ALTERATIONS OF CARDIOVASCULAR FUNCTION



Cardiovascular disease is the leading cause of death in the United States (Table 30-1) and in the world.1 The pathophysiology of heart disease is now known to be much more complicated than just structural and hemodynamic changes. Today the focus is on the genetic, neurohumoral, and inflammatory mechanisms that underlie tissue and cellular processes, such as endothelial injury, remodeling, stunning, reperfusion injury, and autoimmune disease.
Table 30-1
Death Rates and Percent of Total Deaths for the 15 Leading Causes of Death in the United States (Final Data Report 2006)
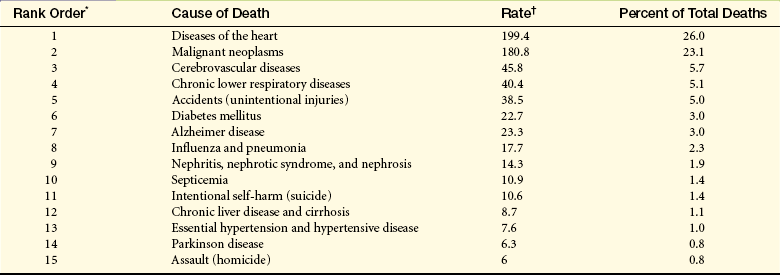
∗Rank based on number of deaths.
†Rates per 100,000 population.
Data from National Center for Health Statistics: National vital statistics report 57, No 14, 2009, National Center for Health Statistics. Available at www.cdc.gov/nchs.
DISEASES OF THE VEINS
Varicose Veins and Chronic Venous Insufficiency
A varicose vein is a vein in which blood has pooled. Varicose veins typically involve the saphenous veins of the legs and are distended, tortuous, and palpable (Figure 30-1). Varicose veins are caused by (1) trauma to the saphenous veins that damages one or more valves, or (2) gradual venous distention caused by the action of gravity on blood in the legs.
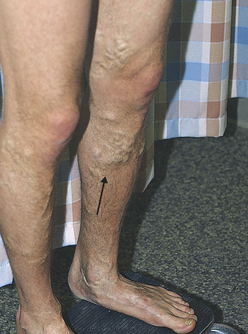
Figure 30-1 Varicose veins of the leg (arrow). (From Kumar V, et al: Robbins basic pathology, ed 8, Philadelphia, 2007, Saunders. Courtesy of Dr. Magruder C. Donaldson, Brigham and Women’s Hospital, Boston.)
Veins are thin-walled, highly distensible vessels. Normally, the muscular pump in the legs moves venous blood up toward the heart and valves prevent backflow and pooling of blood (see Figures 29-30 and 29-31). If a valve is damaged, permitting backflow, a section of the vein is subjected to the pressure exerted by a larger volume of blood under the influence of gravity. The vein swells as it becomes engorged and surrounding tissue becomes edematous because increased hydrostatic pressure pushes plasma through the stretched vessel wall.
In individuals who habitually stand for long periods, wear constricting garments, or cross the legs at the knees, distention progresses until the pressure in the vein damages venous valves, rendering the valves incompetent. Damaged valves cannot maintain normal venous pressure, which causes hydrostatic pressure in the vein to increase. As the vein distends further, it becomes tortuous, and edema develops in the extremity.
Varicose veins and valvular incompetence can progress to chronic venous insufficiency (CVI). CVI is inadequate venous return over a long period. Venous hypertension, circulatory stasis, and tissue hypoxia lead to an inflammatory reaction in vessels and tissue leading to fibrosclerotic remodeling of the skin and then to ulceration.2 Symptoms include chronic pooling of blood in the veins of the lower extremities and hyperpigmentation of the skin of the feet and ankles. Edema in these areas may extend to the knees.
Circulation to the extremities can become so sluggish that the metabolic demands of the cells for oxygen, nutrients, and waste removal are barely met. Any trauma or pressure can therefore lower the oxygen supply and cause cell death and necrosis (venous stasis ulcers). Infection can occur because poor circulation impairs the delivery of the cells and biochemicals for the immune and inflammatory responses. This same sluggish circulation makes infection following reparative surgery a significant risk. Varicose veins and CVI may be associated with deep venous thrombosis (in a deep vein, DVT) in some individuals because of changes in collateral flow and shared risk factors; therefore, anyone with new onset varicose veins should be evaluated for the possibility of underlying DVT.
Treatment of varicose veins and CVI begins conservatively, and excellent wound healing results have followed noninvasive treatments, such as leg elevation, compression stockings, and physical exercise.3 Invasive management includes endovascular ablation or surgical ligation and vein stripping.4
Thrombus Formation in Veins
A thrombus is a blood clot that remains attached to a vessel wall (Figure 30-2). A detached thrombus is a thromboembolus. Venous thrombi are more common than arterial thrombi because flow and pressure are lower in the veins than in the arteries. DVT occurs primarily in the lower extremity. Three factors (triad of Virchow) promote venous thrombosis: (1) venous stasis (e.g., immobility, obesity, prolonged leg dependency [e.g., air travel], age, congestive heart failure [CHF], (2) venous endothelial damage [e.g., trauma, medications], and (3) hypercoagulable states (e.g., inherited disorders, malignancy, pregnancy, oral contraceptives, hormone replacement, hyperhomocysteinemia, antiphospholipid syndrome).5–9 All individuals who are hospitalized are at significant risk and orthopedic trauma or surgery, spinal cord injury, and obstetric/gynecologic conditions can be associated with up to a 100% likelihood of DVT. There are numerous genetic abnormalities associated with an increased risk for venous thrombosis primarily related to states of hypercoagulability. These inherited abnormalities include factor V Leiden mutation; prothrombin mutations; and deficiencies of protein C, protein S, and antithrombin, and are commonly found in individuals who develop thrombi in the absence of the usual risk factors.6,10,11
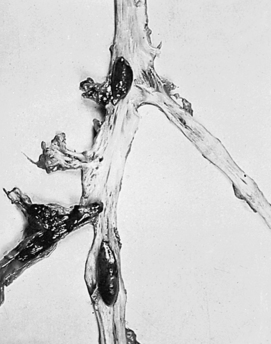
Figure 30-2 Multiple venous thrombi. (From Rosai J: Ackerman’s surgical pathology, ed 8, vol 2, St Louis, 1996, Mosby.)
Accumulation of clotting factors and platelets leads to thrombus formation in the vein, often near a venous valve. Inflammation around the thrombus promotes further platelet aggregation and the thrombus propagates or grows proximally.12 This inflammation may cause local symptoms but because the vein is deep in the leg, it is usually not accompanied by clinical symptoms or signs. If the thrombus creates significant obstruction to venous blood flow, increased pressure in the vein behind the clot may lead to edema of the extremity.
Most thrombi will eventually dissolve without treatment, but untreated DVT is associated with a high risk of thromboembolization of a part of the clot from the leg to the lung (pulmonary embolism) (see Chapter 33).13 Persistent venous outflow obstruction may lead to post-thrombotic syndrome (PTS), a frequent complication of DVT characterized by chronic, persistent pain, swelling, and ulceration of the affected limb.14
Because DVT is usually asymptomatic and difficult to detect clinically, prevention in at-risk individuals is crucial. If possible, individuals should be mobilized as soon as possible after illness, injury, or surgery. Prophylactic treatment can include low-molecular-weight heparin, antithrombin agents, warfarin, or pneumatic devices.15–17 In individuals at high risk for pulmonary embolism but for whom anticoagulation is contraindicated, placement of an inferior vena caval filter may be necessary to prevent pulmonary embolism.13
Diagnosis is most often made by combining measurement of serum D-dimer concentration with lower extremity ultrasonography.18,19 In selected individuals, computed tomography (CT) or magnetic resonance imaging (MRI) may be needed to make the diagnosis.20,21If noninvasive testing is nondiagnostic, a venogram may be indicated. DVT is treated with low-molecular-weight heparin, unfractionated intravenous heparin, antithrombin agents, or adjusted-dose subcutaneous heparin.22,23 Fibrinolytic therapy may be used in selected individuals to dissolve the clot more quickly but is associated with a greater risk for bleeding than heparin.
Superior Vena Cava Syndrome
Superior vena cava syndrome (SVCS) is a progressive occlusion of the superior vena cava (SVC) that leads to venous distention in the upper extremities and head. The leading cause of SVCS is bronchogenic cancer (approximately 75% of cases), followed by lymphomas and metastasis of other cancers.24 Benign causes of SVCS include thrombosis, histoplasmosis, tuberculosis, mediastinal fibrosis, cystic fibrosis, and benign tumors, such as retrosternal goiter.25 Invasive therapies, including pacemaker wires, central venous catheters, and pulmonary artery catheters, can lead to acute and chronic SVCS.
The SVC is a relatively low-pressure vessel that lies in the closed thoracic compartment; therefore, tissue expansion can easily compress the SVC. The right mainstem bronchus abuts the SVC so that cancers occurring in this bronchus may press on the SVC. Additionally, the SVC is surrounded by lymph nodes and lymph chains that commonly become involved in thoracic cancers and compress the SVC during tumor growth. Because onset of SVCS is slow, collateral venous drainage to the azygos vein usually has time to develop.
Clinical manifestations of SVCS include edema and venous distention in the upper extremities and face, including the ocular beds. Individuals may complain of a feeling of fullness in the head, or tightness of shirt collars, necklaces, and rings. Cerebral and central nervous system edema may cause headache, visual disturbance, and impaired consciousness. The skin of the face and arms is purple and taut, and capillary refill time is prolonged. Respiratory distress may be present because of edema of bronchial structures or compression of the bronchus by a carcinoma.
Diagnosis is made by chest roentgenogram, Doppler studies, CT, MRI, and ultrasound. With slow onset and the development of collateral venous drainage, SVCS is generally not a vascular emergency but rather an oncologic emergency. Treatment includes radiation therapy, chemotherapy, and the administration of diuretics, steroids, and anticoagulants, as necessary.24,25 Treatment also may include bypass surgery using various grafts; local and systemic thrombolysis; balloon angioplasty; and placement of intravascular stents.
DISEASES OF THE ARTERIES
An aneurysm is a localized dilation or outpouching of a vessel wall or cardiac chamber. Laplace’s law can provide an understanding of the hemodynamics of an aneurysm (Figure 30-3). (Laplace’s law is discussed in detail in Chapter 29.) Aneurysms most commonly occur in the thoracic or abdominal aorta.
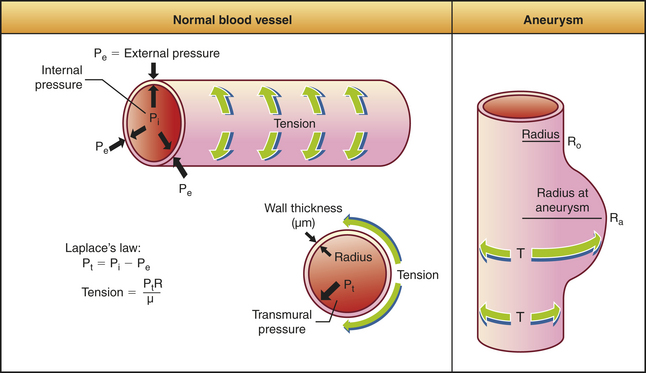
Figure 30-3 Pressure-tension and wall thickness relations in blood vessels or cardiac chambers (Laplace’s law).
The aorta is particularly susceptible to aneurysm formation because of constant stress on the vessel wall and the absence of penetrating vasa vasorum in the media layer (Figure 30-4, A). It is estimated that up to 10% of older individuals have an aortic aneurysm, and about 15,000 persons in the United States die from aortic aneurysm rupture annually.26 Arteriosclerosis and hypertension are found in more than half of all individuals with aneurysms. Chronic hypertension results in mechanical and shear forces that contribute to remodeling and weakening of the vessel wall. Atherosclerosis is a common cause of aneurysms because plaque formation erodes the vessel wall. Infections, such as syphilis, collagen disorders (such as Marfan syndrome), and traumatic injury to the chest or abdomen, also can cause aortic aneurysms. For those aortic aneurysms not clearly related to atherosclerosis, infection, Marfan syndrome or trauma, or numerous genetic susceptibilities have been identified including genes polymorphisms for the production of growth factors, myosin, and proteases.26,27 Inflammation, with the production of toxic oxygen radicals, activates matrix degrading proteins and smooth muscle cell apoptosis resulting in loss of medial elastic lamellae and thinning of the tunica media. Autoimmunity and the production of metalloproteinases and elastases further contribute to the degradation of the vessel wall.26,28
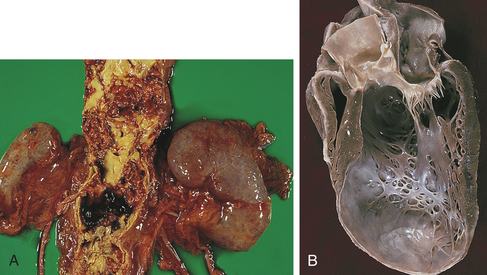
Figure 30-4 Aneurysms. A, Abdominal aortic atherosclerotic aneurysm. B, In a long-axis view of the left ventricle there is a large thin-walled apical aneurysm that does not contain thrombus. (From Damjanov I, Linder J, editors: Anderson’s pathology, ed 10, St Louis, 1996, Mosby.)
True aneurysms involve all three layers of the arterial wall and are best described as a weakening of the vessel wall. Most are fusiform and circumferential (Figure 30-5). False aneurysm is an extravascular hematoma that communicates with the intravascular space. A common cause of this type of lesion is a leak between a vascular graft and a natural artery. Saccular aneurysms are basically spherical.
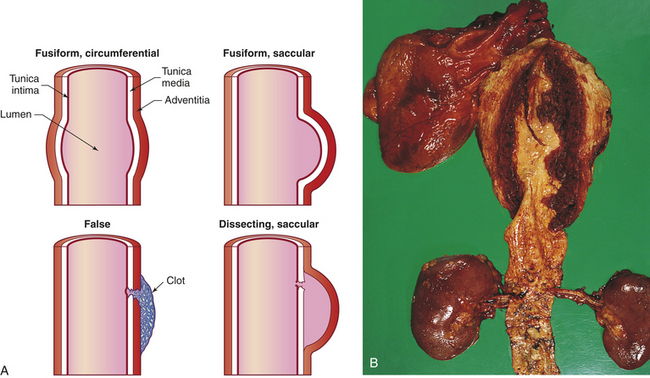
Figure 30-5 Longitudinal sections showing types of aneurysms. A, The fusiform circumferential and fusiform saccular aneurysms are true aneurysms, caused by weakening of the vessel wall. False and saccular aneurysms involve a break in the vessel wall, usually caused by trauma. B, Dissecting aneurysm of thoracic aorta. (B from Damjanov I, Linder J, editors: Anderson’s pathology, ed 10, St Louis, 1996, Mosby.)
Presumably, formation of a ventricular wall aneurysm occurs when intraventricular tension stretches the noncontracting infarcted muscle (see Figure 30-4, B). The stretching produces infarct expansion, a weak and thin layer of necrotic muscle, and fibrous tissue that bulges with each systole. With time, the aneurysm becomes more fibrotic but continues to bulge with each systole, thus acting as a “reservoir” for some of the stroke volume.
Clinical manifestations depend on where the aneurysm is located. Aneurysms in the heart present with dysrhythmias, heart failure, and embolism of clots to the brain or other vital organs. Aortic aneurysms often are asymptomatic until they rupture, when they become painful. Symptoms of dysphagia (difficulty in swallowing) and dyspnea (breathlessness) are caused by the pressure of a thoracic aneurysm on surrounding organs. An abdominal aneurysm can impair flow to an extremity and cause symptoms of ischemia. Aneurysms that occur elsewhere in the body have variable symptoms and signs related to the size of the aneurysm and the potential for rupture and hemorrhage.
The diagnosis of an aneurysm is usually confirmed by ultrasonography, CT, MRI, or angiography. The goals of medical treatment of aneurysms are to maintain a low blood volume and low blood pressure to decrease mechanical forces thought to contribute to vessel wall dilation. Medical treatment is indicated for slow-growing aortic aneurysms, particularly in early stages, and includes smoking cessation, reducing blood pressure and blood volume, and β-adrenergic blockage.29 For those aneurysms that are dilating rapidly, surgical treatment often is indicated. Surgery should be done when aortic aneurysms reach 5 cm in diameter and usually includes replacement with a prosthetic graft. New endovascular surgical techniques with placement of a stent make aneurysm repair possible for more individuals.30
Aortic aneurysms can be complicated by the acute aortic syndromes that include aortic dissection, hemorrhage into the vessel wall, or vessel rupture. Aortic dissection is a devastating complication that can involve any part of the aorta (ascending, arch, or descending) and can disrupt flow through arterial branches, thus creating a surgical emergency. Dissection of the layers of the arterial wall occurs when there is a tear in the intima and blood enters the wall of the artery (see Figure 30-5). This occurs most commonly when there is trauma to the aorta or when there is tissue ischemia and necrosis at the edge of an atherosclerotic plaque that weakens the intima. Persistent chronic hypertension and inflammation contribute to further degradation of the vessel wall with fibrotic obstruction of vessels that feed the arterial wall. Emergent evaluation and surgical intervention are indicated.31
Thrombus Formation
As in venous thrombosis, arterial thrombi tend to develop wherever intravascular conditions promote activation of the coagulation, or clotting, cascade. These conditions include intimal irritation and roughening, inflammation, traumatic injury, infection, and low blood pressures or obstructions that cause blood stasis and pooling within the vessels. (Mechanisms of coagulation are described in Chapter 25.) In the arteries, activation of the coagulation cascade usually is caused by roughening of the tunica intima by atherosclerosis. Invasion of the tunica intima by an infectious agent also roughens the normally smooth lining of the artery, causing platelets to adhere readily. Anatomic changes of an artery can stimulate thrombus formation, particularly if the change results in pooling of arterial blood. This can occur, for example, in blood that is pooled within an aneurysm. Thrombi form also on heart valves altered by calcification or bacterial vegetation. Valvular thrombi are associated most commonly with inflammation of the endocardium (endocarditis) and rheumatic heart disease.
Widespread arterial thrombus formation can occur in shock. Shock, particularly the type resulting from septicemia, can activate the intrinsic and extrinsic pathways of coagulation. The impaired cellular metabolism that occurs with all types of shock activates the extrinsic pathway of coagulation, whereas blood stasis caused by very low blood pressures activates the intrinsic pathway (see Chapter 25). Thrombus formation may be confined to one area or may progress to diffuse coagulopathy, such as disseminated intravascular coagulation (see Chapter 27).
Arterial thrombi pose two potential threats to the circulation. First, the thrombus may grow large enough to occlude the artery, causing ischemia in tissue supplied by the artery. Second, the thrombus may dislodge, becoming a thromboembolus that travels through the vascular system until it occludes flow into a distal systemic vascular bed.
Diagnosis of arterial thrombi is usually accomplished through the use of Doppler ultrasonography and angiography. Pharmacologic treatment involves the administration of heparin, warfarin derivatives, thrombin inhibitors, or thrombolytics. A balloon-tipped catheter also can be used to remove or compress an arterial thrombus. Various combinations of drug and catheter therapies are sometimes used concurrently.
Embolism
Embolism is the obstruction of a vessel by an embolus—a bolus of matter that is circulating in the bloodstream. The embolus may consist of a dislodged thrombus; an air bubble; an aggregate of amniotic fluid; an aggregate of fat, bacteria, or cancer cells; or a foreign substance. An embolus travels in the bloodstream until it reaches a vessel through which it cannot fit. No matter how tiny it is, an embolus eventually will lodge in a systemic or pulmonary vessel. The source of the embolus determines whether the embolus will lodge in a vessel of the pulmonary or systemic circulation. Pulmonary emboli originate in the venous circulation (mostly from the deep veins of the legs) or in the right heart (see p. 1143). Arterial emboli most commonly originate in the left heart and are associated with thrombi after myocardial infarction, valvular disease, left heart failure, endocarditis, and dysrhythmias.
Embolism causes ischemia or infarction in tissues distal to the obstruction. A limb that is ischemic because of arterial occlusion is characterized (1) by an almost waxy whiteness of the skin because the vasculature is devoid of erythrocytes, and (2) by numbness and pain resulting from neural ischemia.
Embolism of a central organ causes organic dysfunction and pain. For example, pulmonary artery embolism causes chest pain and dyspnea; renal artery embolism causes abdominal pain and oliguria; and mesenteric artery embolism causes abdominal pain and a paralytic, ischemic bowel. Infarction and subsequent necrosis of a central organ are life threatening, not only because of organ dysfunction but also because of sepsis. Necrotic tissue is a rich medium for the growth of bacteria from the lungs, bowel, and occasionally, bladder. Necrosis of the bladder, in particular, can quickly lead to peritonitis or septicemia.
Embolism of a coronary or cerebral artery is an immediate threat to life if the embolus severely obstructs a major vessel. Occlusion of a coronary artery will cause a myocardial infarction (see p. 1171), whereas occlusion of a cerebral artery causes a stroke (see Chapter 17).
Thromboembolism
Thromboembolism is a vascular obstruction resulting from a dislodged thrombus. The most common source of arterial thromboemboli to the systemic circulation is the heart. Mitral or aortic valvular disease, especially that associated with abnormal heart rhythms (atrial fibrillation and flutter), causes thrombus formation on roughened vascular surfaces and in atrial blood as a result of stasis. More than half of these thromboemboli lodge in the lower extremities (in the femoral and popliteal arteries). Others lodge in the coronary arteries and the cerebral vasculature. Heart failure also is associated with an increased risk of thrombotic complications, although the mechanism for this increased risk is unclear.
Air Embolism
Room air that enters the circulation through intravenous lines is probably the most common cause of air embolism. Room air is about 70% nitrogen. Although nitrogen dissolves quickly in blood, large amounts of air cannot be dissolved rapidly enough to prevent the displacement of blood in the arterioles and capillary beds. Ischemia and necrosis occur when air totally blocks a vessel.
Air also can be introduced into the bloodstream if trauma to the chest causes air from the lungs to enter the vascular space. For example, gunshot wounds and puncture wounds of the thorax sometimes introduce air emboli. Treatment for air embolism is supportive, including bed rest and supplemental oxygen, once the connection between the source of air and the vascular system is eliminated.
Amniotic Fluid Embolism
The great intra-abdominal pressures generated during labor and delivery may force amniotic fluid into the mother’s bloodstream through the highly vascular uterine wall. Amniotic fluid not only displaces blood, reducing oxygen, nutrient, and waste exchange, but also introduces antigens, cells, and protein aggregates that trigger inflammation, coagulation, and the immune response within the bloodstream. Capillary beds usually are affected by amniotic fluid emboli, especially the capillary beds of the lungs and kidneys. Treatment is supportive and may include dialysis, particularly after a cesarean delivery or hysterectomy.
Bacterial Embolism
Isolated bacteria in the bloodstream do not cause embolism, but aggregates of bacteria may be large enough to do so. The most common cause of bacterial embolism is subacute bacterial endocarditis, during which clumps of vegetation are dislodged from infected cardiac valves and ejected into the pulmonary or systemic circulation. A less common cause is erosion of an artery or vein by bacteria at a source of infection, such as an abscess. Treatment for bacterial embolism includes bed rest, supplemental oxygen, and antibiotics to eradicate the source of infection.
Fat Embolism
Trauma to the long bones is associated with fat embolism, particularly in the lungs. Two mechanisms have been proposed to account for the generation of fat emboli after skeletal trauma. The first is that trauma to the bones initiates defective fat metabolism, causing globules of fat to form in the blood. Platelets adhere to these globules until the conglomerate is large enough to lodge in a capillary bed. The second possible explanation is that globules of fat are released from fatty bone marrow exposed by fracture. Again, platelets adhere to the fat globules and embolism occurs.
Treatment for fat embolism consists of prompt immobilization of fractures and supportive measures that include administration of supplemental oxygen, steroids, and glucose. Steroid administration may decrease the inflammation that occurs with vascular occlusion. Inflammation in the pulmonary bed is especially dangerous because it can cause acute respiratory distress syndrome (ARDS) (see Chapter 33).
Foreign Matter
Foreign matter can enter the bloodstream during trauma or through an intravenous or intra-arterial line. Small particles, such as drug precipitates, small glass shards, or fibers from linen, are sometimes introduced unintentionally into a vessel through intravenous injections or manipulation of monitoring lines. Once in the blood, these small particles initiate the coagulation cascade. The thromboemboli that form around the particles are large enough to occlude a vessel and result in ischemia. Treatment is aimed at preventing thrombus formation around the particle, dissolution of the particle, and supportive measures to alleviate ischemia. If the bolus of foreign matter is relatively large, it usually is removed surgically.
Peripheral Arterial Diseases
Thromboangiitis Obliterans (Buerger Disease)
Thromboangiitis obliterans (Buerger disease) is an inflammatory disease of the peripheral arteries. It is associated with smoking in approximately 95% of cases—the other 5% are related to frostbite, trauma, or the use of sympathomimetic drugs.32 The incidence of Buerger disease has been steadily declining, presumably because of a decrease in cigarette smoking in men. The inflammatory lesions are accompanied by thrombi and sometimes by vasospasm of arterial segments.32 Inflammation, thrombus formation, and vasospasm eventually can occlude and obliterate portions of small and medium-size arteries. Typically affected are the digital, tibial, and plantar arteries of the feet and the digital, palmar, and ulnar arteries of the hands. The pathogenesis of thromboangiitis obliterans is still being explored. There is evidence of significant T-cell activation and autoimmunity.32
The chief symptoms of thromboangiitis obliterans are pain and tenderness of the affected part. Clinical manifestations are caused by sluggish blood flow and include rubor (redness of the skin), which is caused by dilated capillaries under the skin, and cyanosis, which is caused by blood that remains in the capillaries after its oxygen has diffused into the interstitium. Chronic ischemia causes the skin to thin and become shiny and the nails to become thickened and malformed. In advanced disease, ischemia resulting from vessel obliteration can cause gangrene. Buerger disease has been associated with cerebrovascular disease and rheumatic symptoms (joint pain).
The most important part of treatment is cessation of cigarette smoking. All other measures are aimed at improving circulation to the foot or hand. Vasodilators are prescribed to alleviate vasospasm, and exercises are taught that use gravity to improve blood flow.32,33 If vasospasm persists, sympathectomy may be performed.33 Gangrene necessitates amputation.
Raynaud Phenomenon and Disease
Raynaud phenomenon and Raynaud disease are characterized by attacks of vasospasm in the small arteries and arterioles of the fingers and, less commonly, the toes. Although the clinical manifestations of the phenomenon and the disease are the same, their causes differ.
Raynaud phenomenon is secondary to systemic diseases, particularly collagen vascular disease (scleroderma), chemotherapy, cocaine use, hypothyroidism, pulmonary hypertension, thoracic outlet syndrome, serum sickness, vasculitis, malignancy, or long-term exposure to environmental conditions, such as cold or vibrating machinery in the workplace.34 Raynaud phenomenon associated with malignancy can be especially severe and is an important clue to finding a previously undiagnosed cancer.
Raynaud disease is a primary vasospastic disorder of unknown origin. Raynaud disease tends to affect young women and to consist of vasospastic attacks triggered by brief exposure to cold or by emotional stress. It is estimated that the prevalence of Raynaud disease is 11% of women and 8% of men in the United States.34 Blood vessels in these individuals demonstrate endothelial dysfunction with decreased nitric oxide production and increased endothelin-1 activity. Genetic predisposition may play a role in its development.
The clinical manifestations of the vasospastic attacks of either disorder are changes in skin color and sensation caused by ischemia. Vasospasm occurs with varying frequency and severity and causes pallor, numbness, and the sensation of cold in the digits. Attacks tend to be bilateral, and manifestations usually begin at the tips of the digits and progress to the proximal phalanges. Sluggish blood flow resulting in ischemia may cause the skin to appear cyanotic. Rubor follows as vasospasm ends and the capillaries become engorged with oxygenated blood. Rubor often is accompanied by throbbing and paresthesias. Skin color returns to normal after the attack, but frequent, prolonged attacks interfere with cellular metabolism, causing the skin of the fingertips to thicken and the nails to become brittle. In severe, chronic Raynaud phenomenon or disease, ischemia eventually can cause ulceration and gangrene. This outcome is rare, however.
The diagnostic criteria for Raynaud disease include not only the characteristic clinical manifestations described previously but also the absence of necrosis, no detectable underlying cause, normal capillaroscopy findings, normal laboratory tests for inflammation, and negative tests for antinuclear factors. Any condition causing Raynaud phenomenon will usually be detected if one of these evaluative tests is abnormal.
Treatment for Raynaud phenomenon consists of removing the stimulus or treating the primary disease process. When Raynaud phenomenon is associated with malignancy, surgical removal of the tumor may resolve the ischemia. For Raynaud phenomenon not associated with malignancy, treatment is limited to amelioration of symptoms with medications such as calcium channel blockers or other vasodilators. Attacks of vasospasm sometimes can be alleviated at their onset by an exercise in which the arms are swung forward and backward. This maneuver increases hydrostatic pressure (and perfusion pressure) in the arteries by means of centrifugal force.
Treatment of Raynaud disease is limited to prevention or alleviation of vasospasm itself, because no underlying disorder has been identified. Stimuli that trigger attacks (e.g., emotional stress, cold) are avoided, and cigarette smoking is stopped to eliminate the vasoconstricting effects of nicotine. Exercises that build centrifugal force in the extremities also are helpful in the early stages of vasospasm. If attacks of vasospasm become frequent or prolonged, pharmacologic management can include calcium channel blockers, nitric oxide agonists, alpha-blockers, prostaglandin analogs, and selective serotonin reuptake inhibitors.34,35 Biofeedback may be helpful. Sympathectomy may be recommended in severe cases but is not always effective and has a high rate of recurrence. If ischemia leads to ulceration and gangrene, amputation is necessary.
Hypertension
Hypertension is consistent elevation of systemic arterial blood pressure. Hypertension is the most common primary diagnosis in the United States. Approximately 65% of Americans older than the age of 60 have hypertension and less than two thirds of those have adequately controlled hypertension.36 Hypertension is defined by the Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure (JNC7)37 as a sustained systolic blood pressure of 140 mmHg or greater or a diastolic pressure of 90 mmHg or greater (Table 30-2). Normal blood pressure is associated with the lowest cardiovascular risk, whereas those who fall into the prehypertension category are at risk for developing hypertension unless lifestyle modification is instituted (more than 90% will develop hypertension).36,37 The prevalence of hypertension increases with age and is higher for blacks than for whites.36
Table 30-2
Classification of Blood Pressure for Adults Age 18 Years or Older

Data from the JNC 7 Report, JAMA 289(19):2560-2572, 2003.
Individuals with hypertensive disease may have combined systolic and diastolic hypertension or isolated systolic hypertension. Isolated systolic hypertension is elevated systolic blood pressure accompanied by normal diastolic blood pressure (less than 90 mmHg). Most cases of hypertension have no known cause and therefore are diagnosed as primary hypertension. Primary hypertension, also called essential or idiopathic hypertension, affects 90% to 95% of hypertensive individuals.36,37 Secondary hypertension is caused by altered hemodynamics associated with a primary disease, such as renal disease. Although many diseases can cause secondary hypertension, this form of hypertension accounts for only 5% to 8% of cases. Hypertension is a complex disorder that affects the entire cardiovascular system, and all types and stages of hypertension are associated with increased risk for target organ disease events, such as myocardial infarction, kidney disease, and stroke.36–38
Factors Associated with Primary Hypertension
Hypertension is caused by increases in cardiac output or total peripheral resistance, or both. (The many factors affecting cardiac output and peripheral resistance are described in Chapter 29.) Cardiac output is increased by any condition that increases heart rate or stroke volume, whereas peripheral resistance is increased by any factor that increases blood viscosity or reduces vessel diameter, particularly arteriolar diameter.
A specific cause for primary hypertension has not been identified, and a combination of genetic and environmental factors is thought to be responsible for its development. Genetic predisposition to hypertension is thought to be polygenic. The inherited defects are associated with renal sodium excretion, insulin and insulin sensitivity, activity of the renin-angiotensin-aldosterone system (RAAS), cell membrane sodium or calcium transport, and sympathetic response to neurogenic hormones39,40 (see What’s New? Genes and the Risk of Hypertension). Risk factors associated with primary hypertension include (1) family history of hypertension; (2) advancing age; (3) gender (men younger than 55 and women older than 70 years); (4) black race; (5) high dietary sodium intake; (6) glucose intolerance (diabetes mellitus); (7) cigarette smoking; (8) obesity; (9) heavy alcohol consumption; and (10) low dietary intake of potassium, calcium, and magnesium.36,37 Many of these factors are also risk factors for other cardiovascular disorders. In fact, hypertension, dyslipidemia, and glucose intolerance often are found together.
Although populations with high dietary sodium intake have long been shown to have an increased incidence of hypertension, recent studies indicate that low dietary potassium, calcium, and magnesium intakes are also risk factors because without their intake sodium is retained.41–43 The nicotine in cigarette smoke is a vasoconstrictor that can elevate systolic and diastolic blood pressure acutely. In habitual smokers an individual cigarette may not raise blood pressure, yet habitual smoking is associated with a high incidence of severe hypertension, myocardial hypertrophy, and death resulting from coronary artery disease (CAD). The incidence of hypertension is higher among heavy drinkers of alcohol (more than three drinks per day) than among abstainers, but moderate drinkers (two to four drinks per week) appear to have lower blood pressures, as well as lower cardiovascular mortality, than either abstainers or heavy drinkers. Obesity is recognized as an important risk factor for hypertension, even in children and adolescents.44
Primary Hypertension: Primary hypertension is the result of a complicated interaction between genetics and the environment that increase vascular tone (increased peripheral resistance) and blood volume, thus causing sustained increases in blood pressure. Multiple pathophysiologic mechanisms mediate these effects including the sympathetic nervous system (SNS), the RAAS, and natriuretic peptides. Inflammation, endothelial dysfunction, obesity-related hormones, and insulin resistance also contribute to both increased peripheral resistance and increased blood volume. Increased vascular volume is related to a decrease in renal excretion of salt, often referred to as a shift in the pressure-natriuresis relationship. This means that for a given blood pressure, individuals with hypertension tend to secrete less salt in their urine. The pathophysiology of primary hypertension is summarized in Figure 30-6.
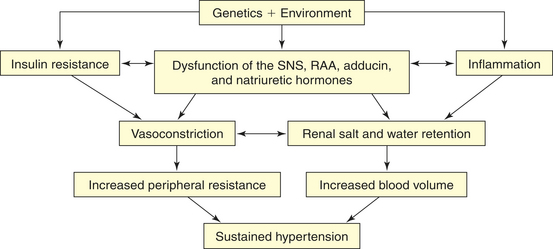
Figure 30-6 Pathophysiology of hypertension. Numerous genetic vulnerabilities have been linked to hypertension and these, in combination with environmental risks, cause neurohumoral dysfunction (sympathetic nervous system [SNS], renin-angiotensin-aldosterone system [RAAS], adducin, and natriuretic hormones) and promote inflammation and insulin resistance. Insulin resistance and neurohumoral dysfunction contribute to sustained systemic vasoconstriction and increased peripheral resistance. Inflammation contributes to renal dysfunction, which, in combination with the neurohumoral alterations, results in renal salt and water retention and increased blood volume. Increased peripheral resistance and increased blood volume are two primary causes of sustained hypertension.
The SNS contributes to the pathogenesis of hypertension in many persons. In the healthy individual, the SNS contributes to the maintenance of adequate blood pressure and tissue perfusion by promoting cardiac contractility and heart rate (maintenance of adequate cardiac output) and by inducing arteriolar vasoconstriction (maintenance of adequate peripheral resistance). In individuals with hypertension, overactivity of the SNS can result from increased production of catecholamines (epinephrine and norepinephrine) or from increased receptor reactivity involving these neurotransmitters. Increased SNS activity causes increased heart rate and systemic vasoconstriction, thus raising the blood pressure. Additional mechanisms of SNS-induced hypertension include structural changes in blood vessels (vascular remodeling), renal sodium retention, insulin resistance, increased renin and angiotensin levels, and procoagulant effects.36,45 The role of the SNS in the pathogenesis of cardiovascular disease is summarized in Figure 30-7.
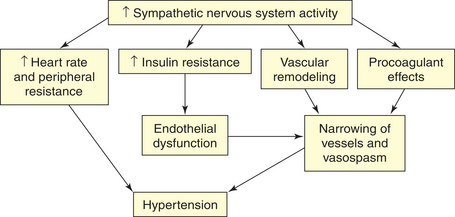
Figure 30-7 Role of the sympathetic nervous system in the pathogenesis of hypertension. Increased activity of the sympathetic nervous system (SNS) not only increases heart rate and peripheral resistance but also causes vascular remodeling with narrowing and vasospasm of arteries. The SNS contributes to insulin resistance, which is associated with endothelial dysfunction and decreased production of vasodilators, such as nitric oxide. The SNS also has procoagulant properties, making vascular spasm and thrombosis more likely. All of these factors contribute to sustained increases in blood pressure.
In the healthy individual, the RAAS provides an important homeostatic mechanism for maintaining adequate blood pressure and therefore tissue perfusion (see Chapter 29). Dysfunction of this system in the hypertensive individual can lead to persistent increases in peripheral resistance and renal salt retention.46 Angiotensin II also causes structural changes in blood vessels (remodeling) that contribute to permanent increases in peripheral resistance and make vessels more vulnerable to endothelial dysfunction and platelet aggregation.36,46–49 Angiotensin II is also responsible for the hypertrophy of the myocardium and much of the renal damage associated with hypertension.46,50,51 Aldosterone not only contributes to sodium retention by the kidney but also has further deleterious effects on the cardiovascular system.52 An important effect of the RAAS is to contribute to insulin resistance.53 Drugs that block renin, angiotensin-converting enzyme (ACE), or angiotensin and aldosterone receptors are used widely in the treatment of hypertension and have been shown to improve vascular, cardiac, and renal function and insulin sensitivity in select populations.36,54–56
Natriuretic peptides modulate renal sodium (Na+) excretion and include atrial natriuretic peptide (ANP), brain natriuretic peptide (BNP), C-type natriuretic peptide (CNP), and urodilantin.57,58 The function of these hormones can be affected by excessive sodium intake; inadequate dietary intake of potassium, magnesium, and calcium; and obesity.41,57 Dysfunction of these hormones, along with alterations in the RAAS and the SNS, cause an increase in vascular tone and a shift in the pressure-natriuresis relationship.42,47 Salt retention leads to water retention and increased blood volume, which contributes to increased blood pressure. Renal injury can result, with renal vasoconstriction and tissue ischemia. Tissue ischemia causes inflammation of the kidney and contributes to dysfunction of the glomeruli and tubules and promotes additional sodium retention.43,47 Elevated levels of natriuretic peptides in hypertension have been linked to an increased risk for ventricular hypertrophy, atherosclerosis, and heart failure.58–62 Drugs that increase the effectiveness of natriuretic peptides are now used for the treatment of hypertension and heart failure.
Inflammation plays a role in the pathogenesis of hypertension. Endothelial injury and tissue ischemia result in oxidative stress and the release of vasoactive inflammatory cytokines. Although many of these cytokines (e.g., histamine, prostaglandins) have vasodilatory actions in acute inflammatory injury, chronic inflammation contributes to vascular remodeling and smooth muscle contraction.63,64 In the kidney vasoconstriction and resultant decreased renal perfusion cause tubular ischemia and preglomerular arteriopathy, leading to decreased sodium filtration and increased sodium retention, thus shifting the pressure-natriuresis curve and contributing to sustained hypertension.42,43,47
Endothelial dysfunction in primary hypertension is characterized by a decreased production of vasodilators, such as nitric oxide, and increased production of vasoconstrictors, such as endothelin (see Chapter 29, Box 29-1).65–68 Increased expression of adhesion molecules and decreased endothelial production of anticoagulant factors contribute to the pathophysiology of hypertension,69,70 and dysfunction of the endothelium contributes to vascular remodeling. Drugs that block the RAAS improve endothelial function.56
Obesity contributes to many of the neurohumoral, metabolic, renal, and cardiovascular processes that cause hypertension, especially those factors that contribute to endothelial
dysfunction and renal sodium retention (see What’s New? Obesity and Hypertension).71 The relationship among obesity, hypertension, and other cardiovascular risks is discussed later (see Metabolic Syndrome, p. 1164 and Chapter 21).
Insulin resistance is associated with endothelial dysfunction and can be detected in approximately 50% of individuals with primary hypertension even without overt diabetes.53 Insulin has several important roles in vascular protection including increased endothelial cell production of nitric oxide. Although hyperinsulinemia may serve as a growth factor and contribute to vascular remodeling, it is believed that it is the resistance to insulin that results in endothelial dysfunction and contributes to the pathogenesis of hypertension and atherosclerosis. Diabetes and insulin resistance also cause changes in SNS and RAAS activity, cause renal glomerular dysfunction, and contribute to the target organ effects of hypertension.53,72–74 It is interesting to note that blood pressure often declines in many diabetic individuals treated with drugs that increase insulin sensitivity, even in the absence of antihypertensive drugs, and insulin sensitivity improves in hypertensive individuals treated with drugs that inhibit the RAAS.53,75
Primary hypertension is the result of an interaction between many of these described processes. The majority of these factors influence renal sodium excretion and shift the pressure-natriuresis relationship as summarized in Figure 30-8. Other effects of this complex group of mechanisms include alterations in endothelial function, vasomotor tone, vascular remodeling, and target organ injury. Our increasing understanding of these complex mechanisms contributes to the development of new and more effective treatments.
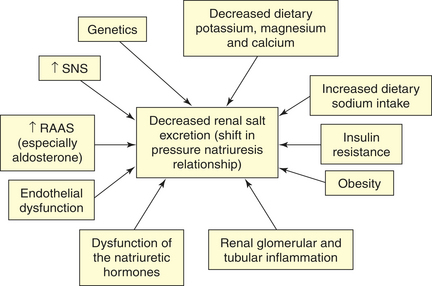
Figure 30-8 Shift in the pressure-natriuresis relationship. Numerous factors have been implicated in the pathogenesis of sodium retention in individuals with hypertension. These factors cause less renal excretion of salt than would normally occur with increased blood pressure. This is called a shift in the pressure-natriuresis relationship and is believed to be a central process in the pathogenesis of primary hypertension. RAAS, Renin-angiotensin-aldosterone system; SNS, sympathetic nervous system.
Secondary Hypertension: Secondary hypertension is caused by an underlying disease process or a medication that raises peripheral vascular resistance or cardiac output. If the cause is identified and removed before permanent structural changes occur, blood pressure returns to normal. Table 30-3 summarizes the pathogenesis of major forms of secondary hypertension.
Table 30-3
Pathogenesis of Major Forms of Secondary Hypertension by Cause
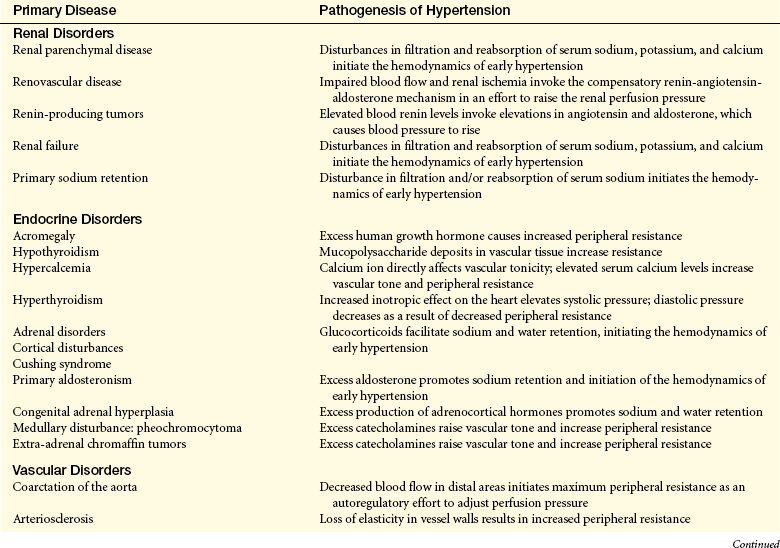
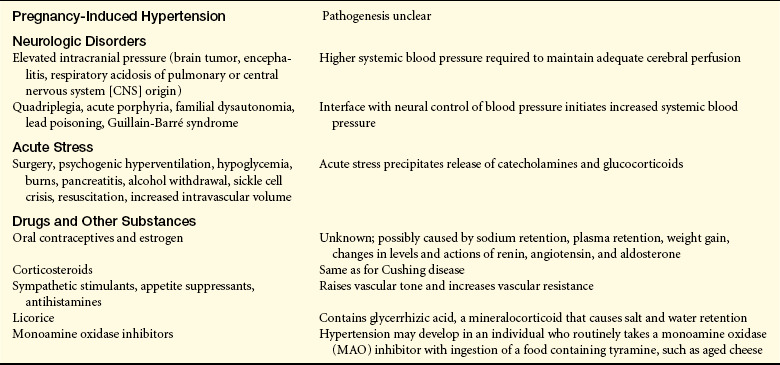
From Kaplan NM: Clinical hypertension, ed 8, Baltimore, 2002, Lippincott Williams & Wilkins; Cheng J et al: Unclear is the role of viral infections, for example, recent data reveal CMV causes an increase in blood pressure, PLoS Pathog May 5(5), 2009. Epub ahead of print.
Complicated Hypertension: Chronic hypertension damages the walls of systemic blood vessels. Within the walls of arteries and arterioles, smooth muscle cells undergo hypertrophy and hyperplasia with associated fibrosis of the tunica intima and media in a process called vascular “remodeling” (Figure 30-9). Endothelial dysfunction, angiotensin II, catecholamines, insulin resistance, and inflammation contribute to this process. Once significant fibrosis has occurred, reduced blood flow and dysfunction of the organs perfused by these affected vessels is inevitable. Target organs for hypertension include the kidney, brain, heart, extremities, and eyes—these effects are summarized in Table 30-4.
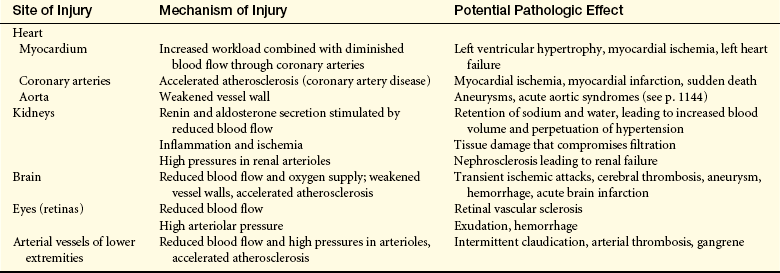
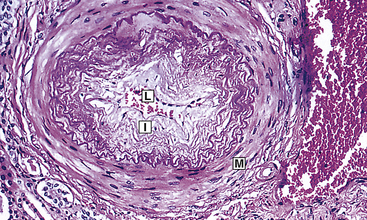
Figure 30-9 Dramatic hypertension change in small arterioles. Fibrous intimal proliferation (I) with reduction in lumen vessel caliber (radius) (L) and normal media (M). (From Stevens A, Lowe J: Pathology, ed 2, St Louis, 2000, Mosby.)
Cardiovascular complications include left ventricular hypertrophy, angina pectoris, congestive heart failure (left heart failure), coronary artery disease, myocardial infarction, and sudden death. Myocardial hypertrophy in response to hypertension is mediated by several neurohormonal substances, including the SNS and angiotensin II. This results in changes in the myocyte proteins, apoptosis of myocytes, and deposition of collagen into the heart muscle. In addition, the increased size of the heart muscle increases demand for coronary perfusion, such that over time contractility of the heart is impaired and the individual is at increased risk for heart failure. Vascular complications include the formation, dissection, and rupture of aneurysms (outpouchings in vessel walls); intermittent claudication; and gangrene resulting from vessel occlusion. Renal complications are parenchymal damage, nephrosclerosis, renal arteriosclerosis, and renal insufficiency or failure.51 Microalbuminuria (small amounts of protein in the urine) is an early sign of impending renal dysfunction and significantly increased risk for cardiovascular events.51,76
Changes in the vascular beds can be estimated by viewing the arterioles of the retina. Complications specific to the retina include retinal vascular sclerosis, exudation, and hemorrhage. Cerebrovascular complications are similar to those of other arterial beds and include transient ischemia, stroke, cerebral thrombosis, aneurysm, and hemorrhage. Chronic hypertension also has been linked to cognitive decline in older adults.77
Malignant hypertension (rapidly progressive hypertension in which diastolic pressure is usually above 140 mmHg) has been linked to dysfunction of renin and angiotensin genes and can cause encephalopathy, a profound cerebral edema that disrupts cerebral function and causes loss of consciousness.78 Encephalopathy occurs because high arterial pressure renders the cerebral arterioles incapable of regulating blood flow to the cerebral capillary beds. Capillary permeability is increased by high hydrostatic pressures in the capillaries, and vascular fluid exudes into the interstitial space.78 If blood pressure is not reduced, cerebral edema and cerebral dysfunction increase until death occurs. Organ damage resulting from malignant hypertension is life threatening. Besides encephalopathy, malignant hypertension can cause papilledema, cardiac failure, uremia, retinopathy, and cerebrovascular accident. This should be considered a hypertensive emergency and managed with rapid administration of parenteral vasodilators, such as nitrates or beta-blockers, with the goal of lowering the blood pressure by 10% to 25% within 2 hours.79
CLINICAL MANIFESTATIONS The early stages of hypertension have no clinical manifestations other than elevated blood pressure. Most important, no signs and symptoms cause the individual to seek healthcare; thus hypertension is called a lanthanic (silent) disease. Some hypertensive individuals never have signs, symptoms, or complications, whereas others become very ill, in which case hypertension can cause death. Still others have anatomic and physiologic damage caused by past hypertensive disease despite having current blood pressures within normal ranges.
The chance of developing primary hypertension increases with age, over and above the natural rise in blood pressure associated with aging. Although hypertension usually is thought to be an adult health problem, it is important to remember that hypertension does occur in children and is being diagnosed with increasing frequency. Usually, however, increased peripheral resistance and early hypertension develop in the second, third, and fourth decades of life. If elevated blood pressure is not detected and treated, it becomes established and may begin to accelerate its effect on tissues when the individual is 30 to 50 years of age. This sets the stage for the complications of hypertension that begin to appear during the fourth, fifth, and sixth decades of life.
The clinical manifestations of chronic hypertension tend to be specific for the organs or tissues affected. Evidence of heart disease, renal insufficiency, central nervous system dysfunction, impaired vision, impaired mobility, vascular occlusion, or edema can be caused by sustained hypertension. (See appropriate chapters for specific clinical manifestations of organ dysfunction.)
EVALUATION AND TREATMENT A single elevated blood pressure reading does not mean that a person has hypertension. Diagnosis requires the measurement of blood pressure on at least two separate occasions averaging two readings at least 2 minutes apart, with the individual seated, the arm supported at heart level, after 5 minutes rest, with no smoking or caffeine intake in the past 30 minutes.36 The American Heart Association updated recommendations for the diagnosis of hypertension in 2005 to include 24-hour ambulatory blood pressure monitoring in selected individuals because of better correlation with end-organ damage and the ability to screen out “white coat hypertension (HTN)” (elevated blood pressure that occurs only in a clinic setting).80,81 Ambulatory measurement also detects those who fail to have a nocturnal decrease in blood pressure and who may be at higher cardiovascular risk. It is especially recommended for individuals with drug resistance, hypotensive symptoms with medications, episodic HTN, and autonomic dysfunction.81 Home blood pressure monitoring with approved devices can help manage hypertension.
Evaluation of the hypertensive individual should include a complete medical history and assessment of lifestyle and other risk factors for hypertension and cardiovascular disease, as well as evidence of possible secondary causes of hypertension. Physical examination should include examination of the optic fundi; calculation of body mass index; auscultation for carotid, abdominal, and femoral bruits; examination of the heart and lungs; palpation of the abdomen; assessment of lower extremity pulses and edema; and neurologic examination.36 Further routine diagnostic tests for the evaluation of hypertension include hematocrit, urinalysis, biochemical blood profile (fasting glucose, sodium, potassium, calcium, creatinine, total cholesterol, high-density cholesterol, triglycerides), and an electrocardiogram (ECG). Optional tests include urinary albumin excretion or albumin/creatinine ratio.36 Individuals who have elevated blood pressure are assumed to have primary hypertension unless their history, physical examination, or initial diagnostic screening indicates secondary hypertension.
Treatment of primary hypertension depends on its severity. Figure 30-10 illustrates an overview of the JNC7 recommendations.37 Treatment begins with reducing or eliminating risk factors. Lifestyle modification can prevent hypertension from developing in those individuals who fall into the prehypertension category, may control the blood pressure in stage I hypertension, and can enhance the effects of drug treatment for those with more significant blood pressure elevation. The usual dietary recommendations are to restrict sodium intake to 2.4 g/day, to increase potassium intake, to restrict saturated fat intake, and to adjust calorie intake as required to maintain optimum weight. The Dietary Approaches to Stop Hypertension (DASH) diet is recommended. An exercise program that promotes endurance and relaxation usually is recommended. Physical training increases stroke volume, which has the effect of lowering heart rate and hence systolic blood pressure, and should consist of regular aerobic physical activity at least 30 minutes most days of the week.37 Relaxation is expected to reduce levels of circulating catecholamines, which has the effect of reducing vascular tone and blood pressure. Individuals are counseled to stop smoking to eliminate vasoconstrictor effects of nicotine.
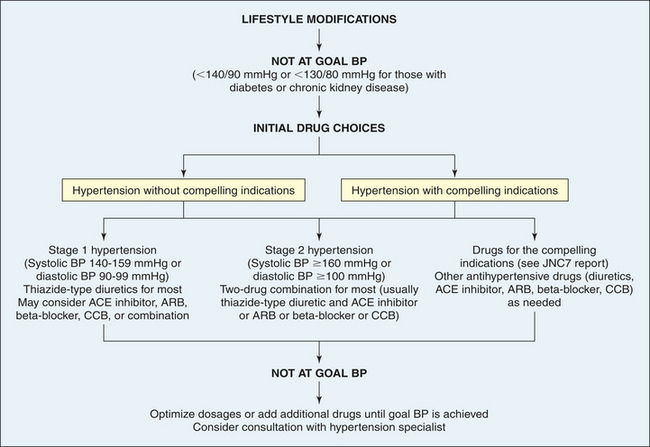
Figure 30-10 Summary of treatment for hypertension. ACE, Angiotensin-converting enzyme; ARB, angiotensin-receptor blocker; BP, blood pressure; CCB, calcium channel blocker. (Data from Chobanian AV et al: The Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure, JAMA 289:2560-2572, 2003.)
Pharmacologic treatment of hypertension reduces the risk of end-organ damage and prevents major diseases, such as myocardial ischemia and stroke. Thiazide diuretics have been shown to be the safest and most effective medications for lowering blood pressure and preventing the cardiovascular complications of hypertension.37 Some individuals will have “compelling indications” for choosing a particular antihypertensive as a first-line medication. For example, individuals with heart failure, with chronic kidney disease, or who are post–myocardial infarction, or who have had recurrent stroke should begin antihypertensive treatment with an ACE inhibitor, angiotensin receptor blocker (ARB), or aldosterone antagonist.37,82 If the individual requires two drugs for blood pressure control, the recommendation is combinations of thiazide diuretics and other antihypertensives, such as beta blockers and ACE inhibitors.
Orthostatic (Postural) Hypotension
The term orthostatic (postural) hypotension means a decrease in systolic and diastolic arterial blood pressure on standing. The American Autonomic Society (AAS) and the American Academy of Neurology (AAN) define orthostatic hypertension as a systolic blood pressure decrease of at least 20 mmHg or a diastolic blood pressure decrease of at least 10 mmHg within 3 minutes of standing up.83 It can be categorized as arteriolar, venular, or mixed.84 When a normal individual stands up, the resultant gravitational changes on the circulation are compensated for by several mechanisms that include reflex arteriolar and venous constriction, increased heart rate, and mechanical factors, such as the closure of valves in the venous system, pumping of the leg muscles, and a decrease in intrathoracic pressure. The normally increased sympathetic activity during upright posture is mediated through stretch receptors (baroreceptors) in the carotid sinus and the aortic arch (see Chapter 29). Their reflex response to shifts in volume caused by postural changes leads to a prompt increase in heart rate and constriction of the systemic arterioles, which maintains a stable blood pressure. These compensatory mechanisms are not effective in maintaining a stable blood pressure in individuals with orthostatic hypotension.
Orthostatic hypotension may be acute and temporary or chronic. Acute orthostatic hypotension, or temporary type, is caused when the normal regulatory mechanisms are sluggish. This delay may be the result of (1) anatomic variation, (2) altered body chemistry, (3) drug action (e.g., antihypertensives or antidepressants), (4) prolonged immobility caused by illness, (5) starvation, (6) physical exhaustion, (7) any condition that produces volume depletion (e.g., massive diuresis, potassium or sodium depletion), and (8) venous pooling (e.g., pregnancy, extensive varicosities of the lower extremities). Older adults are susceptible to this type of orthostatic hypotension, in which postural reflexes are slowed as part of the aging process.
The two forms of chronic orthostatic hypotension are (1) secondary to a specific disease and (2) idiopathic or primary. The diseases that cause secondary orthostatic hypotension are endocrine disorders (e.g., adrenal insufficiency, diabetes mellitus), metabolic disorders (e.g., porphyria), or diseases of the central or peripheral nervous system (e.g., intracranial tumors, cerebral infarcts, Wernicke encephalopathy, peripheral neuropathies). Cardiovascular autonomic neuropathy is a common cause of orthostatic hypotension in diabetes and is a serious and often overlooked complication.
Idiopathic, or primary, orthostatic hypotension is the term for hypotension in which there is no known initial cause. It affects men more often than women and usually occurs between the ages of 40 and 70 years. Up to 18% of the older adult population may be affected by chronic orthostatic hypotension.84 It is a significant risk factor for falls and associated injuries and has been associated with an increased risk for cardiovascular events. In addition to cardiovascular symptoms, impotence and bowel and bladder dysfunction often are found in this type. Orthostatic hypotension is also a feature of multiple system atrophy (MSA), in which there are multiple central nervous system degenerative changes.
Orthostatic hypotension often is accompanied by dizziness, blurring or loss of vision, and syncope or fainting. To assess hypotensive episode frequency, severity, and correlation with symptoms, 24-hour blood pressure monitoring is recommended.85 No curative treatment is available for idiopathic orthostatic hypertension. In the secondary form, syncope ceases when the underlying disorder is corrected. Several treatments can help acute and chronic orthostatic hypotension, including liberalization of salt intake, raising the head of the bed, thigh-high stockings, volume expansion with mineralocorticoids, and vasoconstrictors such as midodrine.84
Atherosclerosis
Atherosclerosis is a form of arteriosclerosis in which thickening and hardening of the vessel are caused by the accumulation of lipid-laden macrophages within the arterial wall, which leads to the formation of a lesion called a plaque. Atherosclerosis is not a single disease but rather a pathologic process that can affect vascular systems throughout the body, resulting in ischemic syndromes that can vary widely in their severity and clinical manifestations. It is the leading contributor to coronary artery and cerebrovascular disease. (Atherosclerosis of the coronary arteries is described on p. 1160; atherosclerosis of the cerebral arteries is discussed in Chapter 17.)
PATHOPHYSIOLOGY Although there remains considerable controversy as to the pathophysiology of atherosclerosis and many questions remain to be answered, the most widely accepted theories of the mechanisms of atherosclerosis are based on the finding that atherosclerosis is an inflammatory disease.86,87 Pathologically, the lesions progress from endothelial injury and dysfunction to fatty streak to fibrotic plaque to complicated lesion (Figures 30-11 and 30-12). Atherosclerosis begins with injury to the endothelial cells that line artery walls.86–88 Possible causes of endothelial injury include the common risk factors for atherosclerosis, such as smoking, hypertension, diabetes, increased levels of low-density lipoprotein (LDL), decreased levels of high-density lipoprotein (HDL), and autoimmunity. Other causes of endothelial injury are called the nontraditional risk factors, such as elevated CRP, increased serum fibrinogen, insulin resistance, oxidative stress, infection, and periodontal disease. These risk factors are discussed in more detail in the following section on coronary artery disease (see p. 1160). Recent evidence indicates that individuals with a defect in the production of precursor endothelial cells in the bone marrow are at greater risk for atherosclerotic disease because these precursor cells are not available to repair injured endothelium.89
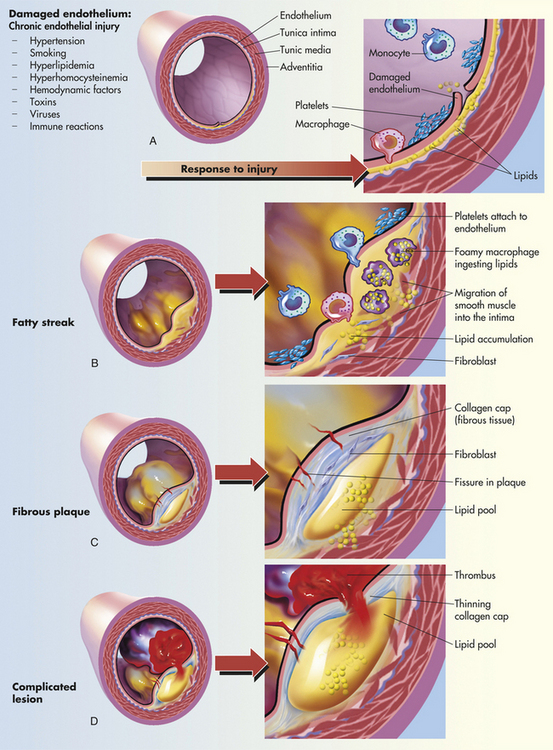
Figure 30-11 Progression of atherosclerosis. A, Damaged endothelium. B, Diagram of fatty streak and lipid core formation (see Figure 30-12 for a diagram of oxidized low-density lipoprotein [LDL]). C, Diagram of fibrous plaque. Raised plaques are visible: some are yellow; others are white. D, Diagram of complicated lesion; thrombus is red; collagen is blue. Plaque is complicated by red thrombus deposition.
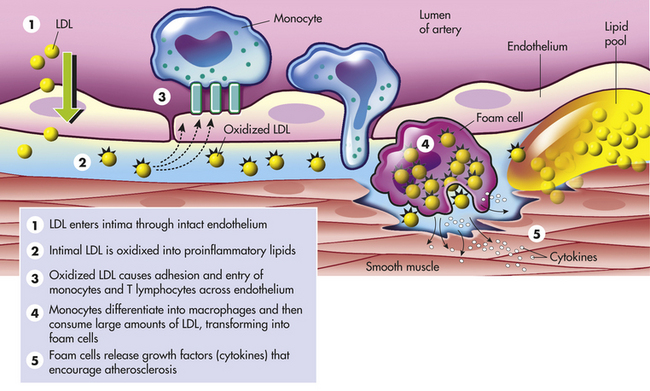
Figure 30-12 Low-density lipoprotein oxidation. (1) Low-density lipoprotein (LDL) enters the arterial intima through an intact endothelium. In hypercholesterolemia, the influx of LDL exceeds the eliminating capacity and an extracellular pool of LDL is formed. This is enhanced by association of LDL with the extracellular matrix. (2) Intimal LDL is oxidized through the action of free oxygen radicals formed by enzymatic or nonenzymatic reactions. (3) This generates proinflammatory lipids that induce endothelial expression of the adhesion molecule; vascular cell adhesion molecule-1 activates complement and stimulates chemokine secretion. All of these factors cause adhesion and entry of mononuclear leukocytes, particularly monocytes and T lymphocytes. (4) Monocytes differentiate into macrophages. Macrophages up-regulate and internalize oxidized LDL and transform into foam cells. Macrophage update of oxidized LDL also leads to presentation of fragments of it to antigen-specific T cells. (5) This induces an autoimmune reaction that leads to production of proinflammatory cytokines. Such cytokines include interferon-γ, tumor necrosis factor-α, and interleukin-1, which act on endothelial cells to stimulate expression of adhesion molecules and procoagulant activity; on macrophages to activate proteases, endocytosis, nitric oxide (NO), and cytokines; and on smooth muscle cells (SMCs) to include NO production and inhibit growth, collagen, and actin expression. (Modified from Crawford MH, DiMarco JP, editors: Cardiology, London, 2001, Mosby-Wolfe.)
Once injury has occurred, endothelial dysfunction and inflammation lead to the following pathophysiologic events:
1. Injured endothelial cells become inflamed and cannot make normal amounts of antithrombotic and vasodilating cytokines90–92 (see Figure 29-29).
2. Numerous inflammatory cytokines are released, including tumor necrosis factor-alpha (TNF-α), interferon-gamma (IFN-γ), interleukin-1 (IL-1), toxic oxygen radicals, and heat shock proteins.86–88
3. Macrophages adhere to injured endothelium by way of adhesion molecules, such as vascular cell adhesion molecule-1 (VCAM-1).86–88
4. These macrophages then release enzymes and toxic oxygen radicals that create oxidative stress, oxidize LDL, and further injure the vessel wall.86–88,93
5. Growth factors also are released, including angiotensin II, fibroblast growth factor, and platelet-derived growth factor, which stimulate smooth muscle cell proliferation in the affected vessel.86–88,94
The oxidation of LDL is an important step in atherogenesis. Inflammation with oxidative stress and activation of macrophages is the primary mechanism.86,93 Diabetes, smoking, and hypertension (especially with increased levels of angiotensin II) are associated with increased LDL oxidation. Oxidized LDL is toxic to endothelial cells, causes smooth muscle proliferation, and activates further immune and inflammatory responses.86–88,95 The oxidized LDL penetrates into the intima of the arterial wall and is engulfed by macrophages. Macrophages filled with oxidized LDL are called foam cells (see Figure 30-12).
Once these lipid-laden foam cells accumulate in significant amounts, they form a lesion called a fatty streak (see Figure 30-11). These lesions can be found in the walls of arteries of most people, even young children. Once formed, fatty streaks produce more toxic oxygen radicals and cause immunologic and inflammatory changes, resulting in progressive damage to the vessel wall. Decreasing levels of LDL can cause regression of atherosclerotic lesions and can improve endothelial function. Increasing attention is being given to the evaluation of children for dyslipidemia so that early dietary intervention to prevent atherosclerosis can be initiated.
At this point, smooth muscle cells proliferate, produce collagen, and migrate over the fatty streak forming a fibrous plaque (see Figure 30-11). This process is mediated by many inflammatory cytokines, including growth factors (e.g., transforming growth factor-beta [TGF-β]).96 The fibrous plaque may calcify, protrude into the vessel lumen, and obstruct blood flow to distal tissues, especially during exercise, which may cause symptoms (e.g., angina or intermittent claudication).
Many plaques, however, are “unstable,” meaning they are prone to rupture even before they affect blood flow significantly, and are clinically silent until they rupture. Plaque rupture occurs because of the inflammatory activation of proteinases, such as the matrix metalloproteinases and the cathepsins, and can be accelerated by bleeding within the lesion (plaque hemorrhage).97,98 Plaques that have ruptured are called complicated plaques (see Figure 30-11). Once rupture occurs, exposure of underlying tissue results in platelet adhesion, initiation of the clotting cascade, and rapid thrombus formation.86,87,91,92 The thrombus may suddenly occlude the affected vessel, resulting in ischemia and infarction. Aspirin or other antithrombotic agents are used to prevent this complication of atherosclerotic disease.99
CLINICAL MANIFESTATIONS Atherosclerosis presents with symptoms and signs that result from inadequate perfusion of tissues because of obstruction of the vessels that supply them. Partial vessel obstruction may lead to transient ischemic events, often associated with exercise or stress. Once the lesion becomes complicated, increasing obstruction with superimposed thrombosis may result in tissue infarction. CAD caused by atherosclerosis is the major cause of myocardial ischemia and is one of the most important health issues in the United States. Atherosclerotic obstruction of the vessels supplying the brain is the major cause of stroke. Similarly, any part of the body may become ischemic when its blood supply is compromised by atherosclerotic lesions. Often more than one vessel will become involved with this disease process; consequently, an individual may present with symptoms from several ischemic tissues at the same time, and disease in one area may indicate that the individual is at risk for other ischemic complications elsewhere.
EVALUATION AND TREATMENT In evaluating individuals for the presence of atherosclerosis, a complete health history including the presence of risk factors and symptoms of cardiovascular disease is essential. Physical examination may detect arterial bruits and evidence of decreased blood flow to tissues. Serum should be tested for risk indicators, such as lipid profile and the highly sensitive C-reactive protein (hs-CRP). If coronary disease is suspected, evaluation of plaques in affected vessels can include roentgenography, electrocardiography, intravascular ultrasonography, CT, MRI, nuclear scanning, and angiography.100–102
Current management of atherosclerosis includes detection of “preclinical” lesion treatment with drugs aimed at stabilizing plaques before they rupture.101,102 Once a lesion obstructs blood flow, the primary goal of management is to restore adequate flow to affected tissues. In situations in which the disease process does not require immediate intervention, management focuses on the reduction of risks to prevent continued endothelial injury and the prevention of plaque progression. Risk reduction includes smoking cessation, weight loss, and the control of hypertension, diabetes, and dyslipidemia through diet, exercise, and medication. Management of atherosclerotic risk factors is discussed further starting on p. 1161. If an individual presents with acute ischemia, such as myocardial infarction (MI) or stroke, interventions are specific to the diseased area (see Myocardial Infarction on p. 1191; see Stroke on p. 600).
Peripheral Artery Disease
Peripheral artery disease (PAD) refers to atherosclerotic disease of arteries that perfuse the limbs, especially the lower extremities. It is estimated that 12 million people in the United States have significant PAD.36 The risk factors for PAD are the same as those for atherosclerotic disease, and it is especially prevalent in individuals with diabetes.
Lower-extremity ischemia, resulting from arterial obstruction in PAD, can be gradual or acute. In most individuals, gradually increasing obstruction to arterial blood flow to the legs caused by atherosclerosis in the iliofemoral vessels results in pain with ambulation called intermittent claudication. If a thrombus forms over the atherosclerotic lesion, perfusion can cease acutely with severe pain, loss of pulses, and skin color changes in the affected extremity.
PAD is often asymptomatic; therefore, evaluation for PAD requires a careful history and physical examination that focuses on looking for evidence of atherosclerotic disease (e.g., bruits) and noninvasive Doppler measurement of blood flow. Treatment includes risk factor reduction (smoking cessation and treatment of diabetes, hypertension, and dyslipidemia) and antiplatelet therapy. Symptomatic PAD should be managed with vasodilators in combination with antiplatelet or antithrombotic medications (aspirin, cilostazol, ticlopidine, or clopidogrel) and exercise rehabilitation.103 If acute or refractory symptoms occur, emergent percutaneous or surgical revascularization may be indicated.
Coronary Artery Disease, Myocardial Ischemia, and Acute Coronary Syndromes
Coronary artery disease, myocardial ischemia, and myocardial infarction form a pathophysiologic continuum that impairs the pumping ability of the heart by depriving the heart muscle of blood-borne oxygen and nutrients. The earliest lesions of the continuum are those of coronary artery disease (CAD)—virtually any vascular disorder that narrows or occludes the coronary arteries. By far the most common cause of coronary artery obstruction is atherosclerosis (Figure 30-13). CAD can diminish the myocardial blood supply until deprivation impairs myocardial metabolism enough to cause ischemia, a local state in which the cells are temporarily deprived of blood supply. They remain alive but cannot function normally. Persistent ischemia or the complete occlusion of a coronary artery causes acute coronary syndrome, including infarction, or irreversible myocardial damage. Infarction constitutes the often-fatal event known as a heart attack.
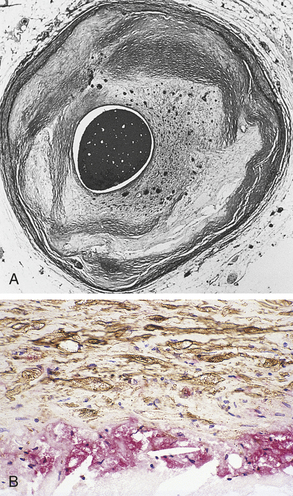
Figure 30-13 Atherosclerosis. A, Concentric coronary plaque. The lumen is central. Multiple new small blood vessels are shown within the plaque, the late result of disruption. B, Cell types in a fibrolipid plaque. The plaque cap (brownish) contains numerous elongated smooth muscle cells; some contain lipid. Macrophages are clustered on the edge of the core. (From Damjanov I, Linder J, editors: Anderson’s pathology, ed 10, St Louis, 1996, Mosby.)
Development of Coronary Artery Disease
More than 16 million people in the United States suffer from coronary artery disease—it is estimated that between 770,000 and 1 million people have a heart attack each year.36 Despite a dramatic decline in mortality in the past decade, CAD causes one third of all deaths in the United States. More than half of men and nearly one third of women older than age 40 will develop CAD.36 The primary cause of CAD is atherosclerosis of the coronary vessels; therefore, those factors that contribute to the development of atherosclerosis also are risk factors for CAD.
Risk factors for CAD can be categorized as conventional (major) versus nontraditional (novel) and modifiable versus nonmodifiable. Much new information has been obtained about the conventional risk factors that has markedly improved prevention and management of CAD. In addition, nontraditional risk factors have been identified in recent years that have provided insight in to the pathogenesis of CAD and may lead to more effective interventions.
Conventional or major risk factors for CAD that are nonmodifiable include (1) advanced age, (2) male gender or women after menopause, and (3) family history. Aging is associated with increased vulnerability to endothelial injury and decreased endothelial repair. The risk for CAD increases dramatically in women after menopause (see What’s New? Women and Heart Disease). Family history may contribute to the risk for CAD through genetics and shared environmental exposures. Many gene polymorphisms have been identified that are related to the development of atherosclerosis and its many associated underlying risk factors.104
Major modifiable conventional risks include (1) dyslipidemia, (2) hypertension, (3) cigarette smoking, (4) diabetes and insulin resistance, (5) obesity, (6) sedentary lifestyle, and (7) an atherogenic diet (see Nutrition & Disease: The Basics on Fats). In individuals with known CAD, the vast majority have the risk factors of smoking, diabetes, dyslipidemia, or hypertension,
and many have several of these risks. These traditional risk factors are often poorly controlled and are associated with the growing epidemic of obesity in the United States. If individuals receive appropriate preventive care, modification of these factors can dramatically reduce the risk for CAD.101,105 Of great concern is a recent finding that exposure to particulate matter air pollution, which is a worsening global problem, can also cause a significant increase in cardiovascular risk.106
Dyslipidemia: The strong link between CAD and elevated plasma lipoprotein concentrations is well documented.107 Lipoprotein refers to lipids, phospholipids, cholesterol, and triglycerides bound to carrier proteins. Lipids (cholesterol in particular) are required by most cells for the manufacture and repair of plasma membranes. Cholesterol is also a necessary component for the manufacture of such essential substances as bile acids and steroid hormones. Although cholesterol can easily be obtained from dietary fat intake, most body cells also can manufacture cholesterol.
The cycle of lipid metabolism is complex. Dietary fat is packaged into particles known as chylomicrons in the small intestine. Chylomicrons are required for absorption of fat; they function by transporting exogenous lipid from the intestine to the liver and peripheral cells. Chylomicrons are the least dense of the lipoproteins and primarily contain triglyceride.
Some of the triglyceride may be removed and either stored by adipose tissue or used by muscle as an energy source. The chylomicron remnants, composed mainly of cholesterol, are taken up by the liver. A series of chemical reactions in the liver results in the production of several lipoproteins that vary in density and function. These include very-low-density lipoproteins (VLDLs), primarily triglyceride and protein; low-density lipoproteins (LDLs), mostly cholesterol and protein; and high-density lipoproteins (HDLs), mainly phospholipids and protein.
Dyslipidemia (or dyslipoproteinemia) refers to abnormal concentrations of serum lipoproteins as defined by the Third Report of the National Cholesterol Education Program107 (Table 30-5). It is estimated that nearly half of the U.S. population has some form of dyslipidemia, especially among white and Asian populations.36 These abnormalities are the result of a combination of genetic and dietary factors. Primary or familial dyslipoproteinemias result from genetic defects that cause abnormalities in lipid-metabolizing enzymes and abnormal cellular lipid receptors (Table 30-6). Secondary causes of dyslipidemia include several common systemic disorders, such as diabetes, hypothyroidism, pancreatitis, and renal nephrosis, as well as the use of certain medications such as certain diuretics, beta-blockers, glucocorticoids, interferons, and antiretrovirals.108
Table 30-5

Data from Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults, JAMA 285:2486-2497, 2001.
Table 30-6
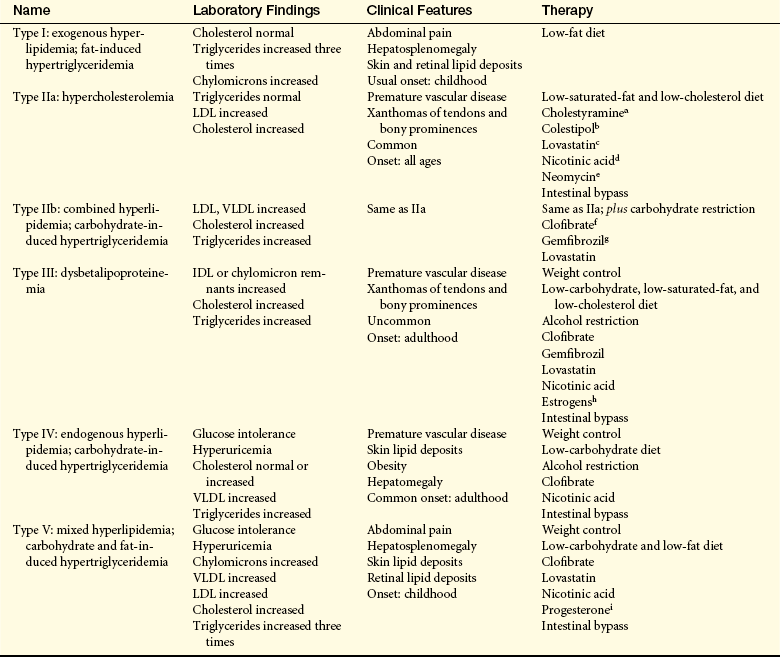
IDL, Intermediate-density lipoprotein; LDL, low-density lipoprotein; VLDL, very-low-density lipoprotein.
aCholestyramine (Questran), anion exchange resin; binds bile acids; enhances cholesterol excretion.
bColestipol (Colestid), same as cholestyramine.
cLovastatin, 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitor; decreases cholesterol synthesis in the liver.
dNicotinic acid (niacin), decreases release of free fatty acids from adipose tissue; increases lipogenesis in liver; decreases glucagon release; most effective for type V disorder.
eNeomycin, experimental medication; questionable mode of action; decreases LDLs.
fClofibrate (Atromid-S), decreases release of free fatty acids from adipose tissue; decreases hepatic secretion of VLDL and increases catabolism of VLDL.
gGemfibrozil (Lopid), similar to clofibrate but increases HDLs more.
hEstrogens, decrease IDL levels in type III disorders; experimental.
iProgesterone, decreases plasma triglycerides in type V disorders; experimental.
An increased serum concentration of LDL is a strong indicator of coronary risk.36,108,109 LDL is responsible for the delivery of cholesterol to the tissues. Serum levels of LDL are normally controlled by hepatic receptors for LDL that bind LDL and limit liver synthesis of this lipoprotein. High dietary intake of cholesterol and fats, often in combination with a genetic predisposition to accumulations of LDL in the serum (e.g., dysfunction of the hepatic LDL receptor), results in high levels of LDL in the bloodstream.108 The term LDL actually describes several types of LDL molecules; the “small dense” LDL particles are the most atherogenic. LDL oxidation, migration into the vessel wall, and phagocytosis by macrophages are key steps in the pathogenesis of atherosclerosis86–88 (see p. 1157 and Figure 30-12). LDL also plays a role in endothelial injury, inflammation, and immune responses that have been identified as being important in atherogenesis. Aggressive reduction of LDL with diet and cholesterol-lowering drugs, such as the statins and ezetimibe, is associated with a dramatic decrease in risk for CAD.108–112
Low levels of HDL cholesterol also are a strong indicator of coronary risk, and high levels of HDL may be more protective for the development of atherosclerosis than low levels of LDL.36,107,113,114 HDL is responsible for “reverse cholesterol transport,” which returns excess cholesterol from the tissues to the liver, where it binds to hepatic receptors (including the LDL receptor) and is processed and eliminated as bile or converted to cholesterol-containing steroids.113 HDL also participates in endothelial repair and decreases thrombosis.113,114 It can be fractionated into several particle densities (HDL-2 and HDL-3) or sizes (large, medium, small) that have different effects on vascular function. HDL-2 is most effective at reverse cholesterol transport and its selective measurement is a better indicator of cardiovascular risk than total HDL levels.113,114 It has been found that inflammation in early atherogenesis results in the production of toxic oxygen radicals that can reduce or eliminate the protective function of HDL.115 Exercise, weight loss, fish oil consumption, and moderate alcohol use can result in modest increases in HDL. Niacin, fibrates, and statins are drugs that can cause modest increases in HDL. Newer drugs aimed specifically at increasing HDL include recombinant apolipoprotein A-I (ApoA-I) mimetics, thiazolidinediones (used to treat diabetes), and cholesteryl ester transfer protein inhibitors, although the safety of these medications is still being evaluated.113,116
Other lipoproteins associated with increased cardiovascular risk include elevated serum VLDL (triglycerides) and increased lipoprotein (a). Triglycerides are associated with an increased risk for CAD, especially in combination with other risk factors such as diabetes.107 Because of this, the measurement of “non-HDL cholesterol” (LDL plus VLDL) is frequently used to assess cardiovascular risk rather than just LDL or HDL levels alone.117 Lipoprotein (a) (Lp[a]) is a genetically determined molecular complex between LDL and a serum glycoprotein called apoprotein (a) that has been shown to be an important risk factor for atherosclerosis, especially in women.117
Hypertension: Hypertension is responsible for a two- to threefold increased risk of atherosclerotic cardiovascular disease. It contributes to endothelial injury, a key step in atherogenesis (see p. 1157), and causes myocardial hypertrophy, which increases myocardial demand for coronary flow. The overactivity of the SNS and RAAS commonly found in hypertension also contributes to the genesis of coronary artery disease. Drugs that block the effects of the SNS and RAAS to treat hypertension have many positive effects on the vasculature.118,119
Cigarette Smoking: Direct and passive (environmental) smoking increase the risk of CAD. The mechanism by which smoking increases atherosclerosis is uncertain. Nicotine stimulates the release of catecholamines (epinephrine and norepinephrine), which increases heart rate and causes peripheral vascular constriction. As a result blood pressure increases, as do cardiac workload and oxygen demand. Cigarette smoking is associated with an increase in LDL and a decrease in HDL, and contributes to vessel inflammation and thrombosis. The risk of CAD increases with heavy smoking and decreases when smoking is stopped.
Diabetes Mellitus: Diabetes mellitus is an extremely important risk factor for CAD.36 Diabetes and insulin resistance have multiple effects on the cardiovascular system. These effects can include endothelial damage, thickening of the vessel wall, increased inflammation and leukocyte adhesion, increased thrombosis, glycation of vascular proteins, and decreased production of endothelial-derived vasodilators such as nitric oxide.120–122 Diabetes is also associated with dyslipidemia because of the resulting alteration of hepatic lipoprotein synthesis and increases in LDL oxidation.122 Aggressive management of this additional risk factor can significantly improve CAD risk in individuals with diabetes.
Obesity and Sedentary Lifestyle: It is estimated that nearly two thirds of the adult population in the U.S. is overweight or obese resulting in a much increased risk for CAD and stroke.36 An estimated 47 million residents have a combination of obesity, dyslipidemia, and hypertension (called the metabolic syndrome) (see Chapter 21), which is associated with an even higher risk for CAD events.123 Obesity is caused by genetics, diet, and inadequate physical exercise. Abdominal obesity has the strongest link with increased CAD risk and is related to insulin resistance, decreased HDL, increased blood pressure, inflammation, and decreased levels of a recently described cardioprotective protein called adiponectin.124–127 (see What’s New? New Serum Markers of Cardiovascular Risk). A sedentary lifestyle not only increases the risk of obesity but also has an independent effect on increasing CAD risk. Physical activity and weight loss offer substantial reductions in risk factors for CAD.
Nontraditional Risk Factors: Nontraditional, or novel, risk factors for CAD include (1) increased serum markers for inflammation and thrombosis, (2) hyperhomocysteinemia, (3) adipokines, and (4) infection. The amount of risk conferred by these relatively newly identified factors is still being explored.
Markers of Inflammation and Thrombosis: Of the numerous markers of inflammation that have been linked to an increase in CAD risk (hs-CRP, fibrinogen, protein C, plasminogen activator inhibitor), the relationship between serum levels of CRP and CAD has been explored in the greatest depth. Highly sensitive C-reactive protein (hs-CRP) is an acute phase reactant or protein mostly synthesized in the liver and is an indirect measure of atherosclerotic plaque-related inflammation.128 Elevated levels of hs-CRP are associated with numerous other CAD risk factors including smoking, obesity, and diabetes. However, as a nonspecific serum marker for inflammation, its utility as a screening tool for cardiovascular risk continues to be debated.101,129 Other markers of inflammation associated with CAD include erythrocyte sedimentation rate, von Willebrand factor concentration, uric acid, IL-6, IL-18, TNF-a, fibrinogen, and CD40 ligand.129
Hyperhomocysteinemia: Hyperhomocysteinemia occurs because of a genetic lack of the enzyme that breaks down homocysteine (an amino acid) or because of a nutritional deficiency of folate, cobalamin (vitamin B12), or pyridoxine (vitamin B6). It has been identified as a risk factor for CAD, although its significance in CAD and stroke continues to be explored.130,131 Mechanisms by which it contributes to coronary disease include associated increases in LDL oxidation, decreases in endogenous vasodilators, increased smooth muscle proliferation, and an increased tendency for thrombosis.131 Routine serum measurement of homocysteine is not currently recommended and prevention and management are focused on increasing the dietary intake of folate and B vitamins; however, the efficacy of vitamin supplementation in improving cardiovascular risk has not been proved.
Adipokines: Adipokines are a group of hormones released from adipose cells. The two that are most studied are adiponectin and leptin. Leptin is primarily implicated in hypertension (see p. 1149) but is also being explored as a factor in diabetes and degenerative joint disease.132 Adiponectin is normally antiatherogenic and is decreased in obesity. It functions to protect the vascular endothelium and is anti-inflammatory. Decreased adiponectin in obese individuals has been linked to a significant increase in cardiovascular risk.127,133,134 A more recently described adipokine is resistin, which has been linked to inflammation in vascular endothelial cells.131 Weight loss, exercise, and treatment with statins improve adipokine levels and are correlated with improved cardiovascular risk.
Infection: Infection may play a role in atherogenesis and CAD risk, although cause and effect has not been proved. Several microorganisms, especially Chlamydia pneumoniae, Helicobacter pylori, and cytomegalovirus, are associated with atherosclerotic lesions and the presence of serum antibodies to microorganisms have been linked to an increased risk for CAD.135,136 Periodontal disease also has been linked to an increased risk for CAD. One hypothesis is that systemic infection results in increased local inflammation of vessels and therefore contributes to vascular disease.137 Unfortunately, the use of antibiotics for the prevention and treatment of CAD has not yielded consistently positive results.
Myocardial Ischemia
PATHOPHYSIOLOGY The coronary arteries normally supply blood flow sufficient to meet the demands of the myocardium as it labors under varying workloads. Oxygen extraction from these vessels occurs with maximal efficiency. If efficient exchange does not meet myocardial oxygen needs, healthy coronary arteries are able to dilate to increase the flow of oxygenated blood to the myocardium. A variety of pathologic mechanisms can interfere with blood flow through the coronary arteries, giving rise to myocardial ischemia. Narrowing of a major coronary artery by more than 50% impairs blood flow sufficiently to hamper cellular metabolism under conditions of increased myocardial demand (see Figure 30-13).
Myocardial ischemia develops if the supply of coronary blood cannot meet the demand of the myocardium for oxygen and nutrients. Imbalances between myocardial demand and coronary blood supply can result from a number of conditions. Common causes of increased myocardial demand for blood include tachycardia, exercise, hypertension (hypertrophy), and valvular disease. The most common cause of decreased coronary blood flow and resultant myocardial ischemia is the formation of atherosclerotic plaques in the coronary circulation. As the plaque increases in size, it may partially occlude the vessel lumina, thus limiting coronary flow and causing ischemia especially during exercise. Some plaques are “unstable,” meaning they are prone to ulceration or rupture (see p. 1159 and Figure 30-20). When this occurs, underlying tissues of the vessel wall are exposed resulting in platelet adhesion and thrombus formation. This can suddenly cut off blood supply to the heart muscle, resulting in acute myocardial ischemia, and if the vessel obstruction cannot be reversed rapidly, ischemia will progress to infarction. Myocardial ischemia also can result from other causes of decreased blood and oxygen delivery to the myocardium, such as coronary spasm, hypotension, dysrhythmias, and decreased oxygen-carrying capacity of the blood (anemia, hypoxemia).
Myocardial cells become ischemic within 10 seconds of coronary occlusion. After several minutes the heart cells lose the ability to contract, and cardiac output decreases. Ischemia also causes conduction abnormalities that lead to changes in the electrocardiogram and may initiate dysrhythmias. Anaerobic processes take over, and lactic acid accumulates. Cardiac cells remain viable for approximately 20 minutes under ischemic conditions. If blood flow is restored, aerobic metabolism resumes, contractility is restored, and cellular repair begins. If the coronary artery occlusion persists beyond 20 minutes, myocardial infarction occurs (Figure 30-14).
CLINICAL MANIFESTATIONS Individuals with reversible myocardial ischemia present clinically in several ways. Chronic coronary obstruction usually results in recurrent predictable chest pain called stable angina. Abnormal vasospasm of coronary vessels results in unpredictable chest pain called Prinzmetal angina. Myocardial ischemia that does not cause detectable symptoms is called silent ischemia.
Stable Angina: Angina pectoris is chest pain caused by myocardial ischemia. The discomfort is usually transient, lasting approximately 3 to 5 minutes. If blood flow is restored, no permanent change or damage results. Angina pectoris is typically experienced as substernal chest discomfort, ranging from a sensation of heaviness or pressure to moderately severe pain. Individuals often describe the sensation by clenching a fist over the left sternal border. Discomfort may radiate to the neck, lower jaw, left arm, and left shoulder or, occasionally, to the back or down the right arm. Discomfort is commonly mistaken for indigestion. The pain is caused by the buildup of lactic acid or abnormal stretching of the ischemic myocardium that irritates myocardial nerve fibers. These afferent sympathetic fibers enter the spinal cord from levels C3 to T4, accounting for the variety of locations and radiation patterns of anginal pain.138 Pallor, diaphoresis, and dyspnea may be associated with the pain. Stable angina is caused by gradual luminal narrowing and hardening of the arterial walls, so that affected vessels cannot dilate in response to increased myocardial demand associated with physical exertion or emotional stress. The pain is usually relieved by rest and nitrates; lack of relief indicates an individual may be developing infarction.
Myocardial ischemia in women may not present with typical anginal pain. Common symptoms in women include atypical chest pain, palpitations, sense of unease, and severe fatigue. Similarly, in individuals who have autonomic dysfunction, such as older adults or those with diabetes, angina may be mild, atypical, or even silent (see following).
Prinzmetal Angina: Prinzmetal angina (also called variant angina) is chest pain attributable to transient ischemia of the myocardium that occurs unpredictably and almost exclusively at rest. Pain is caused by vasospasm of one or more major coronary arteries with or without associated atherosclerosis. The pain often occurs at night during rapid eye movement sleep and may have a cyclic pattern of occurrence. The angina may result from hyperactivity of the SNS, increased calcium reflux in arterial smooth muscle, or impaired production or release of serotonin, histamine, endothelin, or thromboxane.139 If the spasm persists long enough, infarction results. Angina is usually a benign condition, but can occasionally cause serious dysrhythmias.140
Silent Ischemia and Mental Stress (Induced Ischemia):
Myocardial ischemia does not always cause angina and may be associated only with nonspecific symptoms such as fatigue, dyspnea, or feeling of unease141 (see What’s New? Women and Heart Disease, p. 1161). Ischemia can be totally asymptomatic and referred to as silent ischemia. Some individuals only have silent ischemia, and episodes of silent myocardial ischemia are common in individuals who also experience angina. One proposed mechanism for the absence of angina in silent myocardial ischemia is the presence of a global or regional abnormality in left ventricular sympathetic afferent innervation. Such an abnormality might occur as part of a metabolic dysfunction in diabetes mellitus, following surgical denervation during coronary artery bypass grafting (CABG) or cardiac transplantation, or following ischemic local nerve injury by myocardial infarction.
Another area that is receiving renewed interest is the lack of angina, even though an artery is occluded, in some individuals during mental stress (Figures 30-15, 30-16, and 30-17). Rozanski documented myocardial ischemia by radionuclide angiography (RNA) during mental stress; the majority of these cases (83%) were silent ischemias.142 They also noted a smaller increase in heart rate during mental stress than during exercise, although the systolic blood response was comparable and the diastolic blood pressure response is even greater with mental stress. These observations suggest that increases in blood pressure and myocardial oxygen demand induced by mental stress play a role in the pathophysiology of myocardial ischemia. In addition, acute stress has been shown to increase markers of inflammation, such as CRP, and chronic stress has been linked to a hypercoagulable state that may contribute to acute ischemic events.143,144 Although stress management has been associated with a reduction in CAD events in men, further research into the brain-heart pathways is under way to better elucidate appropriate interventions.145
Figure 30-15 Angiogram. A, Baseline (arrow points to narrowing). B, Transient total occlusion of left anterior descending branch of the left coronary artery after mental stress (arrow). C, After administration of nitrates and nifedipine, artery reopened to same diameter as baseline (arrow). (From Stern S, editor: Silent myocardial ischemia, St Louis, 1998, Mosby.)
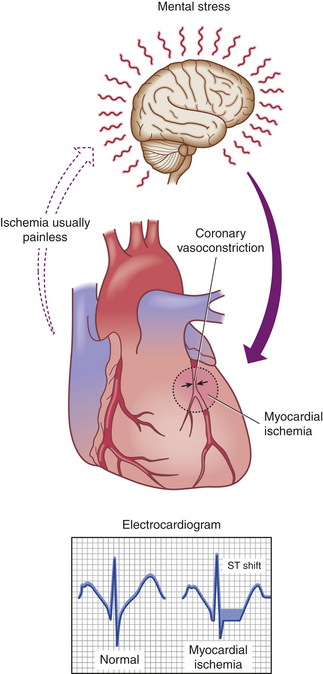
Figure 30-16 The ischemic cost of aggravation. Linkages among daily mental and emotional stimuli, brain activity, and coronary and myocardial physiology. (Modified from Papodemetrion V et al: Am Heart J 132:1299, 1996.)
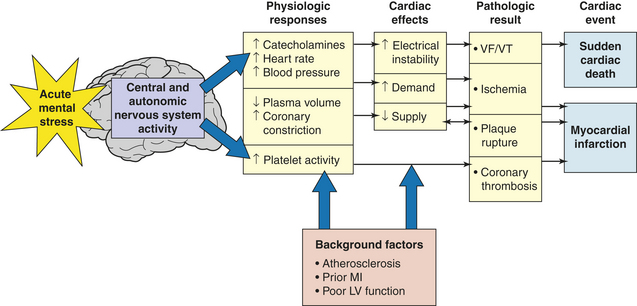
Figure 30-17 Pathophysiologic model of acute stress effects triggering cardiac clinical events. Acting via the central and autonomic nervous systems, stress can produce a cascade of physiologic responses that may lead to myocardial ischemia, potentially fatal dysrhythmia, plaque rupture, or coronary thrombosis. LV, Left ventricular; MI, myocardial infarction; VF, ventricular fibrillation; VT, ventricular tachycardia. (From Kranz DS et al: Mental stress as a trigger of myocardial ischemia and infarction. In Deedwania PC, Tofler GH, editors: Triggers and timing of cardiac events, ed 2, London, 1996, Saunders.)
Screening for silent ischemia is based on the presence of risk factors.146 Silent ischemia is detected with greater sensitivity and specificity using stress radionucleotide imaging than by exercise electrocardiogram testing alone. Detection of silent ischemia is important because it is an indicator of increased risk for serious CVD events, and aggressive treatment may be indicated.147
EVALUATION AND TREATMENT Many individuals with reversible myocardial ischemia exhibit a normal physical examination between episodes. Physical examination of an individual experiencing myocardial ischemia may disclose tachycardia, extra heart sounds (gallops or murmurs), and pulmonary congestion indicating impaired left ventricular function. The presence of xanthelasmas (small fat deposits) around the eyelids or arcus senilis of the eyes (a yellow lipid ring around the cornea) suggests dyslipidemia and possible atherosclerosis. The presence of peripheral or carotid arterial bruits suggests probable atherosclerotic disease and increases the likelihood that CAD is present.
Electrocardiography is a critical tool for the diagnosis of myocardial ischemia. Because many individuals have normal ECGs in the absence of pain, diagnosis requires that electrocardiography be performed during an attack of angina. Transient ST segment depression and T wave inversion are characteristic signs of subendocardial ischemia frequently seen in angina. ST elevation, indicative of transmural ischemia, can be seen in individuals with variant angina but is more common in infarction (Figure 30-18). The ECG also can give some indication of which coronary artery is involved. Approximately 30% of individuals with angina have nondiagnostic ECG tracings and require other diagnostic studies.
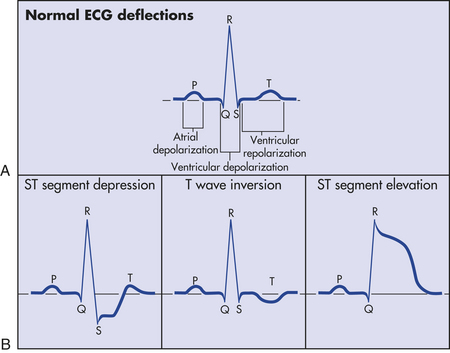
Figure 30-18 Electrocardiogram (ECG) and ischemia. A, Normal ECG. B, Electrocardiographic alterations associated with ischemia.
Exercise stress testing is useful in differentiating angina from other types of chest pain, as well as detecting ischemic changes that occur in the absence of anginal pain (silent ischemia). Stress testing is made more sensitive when radioisotope imaging is added to the ECG as an indicator of myocardial ischemia. Currently, the modality of choice for the diagnosis of myocardial ischemia is single-photon emission computed tomography (SPECT), which is effective at identifying ischemia and estimating risk for coronary events. Radioisotope imaging with thallium-201 and stress echocardiography also are used.
Imaging the coronary arteries for the evaluation of atherosclerotic plaques involves the use of CT with and without angiography, MRI, or intravascular ultrasound.148 Coronary angiography is useful in determining the anatomic extent of CAD. The procedure is expensive and carries some risk; thus it is used primarily to evaluate for possible percutaneous coronary intervention (PCI) or CABG surgery for individuals whose noninvasive studies suggest severe disease.
The primary aim of therapy for myocardial ischemia and angina is to increase delivery of oxygen by improving coronary artery blood flow and to reduce myocardial oxygen consumption by favorably altering its various determinants. Coronary blood flow is improved by reversing vasoconstriction, preventing clotting, and reducing plaque growth and rupture. Myocardial oxygen consumption is reduced by manipulation of blood pressure, heart rate, contractility, and left ventricular volume. Several classes of drugs are useful for increasing coronary flow and decreasing myocardial demand, especially nitrates, beta blockers, and calcium channel blockers.149,150
Nitrates improve coronary blood flow by reducing coronary artery spasm and thereby increase myocardial blood supply, but their actions are impaired in vessels with significant atherosclerosis. Nitrates also reduce myocardial demand by causing peripheral veins and, to a lesser extent, peripheral arteries to dilate. Dilation reduces peripheral vascular resistance and venous return to the heart (preload) and thereby reduces left ventricular filling pressure and decreases workload for the heart.
β-Adrenergic blockade also helps restore the balance between oxygen supply and myocardial demand. Beta blockers diminish catecholamine-induced elevations of heart rate, myocardial contractility, and blood pressure. Reduction in heart rate provides additional diastolic filling time for coronary perfusion, leading to enhanced oxygen delivery to the heart.
Calcium plays a key role in the electrical excitation of cardiac cells and in mechanical contraction of the myocardial and vascular smooth muscle cells (see Chapter 29). By blocking the influx of calcium into myocardial cells and vascular smooth muscle cells, the pacemaker activity of the sinoatrial (SA) node and conduction properties of the atrioventricular (AV) node can be modified so that myocardial oxygen demand is reduced. Thus calcium channel blockers can improve myocardial ischemia and reduce anginal symptoms; however, short-acting formulations should be used with caution because of potentially harmful effects on cardiac contractility and heart rate.149
Combinations of nitrates, beta-blockers, and calcium antagonists may provide dramatic relief from clinical manifestations of ischemic heart disease and make more invasive interventions unnecessary. However, they do not reverse the atherosclerotic process; thus the individual remains at risk for persistent or worsening CAD. Recommendations for appropriate diet, exercise, and risk reduction strategies have been widely distributed. Medications aimed at plaque stabilization, regression, and prevention of clotting also are indicated in the majority of individuals.150 These include statins, ACE inhibitors or receptor blockers, and antithrombotics. Statins have been shown to significantly improve vascular function, help stabilize plaques, promote plaque regression, and reduce the risk of coronary events.109-112,150,151 Angiotensin blockade lowers blood pressure, reduces myocardial hypertrophy, and improves vascular function.149,150 Antithrombotics are important for reducing the risk of clot formation on vulnerable plaques and therefore reduce the risk for developing the acute coronary syndromes. Currently recommended antiplatelet agents include aspirin and clopidogrel, although some individuals may require warfarin.150,152
Percutaneous coronary intervention (PCI) is a procedure whereby stenotic (narrowed) coronary vessels are dilated with a catheter. Several different types of catheters can be used to open the blocked vessel. PCI is generally used to treat single-vessel disease, but it can be effective with multiple-vessel disease or restenosis of a CABG in selected individuals.150,153 Restenosis of the artery is the major complication of the procedure; however, placement of a coronary stent can reduce this risk. Antithrombotic treatment with glycoprotein (GP) IIb-IIIa receptor antagonists after stenting also can greatly improve outcomes.153
Ischemic heart disease can be surgically treated by a coronary artery bypass graft. A saphenous vein from the lower leg is most commonly used to bypass the obstructed coronary artery. A technique using the left internal mammary artery (LIMA) rather than the saphenous vein has shown significant improvement in long-term graft patency. In selected individuals a procedure called minimally invasive direct coronary artery bypass (MIDCAB) can allow for effective bypass grafting of the heart, but with minimal disruption of the chest wall and without cardiopulmonary bypass or cardioplegia; thus recovery times are much shorter. One of the most common indications for bypass surgery is incapacitating angina in an individual who has good left ventricular function and technically operable coronary arteries but who has not responded to medical therapy and is not a candidate for PCI. When compared with PCI, CABG is more effective in relieving anginal symptoms but has more risks.154 Although surgery has been shown to relieve angina, it does not halt the progression of atherosclerosis. Surgery also does not prolong life except when multiple coronary vessels are obstructed or when the left main coronary artery is blocked. A successful coronary artery bypass graft can, however, diminish the probability of lethal insult to the coronary tissues and can markedly improve quality of life.
Newer therapies for refractory angina include transmyocardial laser revascularization (TMR), gene therapy for myocardial angiogenesis, enhanced external balloon counterpulsation, spinal cord stimulation, laser revascularization, and percutaneous in situ coronary venous arterialization. Drugs being evaluated in the medical management of myocardial ischemic syndromes include ranolazine, trimetazidine, nicorandil, ivabradine, fasudil, and molsidomine.149
Acute Coronary Syndromes
The process of atherosclerotic plaque progression can be gradual. However, when there is sudden coronary obstruction caused by thrombus formation over a ruptured or ulcerated atherosclerotic plaque, acute coronary syndromes result (Figure 30-19). Unstable angina is the result of reversible myocardial ischemia and is a harbinger of impending infarction. Myocardial infarction (MI) results when there is prolonged ischemia causing irreversible damage to the heart muscle. MI can be further subdivided into non–ST elevation MI (non-STEMI) and ST elevation MI (STEMI). Sudden cardiac death can occur as a result of any of the acute coronary syndromes.
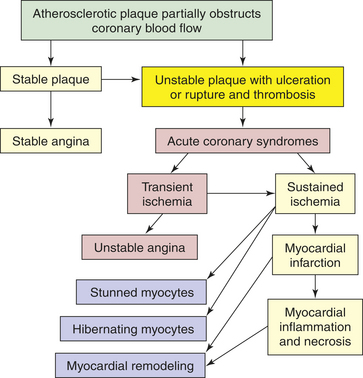
Figure 30-19 Pathophysiology of acute coronary syndromes. The atherosclerotic process can lead to stable plaque formation and stable angina or can result in unstable plaques that are prone to rupture and thrombosis. Thrombus formation on a ruptured plaque that disperses in less than 20 minutes leads to transient ischemia and unstable angina. If the vessel obstruction is sustained, myocardial infarction with inflammation and necrosis of the myocardium result. In addition, myocardial infarction is associated with other structural and functional changes, including myocyte stunning and hibernation and myocardial remodeling.
An atherosclerotic plaque that is prone to rupture is called unstable and has a core that is especially rich in deposited oxidized LDL and a thin fibrous cap (Figure 30-20.) Plaque disruption (ulceration or rupture) occurs because of shear forces, inflammation with release of multiple inflammatory mediators, secretion of macrophage-derived degradative enzymes, and apoptosis of cells at the edges of the lesions.85-88,98,155,156 Exposure of the plaque substrate activates the clotting cascade.91,92 In addition, platelet activation results in the release of coagulants and exposure of platelet GPIIb-IIIa surface receptors, resulting in further platelet aggregation and adherence.91,92 The resulting thrombus can form very quickly. Vessel obstruction is further exacerbated by the release of vasoconstrictors such as thromboxane A2 and endothelin. The thrombus may break up before permanent myocyte damage has occurred (unstable angina), or it may cause prolonged ischemia with infarction of the heart muscle (myocardial infarction). Some individuals have sudden cardiac death without underlying histologic evidence of infarction most often caused by ischemia-initiated dysrhythmias.
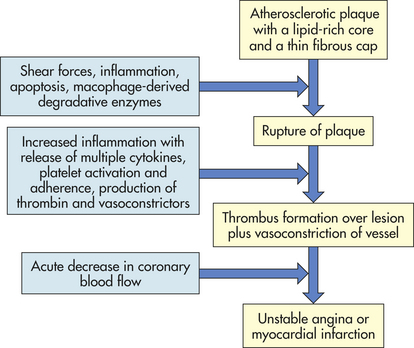
Unstable Angina: Unstable angina is a form of acute coronary syndrome that results in reversible myocardial ischemia. Important, however, is that it signals that the atherosclerotic plaque has become complicated, and infarction may soon follow.
PATHOPHYSIOLOGY A fairly small fissuring or superficial erosion of the plaque leads to transient episodes of thrombotic vessel occlusion and vasoconstriction at the site of plaque damage.156 This thrombus is labile and occludes the vessel for no more than 10 to 20 minutes, with return of perfusion before significant myocardial necrosis occurs.
CLINICAL MANIFESTATIONS Unstable angina presents as new-onset angina, angina that is occurring at rest, or angina that is increasing in severity or frequency (Box 30-1). Individuals may experience increased dyspnea, diaphoresis, and anxiety as the angina worsens. Those with unstable angina at rest have the greatest risk of subsequent infarction or death.
EVALUATION AND MANAGEMENT Physical examination may reveal evidence of ischemic myocardial dysfunction such as tachycardia, S3 gallop, or pulmonary congestion. The ECG most commonly reveals ST segment depression and T wave inversion during pain that resolves as the pain is relieved. The ECG may be inconclusive in up to one third of individuals with unstable angina, for whom further evaluation is necessary. The cardiac biomarkers (troponins, creatine phosphokinase-myocardial bound [CPK-MB], and lactate dehydrogenase [LDH1]) remain normal. Emergency echocardiography may reveal abnormal cardiac contraction. Approximately 20% of individuals with unstable angina progress to myocardial infarction or death within 30 days.156 Management of unstable angina requires immediate hospitalization with administration of oxygen, aspirin (if not contraindicated), nitrates, and morphine if pain is still present. Additional antithrombotic therapy with clopidogrel or GP IIb-IIIa platelet receptor antagonists may be indicated. Beta-blockers and ACE inhibitors, anticoagulants such as low-molecular-weight heparin, or direct thrombin inhibitors (e.g., fondaparinux) also can be given. Individuals with refractory angina and those with electrical or hemodynamic instability require immediate intervention with PCI or CABG.156,157 Those who are stabilized with medications are then assessed for potential PCI or CABG. After discharge, individuals will continue on antithrombotics and also should be managed with risk factor reduction, including the use of statins.156–159
Myocardial Infarction: When coronary blood flow is interrupted for an extended period, myocyte necrosis occurs.160 This results in MI. In the majority of cases of MI, the decrease in coronary flow is the result of atherosclerotic CAD; other causes include coronary spasm and coronary artery embolism.161 Pathologically there are two major types of myocardial infarction: subendocardial infarction and transmural infarction. Clinically, however, myocardial infarction is categorized as non-STEMI or STEMI.
PATHOPHYSIOLOGY Plaque progression, disruption, and subsequent clot formation is the same for myocardial infarction as it is for unstable angina (see Figures 30-19, 30-20, and 30-21). In this case, however, the thrombus is less labile and occludes the vessel for a prolonged period, such that myocardial ischemia progresses to myocyte necrosis and death. If the thrombus breaks up before complete distal tissue necrosis has occurred, the infarction will involve only the myocardium directly beneath the endocardium (subendocardial MI). This infarction usually presents with ST depression and T wave inversion without Q waves, so is termed non-STEMI. It is especially important to recognize this form of acute coronary syndrome because recurrent clot formation on the disrupted atherosclerotic plaque is likely unless some intervention is undertaken as soon as possible. If the thrombus lodges permanently in the vessel, the infarction will extend through the myocardium all the way from endocardium to epicardium (transmural MI), resulting in severe cardiac dysfunction. Clinically it is important to identify those individuals with transmural infarction who are at highest risk for serious complications and who should receive definitive intervention without delay. Those individuals usually have marked elevations in the ST segments on ECG and are categorized as having STEMI.
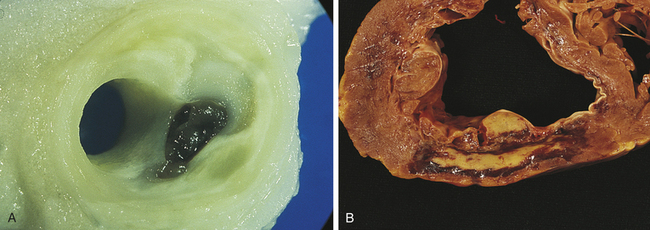
Figure 30-21 Plaque disruption and myocardial infarction. A, Plaque disruption. The cap of the lipid-rich plaque has become torn with the formation of a thrombus, mostly inside the plaque. B, Myocardial infarction. This infarct is 6 days old. The center is yellow and necrotic with a hemorrhagic red rim. The responsible coronary artery occlusion is probably in the right coronary artery. The infarct is on the posterior wall. (From Damjanov I, Linder J, editors: Anderson’s pathology, ed 10, St Louis, 1996, Mosby.)
Cellular Injury: Cardiac cells can withstand ischemic conditions for about 20 minutes before cellular death takes place. After only 30 to 60 seconds of hypoxia, ECG changes are visible. Yet even if cells are metabolically altered and nonfunctional, they can remain viable if blood flow returns within 20 minutes. Reports suggest previous recurrent episodes of myocardial ischemia can result in myocyte adaptation to oxygen deprivation and preservation of myocardium. This process, termed ischemic preconditioning, is being studied to determine whether it has potential prophylactic or therapeutic uses. Although this phenomenon is now well described in other organs, such as the liver, its clinical utility in heart disease and surgery has yet to be determined.161,162
After 8 to 10 seconds of decreased blood flow, the affected myocardium becomes cyanotic and cooler. Myocardial oxygen reserves are used very quickly (within about 8 seconds) after complete cessation of coronary flow. Glycogen stores decrease as anaerobic metabolism begins. Unfortunately, glycolysis can supply only 65% to 70% of the total myocardial energy requirement and produces much less adenosine triphosphate (ATP) than aerobic processes. Hydrogen ions and lactic acid accumulate. Because myocardial tissues have poor buffering capabilities and myocardial cells are very sensitive to low cellular pH, accumulation of these products further compromises the myocardium.161 Acidosis may make the myocardium more vulnerable to the damaging effects of lysosomal enzymes and may suppress impulse conduction and contractile function, thereby leading to heart failure.
Oxygen deprivation also is accompanied by electrolyte disturbances—specifically, loss of potassium, calcium, and magnesium from cells. Myocardial cells deprived of necessary oxygen and nutrients lose contractility, thereby diminishing the pumping ability of the heart. Ischemic myocardial cells release catecholamines (epinephrine and norepinephrine), predisposing the individual to serious imbalances of sympathetic and parasympathetic function, irregular heartbeats (dysrhythmia), and heart failure. Catecholamines mediate the release of glycogen, glucose, and stored fat from body cells. Therefore, plasma concentrations of free fatty acids and glycerol rise within 1 hour after onset of acute myocardial infarction. Excessive levels of free fatty acids can have a harmful detergent effect on cell membranes. Norepinephrine elevates blood sugar levels through stimulation of liver and skeletal muscle cells. It also suppresses pancreatic B-cell activity, which reduces insulin secretion and elevates blood glucose further. Not surprisingly, hyperglycemia is noted approximately 72 hours after an acute myocardial infarction.
Angiotensin II is released during myocardial ischemia and contributes to the pathogenesis of MI in several ways. First, it results in the systemic effects of peripheral vasoconstriction and fluid retention. These homeostatic responses are counterproductive in that they increase myocardial work and thus exacerbate the effects of the loss of myocyte contractility. Angiotensin II is also released locally, where it is a growth factor for vascular smooth muscle cells, myocytes, and cardiac fibroblasts; promotes catecholamine release; and causes coronary artery spasm.
Cellular Death: After about 20 minutes of myocardial ischemia, irreversible hypoxic injury causes cellular death and tissue necrosis.161 (Types of necrosis are described in Chapter 2.) Necrosis of myocardial tissue results in the release of certain intracellular enzymes through the damaged cell membranes into the interstitial spaces. The lymphatics pick up the enzymes and transport them into the bloodstream, where they can be detected by serologic tests.
Structural and Functional Changes: Myocardial infarction results in structural and functional changes of cardiac tissues (Figure 30-22). Table 30-7 outlines the tissue changes that may follow myocardial infarction. Gross tissue changes in the area of infarction may not become apparent for several hours, despite almost immediate onset (within 30 to 60 seconds) of ECG changes. The infarcted myocardium is surrounded by a zone of hypoxic injury, which may progress to necrosis, undergo remodeling, or return to normal. Cardiac tissue surrounding the area of infarction also undergoes changes that can be categorized into (1) myocardial stunning, a temporary loss of contractile function that persists for hours to days after perfusion has been restored161,163; (2) hibernating myocardium, tissue that is persistently ischemic and undergoes metabolic adaptation to prolong myocyte survival until perfusion can be restored161,163; and (3) myocardial remodeling, a process mediated by angiotensin II, aldosterone, catecholamines, adenosine, and inflammatory cytokines, which causes myocyte hypertrophy and loss of contractile function in the areas of the heart distant from the site of infarction.161,164 All these changes can be limited through rapid restoration of coronary flow and the use of ACE inhibitors and beta-blockers after MI.165
Table 30-7
Tissue Changes After Myocardial Infarction
| Time After Myocardial Infarction | Tissue Changes | Stage of Healing Process |
| 6-12 hours | No gross changes; subcellular cyanosis with decreased temperature | Not begun |
| 18-24 hours | Pale to gray-brown; slight pallor | Inflammatory response; intercellular enzyme release |
| 2-4 days | Visible necrosis: yellow-brown in center and hyperemic around edges | Proteolytic enzymes remove debris; catecholamines, lipolysis, and glycogenolysis elevate plasma glucose and increase free fatty acids to assist depleted myocardium recovery from anaerobic state |
| 4-10 days | Area soft, with fatty changes in center, regions of hemorrhage in infarcted area | Debris cleared; collagen matrix laid down |
| 10-14 days | Weak, fibrotic scar tissue with beginning revascularization | Healing continues but area very mushy, vulnerable to stress |
| 6 weeks | Scarring usually complete | Tough inelastic scar replaces necrotic myocardium |
NOTE: Processes of tissue healing are described and illustrated in Chapter 6.
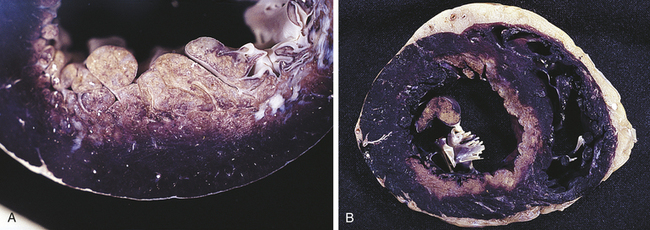
Figure 30-22 Myocardial infarction. A, Local infarct confined to one region. B, Massive large infarct caused by occlusion of three coronary arteries. (From Damjanov I, Linder J, editors: Anderson’s pathology, ed 10, St Louis, 1996, Mosby.)
The severity of functional impairment depends on the size and the site of infarction. Functional changes can include (1) decreased cardiac contractility with abnormal wall motion, (2) altered left ventricular compliance, (3) decreased stroke volume, (4) decreased ejection fraction, (5) increased left ventricular end-diastolic pressure, and (6) SA or AV node malfunction. Life-threatening dysrhythmias and heart failure often follow MI.
Repair: Myocardial infarction causes a severe inflammatory response that ends with wound repair (see Chapter 6). Repair consists of degradation of damaged cells, proliferation of fibroblasts, and synthesis of scar tissue. Many cell types, hormones, and nutrient substrates must be available for optimal healing to proceed. Within 24 hours, leukocytes infiltrate the necrotic area and proteolytic enzymes from scavenger neutrophils degrade necrotic tissue. A pseudodiabetic state often develops as catecholamines released from damaged cells stimulate release of glucose and free fatty acids. By the second week, insulin secretion increases to mobilize glucose from the repair processes. The collagen matrix that is deposited is initially weak, mushy, and vulnerable to reinjury. Unfortunately it is at this time in the recovery period (10 to 14 days after infarction) that individuals feel more capable of increasing activities and thus may stress the newly formed scar tissue. After 6 weeks the necrotic area is completely replaced by scar tissue, which is strong but unable to contract and relax like healthy myocardial tissue.
CLINICAL MANIFESTATIONS The first symptom of acute MI is usually sudden, severe chest pain. It is not possible to distinguish between angina and MI by symptoms alone, although the pain associated with MI tends to be more severe and prolonged. It may be described as heavy and crushing, such as a “truck sitting on my chest.” Radiation to the neck, jaw, back, shoulder, or left arm is common. Some individuals (especially older adults or those with diabetes) experience no pain, thereby having a “silent” infarction. Infarction often stimulates a sensation of unrelenting indigestion. Nausea and vomiting may occur because of reflex stimulation of vomiting centers by pain fibers. Vasovagal reflexes from the area of the infarcted myocardium also may affect the gastrointestinal tract. Catecholamine release results in sympathetic stimulation, producing diaphoresis and peripheral vasoconstriction that cause the skin to become cool and clammy.
EVALUATION AND TREATMENT The diagnosis of acute myocardial infarction is made on the basis of history, physical examination, ECG, and serial cardiac biomarker alterations (Box 30-2). A variety of cardiovascular changes may be found on physical examination. With an acute myocardial infarction, blood pressure may initially decrease. The drop in blood pressure reflexively activates the sympathetic nervous system to compensate and increase the heart rate in an effort to restore the blood pressure. Blood pressure may remain low (hypotension) or may become elevated depending on the severity of myocardial damage. Abnormal extra heart sounds (S3, S4) reflect left ventricular dysfunction. Inflammation can cause pericardial friction rub. Cardiac murmurs may indicate acute valvular insufficiency. The pulmonary examination may reveal inspiratory crackles consistent with pulmonary edema.
Cardiac troponins (troponin I and troponin T) are the most specific indicators of MI. A transient rise in these plasma biomarker levels can confirm the occurrence of MI and indicate its severity. Other biomarkers released by myocardial cells include CPK-MB and LDH. These isoenzymes exist in several different active molecular forms called isoenzymes and are present in different amounts within particular tissues. Blood is drawn for troponin and isoenzyme determinations as soon as possible after the onset of symptoms; serial serum levels of these biomarkers are assessed for several days. If serologic tests show abnormally high levels of troponin and isoenzymes, acute myocardial infarction probably has occurred. CPK-MB is less specific than troponins and may increase in individuals with certain other conditions (e.g., muscular dystrophy, hypothermia, chronic obstructive pulmonary disease [COPD], pulmonary embolism, extensive third-degree burns, small bowel infarction). Elevation of troponin, CPK-MB, and LDH1 may not increase immediately after infarction and laboratory confirmation that an infarction has occurred may be delayed up to 12 hours.
Myocardial infarction can occur in various regions of the heart wall and may be described as anterior, inferior, posterior, lateral, subendocardial, or transmural, depending on its location and extent of tissue damage from infarction. Twelve-lead ECGs help localize the affected area through identification of Q waves and changes in ST segments and T waves (Figure 30-23). The infarcted myocardium is surrounded by a zone of hypoxic injury, which may progress to necrosis or return to normal, and adjacent to this zone of hypoxic injury is a zone of reversible ischemia (Figure 30-24). If the thrombus breaks up before complete distal tissue necrosis has occurred, the infarction will involve only the myocardium directly beneath the endocardium. This type of myocardial infarction most often presents with no elevation of the ST segment on ECG and therefore is non-STEMI. In addition, this form of infarction will not be associated with the classic Q-wave tracing on the ECG (non–Q-wave MI). It is especially important to recognize this form of acute coronary syndrome because recurrent clot formation on the disrupted atherosclerotic plaque is likely, with resultant infarct expansion. If the thrombus lodges more permanently in the vessel, the infarction will extend through the myocardium from endocardium to epicardium resulting in severe cardiac dysfunction. This usually presents with significant ST-segment elevation on ECG (STEMI). A characteristic Q wave often develops on ECG some hours later (Q-wave MI). STEMI requires rapid intervention to prevent serious complications and sequelae.
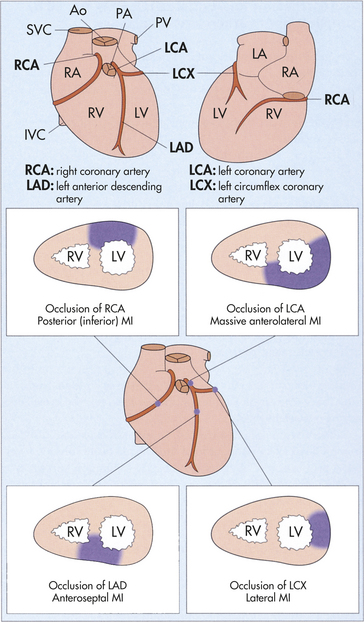
Figure 30-23 Site of myocardial infarction (MI) and vessel involvement. Ao, Aorta; IVC, inferior vena cava; LA, left atrium; LV, left ventricle; PA, pulmonary artery; PV, pulmonary vein; RA, right atrium; RV, right ventricle; SVC, superior vena cava. (Modified from Stevens A, Lowe J: Pathology, St Louis, 1995, Mosby.)
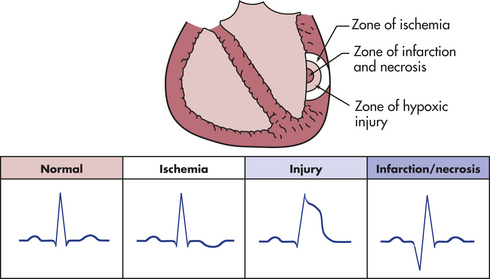
Figure 30-24 Electrocardiographic alterations associated with the three zones of myocardial infarction.
Additional laboratory data may reveal leukocytosis and elevated sedimentation rate, both of which indicate inflammation. The individual’s blood sugar is usually elevated and the glucose tolerance level may remain abnormal for several weeks. Hypoxemia may accompany heart failure.
Acute myocardial infarction requires admission to the hospital, often directly into a coronary care unit. The individual should be placed on supplemental oxygen and given an aspirin immediately (ticlopidine if allergic to aspirin).156–158,166 Pain relief is of utmost importance and involves the use of sublingual nitroglycerin or morphine sulfate. Continuous monitoring of cardiac rhythms and biomarker changes is essential because the first 24 hours after onset of symptoms is the time of highest risk for sudden death. Both non-STEMI and STEMI are managed with the urgent administration of thrombolytics or by PCI along with antithrombotics.156,166–169 Further management may include ACE inhibitors and beta-blockers. Individuals who are in shock require aggressive fluid resuscitation, ionotropic drugs, and possible emergent invasive procedures.
Bed rest, followed by gradual return to activities of daily living, reduces the myocardial oxygen demands of the compromised heart. Individuals not receiving thrombolytic or heparin infusion must receive DVT prophylaxis as long as their activity is significantly limited. Stool softeners are given to eliminate the need for straining, which can precipitate bradycardia and can be followed by increased venous return to the heart, causing possible cardiac overload.
Treatment of dyslipidemia with 3-hydroxy-3-methyl-glutaryl-CoA (HMG-CoA) reductase inhibitors (statins) can reduce the risk of cardiovascular events. The National Cholesterol Education Program (NCEP) Expert Panel107 has recommended target total blood cholesterol levels of less than 200 mg/dl with LDLs less than 100 mg/dl and HDLs more than 40 mg/dl. Drugs that decrease lipidemia should be administered prior to discharge from the hospital.169 Education on diet, caffeine, smoking cessation, exercise, and other aspects of risk factor reduction is crucial for secondary prevention of recurrent myocardial ischemia.
COMPLICATIONS The number and severity of postinfarction complications depend on the location and extent of necrosis, the individual’s physiologic condition before the infarction, and the availability of swift therapeutic intervention.
Dysrhythmias (arrhythmias), which are disturbances of cardiac rhythm, are the most common complication of acute myocardial infarction, affecting more than 90% of individuals. Dysrhythmias can be caused by ischemia, hypoxia, autonomic nervous system (ANS) system imbalances, lactic acidosis, electrolyte abnormalities, alterations of impulse conduction pathways or conduction defects, drug toxicity, or hemodynamic abnormalities. Dysrhythmias may originate from the atria, ventricles, nodal regions, or conduction tissues. The seriousness of dysrhythmias depends on the hemodynamic consequences. For example, there is no ventricular contraction in ventricular fibrillation; consequently, there is no cardiac output. Atrial fibrillation, however, does not affect ventricular contraction and thus can be tolerated by most individuals. (Dysrhythmias are described in Table 30-12.) Prophylactic use of antiarrhythmics, such as lidocaine and amiodarone, do not improve mortality; however, individuals at high risk should be considered for implantable cardioverter-defibrillators (ICDs).
Acute MI usually is accompanied by a reduction in cardiac output and some degree of left ventricular failure (congestive heart failure), which is characterized by pulmonary congestion, reduced myocardial contractility, and abnormal heart wall motion (see p. 1171). Anterior infarction is associated with more severe left heart failure than is inferior infarction. If cardiac output is insufficient to maintain normal arterial pressure and to perfuse the kidneys and other organs adequately, cardiogenic shock develops. Cardiogenic shock characteristically develops if 40% or more of the left ventricular myocardium is infarcted. (Cardiogenic shock is discussed in Chapter 46.)
Inflammation of the pericardium (pericarditis) is a common complication of acute myocardial infarction. Pericardial friction rubs often are noted 2 to 3 days after MI and are associated with anterior chest pain that worsens with respiratory effort. Specific treatment is not required; however, corticosteroids dramatically relieve symptoms. Dressler postinfarction syndrome, which is a delayed form of acute pericarditis, can occur from 1 week to several months after acute myocardial infarction. Although poorly understood, the syndrome is thought to be an immunologic (antigen-antibody) response to the necrotic myocardium. Pain, fever, friction rub, pleural effusion, and arthralgias may accompany this syndrome. Steroids may alleviate symptoms.
Organic brain syndrome may occur in acute or chronic form if blood flow to the brain is impaired secondary to MI. Transient ischemic attacks or an outright cerebrovascular accident may result from thromboemboli that have broken loose from the wall of the left ventricle or from cardiac valves.
Cardiac complications of MI can include rupture of heart structures. Necrosis of tissue in or around the papillary muscles can cause rupture of these muscles or of the chordae tendineae. Factors that lead to rupture of the free wall of the infarcted ventricle include thinning of the wall, poor collateral flow, shearing effect of muscular contraction against the stiffened necrotic area, marked necrosis at the terminal end of the blood supply, and aging of the myocardium with laceration of the myocardial microstructure. Infarctions around septal structures that separate the heart chambers can lead to septal rupture. Ruptures are associated with audible, harsh cardiac murmurs; increased left ventricular end-diastolic pressure; and decreased systemic blood pressure.
Weakening of the wall of the infarcted ventricle can cause ventricular aneurysm formation. According to Laplace’s law, with decreased muscle mass at the infarcted site, the wall is weakened and tension stretches the noncontracting infarcted heart muscle, thus producing infarct expansion or aneurysm formation (see p. 1144 for a discussion of aneurysm). Decreased muscle mass causes an increase in the radius of the ventricle, and because the radius is directly proportional to pressure and tension, both increase with time. The wall of the aneurysm becomes more fibrotic but continues to bulge with systole. The bulge results in impaired pump function. Although rare, rupture may occur when the tension becomes too great. Death in individuals with a left ventricular aneurysm is usually related to ventricular tachydysrhythmias and not to ventricular rupture. Left ventricular aneurysm is a late complication of MI, occurring months or years after the acute event.
Thromboembolism is commonly found during postmortem examinations of individuals who have died of MI. Thromboemboli may disseminate from debris and clots that collect inside dilated aneurysmal sacs or from the infarcted endocardium and travel to the pulmonary or systemic vascular systems. Pulmonary emboli also may result from the breaking loose of deep venous thrombi of the legs in individuals who are confined to bed (see p. 1143). Early mobilization and prophylactic anticoagulation therapy are essential to reduce the incidence of this complication.
Sudden death resulting from cardiac arrest is often caused by dysrhythmias, particularly ventricular fibrillation. Other dysrhythmias may be equally lethal. Widespread knowledge of cardiopulmonary resuscitation has increased the probability of survival during the first few hours after cardiac insult. Immediate intervention and careful monitoring also have reduced mortality and have improved chances for long-term survival. Several factors, however, contribute to the risk of death during acute infarction or reduce the chances of long-term survival, despite the best possible treatment. They are (1) degree of left ventricular dysfunction, (2) degree of left ventricular ischemia, (3) potential for ventricular dysrhythmias, and (4) age of the individual.