressure is low in the full-term newborn (approximately 39 to 59 mmHg), reflecting the decreased LV strength. As the systemic vascular resistance increases, the LV becomes more developed and the systolic pressure rises steadily until it equals adult levels once the child reaches puberty.
The heart rate of the newborn ranges from 100 to 180 beats/minute, which gradually decreases as the child grows. Similarly, the newborn’s cardiac output is high, which is a reflection of the fetal circulation described earlier. Oxygen consumption doubles at birth; to maintain adequate oxygen delivery, the cardiac output also remains high. These changes, however, cause minimal cardiac reserve in the newborn. Additional stressors could increase oxygen demands and result in acute deterioration. By 2 months of age, oxygen consumption decreases by half. As the newborn grows, stroke volume steadily increases while the heart rate decreases.2
Postnatal Circulation
Postnatal circulation allows the lungs to oxygenate the venous blood and allows saturated blood to be delivered to the systemic circulation. Desaturated blood returning from the superior vena cava, inferior vena cava, and coronary veins enters the RA and is pumped to the RV through the tricuspid valve. The RV then pumps the blood through the pulmonic valve to the PA; the blood flows to the lungs, where it is oxygenated. The oxygenated blood returns from the lungs through the pulmonary veins and enters the LA, which pumps blood to the LV through the mitral valve. The LV then pumps blood through the aortic valve and into the aorta. The coronary arteries receive the saturated blood along with delivery to the systemic circulation.
CONGENITAL HEART DEFECTS
Congenital heart disease is the leading cause of death, excluding prematurity, during the first year of life.5 It is estimated that as many as 35% of deaths caused by congenital heart defects occur in the first year of life and that one third of children born with congenital heart disease will die as a result of their cardiac disease (Box 31-1). There are more than 35 documented types of congenital heart defects, and the frequency of occurrence in the United States is on the rise. Although researchers have not determined the reason for this increase, one explanation is that it may be the result of improved methods of detection.6
The underlying cause of congenital heart disease is known in only 10% of cases. Several factors place the fetus at risk for developing congenital heart disease, including prenatal, environmental, and genetic factors. Among the prenatal factors are maternal rubella, maternal insulin-dependent diabetes, maternal alcoholism, maternal age (older than 40 years), maternal phenylketonuria, and maternal hypercalcemia (Table 31-1). The use of some drugs during pregnancy is associated with an above-average incidence of congenital heart disease. Examples of these drugs include thalidomide, lithium, phenytoin (Dilantin), and warfarin. The incidence of heart defects also has been found to be higher in stillbirths, spontaneous abortions, and low-birth-weight or small-for-gestational-age infants.2 In general, the likelihood of unaffected parents having a child with congenital heart disease is about 1% with a recurrence risk of 2% to 6%.

Genetic factors also have been implicated in the development of congenital heart disease, although the mechanism of causation is often multifactorial. Recent progress, accelerated through the Human Genome Project, has resulted in the rapid identification of some genes causing congenital heart disease.5
ETIOLOGY The etiology of congenital heart disease is unknown. Early epidemiologic studies report a multifactorial influence to be the cause of up to 90% of cardiac anomalies, with a recurrence rate of 2% to 6%.2 Associated risk factors include maternal, gestational, and familial conditions. (Maternal risk factors are discussed in the previous section.) Exposure to teratogens in utero also may be a risk factor. Likewise, fetal exposure to active maternal infections, such as rubella, herpesvirus, coxsackievirus B5, and cytomegalovirus, may be a risk.
Chromosomal aberrations account for about 6% of all congenital heart defects (Table 31-2). Many genetic and hereditary diseases are associated with congenital heart defects, although the mechanism of causation is unknown (Table 31-3). As many as 50% of infants with trisomy 21 have a congenital heart defect, either an AV canal defect or a VSD. Extracardiac defects are noted in as many as 35% of infants with cardiac lesions. Prospective studies using chromosomal analysis have suggested that congenital cardiac malformations may be the result of a single gene defect.5
Table 31-2
Genetic Factors and Congenital Heart Defects
| Chromosomal Aberrations or Syndrome | Incidence of Defects | Type of Defect |
| Trisomy 13 | 80% | Ventricular septal defect (VSD), atrial septal defect (ASD), patent ductus arteriosus (PDA), anomalous pulmonary venous connection, bicuspid aorta, overriding aorta |
| Trisomy 18 | 90% | VSD, PDA, patent foramen ovale, bicuspid aortic valve, dextrocardia |
| Down syndrome | 12%-44% | Endocardial cushion defects, VSD, PDA, ASD, transposition of great vessels, tetralogy of Fallot, persistent truncus arteriosus, coarctation of aorta, endocardial fibroelastosis |
| Cri du chat syndrome | 20% | PDA, mixed defects |
| Turner syndrome | 20%-40% | Coarctation of aorta, pulmonary stenosis, subaortic and aortic stenosis, PDA, septal defects |
Data from Doyle EF, Rutkowski M: Etiology of congenital heart disease, Cardiovasc Clin 2:1, 1970.
Table 31-3
Disorders Coexistent with Congenital Heart Defects
| Disorder | Associated Cardiovascular Defect |
| Connective Tissue Disorders | |
| Marfan syndrome | Aortic or mitral regurgitation, aortic aneurysm |
| Hurler syndrome | Pseudoatherosclerosis |
| Hunter syndrome | Pseudoatherosclerosis, hypertension |
| Osteogenesis imperfecta | Incompetent aortic valve |
| Complex Syndromes | |
| Kartagener syndrome | Dextrocardia |
| Holt-Oram syndrome | Atrial septal defect (ASD), ventricular septal defect (VSD) |
| Ellis–van Creveld syndrome | Defect or absence of atrial septum |
| Laurence-Moon-Biedl syndrome | Tetralogy of Fallot, single ventricle, transposition of aorta |
| Inborn Errors of Metabolism | |
| Pompe disease | Cardiomegaly, left heart failure, supraventricular tachycardia |
| Homocystinuria | Thromboembolic episodes, pulmonic and aortic regurgitation |
| Phakomatosis | |
| Neurofibromatosis (von Recklinghausen disease) | Hypertension, pheochromocytoma |
| von Hippel–Lindau disease | Hypertension, pheochromocytoma |
| Sturge-Weber-Dimitri disease | Anomalies of carotid and meningeal arteries |
| Vascular Malformations | |
| Osler-Weber-Rendu disease (hereditary hemorrhagic telangiectasia) | Atrioventricular fistula, telangiectasia |
| Milroy disease (lymphedema) | Hypoplasia or lymphatic vessels |
Data from Doyle EF, Rutkowski M: Etiology of congenital heart disease, Cardiovasc Clin 2:1, 1970.
Because of improved screening methods, surgical interventions, and management, children with congenital heart defects are now surviving into adulthood and bearing children of their own. Studies report a 5% to 15% incidence of congenital heart disease in offspring of a parent having a congenital heart lesion. If two siblings have a congenital cardiac anomaly, the recurrence risk is 9%, and if three siblings have a congenital cardiac anomaly, the rate jumps to a 50% chance that the next child also will have a cardiovascular malformation.
Classification of Congenital Heart Defects and Associated Conditions
There are more than 35 different types of congenital anomalies that can be classified into four categories based on blood flow pattern: (1) lesions increasing pulmonary blood flow; (2) lesions decreasing pulmonary blood flow; (3) obstructive lesions, in which right- or left-sided outflow tract obstructions curtail or prohibit blood flow out of the heart; and (4) mixing lesions, in which desaturated blood and saturated blood mix within the chambers or great arteries of the heart (Table 31-4). By classifying lesions in this way, the clinical manifestations, as well as associated sequelae, are more predictable.
Table 31-4
Classification of Congenital Heart Defects
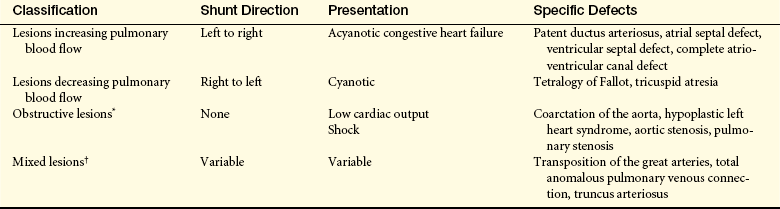
∗If patent ductus arteriosus closes, newborns with hypoplastic left heart syndrome, coarctation of the aorta, or critical aortic stenosis will present with shock. Newborns with aortic stenosis or pulmonary stenosis may have only mild symptoms depending on severity of stenosis.
†Transposition of the great arteries and truncus arteriosus will present with cyanosis as patent ductus arteriosus closes. Total anomalous pulmonary venous connection usually presents with congestive heart failure.
Associated conditions and their clinical manifestations are lesion dependent. The two most common conditions associated with congenital heart disease are heart failure (HF) and hypoxemia. Lesions increasing pulmonary blood flow include defects that allow blood flow to shunt from the high-pressure left side to the lower-pressure right side, resulting in pulmonary congestion. Lesions that cause decreased pulmonary blood flow are generally complex and result in cyanosis. Obstructive lesions increase the pressure needed to eject the blood from the ventricle. The two types of obstructive lesions are right-sided lesions that may result in hypoxemia and cyanosis, and left-sided lesions that may result in HF. Mixing lesions are variable in their physiology and clinical manifestation.
Heart Failure
Heart failure (HF), sometimes called congestive heart failure (CHF), is classified as an acquired condition. HF occurs when the heart is unable to maintain sufficient cardiac output to meet the metabolic demands of the body. HF can occur as the result of decreased myocardial function or excessive metabolic demands. The most common causes of HF in infancy and childhood are cardiomyopathy or the result of poor ventricular function. Table 31-5 lists the congenital heart defects that cause HF by age. Pulmonary overcirculation from large left-to-right shunts (mixing) is often called CHF but is not usually associated with decreased ventricular function and failure to meet metabolic demands. However the clinical manifestations are similar, such as failure to thrive (FTT), tachypnea, tachycardia, and respiratory infections.
Table 31-5
Congenital Heart Defects Causing Heart Failure
| Age | Congenital Heart Defect |
| Time of birth | Hypoplastic left heart syndrome |
| Volume overload caused by tricuspid regurgitation (rare) | |
| Arteriovenous fistula | |
| Birth to 1 week | Hypoplastic left heart syndrome |
| Aortic atresia | |
| Transposition of the great vessels with ventricular septal defect (VSD) | |
| Coarctation of the aorta | |
| Total anomalous pulmonary venous connection (TAPVC) with obstruction | |
| Patent ductus arteriosus (PDA) in premature infants | |
| First 4 weeks | Coarctation of the aorta |
| TAPVC | |
| Large left-to-right shunt caused by VSD, PDA in premature infants | |
| Tricuspid atresia | |
| All previously mentioned defects | |
| 4 to 6 weeks | Transposition of the great vessels with VSD |
| Large left-to-right shunt caused by endocardial cushion defect | |
| 6 weeks to 6 months | VSD |
| 6 months | Endocardial fibroelastosis |
| Persistent truncus arteriosus with large left-to-right shunt |
PATHOPHYSIOLOGY In general, the pathophysiologic mechanisms of HF in infants and children are very similar to those in adults. The same compensatory mechanisms are activated in the face of inadequate cardiac output. An acute decrease in blood pressure stimulates stretch receptors and baroreceptors in the aorta and carotid arteries, which in turn stimulate the sympathetic nervous system. With the release of catecholamines and the stimulation of β-receptors, heart rate and the force of myocardial contraction increase. Venous smooth muscle tone also increases, which increases return of venous blood to the heart. Sympathetic stimulation also decreases blood flow to the kidneys, skin, spleen, and extremities so that maximum flow to the brain, heart, and lungs can be maintained. Decreased blood flow to the kidneys causes the release of renin, angiotensin, and aldosterone. If chronic, this cycle results in retention of sodium and fluid by the kidneys, which in turn increases volume in the circulatory system.
These neurohumoral and hemodynamic changes create abnormal ventricular wall stress and cause the myocardium to hypertrophy. The myocardial fibers also stretch to accommodate the increased volume. Hypertrophy and fiber stretch temporarily increase contractility and hence the force of ventricular contraction. These mechanisms eventually fail to maintain cardiac output as HF progresses. A review of the Frank-Starling law of the heart (see Chapter 29) is useful for an understanding of the cycle of compensation and decompensation that occurs in HF.
CLINICAL MANIFESTATIONS Symptomatic HF in children has many causes. It is not usually necessary to determine if it is right- or left-sided HF. When assessing a child with HF, a combination of symptoms generally is present. Pulmonary overcirculation is the predominant cause associated with congenital defects.
HF in infants is manifested as poor feeding and sucking, often leading to FTT. Dyspnea, tachypnea, and diaphoresis may be accompanied by retractions, grunting, and nasal flaring. Wheezing, coughing, and rales are rare even with significant HF. Common skin changes, such as pallor or mottling, are often present.
Hepatomegaly (enlargement of the liver) is atypically attributable to systemic venous congestion. In infants the normal liver is soft, sharp-edged, and palpable 1 to 2 cm below the costal margin. However, the absence of hepatomegaly does not rule out HF.
Periorbital edema and weight gain without caloric increase are uncommon manifestations of right ventricular failure in infants. Peripheral edema, which is a common finding in adults, is rare in infants and young children and more often signifies renal disease rather than cardiac disease. The clinical manifestations of HF are listed in Box 31-2.
EVALUATION AND TREATMENT A thorough physical examination with an emphasis on cardiac and pulmonary findings often will reveal the degree of HF. Plotting the child’s growth (height, weight, head circumference) is an important method for monitoring a child’s health. Infants with HF and pulmonary overcirculation usually have low weight with normal length and head circumference. Failure to thrive (FTT) is usually the result of increased metabolic expenditure relative to caloric intake. An electrocardiogram (ECG) should be performed to determine the presence of dysrhythmias or hypertrophy. A chest radiograph is useful in assessing the presence of cardiomegaly and signs of increased pulmonary circulation.
Treatment is aimed at decreasing cardiac workload and increasing the efficiency of heart function. Medical management initially consists of diuretics, such as furosemide. Depending on the degree of HF, other diuretics can be used in combination with furosemide to counteract potassium losses. Agents that reduce afterload, such as angiotensin-converting enzyme (ACE) inhibitors and beta-blockers, have recently been used to further manage severe HF.2,3 Caloric supplementation is routinely prescribed.
Hypoxemia
Heart defects that allow desaturated blood to enter the systemic system without passing through the lungs result in hypoxemia and cyanosis. Hypoxemia occurs when arterial oxygen tension is below normal and results in low oxygen arterial saturations and cellular function alteration. Cyanosis, a blue discoloration of the mucous membranes and nail beds, results from deoxygenated hemoglobin in a concentration of at least 5 g/dl of blood or from arterial saturations less than 85%.2,4 Anemia may mask the signs of hypoxemia, whereas children who are polycythemic with a normal arterial saturation may appear cyanotic. Older children who have an unrepaired septal defect with a left-to-right shunt may become cyanotic because of pulmonary vascular changes secondary to increased pulmonary blood flow. Because of these progressive pulmonary vascular changes, pulmonary vascular resistance increases to exceed or equal vascular resistance, resulting in a reversal of shunting known as Eisenmenger syndrome. Three types of defects cause hypoxemia and cyanosis:
1. Lesions that cause right ventricular outflow tract obstruction and shunting from the right side of the heart to the left side, as in tetralogy of Fallot (see p. 1223)
2. Defects involving the mixing of saturated and unsaturated blood within the heart chambers, as in a univentricular heart (also referred to as a single ventricle)
3. Defects in children with transposition of the great arteries (see p. 1231), in which two parallel circulations exist and survival depends on the existence of a patent ductus arteriosus or septal defect
CLINICAL MANIFESTATIONS Infants with mild hypoxemia may show signs of cyanosis only occasionally when stressed; otherwise they may exhibit near-normal age-projected growth and development. Infants with severe hypoxemia may display signs of feeding intolerance, poor weight gain, tachypnea, and dyspnea. Children with chronic hypoxemia are small for their age, may display cognitive and motor skill delays, experience shortness of breath with exertion, fatigue easily, and have exercise intolerance. Acute, severe hypoxemia will lead to tissue hypoxia, metabolic acidosis, hyperventilation, poor perfusion, and eventually shock.
In response to chronic hypoxemia, polycythemia occurs as the body generates additional red blood cells to increase the oxygen-carrying capacity of the blood. In some infants, however, microcytic anemia may result because of limited stores of iron. Polycythemia and the associated platelet dysfunction also places children at risk for thromboembolic events, especially infants with severe cyanosis and iron deficiency anemia. In addition to the 2% risk of cerebrovascular accidents, there is a small chance that children with right-to-left shunting will develop a brain abscess.4 Clubbing of the nail beds occurs because of chronic tissue hypoxemia and polycythemia.
Defects Increasing Pulmonary Blood Flow
Cardiac lesions that increase pulmonary blood flow include defects that involve septal abnormalities or communications between the great arteries. These allow the shunting of blood from the high-pressure left side to the lower-pressure right side. Infants with left-to-right shunts are acyanotic and, depending on the degree of shunting, will often develop signs and symptoms of CHF. Children with significant left-to-right shunts left untreated are at risk for development of irreversible pulmonary hypertension.
Patent Ductus Arteriosus
The patent ductus arteriosus (PDA) is a vessel located between the junction of the main and left pulmonary arteries and the lesser curvature of the descending aorta, usually just distal to the left subclavian artery. During fetal circulation the PDA allows blood to shunt from the PA to the aorta. At birth, once the placenta is removed and the lungs are expanded, the PDA will start to constrict within the first hours of life. Closure of the PDA in full-term infants is usually noted between 15 hours of life and 2 weeks of age.7 As an isolated defect, PDA occurs in 5% to 10% of all congenital cardiac defects. In premature infants, studies have shown that the incidence of PDA is as high as 45% in newborns less than 1750 g.8
PATHOPHYSIOLOGY Failure of the PDA to close results in persistent patency of the ductus arteriosus. The hemodynamic effects of PDA depend on the size of the lumen and the resistance in the pulmonary and systemic circulations. At birth the pulmonary and systemic vascular resistances are almost equal and are reflected in the PA and aorta, respectively; therefore, shunting is minimal. However, as pulmonary vascular resistance falls, a reversal of fetal shunting occurs. Blood now begins to shunt left to right, from the aorta to the PA. The hemodynamic effect is increased pulmonary blood flow, resulting in increased pulmonary venous return to the LA and LV with increased workload on the left side of the heart. The increased workload is caused by increased pulmonary venous return to the LA and, potentially, an increase in right ventricular pressure if pulmonary vascular changes occur in response to the increased blood flow, leading to an increase in pulmonary vascular pressure (Figure 31-5, C).
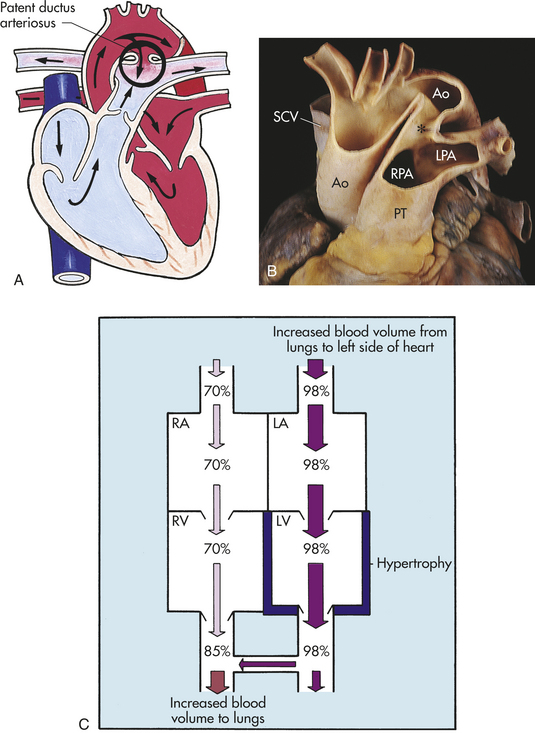
Figure 31-5 Patent ductus arteriosus (PDA). A, PDA with left-to-right shunt. B, PDA (∗) in an adult with pulmonary hypertension. C, Changes in oxygen saturation, left ventricular volume, and the myocardium caused by left-to-right shunt through a PDA. Ao, Aorta; LA, left atrium; LPA, left pulmonary artery; LV, left ventricle; PT, pulmonary trunk; RA, right atrium; RPA, right pulmonary artery; RV, right ventricle; SCV, subclavian vein. (A from Hockenberry MJ et al: Wong’s essentials of pediatric nursing, ed 8, St Louis, 2009, Mosby; B from Damjanov I, Linder J, editors: Anderson’s pathology, ed 10, St Louis, 1996, Mosby.)
CLINICAL MANIFESTATIONS If pulmonary vascular resistance has fallen, infants with PDA will characteristically have a continuous-machinery type murmur heard best at the left upper sternal border throughout systole and diastole. If the PDA is significant, the infant also will have bounding pulses, an active precordium, a thrill upon palpation, and signs and symptoms of pulmonary overcirculation. Infants with a small PDA will usually remain asymptomatic.
EVALUATION AND TREATMENT Chest radiograph will reveal cardiomegaly and increased pulmonary vascular markings. An ECG may demonstrate ventricular enlargement, particularly on the left, but in most cases it is within the normal range. Echocardiography and auscultation confirm the diagnosis based on the characteristic continuous-machinery type of murmur.
PDA closure in asymptomatic children is recommended by 2 years of age because of the risk of subacute bacterial endocarditis. Premature infants who develop respiratory distress are initially given indomethacin, a prostaglandin inhibitor, to close the duct. If this is unsuccessful, surgical ligation and division may be warranted.
Historically the most widely used method for PDA closure is surgical closure involving ligation and division of the ductus with complete closure in nearly 100% of cases. Mortality associated with surgical intervention nears 0%; however, there continues to be some morbidity associated with the approach through a left thoracotomy incision.
Several other options for PDA closure are available depending on the size of the child and the PDA. Many specialists perform interventional closure of the PDA during catheterization. The catheter is advanced into the ductal opening whereby multiple coils or other devices are placed into the lumen that prohibit flow through the duct. The greatest advantages to this procedure are the avoidance of a surgical procedure and thoracotomy pain and a brief observation stay in the hospital.
Another option is closure through video-assisted thoracoscopic surgery. This procedure involves making three small incisions in the left lateral wall, through which a probe is inserted. A clip is then placed around the vessel to occlude it. An advantage of this procedure over surgery is that there is less associated morbidity because of the avoidance of a thoracotomy incision.7
Atrial Septal Defect
An atrial septal defect (ASD) is an abnormal communication between the atria (Figure 31-6, A and B). Although it is an isolated lesion, it is the fourth most common congenital heart defect, occurring in 5% to 10% of all congenital cardiac defects. The three major types are an ostium primum defect, an opening found low in the septum that may be associated with AV valve abnormalities, especially mitral insufficiency; an ostium secundum defect, an opening in the center of the septum (this is the most common type of atrial defect); and a sinus venosus defect, an opening that occurs high up in the atrial septum near the superior vena cava and RA junction. This defect is often associated with partial anomalous pulmonary venous connection.
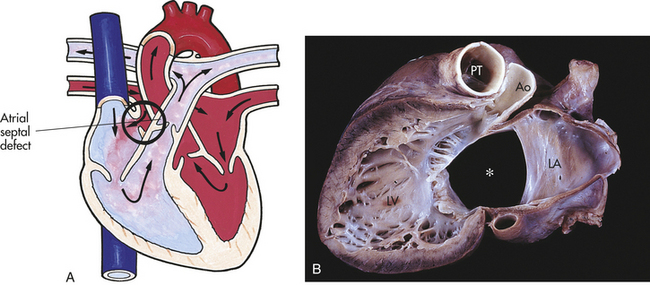
Figure 31-6 Atrial septal defect (ASD). A, Abnormal opening between the atria causing blood from the higher-pressure left atrium to flow into the lower-pressure right atrium. B, Complete ASD (∗) form in children. Ao, Aorta; LA, left atrium; LV, left ventricle; PT, pulmonary artery trunk. (A from Hockenberry MJ et al: Wong’s essentials of pediatric nursing, ed 8, St Louis, 2009, Mosby; B from Damjanov I, Linder J, editors: Anderson’s pathology, ed 10, St Louis, 1996, Mosby.)
PATHOPHYSIOLOGY Although the pressure difference between the two atria is minimal, the ASD allows blood to be shunted from left to right because of the slightly higher pressure of the left atrial chamber and lower pulmonary vascular resistance as compared with systemic vascular resistance. Right atrial and ventricular enlargement develops as a result of left-to-right shunting. Children with ASD are generally asymptomatic and rarely display signs of pulmonary overcirculation. Moderate to large ASDs allow an increase in pulmonary blood flow and, over time, pulmonary vascular changes can occur that may, although rarely, result in pulmonary hypertension.
CLINICAL MANIFESTATIONS Because most children with ASD are asymptomatic, diagnosis usually is made during a routine physical examination by the auscultation of a crescendo-decrescendo systolic ejection murmur that reflects increased blood flow through the pulmonary valve. The location of the murmur is between the second and third intercostal spaces along the left sternal border. A wide fixed splitting of the second heart sound is also characteristic of ASD, reflecting volume overload to the RV, causing prolonged ejection time and delay of pulmonic valve closure.
EVALUATION AND TREATMENT In most cases an echocardiogram is sufficient to confirm the diagnosis of an ASD. A chest radiograph may reveal cardiomegaly and increased pulmonary vascular markings in an asymptomatic child. An ECG may demonstrate right axis deviation and diastolic overload of the RV manifested as right ventricular hypertrophy.2
ASD closure, generally before the child reaches school age, results in improved health later in life. If left unrepaired, right ventricular compliance decreases with age, and pulmonary hypertension and right ventricular hypertrophy may occur, placing the person at risk for the development of HF, atrial dysrhythmias, or embolic events later in life. Surgical closure is the corrective method of choice and involves a pericardial patch or suture closure of the defect, depending on the size of the opening. Repair is done through a midsternal approach with the use of cardiopulmonary bypass. Minimally invasive techniques are being used on near-adult sized patients. Sinus venosus defects require a slightly different approach that consists of a synthetic patch to close the opening and baffle the anomalous right pulmonary venous drainage to the LA. Operative mortality associated with ASD closure is near 0%, with minimal morbidity.7,9,10 Device closure performed in the catheterization laboratory is becoming a routine alternative to surgical closure.7
Ventricular Septal Defect
A ventricular septal defect (VSD) is an abnormal communication between the ventricles (Figure 31-7, A). VSDs are the most common type of congenital heart lesion and account for 25% to 33% of all congenital heart defects. The four types of VSDs are based on location in the septum. The perimembranous type, which occurs in the outflow tract on the LV immediately below the aortic valve, is the most common type, accounting for up to 80% of all VSDs that require treatment. Muscular VSDs, which occur low in the ventricular septum between the trabeculae, are most likely to close spontaneously and are difficult to close surgically because of their location low in the ventricular apex. Most muscular VSDs are hemodynamically insignificant and require no medical or surgical treatment. Supracristal VSDs occur in the right ventricular outflow tract or infundibulum, below the pulmonary valve. AV canal or inlet VSDs occur posterior and inferior to the membranous system, beneath the septal cusp of the tricuspid valve and inferior to the papillary muscles of the conus.
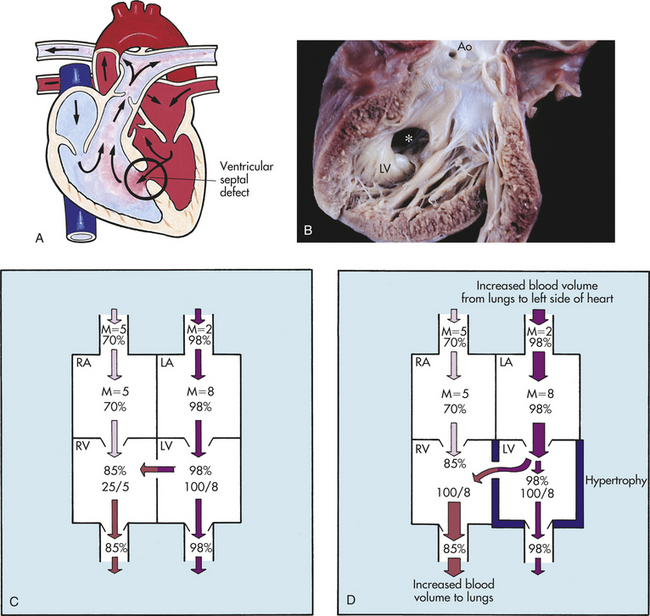
Figure 31-7 Ventricular septal defects (VSDs). A, VSD with left-to-right shunt. B, Muscular (∗) defect (opened left ventricle). C, Hemodynamics of a small VSD with left-to-right shunt. Mean (M) indicates mean of pressure; systolic/diastolic pressures are in mmHg; and percentages indicate oxygen saturation. D, Hemodynamics of a large VSD with left-to-right shunt. Like the shunting that occurs in preductal coarctation of the aorta, the shunting pictured here causes left ventricular overload and hypertrophy. Ao, Aorta; LA, left atrium; LV, left ventricle; RA, right atrium; RV, right ventricle. (A from Hockenberry MJ et al: Wong’s essentials of pediatric nursing, ed 8, St Louis, 2009, Mosby; B from Damjanov I, Linder J, editors: Anderson’s pathology, ed 10, St Louis, 1996, Mosby.)
PATHOPHYSIOLOGY The direction of shunting in a child with a VSD is from the high-pressure left side to the lower-pressure right side. The amount of shunting depends on the size of the defect and the degree of pulmonary vascular resistance. Small VSDs present increased resistance to shunting and limit blood flow through the defect; thus the degree of pulmonary vascular congestion and ventricular chamber enlargement is minimal (Figure 31-7,C).
After 1 to 2 weeks of life, when pulmonary vascular resistance has decreased, moderate-size to large VSDs allow a large amount of shunting from left to right. The shunted blood goes directly out the RV outflow tract and into the PA rather than remaining in the RV cavity (Figure 31-7,D). Therefore, the main PA, LA, and LV all enlarge. LV hypertrophy occurs to effectively pump the additional volume. Pulmonary overcirculation accounts for the symptoms associated with a large VSD in most cases.
Over time the pulmonary bed also undergoes changes because of increased pulmonary blood flow caused by the left-to-right shunting. The smooth muscle layer in the arteriolar walls thickens and proliferation of the intimal layer occurs. The effect of these changes is a decrease in the diameter of the pulmonary vessels, which increases the resistance to blood flow. If the pulmonary vascular resistance is severely increased these changes eventually become irreversible, and pulmonary vascular resistance continues to rise. In some cases it exceeds systemic vascular resistance, causing the shunt through the VSD to reverse direction. Deoxygenated blood now flows into the systemic circulation, and cyanosis occurs, a phenomenon known as Eisenmenger syndrome.
CLINICAL MANIFESTATIONS Clinical manifestations in children with VSDs depend on the age of the child, size of the defect, and level of pulmonary vascular resistance. Newborns with small VSDs are relatively asymptomatic. Initially no murmur is present because the newborn’s high pulmonary vascular resistance causes equalization of the pressures between both ventricles. Once pulmonary vascular resistance has dropped, left-to-right shunting occurs, creating a murmur. Infants with large VSDs display symptoms of HF and poor weight gain. Adults who develop pulmonary vascular obstructive disease as a result of unrepaired VSD will be cyanotic and have clubbing.
On physical examination a loud, harsh, holosystolic murmur and systolic thrill can be detected at the left lower sternal border. The intensity of the murmur reflects the pressure gradient across the VSD. An apical diastolic rumble may be present with a moderate to large defect, reflecting increased flow across the mitral valve.
EVALUATION AND TREATMENT ECG and chest radiographs reflect the amount of shunting through the defect. The ECG of an individual with a small VSD may be normal, whereas an ECG of an individual with a large VSD may reveal biventricular hypertrophy and LA enlargement. Chest roentgenographic findings are significant for cardiomegaly and increased pulmonary vascular markings; again, the severity is directly related to the magnitude of shunting. An echocardiogram identifies the position, size, direction of shunting, and dimensions of the LA, LV, and RV chambers. It also can provide an estimate of PA and RV pressures. Cardiac catheterization may be performed to determine hemodynamics and, in some instances, the location of other VSDs.
Many VSDs spontaneously close during the first year of life.2 Infants with symptoms of HF and poor weight gain despite medical management should have their VSD corrected as soon as possible. Left-to-right shunting with a pulmonary flow/systemic flow (Qp:Qs) ratio of greater than 2:1 or evidence of elevated pulmonary vascular resistance are indications for closure. Closure of the VSD at this time is to prevent the development of pulmonary vascular obstructive disease.
Placement of a PA band to decrease the amount of pulmonary blood flow was initially used as a palliative procedure but is now rarely used unless the presence of an additional lesion makes complete repair difficult. Patch closure, using a synthetic material such as Dacron, is accomplished through a sternotomy and with the use of cardiopulmonary bypass. A transatrial approach is preferable to a right ventriculotomy because of the increased incidence of conduction disturbances associated with ventriculotomy. Contraindications for VSD closure include evidence of pulmonary vascular obstructive disease or Eisenmenger syndrome.2,7 Occlusion devices for VSD closure performed in the cardiac catheterization laboratory are now in early clinical use.7,11
Atrioventricular Canal Defect
An atrioventricular canal (AVC) defect results from nonfusion of the endocardial cushions during fetal life, yielding abnormalities in both the atrial and ventricular septa and AV valves (Figure 31-8). This defect accounts for as many as 5% of all congenital heart defects, and approximately 30% of AVC defects occur in children with Down syndrome.7 The three types of AVC defects are based on the cardiac components involved. Complete AVC (CAVC) defects consist of an inlet VSD, a primum type of ASD, and defects in both the mitral and tricuspid valves. Partial AVC (PAVC) defects consist of a primum type of ASD and a cleft in the septal or anterior leaflet of the mitral valve. Transitional AVC (TAVC) defects involve partial fusion of the endocardial cushions, resulting in variable AV valve abnormalities.7
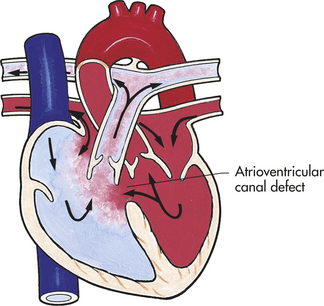
Figure 31-8 Atrioventricular canal defect. (From Hockenberry MJ et al: Wong’s essentials of pediatric nursing, ed 8, St Louis, 2009, Mosby.)
PATHOPHYSIOLOGY Hemodynamic abnormalities seen in AVC defects depend on the components of the lesion and the level of pulmonary vascular resistance. Shunting is minimal during the neonatal period when pulmonary vascular resistance is high. However, once pulmonary vascular resistance drops, left-to-right shunting occurs through the septal defects, resulting in increased pulmonary blood flow and HF.
PAVC defects mimic the hemodynamics of secundum ASD in which the left-to-right shunting through the primum ASD causes RA and RV dilation and increased pulmonary blood flow. The mitral regurgitation that occurs, caused by the cleft mitral valve, is not usually hemodynamically significant.
CAVC defects reflect the hemodynamics of an ASD and a VSD, resulting in biatrial and biventricular enlargement. RA and RV volume overload occurs because of shunting through the primum ASD and tricuspid regurgitation. Likewise, LA and LV volume overload occurs because of shunting through the VSD, increased pulmonary venous return, and mitral regurgitation.
CLINICAL MANIFESTATIONS Children with PAVC defects are generally asymptomatic. Findings on physical examination are similar to those of secundum ASD except for the systolic regurgitant murmur of mitral regurgitation at the apex. At 4 to 12 weeks of age, when pulmonary vascular resistance drops, children with CAVC defects usually begin to show symptoms of HF. Physical findings are similar to those found in individuals with VSDs with the addition of a holosystolic murmur radiating to the back and apex, reflecting mitral regurgitation. A mid-diastolic rumble at the left lower sternal border or apex reflects relative stenosis of the mitral or tricuspid valve from increased flow. Infants with CAVC may have signs of HF and frequent respiratory infections.
EVALUATION AND TREATMENT The ECG generally demonstrates a superior left axis deviation, first-degree AV block, and RV hypertrophy or right bundle branch block. The ECG of CAVC defects also may show LV hypertrophy. Chest radiograph shows cardiomegaly, increased pulmonary vascular markings, and a prominent main PA. Echocardiography allows visualization of the components of the defect, including continuity between the AV valves, their sizes, and chordal attachments. Cardiac catheterization may be electively performed and can confirm the location of septal defects, AV valve abnormalities, degree of left-to-right shunting, and presence of pulmonary hypertension.
Timing of surgical repair depends on the severity of symptoms, degree of shunting, and level of pulmonary vascular resistance. The trend is to perform complete repair between 3 and 6 months of life to avoid the development of pulmonary vascular changes. Surgical repair is performed through a midsternotomy implementing a one- or two-patch repair to close the septal defects and repair the involved AV valves. Mortality has declined below 10% unless the child is a newborn, has severe AV valve incompetence, or has a small LV (unbalanced AV canal). Postoperative complications include heart block, dysrhythmias, or mitral regurgitation requiring further surgical intervention or valve replacement.7,12
Defects Decreasing Pulmonary Blood Flow
Defects decreasing pulmonary blood flow involve obstruction to pulmonary blood flow and septal communications. Because of RV outflow tract obstruction, right-sided pressures exceed left-sided pressures, resulting in right-to-left shunting. Children with these defects have hypoxemia and cyanosis.
Tetralogy of Fallot
Tetralogy of Fallot (TOF) consists of four defects: a large VSD that is high in the septum, an overriding aorta that straddles the VSD, pulmonary stenosis (PS), and RV hypertrophy (Figure 31-9,A). It is the most common cyanotic congenital heart defect and accounts for 10% of all defects.2
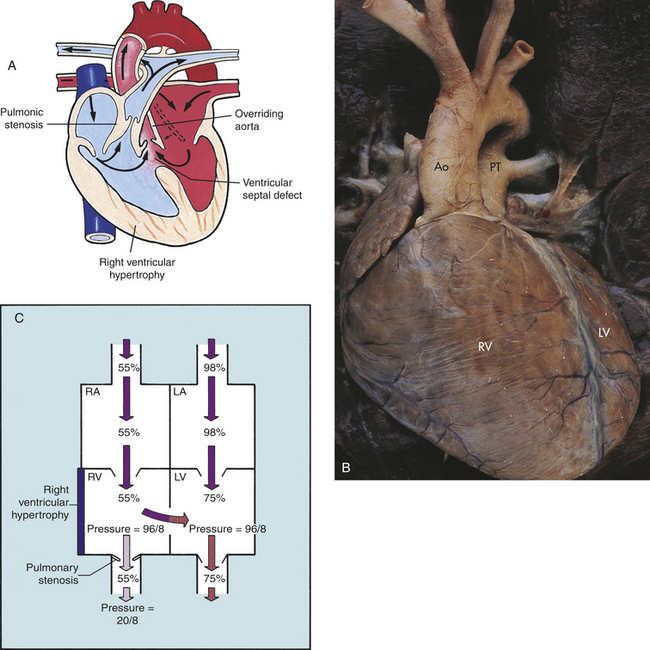
Figure 31-9 Tetralogy of Fallot. A, Anatomic defects in tetralogy of Fallot. B, Complete transposition of the aorta and pulmonary artery. C, Hemodynamics of tetralogy of Fallot with right-to-left shunt. Ao, Aorta; LA, left atrium; LV, left ventricle; PT, pulmonary artery trunk; RA, right atrium; RV, right ventricle. (A from Hockenberry MJ et al: Wong’s essentials of pediatric nursing, ed 8, St Louis, 2009, Mosby; B from Damjanov I, Linder J, editors: Anderson’s pathology, ed 10, St Louis, 1996, Mosby.)
PATHOPHYSIOLOGY TOF develops during two phases of embryologic growth: (1) during the division of the truncus arteriosus by the spiral septum in the third or fourth week of gestation and (2) during the division of the ventricles between the fourth and eighth weeks of gestation. Normally as these events progress, the truncal septum fuses with the bulbar ridges and in turn with the endocardial cushions. The membranous portion of the interventricular septum grows upward to meet the endocardial cushions, and ultimately all of these tissues come together to complete the interventricular septum.
The embryologic error that causes TOF is not known for certain, but two theories have been proposed.1 The first is that the truncus arteriosus divides unevenly, resulting in great vessels of unequal size. The second theory proposes that infundibular overgrowth in the RV is the major developmental anomaly. Defects in ventricular septation also occur, producing the large VSD, which allows the aorta to override the VSD.
The pathophysiology associated with TOF varies widely, depending primarily on the degree of pulmonary stenosis, the size of the VSD, and the pulmonary and systemic resistance to flow. Because the VSD is usually large, pressures are equal in the RV and LV. Therefore, the major determinant of shunt direction through the VSD is the difference between pulmonary and systemic vascular resistance (see Figure 31-9,C). Infants who have little or no right-to-left shunting are acyanotic and are known as “pink tets.” They may have a net left-to-right shunt similar to a large VSD. If pulmonary vascular resistance is higher than systemic resistance, the shunt is from right to left. Because many factors can alter the balance between pulmonary and systemic resistance, shunt direction is not necessarily constant.
Pulmonary stenosis decreases blood flow to the lungs and, consequently, the amount of oxygenated blood that returns to the left heart. If blood also shunts from right to left through the VSD, deoxygenated blood mixes with the oxygenated blood returning from the lungs. The result is low oxygen saturation (hypoxemia) in the systemic circulation. The body attempts to compensate for chronic hypoxemia by producing more red blood cells (thereby causing polycythemia) and by increasing blood flow to the lungs through collateral bronchial vessels in long-standing cases.
CLINICAL MANIFESTATIONS In cases with decreased pulmonary flow through the right ventricular outflow tract as long as the ductus arteriosus remains open, the newborn’s pulmonary blood flow may be adequate. As the ductus closes, however, cyanosis becomes apparent. Chronic hypoxemia causes clubbing of the fingers and toes (see Chapter 33).
A rare manifestation of TOF is the sudden onset of dyspnea, cyanosis, and restlessness, sometimes called a hypercyanotic spell or a “tet spell,” that generally occurs with crying and exertion. The cause of these episodes is unknown, but it is theorized that the RV outflow tract goes into spasm or the systemic resistance drops suddenly. In either case the relative or actual increase in pulmonary vascular resistance increases the right-to-left shunt and the cyanosis. Hypercyanotic spells are often the event that initiates surgical intervention. If the spells are frequent or do not terminate spontaneously, they are considered a medical-surgical emergency.7
Infants with TOF may have difficulty with feeding because the exertion required increases hypoxia, and therefore they experience slow growth and FTT. Most infants with TOF grow normally.
Squatting is a spontaneous compensatory mechanism used by older children to alleviate hypoxic spells. Squatting and its variants increase systemic resistance while decreasing venous return to the heart from the inferior vena cava. The decrease of systemic return makes relatively more oxygenated blood available to the body. The increase of systemic resistance also reverses the shunt through the VSD to a left-to-right shunt, which has the effect of increasing pulmonary blood flow. Through both of these mechanisms, squatting temporarily decreases the degree of hypoxemia. It is uncommon to witness this because most cases are surgically corrected in early infancy.
The typical heart murmur of TOF is a pulmonary systolic ejection murmur caused by the obstruction in the outflow tract, which creates turbulence during systole. More obstruction to flow (e.g., smaller orifice for blood to flow through) produces a louder murmur. This explains why the murmur often disappears during a hypoxic spell, when obstruction increases and pulmonary blood flow decreases to a minimal amount. The second heart sound seems to be single, but in fact it is not. The pulmonary component is very soft and delayed and usually is not heard, although it is present. The enlarged RV may cause the left side of the chest to be more prominent, and a “heave” also may be palpated.
EVALUATION AND TREATMENT The ECG indicates RV hypertrophy. Chest radiographic examination shows that the heart is shaped like a boot (upturned apex because of a small main PA) and that pulmonary vascular markings are decreased. Echocardiograms and angiograms enable the clinician to see the size and position of the VSD, the stenotic pulmonary infundibulum or valve, the smaller-than-normal PA, and the overriding aorta. Measurements made during cardiac catheterization (electively done) demonstrate equal systemic pressure in the RV and LV, decreased pressure in the PA distal to the obstruction, and low oxygen saturation in the aorta if there is right-to-left shunting.
The current practice is to repair TOF before 1 year of life. Triggers for repair include increasing cyanosis and hypercyanotic spells. Palliative procedures include the placement of a pulmonary-to-systemic artery shunt known as the Blalock-Taussig shunt to increase pulmonary blood flow or a modification of the shunt using prosthetic graft material placed from either the subclavian or innominate artery to the PA. These shunts may cause PA distortion but may be necessary in a very small symptomatic child. Corrective repair involves patch closure of the VSD, resection of infundibular or valvular stenosis, and patch augmentation of the RV outflow tract. The procedure is done through a median sternotomy on cardiopulmonary bypass. The operative mortality is less than 5%. Complications include dysrhythmias and occasionally heart block.13
Tricuspid Atresia
Tricuspid atresia consists of an imperforate tricuspid valve, resulting in no communication between the RA and RV (Figure 31-10). This defect accounts for 2% to 3% of congenital heart defects and is the third most common cyanotic heart defect. Tricuspid atresia is a combination of defects, including the imperforate tricuspid valve as well as a septal defect, hypoplastic or absent RV, enlarged mitral valve and LV, and varying degrees of pulmonic stenosis. Tricuspid atresia also may be associated with transposition of the great vessels. The most common type of tricuspid atresia involves a hypoplastic RA with decreased pulmonary blood flow, ASD, VSD, and normally related great vessels.2
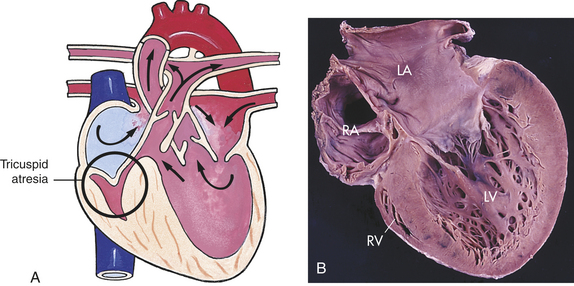
Figure 31-10 Tricuspid atresia. A, No communication from the right atrium to the right ventricle. B, Tricuspid atresia with absent right atrioventricular connection with a hypoplastic right ventricle (four-chamber view). LA, Left atrium; LV, left ventricle; RA, right atrium; RV, right ventricle. (A from Hockenberry MJ et al: Wong’s essentials of pediatric nursing, ed 8, St Louis, 2009, Mosby; B from Damjanov I, Linder J, editors: Anderson’s pathology, ed 10, St Louis, 1996, Mosby.)
PATHOPHYSIOLOGY Systemic blood returns through the superior and inferior venae cavae to the RA. Venous return flows through the ASD into the LA, mixing with blood returning from the pulmonary circulation. The blood then enters the LV. Most of this blood goes out into the systemic circulation through the aorta, but varying amounts pass through the VSD into the hypoplastic RV and to the lungs. Pulmonary circulation depends on the presence of a VSD and the presence of a functioning RV of reasonable capacity. If the RV is absent, the pulmonary valve is usually imperforate as well. If this is the case, a PDA is necessary to ensure that some blood flows into the pulmonary circulation.7
Pulmonary circulation also depends on the relationship between pulmonary and systemic vascular resistance. As long as pulmonary resistance is lower than systemic resistance, blood flows through the VSD from left to right, feeding the pulmonary circulation. If pulmonary resistance rises above systemic resistance, pulmonary blood flow will be significantly diminished.
CLINICAL MANIFESTATIONS Some degree of central cyanosis is common in tricuspid atresia, depending on the amount of pulmonary blood flow. Growth failure also is common. Children experience exertional dyspnea, tachypnea, and hypoxemia. Long-term effects of hypoxia are polycythemia and clubbing. These children also may display hypercyanotic spells. Hepatomegaly may be present if the ASD is restrictive or CHF occurs as a result of increased pulmonary blood flow.
The murmur heard with tricuspid atresia may have several components. The VSD causes a systolic regurgitant murmur; the larger the VSD, the softer and shorter the murmur is likely to be. A narrowly split second heart sound caused by decreased pulmonary blood flow may be present, or the second heart sound may be single if there is pulmonary atresia.
EVALUATION AND TREATMENT Chest radiographic examination shows a heart size that is normal or slightly increased. ECG usually shows RA, LA, and LV hypertrophy with left axis deviation. Echocardiography and cardiac catheterization, if performed, depict left-to-right shunting at the ventricular level, inability of blood flow to enter the RV, and the presence of associated defects.
Newborns with ductal dependent pulmonary blood flow are immediately given prostaglandins to maintain adequate pulmonary perfusion. Initial surgical intervention involves the placement of a Blalock-Taussig shunt (or its modification). If the ASD is restrictive, a Rashkind procedure (balloon atrial septostomy) may be performed during catheterization. Children who experience increased pulmonary blood flow may require the placement of a PA band. Corrective repair involves closing the septal defects, taking down the previous shunts or band, and connecting the superior and inferior venae cavae to the PA to separate the pulmonary systemic circulation (Fontan procedure and its modifications). Postoperative complications include pleural effusions, elevated pulmonary vascular resistance, LV dysfunction, and dysrhythmias.13
Obstructive Defects
Obstructive defects are conditions in which anatomic stenosis (narrowing) in either the right or left outflow tract causes obstruction to blood flow and results in a pressure load on the ventricles. The difference between the obstruction is the gradient that reflects the severity of the narrowing; the higher the gradient is the more obstruction to flow and increased afterload on the ventricle. The location is classified according to the location of the narrowing in relation to the valve. Valvular stenosis refers to stenosis of the valve itself; subvalvular indicates that the stenotic area is below the valve or in the ventricular outflow tract; and supravalvular is the area above the valve in the great artery. The obstructive defects include coarctation of the aorta, aortic stenosis, pulmonary stenosis, and hypoplastic left heart syndrome. Symptoms associated with the defect depend on the site and severity of stenosis.
Coarctation of the Aorta
Coarctation of the aorta (COA) is a narrowing of the lumen of the aorta that impedes blood flow. This defect accounts for 8% to 10% of all congenital heart defects. COA is almost always in a juxtaductal position, although it can occur anywhere between the origin of the aortic arch and the bifurcation of the aorta in the lower abdomen. About 50% of individuals with COA have a bicuspid aortic valve (Figure 31-11).2
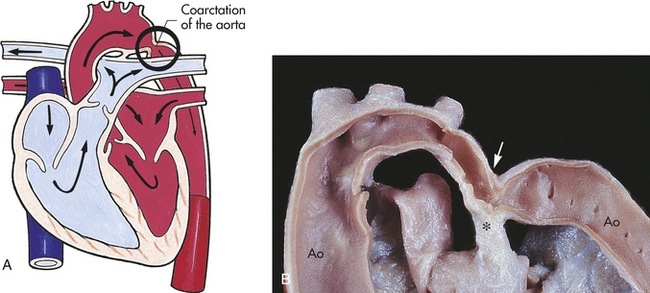
Figure 31-11 Postductal and preductal coarctation of the aorta. A, Postductal coarctation occurs distal to (“after”) the insertion of the closed ductus arteriosus into the aortic arch. Preductal coarctation occurs proximal to (“before”) insertion of the patent ductus arteriosus. The coarctation consists of a flap of tissue that protrudes from the tunica media of the aortic wall. B, Coarctation of the aorta with typical indentation of the aortic wall (arrow) opposite the ductal arterial ligament (∗). Ao, Aorta. (A from Hockenberry MJ et al: Wong’s essentials of pediatric nursing, ed 8, St Louis, 2009, Mosby; B from Damjanov I, Linder J, editors: Anderson’s pathology, ed 10, St Louis, 1996, Mosby.)
PATHOPHYSIOLOGY COA may develop because of an abnormal contractile ductal tissue that constricts at the time of ductal closure. COA causes a condition in which there are higher pressures proximal to the site of stenosis and lower pressures distal to the site. In preductal COA the RV acts as a systemic pump, sending unoxygenated blood through the ductus into the descending aorta below the coarctation (Figure 31-12). In postductal COA the RV cannot pump enough blood through the ductus to the descending aorta because of pressure caused by the narrowed aorta. Systolic pressures increases in the ascending aorta and LV and decreases in the descending aorta beyond the COA (Figure 31-13). Long-standing COA, collateral circulation, which involves small arteries arising from the subclavian arteries, joins intercostal arteries that flow into the descending aorta. These collateral vessels bypass the COA and supply blood to the lower extremities. The direction of shunting through the ductus, if present, depends on the pressure difference between the PA and aorta and the location of the ductus. When blood pressure is greater in the aorta than in the PA, blood flow through the ductus will be left to right toward the lungs, resulting in increased pulmonary venous return to the left side of the heart. This may place an additional strain on the LA and LV, leading to increased volume and work. With time LV hypertrophy develops because of increased afterload and obstruction to flow caused by the coarctation. HF also may develop.
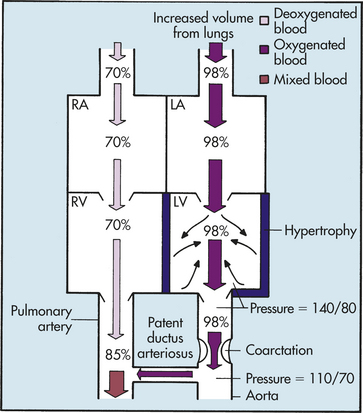
Figure 31-12 Hemodynamics of preductal coarctation of the aorta with a patent ductus arteriosus. The left-to-right shunt through the ductus arteriosus increases the volume of blood in the pulmonary circulation. Afterload (small black arrows) is increased in the left heart by (1) increased return from the lungs and (2) decreased ventricular outflow caused by the coarctation. The outcome is heart failure. LA, left atrium; LV, left ventricle; RA, right atrium; RV, right ventricle.
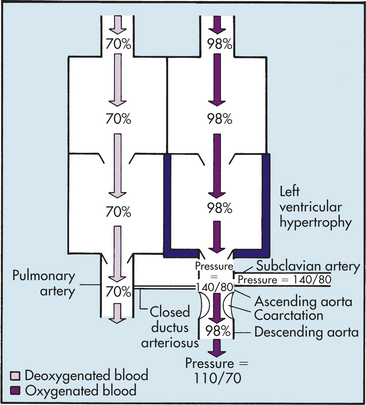
Figure 31-13 Hemodynamics of postductal coarctation of the aorta. Blood pressure increases in the ascending aorta and subclavian artery and decreases in the descending aorta. These pressure changes eventually occur in the parts of the systemic circulation served by arteries that branch from the aorta before and after the coarctation.
CLINICAL MANIFESTATIONS Clinical manifestations vary depending on the severity of the coarctation and age of presentation. In newborns the onset of symptoms depends on the timing of ductal closure after a fall in pulmonary vascular resistance, the location of the COA, and the presence of associated defects. The newborn usually presents with CHF symptoms. Once the ductus closes, these infants will deteriorate rapidly from the development of hypotension, acidosis, and shock. Older children may not be diagnosed until hypertension is noted. Hypertension is noted in the upper extremities with decreased or absent pulses in the lower extremities. Children may have cool mottled skin and occasionally leg cramps during exercise. A systolic ejection murmur, heard best at the left interscapular area, is caused by rapid blood flow through the narrowed area.
EVALUATION AND TREATMENT A chest radiograph shows an enlarged heart with congested lung fields in newborns. Rib notching between the fourth and eighth ribs may be seen in children older than 5 years, reflecting erosion of the ribs from enlarged collateral vessels from the ascending aorta to the descending aorta, bypassing the coarctation. An ECG may be normal or reveal LV hypertrophy. An echocardiogram will confirm the diagnosis and rule out other intracardiac defects. Cardiac catheterization and/or magnetic resonance imaging (MRI) is performed only if the echocardiogram is inconclusive.
The first step in treatment of the symptomatic infant is stabilization, which may require prostaglandin administration, mechanical ventilation, and inotropic support to maintain adequate cardiac output. Once this is achieved, surgical intervention is indicated. Surgical repair for infants younger than 1 year consists of either a subclavian flap aortoplasty technique to enlarge the constricted area or resection with end-to-end anastomosis of the arch segments. Depending on the arch morphology, a modification of this procedure enlarges the aorta beyond the area of constriction. For children older than 1 year, surgical repair consists of resection with end-to-end anastomosis.13 Cardiopulmonary bypass is not required because of the extracardiac nature of the lesion, and the approach is accomplished through a left thoracotomy.
Postoperative complications include recoarctation and paradoxical postoperative hypertension. Residual permanent hypertension requiring continued medical therapy is related to age at repair; therefore, surgical intervention is recommended at the time of diagnosis. Operative mortality for infants is less than 5%, and for children older than 1 year, it is less than 1%. Balloon dilation angioplasty in newborns has been successfully performed. However, aortic aneurysm formation and restenosis have been noted; therefore, surgical repair remains the correction of choice for the newborn.13–16
Aortic Stenosis
Aortic stenosis (AS) is a narrowing of the aortic outflow tract (Figure 31-14). The lesion accounts for 5% of all congenital heart defects.2 Valvular stenosis is caused by malformation or fusion of the cusps. It is the most common type of AS, tends to be progressive, and, in rare cases, can lead to sudden death as a result of low cardiac output or myocardial ischemia. For children with mild AS, no exercise restrictions may be needed. For those with moderate AS, some exercise limitations may be advised. Severe AS is an indication for limiting exercise until the repair is accomplished. Less common forms of AS are subvalvular stenosis caused by a constricting fibrous ring below the valve and supravalvular stenosis that occurs above the valve.7
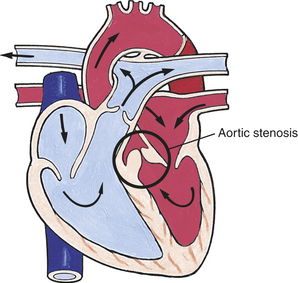
Figure 31-14 Aortic stenosis. Narrowing of the aortic valve causing resistance to blood flow in the left ventricle, decreased cardiac output, left ventricular hypertrophy, and pulmonary congestion. (From Hockenberry MJ et al: Wong’s essentials of pediatric nursing, ed 8, St Louis, 2009, Mosby.)
PATHOPHYSIOLOGY Obstruction to blood flowing out of the aorta causes an increased workload on the LV, resulting in left ventricle hypertrophy (LVH). LV failure may develop, leading to an increase in LA pressure and a backup in the system, eventually resulting in pulmonary vascular congestion and pulmonary arterial hypertension. LVH can decrease coronary artery perfusion, resulting in myocardial ischemia and it can alter the LV papillary muscle, causing mitral insufficiency.
CLINICAL MANIFESTATIONS Most children with mild to moderate AS are asymptomatic. Signs of exercise intolerance may not appear until preadolescence. Syncopal episodes, epigastric pain, and exertional chest pain may occur in more severe forms of AS. A systolic ejection murmur at the right upper sternal border that transmits to the neck and left lower sternal border is produced by blood flow through the stenotic area. An ejection click may be heard with valvular AS. Severe forms of AS, especially critical AS in the newborn, result in shock and require immediate intervention.
EVALUATION AND TREATMENT Diagnosis may be made based on previous medical history and physical findings. Chest radiographic examination may reveal a dilated ascending aorta or LV enlargement. Increased pulmonary vascular markings may be seen in severe forms. In mild cases the ECG is normal. LVH with strain pattern (features of LVH with inverted T waves in the lateral precordial leads) may be seen in severe forms. An echocardiogram may reveal a thickened and poorly functioning LV with abnormal closure of the damaged bicuspid aortic valve. Cardiac catheterization may be performed to augment echocardiographic data or intervention and can determine the location, cause, and severity of obstruction.
The presence of ST-segment changes on ECG, severe CHF, and evidence of discrete severe stenosis at the aortic outflow tract are indications for intervention. Balloon aortic valvuloplasty is a palliative procedure performed for valvular AS; however, it is associated with complications, including aortic insufficiency and dysrhythmia.17 Aortic valvotomy, under inflow occlusion or cardiopulmonary bypass, is performed for valvular AS. Operative mortality remains high in infants (up to 20%), although older children have a mortality close to 0%.13 As many as 25% of individuals require a second surgery within 10 years for restenosis, at which time valve replacement may be the procedure of choice.
Subvalvular AS and supravalvular AS require surgical repair involving excision of the area causing the constriction. For subvalvular AS involving a small LV outflow tract and aortic annulus, a Konno procedure may be done to enlarge the LV outflow tract and aortic annulus with a patch.13,18
Pulmonary Stenosis
Pulmonary stenosis (PS) is the narrowing of the pulmonary outflow tract. This may be in the form of abnormal thickening of the valve leaflets or narrowing of the arterial (supravalvular) or ventricular (subvalvular) side of the valve (Figure 31-15,A). Pulmonary atresia is the severe form of PS and involves complete fusion of the commissures, allowing no blood flow out of the RV to the PA. PS accounts for 5% to 8% of all congenital heart defects.2
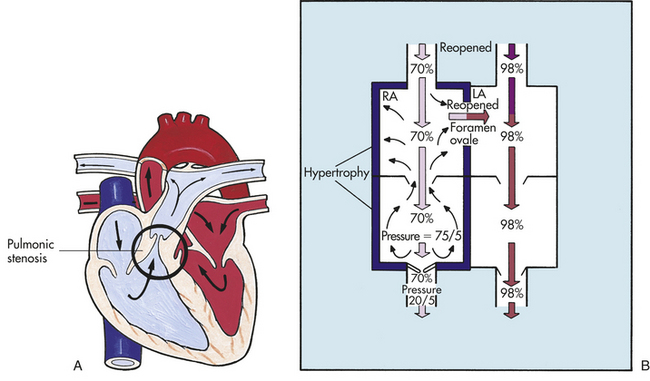
Figure 31-15 Pulmonary stenosis. A, Obstruction of right ventricular outflow caused by pulmonary stenosis. Pressure on the ventricular side of the pulmonic semilunar valve (pulmonary valve) is much greater than that on the pulmonary arterial side. This difference disrupts the normal pressure gradient across the valve. Pulmonary stenosis increases ventricular afterload by decreasing blood flow through the valve, which causes ventricular hypertrophy. B, The backup of ventricular afterload into the right atrium reopens the foramen ovale. Venous blood then flows from the area of higher pressure (the right atrium) to the area of lower pressure (the left atrium), causing a left-to-right shunt. Cyanosis occurs if enough venous blood shunts from right to left to reduce oxygen saturation in the systemic circulation by 3% to 5%. LA, left atrium; RA, right atrium. (A from Hockenberry MJ et al: Wong’s essentials of pediatric nursing, ed 8, St Louis, 2009, Mosby.)
PATHOPHYSIOLOGY PS creates resistance to blood flow from the RV to the PA. The narrowed orifice (valve) produces increased resistance (afterload) to ejection. In order for the RV to maintain adequate cardiac output, the myocardium hypertrophies. If the RV outflow tract obstruction is severe, blood may back up into the RA, causing dilation. This may result in reopening of the foramen ovale with resultant unoxygenated blood shunting to the LA, causing cyanosis (see Figure 31-15,B).
CLINICAL MANIFESTATIONS Clinical manifestations depend on the severity of PS. A systolic ejection murmur at the left upper sternal border reflects obstruction to flow through the narrowed pulmonary valve. A systolic ejection click is present with valvular stenosis at the upper left sternal border. A thrill also may be palpated at the upper left sternal border. Children with moderate PS may have exertional dyspnea and fatigability because of the inability of the body to increase pulmonary blood flow to meet demands for increased cardiac output. Severe PS will produce cyanosis and HF.
EVALUATION AND TREATMENT A chest radiograph shows a normal-size heart with a prominent main PA caused by poststenotic dilation. An ECG is usually normal but may reveal right axis deviation and RV hypertrophy with moderate PS. Echocardiography confirms the diagnosis and detects associated defects. Cardiac catheterization, if performed for interventional purposes, further demonstrates PA anatomy.
Mild to moderate PS will not likely require intervention but should be observed closely with prophylaxis for subacute bacterial endocarditis. Most mild PS is not progressive. Treatment is indicated when a significant pressure gradient is detected across the RV outflow tract.
Critical (severe) PS must be addressed immediately. The treatment of choice is balloon angioplasty. This procedure is considered highly effective in decreasing the pressure gradient across the pulmonic valve and is noted to have few associated complications.19 Surgical correction involves a pulmonary valvotomy incising the fused commissures. Operative mortality is less than 1%.2 Both valvotomy and balloon angioplasty may result in some pulmonary valve incompetence, and long-term follow-up may reveal the need for further intervention.
Hypoplastic Left Heart Syndrome
Hypoplastic left heart syndrome (HLHS) refers to the abnormal development of the left-sided cardiac structures, resulting in obstruction to blood flow from the LV outflow tract. HLHS involves underdevelopment of the LV, aorta, and aortic arch, as well as mitral atresia or stenosis (Figure 31-16). Therefore, infants with HLHS must have a well-functioning RV and the presence of a PDA and atrial septal communication for survival. HLHS accounts for 1% of all congenital heart defects and is considered the most complex congenital defect.3
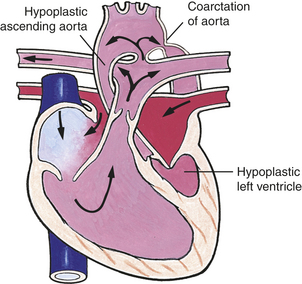
Figure 31-16 Hypoplastic left heart syndrome. (From Hockenberry MJ et al: Wong’s essentials of pediatric nursing, ed 8, St Louis, 2009, Mosby.)
PATHOPHYSIOLOGY Because of the high pressures caused by LV outflow tract obstruction, saturated blood enters the LA and mixes with desaturated blood in the RA through an atrial septal communication. Blood flow follows the normal pathways through the right side of the heart. Exiting the PA, the mixed-saturation blood flows through the ductus and to the descending aorta. The amount of blood flow that travels to the pulmonary and systemic circulations depends on vascular resistance in the respective systems. Retrograde blood flow through the hypoplastic ascending aorta provides coronary and cerebral blood flow.
CLINICAL MANIFESTATIONS Newborns with HLHS generally are born full term and initially appear healthy. As the ductus closes, systemic perfusion is decreased, resulting in hypoxemia, acidosis, and shock. Usually no heart murmur is detected. The second heart sound is loud and single because of aortic atresia.
EVALUATION AND TREATMENT A chest radiograph shows cardiomegaly and increased pulmonary venous congestion. ECG shows RV hypertrophy and diminished left-sided forces. Echocardiography reveals the components of the defect with a diminutive LV cavity, hypoplastic aortic valve and arch, and hypoplastic or absent mitral valve with an enlarged RV cavity. Interventional cardiac catheterization with balloon septostomy may be necessary if the atrial communication is inadequate. This procedure is very high risk.7
Prostaglandin infusion to maintain patency of the ductus arteriosus is essential for newborn infant survival. Immediate correction of acidosis, inotropic support for adequate cardiac output, and ventilatory manipulation to balance systemic and pulmonary blood flow prevent further deterioration and achieve stabilization.
Surgical intervention includes a three-stage approach that classically begins with a Norwood procedure. The Norwood procedure consists of an atrial septectomy, placement of a pulmonary-to-systemic artery shunt to maintain adequate pulmonary blood flow, creation of a permanent communication between the RV and aorta, and patch augmentation of the hypoplastic aorta. The Sano modification utilizes a graft between the RV and the PA to provide stable pulmonary blood flow rather than a pulmonary to systemic arterial shunt.20 Postoperative complications include imbalance of systemic and pulmonary blood flow, leading to inadequate cardiac output and persistent HF. In most centers, survival after the initial procedure is now averaging greater than 70%.
The second stage is the bidirectional Glenn procedure, which is performed between 2 and 9 months of age, depending on the child’s clinical status, pulmonary vascular resistance, and ventricular function. This involves joining the superior vena cava to the PA and take-down of the shunt to the lungs. Complications include superior vena cava obstruction, pleural effusion, and low cardiac output.
The third stage is the Fontan procedure, described earlier, which separates the systemic from the pulmonary circulation. Timing for surgical repair depends on the child’s ventricular function, presence of AV valve regurgitation, and pulmonary vascular resistance. Most surgeons perform the Fontan procedure when the child is approximately 2 to 4 years of age.7,13
Cardiac transplant also may be an option for these newborns. Most surgical centers offer the three-staged palliative surgeries as an initial approach, with an option for cardiac transplantation later in life.7,21
Mixing Defects
Many complex defects are classified as mixing defects because of their dependence on the mixing of pulmonary and systemic circulations for survival during the postnatal period. This mixing results in desaturated systemic blood flow and cyanosis. Pulmonary congestion occurs because of preferential pulmonary blood flow. Clinically each defect has varying degrees of cyanosis and HF depending on the various components of the lesion.
Transposition of the Great Arteries
Transposition of the great arteries (TGA) refers to a condition in which the aorta arises from the RV and the PA from the LV (Figure 31-17,A). The result is two separate, parallel circuits in which unoxygenated blood circulates continuously through the systemic circulation and oxygenated blood circulates repeatedly through the pulmonary circulation. This condition is incompatible with extrauterine life unless a communication exists between the two circuits to provide the necessary oxygen to the body. Communication is accomplished through mixing of pulmonary and systemic circulations through a PDA, ASD, or VSD (see Figure 31-17, B). Dextro-transposition of the great arteries (d-TGA) is the most common cyanotic congenital heart defect and accounts for 10% of all congenital heart defects; “dextro” refers to the aorta remaining to the right of the PA.2
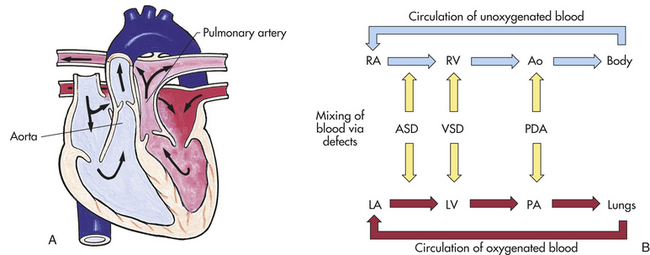
Figure 31-17 Hemodynamics in transposition of the great arteries (TGA). A, Complete transposition of the great vessels with an intact interventricular septum. The aorta arises from the right ventricle and the pulmonary artery from the left. B, Oxygen saturation in the two parallel circuits. Ao, Aorta; ASD, atrial septal defect; LA, left atrium; LV, left ventricle; PA, pulmonary artery; PDA, patent ductus arteriosus; RA, right atrium; RV, right ventricle; VSD, ventricular septal defect. (A from Hockenberry MJ et al: Wong’s essentials of pediatric nursing, ed 8, St Louis, 2009, Mosby.)
Two factors allow newborns with complete transposition to survive long enough to be treated. First, blood from the two closed systems can mix through the ductus arteriosus for a short time after birth if pulmonary vascular resistance remains high. Some mixing also may occur through the foramen ovale. If the child has a VSD, mixing occurs through that opening as well.
PATHOPHYSIOLOGY It is not known precisely which embryologic events lead to transposition, but researchers have proposed that the fault lies in the development of conal tissue in the fibrous skeleton of the heart.1 The conus is a segment of muscle that separates the AV (tricuspid and mitral) valves from the semilunar (aortic and pulmonic) valves. (The fibrous skeleton and heart valves are described and illustrated in Chapter 29; see Figure 29-4.) The interventricular septum is intact in about 60% of cases of transposition; a VSD is present in the remaining 40%. PS is associated with the transposition in about 4% to 6% of children with intact septa and in 28% to 31% of children with VSDs.2,7
The discussion that follows is limited to the pathophysiology of complete transposition with an intact interventricular septum.
CLINICAL MANIFESTATIONS The degree of mixing permitted by fetal structures determines the type and severity of clinical manifestations. Cyanosis may be mild shortly after birth and worsen during the first day because of functional closure of the ductus arteriosus. Low oxygen levels in the blood (hypoxemia) cause metabolic acidosis, tachycardia, and tachypnea. The presence of a PDA or large ASD allows for more mixing and results in only mild cyanosis, but the infant may develop CHF.
The first heart sound is normal, and the second sound may be heard as a single sound even though both the aortic and pulmonic valves are functioning. The single S2 may occur because transposition places the aortic valve closer to the chest wall than the pulmonic valve. No murmur is noted with transposition of the great arteries with an intact ventricular septum.
EVALUATION AND TREATMENT On chest radiograph the heart has a characteristic shape—like an egg on its side—and pulmonary vascular markings are increased. The heart may be enlarged if the infant is a few weeks old and has a VSD. ECG findings reveal a right-axis deviation and some RV hypertrophy. Echocardiography confirms the diagnosis of transposition of the great arteries. Cardiac catheterization may be necessary to define the coronary anatomy; measure ventricular ratios; and, if need be, perform a balloon septostomy. Recent reports have raised concerns about the relationship between balloon septostomy and stroke; therefore, balloon septostomy is done only if the situation is critical.22
Surgical repair during the newborn period involves the arterial switch operation that moves the great arteries. The coronary arteries are removed from the aorta before the arterial switch is performed and reimplanted without torsion or kinking into the aorta. This establishes normal blood flow with the LV as the systemic pump. Results are approaching 100% survival. Complications include narrowing at the sites of the great artery anastomoses and, rarely, coronary insufficiency.7,22
Mustard and Senning operations (the creation of an intra-atrial tunnel to baffle the systemic venous blood flow to the mitral valve and the pulmonary venous blood flow to the tricuspid valve) are no longer the procedures of choice because the RV must perform as the systemic pump. Long-term follow-up of children with Mustard and Senning operations revealed significant rates of RV failure and dysrhythmias.7
The Rastelli procedure is used with children with transposition, VSD, and severe PS. This procedure involves closing the VSD with a baffle by rerouting LV blood through the VSD to the aorta. The pulmonary valve is closed, and an RV-to-PA prosthetic or homograft valve conduit is placed. This procedure requires prosthetic conduit replacement as the child grows and is associated with ventricular failure and dysrhythmias in the postoperative period.
Total Anomalous Pulmonary Venous Connection
Total anomalous pulmonary venous connection (TAPVC), or total anomalous pulmonary venous return, occurs when the pulmonary veins abnormally connect to the right side of the heart either directly or through one or more systemic veins that drain into the RA (Figure 31-18). An ASD generally is present also. This defect is extremely rare, accounting for only 1% of all congenital heart defects. The four types of TAPVC are based on the site of drainage. Supracardiac TAPVCs are the most common form (50%) and drain to the superior vena cava through the vertical or innominate vein. Cardiac TAPVCs (20%) drain directly into the RA or through the coronary sinus. Infracardiac TAPVCs (20%) traverse the diaphragm and drain into the portal or hepatic vein or the inferior vena cava. Mixed TAPVCs (10%) are a combination of the various types. Partial anomalous venous connection is a condition in which only one or a few of the pulmonary veins, usually the right-sided veins, drain into the RA or one of its tributaries.2
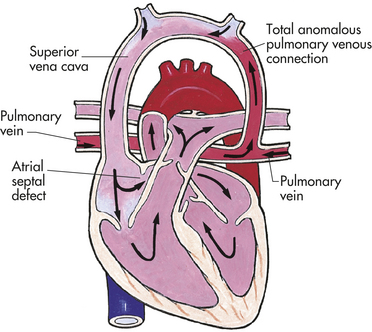
Figure 31-18 Hemodynamics of total anomalous pulmonary venous connection (TAPVC). In the form of TAPVC represented here, the pulmonary veins enter the left anomalous vertical vein instead of the left atrium. From the left anomalous vertical vein, the mixed blood from the lungs flows into the superior vena cava through an innominate vein (literally, a “vein without a name”). Oxygen saturation within the four heart chambers, the pulmonary artery, and the aorta is the same. Blood pressure in the right heart exceeds that in the left heart because the right heart is receiving blood from both the pulmonary and systemic circulatory systems. (Abnormal vessels are shaded.) (From Hockenberry MJ et al: Wong’s essentials of pediatric nursing, ed 8, St Louis, 2009, Mosby.)
PATHOPHYSIOLOGY Physiologically TAPVC can be differentiated into two groups: nonobstructive and obstructive, depending on the absence or presence of obstruction to pulmonary venous drainage. The hemodynamics of the nonobstructive group involve the RA receiving the oxygenated blood that would normally flow into the LA. The amount of blood shunted into the LA versus the volume entering the RV depends on the size of the ASD and compliance of the RV. Therefore, if the ASD is restrictive and RV compliance approaches normal, more blood will enter the RV than the LA, resulting in RA and RV enlargement, as well as increased pulmonary blood flow. This causes increased pulmonary venous blood return and larger amounts of saturated blood. If the ASD is unrestrictive and the RV does not thin out to increase compliance, the majority of mixed saturated blood is shunted from the higher pressure RA to the LA.
The hemodynamics of obstructed TAPVC cause pulmonary venous hypertension because of resistance caused by the obstruction resulting in an elevation in pulmonary vascular and RV pressures. Pulmonary edema occurs from hydrostatic capillary pressure exceeding the osmotic pressure of the blood and eventually contributing to the development of HF. This group has a strong association with the infracardiac type of TAPVC and is a surgical emergency.
CLINICAL MANIFESTATIONS The predominant clinical manifestation in infants with TAPVC is cyanosis caused by mixture of oxygenated and deoxygenated blood entering the systemic circulation. The degree of cyanosis is inversely related to the amount of pulmonary blood flow. Children with unobstructed TAPVC may be asymptomatic until pulmonary vascular resistance drops, at which time pulmonary blood flow will increase, resulting in signs of pulmonary overcirculation, particularly growth retardation and frequent pulmonary infections, in addition to mild cyanosis. Obstructed TAPVC results in cyanosis and rapid deterioration necessitating immediate surgical correction, or death will occur.
Physical examination may reveal a systolic murmur at the left upper sternal border and a mid-diastolic murmur at the left lower sternal border. A murmur may be absent in obstructed TAPVC. A characteristic quadruple rhythm, consisting of S1, widely split S2, and S3 or S4, or a gallop rhythm also is present.
EVALUATION AND TREATMENT The ECG shows a right-axis deviation, RV hypertrophy, and occasionally RA hypertrophy. The chest radiograph of unobstructed TAPVC reveals cardiomegaly, increased pulmonary vascular markings, and a snowman or figure-eight appearance in the supracardiac type. A chest roentgenogram of obstructed TAPVC shows a normal-size heart and a ground-glass appearance of the lung fields, reflecting pulmonary venous congestion or edema. The echocardiogram reveals the abnormal pulmonary venous connections.
Surgical repair varies with the type of TAPVC and whether the defect is obstructed or unobstructed. Obstructed lesions are repaired at the time of diagnosis, whereas the unobstructed type generally is repaired during infancy. The procedure is performed on cardiopulmonary bypass and involves anastomosis of the common pulmonary vein to the LA; ligating the common pulmonary vein; and closing the ASD, as in the supracardiac and infracardiac types. Repair of the supracardiac type involves baffling the pulmonary venous drainage to the LA. This repair has the highest success rate because of the low technical difficulty, whereas infracardiac repair is associated with a high mortality (up to 25%) and morbidity. Potential complications include reobstruction; atrial dysrhythmias, including sick sinus syndrome; PA hypertension; and LV dysfunction.13
Truncus Arteriosus
Truncus arteriosus is the failure of the large embryonic artery, the truncus arteriosus, to divide into the PA and the aorta. This results in a single vessel arising from both ventricles, providing blood flow to the pulmonary and systemic circulations (Figure 31-19,A). This common trunk straddles the VSD (always present) and has a single valve with three or four leaflets, which may result in stenosis or regurgitation, or both. The incidence is 2% of all congenital heart defects, and a right aortic arch is present 50% of the time. There are four types of truncus arteriosus. Type I is the most common (60%) and involves the main PA arising from the truncus and then dividing into the right and left PAs. Type II is less common (20%) and involves the PAs arising from the posterior aspect of the truncus. Type III is the least common (10%) and involves the PAs arising from the lateral aspect of the truncus. Type IV, also known as pseudotruncus, is now considered a severe form of TOF with the bronchial arteries arising from the descending aorta to supply the lungs.2
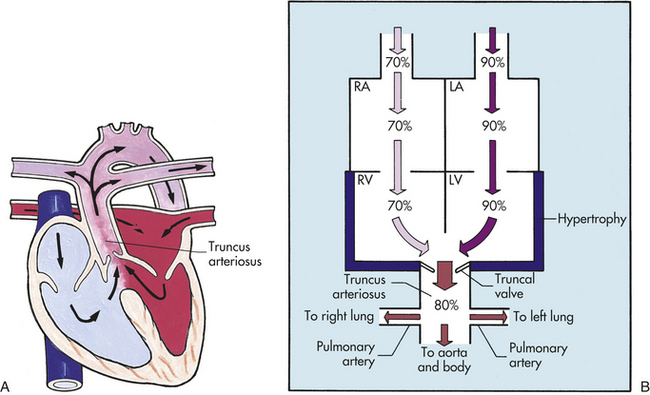
Figure 31-19 Truncus arteriosus. A, Persistent truncus arteriosus. The truncus arteriosus fails to divide into the pulmonary artery and aorta, and the interventricular septum fails to close at the top. Blood from both ventricles mixes in the truncus arteriosus and then enters the pulmonary and systemic circuits. B, Alterations of hemodynamics and oxygen saturation by persistent truncus arteriosus. LA, Left atrium; LV, left ventricle; RA, right atrium; RV, right ventricle. (A from Hockenberry MJ et al: Wong’s essentials of pediatric nursing, ed 8, St Louis, 2009, Mosby.)
PATHOPHYSIOLOGY Blood flow from the RV and LV is pumped into the main truncus, resulting in mixing of the pulmonary and systemic circulations (see Figure 31-19,B). The differential flow out to either the pulmonary bed or the systemic circulation depends on the pulmonary and systemic vascular resistances. Generally the pulmonary vascular resistance is less than the systemic vascular resistance, resulting in the majority of blood flow traveling to the lungs. This may be altered, however, because of PS, small pulmonary arteries, or increased pulmonary vascular resistance. Pulmonary vascular disease develops early with this defect because of increased pulmonary blood flow.
CLINICAL MANIFESTATIONS Physical findings depend on the amount of pulmonary blood flow and the presence of other cardiac anomalies. If PS is present, a newborn will present with cyanosis, caused by already elevated pulmonary vascular resistance, but no HF. Conversely, if PS is not present, the newborn initially will have mild to moderate cyanosis that worsens with activity. Once pulmonary vascular resistance drops, the pulmonary bed will receive preferential flow and the infant will have signs of HF. A wide pulse pressure with bounding pulses also may be present, caused by increased pulmonary blood flow. A harsh systolic regurgitant murmur is present along the left sternal border as a result of the VSD, and a systolic click at the apex and left upper sternal border may be present, reflecting opening of the truncal valve. An apical rumble with or without a gallop rhythm also may be present because of increased pulmonary blood flow. If truncal valve insufficiency exists, an early diastolic, high-pitched decrescendo murmur may be present.
EVALUATION AND TREATMENT An ECG generally reveals biventricular hypertrophy and occasionally LA enlargement. A chest radiograph reveals cardiomegaly with biventricular and LA enlargement, as well as increased pulmonary vascular markings. When PS is present, the heart size is normal and the pulmonary vascular markings are decreased. Echocardiography and cardiac catheterization, if performed, determine the type of truncal defect, competency of the truncal valve, and differential blood flow.
Surgical repair in early infancy is recommended to prevent the sequelae of severe HF and pulmonary vascular disease. The definitive repair consists of a modified Rastelli procedure involving VSD patch closure to divert the blood flow from the LV outflow tract into the truncus. The pulmonary arteries are excised from the aorta and connected to the RV through a tissue homograft—namely, aortic and PA segments or cadaver tissue that is specially preserved. Synthetic conduits may be used but tend to calcify and develop narrowing within the lumen, leading to obstruction and the need for early replacement. Mortality varies depending on the type of truncal anomaly (20% to 50%). Postoperative complications include HF, residual VSD, dysrhythmias, and pulmonary hypertension. The RV to PA homograft requires replacement because it becomes inadequate for somatic growth.13
ACQUIRED CARDIOVASCULAR DISORDERS
Acquired heart diseases are those disease processes or abnormalities that occur after birth. They result from various causes, such as infection, genetic disorders, autoimmune processes in response to infection, environmental factors, or autoimmune diseases. Examples of acquired heart diseases include Kawasaki disease, myocarditis, rheumatic heart disease, cardiomyopathy, and systemic hypertension. This chapter discusses Kawasaki disease and systemic hypertension. Myocarditis, rheumatic heart disease, and cardiomyopathy are discussed in Chapter 30.
Kawasaki Disease
Kawasaki disease, otherwise known as mucocutaneous lymph node syndrome, is an acute, self-limiting systemic vasculitis that may result in cardiac sequelae. Although Kawasaki disease occurs throughout the world, the greatest number of cases are reported in Japan.
Kawasaki disease is primarily a condition of young children: 80% of cases are seen in children younger than 5 years of age, with the incidence peaking in the toddler age group. Males are affected slightly more than females. Its peak incidence is in winter and spring.
The etiology of Kawasaki disease remains unknown. Theories center on an immunologic response to an infectious, toxic, or antigenic substance (including superantigen).2,7,23
PATHOPHYSIOLOGY Kawasaki disease progresses pathologically and clinically in the following stages:
Stage I (days 1 to 12): Small capillaries, arterioles, and venules become inflamed, as does the heart itself.
Stage II (days 13 to 25): Inflammation spreads to larger vessels, and aneurysms of the coronary arteries develop.
Stage III (days 26 to 40): Medium-size arteries begin the granulation process, causing coronary artery thickening; inflammation resolves in the microcirculation; and there is increased formation of thrombi.
Stage IV (day 41 and beyond): Vessels develop scarring, intimal thickening, calcification, and stenosis of coronary arteries.
CLINICAL MANIFESTATIONS The clinical course of the disease progresses in three stages: acute, subacute, and convalescent. In the acute phase the child has fever, conjunctivitis, oral changes (“strawberry” tongue), rash, and lymphadenopathy and is often irritable. During this phase myocarditis may develop. The subacute phase begins when the fever ends and continues until the clinical signs have resolved. It is at this time that the child is most at risk for coronary artery aneurysm development. Desquamation of the palms and soles occurs at this time, as well as marked thrombocytosis. The convalescent phase is marked by the continued elevation of the erythrocyte sedimentation rate and platelet count. Arthritis still may be present. This phase continues until all laboratory values return to normal—usually about 6 to 8 weeks after onset.
EVALUATION AND TREATMENT The diagnosis is based on the diagnosis criteria for Kawasaki disease, which state that the child must exhibit five of six criteria, including fever (Box 31-3). These children usually have leukocytosis, increased erythrocyte sedimentation rates, marked thrombocytosis, and elevated liver enzymes. An echocardiogram is obtained at the time of diagnosis as a baseline to assess for coronary aneurysms or inflammation. Serial echocardiograms are obtained after treatment to assess for future development of coronary aneurysms.
The use of aspirin and intravenous immunoglobulin during the acute phase has decreased the mortality of Kawasaki disease and has reduced the incidence of coronary abnormalities from approximately 20% to less than 2% at 6 to 8 weeks after initiation of therapy. Most children recover completely from Kawasaki disease, including the regression of aneurysms. The most common cardiovascular sequela is coronary thrombosis. Studies are investigating long-term results of the disease.23
Systemic Hypertension
Hypertension (HTN) in children differs from adult HTN in etiology and presentation. Children diagnosed with HTN are often found to have some underlying disease, such as renal disease or COA (Box 31-4). In recent years an increased prevalence of primary HTN in older children has been noted. Researchers are now focusing on primary HTN in older children in relation to morbidity and mortality and the presence of early atherosclerotic disease.2,7,24,25
Systemic hypertension in children is defined as systolic and diastolic blood pressure levels greater than the 95th percentile for age and gender on at least three occasions. The Fourth Task Force on Blood Pressure Control in Children uses height as an additional criterion to the blood pressure guide.26
PATHOPHYSIOLOGY Hypertension is classified as (1) primary (or essential) hypertension, in which a specific cause cannot be identified; or (2) secondary hypertension, in which a cause is secondary to another alteration (see Box 31-4). In infants and children a cause of HTN is almost always found. In general, the younger the child with significant hypertension, the more likely that a correctable cause can be found. Therefore, a thorough evaluation needs to be done.27
The pathophysiology of primary HTN in children is not clearly understood but may result from a complex interaction of a strong disposing genetic component with disturbances in sympathetic vascular smooth muscle tone, humoral agents (angiotensin, catecholamines), renal sodium excretion, and cardiac output (Figure 31-20). Ultimately these factors impair the ability of the peripheral vascular bed to adjust its own resistance to meet tissue perfusion needs.24
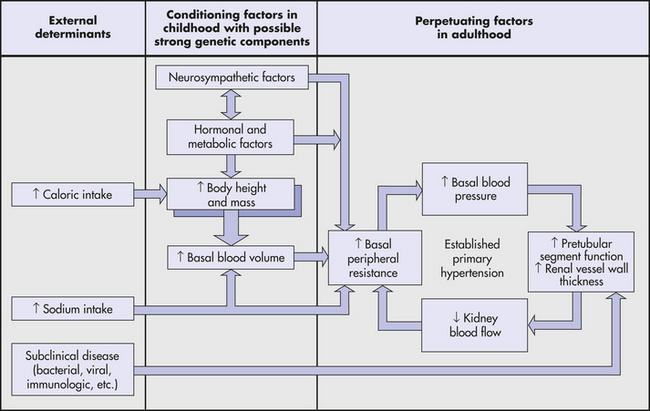
Figure 31-20 Mechanisms believed to influence blood pressure in children. According to this model, a critical factor in the development of hypertension is obesity during childhood. Increased body mass, coupled with excessive sodium intake, can cause primary hypertension in children or set the stage for its development later in life.
CLINICAL MANIFESTATIONS Most children with systemic HTN are asymptomatic. It is necessary that a thorough history and physical examination be obtained. The examination should include an accurate blood pressure measurement on three separate occasions using a cuff of appropriate size (Tables 31-6 and 31-7).
Table 31-6
Suggested Normal BP Values (mmHg) by Auscultatory Method (Systolic/Diastolic K5)
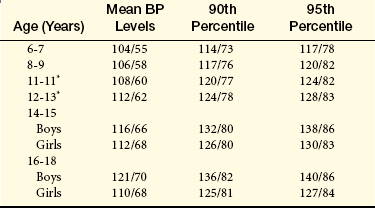
BP, Blood pressure; K5, phase V of Korotkoff sound.
∗Values for ages 10 to 13 years have been extrapolated from these two studies using age-related increments from other studies.
From Park MK: Pediatric cardiology for practitioners, ed 4, St Louis, 2002, Mosby; modified from Goldring D et al: J Pediatr 91:884, 1977; Prineas RJ et al: Hypertension 1 (Suppl):18, 1980.
Table 31-7
Normative BP Levels (Systolic/Diastolic [Mean]) by Dinamap Monitor in Children 5 Years Old and Younger
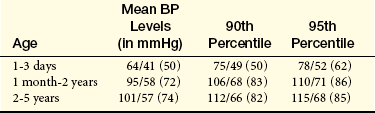
From Park MK: Pediatric cardiology for practitioners, ed 4, St Louis, 2002, Mosby; modified from Park MK, Menard SM: Am J Dis Child 143:860, 1989.
Certain factors influence blood pressure in children. Smoking is also associated with an increased risk for HTN. Obesity is emerging as one of the most important factors in the increasing prevalence of HTN in children.
EVALUATION AND TREATMENT In children the history and physical examination should be directed at determining the etiology of HTN, such as COA or renal disease (Table 31-8). If COA is found, surgical or interventional correction is initiated. A complete blood count, serum chemistry levels, urinalysis, urine culture, lipid profile, and renal ultrasound are part of the routine evaluation for renal disease (Table 31-9).
Table 31-8
Most Common Causes of Chronic Sustained Hypertension
| Age Group | Causes |
| Newborn | Renal artery thrombosis, renal artery stenosis, congenital renal malformation, COA, bronchopulmonary dysplasia |
| <6 years | Renal parenchymal disease, COA, renal artery stenosis |
| 6-10 years | Renal artery stenosis, renal parenchymal disease, primary hypertension |
| >10 years | Primary hypertension, renal parenchymal disease |
COA, Coarctation of the aorta.
From Park MK: Pediatric cardiology for practitioners, ed 4, St Louis, 2002, Mosby; modified from Report of the Second Task Force on Blood Pressure Control in Children. Pediatrics 79:1, 1987.
Table 31-9
Routine and Special Laboratory Tests for Hypertension

COA, Coarctation of the aorta; ECG, electrocardiogram.
Adapted from Park MK: Pediatric cardiology for practitioners, ed 4, St Louis, 2002, Mosby.
Although the criteria for the diagnosis of hypertension are based on the use of a standard blood pressure cuff, some children have what is termed “white coat hypertension.” Elevated blood pressure readings in these children occur only when measured in the clinic and may be caused by fear and anxiety. The use of ambulatory blood pressure monitoring (ABPM) records the blood pressure over a 24-hour period. In addition to identifying children with white coat hypertension, ABPM has been found to be useful in children with hypertension that is resistant to treatment (see What’s New? Ambulatory Blood Pressure Monitoring in Children).26
If HTN is found to be essential, or primary, in nature, nonpharmacologic therapy is used initially. Moderate weight loss can decrease systolic and diastolic pressures in many children. Appropriate diet, regular physical activity, and avoidance of smoking have been shown to be effective in reducing blood pressure.28
Drug therapy is controversial in children with primary hypertension; however, when nonpharmacologic therapy fails, a staged approach with the use of diuretics and/or beta-blockers and afterload reduction is indicated. The emphasis on preventive cardiology, especially for children, is significant because many investigators believe signs of atherosclerosis and other cardiovascular risk factors are present from childhood.29–32
Childhood Obesity
Childhood obesity is considered an epidemic in not only the United States but also in other countries such as Australia.33–35 Despite attention from U.S. federal and state initiatives, the prevalence of obesity in children and young adults has steadily increased over the past four decades, with estimates as high as 25% of children considered obese.33 Although not without controversy, percentile of body mass index (BMI) expressed as weight/height2 (BMI; kg/m2) is used to identify overweight and obesity in children and adolescents. The Centers for Disease Control and Prevention (CDC), the supplier of national growth charts and prevalence data, avoids characterizing children and adolescents as “obese”; instead, the CDC suggests two levels of overweight: (1) the 85th percentile, an “at risk” level; and (2) the 95th percentile, the more severe level.36
Causes of obesity in young children and adolescents are multivariable and multidimensional. Risk factors associated with developing childhood obesity include race, socioeconomic status, and lack of health insurance. Children of black and Hispanic race are at higher risk, as well as children with no insurance.34 The presence of parental obesity also is associated with childhood obesity.37 In addition, early childhood nutrition, level of physical activity, and engagement of sedentary activities, such as watching television and computer use, is associated with the development of overweight and obese children.38–40
Similar to obese adults, overweight and obese children are at risk for acquiring numerous other serious and potentially life-threatening illnesses such as asthma, sleep apnea, hypertension, type 2 diabetes, dyslipidemia, and cardiovascular disease.7 Researchers also have reported a multitude of social and economic consequences in adolescents as a result of being overweight. Overweight adolescents are more likely to complete fewer years of education, are less likely to marry, and have a lower household income in adulthood, independent of familial socioeconomic status.41
As in other acquired diseases, efforts should be focused on prevention. The initial approach is a combined program of physical activity with nutritional improvements. Healthcare professionals play a vital role in recognizing the need for intervention, immediate referral, and support. Successful outcomes for most overweight and obese children require support, change in lifestyle at home, and involvement of family members. Researchers are involving school-based programs in promoting and preventing obesity in the young.42
Aortic stenosis (AS) 1228
Atrial septal defect (ASD) 1219
Atrioventricular canal (AVC) defect 1222
Bulbus cordis 1211
Coarctation of the aorta (COA) 1226
Complete AVC (CAVC) defect 1222
Conus 1231
Cyanosis 1217
Ductus arteriosus 1211
Eisenmenger syndrome 1217
Endocardial cushion 1209
Foramen ovale 1211
Heart failure (HF) 1216
Hypoplastic left heart syndrome (HLHS) 1230
Kawasaki disease 1234
Ligamentum venosum 1212
Ostium primum 1210
Ostium secundum 1210
Partial AVC (PAVC) defect 1222
Patent ductus arteriosus (PDA) 1218
Pulmonary atresia 1229
Pulmonary stenosis (PS) 1229
Septum primum 1210
Septum secundum 1210
Systemic hypertension 1235
Tetralogy of Fallot (TOF) 1223
Total anomalous pulmonary venous connection (TAPVC) 1232
Transitional AVC (TAVC) defect 1222
Transposition of the great arteries (TGA) 1231
Tricuspid atresia 1225
Truncus arteriosus 1233
Ventricular septal defect (VSD) 1220
REFERENCES
1. Clark E.B., Nakazawa M., Takao A., eds. Etiology and morphogenesis of congenital heart disease: twenty years of progress in genetics and developmental biology. Mount Kisco, NY: Futura Publishing Company, 2000.
2. Park, M.K. Pediatric cardiology for practitioners, ed 5. Philadelphia: Mosby; 2008.
3. Wilson, D., Hockenberry, M.J. Wong’s clinical manual of pediatric nursing, ed 7. St Louis: Mosby; 2008.
4. Hazinski, M.F. Nursing care of the critically ill child, ed 2. St Louis: Mosby; 1991.
5. Gelb, D.B. Genetic basis of congenital heart disease. Curr Opin Cardiol. 2004;19(2):110–115.
6. American Heart Association. Heart disease and stroke statistics, 2004 update. Dallas: Author; 2004.
7. Allen, H.D. Moss and Adam’s heart disease in infants, children, and adolescents: including the fetus and young adult, ed 7. Philadelphia: Wolters Kluwer Health/Lippincott Williams & Wilkins; 2008.
8. Dimenna, L., et al. Management of the neonate with patent ductus arteriosus. J Perinat Neonat Nurs. 2006;20(4):333–340.
9. Bichell, D.P., et al. Minimal access approach for the repair of atrial septal defect: the initial 135 patients. Ann Thorac Surg. 2000;70(1):115–118.
10. Moodie, D.S., Sterba, R. Long-term outcomes excellent for atrial septal defect repair in adults. Cleve Clin J Med. 2000;67(8):591–597.
11. Knauth, A.L., et al. Transcatheter device closure of congenital and postoperative residual ventricular septal defects. Circulation. 2004;110(5):501–507.
12. El-Najdawi, E.K., et al. Operation for partial atrioventricular septal defect: a forty-year review. J Thorac Cardiovasc Surg. 2000;119(5):880–889.
13. Mavroudis C., Backer C.L., eds. Pediatric cardiac surgery, ed 3, St Louis: Mosby, 2003.
14. Fawzy, M.R., et al. Long-term outcome (up to 15 years) of balloon angioplasty of discrete native coarctation of the aorta in adolescents and adults. J Am Coll Cardiol. 2004;43(6):1062–1067.
15. Walhout, R.J., et al. Comparison of surgical repair with balloon angioplasty for native coarctation in patients from 3 months to 16 years of age. Eur J Cardiothorac Surg. 2004;25(5):722–727.
16. Ricci, M. Repair of coarctation of the aorta. J Thorac Cardiovasc Surg. 2004;127(4):1224–1225.
17. Hasaniya, N., et al. Outcome of aortic valve repair in children with congenital aortic valve insufficiency. J Thorac Cardiovasc Surg. 2004;127(4):970–974.
18. Hrasja, V., et al. Ross and Ross-Konno procedure in children and adolescents: mid-term results. Eur J Cardiothorac Surg. 2004;25(5):742–747.
19. Poon, L.K., Menahem, S. Pulmonary regurgitation after percutaneous balloon valvoplasty for isolated pulmonary valvar stenosis in childhood. Cardiol Young. 2003;13(5):444–450.
20. Wernovsky, G. Hypoplastic left heart syndrome: consensus and controversies in 2007. Cardiol Young. 2007;17(Suppl 2):75–86.
21. Gutgesell, H.P., Massaro, T.A. Management of hypoplastic left heart syndrome in a consortium of university hospitals. Am J Cardiol. 2000;76(11):809–811.
22. McQuillen, P.S. Balloon atrial septostomy is associated with preoperative stroke in neonates with transposition of the great arteries. Circulation. 2006;113(2):280–285.
23. Newburger, J.W. Diagnosis, treatment, and long term management of Kawasaki disease: statement for health professionals from the Committee on Rheumatic Fever, Endocarditis and Kawasaki Disease, Council on Cardiovascular Disease in the Young, and American Heart Association. Circulation. 110, 2004. [2747–2271].
24. Sinha, M.D., Reid, C. Evaluation of blood pressure in children. Curr Opin Nephrol Hypertens. 2007;16(6):577–584.
25. Munter, R., et al. Trends in blood pressure among children and adolescents. JAMA. 2004;291:2107–2113.
26. National High Blood Pressure Education Program Working Group on High Blood Pressure in Children. The fourth report on the diagnosis, evaluation, and treatment of high blood pressure in children and adolescents,. Pediatrics. 2004;114:555–576.
27. Nguyen, M., Mitsnefes, M. Evaluation of hypertension by the general pediatrician. Curr Opin Pediatr. 2007;19(2):165–169.
28. Seikaly, M.G. Hypertension in children: an update on treatment strategies. Curr Opin Pediatr. 2007;19(2):170–177.
29. Feld, L.G., Corey, H. Hypertension in childhood. Pediatr Rev. 2007;28(8):283–298.
30. McNiece, K.L., et al. Prevalence of hypertension and pre-hypertension among adolescents. J Pediatr. 2007;150(6):640–644. [644e1].
31. Hansen, M.L., et al. Underdiagnosis of hypertension in children and adolescents. JAMA. 2007;298(8):874–879.
32. Din-Dzietham, R., et al. High blood pressure trends in children and adolescents in national surveys, 1963-2002. Circulation. 2007;116(13):1488–1496.
33. Beilin, L., Huang, R.C. Childhood obesity, hypertension, the metabolic syndrome and adult cardiovascular disease. Clin Exp Pharmacol Physiol. 2008;35(4):409–411.
34. Haas, J.S., et al. The association of race, socioeconomic status, and health insurance status with the prevalence of overweight among children and adolescents. Am J Public Health. 2003;93(12):2105–2110.
35. Harding, S., et al. Overweight, obesity and high blood pressure in an ethnically diverse sample of adolescents in Britain: the Medical Research Council DASH study. Int J Obesity. 2008;32(1):82–90.
36. Kuczmarski, R.J., et al, CDC growth charts: United States. Adv Data. 2000(314):1–27.
37. Whitaker, R.C., et al. Predicting obesity in young adulthood from childhood and parental obesity. N Engl J Med. 1997;337(13):869–873.
38. Crespo, C.J., et al. Television watching, energy intake, and obesity in US children: results from the third National Health and Nutrition Examination Study, 1988-1994. Arch Pediatr Adolesc Med. 2001;155(3):360–365.
39. Gordon-Larsen, P., McMurray, R.G., Popkin, B.M. Adolescent physical activity and inactivity vary by ethnicity: The National Longitudinal Study of Adolescent Health. J Pediatr. 1999;135(3):301–306.
40. Kimm, S.Y., et al. Racial divergence in adiposity during adolescence: the NHLBI Growth and Health Study. Pediatrics. 2001;107(3):E34.
41. Rome, E.S., et al. Children and adolescents with eating disorders: the state of the art. Pediatrics. 2003;111(1):e98–e108.
42. Centers for Disease Control and Prevention. Guidelines for school health programs to promote lifelong healthy eating: Centers for Disease Control. MMWR Recomm Rep. 1996;45(RR-9):1–41.