GENES AND GENETIC DISEASES



In the nineteenth century, microscopic studies of cells led scientists to suspect that the nucleus of the cell contained the important mechanisms of inheritance. Scientists found that chromatin, the substance that gives the nucleus a granular appearance, is observable in nondividing cells. Just before the cell divides, the chromatin condenses to form discrete, dark-staining organelles called chromosomes. (Cell division is discussed in Chapter 1.) With the rediscovery of Gregor Mendel’s important breeding experiments at the turn of the twentieth century, it soon became apparent that the chromosomes contained genes, the basic units of inheritance. Chromosomes were the subject of much study, but because of poorly developed laboratory techniques, progress was slow. Since the mid-1950s, however, technologic advances have permitted a rapid increase in scientific knowledge of the form, composition, and function of chromosomes.
The primary constituent of the chromatin is deoxyribonucleic acid (DNA). Genes are composed of sequences of DNA. By serving as the blueprints of proteins in the body, genes ultimately influence all aspects of body structure and function. Estimates suggest that there are approximately 20,000 to 25,000 genes. An error in one of these genes can lead to a recognizable genetic disease.
To date, more than 15,000 genetic conditions have been identified and cataloged.1 As infectious diseases come under increasingly effective control, the proportion of beds in pediatric hospitals occupied by children with genetic diseases has risen to one third.2 In addition, many common diseases that affect primarily adults, such as hypertension, coronary heart disease, diabetes, and cancer, are now known to have important genetic components. (These diseases are also affected by environmental factors. The interaction between genetic and environmental components is discussed in Chapter 5.)
Great progress is being made in the diagnosis of genetic diseases and the understanding of genetic mechanisms underlying them. With the huge strides being made in molecular genetics, gene therapy—the direct alteration of genes in cells—has begun. Genetics is now one of the most rapidly advancing fields of medicine (Box 4-1).
Box 4-1 Genetic Engineering and Gene Therapy: The “New Genetics”
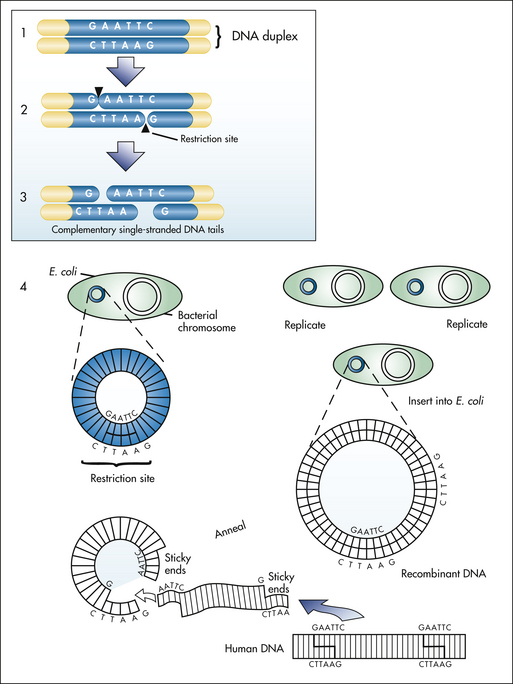
Recombinant DNA technology. Human DNA and circular plasmid DNA are both cleaved by a restriction enzyme, producing sticky ends (1 to 3). This allows the human DNA to anneal and recombine with the plasmid DNA. Inserted into the plasmid DNA, the human DNA is now replicated when plasmid is inserted into the bacterium, such as E. coli (4). G, Guanine; A, adenine; T, thymine; C, cytosine. (From Jorde LB et al: Medical genetics, ed 3, St Louis, 2003, Mosby.)
Terms such as cloning, genetic engineering, and recombinant DNA have received much exposure in the popular press during the past several years as the news media have recognized the potential importance of these techniques. Indeed they are part of the scientific revolution sometimes known as the new genetics.
Genetic engineering refers to laboratory alteration of genes. Most alterations are accomplished by using recombinant DNA techniques, which involve combining the DNA of two or more different organisms. A number of sophisticated methods have been invented to do this; described here is a common approach that is similar in principle to most other approaches.
Among the key components of recombinant DNA research are bacterial plasmids—small, circular pieces of self-replicating DNA that reside in many bacteria but often are not essential to the growth or survival of the bacteria. Plasmids can therefore be extracted from or inserted into bacteria without seriously disrupting bacterial growth or reproduction. Once they are extracted from their bacterial hosts, the plasmids are exposed to restriction endonucleases, which are enzymes that cleave, or cut, the plasmid DNA at a specific nucleotide sequence, called a restriction site.
Different restriction endonucleases have different restriction sites. A commonly used restriction endonuclease is called EcoRI (from the bacteria that produce it, Escherichia coli). EcoRI cleaves DNA only when the sequence GAATTC is found on one DNA strand and the complementary sequence is found on the other strand. The DNA of another organism, such as a human, also can be exposed to EcoRI and can be cleaved at the same restriction sites. The resulting human restriction fragments, which are pieces of DNA, have exposed ends that have base sequences complementary to those of the cleaved plasmid DNA. The human DNA and plasmid DNA, if mixed together, undergo complementary base pairing (i.e., they recombine).
The result is that the human DNA is incorporated within the plasmid. The plasmids, which now contain human genes in addition to their own, are allowed to reenter bacteria. Selection processes can be applied to pick out the bacteria that contain the desired human genes. These are cultured and allowed to form clones (or genetically identical copies) through normal cell division. Through continued cell division, millions of bacterial clones are formed, all containing the same human gene. Like any other gene, the human gene directs protein synthesis in the bacteria, resulting in the production of human proteins by bacteria.
Because bacteria multiply rapidly, large amounts of a given human protein can be manufactured by using this procedure. It has already been used successfully to produce human insulin in mass quantities. Because the insulin produced this way is actually human insulin, it produces fewer allergic reactions than the insulin taken from animal pancreases. Interferon, a substance that may help the body fight cancer and viral infections, also has been produced this way, as has human growth hormone, a substance that can be used to cure pituitary dwarfism.
In trying to isolate a particular gene, it is often more convenient to begin work with the messenger ribonucleic acid (mRNA) that codes for the gene product. The mRNA can be purified from body cells, and then an enzyme called reverse transcriptase can be used to generate the DNA sequence that is complementary to the mRNA. This complementary DNA (cDNA) can be inserted into plasmids and cloned by using the same recombinant techniques, so that virtually unlimited quantities of the desired gene product can be manufactured.
Recombinant DNA methods have been applied toward the understanding of the single-gene disorder phenylketonuria (PKU), which is the result of a lack of the enzyme phenylalanine hydroxylase. First, mRNA coding for this enzyme was purified from rat liver cells. After attachment of a radioactive “label” to cDNA produced from this mRNA, the cDNA was used as a probe. The probe was exposed to a series of cells that had been manipulated in the laboratory so that each cell line contained only one or a few chromosomes. When the probe hybridized consistently with only the cells containing chromosome 12, it proved that the gene that produces phenylalanine hydroxylase and thus causes PKU is located on this chromosome. Knowing the chromosome location of a gene is a very important step in the diagnosis and understanding of a genetic disease. Ultimately, therapeutic techniques might be developed to correct such disorders by replacing or repairing the abnormal gene.
The use of recombinant DNA techniques to clone DNA sequences has been (and continues to be) of great importance in genetics. However, the cloning process can take a great deal of time, even for well-studied genes. When doing genetic diagnoses, it is often necessary to obtain results very quickly. A newer technique, the polymerase chain reaction (PCR), provides a very rapid means of making millions of copies of a DNA sequence in only a few hours (as opposed to 1 week or more using cloning techniques). PCR basically involves the artificial replication of a DNA sequence, achieved by exposing the DNA strand to alterations in temperature in the presence of free DNA bases. At lower temperatures the DNA undergoes complementary base pairing, and at higher temperatures the DNA strands separate to form new templates for another cycle of replication when the temperature is again lowered. By repeating this temperature cycling over and over, DNA copies can be produced rapidly. This technique is very useful for diagnostic purposes because it requires only a very small sample of blood or other tissue and because a large number of copies can be made in a very short time. In theory, even a single DNA molecule can be copied millions of times using PCR. It is also used extensively in forensic medicine (e.g., in identifying the DNA of criminal suspects by using blood, semen, or hair samples left at the scene of a crime).
The advent of this technology has led to fears that organisms that could pose grave threats to the human species might be created. In 1974 a group of molecular geneticists themselves called for a moratorium on recombinant DNA research when its implications began to be realized; however, after much study and the introduction of rules regarding laboratory containment, research was resumed. Because of the elaborate precautions taken to prevent inadvertent creation of harmful organisms and because of the very low probability that such organisms could survive outside the laboratory, the possibility of such an occurrence is now considered to be extremely remote.
An area in which recombinant DNA techniques have generated much interest is gene therapy, which essentially involves the insertion of normal genes. For example, by recombinant DNA methods, a normal gene might be inserted into a human chromosome to counteract the effects of an abnormal or missing gene.
Gene therapy can be applied in two ways. The less controversial approach is somatic cell therapy, which consists of inserting normal genes into the cells of an individual who has a genetic disease. Here a particular tissue, such as bone marrow cells that produce abnormal erythrocytes, would be treated. More controversial is the application of gene therapy very early in embryonic development. By inserting genes into the embryos, all body cells could be altered, including the germ cells. Thus not only would the genetic constitution of the embryo and resulting individual be changed, but also all the descendants of that individual would have altered genetic constitutions. This procedure is sometimes referred to as germ cell therapy.
Somatic cell therapy has now been initiated for a number of human diseases, including hemophilia, cystic fibrosis, familial hypercholesterolemia, and several types of cancer.∗ More than 1300 somatic cell gene therapy protocols are being tested. Although significant setbacks have occurred, considerable technologic progress is being made and somatic cell therapy is beginning to demonstrate therapeutic effects for some diseases. Because of several important technical and ethical considerations, germline therapy will not be attempted in humans in the foreseeable future.
∗Kootstra NA, Verma IM: Annu Rev Pharmacol Toxicol 43:413-439, 2003; Thomas CE, Ehrhardt A, Kay MA: Nat Rev Genet 4:346-358, 2003.
DNA, RNA, AND PROTEINS: HEREDITY AT THE MOLECULAR LEVEL
Composition and Structure
Genes are composed of DNA, which has three basic components: the pentose sugar molecule, deoxyribose; a phosphate molecule; and four types of nitrogenous bases (Figure 4-1). Two of the bases, cytosine and thymine, are single carbon-nitrogen rings called pyrimidines. The other two bases, adenine and guanine, are double carbon-nitrogen rings called purines. The four bases are commonly represented by their first letters: A, C, T, and G.
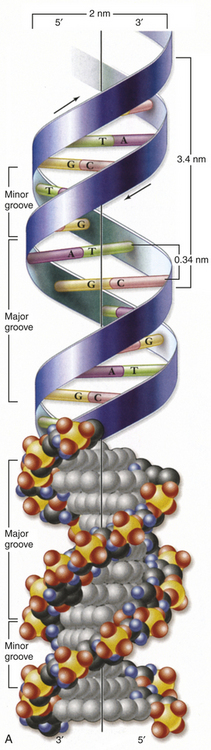
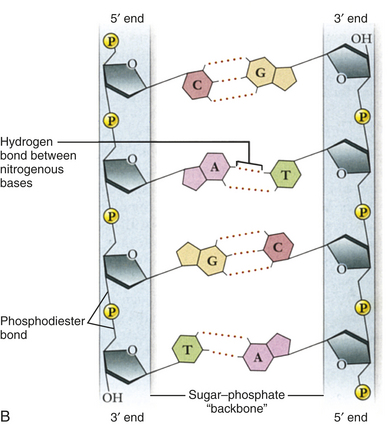
Figure 4-1 Structure of DNA. A, Double helix. Shown with the phosphodiester backbone as a ribbon on top and a space-filling model on the bottom. The bases protrude into the interior of the helix where they hold it together by base pairing. The backbone forms two grooves, the larger major groove and the smaller minor groove. B, Base pairing holds strands together. The H-bonds that form between A and T and between G and C are shown with dashed lines. These produce AT and GC base pairs that hold the two strands together. This always pairs a purine with a pyrimidine, keeping the diameter of the double helix constant. (From Raven PH et al: Biology, ed 8, New York, 2008, McGraw-Hill.)
One of Watson and Crick’s contributions was to demonstrate how these molecules are physically assembled together as DNA. They proposed the now-famous double-helix model, in which DNA can be envisioned as a twisted ladder with chemical bonds as its rungs (see Figure 4-1). The two sides of the ladder are composed of the sugar and phosphate molecules, held together by strong phosphodiester bonds. Projecting from each side of the ladder, at regular intervals, are the nitrogenous bases. The base projecting from one side is bound to the base projecting from the other by a weak hydrogen bond. Therefore, the nitrogenous bases form the rungs of the ladder; adenine pairs with thymine, and guanine pairs with cytosine. Each DNA subunit—consisting of one deoxyribose molecule, one phosphate group, and one base—is called a nucleotide.
DNA as the Genetic Code
To serve as the basis of genetic inheritance DNA must be able to direct the synthesis of all the body’s proteins. Proteins are composed of one or more polypeptides (intermediate protein compounds), which are in turn composed of sequences of amino acids (organic acids containing NH2). The body contains 20 different types of amino acids, and the amino acid sequences that make up polypeptides must in some way be specified by the DNA molecule.
Because there are 20 possible amino acids and only four possible bases, each single nucleotide cannot specify an amino acid. Similarly, the amino acids cannot be specified by couplets of bases (e.g., adenine-guanine, thymine-guanine, guanine-cytosine) because there are only 4 × 4, or 16, possible couplets. If series of three bases are translated into amino acids, however, there are 4 × 4 × 4, or 64, possible combinations—more than enough to specify each different amino acid. By manufacturing synthetic nucleotide sequences and allowing them to direct the formation of amino acids in the laboratory, it was proved that amino acids were specified by these triplets of bases, or codons.
Of the 64 possible codons, three signal the end of a gene and are known as termination, or nonsense, codons. The remaining 61 all specify amino acids, which means that most amino acids can be specified by more than one codon. The genetic code is thus said to be redundant, although each codon can specify only one amino acid.
Another significant feature of the genetic code is that it is universal: all living organisms use precisely the same DNA codes to specify proteins. The one known exception to this rule occurs in mitochondria—cytoplasmic organelles that are the sites of cellular respiration (see Chapter 1). The mitochondria have their own extranuclear DNA. Several codons of mitochondrial DNA encode different amino acids than do the same nuclear DNA codons.
Replication
In addition to having the ability to specify amino acid sequences, DNA must be able to replicate itself accurately during cell division if it is to serve as the basic genetic material. DNA replication consists of the breaking of the weak hydrogen bonds between the bases, leaving a single strand with each base unpaired. The consistent pairing of adenine with thymine and of guanine with cytosine, known as complementary base pairing, is the key to accurate replication. The principle of complementary base pairing dictates that the unpaired base will attract a free nucleotide only if the nucleotide has the proper complementary base. Thus a portion of a single strand with a sequence of bases labeled ATTGCT will bond with a series of free nucleotides with the bases TAACGA. When replication is complete, a new double-stranded molecule identical to the original is formed (Figure 4-2, A). The single strand is said to be a template, or molecule on which a complementary molecule is built, and is the basis for synthesizing the new double strand.
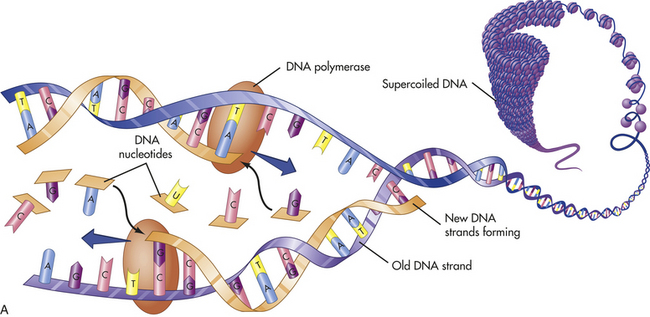
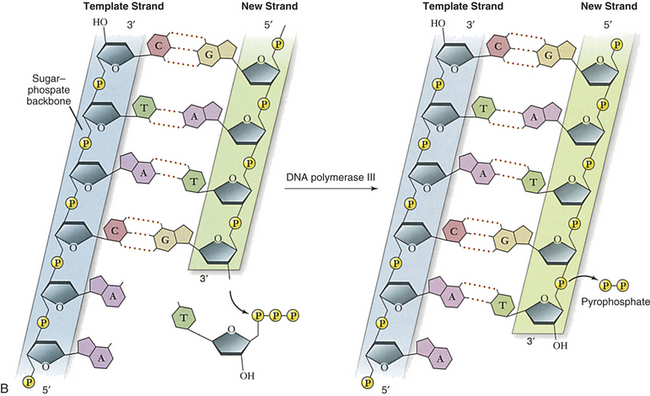
Figure 4-2 Replication and action of DNA. A, Replication of DNA. B, Action of DNA polymerase. DNA polymerases add nucleotides to the 3¢ end of a growing chain. The nucleotide added depends on the base that is in the template strand. Each new base must be complementary to the base in the template strand. With the addition of each new nucleotide, triphosphate, two of its phosphates are cleaved off as pyrophosphate. A, Adenine; T, thymine; G, guanine; C, cytosine. (A from Thibodeau GA, Patton KT: Anatomy & physiology, ed 6, St Louis, 2007, Mosby; B adapted from Raven PH et al: Biology, ed 8, New York, 2008, McGraw-Hill.)
Several different proteins are involved in DNA replication. One protein unwinds the double helix, one holds the strands apart, and others perform different distinct functions. The most important of these proteins is an enzyme known as DNA polymerase. This enzyme travels along the single DNA strand, adding the correct nucleotides to the free end of the new strand (see Figure 4-2, B). Besides adding the new nucleotides, the DNA polymerase performs a proofreading procedure. After the new nucleotide has been added to the chain, the DNA polymerase checks to make sure that its base is actually complementary to the template base. If it is not, the incorrect nucleotide is excised and replaced with a correct one. This procedure, one of the mechanisms of DNA repair, substantially enhances the accuracy of DNA replication.
Mutation
A mutation is any inherited alteration of genetic material. Chromosome aberrations that cause congenital defects are examples of mutations. Other mutations are subtle and are not observable as chromosome aberrations. One such mutation is the base pair substitution, in which one base pair is replaced by another. This mutation is sometimes called missense mutation as the “sense” of the codon produced after transcription of the mutant gene is altered (Figure 4-3). This substitution sometimes results in a change in amino acid sequence, but because of the redundancy of the genetic code, it may have no consequence. If an amino acid change does not occur, the mutation is termed a silent substitution. Profound consequences can result, however, when an amino acid sequence is altered by a base pair substitution. (Many of the serious genetic diseases discussed later are the result of base pair substitutions.)
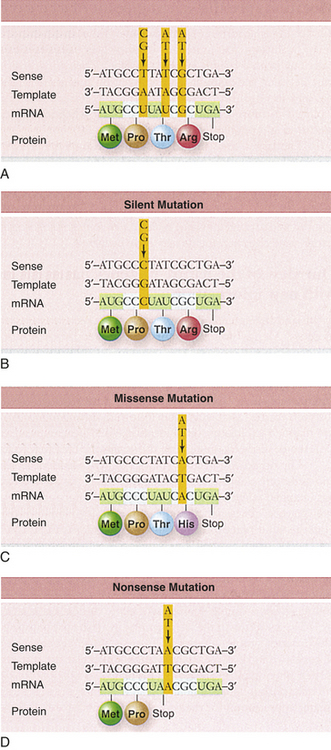
A second major type of mutation is the frameshift mutation. This alteration involves the insertion or deletion of one or more base pairs to the DNA molecule. As Figure 4-4 shows, these mutations can change the entire “reading frame” of the DNA sequence because codons consist of groups of three base pairs. A frameshift mutation thus can greatly alter the resulting amino acid sequence.
Figure 4-4 Different kinds of mutations. (From Patton KT, Thibodeau GA: Anatomy & Physiology, ed 7, 2010, St Louis Mosby.)
A large number of agents are known to increase the frequency of mutations. These agents are known collectively as mutagens. Radiation, such as that produced by x-rays and nuclear fallout, is an important mutagen and is known to cause cell damage (see Chapter 11) Radiation forms electrically charged ions that can produce chemical reactions, which in turn change DNA bases. A variety of chemicals also can induce mutations, often because they are chemically similar to DNA bases. Other chemicals mimic the effects of ionizing radiation, and still others interfere with the process of base pairing. Hundreds of chemicals are now known to be mutagenic in humans or laboratory animals, such as nitrogen mustard, vinyl chloride, alkylating agents, formaldehyde, and sodium nitrite. Some of these chemicals, however, are much more potent mutagens than others. Nitrogen mustard, for example, is extremely mutagenic, whereas sodium nitrate is a weak mutagen.
Measurement of the mutation rate in humans is difficult, in part because mutations are very rare events. Current estimates are that the rate of spontaneous mutation (a mutation that occurs in the absence of exposure to known mutagens) in humans is about 10−4 to 10−7 per gene per generation. This rate appears to vary from one gene to another. Certain areas of some chromosomes have particularly high mutation rates and are known as mutational hot spots. In particular, sequences consisting of a cytosine base followed by a guanine base (CG) are highly susceptible to mutation and are known to account for a disproportionately large percentage of disease-causing mutations.3
From Genes to Proteins
Whereas DNA is formed and replicated in the cell nucleus, protein synthesis takes place in the cytoplasm. The transport of the DNA code from nucleus to cytoplasm and subsequent protein formation involves two basic processes: transcription and translation. Both of these processes are mediated by ribonucleic acid (RNA), a type of nucleic acid that is chemically very similar to DNA (Figure 4-5). RNA is also composed of sugar molecules, phosphate groups, and nitrogenous bases. RNA differs from DNA in that the sugar molecule is ribose rather than deoxyribose and that uracil rather than thymine is one of the four bases. The other bases of RNA, as in DNA, are adenine, cytosine, and guanine. Uracil is structurally very similar to thymine, so it also can pair with adenine. The final difference between RNA and DNA is that whereas DNA usually occurs as a double strand, RNA usually occurs as a single strand.
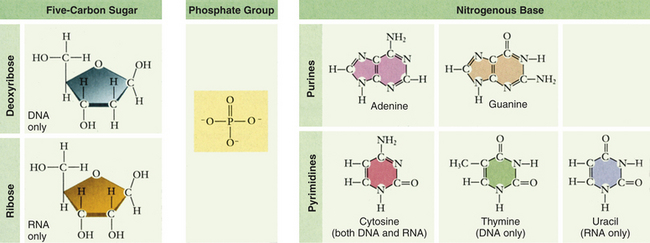
Figure 4-5 Nucleotide subunits of DNA and RNA. (From Raven PH et al: Biology, ed 8, New York, 2008, McGraw-Hill.)
Transcription
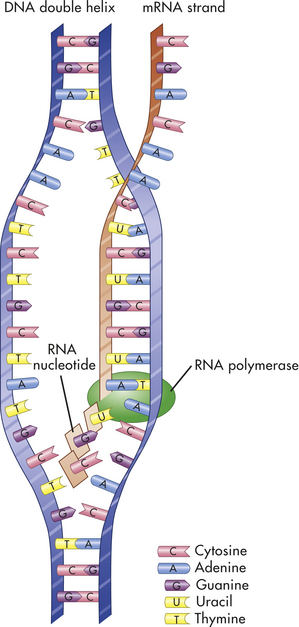
Transcription is the process by which RNA is synthesized from a DNA template. The result is the formation of messenger RNA (mRNA) from the base sequence specified by the DNA molecule. An enzyme called DNA-dependent RNA polymerase, or RNA polymerase, binds to a promoter site on the DNA. A promoter site is a sequence of DNA that specifies the beginning of a gene. The RNA polymerase then pulls a portion of the DNA strands apart from one another, allowing unattached DNA bases to be exposed. One of the DNA strands then provides the template for the sequence of mRNA nucleotides.
The sequence of bases in the mRNA is thus complementary to that of the template strand, and with the exception of the presence of uracil instead of thymine, the mRNA sequence is identical to that of the other DNA strand. Transcription continues until a DNA sequence called a termination sequence is reached. Then the RNA polymerase detaches from the DNA, and the transcribed mRNA is freed to move out of the nucleus and into the cytoplasm. Figure 4-6 summarizes the process of transcription.
Gene Splicing
After the mRNA first has been transcribed from the DNA template, it reflects exactly the base sequence of the DNA. The RNA in this state is sometimes called heterogeneous nuclear RNA (hnRNA). In eukaryotes an important step takes place before this RNA leaves the nucleus. Many of the RNA sequences are removed by nuclear enzymes, and the remaining sequences are spliced together to form the functional mRNA that will migrate to the cytoplasm.
The excised sequences are called introns, and the sequences that are left to code for proteins are called exons. The function, if any, of introns is not yet understood.
Translation
Translation is the process by which RNA directs the synthesis of a polypeptide (Figure 4-7). However, mRNA cannot code directly for amino acids. Instead, it interacts with transfer RNA (tRNA), a cloverleaf-shaped strand of about 80 nucleotides. The tRNA molecule has a site for the attachment of an amino acid. At the opposite side of the cloverleaf is a sequence of three nucleotides called the anticodon. The anticodon undergoes complementary base pairing with an appropriate codon in the mRNA. The mRNA thus specifies the sequence of amino acids by acting through the tRNA.
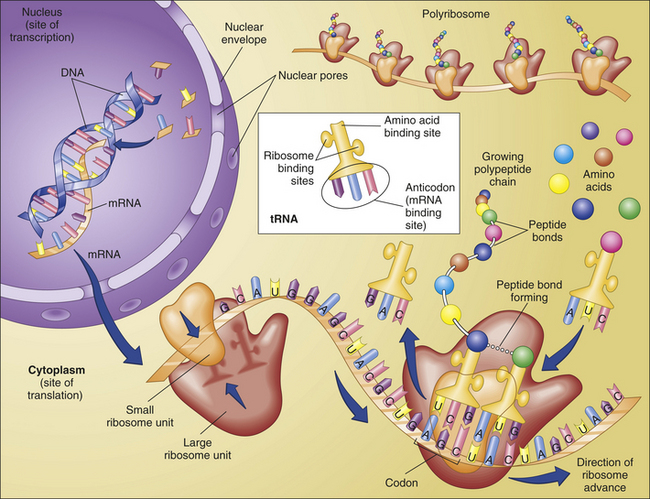
Figure 4-7 Protein synthesis. Protein synthesis begins with transcription, a process in which an mRNA molecule forms along one gene sequence of a DNA molecule within the cell’s nucleus. As it is formed, the mRNA molecule separates from the DNA molecule and leaves the nucleus through the large nuclear pores. Outside the nucleus, ribosome subunits attach to the beginning of the mRNA molecule and begin the process of translation. In translation, transfer RNA (tRNA) molecules bring specific amino acids—encoded by each mRNA codon—into place at the ribosome site. As the amino acids are brought into the proper sequence, they are joined together by peptide bonds to form long strands called polypeptides. Several polypeptide chains may be needed to make a complete protein molecule. A, Adenine; C, cytosine; G, guanine; U, uracil. (From Thibodeau GA, Patton KT: Anatomy & Physiology, ed 6, St Louis, 2007, Mosby.)
The site of actual protein synthesis is the ribosome, which consists of roughly equal parts of protein and ribosomal RNA (rRNA). During translation (Figure 4-8) the ribosome first binds to an initiation site on the mRNA sequence. The ribosome then binds the tRNA to its surface so that base pairing can occur between tRNA and mRNA. The ribosome then moves along the mRNA sequence, codon by codon. As each codon is processed, an amino acid is translated by the interaction of mRNA and tRNA.
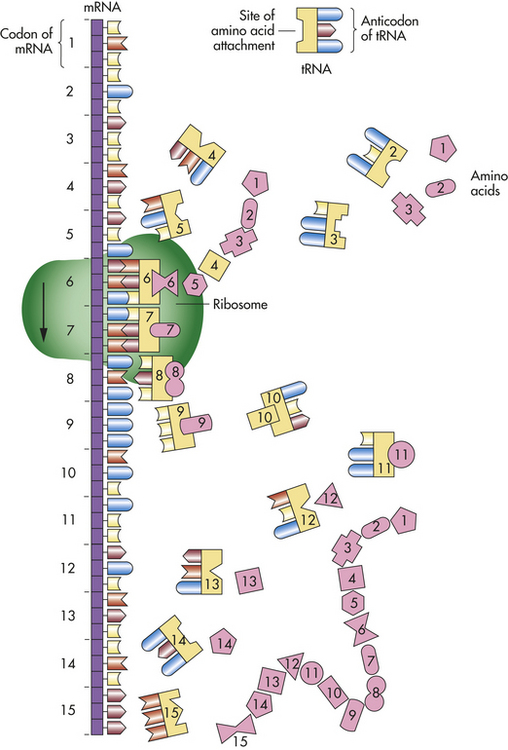
In this process the ribosome provides an enzyme that catalyzes the formation of covalent peptide bonds between the adjacent amino acids, resulting in a growing polypeptide. When the ribosome arrives at a termination signal on the mRNA sequence, translation and polypeptide formation cease. The mRNA, ribosome, and polypeptide separate from one another, and the polypeptide is released into the cytoplasm to perform its required function.
CHROMOSOMES
Human cells can be categorized into two types: gametes (sperm and egg cells) and somatic cells, which include all cells other than gametes. Each somatic cell has 46 chromosomes in its nucleus. These are diploid cells, meaning that the chromosomes occur in pairs. Thus each cell actually contains 23 pairs of chromosomes. One member of each pair comes from an individual’s mother, and one comes from the father. New somatic cells are formed through mitosis and cytokinesis, through which the cell nucleus and cytoplasm are replicated. (The division process that creates new copies of somatic cells is described in Chapter 1.) Gametes are haploid cells: they have only one member of each chromosome pair, giving them a total of 23 chromosomes. The process by which these haploid cells are formed from diploid cells is called meiosis (Figure 4-9).
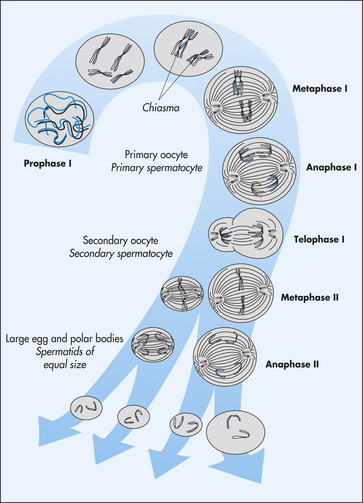
Figure 4-9 Stages of meiosis. From these stages, haploid gametes are formed from a diploid stem cell. For brevity, prophase II and telophase II are not shown. Note the relationship between meiosis and spermatogenesis and oogenesis. (From Jorde LB et al: Medical genetics, ed 3, St Louis, 2003, Mosby.)
In 22 of the 23 chromosome pairs, the two members of each pair are virtually identical in microscopic appearance and are thus said to be homologous to one another. These 22 chromosome pairs are homologous in both males and females and are termed autosomes. The remaining pair of chromosomes, the sex chromosomes, consists of two homologous X chromosomes in females and a nonhomologous pair, X and Y, in males.
Figure 4-10, A, illustrates a metaphase spread, which is a photograph of the chromosomes as they appear in the nucleus of a somatic cell during metaphase. (Chromosomes are easiest to visualize during this stage of mitosis.) A karyotype is an ordered display of chromosomes. In Figure 4-10, B, the chromosomes are arranged according to size, with the homologous chromosomes paired together. The 22 autosomes are numbered according to length, with chromosome 1 as the longest and chromosome 22 as the shortest. Some natural variation in relative chromosome length can be expected from person to person, however, so it is not always possible to distinguish each chromosome by its length. Therefore, the position of the centromere is also used to classify the chromosomes (Figure 4-11).
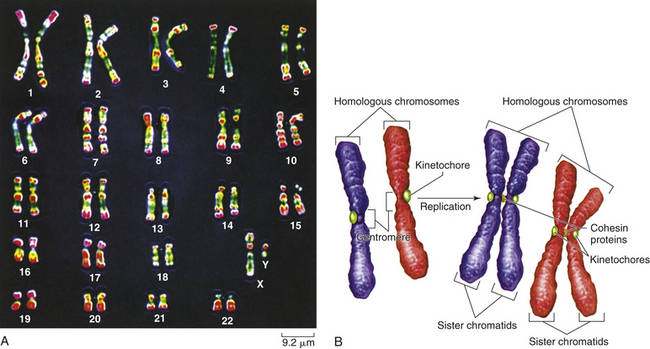
Figure 4-10 Karyotype of chromosomes. A, Human karyotype. B, Homologous chromosomes and sister chromatids. (From Raven PH et al: Biology, ed 8, New York, 2008, McGraw-Hill.)
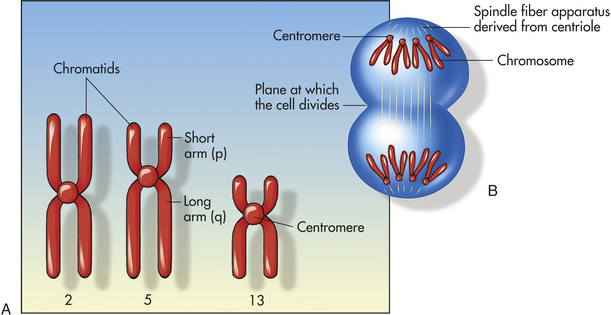
Figure 4-11 Structure of chromosomes. A, Human chromosomes 2, 5, and 13. Each is replicated and consists of two chromatids. Chromosome 1 is a metacentric chromosome because the centromere is close to middle; chromosome 5 is submetacentric because the centromere is set off from middle; chromosome 13 is acrocentric because the centromere is at or very near the end. B, During mitosis, the centromere divides and chromosomes move to opposite poles of the cell. At the time of centromere division, the chromatids are designated chromosomes.
The chromosomes in Figure 4-10, A, were stained with a substance that penetrates all areas of the chromosome (a “solid stain”). In the late 1960s and early 1970s, several staining materials were found to bind preferentially to certain areas of chromosomes. The resulting distinctive chromosome bands are evident in various patterns in the different chromosomes so that each chromosome can be distinguished easily. One of the most commonly used stains is Giemsa stain. By using banding techniques, chromosomes can be unambiguously numbered, and individual variation in chromosome composition can be studied. Missing or duplicated portions of chromosomes, which often result in serious diseases, also can be readily identified.
Chromosome Aberrations and Associated Diseases
Chromosome abnormalities are the leading known cause of mental retardation and miscarriage. Estimates indicate that a major chromosome aberration occurs in at least 1 in 12 conceptions. Most of these fetuses do not survive to term; in fact, about 50% of all recovered first-trimester spontaneous abortuses have major chromosomal aberrations.4 The number of live births affected by these abnormalities is significant; about 1 in 150 has a major diagnosable chromosome abnormality5 (Box 4-2).
Box 4-2 Prenatal Diagnosis of Chromosome Abnormalities
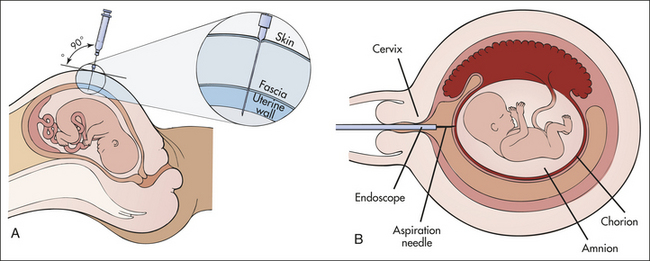
All the chromosome abnormalities discussed here can be detected prenatally, using a procedure called amniocentesis (A). At about the sixteenth week of gestation, a sufficient amount of amniotic fluid is available to enable the withdrawal of a small amount of fluid (2 to 20 ml). This fluid contains live skin cells (fibroblasts) shed by the fetus. These cells can be cultured and karyotyped, and chromosome abnormalities can be detected.
Other disorders can be detected with this procedure. These include most neural tube defects, which cause an elevation of α-fetoprotein in the amniotic fluid, and hundreds of diseases caused by mutations of single genes. The procedure involves a risk of losing the fetus, estimated to be about 0.5% or less. Thus amniocentesis is recommended only for pregnancies known to have an elevated risk for a genetic disease. These include pregnancies of women older than 35 years, in which the risk for Down syndrome and other aneuploidies is elevated, and pregnancies in which parents are known to carry translocations or certain disease genes.
One problem with prenatal diagnosis by amniocentesis is that by the time the sixteenth week of gestation is reached and another 2 or 3 weeks to culture the fibroblasts and test for genetic disease elapse, the mother is near the twentieth week of pregnancy. Pregnancy termination of an affected fetus at this stage can present serious emotional and personal dilemmas as well as some medical risk. For many parents, abortion would be more acceptable for a fetus at an earlier gestational age. A newer technique, chorionic villus sampling (B), consists of extracting a small amount of villous tissue directly from the chorion. This procedure can be performed at 10 weeks’ gestation and does not require in vitro culturing of cells because sufficient numbers are directly available in the extracted tissue. Thus the procedure allows prenatal diagnosis at about 3 months’ gestation rather than at nearly 5 months’ gestation. Chorionic villus sampling involves a slightly higher fetal loss rate than amniocentesis, approximately 1%.
Data from Wang BT et al: Am J Med Genet 53:307, 1994; illustrations from Pagana KD, Pagana TJ: Mosby’s manual of diagnostic and laboratory tests, ed 2, St Louis, 2002, Mosby.
Polyploidy
Cells that have a multiple of the normal number of chromosomes are said to be euploid cells (Greek eu = good or true). Because normal gametes are haploid and most normal somatic cells are diploid, they are both euploid forms. When a euploid cell has more than the diploid number of chromosomes, it is said to be a polyploid cell. Several types of body tissues, including some liver, bronchial, and epithelial tissues, are normally polyploid. A zygote having three copies of each chromosome, rather than the usual two, has a form of polyploidy called triploidy. Tetraploidy, a condition in which euploid cells have 92 chromosomes, also has been observed. Both of these conditions are incompatible with postnatal survival. Nearly all triploid fetuses are spontaneously aborted or stillborn. A few have survived to term but have died shortly after birth. Tetraploidy has been found primarily in early abortuses, although occasionally affected infants have been born alive. Like triploid infants, however, they do not survive. Triploidy and tetraploidy are relatively common conditions, accounting for approximately 10% of all known miscarriages.4
Aneuploidy
A somatic cell that does not contain a multiple of 23 chromosomes is an aneuploid cell. A cell containing three copies of one chromosome is said to be trisomic (a condition termed trisomy) and is aneuploid. Monosomy, the presence of only one copy of a given chromosome in a diploid cell, is the other common form of aneuploidy. Among the autosomes, monosomy of any chromosome is lethal, but newborns with trisomy of some chromosomes can survive. This difference illustrates an important principle: in general, loss of chromosome material has more serious consequences than duplication of chromosome material.
Aneuploidy of the sex chromosomes is less serious than that of the autosomes. For the Y chromosome, this is true because very little genetic material is located on this chromosome. For the X chromosome, inactivation of extra chromosomes largely diminishes their effect. A zygote bearing no X chromosome, however, will not survive.
Aneuploidy is usually the result of nondisjunction, an error in which homologous chromosomes or sister chromatids fail to separate normally during meiosis or mitosis (Figure 4-12). Nondisjunction during either stage of meiosis produces some gametes that have two copies of a given chromosome and others that have no copies of the chromosome. When such gametes unite with normal haploid gametes, the resulting zygote is monosomic or trisomic for that chromosome. Occasionally a cell can be monosomic or trisomic for more than one chromosome.
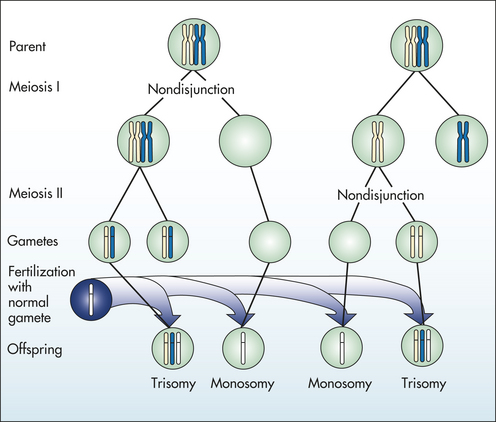
Figure 4-12 Nondisjunction causes aneuploidy when chromosomes or sister chromatids fail to divide properly. (From Jorde LB et al: Medical genetics, ed 3, St Louis, 2003, Mosby.)
Autosomal Aneuploidy: Trisomy can occur for any chromosome, but the only forms seen with an appreciable frequency in live births are trisomies of the thirteenth, eighteenth, or twenty-first chromosome. Fetuses with most other chromosomal trisomies do not survive to term. Trisomy 16, for example, is the most commonly known trisomy among abortuses, but it is not seen in live births.4
Partial trisomy, in which only an extra portion of a chromosome is present in each cell, also can occur. The consequences of partial trisomies are not as severe as those of complete trisomies. Trisomies also may occur in only some cells of the body. Individuals thus affected are said to be chromosomal mosaics, meaning that the body has two or more different cell lines, each of which has a different karyotype. Mosaics are usually formed by early mitotic nondisjunction occurring in one embryo cell but not in others.
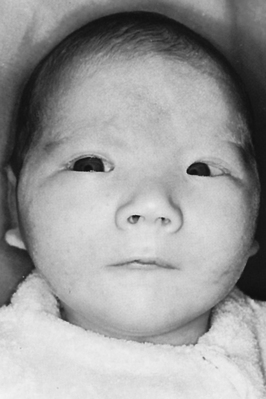
The best-known example of aneuploidy in an autosome is trisomy of the twenty-first chromosome, which causes Down syndrome (named after J. Langdon Down, who first described the disease in 1866). Down syndrome was formerly called mongolism, but this inappropriate term is no longer used. Down syndrome is seen in 1 in 800 live births.4 Individuals with this disease are mentally retarded, with IQs usually ranging from 25 to 70. The facial appearance is distinctive (Figure 4-13), with a low nasal bridge, epicanthal folds (which produce a superficially Asian appearance), protruding tongue, and flat, low-set ears. Poor muscle tone (hypotonia) and short stature are both characteristic. Congenital heart defects affect about one third to one half of live-born children with Down syndrome; a reduced ability to fight respiratory infections and an increased susceptibility to leukemia also contribute to reduced survival rate. By 40 years of age, individuals with Down syndrome virtually always develop symptoms that are nearly identical to those of Alzheimer disease. About three fourths of fetuses known to have Down syndrome are spontaneously aborted or stillborn. About 20% of infants born with Down syndrome die during their first 10 years of life. For those who survive beyond 10 years, average life expectancy is now about 60 years.
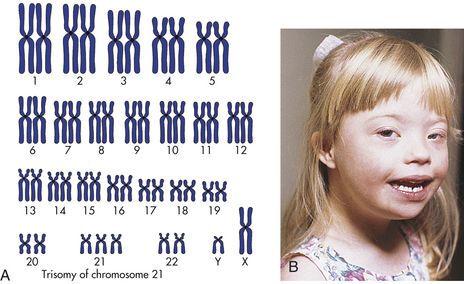
Figure 4-13 Down syndrome. A, The karyotype of Down syndrome consists of 47 chromosomes and shows trisomy 21. B, A child with Down syndrome. (A from Damjanov I: Pathology for the health-related professions, ed 3, Philadelphia, 2006, Saunders; B courtesy Olney A and MacDonald M, University of Nebraska Medical Center, Omaha.)
About 97% of Down syndrome cases are caused by nondisjunction during the formation of one of the parent’s gametes or during early embryonic development. The remaining 3% result from translocations (discussed later). In approximately 90% to 95% of cases, the nondisjunction occurs in the formation of the mother’s egg cell. Paternal nondisjunction is responsible for the remaining cases. Among individuals with Down syndrome, about 1% are known to be mosaics. Because mosaics have a large number of normal cells, the effects of the trisomic cells are attenuated and symptoms are often less severe.
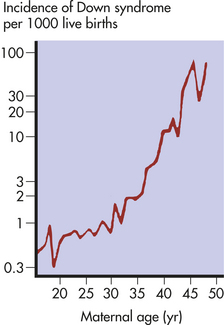
The risk of having a child with Down syndrome increases greatly with maternal age. As Figure 4-14 demonstrates, women younger than 30 years have a risk ranging from about 1 in 1000 births to 1 in 2000 births. The risk begins to rise substantially after 35 years of age, and it reaches 3% to 5% for women older than 45 years of age. This dramatic increase in risk may be caused by the age of maternal egg cells, which are held in an arrested state of prophase I from the time they are formed in the female embryo until they are shed in ovulation. Thus an egg cell formed by a 45-year-old woman is itself 45 years old. This long suspended state may allow for the accumulation of errors leading to nondisjunction. The risk of Down syndrome, as well as other trisomies, does not appear to increase with paternal age.6
Sex Chromosome Aneuploidy: Among live births, about 1 in 400 males and 1 in 650 females have a form of sex chromosome aneuploidy.7 Because these conditions are generally less severe than autosomal aneuploidies, all forms except complete absence of an X chromosome allow at least some individuals to survive.
One of the most common sex chromosome aneuploidies, affecting about 1 in 1000 newborn females, is trisomy X. Instead of two X chromosomes, these females have three X chromosomes in each cell. Most of them have no overt physical abnormalities, although sterility, menstrual irregularity, or mental retardation is sometimes seen. Some females have four X chromosomes, and they are more often mentally retarded. Those with five or more X chromosomes generally have more severe mental retardation and various physical defects.
A condition that leads to somewhat more serious problems is the presence of a single X chromosome and no homologous X or Y chromosome, so the individual has a total of 45 chromosomes. The karyotype is designated 45,X, and it causes a set of symptoms known as Turner syndrome (Figure 4-15). Because they have no Y chromosomes, people with Turner syndrome are females. They are usually sterile, however, and have gonadal streaks rather than ovaries. These streaks of connective tissue are susceptible to cancer in mosaics who have some cells containing a Y chromosome. Other features of the disorder include short stature, webbing of the neck in about half of cases, widely spaced nipples, coarctation (narrowing) of the aorta (in 15% to 20% of cases), edema of the feet in newborns, reduced carrying angle at the elbow (cubitus valgus), and sparse body hair. They are not considered retarded, although evidence indicates some impairment of spatial and mathematical reasoning ability. About three fourths of recognized 45,X conceptions inherit their X chromosome from the mother. Thus most cases are caused by a loss of the paternal X chromosome.
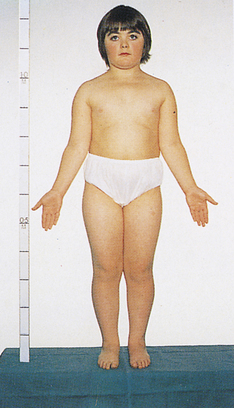
Figure 4-15 Turner syndrome. A sex chromosome is missing, and the person’s chromosomes are 45,X. Characteristic signs are short stature, female genitalia, webbed neck, shieldlike chest with underdeveloped breasts and widely spaced nipples, and imperfectly developed ovaries. (From Patton KT, Thibodeau GA: Anatomy & physiology, ed 7, St Louis, 2010, Mosby.)
The frequency of Turner syndrome is low compared with that of other sex chromosome aneuploidies: only about 1 in 3000 newborn females is affected.8 About half of individuals with Turner syndrome have simple monosomy of the X chromosome; others have one of several more complex X chromosome abnormalities. The 45,X karyotype is more common among conceptions, however, and about 15% to 20% of spontaneous abortions with chromosome abnormalities have this karyotype, making it one of the most common single-chromosome aberrations. Thus the condition is highly lethal during gestation: less than 1% of 45,X conceptions survive to term. Most fetuses that survive to term are mosaics, with combinations of 45,X cells and XX, XXX, or XY cells. It is likely that the presence of some normal cells in mosaic fetuses enhances fetal survival.
Teenagers with Turner syndrome are typically treated with estrogen to promote the development of secondary sexual characteristics. The dose is then continued at a reduced level to maintain these characteristics and to help avoid osteoporosis. Human growth hormone is sometimes administered to increase stature.
Individuals with at least two X chromosomes and a Y chromosome in each cell (47,XXY karyotype) have a disorder known as Klinefelter syndrome (Figure 4-16). Because of the presence of a Y chromosome, these individuals have a male appearance, but they are usually sterile, and about half develop female-like breasts (a condition called gynecomastia). The testes are small, body hair is sparse, the voice is often somewhat high pitched, stature is elevated, and a moderate degree of mental impairment may be present. Klinefelter syndrome is found in about 1 in 1000 male births. About two thirds of the cases are caused by nondisjunction of the X chromosomes in the mother, and the frequency of the disorder rises with maternal age. Individuals with the XXXY and XXXXY karyotypes also are considered to have Klinefelter syndrome, and the degree of physical and mental impairment increases with each additional X chromosome. Regardless of the number of X chromosomes, however, these individuals have a male appearance. The presence of a single Y chromosome, which causes the undifferentiated gonads to become testes, always produces a male. Mosaicism is sometimes seen in Klinefelter syndrome and results in less severe disease; the most prevalent combination is XXY and XY cells.
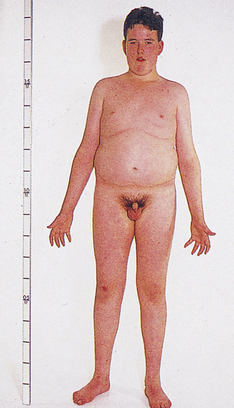
Figure 4-16 Klinefelter syndrome. This young man exhibits many characteristics of Klinefelter syndrome: small testes, some development of the breasts, sparse body hair, and long limbs. This syndrome results from the presence of two or more X chromosomes with one Y chromosome (genotypes XXY or XXXY, for example). (From Patton KT, Thibodeau GA: Anatomy & physiology, ed 7, St Louis, 2010, Mosby.)
The other sex chromosome aneuploidy that affects males is the 47,XYY karyotype. Individuals with this karyotype tend to be taller than average, and they have a 10- to 15-point reduction in average IQ. This condition, which causes few serious physical problems, achieved notoriety when it was found that its incidence in prison populations was about 1 in 30 (compared with 1 in 1000 in the general male population). This discovery led to the suggestion that this chromosome might predispose affected individuals to violent, criminal behavior. Several dozen studies have addressed this issue, and they have shown that 47,XYY males are not inclined to commit violent crimes. However, even after adjusting for the effects of decreased IQ, some evidence exists for an increased incidence of behavioral disorders.
Abnormalities of Chromosome Structure
In addition to the loss or gain of whole chromosomes, parts of chromosomes can be lost or duplicated as gametes are formed, and the arrangement of genes on chromosomes can be altered. Unlike aneuploidy and polyploidy, these changes sometimes do not have serious consequences for an individual’s health. Some of them can even go entirely unnoticed, especially when very small pieces of chromosomes are involved. Nevertheless, abnormalities of chromosome structure also can produce serious disease in individuals or their offspring.
During meiosis and mitosis, chromosomes usually maintain their structural integrity very well, but chromosome breakage occasionally does occur. Mechanisms exist to “heal” these breaks, and generally the break is repaired perfectly with no damage resulting to the daughter cell. Sometimes, however, the breaks remain, or they heal in a fashion that alters the structure of the chromosome. The extent of chromosome breakage is increased in the presence of certain harmful agents, called clastogens. Identified clastogens include ionizing radiation, some viral infections, and certain chemicals.
Deletions: Broken chromosomes and loss of DNA cause deletions (Figure 4-17). Usually a gamete with a deletion unites with a normal gamete to form a zygote. The zygote thus has one chromosome with the normal complement of genes and one with some missing genes. Because a fairly large number of genes can be lost in a deletion, serious consequences can result even though one copy of the chromosome is normal. An often cited example of a disease caused by a chromosomal deletion is the cri du chat syndrome (Figure 4-18). The term, which literally means “cry of the cat,” describes the characteristic cry of the affected child. Other symptoms include low birth weight, severe mental retardation, microcephaly (smaller than normal head size), heart defects, and the typical facial appearance shown in Figure 4-18. The disease is caused by a deletion of part of the short arm of chromosome 5.
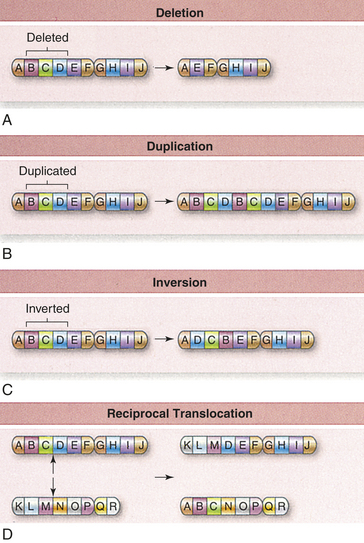
Figure 4-17 Chromosomal mutations. Larger-scale changes in chromosomes are also possible. Material can be deleted (A), duplicated (B), and inverted (C). Translocations occur when one chromosome is broken and becomes part of another chromosome. This often occurs where both chromosomes are broken and exchange material, an event called a reciprocal translocation (D). (From Raven PH et al: Biology, ed 8, New York, 2008, McGraw-Hill.)
Duplications: Duplications of chromosome material are, like deletions, a form of chromosome aberration (see Figure 4-17). Because a deficiency of genetic material is more harmful than an excess, duplications usually have less serious consequences than deletions. For example, a deletion of a region of chromosome 5 causes cri du chat syndrome, but a duplication of the same region causes less severe disease.
Inversions: An inversion is the occurrence of two breaks on a chromosome, followed by the reinsertion of the missing fragment at its original site but in inverted order (see Figure 4-17). Thus a chromosome symbolized as ABCDEFG might become ABEDCFG after an inversion.
Unlike deletions and duplications, inversions result in no loss or gain of genetic material. They are thus said to be a “balanced” alteration of chromosome structure, and they often have no apparent physical effect. Genes are sometimes influenced by neighboring DNA sequences, however, and this position effect, a change in a gene’s expression caused by its position, does sometimes result in physical defects in persons with inversions.
The serious problems caused by inversions usually occur in the offspring of individuals carrying the inversion. Because chromosomes must line up in perfect order during prophase I, a chromosome with an inversion must form a loop to line up with its normal homolog. Crossing over within this loop can result in duplications or deletions in the chromosomes of daughter cells. Thus the offspring of individuals who carry inversions often have chromosome deletions or duplications.
Translocations: The interchanging of genetic material between nonhomologous chromosomes is called translocation. The clinically most important type of translocation is termed a Robertsonian translocation. In this translocation the long arms of two nonhomologous chromosomes fuse at the centromere, forming a single chromosome (Figure 4-19). Robertsonian translocations are confined to chromosomes 13, 14, 15, 21, and 22 because the short arms of these chromosomes are very small and contain no essential genetic material. When a Robertsonian translocation takes place, the short arms are usually lost during subsequent cell divisions. Because the carriers of Robertsonian translocations lose no important genetic material, they are normal, although they have only 45 chromosomes in each cell. Their offspring, however, may have serious deletions or duplications (see Figure 4-19). For example, a common Robertsonian translocation involves the fusion of the long arms of chromosomes 21 and 14. An offspring who inherits a gamete carrying the fused chromosome receives an extra copy of the long arm of chromosome 21 and thus develops Down syndrome. Robertsonian translocations are responsible for approximately 3% to 5% of Down syndrome cases. Parents who carry a Robertsonian translocation involving chromosome 21 have an increased risk for producing multiple offspring with Down syndrome.
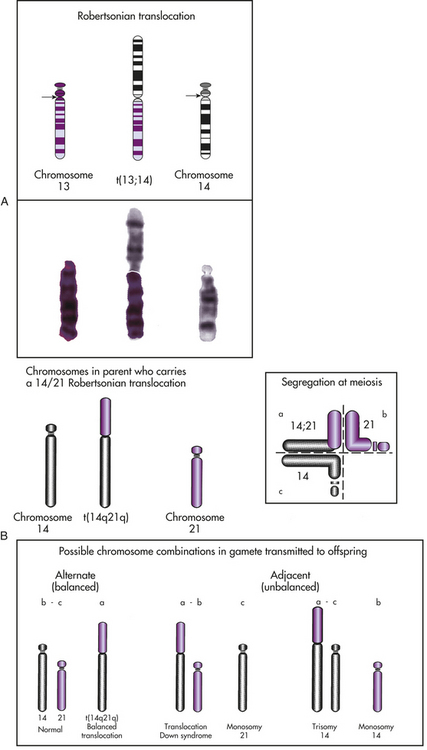
Figure 4-19 Translocation. A, In a Robertsonian translocation, shown here, the long arms of two acrocentric chromosomes (13 and 14) fuse, forming a single chromosome. B, The possible segregation patterns for gametes formed by a carrier of a Robertsonian translocation. Alternate segregation (quadrant a alone, or quadrant b with quadrant c) produces either a normal chromosome constitution or a translocation carrier with a normal phenotype. Adjacent segregation (quadrant a with c, quadrant c alone, quadrant a with b, or quadrant b alone) produces unbalanced gametes and results in conceptions with translocation Down syndrome, monosomy 21, trisomy 14, or monosomy 14, respectively. For example, monosomy 14 is produced when the parent who carries the translocation transmits a copy of chromosome 21 but does not transmit a copy of chromosome 14 (as in the lower right corner). (From Jorde LB et al: Medical genetics, ed 3, St. Louis, 2003, Mosby.)
A reciprocal translocation occurs when breaks take place in two different chromosomes and the material is exchanged (see Figure 4-17). As with Robertsonian translocations, the carrier of a reciprocal translocation is usually normal because the individual has a normal complement of genetic material. However, the carrier’s gametes can be normal, can carry the translocation, or can have duplications and deletions.
Fragile Sites: For reasons not yet fully understood, a number of areas on chromosomes develop distinctive breaks and gaps (observable microscopically) when the cells are cultured in a folate-deficient medium. Most of these fragile sites have no apparent relationship to disease. However, one fragile site, located on the long arm of the X chromosome, is associated with a disorder of considerable importance, both clinically and genetically. This disorder is known as the fragile X syndrome. The most important feature of this syndrome is mental retardation. With a relatively high population prevalence (affecting approximately 1 in 4000 males and 1 in 8000 females), the fragile X syndrome is the second most common genetic cause of mental retardation (after Down syndrome).
Fragile X syndrome involves a puzzling pattern of inheritance. In particular, males who inherit the mutation do not necessarily express the disease condition but they can pass it on to descendants who do express it. Ordinarily, a male who inherits a disease gene on the X chromosome expresses the condition because he has only one X chromosome. Another uncommon feature of this disease is that about one third of carrier females are affected, although less severely than males. Many mechanisms have been proposed to account for the complex mode of inheritance of the fragile X syndrome. It has been shown that unaffected transmitting males have an elevated number (more than about 50) of repeated DNA sequences in the first exon of the fragile X gene. These “repeats” consist of CGG sequences that are duplicated again and again. Affected males have a much larger number of these repeats—200 or more9 (Figure 4-20). An increase in the number of these repeated sequences in successive generations can lead to expression of the fragile X syndrome. More than 20 other genetic diseases also are caused by this mechanism.10,11
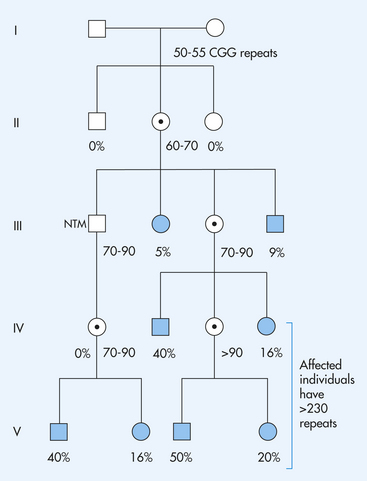
Figure 4-20 A pedigree showing the inheritance of the fragile X syndrome. Females who carry a premutation (50 to 320 CGG repeats) are represented with a  . Affected individuals are represented by solid symbols. A normal transmitting male (NTM), who carries a premutation of 70 to 90 repeats increases each time the mutation is passed through another female. Also, only 5% of the NTM’s sisters are affected, and only 9% of his brothers are affected, but 40% of his grandsons and 16% of his granddaughters are affected. This is the Sherman paradox. (From Jorde LB et al: Medical genetics, ed 3, St Louis, 2003, Mosby.)
. Affected individuals are represented by solid symbols. A normal transmitting male (NTM), who carries a premutation of 70 to 90 repeats increases each time the mutation is passed through another female. Also, only 5% of the NTM’s sisters are affected, and only 9% of his brothers are affected, but 40% of his grandsons and 16% of his granddaughters are affected. This is the Sherman paradox. (From Jorde LB et al: Medical genetics, ed 3, St Louis, 2003, Mosby.)
ELEMENTS OF FORMAL GENETICS
The mechanisms by which an individual’s set of paired chromosomes produces traits are the principles of genetic inheritance. Mendel’s work with garden peas first defined these principles. Later geneticists have refined Mendel’s work to explain patterns of inheritance for traits and diseases that appear in families.
Analysis of traits that occur with defined, predictable patterns has helped geneticists link the pieces of the human gene map. Research focuses on assigning genes to specific locations on chromosomes. Eventually, diseases and defects caused by single genes can be traced, and therapies to prevent and treat such diseases can be developed.
Many traits are caused by single genes and are often called mendelian traits (after Gregor Mendel). Each gene occupies a position along a chromosome known as a locus. The genes at a particular locus can take different forms (i.e., they can be composed of different nucleotide sequences). These different forms are called alleles. For example, most people have a type of hemoglobin known as hemoglobin A. A few individuals have an alternative form of hemoglobin, termed hemoglobin S, which differs from hemoglobin A by a single amino acid substitution in the beta-globin component of the molecule. The β-globin locus thus has two different alleles, one that encodes hemoglobin A and another that encodes hemoglobin S. A locus that has two or more alleles that occur with an appreciable frequency in a population is said to be polymorphic or a polymorphism.
Because humans are diploid organisms, each chromosome is represented twice, with one member of the chromosome pair contributed by the father and one by the mother. At a given locus an individual has one gene whose origin is paternal and one whose origin is maternal. When the two genes are identical, the individual is homozygous at that locus. When the genes are not identical, the individual is heterozygous at the locus.
Phenotype and Genotype
The composition of genes at a given locus is known as the genotype. The outward appearance of an individual, which is the result of both genotype and environment, is the phenotype. For example, an infant who is born with an inability to metabolize the amino acid phenylalanine has the single-gene disorder known as phenylketonuria (PKU) and thus has the PKU genotype. If the condition is left untreated, abnormal metabolites of phenylalanine will begin to accumulate in the infant’s brain and irreversible mental retardation will occur. Mental retardation is thus one aspect of the PKU phenotype. By imposing dietary restrictions to limit the intake of food containing phenylalanine, however, retardation can be prevented. Although the child still has the PKU genotype, a modification of the environment (in this case the child’s diet) produces an outwardly normal phenotype.
Dominance and Recessiveness
In many loci the effects of one allele mask those of another when the two are found together in a heterozygote. The allele whose effects are observable is said to be dominant. The allele whose effects are hidden is said to be recessive (from the Latin root for “hiding”). Traditionally, for loci having two alleles, the dominant allele is denoted by an uppercase letter and the recessive allele is denoted by a lowercase letter. When one allele is dominant over another, the heterozygote genotype Aa has the same phenotype as the dominant homozygote AA. For the recessive allele to be expressed, it must exist in the homozygote form, aa.
When the heterozygote is distinguishable from both homozygotes, the locus is said to exhibit codominance. For example, in the MN blood group, both alleles, M and N, of the heterozygote are detectable and therefore codominant. Another example is the ABO blood group, in which heterozygotes having the A and B alleles express both of them as A and B antigens on their red cells (forming blood group AB).
A carrier is an individual who has a disease gene but is phenotypically normal. Most genes for recessive diseases occur in heterozygotes who carry one copy of the gene but do not express the disease. Because many recessive genes are lethal in the homozygous state, they are eliminated from the population when they occur in homozygotes. By “hiding” in carriers, however, most recessive genes for diseases survive to be passed on to the next generation.
TRANSMISSION OF GENETIC DISEASES
An important aspect of a genetic disease is the pattern in which it is inherited through the generations of a family, or its mode of inheritance. Once the mode of inheritance is known, much can be learned about the disease gene itself, and reliable genetic counseling can be given to members of families in which the disease is present.
Modes of inheritance were systematically studied by Mendel, who formulated two basic laws of inheritance. His principle of segregation states that homologous genes separate from one another during reproduction and that each reproductive cell carries only one of the homologous genes. Mendel’s second law, the principle of independent assortment, states that the hereditary transmission of one gene has no effect on the transmission of another. Mendel discovered these laws in the mid-nineteenth century by performing breeding experiments with garden peas. He had no knowledge of chromosomes. Early in the twentieth century geneticists found that the behavior of chromosomes does essentially correspond to Mendel’s laws, which now form the basis for the chromosome theory of inheritance.
The known single-gene diseases can be classified into four major modes of inheritance: autosomal dominant, autosomal recessive, X-linked dominant, and X-linked recessive. The first two types involve genes known to occur on the 22 pairs of autosomes. The last two types occur on the X chromosome; no good documentation exists of disease genes occurring on the Y chromosome. The number of diseases assigned to each category is growing rapidly. Current catalogs of single-gene traits, which include disease-producing and nonclinical traits (e.g., attached earlobes), list more than 17,000 known autosomal traits and 1027 X-linked traits.1
An important tool in the analysis of modes of inheritance is the pedigree chart. It summarizes family relationships and shows which members of a family are affected by a genetic disease (Figure 4-21). Generally, the pedigree begins with one individual in the family, the proband, also termed the propositus (male) or proposita (female). This individual is usually the first person in the family diagnosed or seen in a clinic.
Autosomal Dominant Inheritance
Diseases caused by autosomal dominant genes are rare. The most common occur in fewer than 1 in 500 individuals, so it is uncommon for two individuals both affected by the same autosomal dominant disease to produce offspring together. Figure 4-22, A, illustrates this unusual pattern. More often, affected offspring are produced by the union of a normal parent with an affected heterozygous parent. The diagram (Punnett square) in Figure 4-22 illustrates this mating. The affected parent can pass either a disease gene or a normal gene to his or her children. Each event has a probability of 0.5; thus on the average, half of the children will be heterozygous and will express the disease and half will be normal.
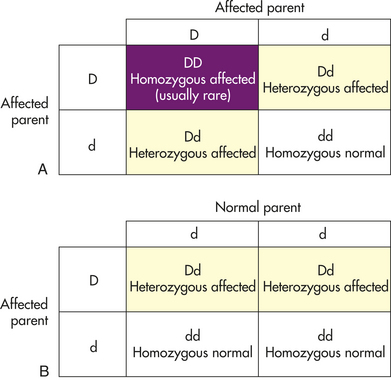
Figure 4-22 Punnett square and autosomal dominant traits. A, Punnett square for the mating of two individuals with an autosomal dominant gene. Here both parents are affected by the trait. B, Punnett square for the mating of a normal individual with a carrier for an autosomal dominant gene.
Figure 4-23, A, is a typical pedigree showing the transmission of an autosomal dominant gene. The gene shown here causes achondroplasia (Figure 4-23, B). Several important characteristics of this pedigree support the conclusion that the trait is caused by an autosomal dominant gene:
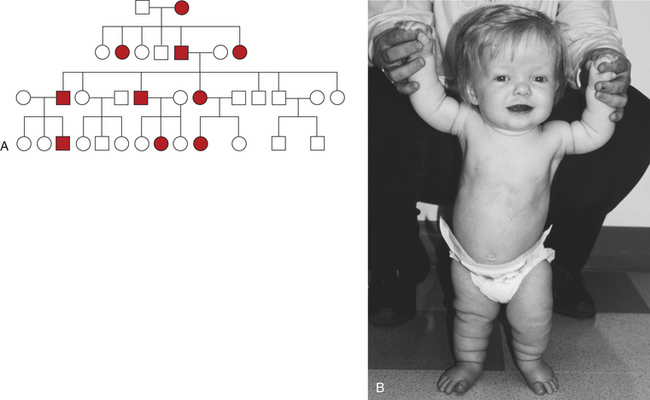
Figure 4-23 Pedigree for achondroplasia. A, Pedigree showing the transmission of an autosomal dominant disease. B, Achondroplasia. This girl has short limbs relative to trunk length. She also has a prominent forehead, low nasal root, and redundant skin folds in the arms and legs. (B from Jorde LB et al: Medical genetics, ed 3, St Louis, 2003, Mosby.)
1. The two sexes exhibit the trait in approximately equal proportions, and males and females are equally likely to transmit the trait to their offspring.
2. There is no skipping of generations. If an individual has achondroplasia, one parent must also have it. If neither parent has the trait, none of the children has it (with the exception of new mutations, as discussed later).
3. Affected heterozygous individuals transmit the trait to approximately half of their children, but because gamete transmission is subject to chance fluctuations, it is possible that all or none of the children of an affected parent may have the trait. When large numbers of matings of this type are studied, however, the proportion of affected children will closely approach one half.
Recurrence Risks
Parents at risk for producing children with a genetic disease nearly always ask the question, “What is the chance that our child will have this disease?” When one child has already been born with a genetic disease, the parents can be given a recurrence risk, which is the probability that subsequent children also will have the disease. When one parent is affected by an autosomal dominant disease (and is a heterozygote) and the other is normal, the recurrence risks for each child are one half.
An important principle is that each birth is an independent event, much like a coin toss. Thus, even though parents may already have had a child with the disease, their recurrence risk remains one half. If they have had several children, all affected (or all unaffected) by the disease, the law of independence dictates that the probability that their next child will have the disease is still one half. Parents’ misunderstanding of this principle is a common problem encountered in genetic counseling.
If a child has been born with an autosomal dominant disease and there is no history of the disease in the family, the child is probably the product of a new mutation. The gene transmitted by one of the parents has thus undergone a mutation from a normal to a disease-causing allele. The genes at this locus in most of the parent’s other germ cells would still be normal. In this situation the recurrence risk for the parent’s subsequent offspring is not greater than that of the general population. The offspring of the affected child, however, will have an occurrence risk of one half. Because these diseases often reduce the potential for reproduction, a large proportion of the observed cases of many autosomal dominant diseases are the result of new mutations. For example, approximately seven eighths of all cases of achondroplasia are caused by new mutations.
Occasionally, two or more offspring will present symptoms of an autosomal dominant disease when there is no family history of the disease. Because mutation is a rare event, it is unlikely that this disease would be a result of multiple mutations in the same family. The mechanism most likely to be responsible is termed germline mosaicism. During the embryonic development of one of the parents, a mutation occurred that affected all or part of the germline but few or none of the somatic cells of the embryo. Thus the parent carries the mutation in his or her germline but does not actually express the disease. As a result, the unaffected parent can transmit the mutation to multiple offspring. This phenomenon, although relatively rare, can have significant effects on recurrence risks.12
Penetrance and Expressivity
An important variation seen in some autosomal dominant diseases is incomplete penetrance. The penetrance of a trait is the percentage of individuals with a specific genotype who also exhibit the expected phenotype. Incomplete penetrance means that individuals who have the gene for a disease may not exhibit the disease phenotype at all, even though the gene and the associated disease may be transmitted to the next generation. A pedigree illustrating the transmission of an autosomal dominant gene with incomplete penetrance is given in Figure 4-24. Retinoblastoma, the most common malignant eye tumor affecting children, is one disease that typically exhibits incomplete penetrance. About 10% of the individuals who are obligate carriers of the gene (i.e., those who have an affected parent and affected children and therefore must themselves carry the gene) do not have the disease. The penetrance of the gene is then said to be 90%.
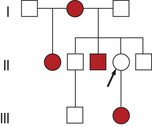
Figure 4-24 Pedigree for retinoblastoma showing incomplete penetrance. The female with marked arrow in line II must be heterozygous, but she does not express the trait.
The gene responsible for retinoblastoma has been mapped to the long arm of chromosome 13, and its DNA sequence has been studied extensively. This gene is known as a tumor-suppressor gene: the normal function of its protein product is to regulate the cell cycle so that cells do not grow uncontrollably. When a mutation alters the protein, its tumor-suppressing capacity is lost and a tumor can form13,14 (see Chapters 11 and 19).
Another well-known autosomal dominant diseases is Huntington disease, a neurologic disorder whose main features are progressive dementia and increasingly uncontrollable movements of the limbs (discussed further in Chapter 17). The latter is known as chorea (Greek khoreia = dance; the disease was formerly called Huntington chorea).
One of the key features of this disease is that symptoms are not usually seen until age 40 years or later, a pattern known as age-dependent penetrance. Thus people who develop the disease often have had children before they are aware that they have the gene. If the disease were present at birth, nearly all those affected would die before reaching reproductive age, and the occurrence of the gene in the population would be much lower. From the gene’s “point of view,” a delayed age of onset is quite advantageous. An individual whose parent has the disease has a 50% chance of developing it during middle age. He or she is thus confronted with a torturous question: “Should I have children, knowing that there is a 50-50 chance that I may have this disease gene and pass it to half my children?” Age-dependent penetrance characterizes a number of important genetic diseases, including familial breast cancer, hemochromatosis, and polycystic kidney disease.
Most genetic diseases exhibit variable expressivity. Expressivity is the extent of variation in phenotype associated with a particular genotype. If expressivity of a disease is variable, the penetrance may be complete but the severity of the disease can vary greatly. A well-known example of variable expressivity in an autosomal dominant disease is type 1 neurofibromatosis, or von Recklinghausen disease. The gene that causes neurofibromatosis has been mapped to the long arm of chromosome 17, and studies of its DNA sequence indicate that it, like the retinoblastoma gene, is a tumor-suppressor gene.15 The expression of this gene can vary from a few harmless café-au-lait spots (“coffee with milk,” describing the light brown color) on the skin to numerous malignant neurofibromas, scoliosis, seizures, gliomas, neuromas, hypertension, and learning disabilities (Figure 4-25).
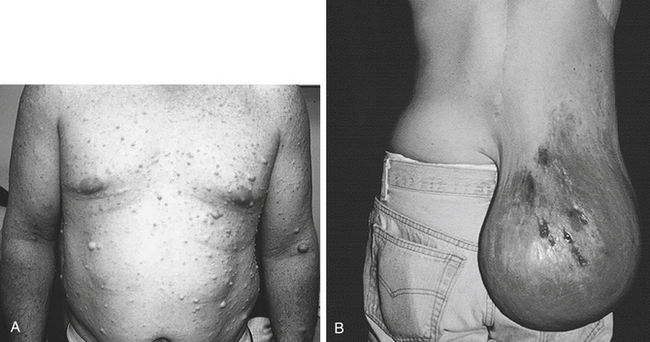
Figure 4-25 Neurofibromatosis. A, Young adult with multiple dermal neurofibromas of the trunk. B, Individual has a large plexiform neurofibroma hanging from lower right back, causing considerable inconvenience and discomfort (substantially improved by surgical removal of tumor). (From Jorde LB et al: Medical genetics, ed 3, St Louis, 2003, Mosby. B courtesy Dr. D. Viskochil, University of Utah Health Sciences Center.)
A parent with mild expression of the disease—so mild that he or she is not aware of it—can transmit the gene to a child, who can then exhibit severe expression of the disease. As with incomplete penetrance, variable expressivity provides a mechanism by which autosomal dominant genes can be maintained at higher prevalence rates in populations.
Several factors can cause variation in expressivity. Genes at other loci can sometimes modify the expression of a disease gene (these are termed modifier genes). Environmental factors also can influence the expression of a disease gene. Finally, different types of mutations at a locus can cause variation in severity. For example, a base substitution resulting in a single amino acid change usually produces a mild form of the clotting disorder hemophilia A (Box 4-3). A base substitution resulting in a “stop” codon (and thus premature termination of translation) usually produces a more severe form of hemophilia A.
Box 4-3 Hemophilia A and the Russian Revolution
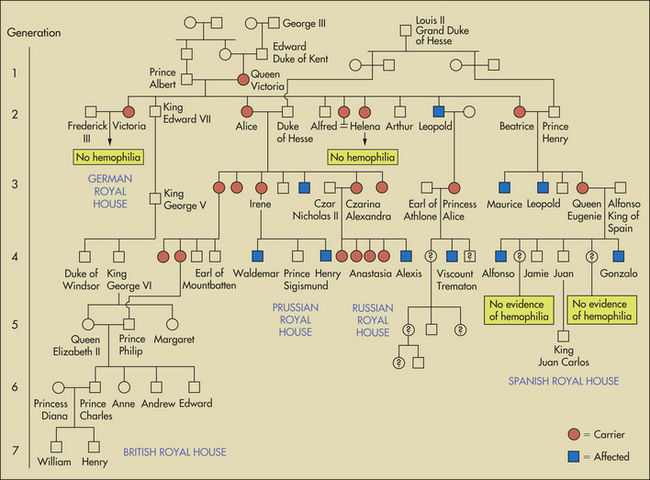
Partial pedigree for descendants of Queen Victoria, showing appearance of hemophilia A in one of her sons and in his descendants and in descendants of her daughters and granddaughters. Royal families of Prussia, Hesse, Battenberg (Mountbatten), Russia, and Spain were thus affected with the disease. The present royal family of England, however, is free of the disease, in spite of inbreeding.
The figure (partial pedigree for descendants of Queen Victoria) is one of the best-known disease pedigrees in existence. It shows the transmission of hemophilia A in the European royal families. This disease, often called a bleeder syndrome, is caused by a defect in one of the blood-clotting factors, factor VIII, and can cause severe hemorrhages. In this pedigree Queen Victoria of England was the first known carrier of the disease, and several of her male descendants were affected by it. One of the most historically significant consequences of this pedigree involves the hemophiliac Czarevich Alexis, son of Czar Nicholas II of Russia. Gregori Rasputin, the “mad monk,” was reputedly the only person able to prevent the young boy’s bleeding episodes and was thus able to gain considerable power over the royal family. Rasputin’s destabilizing influence is thought to have hastened the 1917 Bolshevik revolution.
The Russian royal family was again touched by genetics. Modern DNA “fingerprints” and mitochondrial DNA sequences were used to prove that a mass burial near Ekaterinburg, Russia, contained the remains of most of the executed members of the czar’s family.
Epigenetics and Genomic Imprinting
Although the emphasis of this chapter is on DNA sequence variation and its consequence for disease, there is increasing evidence that the same DNA sequence can produce dramatically different phenotypes, depending on chemical modifications that alter the expression of genes (these modifications are collectively termed epigenetic). An important example of such a modification is DNA methylation, the attachment of methyl groups to cytosine bases in the DNA sequence (Figure 4-26). When the DNA sequence near a gene becomes heavily methylated, the DNA is less likely to be transcribed into mRNA. In other words, the gene becomes transcriptionally inactive or silenced (also see Chapters 11 and 12). A study showed that identical (monozygotic) twins accumulate different methylation patterns in the DNA sequences of their somatic cells as they age, causing increasing numbers of phenotypic differences. Intriguingly, twins with more differences in their lifestyles (e.g., smoking versus nonsmoking) accumulated larger numbers of differences in their methylation patterns. The twins, despite having identical DNA sequences, become more and more different as a result of epigenetic changes, which in turn affect the expression of genes.
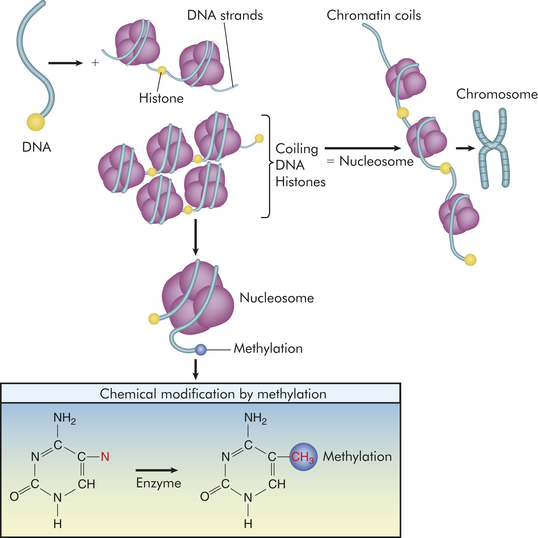
Figure 4-26 Epigenetic modifications. Because DNA is a long molecule, it needs packaging to fit into the tiny nucleus. Packaging involves coiling of the DNA in a “left-handed” spiral around spools, made of four pairs of proteins individually known as histones and collectively as the histone octamer. The entire spool is called a nucleosome. Nucleosomes are organized into chromatin, the repeating building blocks of a chromosome. Histone modifications are correlated with methylation, are reversible, and occur at multiple sites. Methylation occurs at the 5 position of cytosine and provides a “footprint” or signature as a unique epigenetic alteration (red). When genes are expressed, chromatin is open or active; however, when chromatin is condensed because of methylation and histone modification, genes are inactivated.
Epigenetic alteration of gene activity can have important disease consequences. For example, a major cause of one form of inherited colon cancer (termed hereditary nonpolyposis colorectal cancer, or HNPCC) is the methylation of a gene whose protein product repairs damaged DNA. When this gene becomes inactive, damaged DNA accumulates, resulting eventually in colon tumors.
Approximately 100 human genes are thought to be methylated differentially, depending on which parent transmits the gene. This epigenetic modification, characterized by methylation and other changes, is termed genomic imprinting. For each of these genes, one of the parents “imprints” the gene (inactivates it) when it is transmitted to the offspring. An example is the insulin-like growth factor 2 gene (IGF-II) on chromosome 11, which is transmitted by both parents. But the copy inherited from the mother is normally methylated and inactivated (imprinted). Thus, only one copy of IGF-II is active in normal individuals. However, the maternal “imprint” is occasionally lost, resulting in two active copies of IGF-II. This causes excess fetal growth and a condition known as Beckwith-Wiedemann syndrome.
A second example of genomic imprinting is a deletion of part of the long arm of chromosome 15 (15q11-q13) which, when inherited from the father, causes the offspring to manifest a disease known as Prader-Willi syndrome (short stature, obesity, hypogonadism). When the same deletion is inherited from the mother, the offspring develop Angelman syndrome (mental retardation, seizures, ataxic gait). The two different phenotypes reflect the fact that different genes are normally active in the maternally and paternally transmitted copies of this region of chromosome 15.
Autosomal Recessive Inheritance
Like autosomal dominant diseases, those caused by autosomal recessive genes are rare in populations, although the number of carriers for recessive diseases can be high. The most common lethal recessive disease in white children, cystic fibrosis, occurs in about 1 in 2500 births. Approximately 1 in 25 whites carries one copy of the gene for cystic fibrosis (see Chapter 34). Because an individual must be homozygous for a recessive gene to express the disease, the carriers are phenotypically normal. Because most genes for recessive diseases are maintained in normal carriers, they are able to survive in the population from one generation to the next. As with many autosomal dominant diseases, many autosomal recessive diseases are characterized by delayed age of onset, incomplete penetrance, and variable expressivity.
Figure 4-27 shows a pedigree for cystic fibrosis. The cystic fibrosis gene, which has been mapped to the long arm of chromosome 7, encodes a protein product that forms chloride channels in the membranes of specialized epithelial cells.16 Defective transport of chloride ions leads to a salt imbalance that results in secretions of abnormally thick, dehydrated mucus. Some of the digestive organs, particularly the pancreas, become obstructed, causing malnutrition, and the lungs become clogged with mucus, making them highly susceptible to bacterial infections (especially Pseudomonas). Death from lung disease or heart failure occurs on average by about 37 years of age. In the pedigree shown, the two affected individuals are the offspring of the marriage of two first cousins. Marriage between related individuals, termed consanguinity (from the Latin root meaning “with blood”), is often a factor in producing children with recessive diseases because related individuals are more likely to share the same recessive genes. Consanguinity is seen most often in rare recessive diseases because carriers of common recessive diseases have a fairly high probability of encountering one another just by chance.
Important criteria for discerning autosomal recessive inheritance include the following:
Recurrence Risks
In most cases of recessive disease, both parents of affected individuals are heterozygous carriers. On the average, one fourth of their offspring will be normal homozygotes, one half will be phenotypically normal carrier heterozygotes, and one fourth will be homozygotes with the disease (Figure 4-28). Thus the recurrence risk for the offspring of carrier parents is 25%. As stated, these are the average figures. In any given family, chance fluctuations are likely, but a study of a large number of families would yield figures close to these proportions.
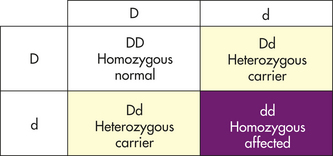
Figure 4-28 Punnett square for the mating of heterozygous carriers. This is typical of most cases of recessive disease.
If two parents have a recessive disease, they each must be homozygous for the disease. Therefore, when two parents are affected by a recessive disease, all their children also must be affected. This observation helps to distinguish recessive from dominant inheritance, because two parents both affected by a dominant gene are nearly always both heterozygotes and thus one fourth of their children will be unaffected.
Because carrier parents usually are unaware that they both carry the same recessive gene, they often produce an affected child before knowing of their condition. Carrier detection tests can identify heterozygotes by measuring the reduced amount of a critical enzyme available. The critical enzyme is totally lacking in a homozygous recessive individual, but an essentially normal phenotype is seen when it is present in a reduced quantity in the carrier. Often carriers can be detected by direct examination of the disease locus for a mutation. Such testing is especially valuable for siblings of known carriers, who may themselves be carriers. Some recessive diseases for which carrier detection tests are now available are PKU, sickle cell disease, cystic fibrosis, Tay-Sachs disease, hemochromatosis, and galactosemia.
Consanguinity
Consanguinity and inbreeding are related concepts. Consanguinity refers to the mating of two related individuals, and the offspring of such matings are said to be inbred. Consanguinity is often an important characteristic of pedigrees for recessive diseases because relatives share a certain proportion of genes received from a common ancestor. The proportion of shared genes depends on the closeness of their biologic relationship. For example, siblings share one half of their genes on average. With each decreasing degree of relationship, this proportion is reduced by one half. Uncles share one fourth of their genes with nephews and nieces; first cousins share one eighth; first cousins once removed∗ share one sixteenth; second cousins share one thirty-second; and so on. With consanguineous matings, recessive disorders are significantly increased. Most empirical studies show that the proportion of offspring of marriages of first cousins who are affected by genetic diseases is approximately double that of the general population.17 Marriages between first cousins are prohibited in most states of the United States. Marriages between closer relatives (except between double first cousins†) are prohibited throughout the United States.
X-Linked Inheritance
Not all genetic diseases are caused by genes located on the 22 autosomes. Some conditions are instead caused by genes located on the sex chromosomes, and that mode of inheritance is referred to as sex-linked. The Y chromosome contains only a few dozen genes, so most sex-linked traits are located on the X chromosome and are said to be X-linked. Only a few diseases are known to be inherited as X-linked dominant traits. Because these diseases are so seldom encountered, only the much more common X-linked recessive diseases are discussed here.
Because females receive two X chromosomes, one from the father and one from the mother, they can be homozygous for a disease allele at a given locus, homozygous for the normal allele at the locus, or heterozygous. Males, having only one X chromosome, are said to be hemizygous for genes on this chromosome. A male who inherits a recessive disease gene on the X chromosome will be affected by the disease because the Y chromosome does not carry a normal allele to counteract the effects of the disease gene. Males are always more frequently affected by X-linked recessive diseases, with the difference becoming more pronounced as the disease becomes rarer.
X Inactivation
In the late 1950s Mary Lyon proposed that one X chromosome in the somatic cells of females is permanently inactivated, a process termed X inactivation.18 This proposal, known as the Lyon hypothesis, explains why most gene products coded by the X chromosome are present in equal amounts in males and females, even though males have only one X chromosome and females have two X chromosomes. This phenomenon is called dosage compensation. The inactivated X chromosomes are observable in many interphase cells as highly condensed intranuclear chromatin bodies, termed Barr bodies (after Barr and Bertram, who discovered them in the late 1940s). Normal females have one Barr body in each somatic cell, whereas normal males have no Barr bodies.
The actual process of inactivation occurs very early in embryonic development—approximately 7 to 14 days after fertilization. In each somatic cell one of the two X chromosomes is inactivated. In some cells the X chromosome contributed by the father is inactivated; in others the maternal X chromosome is inactivated. Because the inactivation process is random, the maternal X chromosome is inactivated in approximately half the cells and the paternal X chromosome is inactivated in approximately half the cells. Once the X chromosome has been inactivated in a cell, all the descendants of that cell have the same chromosome inactivated. Thus inactivation is said to be random but fixed.
Some individuals do not have the normal number of X chromosomes in their somatic cells. For example, males with Klinefelter syndrome typically have two X chromosomes and one Y chromosome. These males do have one Barr body in each cell. Females whose cell nuclei have three X chromosomes have two Barr bodies in each cell, and females whose cell nuclei have four X chromosomes have three Barr bodies in each cell. Females with Turner syndrome have only one X chromosome and no Barr bodies. Thus the number of Barr bodies is always one less than the number of X chromosomes in the cell. All but one X chromosome are always inactivated.
People with abnormal numbers of X chromosomes, such as those with Turner syndrome or Klinefelter syndrome, are not physically normal. This situation presents a puzzle because they presumably have only one active X chromosome, just as individuals with normal numbers of chromosomes do. However, the distal portions of the short and long arms of the X chromosome, as well as several other regions on the chromosome, are not inactivated. Thus X inactivation is also known to be incomplete.
Although the mechanism underlying X inactivation is still incompletely understood, the gene responsible for initiating X inactivation, XIST, has been located.19 This gene encodes an mRNA that coats one of the X chromosomes, which is then inactivated. Methylation of X chromosome DNA, a process in which DNA is inactivated when cytosine bases are enzymatically converted to 5-methylcytosine, occurs on the inactivated X chromosome. Inactive X chromosomes can be at least partially reactivated in vitro by administering 5-azacytidine, a demethylating agent.
Sex Determination
The process of sexual differentiation, in which the embryonic gonads become either testes or ovaries, begins during the sixth week of gestation. A key principle of sex determination in the human is that one copy of the Y chromosome is sufficient to initiate the process of gonadal differentiation that produces a male fetus (Figure 4-29, B). The number of X chromosomes does not alter this process. For example, an individual with two X chromosomes and one Y chromosome in each cell is still phenotypically a male. Thus it is logical that the Y chromosome must contain a gene that begins the process of male gonadal development.
Figure 4-29 The distal short arms of the X and Y chromosomes exchange material during meiosis in the male. A, The region of the Y chromosome in which this crossover occurs is called the pseudoautosomal region. The SRY gene, which triggers the process leading to male gonadal differentiation, is located just outside the pseudoautosomal region. Occasionally, the crossover occurs on the centromeric side of the SRY gene, causing it to lie on an X chromosome instead of a Y chromosome. An offspring receiving this X chromosome will be an XX male, and an offspring receiving the Y chromosome will be an XY female. B, X and Y chromosomes. (A from Jorde LB et al: Medical genetics, ed 3, St Louis, 2003, Mosby. B from Raven PH et al: Biology, ed 8, New York, 2008, McGraw-Hill.)
This gene, termed SRY (for “sex-determining region on the Y”) has been located on the short arm of the Y chromosome.20,21 The SRY gene lies immediately proximal to the distal tip of the Y chromosome, known as the pseudoautosomal region (Figure 4-29, A). This portion of the Y chromosome is so named because it pairs with the distal tip of the short arm of the X chromosome during meiosis and exchanges genetic material with it (crossover), just as autosomes do. The DNA sequences of these regions on the X and Y chromosomes are highly similar. The remainder of the X and Y chromosomes, however, do not exchange material and are not similar in DNA sequence. An important piece of evidence that supports SRY as the male-determining gene is that female mouse embryos injected with this gene develop as phenotypic males.
Although the SRY gene is located on the Y chromosome, the other genes that contribute to male differentiation are located on other chromosomes. Thus SRY appears to act as a trigger that initiates the action of genes on other chromosomes (e.g., those that control Sertoli cell differentiation or secretion of müllerian-inhibiting substance). This concept is supported by the fact that the SRY gene is similar in sequence to other genes that are known to regulate the transcription of DNA (i.e., they turn other genes on and off).
Occasionally the crossover between X and Y occurs closer to the centromere than it should, placing the SRY gene on the X chromosome after crossover. This variation can result in offspring with an apparently normal XX karyotype but a male phenotype. Such XX males are seen in about 1 in 20,000 live births and closely resemble males with Klinefelter syndrome, although their stature is normal. Conversely, it is possible to inherit a Y chromosome that has lost the SRY gene (because of either a crossover error or a deletion of the gene). This situation produces an XY female. Such females have gonadal streaks rather than ovaries and have poorly developed secondary sex characteristics.
Characteristics of Pedigrees
X-linked pedigrees show distinctive modes of inheritance. The most striking characteristic is that females are seldom affected. To express an X-linked recessive trait, a female must be homozygous: either both her parents are affected or her father is affected and her mother is a carrier. Such matings are rare.
An important example of an X-linked recessive disease is hemophilia A. The pedigree shown in Box 4-3 demonstrates the following principles of X-linked recessive inheritance:
1. The trait is seen much more often in males than in females.
2. Because a father can give a son only a Y chromosome, the trait is never transmitted from father to son.
3. The gene can be transmitted through a series of carrier females, causing the appearance of a “skipped generation.”
4. The gene is passed from an affected father to all his daughters, who, as phenotypically normal carriers, transmit it to approximately half their sons, who are affected.
The most common and severe of all X-linked recessive disorders is Duchenne muscular dystrophy (DMD), which affects approximately 1 in 3500 males. As its name suggests, this disorder is characterized by progressive muscle degeneration. Affected individuals are usually unable to walk by 10 to 12 years of age. The disease affects the heart and respiratory muscles, and death caused by respiratory or cardiac failure usually occurs before 20 years. Until recently, the underlying pathologic origin of this disorder was a mystery. However, mapping and cloning of the disease gene (on the short arm of the X chromosome) have greatly increased our understanding of the disorder.22 The DMD gene is the largest gene ever found in the human, spanning more than 2 million DNA bases. It encodes a previously undiscovered muscle protein, termed dystrophin. Extensive study of dystrophin indicates that it plays an essential role in maintaining the structural integrity of muscle cells: one end of the protein binds to actin filaments in the cytoplasm of the cell, and the other end binds to a group of membrane-spanning proteins known as the dystrophin-associated glycoproteins. When dystrophin is absent, as in individuals with DMD, the cell cannot survive, and muscle deterioration ensues.
Most cases of Duchenne muscular dystrophy are caused by deletions of portions of the DMD gene. They generally involve frameshift deletions in which all the amino acids following the deletion are altered. It is interesting that an “in frame” deletion (in which a multiple of three bases is deleted, and the amino acids following the deletion are not altered) produces a milder form of muscular dystrophy, the Becker type. These two types of dystrophy are examples of a disease in which different types of mutations at the same locus produce variable expression of the disease.
Recurrence Risks
The most common mating type involving X-linked recessive genes is the combination of a carrier female and a normal male. On the average, the carrier mother will transmit the disease gene to half her sons and half her daughters. As Figure 4-30, A, shows, half the daughters in such a mating will be carriers, whereas half will be normal. Half the sons will be normal, whereas half will have the disease. These are probabilities that indicate what risks can be expected on the average (see Box 4-3).
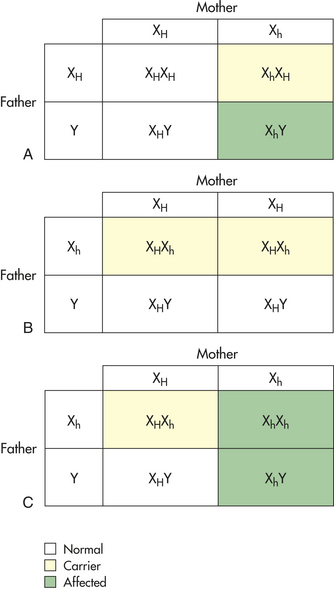
Figure 4-30 Punnett square and X-linked recessive traits. A, Punnett square for the mating of a normal male (XHY) and a female carrier of an X-linked recessive gene (XHXh). B, Punnett square for the mating of a normal female (XHXH) with a male affected by an X-linked recessive disease (XhY). C, Punnett square for the mating of a female who carries an X-linked recessive gene (XHXh) with a male who is affected with the disease caused by the gene (XhY).
The other common mating type is an affected father and a normal mother (see Figure 4-30, B). In this situation all the sons must be normal because the father can transmit only his Y chromosome to them. Because all the daughters must receive the father’s X chromosome, they will all be heterozygous carriers. Because the sons must receive the Y chromosome and the daughters must receive the X with the disease gene, these are predictions and not probabilities. None of the children will express the disease.
The final mating pattern, less common than the other two, involves an affected father and a carrier mother (see Figure 4-30, C). With this pattern, on average, half the daughters will be heterozygous carriers and half will be homozygous for the disease gene and thus affected. Half the sons will be normal, and half will be affected. Some X-linked recessive diseases, such as DMD, are fatal or incapacitating before the affected individual reaches reproductive age, and therefore affected fathers are rare or nonexistent.
Sex-Limited and Sex-Influenced Traits
Confusion sometimes exists regarding the difference between traits that are sex-linked and those that are sex-limited or sex-influenced. A sex-limited trait is one that can occur in only one of the sexes, often because of anatomic differences. Inherited uterine and testicular defects are two obvious examples.
A sex-influenced trait is one that occurs much more often in one sex than in the other. A good example of a sex-influenced trait is male-pattern baldness, which occurs in both males and females but is much more common in males. Another example is autosomal dominant breast cancer, which is approximately 70 times more common in females than males.
Evaluation of Pedigrees
With complications such as incomplete penetrance, variable expressivity, delayed age of onset, and sex-influenced traits, it is not always possible simply to look at a disease pedigree and determine the mode of inheritance. A sophisticated statistical methodologic approach has evolved to deal with such complications. Incorporated into computer programs, these statistical techniques assess the probability of observing a certain pedigree if a particular mode of inheritance (e.g., autosomal dominant with incomplete penetrance) is in effect.
LINKAGE ANALYSIS AND GENE MAPPING
Locating genes on chromosomes and on specific areas of chromosomes is one of the most important endeavors in human genetics. The location of a gene can tell much about the function of the gene, its interaction with other genes, and the likelihood that certain individuals will develop a genetic disease.
Classical Pedigree Analysis
Mendel’s second law, the principle of independent assortment, states that an individual’s genes will be transmitted to the next generation independently of one another. This law is only partly true, however, because genes located close together on the same chromosome do tend to be transmitted together to the offspring. Thus Mendel’s principle of independent assortment holds true for most pairs of genes but not those that occupy the same region of a chromosome. Such loci demonstrate linkage and are said to be linked.
During the first meiotic stage, the arms of homologous chromosome pairs intertwine and sometimes exchange portions of their DNA (Figure 4-31) in a process known as crossing over. During crossing over, new combinations of alleles can be formed. For example, two loci on a chromosome have alleles A and a and alleles B and b. Alleles A and B are located together on one chromosome arm, and alleles a and b are located on the other arm. The genotype of this individual is denoted as AB/ab.
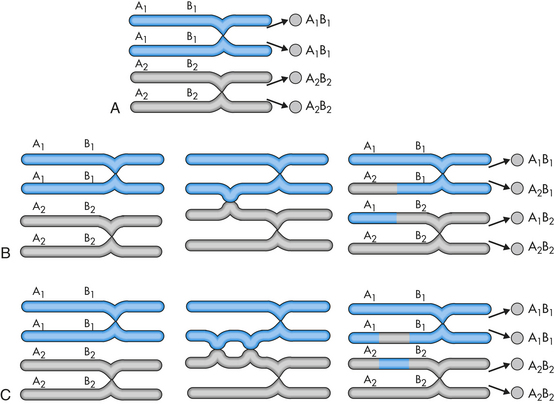
Figure 4-31 The genetic results of crossing over. A, No crossing over: A1 and B1 remain together after meiosis. B, Crossing over between A and B results in a recombination: A1 and B2 are inherited together on one chromosome, and A2 and B1 are inherited together on another chromosome. C, A double crossover between A and B results in no recombination of alleles.
As Figure 4-31, A, shows, the allele pairs AB and ab would be transmitted together when no crossing over occurs. However, when crossing over does occur (Figure 4-31, B), all four possible pairs of alleles can be transmitted to the offspring: AB, aB, Ab, and ab. The process of forming such new arrangements of alleles is called recombination. Crossing over does not necessarily lead to recombination, however, because double crossing over between two loci can result in no actual recombination of the alleles at the loci (Figure 4-31, C).
The rate of crossing over can be used to infer the distance between two loci on a chromosome because the probability of crossovers occurring between two loci increases as the loci become more distant. For example, if an individual with genotype AB/ab produces recombinant offspring gametes (composition of Ab and aB) 2% of the time, it is said that the two loci are two map units apart. One map unit equals a 1% recombination rate between two loci. When loci on the same chromosome are 50 or more map units apart, they are considered unlinked because their recombination frequency is just as great as it would be if they were on different chromosomes (where the probability of being transmitted together must equal one half). Because they are on the same chromosome, they are said to be unlinked but syntenic loci. Recombination frequencies provide a good estimate of actual physical distance between loci at smaller distances, but because of double crossovers, they tend to yield underestimates at larger distances. On average, each map unit is equal to approximately 1 million DNA base pairs.
Pedigrees can be used to determine recombination rates between loci. Figure 4-32 shows a pedigree in which the rare disease nail-patella syndrome (an autosomal dominant disease consisting of malformed patellae and nails) is being transmitted. The individuals in this pedigree have been typed for the ABO blood group, whose locus is also located on chromosome 9. Examination of generations I and II shows that the nail-patella gene must be on the same chromosome arm as the gene for blood type A because the mother, whose blood type was B, was unaffected with the disease. The daughter’s genotype would then be AN/Bn, in which N indicates the disease allele and n indicates the normal allele. The daughter’s husband (individual II-1) must have the genotype On/On. If the loci for nail-patella syndrome and the ABO blood group are linked, the children of this union who are affected with nail-patella syndrome should have blood type A; those who are unaffected should have blood type B. In six of seven cases we find this to be true. In one case a recombination occurred (individual III-6), indicating a recombination rate of 1 in 7, or 14%. The two loci are therefore 14 map units apart.
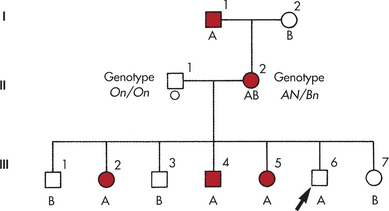
Figure 4-32 The ABO nail-patella linkage in three generations of a family. Letters below symbols indicate ABO blood groups. Individual III-6 shows recombination.
In practice, a much larger sample of families would be used to ensure against statistical artifacts. Also, as with the determination of mode of inheritance, the situation is not always as clear as that pictured in Figure 4-32. Elaborate statistical procedures have been devised to evaluate the probabilities that two loci are linked at a given map distance.
Once a close linkage has been established between a disease locus and a marker locus (a DNA variant that can easily be assayed in the laboratory) and once the alleles of the two loci that are inherited together within a family have been determined, reliable predictions of whether a member of a family will develop the disease can be made. If, for example, the recombination rate between a disease locus and a marker locus, such as the ABO blood group, is less than 1%, family members can simply have their ABO blood type assayed to find out, with 99% or greater certainty, whether each member carries the disease gene.
This capability is especially important for diseases with delayed age of onset. Linkage has been established between several DNA polymorphisms and the gene for Huntington disease. Determining this kind of linkage means that it is possible for offspring of an individual with Huntington disease to know whether they also carry the gene and thus could pass it on to their own children. The difficult decision of whether to have children will be made easier for these individuals, although some individuals may prefer to remain uninformed of their genotypes. Other delayed-onset diseases for which linked markers have been found include adult polycystic kidney disease, familial Alzheimer disease, and two forms of autosomal dominant breast cancer (about 5% of breast cancer cases are caused by an autosomal dominant gene). Pinpointing specific mutations in these genes also has made direct genetic diagnosis possible. The advantage of direct diagnosis is that it is more accurate because it tests for the disease-causing mutation itself.
For some genetic diseases, prophylactic treatment is available if the condition can be diagnosed in time. An example of this is hemochromatosis—a recessive genetic disease in which excess iron is retained, causing degeneration of the heart, liver, brain, and other vital organs. Diagnosis is usually made at about 40 years of age in males, after which most individuals survive only a few years. If earlier tests could determine whether an individual had the disease, preventive treatment, consisting of phlebotomies to remove blood and thus excess iron, could be administered before degeneration began. This has been made easier by mapping the hemochromatosis gene to a specific region of chromosome 6 and subsequently identifying the major disease-causing mutations. Individuals at risk for developing the disease can be identified by testing for presence of the mutations, and if necessary, preventive therapy can be given, ensuring an ordinary life span. This example is one instance in which genetics contributes to preventive medicine in its best sense.
Assigning Loci to Specific Chromosomes
With completion of the human DNA sequence (see following), computer analysis of the published sequence has become an effective and popular approach for identifying genes. Computerized databases of known DNA sequences play an important role in gene identification. When studying a specific region of DNA to find a gene, it is common to search for similarity between DNA sequences from the region and DNA sequences in the database. The sequences in the database may derive from genes with known function or tissue-specific expression patterns. Suppose, for example, that we have used linkage analysis to identify a region containing a gene that causes a developmental disorder such as a limb malformation. As we evaluate DNA sequences in the region, we would look for similarity between a DNA sequence from this region and a plausible sequence from the database (e.g., sequence from a gene that encodes a protein involved in bone development, such as a fibroblast growth factor). Because genes that encode similar protein products usually have similar DNA sequences, a match between the sequence from our region and a sequence in the database could be a vital clue that this particular DNA sequence is actually part of the gene that causes the limb malformation.
Complete Human Gene Map: Prospects and Benefits
Rapid progress is being made in assigning genes to their chromosomal locations. A number of important genetic diseases have been located on specific areas of individual chromosomes: these include Huntington disease, retinoblastoma, DMD, hemophilia A, cystic fibrosis, PKU, neurofibromatosis, familial breast cancer, and familial Alzheimer disease23 (Figure 4-33). Table 4-1 contains a partial list of mapped diseases. The development of thousands of new DNA markers is especially helpful in this effort. A marker map of the human genome has been completed, and completion of the entire sequence of the human genome was announced in April 2003. Achievement of this goal serves several purposes:
Table 4-1
Examples of Disease Genes That Have Been Mapped and Cloned∗


∗Additional disease-causing loci have been mapped and/or cloned.
Modified from Jorde LB et al: Medical genetics, ed 3, St Louis, 2003, Mosby.
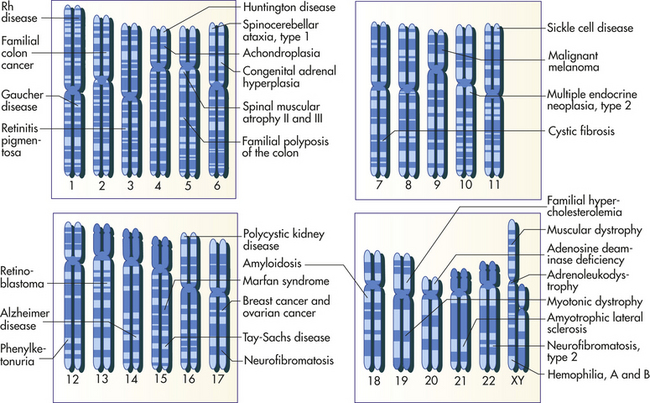
1. Markers are available to establish close linkages for genetic diseases. With the establishment of a comprehensive marker map, accurate predictions can be made for the inheritance of most genetic diseases.
2. Knowing the location of genes often yields valuable information about the way genes function and interact with one another. A number of genes with similar functions (e.g., some of the globin genes) are located close to one another on the same chromosome. This characteristic can have important implications for the diseases caused by these genes.
3. Mapping a disease gene is an important step toward isolating and cloning the gene (clones are identical copies of genes). Once a gene can be cloned, its DNA sequence can be studied to determine the nature and function of the protein encoded by the gene. Cloning the genes that cause diseases such as cystic fibrosis and DMD has contributed immensely to our understanding of the pathophysiologic aspect of these disorders. In addition, the ability to clone a gene opens up the possibility of gene therapy for the disorder (Box 4-4).
Adenine 129
Age-dependent 148
Allele 145
Amino acid 129
Aneuploid cell 136
Anticodon 134
Autosome 134
Barr body 153
Base pair substitution 129
Carrier 145
Carrier detection test 152
Chromosomal mosaic 137
Chromosome band 135
Chromosome breakage 142
Chromosome theory of inheritance 146
Clastogen 142
Cloning 159
Codominance 145
Codon 129
Complementary base pairing 129
Consanguinity 151
Cri du chat syndrome 142
Crossing over 155
Cytosine 129
Deletion 142
Deoxyribonucleic acid (DNA) 126
Diploid cell 134
DNA methylation 149
DNA polymerase 129
Dominant 145
Dosage compensation 153
Double helix 129
Down syndrome 137
Duplication 142
Dystrophin 154
Epigenetic 149
Euploid cell 135
Exon 134
Expressivity 148
Fragile site 143
Frameshift mutation 132
Gamete 134
Gene 126
Genomic imprinting 149
Genotype 145
Germline mosaicism 148
Giemsa stain 135
Guanine 129
Haploid cell 134
Hemizygous 153
Heterogeneous nuclear RNA (hnRNA) 133
Heterozygote 145
Heterozygous 145
Homologous 134
Homologous chromosome 134
Homozygote 145
Homozygous 145
Inbreeding 152
Intron 134
Inversion 143
Karyotype 134
Klinefelter syndrome 141
Linkage 155
Locus 143
Map unit 156
Marker locus 156
Meiosis 134
Messenger RNA (mRNA) 132
Metaphase spread 134
Missense mutation 129
Mode of inheritance 145
Monosomy 136
Mutagen 132
Mutation 129
Mutational hot spot 132
Nondisjunction 137
Nucleotide 129
Obligate carrier 148
Partial trisomy 137
Pedigree 146
Penetrance 148
Phenotype 145
Polymorphic (polymorphism) 145
Polypeptide 129
Polyploid cell 135
Position effect 143
Principle of independent assortment 146
Principle of segregation 146
Proband (propositus/proposita) 146
Promoter site 132
Pseudoautosomal 153
Purine 129
Pyrimidine 129
Recessive 145
Reciprocal translocation 143
Recombination 156
Recurrence risk 148
Ribonucleic acid (RNA) 132
Ribosomal RNA (rRNA) 134
Ribosome 134
RNA polymerase 132
Robertsonian translocation 143
Sex chromosome 134
Sex-influenced trait 155
Sex-limited trait 155
Sex-linked (inheritance) 152
Silent substitution 132
Somatic cell 134
Spontaneous mutation 132
Syntenic loci 156
Template 129
Termination (nonsense) codon 129
Termination sequence 133
Tetraploidy 135
Thymine 129
Transcription 132
Transcriptionally inactive (silenced) 149
Transfer RNA (tRNA) 134
Translation 134
Translocation 143
Triploidy 135
Trisomy 136
Tumor-suppressor gene 148
Turner syndrome 139
REFERENCES
1. Online: Mendelian inheritance in man, available at www.ncbi.nlm.nih.gov/sites/entrez?db=OMIM
2. Hall, J.G., et al. The frequency and financial burden of genetic disease in a pediatric hospital. Am J Med Genet. 1978;1(1):417–436.
3. Crow, J.F. The origins, patterns and implications of human spontaneous mutation. Nat Rev Genet. 2000;1:40–47.
4. Hall, H., Hunt, P., Hassold, T. Meiosis and sex chromosome aneuploidy: how meiotic errors cause aneupolidy; how aneuploidy causes meiotic errors. Curr Opin Genet Dev. 2006;16(3):323–329.
5. Tolie, J.L., MacFayden, U. Clinical genetics of common autosomal trisomies. In: Rimoin D.L., et al, eds. Emery and Rimoin’s principles and practice of medical genetics. ed 5. Philadelphia: Churchill Livingstone; 2007:1015–1037.
6. Antonarakis, S.E., Epstein, C.J. The challenge of Down syndrome. Trends Molec Med. 2006:473–479.
7. Jorde, L.B., et al. Medical genetics, ed 3. St Louis: Mosby; 2006.
8. Graham, G.E., Allanson, J.E., Gerritsen, J.A. Sex chromosome abnormalities. In: Rimoin D.L., et al, eds. Emery and Rimoin’s principles and practice of medical genetics. ed 5. Philadelphia: Churchill Livingstone; 2007:1038–1057.
9. Garber, K.B., Visootsak, J., Warren, S.T. Fragile X syndrome. Eur J Hum Genet. 2008;16(6):666–672.
10. Ranum, N.L., Cooper, T.A. RNA-mediated neuromuscular disorders. Annu Rev Neurosci. 2006;29:259–277.
11. Orr, H.T., Zoghbi, H.Y. Trinucleotide repeat disorders. Annu Rev Neurosci. 2007;30:575–621.
12. Zlotogora, J. Germ line mosaicism. Hum Mol Genet. 1998;102(4):381–386.
13. Balmain, A., Gray, J., Ponder, B. The genetics and genomics of cancer. Nat Genet. 2003;33:238–244.
14. Knudson, A.G. Cancer genetics. Am J Med Genet. 2002;111(1):96–102.
15. Theos, A., Korf, B.R. Pathophysiology of neurofibromatosis type 1. Ann Intern Med. 2006;144(11):842–849.
16. Rowe, S.M., Miller, S., Sorscher, E.J. Cystic fibrosis. N Engl J Med. 2005;342(19):1992–2001.
17. Jorde, L.B., Inbreeding in human populations. Dulbecco, R., eds. Encyclopedia of human biology, vol 5. New York: Academic Press, 1997.
18. Lyon, M.F. Sex chromatin and gene action in the mammalian X-chromosome. Am J Hum Genet. 1962;14:135–148.
19. Wutz, A., Gribnau, J. X inactivation Xplained. Curr Opin Genet Dev. 2007;17(5):387–393.
20. Fleming, A., Vilain, E. The endless quest for sex determination genes. Clin Genet. 2005;67(1):15–25.
21. Ostrer, H. Sex determination: lessons from families and embryos. Clin Genet. 2001;59(4):207–215.
22. Dalkilic, I., Kunkel, L.M. Muscular dystrophies: genes to pathogenesis. Curr Opin Genet Dev. 2003;13(3):231–238. [Review].
23. Collins, F.S., Morgan, M., Patrinos, A. The Human Genome Project: lessons from large-scale biology. Science. 2003;300(5617):286–290.