ALTERATIONS OF MUSCULOSKELETAL FUNCTION IN CHILDREN


Musculoskeletal alterations in children are very common. They may be congenital, such as clubfoot; hereditary, such as muscular dystrophy; or acquired, such as Legg-Calvé-Perthes disease. Some of these disorders are acute, and the child will recover completely; other disorders are chronic or, in some cases, terminal. An understanding of the pathophysiology of these alterations will aid in providing the best care possible for these children.
MUSCULOSKELETAL DEVELOPMENT IN CHILDREN
Bone formation, which begins at about the sixth week of gestation, involves two phases: (1) the delivery of bone cell precursors to sites of bone formation and (2) the aggregation of these cells at primary centers of ossification, where they mature and begin to secrete osteoid (see Chapter 41). Some of the bone cell precursors are present in fetal connective tissues, whereas others migrate in blood to sites of bone formation after blood vessels have grown into the tissue.
Cellular aggregation and maturation occur in two types of fetal tissue, depending on which bones are being formed. The cranium, facial bones, clavicles, and parts of the jawbone (classically called “flatbones”) arise from a fetal membrane termed the mesenchyme. Bones that develop on or within the mesenchyme grow by the process of intramembranous formation of bone. As the mesenchyme becomes vascularized, the immature bone cells aggregate and mature into osteoblasts, which form the centers of ossification. Osteoblasts secrete osteoid, which surrounds them and quickly ossifies, forming the lacunae and canaliculi of compact bone. Spicules of bone radiate from the ossification centers to form the primary trabeculae characteristic of spongy bone. Later, some of the spongy bone is replaced by compact bone.
Endochondral formation of bone is the development of new bone from cartilage (Figure 43-1). First, mesenchymal tissue forms a cartilage anlage, which defines the shape of the bone. This is usually found by 6 weeks of gestation. Blood vessel invasion to the inside the anlage brings osteoprogenitor cells leading to primary centers of calcification by 8 weeks. Endochondral bone formation begins in the outer layer of the cartilage model, which consists of a layer of dense connective tissue called perichondrium. The perichondrium contains cells that develop into osteoblasts, forming a collar of bone, termed the periosteal collar, around the cartilage model. Cartilage enclosed within the periosteal collar degenerates, and capillaries from outside the perichondrium invade the degenerating cartilage cells, carrying with them osteoblast precursors from the inner layer of the perichondrium and osteoclast precursors from the blood itself.
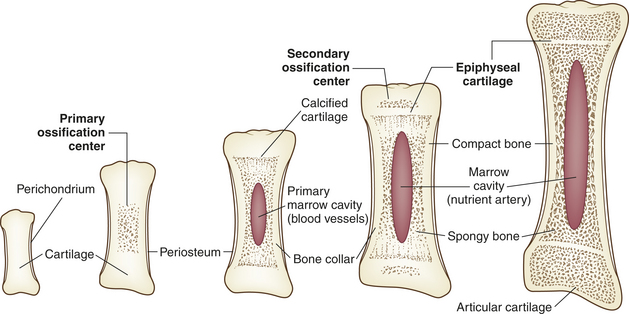
Endochondral bone formation progresses at the primary center of ossification in the middle of the cartilage model and extends toward either end of the developing bone. At the same time, the periosteal collar thickens and becomes wider toward the epiphyses. By the end of gestation secondary centers of ossification (i.e., the epiphyseal centers) begin to lay down bone at both ends of the cartilage model. Here, too, cartilage within the periosteal collar degenerates, and blood vessels grow inward, delivering bone cell precursors. Once the osteoblasts begin to secrete osteoid, ossification spreads from the secondary centers in all directions until all the cartilage within the model is replaced by bone.
Two regions of cartilage remain at the ends of long bones: (1) articular cartilage over the free ends of the bone and (2) the physeal plate, a layer of cartilage between the metaphysis and epiphysis. (These structures are described and illustrated in Chapter 41; see Figure 41-3.) The physeal plate retains the ability to form and calcify new cartilage and deposit bone until the skeleton matures roughly 1 year after sexual maturity (11 to 15 years of age in females, 15 to 18 in males).
Bone Growth
Until adult stature is reached, growth in the length of bone occurs at the physeal plate through endochondral ossification. Cartilage cells at the epiphyseal side of the physeal plate multiply and enlarge. As rapidly as new cartilage cells form, cartilage cells at the metaphyseal side of the plate are destroyed and replaced by bone.
In the shaft of new bone, where growth is relatively slow, the bone produced by accretion is compact and dense. The compact bone is thickest where it has to withstand the maximal stresses, which generally occur in the middle of the shaft.
The two physes of the long bone often have varying activity rates. For example, the distal physis in the femur contributes 80% of the overall length, whereas the proximal physis at the hip contributes only 20%. The more active of the two has more power to remodel deformity but also can be more sensitive to injury. The architecture of the physis also dictates its sensitivity to injury. The distal femur, for example, has an undulating pattern that increases its resistance to sheer force; when injured, however, growth disturbance is highly likely, whereas the distal radius, which contributes 80% of overall radial length, is a flat, smooth physis that is far more resistant to traumatic injury.
Growth in the diameter of bone occurs by deposition of new bone on an existing bone surface. Bone matrix is laid down by osteoblasts on the periosteal surface and subsequently becomes calcified. At the same time, bone resorption occurs on the endosteal surface. Endosteal resorption increases the diameter of the medullary cavity, which contains marrow and spongy bone.
Many factors affect the development, physiology, and rate of growth of the epiphyseal plate. Growth hormone must be secreted by the pituitary gland at a constant rate to stimulate the growth plate consistently. Other known factors affecting growth include peptide regulatory factors (e.g., fibroblast growth factor [FGF]); changes in cell-to-cell interactions through cell adhesion molecules (CAMs) and cell junctions; and complex interactions or changes in extracellular matrix (ECM), nutrition, general health, and other hormones (e.g., thyroid hormone, adrenal and gonadal androgens, estrogens). These factors influence both the rate of bone growth and the time of appearance of the secondary ossification centers. When the skeleton is mature, the epiphyseal plate is replaced by bone. This process, termed physeal closure, unites the metaphysis and the epiphysis. Physeal closure occurs earlier in females than males because of earlier puberty in females.
Throughout life, bone is constantly being destroyed and re-formed (see Chapter 41), but the process is at its maximum in children approximately 2½ years of age. By young adulthood, bone turnover, or remodeling, occurs at a relatively slow rate. Peak bone mass is achieved by the mid- to late 20s and slowly decreases throughout life; therefore, calcium intake, weightbearing lifting and exercise, minimizing caffeine, and phosphorus are especially important for a young female if she is to avoid osteoporosis in later life.
Skeletal Development
The axial skeleton changes shape with growth. (The axial skeleton and appendicular skeleton are described and illustrated in Chapter 41; see Figure 41-5). In a newborn the entire spine is concave anteriorly, or kyphosed. In the first 3 months of life, with the infant’s ability to control the head, the upper (cervical) spine begins to arch, or become lordotic. The normal lordotic curve of the lower (lumbar) spine begins to develop with sitting.
The appendicular skeleton (the extremities) grows faster during childhood than does the axial skeleton (see Figure 41-5). The newborn has a relatively large head and long spine with disproportionately shorter limbs than an adult. By 1 year of age, 50% of the total growth of the spine has occurred and is more than 70% complete by age 8.1 Therefore, failure of the spine to grow (e.g., spinal fusion) does not limit eventual height as much as the premature fusion of the growth plates of the lower extremities. In children with congenital curvature of the spine, growth tends to worsen the deformity rather than to increase the length of the spine.
Besides getting longer, growing bones of the extremities undergo changes in rotation and alignment. In the newborn the proximal femur is rotated forward up to 40 degrees and the tibia is rotated inward. With growth the femur assumes its normal alignment (by 8 years of age) and tibial rotation neutralizes at 5 years of age. Bowlegs and knock knees are normal at certain stages of growth. At birth the newborn’s legs are bowed because of stresses in utero. Genu varum (bowleg) reaches a peak by 30 months of age, whereas genu valgum (knock knee) maximizes by 5 to 6 years of age. If genu varum or genu valgum persists past these ages, a pathologic process rather than a physiologic phase may be present. Pathologic causes of genu varum are Blount disease, rickets, skeletal dysplasias (such as achondroplastic dwarfism), and traumatic injury. Genu valgum may persist also as a result of skeletal dysplasia or genetic predisposition.
Muscle Growth
The composition and size of muscles vary with age. In the fetus, muscle tissue contains a large amount of water and much intercellular matrix. After birth, both are reduced considerably as the muscle fibers (cells) enlarge by accumulating cytoplasm. Little information is available about the numbers of fibers in a given muscle at various ages, but the total mass of muscle in the body can be estimated from the amount of creatinine excreted in the urine, because the conversion of creatine to creatinine takes place only in muscle (see Chapter 41). Between birth and maturity the number of muscle nuclei in the body increases 14 times in boys and 10 times in girls. Muscle fibers reach their maximal size in girls at approximately 10 years of age and in boys by 14 years. Growth in length occurs at the ends of muscles, and the increase in length is accompanied by an increase in number of nuclei in the fibers. Muscle fibers increase in diameter as the fibrils become more numerous. The fibrils themselves do not increase in diameter. Connective tissue components of muscle grow where the tendon and muscle meet.
A potent stimulus to the growth of a muscle is the separation of its attachments as the skeleton grows. The length of a muscle fiber is the direct consequence of the range of movement it is called on to perform. The stimulus for the formation of a tendon is probably the pull of the muscle rudiment on undifferentiated connective tissue. The repair of a tendon from which a segment has been removed does not occur if the muscle is prevented from exercising tension on the damaged tendon. Replacement of muscle by bone (myositis ossificans) sometimes is the result of limitation of movement. If the normal opponents of a muscle are paralyzed, the muscle fails to grow properly, and it may be that the full development of a muscle depends on the progressive rise in the tension exerted on it by its antagonists.
Muscle growth during adolescence is a major factor in weight gain. Gender differences in muscle size and weight are minor in childhood but become considerable with the onset of puberty.
In the infant, muscle accounts for approximately 25% of total body weight, compared with 40% in the adult. In the adult, approximately 55% of muscle weight is in the lower limb muscles, whereas in the infant the majority of the weight is axial musculature. The respiratory and facial muscles are well developed at birth so that the infant can perform the vital functions of breathing and sucking. Other muscle groups, such as the pelvic muscles, take several years to develop fully. Throughout life the weight of the skeletal muscles can be increased by exercise. Less is known about the development of visceral and cardiac muscle. Visceral muscle fibers increase in number and size, but the increase in fiber size is most important. Fiber enlargement alone can increase the bulk of visceral muscle by as many as eight times. Cardiac muscle also grows mainly by enlargement of existing fibers.
MUSCULOSKELETAL ALTERATIONS IN CHILDREN
Syndactyly
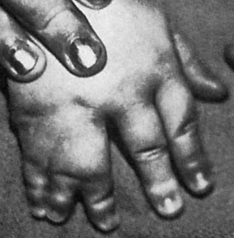
The most common congenital defect of the upper extremity is syndactyly, or webbing of the fingers (Figure 43-2). Simple webbing involves the soft-tissue envelope alone and is best released surgically when the child is 1 to 2 years of age. Complex syndactyly involves fusion of the bones and nails as well as the soft tissues; it may be associated with absence or anomaly of bony or neurovascular units. The primary goal in surgical correction of these defects is to achieve maximal function and appearance. Ideally, corrective surgery is deferred until the child is 6 to 12 months of age and completed before the child enters school. Vestigial tabs, such as an extra digit, however, are best removed during the immediate neonatal period. Anomalies on the medial or radial aspect of the arm are often associated with abnormalities of blood, heart, or kidneys. Lateral or ulnar-sided defects are less often associated with systemic anomalies and are far more rare.
Developmental Dysplasia of the Hip
Developmental dysplasia of the hip (DDH), formerly known as congenital dislocation of the hip, is an abnormality in the development of the proximal femur, acetabulum, or both. Although most often present at birth, it may occur at any time in the newborn or infant period.
The incidence of true dislocation of the hip or a dislocatable hip is 1 in 1000 live births. Some degree of instability of the hip is present in approximately 10 per 1000 live births. The left hip is affected in 60% of cases, whereas the right hip alone is affected only 20% of the time. Bilateral DDH occurs 20% of the time.
Risk factors for DDH include family history, female sex (6:1), metatarsus adductus (20%), torticollis (10%), oligohydramnios, first pregnancy, and breech presentation. First pregnancies and oligohydramnios (deficient volume of amniotic fluid) are thought to limit fetal movement, and breech presentation not only limits movement but also places the hips in a position of flexion and adduction. Although only 2% of births have breech history, as many as 40% of infants with DDH had a breech birth. Maternal hormones that reportedly increase joint laxity also have an effect on DDH, although the exact mechanism is unknown. DDH also is more common in whites and those cultures that swaddle infants with the hips in extension and adduction. It is almost unknown in African cultures where infants are carried, with legs abducted, on the back.
PATHOPHYSIOLOGY The hip can be described as subluxated, dislocatable, or dislocated (Figure 43-3). The subluxated hip maintains contact with the acetabulum but is not well seated within the hip joint. The acetabulum is often dysplastic (or shallow) and the femur is often normal. The dislocatable hip is sometimes located but can be dislocated easily. The dislocated hip has no contact between the femoral head and the acetabulum. Some degree of acetabular dysplasia is present in almost all cases. Typically the acetabulum is shallow or sloping rather than cup shaped.
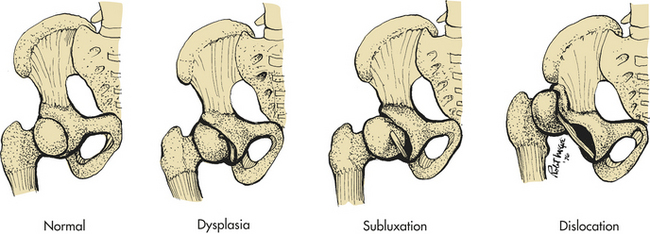
Figure 43-3 Configuration and relationship of structures in developmental dysplasia of the hip. (From Hockenberry MJ: Wong’s nursing care of infants and children, ed 8, St Louis, 2007, Mosby.)
By approximately 10 weeks of gestation, the femur, acetabulum, and hip joint capsule are well developed. It appears that most dysplasias occur within the second and third trimesters and are often the result of positioning factors. Experimentally, DDH can be produced in laboratory animals by placing the developing hip in adduction and extension, replicating the breech position. There is, however, a genetic component that is poorly understood. In addition, 2% of DDH cases are teratologic or caused by a systemic syndrome, such as arthrogryposis or spina bifida, in which muscle contracture or imbalance leads to DDH.
If DDH is left untreated in the growing child, secondary changes occur. If the hip is left subluxated or dislocated, the acetabulum becomes increasingly shallow and the soft tissues shorten about the proximal femur. If the hip is dislocated, the bone acetabulum fills with soft tissue and a false acetabulum forms where the femoral head contacts the iliac crest. An apparent limb length inequity and hip muscle weakness occurs, leading to a waddling gait. The subluxation leads to early osteoarthritis (OA), and it is now estimated that at least 60% of all OA of the hip is related to DDH.2
CLINICAL MANIFESTATIONS The clinical manifestations of DDH vary with the severity of the condition and the age of the child. Signs and symptoms that should be noted include the following:
1. Asymmetry of gluteal or thigh folds
2. Limb length discrepancy (Galeazzi sign)
3. Limitation of hip abduction
4. Positive Ortolani sign (clunk of dislocation)
5. Positive Barlow test (clunk of reduction)
The child also should be examined for other anomalies, such as torticollis or metatarsus adductus, which can be associated with DDH.
EVALUATION AND TREATMENT In the newborn period clinical examination is the most important diagnostic tool. Real-time ultrasound, in which the hip is examined while the ultrasound is performed, also is extremely valuable in the newborn period, especially in high-risk infants. The use of ultrasound allows visualization of the cartilaginous structures of the hip (the femoral head and the outer lip of the acetabulum), which are not seen on plain roentgenogram. Radiographs are used after age 6 months.3
Treatment depends on the age of the child, severity of dysplasia, and duration of dysplasia. The earlier that treatment is begun, the better the result. In children less than 4 months of age, a Pavlik harness can brace the hip in abduction and flexion, and the acetabulum will remodel as the femoral head rests centered in the socket. With this treatment, up to 98% of children will have an excellent result. A “closed” reduction (without opening the joint) followed by spica or body casting for up to 3 months can be done in children up to 12 months of age. After 12 months, surgical intervention—including opening the joint and cutting and realigning the femur and/or acetabulum—may be required. In teratologic dislocations, bracing often is unsuccessful and therefore surgery is more often needed.
Deformities of the Foot
Congenital Deformity: Congenital foot deformity is found in approximately 4% of all newborns, and metatarsus adductus accounts for 75% of these deformities (Table 43-1). Metatarsus adductus is a forefoot adduction deformity associated with a normal, plantigrade hindfoot and is believed to be secondary to intrauterine positioning. Metatarsus adductus is usually classified by two criteria: flexibility (passively correctable or rigid) and degree of deformity. The degree of deformity (mild, moderate, severe) is ascertained by the heel bisection line. A mild deformity is one in which the heel bisection line passes medial to the third toe; moderate, through the third or fourth toes; and severe, lateral to the fourth toe. It should be emphasized that a majority of children are well served by expectant treatment rather than early surgical intervention. Serial casts during the first 6 months of life are suggested for moderate to severe deformities and those deformities that appear less flexible. Casts are changed weekly for 6 to 12 weeks. Eighty-seven percent of children usually correct spontaneously by 6 years of age and 95% by 15 years of age. Even in those children with some residual deformity and an oblique medial cuneiform, they are rarely symptomatic.

Equinovarus Deformity: There are three types of equinovarus (clubfoot): positional equinovarus, idiopathic congenital equinovarus, and teratologic equinovarus (Figure 43-4). The true positional equinovarus lends itself to rapid correction by application of serial casts. The idiopathic variety is treated by attempting cast correction, followed by surgical intervention of resistant deformities. Teratologic equinovarus nearly always requires surgical correction and/or muscle balancing procedures.
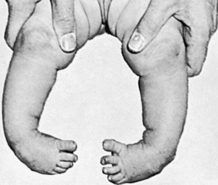
Figure 43-4 Infant with bilateral congenital talipes equinovarus. (From Brashear HR, Raney RB: Shand’s handbook of orthopedic surgery, ed 9, St Louis, 1978, Mosby.)
Positional Equinovarus: Positional equinovarus is a deformity in which an infant’s foot is in equinovarus position but does not have a deep posterior or plantar medial crease. It appears to be secondary only to intrauterine position. The foot can be passively brought to a plantigrade position and is amenable to casting. In general, 1 to 3 months of serial above-knee casting corrects this foot without the need for surgical intervention.
Idiopathic Congenital Equinovarus: The etiology of idiopathic equinovarus (clubfoot) is unknown. In one human fetal study, all clubfeet were associated with identifiable anterior horn cell changes in L5 and S1. Enterovirus infection, known to cause anterior horn cell damage if intrauterine, reaches a peak prevalence in the summer or fall of temperate climates. These two seasons correlate with the peaks of conception in children with clubfeet. Muscle biopsies of both the anterior tibialis long flexors and peroneus brevis muscles in clubfoot reveal that at least 50% of cases show a decreased number of muscle fibers and/or abnormal fiber histology. The soleus often has an increase in type 1 fibers, whereas the peroneus brevis has a fiber type disproportion. The more abnormal the histopathology is, the more severe the deformity, and the greater the chance of recurrent deformity after treatment. In addition to neurologic causes, positional abnormalities (e.g., oligohydramnios) have been implicated. The genetic component is unclear and studies are ongoing.
Idiopathic equinovarus occurs in approximately 1 of every 1000 live births, with males being affected twice as often as females. Although these deformities have been historically treated nonoperatively, surgical intervention became much more common after 1950. Over the past 10 years, nonoperative management has again become the mainstay. Ignacio Ponseti developed a casting technique that has been used for more than 50 years. Although used in Iowa since 1950, it was not well accepted nationally until recently. The technique involves six casts, left on for 5 to 7 days each, followed by a percutaneous tendoachilles lengthening procedure performed with local anesthesia in the clinic. The child then uses braces until 3 years of age. Noncompliance with braces leads to increased recurrence. Nearly 20% of children may need an anterior tibialis transfer around age 3.4 Studies comparing operative posteromedial release with Ponseti techniques show better long-term results with the less invasive Ponseti method5 (see What’s New? Ponseti Casting).
Teratologic Equinovarus: The most common causes of teratologic equinovarus are either neuromuscular (such as spina bifida) or syndromic, as in arthrogryposis or osteochondrodysplasia (such as diastrophic dwarfism). The teratologic clubfoot, unlike the idiopathic type, usually fails to be corrected with casting protocols and requires operative intervention. The surgery is often more extensive than that for an idiopathic clubfoot, and revision surgery is also more common.
Pes Planus (Flatfoot) Deformity: Pes planus (flatfoot) commonly raises parental concern. Despite medical evidence to the contrary, it can be very difficult to convince families that a flexible flatfoot is as functional as the one with a “normal” arch. The majority of babies are born with flat (or “fat”) feet, with the arch becoming more apparent with age. The relatively benign natural history, however, should not overshadow the importance of accurate diagnosis. Significant ankle valgus, vertical talus, tarsal coalition, and skewfoot must be accurately differentiated from flexible pes planus.
Flexible flatfoot deformity appears to be familial, with occasional association of generalized ligamentous laxity. Careful evaluation of possible occult Achilles contracture is done by holding the hindfoot in varus position and dorsiflexing the ankle. Achilles contracture can signify a more severe flatfoot variant. The flexibility of the hindfoot is evaluated by having the child stand on his or her toes or by dorsiflexing the first toe passively with the child in a non-weightbearing position. This “windlass mechanism” tightens the plantar fascia, thereby reconstituting an arch and hindfoot varus if the foot is indeed flexible. If hindfoot flexibility and an “underlying” arch are present, then flexible flatfoot is diagnosed.
By all recent data, the surgical or orthotic treatment of asymptomatic flexible pes planus is unnecessary. Custom orthotics, Helfet heel cups, and corrective orthopedic shoes may
relieve discomfort but will have no influence over the natural history (clinically or radiographically) of flat feet. Adult studies on army recruits have shown that soldiers with flat feet perform just as well as their counterparts without “fallen” arches.
There is a small subset of children with painful flexible flat feet. For these children careful attention to the possibility of Achilles contracture or tarsal coalition (congenital union of the hindfoot bones) must be made. This small group of children is best treated with inexpensive shoe inserts and then expectantly watched. If pain continues into adolescence, requiring more aggressive treatment, calcaneal lengthening will correct the pes planus without decreasing hindfoot motion. In rigid flat feet, a computed tomographic (CT) scan often will reveal coalition—if painful, this can be resected. Heel cord contractures can be surgically lengthened because stretching alone is often inadequate. All surgery carries risk; if a foot is flat but nonpainful, treatment is not required. The painless flatfoot should be viewed as a variation of normal feet.
Abnormal Density or Modeling of the Skeleton
Osteogenesis imperfecta (OI) (brittle bone disease) is a genetic disorder of connective tissues that affects primarily bone. The disorder was first described in 1840 as a syndrome in newborns that consisted of osteoporosis with fractures and skeletal deformities. The Sillence classification is based on both models of inheritance and clinical findings (Table 43-2). In the most severe form of this disorder, the child is usually stillborn or dies soon after birth, although some survive into childhood. OI in its more severe forms is evident at birth because fractures and deformity have occurred in utero. The less severe forms may not become evident until the child begins to walk. Some children with this milder form then experience numerous fractures and can be mistaken for battered children until the diagnosis is made.
Table 43-2
Sillence Classification of Osteogenesis Imperfecta Syndromes
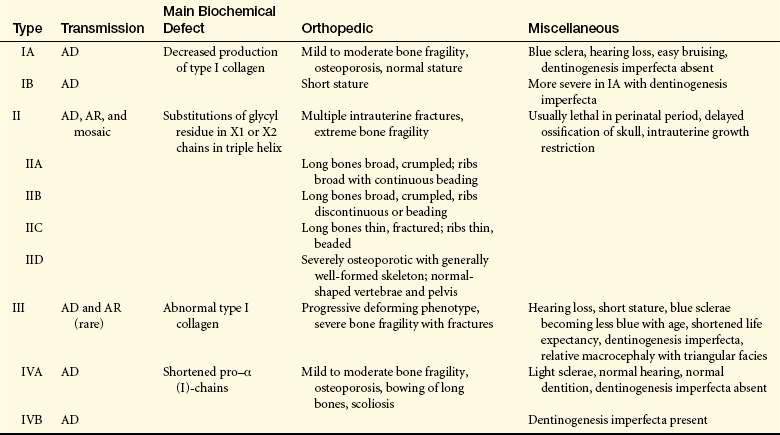
AD, Autosomal dominant; AR, autosomal recessive.
From Orthopaedic knowledge update 8, p 248, Rosemont IL, 2005, American Academy of Orthopaedic Surgeons.
The prevalence rate of the most common form is about 1 in 30,000. Inheritance is usually autosomal dominant but can be autosomal recessive. At least four syndromes have been identified that have various clinical manifestations and prognoses (see Table 43-2).
PATHOPHYSIOLOGY The major errors in OI lie in the synthesis of collagen. Genetic studies have shown that the gene responsible for the encoding of collagen easily mutates. These mutations cause osteogenesis imperfecta. The large range of phenotypes includes all mutants of the two collagen structural genes. (Genes are discussed in Chapter 4.) Abnormalities in collagen include (1) an increase in collagen hydroxylysine residue in bones; (2) a decrease in hydroxylysine-norleucine in skin collagen; and (3) absence of α-polypeptide production in cultured skin fibroblasts.6
A number of metabolic abnormalities have been reported. Some individuals have increased serum thyroxine levels, suggesting hyperthyroidism. This is consistent with the findings of increased sweating, heat intolerance, increased body temperature, a resting tachycardia, and tachypnea. The hyperthyroid findings, however, are not consistent in all individuals with OI. Studies of leukocyte metabolism suggest an uncoupling of oxidative phosphorylation. Reports of alterations of platelet function with defects in adhesion and clot retraction also exist.
CLINICAL MANIFESTATIONS The classic clinical manifestations of OI are osteoporosis and increased rate of fractures, possible bony deformation, triangular facies, possible vascular weakness (i.e., aortic aneurysm), possible blue sclera, and poor dentition. The Sillence classification designated types I through IV based on severity. The most severe, types II and III, are comparable to osteogenesis imperfecta congenita. These two types are characterized by autosomal recessive inheritance and early onset of manifestations. Both can cause stillbirth or severe neonatal deformity and a short life expectancy. Less severe are types I and IV, which are comparable to osteogenesis imperfecta tarda. Type I is slightly more common than types II and III, and type IV is quite rare. Types I and IV are inherited as autosomal dominant traits and vary in age of onset from birth to adulthood. Type IV, especially when the sclera are white, is the least deforming type and is often confused with nonaccidental trauma (child abuse).
EVALUATION AND TREATMENT Evaluation of OI is based on clinical manifestations and serologic tests. Serum alkaline phosphatase is elevated in all forms of the disease. Osteogenesis imperfecta can be diagnosed prenatally by ultrasound or chorionic villi sampling. Quantitative analysis of cultured skin fibroblast collagen by electrophoresis shows a decreased quantity of collagen in the affected individual.
Type II OI is often terminal in the perinatal period, and therefore little is known about appropriate treatment for the few children who survive. For other types of OI, careful positioning and handling of the newborn may prevent fractures. Beyond the neonatal period, various orthopedic measures are applied, such as prompt splinting of fractures and correction of deformities arising from the progressive bowing or bending of the skeleton by intermedullary rodding of the bones (Figure 43-5). Newer, telescoping rods, which grow with the child, have been shown to reduce the reoperative rate by 30%.7 Scoliosis is present in up to 50% of Sillence III and often requires surgery. A multicenter study of a bisphosphonate therapy showed promising results in type III OI, with marked improvements of bone density (up to 30%). Despite these results, there is concern that the healing of fractures and surgical intervention can be more difficult. More study is needed to address the efficacy and safety of these types of drugs. Genetic counseling for affected families should aim at primary prevention.
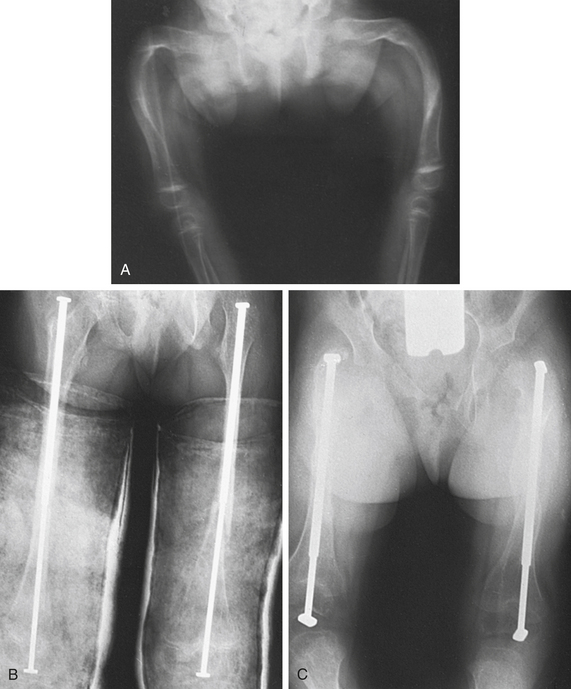
Figure 43-5 Osteogenesis imperfecta treated with osteotomies and telescoping medullary rods A, Severe deformity of both femurs. B, Same individual after multiple osteotomies with telescoping medullary rod fixation. C, Same individual 4 years later demonstrating growth of femurs, no recurrence of deformity, and elongation of rods. (Plaster casts are in place for immobilization of tibial osteotomies.) (From Crenshaw AH, editor: Campbell’s operative orthopaedics, ed 8, vol 3, St Louis, 1992, Mosby.)
Rickets
Rickets is a disorder in which growing bone fails to become mineralized (ossified), resulting in “soft” bones and skeletal deformity. Rickets results from either insufficient vitamin D, insensitivity to vitamin D, wasting of vitamin D by the kidney, or inability to absorb vitamin D and calcium in the gut. In industrialized nations the most common X-linked dominant form is hypophosphatemic rickets. Although in the past few years, as exclusive breast-feeding for a lengthy period has been encouraged, vitamin D deficiency has been increasing. This is especially problematic when the mother is vitamin D deficient.8 Although unprotected exposure to ultraviolet rays is not suggested, children still need 15 to 20 minutes per week of true sun exposure to activate vitamin D, the mineral necessary for absorption and metabolism of calcium and phosphate. Rickets in the immature skeleton leads to broad, irregular growth plates because the rows of cells in the growth plate that are intended to ossify fail to do so as they reach the metaphysis (Figure 43-6).
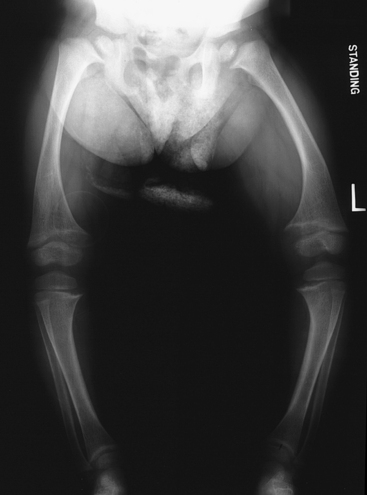
Figure 43-6 Rickets. This standing radiograph of an 8-year-old female with hypophosphatemic rickets shows cupping and widening of the growth plates throughout the lower extremities. Also note the bowing femoral deformity and hip deformity.
Children with rickets are often listless and irritable. They have hypotonia and muscle weakness and may be unable to walk without support. Abnormal parietal flattening and frontal bossing occur in the skull. The calvaria become soft, and the sutures may widen. Cartilaginous attachments of the ribs become prominent, and the long bones of the extremities (tibia, femur, radius, ulna) may be bowed. Growth is restricted, and fractures are common.
Like osteogenesis imperfecta, surgical treatment of bony deformity is often required. However, medical management of calcium, phosphorus, and vitamin D levels must be optimized before surgical intervention. Deformity often improves with normalization of bone metabolism.
Scoliosis
Scoliosis is a rotational curvature of the spine most obvious in the anteroposterior plane (Figure 43-7). It can be classified as nonstructural or structural. Nonstructural scoliosis results from a cause other than the spine itself, such as posture, leg length discrepancy, or pain. Structural scoliosis is curvature of the spine associated with vertebral rotation. Nonstructural scoliosis can become structural if the underlying cause is not found and treated.
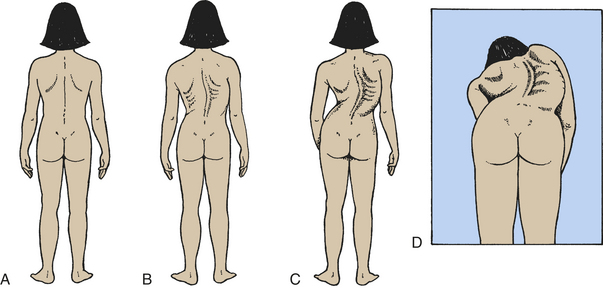
Figure 43-7 Scoliosis in children. Normal spinal alignment and abnormal spinal curvatures associated with scoliosis. A, Normal. B, Mild. C, Severe. D, Rotation and curvature of scoliosis.
Structural scoliosis can be caused by a great variety of conditions. It can result from congenital skeletal abnormalities (15%), neuromuscular diseases (15%), trauma, extraspinal contractures, bone infections that involve the vertebrae, metabolic bone disorders (e.g., rickets, osteoporosis, osteogenesis imperfecta), joint disease, and tumors. Most cases of structural scoliosis, however, have no known cause, although genetic factors are suggested. Structural scoliosis with no known cause, termed idiopathic scoliosis, accounts for at least 65% of cases.
Idiopathic scoliosis is classified as infantile, juvenile, or adolescent, depending on the child’s age at the time of onset. In infantile scoliosis, spinal curvature develops during the first 3 years of life; in juvenile scoliosis, curvature develops between the skeletal age of 4 years and the onset of adolescence; and in adolescent scoliosis, it develops after the skeletal age of 10. Adolescent idiopathic scoliosis is the most common. Scoliosis in its milder forms occurs equally in boys and girls once curves measure more than 15 degrees; however, girls are 5 times more likely to have scoliosis than boys.
PATHOPHYSIOLOGY It has been hypothesized that in individuals with adolescent scoliosis, there is an abnormality of the central nervous system involving the balance mechanism (reticular system) in the midbrain. A genetic component is also suggested because 30% occur within families. A recent study is making exciting gains in this area.
Experimentally it also has been shown that individuals with adolescent idiopathic scoliosis have an abnormality in the function of the posterior columns of the spinal cord. This results in abnormal proprioception and is not evident clinically except in the presence of scoliosis. The exact cause of scoliosis, however, remains elusive.9,10
The earliest pathologic changes, which are probably secondary changes, occur in the soft tissues. The muscles, ligaments, and other soft tissues become shortened on the concave side of the curve. With time, progressive deformities of the vertebral column and ribs develop. In growing children, lateral deviation of the spinal column ceases, and one-sided compression of the vertebral bodies on the concave side of the curve begins. Vertebral deformity occurs as asymmetric forces are applied to the epiphyseal center of the ossification by shortened and tight soft tissues on the concave side of the curve. The degree of compression and twisting varies according to the position of the vertebrae in the curve. The compressive force is greatest on the vertebrae in the apex of the concavity, so that the apical vertebrae become most deformed.
The curves increase most rapidly during periods of rapid skeletal growth. If the curve is less than 40 degrees at skeletal maturity, the risk of progression is quite small. In curves greater than 50 degrees, the spine is biomechanically unstable, and the curve will in all likelihood continue to progress even after the cessation of growth at an average rate of 1 degree per year. Curves in the thoracic spine greater than 80 degrees result in decreased pulmonary function, whereas the most common complication of large curves in the lumbar spine is back pain.
CLINICAL MANIFESTATIONS The clinical manifestations of nonstructural scoliosis are mild spinal curvature with prominence of one hip or rounded shoulders. The curvature disappears with forward flexion of the spine, lying down, or traction of the head. Treatment for nonstructural scoliosis is correction of the underlying disorder. The clinical manifestations of structural scoliosis include asymmetry of hip height, asymmetry of shoulder height, shoulder and scapular (shoulder blade) prominence, and rib prominence.
EVALUATION AND TREATMENT Spinal curvature is usually visible or palpable, and muscles on one side of the lower back (the convex side) may be prominent or bulging. Most cases of idiopathic scoliosis are noticed during school screening programs. In girls the deformity may be noticed because clothing does not “hang” properly on the body. Diagnosis is made by roentgenographic examinations.
Treatment of curves between 25 and 35 degrees in the skeletally immature child is with bracing. In most cases the low-profile brace is used. A brace used only at night, the Charleston bending brace, has shown it to be less effective than the traditional low-profile braces in preventing progression of curves. Occasionally a Milwaukee brace, which has a metal upper structure and neck ring, is needed for curves with an apex higher than midthoracic level. Low-profile and Milwaukee braces are worn for 23 hours daily until skeletal maturity. Bracing will only prevent progression of the curve; it will not correct the curvature. Bracing is not effective in curves greater than 40 degrees or in skeletally mature individuals; the most effective time for bracing is in a young child (less than 12 years of age) with a small curve.11 Extensive chiropractic manipulations and electrical stimulation have not been shown to change natural history. Surgical treatment with spinal fusion with instrumentation is recommended for curves greater than 40 to 50 degrees. If surgery is indicated, it is better performed during the adolescent years while there is greater flexibility of the curves and less risk of complications.
Bone Infection: Osteomyelitis
Osteomyelitis is an infection of the bone. Occurring twice as often in males as females, acute osteomyelitis may affect infants and children of any age, but it occurs most often between 3 and 12 years of age.
Bacteria enter the bone through the bloodstream and lodge in the medullary cavity, where a rich phagocytic mechanism often prevents most of the bacteria from establishing an infectious state. In some cases, however, the bacteria may lodge at the end of the venous loops beneath the epiphyseal plate, and infection then develops because there are no phagocytic cells present to remove the bacteria12–14 (Figure 43-8).
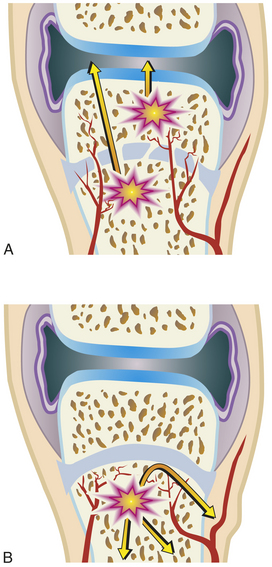
Figure 43-8 Pathogenesis of acute osteomyelitis differs with age A, In infants younger than 1 year the epiphysis is nourished by penetrating arteries through the physis, allowing development of the condition within the epiphysis. B, In children up to 15 years of age the infection is restricted to below the physis because of interruption of the vessels.
The microorganism responsible for osteomyelitis varies and is related to the age of the child (Box 43-1). Osteomyelitis in the newborn is caused primarily by Staphylococcus aureus. Group B streptococcus and Escherichia coli infections are responsible for some cases, especially those of multiple bone involvement and in high-risk infants.14
S. aureus is the responsible microorganism in 80% to 90% of osteomyelitis cases in older children. Haemophilus influenzae, a previously common cause of osteomyelitis in children less than 5 years of age, has become rare with the improvements in immunization. However, cases of methicillin-resistant Staphylococcus aureus (MRSA) have risen alarmingly in the past 5 years. Once an infrequent cause of childhood osteomyelitis, MRSA now causes up to 30% of new cases.15 Gram-negative microorganisms account for an increasing number of infections of the vertebrae,16,17 whereas Salmonella infections are associated with sickle cell disease.
Factors that predispose an individual to the development of osteomyelitis include impetigo, furunculosis, infected lesions of varicella (chickenpox), infected burns, cerebral abscesses, immunization with bacille Calmette-Guérin (BCG) vaccine, prolonged intravenous or central parenteral alimentation, drug addiction, and direct trauma to the area adjacent to the site of osteomyelitis.
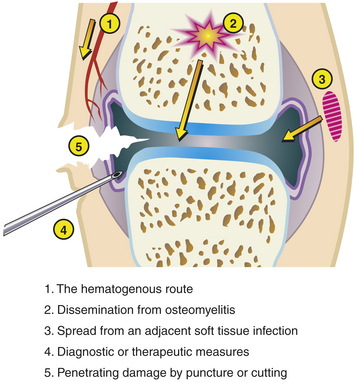
PATHOPHYSIOLOGY Osteomyelitis usually begins as a bloody abscess in the metaphysis of the bone. The abscess ruptures under the periosteum and spreads along the bone shaft or into the bone marrow cavity if untreated. Infection rarely spreads down the medullary cavity of the bone but rather first gains entrance to the subperiosteal space in the metaphysis. This is the path of least resistance because the cortex of the bone in this area is porous or mazelike and the inflammatory response blocks spread within the bone.18 Because of the accumulation of debris caused by the infection, the periosteum may separate and form a shell of new bone around the infected portion of the shaft. Because the periosteum is separated from an adequate blood supply, sections of the bone die; these pieces of dead bone are called sequestra. The periosteum that maintains a blood supply generates new bone and is responsible for the appearance of the periosteal new bone, or involucrum. The presence of the sequestra and involucrum indicates that the disease has progressed to subperiosteal abscess formation.
In cases in which the infection in the metaphysis occurs near the joint, the accumulating pus (bacteria, white blood cells, fluid) creates increasing pressure that may cause a rupture into the joint cavity. If rupture into the joint occurs, the pus causes inflammation and a condition called secondary septic arthritis.16 Although thought uncommon, a recent study shows 40% of children will have adjacent joint involvement with osteomyelitis. The most common joint to be affected is the knee. Osteomyelitis is most commonly caused by bacteria that reach the metaphysis through the bloodstream but may occur through secondary inoculation of microorganisms caused by trauma or contagious spread of infection from cellulitis in adjacent soft tissue.
Osteomyelitis in infants is often associated with septic arthritis because the infant’s bone has blood vessels that perforate the growth plate. Because of the unique nature of blood supply to an infant’s bones, osteomyelitis and septic arthritis commonly occur together. Normal anatomic variations in infants allow infection to spread directly to the epiphysis, which causes both joint disease and permanent epiphyseal disease. Multiple sites of osteomyelitis are also more common in children younger than 2 years, necessitating bone scan to check other bones when infants are infected. This can then lead to other areas of osteomyelitis and possibly septic arthritis.19
Children are susceptible to joint involvement for several reasons (Figure 43-9). In the immature infant, there is no epiphyseal plate or an ossific nucleus at the end of the bone and the cartilage precursor of bone is penetrated by vascular channels. In these infants the infection begins in the vulnerable cartilage precursor of the end bone itself and results in rapid destruction of the joint and arrested growth of the bone. For this reason the early detection and treatment of osteomyelitis are crucial if the infant’s joint is to be saved from destruction. As the child matures and the epiphyseal plate forms, a temporary barrier is established against infection because the arterioles end beneath the epiphyseal plate.18
In children older than 2 years, the epiphyseal plate prevents the spread of a metaphyseal abscess into the epiphysis and the cortex of the metaphysis is thicker. These anatomic differences increase the likelihood that the metaphyseal abscess will extend into the diaphysis, and the blood supply of the bone will be disrupted. The periosteum is also more difficult to perforate in older children; this may lead to a larger subperiosteal abscess that could endanger the periosteal blood supply as well. This process commonly results in extensive sequestrum formation and chronic osteomyelitis.18
Osteomyelitis is much less common after the physeal plates are closed, except in the vertebral body. Infection may develop in any part of a bone, and abscesses spread slowly. Destruction of the cortex in a localized area may result in a pathologic fracture.16,17
Spread of infection to contiguous joints is related to the child’s age. Metaphyseal infection may spread to contiguous joints if the fibrous joint capsule includes the metaphysis and epiphysis. This special situation exists at the hip joint, distal femur, proximal humerus and radius, and lateral ankle. Recent studies have shown, however, that like infants older children may demonstrate up to 42% of contiguous joint involvement. Even in areas where the involved osteomyelitis was extra-articular, joint involvement occurred; this differs from previous reports in the literature.19
CLINICAL MANIFESTATIONS The clinical manifestations of osteomyelitis are age dependent and are related to the differing vascular patterns found in the skeletal system at various ages. Three distinct groups may be identified: (1) infants younger than 1 year, (2) children from 1 year of age to puberty, and (3) adolescents after cessation of bone growth and adults.
Infants: Osteomyelitis may be an acute illness characterized by fever and failure to move the affected limb (pseudoparalysis). Infantile osteomyelitis is characterized by involvement of multiple sites within the same bone or in multiple bones. If untreated, involvement of the adjacent growth plate can result in growth arrest.
Children: Osteomyelitis in children between the ages of 1 year and puberty is characterized by fever and systemic signs of toxicity. The illness is sometimes subacute, with the child complaining of swelling, redness, tenderness, and decreasing ability to bear weight on or move the affected area. Onset can be abrupt. Osteomyelitis during childhood most often affects the long bones but also may be found in the pelvis and spine. Clinical manifestations are usually accompanied by elevated white blood cell counts and elevated erythrocyte sedimentation rates. C-reactive protein (CRP), when elevated, is a sensitive sign of osteomyelitis and can rapidly decrease with appropriate treatment. Evidence of infection using roentgenograms can be delayed but bone scan is positive within 48 hours.
EVALUATION AND TREATMENT White blood cell counts and erythrocyte sedimentation rates are sometimes elevated, but this is not a consistent finding. Monitoring of erythrocyte sedimentation rates is an indication of response to management but can be delayed. CRP is more quickly responsive to appropriate treatment. Blood cultures (positive in 30% to 40%) and aspiration of the soft tissue or bone, or both, should be done to identify the causative microorganism. Appropriate antibiotics should be prescribed after culture and sensitivity studies have been completed. A tuberculin test also is administered because Mycobacterium tuberculosis is sometimes responsible and has had a slight resurgence in incidence. Bone scans can be quite helpful with diagnosis and in children younger than 1 year are absolutely required to define whether multiple sites are involved.
Treatment includes intravenous (IV) antibiotics or, in highly reliable children and families, a combination of IV and oral antibiotics for 6 weeks. Drainage and margination of bone is required if changes are present on radiographs signifying abscess. Immobilization may help with pain control. If a joint is also infected (termed “septic arthritis”), the situation becomes a surgical emergency; surgery on the affected joint can help prevent damage to the articular cartilage by lysozymes released from the involved neutrophils.
Death is rare, but serious sequelae may occur. The course of the disease and prognosis depend on the age of the child, the rapidity with which the diagnosis is established, the initiation of early treatment, and maintenance of the treatment for an adequate time. The most serious complications are growth arrest, osseous necrosis, and recurrence. Recurrence with presently available antibiotic regimens is less than 10%.
Juvenile Rheumatoid Arthritis
The rheumatic diseases are a group of diverse conditions having in common the inflammation of connective tissues. They include rheumatoid arthritis (RA), systemic lupus erythematosus, dermatomyositis, scleroderma, and polyarthritis. Incidence of these disorders in children is estimated in Table 43-3.
Table 43-3
Incidence of Connective Tissue Diseases in Children
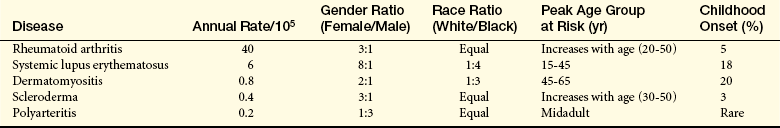
Data from Hollingworth P. In Klippel JH, Dieppe PA, editors: Rheumatology, St Louis, 1994, Mosby.
Juvenile rheumatoid arthritis (JRA) is the childhood form of rheumatoid arthritis (see Chapter 42). Like adult-onset RA, JRA is a syndrome that is often accompanied by systemic manifestations. Approximately 5% of all cases of RA begin in childhood. An estimated quarter of a million children in the United States have JRA.
The basic pathophysiology of JRA is the same as that of adult RA. The clinical manifestations of JRA may differ, however, beginning with mode of onset. Unlike adult RA, which begins insidiously with systemic signs of inflammation and generalized aches, JRA has three distinct modes of onset: arthritis in fewer than five joints (pauciarticular arthritis), arthritis in more than five joints (polyarticular arthritis), and systemic disease. Onset is less gradual in JRA than in adult RA. JRA also differs from the adult form in the following respects20,21:
1. Predominantly the large joints are affected.
2. Subluxation and ankylosis of the cervical spine are common if the disease progresses.
3. Joint pain may not be severe as in the adult type.
4. Serologic tests often detect antinuclear antibody (ANA).
5. Chronic uveitis is common, especially if ANA positive.
6. Serologic tests seldom detect rheumatoid factor.
7. Rheumatoid nodules are not limited to subcutaneous tissue but are found in the heart, lungs, eyes, and other organs.
Treatment for children with JRA is supportive but not curative. Many children with pauciarticular arthritis who are seronegative for ANA will resolve their symptoms over time. However, with systemic onset (Stills disease) or seropositivity, JRA may progress to true adult RA. The aims of treatment are to control inflammation and other clinical manifestations of the disease and to minimize deformity.
Avascular Diseases of the Bone: Osteochondrosis
The avascular diseases of the bone, collectively termed osteochondroses, are caused by insufficient blood supply to growing bones. Disturbances of blood supply to primary and secondary centers of ossification during periods of rapid bone growth results in a variety of skeletal abnormalities.
The cause of the osteochondroses remains obscure. In the past, infection, nutritional deficiencies, and hormonal imbalances were blamed, but these causes have been largely disproved. Currently, vascular impairment and trauma, coupled with an underlying developmental or genetic predisposition, have been identified as probable causes of osteochondroses. The most common osteochondroses are Osgood-Schlatter disease (tibial tubercle), Sinding-Larsen-Johansson syndrome (distal patellar pole), Panner disease (radial head), Kohler disease (the navicular bone of the foot), and Sever disease (calcaneus). All are associated with activity-related pain of the affected region that improves with rest. All are more common in boys than girls and in athletes more than nonathletes.
The osteochondroses involve areas of significant tensile or compressing stress that undergo partial osseous necrosis, progressive bony weakness, and then microfracture. Most of these are associated with trauma and overuse and improve with rest. Anti-inflammatories, modification of activities, and even immobilization are used during active disease. Reparative correction by revascularization is the rule, although this may be a lengthy process.
Legg-Calvé-Perthes Disease
Legg-Calvé-Perthes disease, commonly called Perthes disease, is classically thought to be an osteochondroses like those previously described. This self-limited disease of the hip is presumably produced by recurrent interruption of the blood supply to the femoral head. The ossification center first becomes necrotic, collapses, and then is gradually remodeled by live bone.
Legg-Calvé-Perthes disease is relatively common (1 in 5000 children), usually occurring in children between 3 and 10 years of age, with a peak incidence at 6 years. It is more common in boys than in girls by a ratio of about 5:1. The condition is bilateral in approximately 10% of affected children; in 80% of these children, the effect on the second side is less severe.22
The cause of decreased blood supply to the head of the femur is unknown. Several theories have been proposed, including trauma, infection, and protein C and S deficiencies, which cause a hypercoagulable state or vascular anamolies.23 A plausible theory is that acute synovitis (infection of the synovial membrane) and increased hydrostatic pressure in the hip joint compress blood vessels that supply the femoral head.
Constitutional factors definitely play a role. Skeletal maturation is delayed an average of 2 years in children with Legg-Calvé-Perthes disease, and affected children are between 2.5 and 7 cm shorter than unaffected children of the same age. Familial occurrence is 30% to 40%. The disease is rare in blacks, and it is frequent in children of Japanese and central European ancestry.
PATHOPHYSIOLOGY Legg-Calvé-Perthes disease runs its natural course in 2 to 5 years. In the incipient stage the soft tissues of the hip (synovial membrane and joint capsule) are swollen, edematous, and hyperemic, often with fluid present in the joint (Figure 43-10). The joint space widens, and the joint capsule bulges. The first stage lasts only a few weeks. In the second (or active avascular necrotic) stage, the entire epiphysis or the anterior half of the epiphysis of the femoral head loses blood supply and the metaphyseal bone at the junction of the femoral neck and capital epiphyseal plate is softened because of decalcification. Soon granulation tissue (procallus) and blood vessels invade the dead bone. This stage lasts several months to 1 year.
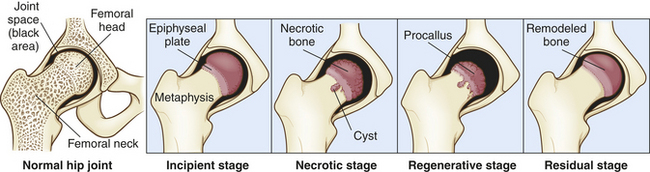
The third (or regenerative healing) stage ordinarily lasts 2 to 4 years. The dead femoral head is replaced by procallus, and new bone is laid down. Collapse and flattening of the femoral head occur, and the femoral neck becomes short and wide (see Figure 43-10).
In the fourth (or residual) stage, remodeling takes place and the newly formed bone is organized into a live spongy bone. Children less than 6 years of age at onset have more time to remodel the damage Perthes has caused and have the best outcome. Recent multicenter studies, using the Herring “lateral pillar” classification,24 have shown that hips younger than 6 years with no involvement of the lateral femoral head (type A) do better than those with involvement of the lateral femoral head. Those children with complete collapse of the lateral femoral head (type C) have the worst prognosis. Long-term studies of type C hips show that 70% to 90% progress to osteoarthritis by 40 years of age.25
CLINICAL MANIFESTATIONS Injury or trauma precedes the onset of clinical manifestations in approximately one third of children with Legg-Calvé-Perthes disease. Onset of symptoms is insidious unless trauma aggravates the disease process. The child often complains of a limp or pain for several months. The pain usually is referred to the knee, inner thigh, and groin, following the path of the obturator nerve. In some children, pain may be absent or minimal. If pain is present, it is usually aggravated by activity and relieved by rest.
The typical physical findings include spasm on inward rotation of the hip and a limitation of internal rotation flexion and adduction. If the child is walking, an abnormal gait, termed a Trendelenburg gait or abductor lurch, is apparent. The child moves the trunk toward the affected side with stance to compensate for weak abductor musculature. If the hip pain or limp has been present for a prolonged period, muscles of the hip and thigh atrophy. A limb length inequity may be present if the proximal femoral physis is involved.
EVALUATION AND TREATMENT Diagnosis is confirmed by radiographic examination. Principles of treatment are containment (keeping the ball completely in the socket) and motion to maintain the articular cartilage. In the past, children were treated with bed rest and a variety of braces, which have now been shown to be ineffective.26 Currently, most children can be managed with anti-inflammatory medications and crutches for episodes of synovitis and activity modification (avoidance of jumping activities that place increased stress on the hip) during the active phase of the disease. Serial roentgenograms monitor the progress of the disease and ensure that the hip remains congruent. Surgery may be necessary if the femoral head becomes subluxated or incongruent with the acetabulum before the reparative process. The ball must be congruent to take on the shape of the socket as remodeling occurs.
Factors affecting the outcome of Legg-Calvé-Perthes disease are the age of the child, the extent of necrosis, the stage of disease at the time treatment is begun, and congruence of the joint with skeletal maturity. Recent studies have shown that girls, despite earlier skeletal maturity, do as well as boys. Outcome is 70% satisfactory with Herring stage A; for Herring stages B and C or age greater than 8 years, outcome is guarded. Present prospective studies are evaluating more aggressive early treatment (i.e., osteotomy of the femur or pelvis) on the more involved hips to change long-term outcome.
Osgood-Schlatter Disease
Osgood-Schlatter disease consists of tendinitis of the anterior patellar tendon, within which the patella (kneecap) is embedded, and associated osteochondrosis of the tubercle of the tibia. Osgood-Schlatter disease occurs most often in preadolescents and adolescents who participate in sports. The incidence is higher in boys than in girls, many of whom have increased outward tibial fusion compared to controls.27
PATHOPHYSIOLOGY The severity of the lesion varies from mild tendinitis to a complete separation of the anterior extension of the tibial epiphysis, which is the part of the epiphysis that contributes to growth of the tibial tubercle. The underlying pathologic alterations also vary. The mildest form of Osgood-Schlatter disease causes ischemic (avascular) necrosis in the region of the bony tibial tubercle, with hypertrophic cartilage formation during the stages of repair. In more severe cases the abnormality involves a true epiphyseal separation of the tibial tubercle, with the characteristics of avascular necrosis that are described in the section on Legg-Calvé-Perthes disease.
CLINICAL MANIFESTATIONS The child experiences pain and swelling in the region of the patellar tendon and tibial tubercle, which becomes prominent and is tender to direct pressure. The pain is most severe after physical activity that involves vigorous quadriceps contraction or direct local trauma to the tibial tubercle area. Often the child experiences sudden acute discomfort referable to the affected region. Sudden onset of pain is caused by a pathologic fracture through an area of ischemic necrosis.
EVALUATION AND TREATMENT Diagnosis is confirmed by roentgenographic examination. The goal of treatment for Osgood-Schlatter disease is to decrease the stress at the tubercle. Often a period of 4 to 8 weeks of restriction from strenuous physical activity, especially activities requiring deep-knee bending, is sufficient. If pain relief is not achieved, a cast or brace is required to immobilize the knee, a situation that is particularly difficult if the condition is bilateral.
Gradual resumption of activity is permitted after 8 weeks, but return to unrestricted athletic participation requires an additional 8 weeks to allow for revascularization, healing, and ossification of the tibial tubercle.
Cerebral Palsy
Cerebral palsy (CP) is a static disorder of muscle tone and balance caused by an ischemic insult to the brain, usually perinatally. The incidence is presently 3% to 5% but is increasing with successful resuscitation of premature infants.
EVALUATION AND DIAGNOSIS The diagnosis of CP is often made when gross motor milestones are not met by predicted ages. In some infants, diagnosis can be made as early as 4 months.28 Cognitive involvement is widely variable and is dependent on the amount of central nervous system (CNS) involvement. There are classic patterns: hemiplegia involves one side of the body, diplegia usually involves the lower extremities only, and quadriplegia involves all four extremities. Quadraplegic involvement is most often associated with cognitive involvement, seizure disorder, and aphasia. Many quadriplegics, however, are of normal intelligence and are “trapped” within aphasia. When given communication devices, these children are sometimes “discovered,” as is their normal intelligence.
TREATMENT Treatment of cerebral palsy is multifaceted and undergoing constant evolution. The use of physical and occupational treatments, orthotics, spasticity reduction (by selected dorsal rhizotomy, oral, or intrathecal baclofen), botulinum-A (Botox) toxin injections, and surgery are often used to maximize a child’s function. In many centers, a multispecialty approach at “CP clinics” occurs so that a family may, within one clinic visit, see neurology, pediatrics, orthotics, orthopedic surgery, and rehabilitation clinicians.
Children with CP should be carefully followed and given all possible opportunities to flourish. Although CP is a static disorder, progressive deformity because of increased muscle tone can occur. Monitoring these children as they grow with a multispecialty approach is essential to their optimal outcome.
Muscular Dystrophy
The muscular dystrophies are a group of familial disorders that cause degeneration of skeletal muscle fibers. The muscular dystrophies are the most prevalent of the muscle diseases in childhood and are characterized by progressive, symmetric weakness and wasting of skeletal muscle groups, with increasing disability and deformity.
Classification of the muscular dystrophies is based on age of onset, rate of progression, distribution of muscular involvement, and inheritance patterns. The major clinically and genetically distinct types are the pseudohypertrophic (Duchenne), facioscapulohumeral, limb girdle, and oculopharyngeal dystrophies (Figure 43-11). Because the clinical findings and genetic inheritance patterns are consistent for each type, some researchers believe that each involves a separate biochemical defect. Genetic research has focused on identifying the site of abnormal gene function for each defect. This will permit more accurate carrier detection and, eventually, description of the biochemical aberration. (Table 43-4 summarizes the types of muscular dystrophy.)
Table 43-4
Major Muscular Dystrophy Syndromes
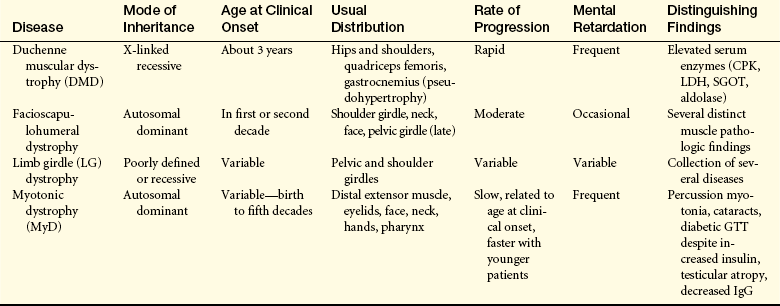
CPK, Creatine phosphokinase; GTT, glucose tolerance test; IgG, immunoglobulin G; LDH, lactate dehydrogenase; SGOT, serum glutamic oxaloacetic transaminase.
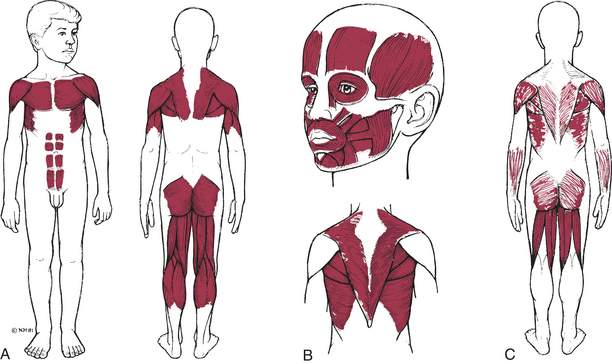
Figure 43-11 Initial muscle groups involved in three types of muscular dystrophy A, Pseudohypertrophic. B, Facioscapulohumeral. C, Limb girdle. (From Hockenberry MJ: Wong’s nursing care of infants and children, ed 8, St Louis, 2007, Mosby.)
Duchenne Muscular Dystrophy
In 1868 the French neurologist G.B.A. Duchenne described a pseudohypertrophic muscular paralysis associated with large amounts of fat and connective tissue. Today this form of muscular dystrophy, called Duchenne muscular dystrophy, is the most common of the muscular dystrophies. Its incidence is approximately 1 in 3500 male births.29 Classic Duchenne muscular dystrophy occurs only in boys and has a history of X-linked inheritance in half of the cases.
PATHOPHYSIOLOGY The X-linked inherited type of Duchenne muscular dystrophy is thought to be caused by a deletion of a segment of deoxyribonucleic acid (DNA)29 or a single-gene defect on the short arm of the X chromosome. A protein encoded by the Duchenne muscular dystrophy gene, called dystrophin, has been identified.
Dystrophin is present in normal muscle cells and absent in Duchenne muscular dystrophy. Dystrophin mediates anchorage of the actin cytoskeleton of skeletal muscle fibers to the basement membrane through a membrane glycoprotein complex. The complete lack of dystrophin in severe Duchenne dystrophy means that poorly anchored fibers tear themselves apart under the repeated stress of contraction. Free calcium then enters the muscle cells, causing cell death and fiber necrosis30 (Figure 43-12).
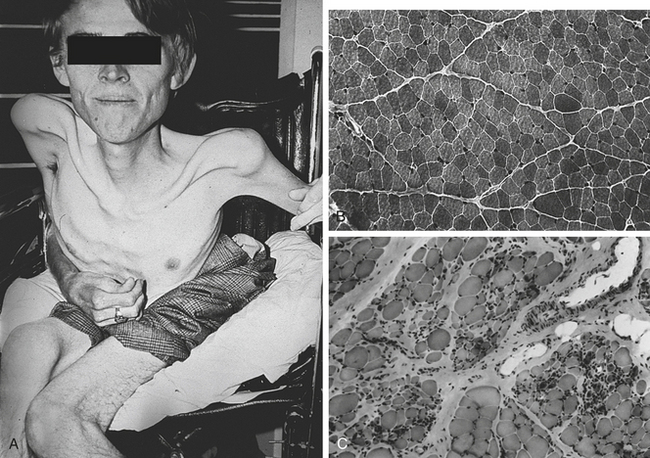
Figure 43-12 Duchenne muscular dystrophy. A, Patient with late-stage Duchenne muscular dystrophy showing severe muscle loss. B, Transverse section of gastrocnemius muscle from a normal boy. C, Transverse section of gastrocnemius muscle from a boy with Duchenne muscular dystrophy. Normal muscle fiber is replaced with fat and connective tissue. (From Jorde LB et al: Medical genetics, ed 3, St Louis, 2003, Mosby.)
There is increased endomysial connective tissue and fat; loss of striations; and concomitant hyaline, granular, and fatty degeneration of fibers. Disorganization of tendinous insertions is associated with fat accumulation in these areas. Although fibers regenerate in the younger child, they are abnormal in many ways and become nonfunctional with time.
CLINICAL MANIFESTATIONS Duchenne muscular dystrophy is usually identified in children at approximately 3 years of age, when the parents first notice slow motor development with progressive weakness and muscle wasting. Sitting, standing, and walking are delayed, and the child is clumsy, falls frequently, and has difficulty climbing stairs.
Muscular weakness always begins in the pelvic girdle, causing a “waddling” gait. Hypertrophy of the calf muscles is apparent in 80% of cases. The method of rising from the floor by “climbing up the legs” (Gower sign) is characteristic and is caused by weakness of the lumbar and gluteal muscles. The foot assumes an equinovarus position (see Figure 43-4), and the child tends to walk on the toes because of weakness of the anterior tibial and peroneal muscles. Within 3 to 5 years, muscles of the shoulder girdle become involved. Contractures and wasting of the muscles contribute to muscular atrophy and deformity of the skeleton.
Duchenne muscular dystrophy has serious complications. Pulmonary function is compromised greatly because of marked kyphoscoliosis (“humped” upper spine combined with scoliosis), which usually develops after the child is confined to a wheelchair (see Figure 43-12). The incidence of cardiac involvement in Duchenne muscular dystrophy is as high as 95%. Chronic heart failure may occur in 50% of children. A moderate degree of mental retardation causes these children to have a mean IQ of approximately 80. Smooth muscle dysfunction may cause megacolon, volvulus, cramping pain, and malabsorption in the gastrointestinal tract. The children usually succumb to other pulmonary or cardiac causes and death ensues by the late teens. Only 25% live to age 21.
EVALUATION AND TREATMENTDiagnosis is confirmed by measurement of serum enzymes, electromyography (EMG), and muscle biopsy. The serum enzymes, especially creatine phosphokinase (CPK), are increased to more than 10 times normal, even during infancy and before the onset of weakness. Histologic changes in muscle include degeneration of muscle fibers, with variation in fiber size and central nuclei. Fat and connective tissue replace muscle fibers.
Although intrauterine diagnosis is not yet possible, work is being done in this area.31 Elevated CPK levels at birth are diagnostic indicators of Duchenne muscular dystrophy. Identification of female carriers of the disease cannot be achieved with certainty, but serum CPK is elevated in 60% to 80% of carriers.
There is no effective treatment for Duchenne muscular dystrophy, but with characterization of the genetic deficit for Duchenne, gene therapy for Duchenne muscular dystrophy may be possible.32 Maintaining function in unaffected muscle groups for as long as possible is the primary goal. Although activity fosters maintenance of muscle function, strenuous exercise may hasten the breakdown of muscle fibers. Range-of-motion exercises, bracing, and surgical release of contracture deformities are used to maintain normal function as long as possible. The recent addition of oral steroids early in the disease has dramatically improved outcome. Children are able to walk an additional 2 to 5 years and life expectancy has increased.33 Scoliotic surgery is suggested when curves reach greater than 20 degrees to prolong respiratory function or walking ability or both. Genetic counseling is recommended. With X-linked inheritance, male siblings of an affected child
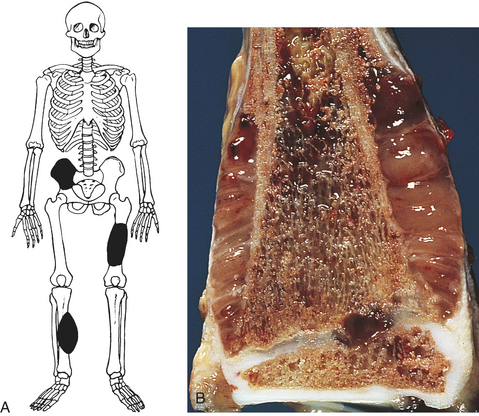
Figure 43-13 Ewing sarcoma A, Most common anatomic sites. B, Close-up view of Ewing sarcoma of the distal end of the tibia. Tumor extends into the soft tissue. (From Damjanov I, Linder J, editors: Anderson’s pathology, ed 10, St Louis, 1996, Mosby.)
have a 50% chance of being affected and female siblings have a 50% chance of being carriers.
Becker Muscular Dystrophy
Becker muscular dystrophy is often called benign Duchenne muscular dystrophy because it shares the X-linked inheritance pattern and similar but milder clinical features. The incidence of Becker dystrophy is one tenth that of Duchenne dystrophy. In Becker dystrophy, mutations in the middle rod region of dystrophin still allow anchorage of muscle to basement membrane. Clinical symptoms often begin between 5 and 15 years of age. Children with Becker muscular dystrophy remain ambulatory into their teens and early 20s; in one study the average age at the time of necessity for a wheelchair was 25 years.
The pattern of muscle weakness for both dystrophies is almost identical, but scoliosis and contractures are rare until the child with Becker muscular dystrophy is permanently wheelchair bound. The changes in creatine kinase levels and EMG and electrocardiogram (ECG) readings are the same as those seen in Duchenne muscular dystrophy. Many individuals live well into middle age. Heart failure is infrequent but can be a cause of premature death and disability.
Maintaining ambulation and careful follow-up for evidence of cardiopulmonary complications are essential for long-term care. Children with Becker muscular dystrophy rarely show the mental changes seen in Duchenne dystrophy. The accurate diagnosis of Becker muscular dystrophy is important. If the affected individual marries and has children, all daughters will be carriers of this X-linked recessive disorder. Genetic counseling should be offered to the mother, female siblings, offspring, and any maternal relatives.
Facioscapulohumeral Muscular Dystrophy
Facioscapulohumeral muscular dystrophy is a mild form of progressive, autosomal dominant muscular dystrophy. Age at onset varies from early childhood to adulthood, and the disease affects males and females equally. As the name implies, clinical manifestations begin with weakness and atrophy of facial and shoulder girdle (scapulohumeral) muscles. The illness progresses slowly. Inability to close the eyes completely may be noted from early childhood. The face is expressionless, and pouting of the lips makes whistling impossible. The first symptoms usually include drooping of the shoulders with difficulty in raising the arms above the head. Onset of weakness in the lower limbs often is delayed for 20 to 30 years, and pseudohypertrophy of muscles is rare. Contractures and skeletal deformities develop less often and are less prominent than in Duchenne muscular dystrophy.
Treatment includes supportive physiotherapy to prevent contractures and prolong ambulation. Lightweight plastic ankle-foot orthoses (AFOs) for footdrop are extremely helpful. Surgery to stabilize the shoulder is sometimes advised.
Some individuals with facioscapulohumeral muscular dystrophy improve with steroid therapy, particularly if the clinical picture includes rapidly progressive weakness. The disease may be arrested for prolonged periods; however, most individuals remain active and have a normal life expectancy. Vocational training and genetic counseling are important to provide the information necessary to plan their future.
Scapuloperoneal Muscular Dystrophy
Scapuloperoneal muscular dystrophy is considered a variant of facioscapulohumeral muscular dystrophy, but distal muscles in the lower extremity are involved early instead of the facial and shoulder muscle weakness that is the early sign in facioscapulohumeral dystrophy. Many individuals seek initial treatment for troublesome footdrop and shoulder weakness. Analysis of inheritance patterns shows that the disease can be inherited either as an autosomal dominant trait or as an X-linked recessive trait.
The initial symptoms may resemble those of several other illnesses, including nemaline myopathy (a congenital muscle disease) and early hypertrophic peripheral neuropathy. A careful diagnostic evaluation therefore is in order. Other clinical findings include hypertrophy of the muscle that extends the toes, brought about by a futile attempt to overcome footdrop, and depressed or absent muscle stretch reflexes. Creatine kinase is elevated 2 to 20 times the normal level; EMG readings show myopathy.
Treatment is directed toward relieving symptoms and preserving ambulation and functional ability. Footdrop is easily treated with AFOs. Individuals with scapuloperoneal muscular dystrophy remain ambulatory for 40 or more years. Occasionally, walking may be hampered by paraspinal muscle contractures; in that case a wheelchair may assist the individual when it is necessary to cover long distances. The life span in this disorder is normal. It is important that genetic counseling be available to all persons affected.
Limb Girdle Muscular Dystrophy
The diagnosis of limb girdle muscular dystrophy is considered when acute causes of proximal weakness are eliminated and the clinical findings and genetic pattern exclude Duchenne and facioscapulohumeral muscular dystrophy. The diagnosis is often determined by exclusion because few consistent clinical features make this type of muscular dystrophy unique. In fact, some researchers think that limb girdle dystrophy actually may be several separate diseases awaiting more sophisticated methods of evaluation to give them precise labels. Most individuals have a negative family history, making sporadic disease or an autosomal recessive pattern of inheritance likely. The prevalence rate for limb girdle muscular dystrophy is set at 20 per million.
The initial symptoms include shoulder and pelvic girdle weakness, which are usually noted in the early 20s but can be seen as late as the 40s. The muscle weakness is often asymmetric and progresses at a much slower pace than in Duchenne muscular dystrophy. The biceps and deltoid muscles can be extremely atrophic and weak. Individuals can remain ambulatory for extended periods, often up to 20 years after initial diagnosis. When confined to wheelchairs, they show few of the severe effects of other dystrophies, such as contractures and scoliosis. Heart involvement and mental retardation also are rare.
The individual will have mild elevation in creatine kinase levels and a myopathic pattern on EMG. Muscle biopsy is often more characteristic, with fiber splitting and fibers that appear profusely “moth-eaten” and whorled. Treatment includes supportive measures to maintain ambulation and functional ability and frequent follow-up to eliminate secondary complications such as cardiopulmonary disease.
Musculoskeletal Tumors in Children
Bone tumors are uncommon childhood tumors and comprise less than 5% of all childhood malignancies. Of the malignant tumors, osteosarcoma and Ewing sarcoma are the most common. Fortunately, the majority of pediatric tumors are benign, most commonly nonossifying fibroma, chondroma, simple bone cyst, aneurysmal bone cyst, osteoid osteoma, and fibrous dysplasia.
Nonossifying Fibroma: The nonossifying fibroma (fibrous cortical deficit), which is believed to be a defect in ossification rather than a true tumor, makes up approximately 50% of benign bone tumors. Most fibrous cortical defects resolve spontaneously or are obliterated by reparative ossification or remodeling. In some cases, however, the fibrous cortical defect persists and proliferates, becoming a fibroma. Fibromas are found primarily in children and adolescents. Ninety percent of these tumors occur in persons younger than 20 years.
The nonossifying fibroma is a sharply demarcated cortical-based tumor surrounded by a dense border of hardened bone. The tumor itself consists of fibrocytes arranged in whorled bundles, fibroblastic tissue, and osteoclast-like giant cells. As the tumor evolves the fibrocytes imbibe lipids and assume a foamy appearance, known as foam cells. The tumor also contains extensive deposits of hemosiderin pigment. The long axis of the tumor parallels the long axis of the bone.
The nonossifying fibroma is usually asymptomatic and is found incidentally on radiographs. In the 1950s, when fluoride was added to drinking water, random skeletal surveys were done on hundreds of children. Nonossifying fibromas were discovered in 20% to 30% of children and were distributed in nearly every bone of the body. The fibroma is generally not treated until it occupies more than 50% of the diameter of the bone or extends more than 3 to 4 cm into the cortex. When the tumor grows to this size, a pathologic fracture may occur, and curettage and bone grafting of the defect is undertaken.
Simple Bone Cyst: Simple bone cysts (SBCs) are cystic lesions of the central region of the metaphyseal region in skeletally mature children. With growth these lesions may appear within the diaphysis. These children are usually asymptomatic until pathologic fracture or incidental discovery occurs. Lesions often heal after a fracture, but large lesions may require treatment. A large prospective, randomized study is comparing steroid injection (the classical treatment for these lesions) with bone marrow injection for treatment. Very large lesions in weightbearing areas may require internal fixation and bone grafting.
Aneurysmal Bone Cyst: Aneurysmal bone cysts (ABCs) are typically eccentric, metaphyseal lesions that occur in a slightly older population than SBCs. The etiology remains controversial; many consider ABC a lesion secondary to another process, such as giant cell tumor. This lesion must be differentiated from telangiectatic sarcoma, thus biopsy is necessary. Once diagnosed, curettage with complete removal of the “pseudolining” must be done with chemical or electrocautery to minimize recurrence. Bone graft is placed in the defect. Even with modern techniques, recurrence can be as high as 21%.34
Osteoid Osteoma: Osteoid osteoma, or the larger counterpart osteoblastoma, presents as painful lesions of the diaphysis or metadiaphysis of long bones. Involvement of the posterior elements of the spine—with resultant “splinting” scoliosis—can occur. Night pain is common, as is relief from symptoms with nonsteroidal anti-inflammatory drugs (NSAIDs), because these tumors release prostaglandins. When pain is too extreme to be controlled medically, resection of the “nidus,” or central portion, of the lesion is uniformly successful. CT guidance to the lesion is often used and, if possible, CT-guided laser ablation can be done.
Fibrous Dysplasia: Fibrous dysplasia (FD) can occur in one bone (monostotic) or in multiple bones (polyostotic). Polyostotic fibrous dysplasia can occur with a triad of Albright syndrome that also includes precocious puberty and cutaneous pigmentation. Although any bone can be affected, the long bones, ribs, and skull are the most common. A radiographic “ground glass” appearance is present primarily in the metaphyseal or metadiaphyseal areas. Deformity can be marked and necessitate operative intervention. When allograft is used to replace fibrous dysplasia bone, it can become involved in the fibrous dysplasia as well. The majority of individuals are simply observed; however, endocrinology management also will be involved if Albright syndrome is present.
Osteosarcoma: Osteosarcoma is the most common bone tumor that occurs during childhood; it originates from bone-producing mesenchymal cells. Osteosarcoma occurs most commonly in the metaphysis of long bones, especially near active physes, such as the distal femur and proximal tibia. It accounts for 60% of all malignant bone tumors and strikes between the ages of 10 and 18.
Molecular analysis has demonstrated deletion of genetic material on the long arm of chromosome 13, leading to the identification of a tumor-suppressor gene as part of the mechanism for tumor development. The oncogene src also has been associated with osteosarcoma.
PATHOPHYSIOLOGY Osteosarcoma occurs mainly in the metaphyses of long bones. Most tumors arise in bones involved with the knee joint at the distal end of the femur or proximal end of the tibia. As a tumor of mesenchymal cells, osteosarcoma demonstrates production of osteoid tissue.
Osteosarcoma is a bulky tumor that extends beyond the bone into the soft tissues. It may encircle the bone and destroy the trabeculae of the diseased bone. Osteosarcoma disseminates through the bloodstream, usually to the lung. As many as 25% of children diagnosed with osteosarcoma exhibit lung metastases at diagnosis. Other sites of metastatic spread include other bones and visceral organs.
CLINICAL MANIFESTATIONS The most common presenting complaint is pain. There may be swelling, warmth, and redness caused by the vascularity of the tumor. Symptoms also may include cough, dyspnea, and chest pain if lung metastasis is present. If a lower extremity is involved, a limp or even pathologic fracture may be present.
Initial evaluation includes roentgenographic examination that shows the osteosarcoma’s characteristic osteoblastic and osteolytic changes. “Staging” studies to determine not only local extent of the tumor but also possible metastatic spread must be done. These include bone scan (to assess bony spread), magnetic resonance imaging (MRI) of the lesion (to plan surgical resection and to compare with postchemotherapy studies), and chest roentgenograms or CT or both. The chest roentgenogram must be done before biopsy because the general anesthetic required for biopsy can give false-positive results on the roentgenogram.
EVALUATION AND TREATMENT Tissue biopsy confirms the diagnosis, although needle biopsy is often sufficient to establish the diagnosis. There are five histologic types of osteosarcoma, each determined by the predominant cell type. The tumor is then graded according to degree of malignancy; the higher the number, the worse the prognosis.
Surgery and chemotherapy are the primary treatments for osteosarcoma. Radiation is occasionally used. The 5-year survival rate with modern protocols is 70% to 80%.35
Chemotherapy is an important component of treatment because as many as 80% of children treated with surgery alone eventually develop metastatic disease. Chemotherapy is used preoperatively to shrink the size of the tumor and minimize metastatic growth. Following chemotherapy, the child is given a short “rest period” to regain strength for surgery. Following adjunctive chemotherapy, the majority of children undergo “limb salvage” rather than amputation procedures. Using preoperative MRI, the extent of the lesion is mapped and a tumor-free margin is left and reconstructed either with allograft bone or arthroplasty (artificial joint). The long-term survival rate of children treated with limb salvage and chemotherapy is now near equal to amputation in 5- and 10-year survival.36
A number of approaches have been used to treat pulmonary metastases. Because pulmonary metastases are generally solitary, thoracotomy with wedge resection has proved the most effective. Investigators have searched for adjuvant treatment to prevent pulmonary metastases, but nothing has proved useful.
Ewing Sarcoma: Ewing sarcoma is a malignant round cell tumor of bone and soft tissue that has a poor prognosis. It is the second most common and most lethal malignant bone tumor that can occur during childhood. This tumor is named after James Ewing, who first identified it as a separate clinical diagnosis in 1921. The most common period of diagnosis is between 5 and 15 years of age, and rare after 30 years of age; however, it may be seen in children younger than 3 years. Like osteosarcoma, Ewing sarcoma is slightly more common in males than females and is linked with periods of rapid bone growth. The incidence of Ewing sarcoma is less than 2% in blacks.37
PATHOPHYSIOLOGY Ewing sarcoma commonly occurs in the midshaft or diaphysis of long bones or in flat bones. The most common sites include the pelvis, femur, and tibia. The femur is involved in most cases, with the pelvis being the second most common site. It can occur in any bone.
Arising from bone marrow, Ewing sarcoma can break through the cortex of the bone to form a soft-tissue mass. It does not form bone, but abundant reactive bone may be present in an attempt to contain this quickly growing lesion. Metastasis occurs early and is usually apparent at diagnosis or within 1 year. The most common sites are the lung, other bones, lymph nodes, bone marrow, liver, spleen, and central nervous system, although invasion of any organ is possible.
CLINICAL MANIFESTATIONS Like osteosarcoma, the most common complaint is pain about the diaphysis that increases in severity. A soft-tissue mass is often present. Additional symptoms may include fever, malaise, and anorexia. Known as “the great imitator,” Ewing sarcoma can appear radiographically identical to infection or even benign lesions such as Langerhans cell granulomatosis. Any pervasive diaphyseal or rib lesion must be regarded with a high index of suspicion.
EVALUATION AND TREATMENT In addition to plain roentgenogram, CT and MRI are needed to help establish the diagnosis and extent of the tumor. Bone scan, chest roentgenogram, and chest CT scan are also used to detect metastases. No specific laboratory test is diagnostic; however, the sedimentation rate will be elevated and lactic dehydrogenase (LDH) often is elevated. An elevated LDH level is a poor prognostic sign. Biopsy is used to conclusively establish the diagnosis. The identification of an 11:22 chromosomal translocation within the tumor cells confirms the diagnosis of Ewing sarcoma.
The use of multidrug chemotherapy has improved survival rates. Treatment protocols call for preoperative chemotherapy followed by radiation or surgical resection or both, with continuation of chemotherapy for 12 to 18 months afterward. Amputation is avoided when possible but may be considered in lower limb tumors of children younger than 8 years because of the serious discrepancy in bone growth that results if the primary treatment used is radiation, which can damage the physis. Secondary malignancies caused by high-dose radiation are also a concern.38
Historically, Ewing sarcoma had a dismal prognosis, with 5-year survival rates no better than 5% to 10%. Combinations of aggressive radiation, chemotherapy, and surgical resection have, however, improved the survival rate for localized disease to more than 60%.39 The major predictor of prognosis appears to be the location of the primary tumor and whether metastases are present at diagnosis. The most favorable sites of involvement are the extremities; the worst prognosis involves tumors of the trunk, particularly the pelvis.
Muscle Tumors
Most soft-tissue tumors in children are benign. Only two malignant soft-tissue tumors occur with any frequency— rhabdomyosarcoma in the younger child and synovial cell sarcoma in the teenager. Both of these occur rarely. The annual incidence is 8 cases per million for white children and 7.7 per million for black children. About 230 U.S. children are diagnosed with a soft-tissue tumor each year. Soft-tissue tumors originate from the primitive mesenchymal cells that normally give rise to muscle, tendons, blood vessels, lymphatic structures, fibrous and connective tissue, and bursa and fascia. Table 43-5 identifies the classification of soft-tissue tumors according to origin. All malignant soft-tissue tumors are characterized as highly aggressive tumors that invade surrounding structures and metastasize early.
Table 43-5
Classification of Tumors by Origin
| Tissue | Tumor |
| Muscle | |
| Striated | Rhabdomyosarcoma |
| Smooth | Leiomyosarcoma |
| Adipose | Liposarcoma |
| Fibrous | Fibrosarcoma |
| Synovial mesothelium | Synovial sarcoma |
| Lymphatic structures | Lymphangiosarcoma |
| Blood vessels | Hemangiopericytoma |
| Nerve sheath | Neurogenic sarcoma |
Rhabdomyosarcoma (RMS) is the most common soft-tissue sarcoma of childhood and accounts for more than 50% of soft-tissue tumors but less than 3% of all childhood cancers. RMS arises from embryonal rhabdomyoblasts that normally differentiate into mature striated muscle.
RMS can develop anywhere striated muscle is located. The primary locations and percentage range of incidence are the head and neck (including the orbit), 36% to 61%; the trunk, 8% to 33%; the extremities, 14% to 24%; and the genitourinary tract, 10% to 17%. Two age ranges (2 to 6 years and 15 to 19 years) are associated with RMS. More than two thirds of children with RMS are diagnosed by 10 years of age, and RMS is slightly more common in males than females.
Recent studies demonstrate an association between TP53 (a tumor-suppressor gene) mutations and sporadic rhabdomyosarcoma.40 Three oncogenes (src, ras, and c-myb) have been associated with this tumor.41
PATHOPHYSIOLOGY RMS generally appears as a firm, fleshy, grayish white mass. It sometimes exhibits variations that appear as a cystic polypoid mass. RMS has various appearances, depending on the phase of differentiation of the rhabdomyoblast. The cells may be round, spindle shaped, tadpole shaped, or multinucleated giant cells.
At least 20% of children with RMS have metastatic disease at diagnosis. The preferred sites of metastases include the lungs, lymph nodes, bone marrow, liver, brain, and bone. Another 30% have disease that is unresectable, although not widely spread. This also becomes a grave prognosis.
CLINICAL MANIFESTATIONS The signs and symptoms of RMS depend on the anatomic location of the primary tumor and presence of symptomatic metastases. The tumors are usually painless, and early detection of RMS is facilitated by the presence of a palpable or visible mass. Deep-seated tumors may cause functional impairment but can be silent until they are very large. The clinical manifestations of RMS are outlined in Table 43-6.

EVALUATION AND TREATMENT Diagnostic studies during the pretreatment phase are used to determine the extent of the primary tumor and presence or absence of distant metastases. Specific diagnostic studies depend on the primary site, but a combination of radiographic, nuclear, and CT scanning or MRI technology and blood studies is used. A biopsy of the primary tumor is necessary to confirm the diagnosis. Chromosomal abnormalities are being investigated for RMS. Although not widely incorporated into clinical management, identification of the DNA content has value as a prognostic factor and a determinant of treatment.
RMS is treated by a combination of surgery, radiation, and chemotherapy. Complete surgical resection provides the greatest assurance that cure can be achieved; however, cure occurs in only 16% of children. If surgical resection leads to serious disfigurement or functional disability (e.g., enucleation for orbital tumors or cystectomy for bladder tumors), chemotherapy and radiation serve as the primary treatment, and surgery is avoided or minimized. For all tumors except stage I disease, local radiation therapy is given. A variety of combination chemotherapies are used for RMS. Chemotherapy for stage I or II disease is given for 1 year if disease does not recur. For stage III or IV, treatment (combined with radiation therapy) continues an additional year. Intrathecal chemotherapy is given to children whose tumor locations favor CNS spread.
The primary prognostic factor in RMS is the degree of residual disease after surgical resection. Children with localized disease (stages I and II) have long-term survival rates of 70% to 80%. With widespread disease, long-term survival rates drop to 20%. Orbital tumors have an overall favorable prognosis, probably because of the lack of lymphatics in the area and early physical signs of disease.
NONACCIDENTAL TRAUMA
Abuse is estimated to occur in more than 1.5 million U.S. children per year. The maltreatment may be psychologic, sexual, or physical. Of children who suffer physical abuse, 30% are initially seen by an orthopedist. Accurate and appropriate referrals to child protection are not only legally mandated but also essential for the well-being of the child; an abused child who returns unmonitored to an abusive situation has a 15% chance of mortality.
ETIOLOGY Children who are not yet ambulatory and present with a long bone fracture have a greater than 75% chance of that fracture being caused by nonaccidental trauma. “Corner” metaphyseal fractures, caused by a twisting force, are nearly pathognomonic of abuse but occur only 25% of the time (Figure 43-14). Fractures at multiple stages of healing are also suggestive; however, osteogenesis imperfecta must be ruled out. The most common presentation is a transverse tibia fracture.42 After walking age, only 2% of long bone fractures are the result of nonaccidental trauma.43
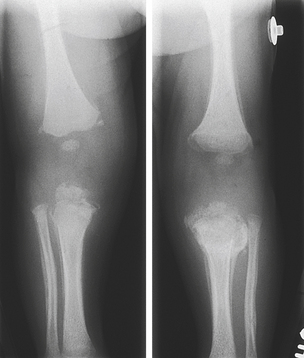
Figure 43-14 Corner fracture. Bilateral knee radiograph showing healing corner fractures of bilateral proximal tibias and distal femurs. Note the varying amount of callus formation signifying fractures at different stages of healing.
EVALUATION If suspected, nonaccidental trauma necessitates early consultation with child protective services. The child should undergo skeletal radiographic survey, especially if less than 2 years of age, and have a complete physical examination to evaluate for patterned bruising, burns, or multiple soft-tissue injuries. Ophthalmologic examination should be used to evaluate for retinal hemorrhage caused by shaking. A thorough history must be obtained for all identified injuries. It is important to remember that social isolation can lead to increased likelihood of abuse, but no social stratum is immune.
When cause is unclear, bone scan can be helpful in diagnosing subtle injuries, especially rib fractures. Posterior rib fractures are especially likely to be caused by abuse. MRI and CT of the brain can help diagnose injuries caused by shaking.
TREATMENT A nonjudgmental attitude on the part of the treating healthcare provider is essential. The child and family involved in nonaccidental trauma are delicate and require not only physical but emotional care. Social workers need to be involved early to ensure appropriate medical care to the child. Fortunately, fractures heal quickly in young children; neurologic injury and social disease, however, are much more difficult to cure.
Aneurysmal bone cyst (ABC) 1637
Becker muscular dystrophy 1636
Cartilage anlage 1618
Cerebral palsy (CP) 1632
“Corner” metaphyseal fracture 1640
Developmental dysplasia of the hip (DDH) 1621
Duchenne muscular dystrophy 1633
Dystrophin 1633
Endochondral formation of bone 1618
Ewing sarcoma 1638
Facioscapulohumeral muscular dystrophy 1636
Fibrous dysplasia (FD) 1637
Genu valgum (knock knee) 1620
Genu varum (bowleg) 1620
Idiopathic equinovarus 1623
Idiopathic scoliosis 1625
Intramembranous formation of bone 1618
Involucrum 1629
Juvenile rheumatoid arthritis (JRA) 1630
Kyphosed 1620
Legg-Calvé-Perthes disease 1631
Limb girdle muscular dystrophy 1636
Lordotic 1620
Metatarsus adductus 1622
Muscular dystrophy 1633
Nonossifying fibroma (fibrous cortical deficit) 1637
Nonstructural scoliosis 1625
Osgood-Schlatter disease 1632
Osteochondrosis 1631
Osteogenesis imperfecta (OI) (brittle bone disease) 1624
Osteoid osteoma 1637
Osteomyelitis 1628
Osteosarcoma 1637
Perichondrium 1618
Periosteal collar 1619
Pes planus (flatfoot) 1623
Physeal closure 1620
Positional equinovarus 1623
Primary centers of ossification 1618
Rhabdomyosarcoma (RMS) 1639
Rickets 1625
Scapuloperoneal muscular dystrophy 1636
Scoliosis 1625
Secondary centers of ossification 1619
Secondary septic arthritis 1629
Sequestra (sing., sequestrum) 1629
Simple bone cyst (SBC) 1637
Structural scoliosis 1625
Syndactyly 1620
Teratologic equinovarus 1623
Vestigial tab 1621
REFERENCES
1. Simkin, P. The musculoskeletal system. In Klippel J.H., Dieppe P.A., eds.: Rheumatology, ed 2, London: Mosby-Wolfe, 1998.
2. Weinstein, S.L., Congenital hip dislocation: long range problems, residual signs, and symptoms after successful treatments. Clin Orthop Relat Res Aug. 1992(281):69–74.
3. Weintraub, S., Grill, F. Ultrasonography in developmental dysplasia of the hip. J Bone Joint Surg Am. 2000;82-A(7):1004.
4. Ippolito, E., et al. Long-term comparative results in patients with congenital clubfoot treated with two different protocols. Arch Gynecol Obstet. 2003;268(4):331–332.
5. Dobbs, M.B., et al. Factors predictive of outcome after use of the Ponseti method for the treatment of idiopathic clubfeet. J Bone Joint Surg Am. 2004;86-A(1):22–27.
6. Zaleske, D.J., Doppelt, S.H., Mankin, H.J. Endocrine abnormalities of the immature skeleton. In Lovell W.W., Winter R.B., eds.: Pediatric orthopedics, ed 2, Philadelphia: Lippincott, 1986.
7. Zeitlin, L., Fassier, F., Glorieux, F.H. Modern approach to children with osteogenesis imperfecta. J Pediatr Orthop B. 2003;12(2):77–87.
8. Wharton, B., Bishop, N. Rickets. Lancet. 2003;362(9393):1389–1400.
9. Barrack, R., et al. Vibratory hypersensitivity in idiopathic sclerosis. J Pediatr Orthop. 1988;8(4):389.
10. Byrd, J.A., III: Current theories on the etiology of idiopathic scoliosis. Clin Orthop Relat Res Apr. 1988(299):114–119.
11. Vijermans, V., et al. Factors determining the final outcome of treatment of idiopathic scoliosis with the Boston brace: a longitudinal study. J Pediatr Orthop B. 2004;13(3):143–149.
12. Caksen, H., et al. Septic arthritis in childhood. Pediatr Int. 2000;42(5):534.
13. Gillespie, W.J., et al. Aspects of the microbe: host relationship in staphylococcal hematogenous osteomyelitis. Orthopedics. 1987;10(3):475.
14. Hedström, S.A., Lidgren, L. Septic arthritis and osteomyelitis. In Klippel J.H., Dieppe P.A., eds.: Rheumatology, ed 2, London: Mosby-Wolfe, 1998.
15. Stanitski, C. Changes in pediatric acute hematogenous osteomyelitis management. J Pediatr Orthop. 2004;24(4):444–445.
16. Przybylski, G.J., Sharan, A.D. Single-stage autogenous bone grafting and internal fixation in the surgical management of pyogenic discitis and vertebral osteomyelitis. J Neurosurg. 2001;94(1 Suppl):1.
17. Ray, N.J., Basset, R.L. Pyogenic vertebral osteomyelitis. Orthopedics. 1985;8(4):504.
18. Mader, J.T., et al. The host and skeletal infection: classification and pathogenesis of acute bacterial bone and joint sepsis. Baillieres Best Pract Res Clin Rheumatol. 1999;13(1):1.
19. Perlman, M., et al. The incidence of joint involvement of adjacent osteomyelitis in pediatric patients. J Pediatr Orthop. 2000;20(1):40.
20. Cassidy, J.T. Textbook of pediatric rheumatology. New York: Wiley & Sons; 1982.
21. Hollingworth, P. Juvenile chronic arthritis. In Klippel J.H., Dieppe P.A., eds.: Rheumatology, ed 2, London: Mosby-Wolfe, 1998.
22. Guille, J.T., et al. Bilateral Legg-Calvé-Perthes disease: presentation and outcome. J Pediatr Orthop. 2002;22(4):458–463.
23. Glueck, C.J., et al. Association of antithrombotic factor deficiencies and hypofibrinolysis with Legg-Perthes disease. J Bone Joint Surg Am. 1996;78(1):3–13.
24. Herring, J.A., et al. The lateral pillar classification of Legg-Calvé-Perthes disease. J Pediatr Orthop. 1992;12(2):143–150.
25. Schoenecker, P.L., et al. Legg-Perthes disease in children under 6 years old. Orthop Rev. 1993;22:201.
26. Askoy, M.C., et al. Comparison between braced and non-braced Legg-Calvé-Perthes-disease: a radiological outcome study. J Pediatr Orthop B. 2004;13(3):153–157.
27. Gigante, A., et al. What is the best treatment for Osgood-Schlatter disease? J Fam Pract. 2004;53(2):153–156.
28. Swanson, M.W., et al. Identification of neurodevelopmental abnormality at 4 and 8 months by the movement assessment of infants. Dev Med Child Neurol. 1992;34(4):321–337.
29. Scott, M.O., et al. Duchenne muscular dystrophy gene expression in normal and diseased human muscle. Science. 1988;239:1418.
30. Stevens, A., Lowe, J. Pathology. London: Mosby-Wolfe; 1995.
31. Nevo, Y., et al. Fetal muscle biopsy as a diagnostic tool in Duchenne muscular dystrophy. Prenat Diag. 1999;19(10):921.
32. Kapsa, R., et al. Novel therapies for Duchenne muscular dystrophy. Lancet Neurol. 2003;2(5):299–310.
33. Moxley, R.T., 3rd., et al. Practice parameter: corticosteroid treatment of Duchenne dystrophy: report of the Quality Standards Subcommittee of the American Academy of Neurology and the Practice Committee of the Child Neurology Society. Neurology. 2005;64(1):13–20.
34. Campanacci, M., Capanna, R., Picci, P., Unicameral and aneurysmal bone cysts. Clin Orthop Relat Res Mar. 1986(204):25–36.
35. Glasser, D.B., et al. Survival, prognosis, and therapeutic response in osteogenic sarcoma: the Memorial Hospital experience. Cancer. 1992;69(3):698–708.
36. Rougraff, B.T., et al. Limb salvage compared with amputation for osteocarcoma of the distal end of the femur. J Bone Joint Surg Am. 1994;76(5):649–656.
37. Ayala, A.G., Ro, J.Y., Raymond, A.K. Bone tumors. In Damjanov I., Linder J., eds.: Anderson’s pathology, ed 10, St Louis: Mosby, 1996.
38. Smith, L.M., Cox, R.S., Donaldson, S.S., Second cancers in long term survivors of Ewing’s sarcoma. Clin Orthop Relat Res Jan. 1992(274):275–281.
39. Ruyman, F.B., Grovas, A.C. Progress in the diagnosis and treatment of rhabdomyosarcoma and related soft tissue sarcomas. Cancer Invest. 2000;18(3):223.
40. Lugo-Vicente, H. Molecular biology and genetics affecting pediatric solid tumors. Bol Asoc Med P R. 2000;92(4-8):72–82.
41. Israel, M.A. Molecular and cellular biology in pediatric malignancies. In: Pizzo P.A., ed. Principles and practice of pediatric oncology. Philadelphia: Lippincott, 1989.
42. King, J., et al. Analysis of 429 fractures in 189 battered children. J Pediatr Orthop. 1988;8(5):585–589.
43. Thomas, S.A., et al. Long-bone fracture in young children: distinguishing accidental injuries from child abuse. Pediatrics. 1991;88(3):471–476.