Oral Aspects of Metabolic Diseases
 . Disturbances in Mineral Metabolism
. Disturbances in Mineral MetabolismLoeb described individual man as a ‘mosaic of many tissues and organs’, each one functioning and metabolizing in its own peculiar way. Each tissue or organ has properties not restricted to it, but common to all parts of the organisms, and it is these common properties which bind the tissues and organs well together into a unit.
Health is largely determined by man’s reaction to his environment, both social and physical, but different individuals behave differently in the same environment and under apparently identical conditions. On this basis we recognize the variations of the substratum upon which environmental factors act. Individual variations in response to the internal and external environment are dependent upon constitution. Each response represents a complex interplay between the genetic and environmental factors acting on the individual.
Certain characteristics of an organism are fixed in the germ cells and give rise to definite metabolic, structural, and functional conditions in the individual. These inherited features represent the core of his constitution, the unchangeable part of it. In actual life; however, it is often difficult to separate this core from effects produced by the environment.
It might be well to visualize man—the organism—as a universe: a universe of cells living together within a restricted framework. Some of the individuals (the cells) of this universe form rather tightly knit communities (the organs) which perform highly specialized tasks. Each individual within the community responds to his inner as well as his outer environment, and each is influenced by his neighbor. The outer environment in this instance is fluid—mostly water. Each individual cell influences the tissue fluid in some manner by removing and/or adding metabolic products. Each cell or organ system reacts to its changing environment within the limits of its inherent capabilities. If an analogy is drawn between the cells as an individual within the organisms and the organisms as an individual within the cosmos, the complexity of the interrelations becomes apparent, even though the nature of the interrelations is nebulous.
Duncan defined metabolism as “the sum total of tissue activity as considered in terms of physicochemical changes associated with and regulated by the availability, utilization, and disposal of protein, fat, carbohydrate, vitamins, minerals, water, and the influences which the endocrines exert on these processes”. Alterations from these normal metabolic processes constitute the disturbances of metabolism. One recognizes immediately that this definition embodies a concept of cellular change as influenced not only by intrinsic factors, but also by such extrinsic factors as food supply, temperature, altitude, society; in other words, by environment.
The volume of literature pertaining to metabolism is rapidly surpassing the ability of investigators to keep pace with it. Obviously it is impossible to provide an in-depth look at any particular area. Excellent references, many recent and some classic, are available. These include Wolbach and Bessey (1942), Schour and Massler (1943 and 1945), Follis (1948 and 1958), Bodansky and Bodansky (1952), Bourne and Kidder (1953), Duncan (1959), Comar and Bronner (1960), Gÿorgy and Pearson (1967), Sebrell and Harris (1967), Greenberg (1967, 1968, 1969, 1970), Vogel (1971), Hokin (1972), Prasad (1976), Underwood (1977), Stanbury, Wyngaarden, and Fredrickson (1978), Alfin-Slater and Kritchevsky (1979), Goodhart and Shills (1980) and Bondy and Rosenberg (1980).
Disturbances in Mineral Metabolism
Although hormones are the primary regulators of metabolism, they are ineffective without minerals and vitamins. Minerals are inorganic elements that are essential for life and provide both the structural and regulatory functions of the body. It is observed that there are at least 29 different elements in our body constituting about 4% of the body weight, concentrated mostly in the skeleton. The elements considered essential for normal growth and development of mammals are calcium, phosphorus, magnesium, potassium, sodium, chlorine, iodine, copper, iron, zinc, manganese, cobalt, chromium, selenium, and fluoride.
Minerals that are present in relatively high amounts in the body are referred to as macrominerals and those that are less than 0.005% of the body weight are called the microminerals. Macrominerals or principal elements are nutritionally important minerals whose daily requirement is more than 100 mg. These include sodium, potassium, chloride, calcium, phosphorus, magnesium and sulfur. The microminerals or trace elements are those found in tissues in minute amounts but are found to be essential to life. Their requirement is less than 100 mg/day and these include chromium, copper, cobalt, iron, iodine, manganese, selenium, fluorine, and zinc. The other trace elements that are possibly essential include cadmium, nickel, silicon, tin, and vanadium.
Inorganic and organic combinations of these elements are active in many physiologic processes. They constitute the basic structure of bone and teeth; help maintain the osmotic relations of the body fluids; regulate the acid-base equilibrium of the tissues; form part of hormones; are an integral part of some enzymes; serve as activators of certain enzymatic reactions; and they are an essential part of the oxygen-carrying pigments. Mertz’s definition of ‘essential’ has been widely accepted. He stated that an element is considered essential when a deficient intake consistently results in suboptimal physiologic function that can be prevented or reversed by supplementation with physiologic levels of the element.
It is interesting that in many physiologic processes one mineral element may be substituted for another. For example, strontium or lead may replace calcium in the inorganic structure of bone. Rubidium may replace potassium in a potassium-deficient diet with the result that, even though the animals maintained on the diet die, the characteristic myocardial necrosis found in potassium deficiency will not occur. A thorough understanding of the normal processes of mineral metabolism and the effects of abnormal mineral metabolism is essential in pointing the way to the solution of many of the problems related to calcification of the teeth and jaws that constantly arise during the practice of dentistry.
Minerals
Although the literature concerned with calcium, phosphorus, and magnesium is voluminous, we still do not have a clear picture of the role of these elements in nutrition. The exact relation of magnesium to calcium and phosphorus metabolism is not known. For convenience, we shall discuss each element separately, trying to combine or integrate our knowledge whenever possible.
Calcium
Calcium is the fifth most abundant element in the body, and in crystalline form, with phosphorus, in a proteinaceous matrix, forms the major structural support of the body (bones). The total calcium in the body is 100–170 gm, about 99% of which is found in bones existing as carbonate or phosphate of calcium while about 0.5% is present in soft tissue and 0.1% in extracellular fluid. The normal serum calcium level is about 9–11 mg/dl. The calcium in plasma is of three types: ionized calcium, protein bound calcium, and complexed calcium. About 40% of the total calcium is in ionized form, which is also physiologically active form of calcium. The level of the blood calcium is largely controlled by the action of the parathyroid glands, which are stimulated by low serum calcium levels and inhibited by high serum calcium levels.
Requirements and Absorption
The Food and Nutrition Board of the National Academy of Sciences, National Research Council, recommends a daily dietary calcium intake of 360 mg for newborn infants and 800 mg for children and adults. Adolescents and pregnant and lactating women are advised to increase their daily dietary calcium intake by 50% to 1,200 mg. Calcium is taken in diet principally as calcium phosphate, carbonate, and tartrate. Unlike sodium and potassium that are readily absorbed, the absorption of calcium in man is an inefficient process. Only about one-third of the daily dietary intake of calcium is absorbed under normal conditions. About 40% of average daily dietary intake of calcium is absorbed from the gut, mainly from the duodenum and first half of jejunum against an electrical and concentration gradient.
Absorption
In well-balanced diets, the ratio of calcium to phosphorus is of little significance, but in less balanced diets, this ratio assumes considerable importance. Phytic acid, which is found in cereals, forms an insoluble calcium phytate with ingested calcium and renders it nonavailable. Since this substance constitutes over 50% of the phosphate of cereals and is hydrolyzed only to the extent of 30–60% in the alimentary tract, the phosphate-calcium ratio is thus upset, interfering with the normal absorption of calcium. Vitamin D increases absorption of calcium from the intestine. Under normal metabolic conditions for fat splitting and fat absorption, the ingestion of fat has been found to aid calcium absorption, but in conditions in which there is excessive fat excretion, such as in sprue or idiopathic steatorrhea, calcium is lost in the feces as calcium soaps.
Citrates, that may lower the pH of the intestinal tract, form calcium citrate which is relatively soluble. The addition of citrates to a rachitogenic diet seems to render the diet nonrachitogenic and also aid calcification. It has therefore been suggested that the lowering of the intestinal pH aids absorption of calcium and that the calcium citrate ion, though relatively soluble, aids the deposition of calcium in bones by raising the pH of the calcifying tissue or of the fluids surrounding the calcifying tissue. High protein diets have also been shown to increase calcium absorption, probably through the formation of soluble calcium compounds with the amino acids produced by the digestion of the protein.
Oxalic acid interferes with calcium absorption by forming an insoluble calcium oxalate. For example, spinach contains sufficient oxalic acid to render all its calcium nonavailable, with some oxalic acid to spare for other calcium which might be present in the diet. The presence of hypochlorhydria or achlorhydria also exerts an adverse influence upon calcium and phosphate absorption, since normal secretion of hydrochloric acid by the stomach is necessary for optimal absorption of calcium and phosphate.
Lactose or milk sugar increases calcium absorption in rats, presumably by increasing intestinal acidity. In humans, lactose increases the retention of calcium without materially affecting absorption.
Many factors affect the utilization of absorbed calcium and phosphorus. Obviously, conditions which produce profound disturbance of any vital metabolism may have an indirect influence upon the metabolism of these minerals. Those factors; however, which appear to have a well-defined effect on calcium and phosphorus metabolism, are the parathyroid hormone, vitamin D, thyroid, calcitonin, and the steroid hormones.
Excretion
Calcium is excreted in both the feces and the urine, with 80% of the total amount being excreted in the feces. Fecal calcium consists not only of unabsorbed calcium, but also of calcium which has been absorbed and re-excreted. Although the small intestine is the predominant site in which the calcium is re-excreted, all segments of the intestinal tract probably excrete some calcium. Unless there is excessive perspiration, the dermal losses do not exceed 50 mg/dl. The normal daily urinary calcium excretion in adults is less than 250 mg for women and 300 mg for men. The calcium in the urine is excreted mainly as calcium chloride and calcium phosphate. The renal threshold for calcium is approximately 7 mg/dl of serum calcium. The urinary excretion of calcium is increased by increased plasma calcium, deprivation of phosphate, excessive vitamin D, increased urinary excretion of sodium, immobilization, corticosteroid administration, increased dietary calcium, metabolic acidosis, hyperthyroidism, and idiopathic; whereas urinary excretion of calcium is decreased by decreased ultrafiltrable plasma calcium, decreased glomerular filtration rate, parathyroid hormone, decreased dietary calcium, increased dietary phosphate, increased calcium utilization as in growth, pregnancy, and lactation.
Function
Calcium plays a large role in the formation of bones and teeth, in the maintenance of skeletal structure, tooth structure, normal membrane permeability, normal heart rhythm and other neuromuscular excitability, in the coagulation of blood, muscle contraction, and as a secondary or tertiary messenger in hormone action. Variations of serum calcium ion concentration from the limited optimal range of 9–11 mg/dl have profound effects. A low concentration of calcium ions (about 8 mg/dl) produces hyperirritability and tetany with characteristic carpopedal spasm and at times laryngospasm and convulsions, while high concentration produces depressed nerve conductivity and muscle rigor.
Hypocalcemia is said to exist when serum calcium is less than 8.5 mg/dl. The commonest cause of hypocalcemia is hypoalbuminemia, closely followed by renal failure. The other common cause of hypocalcemia is surgically induced hypoparathyroidism. Hypercalcemia occurs when serum calcium levels exceed 11.0 mg/dl and the most common cause is primary hyperparathyroidism, malignancy, and endocrine causes such as acute adrenal insufficiency and renal failure.
Experimental calcium deficiency in rats leads to a derangement of blood coagulation and of the integrity of the capillaries. Internal hemorrhages and generalized paralysis of the young born of calcium-deficient females are common. In addition, stomach ulcers have been described in rats, and lens opacities (cataracts) have been described in rabbits deficient in calcium. Hyperplasia and hypertrophy of the parathyroid glands of rats maintained on calcium-deficient diets have also been observed. In adult animals maintained on low calcium diets, sterility and reduction in lactation are frequently found. There are no descriptions of the teeth of animals maintained on a low calcium diet.
Osteoporosis and Calcium Deficiency
The etiology of osteoporosis was once thought to be a lack of adequate bone matrix. But evidences indicate that it may be due to a longterm negative calcium balance. Skeletal mass in old age is proportional to skeletal mass at maturity, indicating that infant and childhood calcium intake may play a major role in the occurrence and severity of the disease in later years. Based on these findings, the treatment of osteoporosis has changed over the years. Androgen and estrogen therapies have been replaced by increased calcium intake and strontium and sodium fluoride ingestion. The role of strontium and fluoride in bone metabolism is not fully known, but they do act to sustain bone mass in elderly osteoporotic patients. Long-term metabolic balance studies indicate that in a majority of osteoporotic patients, calcium balance can be achieved with a high calcium intake. The importance of calcium, strontium, and sodium fluoride in the prevention and treatment of senile osteoporosis have been found encouraging.
Phosphorus
Total body phosphorus is approximately 500–800 gm, of which 85–90% is in the skeleton, leaving approximately 100 gm in soft tissues. There are multiple pools of phosphorus having different turnover rates; bones and teeth have the lowest rates. A major portion of phosphorus is incorporated into organic phosphorus compounds (phospholipids of cell membranes, nucleic acids, etc). The normal inorganic phosphate level of blood in adults ranges from 2–4 mg/dl, while in children its range is from 3–5 mg/dl. These blood levels are maintained by a balance of various factors, such as parathyroid hormone, phosphatase activity, and vitamin D. Phosphorus is; however, not as finely regulated as plasma calcium but is under some hormonal control via PTH and renal production of 1,25 (OH)2D3.
Requirements and Absorption
The suggested daily dietary intake of phosphorus ranges from 240 mg for infants to 800 mg for adults. As with calcium, adolescents, pregnant and lactating women are advised to increase their daily dietary phosphorus intake by 50% to 1,200 mg. 90% of daily dietary phosphate is absorbed. Absorption of phosphorus takes place in the small intestine in the form of soluble inorganic phosphate. Approximately 70% of food phosphorus is absorbed in the form of orthophosphate after intestinal phosphatase releases the food-bound phosphorus during the digestive process. An excess of calcium, iron, or aluminum may interfere with the absorption of phosphorus because of a tendency to form insoluble phosphates in the intestinal tract.
Excretion
Regulation of calcium and phosphorus is under the similar control mechanisms by kidney with respect to parathormone and vitamin D. Excretion of phosphorus occurs primarily in the urine. Phosphate uptake is sodium dependent, about 85% of filtered PO4 is reabsorbed by the proximal tubules. Phosphate reabsorption is increased when dietary intake is reduced by a parathormone-dependent mechanism. Almost two-thirds of the total phosphorus excreted is found in the urine as phosphates of various cations. Fecal phosphorus, which is usually composed of unabsorbed as well as re-excreted phosphate, is usually excreted as calcium phosphate.
Function
Although most of the body phosphorus is intimately associated with calcium in the metabolism of bones and teeth, a much higher proportion of phosphorus than of calcium is concerned in other vital processes. Phosphates form an intermediate stage in the metabolism of fats and carbohydrates by their function in phosphorylation. They are used in building the more permanent organic phosphates, including some catalysts, essential to the structure and function of cells. Phosphates are utilized in the formation of phosphoproteins, such as milk casein, and in the formation of the nerve phosphatides and the nucleoproteins of cells. They provide the energy-rich bonds in such compounds as adenosine triphosphate, which is important in muscle contraction, and they form part of such coenzymes as pyridoxal phosphate, which is necessary in decarboxylation and transamination of certain amino acids, such as tyrosine, tryptophan and arginine.
When young rats are placed on a low phosphorus diet, there is some retardation in growth. The only specific gross or microscopic alterations are found in the skeletal system, where severe rickets is present (Fig. 15-1). This finding appears after the rats have been on the experimental phosphorus-deficient diet for only one week.
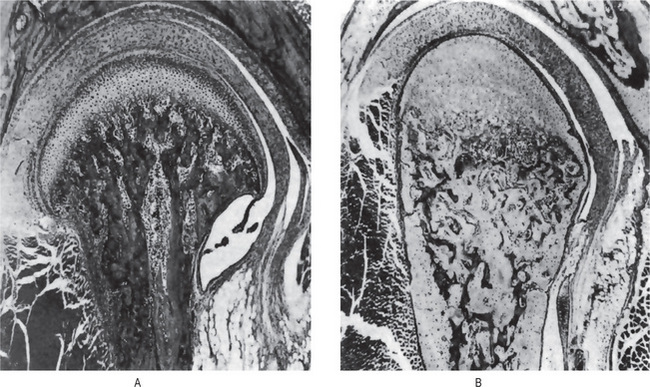
Figure 15-1 Phosphorus deficient diet.
(A) Sagittal section of mandibular joint of a normal rat 70 days of age. (B) Sagittal section of mandibular joint of rat 70 days old, which received a phosphorus-deficient diet since weaning at 21 days of age. Courtesy of Dr Herman Becks
Phosphate depletion in man is nonexistent under most dietary regimens. Long-term antacid use; however, will render phosphate unabsorbable. Lotz and coworkers have described such a condition, which is characterized by weakness, malaise, anorexia, and bone pain. Increased calciuria results in a negative calcium balance with bone demineralization. Rickets and osteomalacia are important dietary deficiency disorders of calcium, phosphorus, or vitamin D. The other causes of hypophosphatemia may be due to decreased intake (starvation, malabsorption, or vomiting) or increased cell uptake as in high dietary carbohydrate, liver disease, or increased excretion due to diuretics, hypomagnesemia, and increased parathormone. Hyperphosphatemia, on the other hand, is due to factitious hemolysis, increased intake of vitamin D, increased release from bone as in malignancy or decreased excretion.
Magnesium
Magnesium is the fourth most abundant and important cation in humans. It is extremely essential for life and is present as intracellular ion in all living cells and tissues. Magnesium appears to participate in practically every phosphorylating mechanism. In addition, this ion is necessary for the activity of certain enzymes, such as phosphatase and cocarboxylase.
Body Distribution
Although the concentration of magnesium in the intracellular fluids is not as great as the concentration of potassium, magnesium is widely distributed in the tissues of the animal body. The body of a 70 kg man contains approximately 25 mg of magnesium. Over half of this amount is found in the bones, and one quarter in the muscles. The remainder is distributed between liver, pancreas, erythrocytes, serum, and cerebrospinal fluid.
Requirements
The recommended daily dietary allowance for magnesium ranges from 50 mg for infants to 400 mg for teenage males. A daily increase of 150 mg is suggested during pregnancy and lactation. Like calcium, magnesium is ingested in inorganic and organic forms. It is also absorbed and excreted in the same manner as calcium. Absorption takes place primarily in the small bowel. Besides other factors, the size of magnesium load is important as absorption is doubled when normal dietary Mg requirement is doubled and vice versa. Since there is a common transport mechanism from intestinal tract for both Ca and Mg, decreased absorption occurs in the presence of excess Ca. The absorption is also affected in hurried bowel and damaged mucosal states. Vitamin D, parathormone, growth hormone, high protein intake, and neomycin therapy increase absorption. High calcium diets raise the requirement for magnesium.
Excretion
Almost 60% of the excreted magnesium is fecal, the rest being urinary; 0.75 mEq and 3–7 mEq of magnesium is lost daily in sweat and urine, respectively.
Hypermagnesemia is rare because of the renal capacity to excrete excess ion. The administration of magnesiumcontaining antacids to patients with renal insufficiency has resulted in central nervous system depression. Somjen and coworkers have also reported severe voluntary muscle paralysis with hypermagnesemia. Controlled human hypomagnesemia was studied by Shils, who noted a concurrent hypocalcemia and hypokalemia despite normal dietary calcium and phosphorus intake. Clinically the patients exhibited personality change, anorexia, nausea and vomiting, and carpopedal spasms.
High magnesium intake will produce rickets in growing animals, especially if the phosphorus and calcium intake is relatively low. The normal serum magnesium level is 1–3 mg/dl. When the level reaches 5 mg/dl, mild sedative or hypnotic effects may occur. Profound coma and even death may result when the serum level reaches 18–21 mg. A distinct but not fully understood relationship exists between magnesium, calcium, parathyroid hormone, and bone metabolism. Buckle and coworkers have shown that hypomagnesemia and hypocalcemia have identical effects on the parathyroid glands, i.e. increased parathyroid hormone production. An apparent contradiction exists in that despite elevated hormone levels, many affected individuals exhibit hypocalcemia. Studies have indicated that the parathyroid hormone produced is defective, although some investigators have described hypomagnesemic patients who were refractory to exogenous parathyroid extract, suggestive of a bone defect rather than a glandular abnormality.
Functions
Magnesium is involved as a cofactor and as an activator to a wide spectrum of enzymatic actions. It is essential for peptidases, ribonucleases, glycolytic enzymes and cocarboxlylation reactions. Magnesium exerts an effect on neuromuscular irritability similar to that of calcium ions. High levels depress nerve conduction and low levels may produce tetany (hypomagnesemic tetany). As constituent of bones and teeth, about 70% of body magnesium is present as apatites in bones, enamel, and dentin.
Deficiency
In humans, ‘overt’ magnesium deficiency occurs rarely. In experimental animals, magnesium deficiency leads to disturbances in the neuromuscular and vascular systems as well as to changes in the teeth, liver, and kidneys. The effects of magnesium-deficient diets on the teeth and their supporting structures have been thoroughly described by Becks and Furuta and by Klein and his associates. Diets containing only 13 ppm of magnesium caused the ameloblasts from the labial side near the apex of the growing incisor tooth in rats to show various stages of localized degeneration with subsequent formation of enamel hypoplasia. The hypoplastic areas increased in size and number with the duration of the experiment, although the changes were noted in all animals after 41 days.
The syndrome of human magnesium-deficiency tetany was first described by Vallee and his associates in 1960. The condition is virtually identical with that of hypocalcemic tetany from which it can be differentiated only by chemical means. Clinically, patients with this deficiency exhibit a semicoma; severe neuromuscular hyperirritability, including carpopedal spasm and a positive Chvostek’s sign; athetoid movements; marked susceptibility to auditory, visual, and mechanical stimuli; a decreased serum magnesium; and a normal serum calcium concentration. Precipitating factors are severe dietary inadequacy of magnesium or excessive losses of this ion due to vomiting, intestinal malabsorption, and the administration of large amounts of magnesium free parenteral fluids which induce a large urine volume. The tetany appears when the serum magnesium level is depressed below 1.30 mEq per liter. Treatment by the intramuscular injection of magnesium sulfate is followed by a prompt rise in serum magnesium and a concomitant disappearance of the tetany and convulsions. Discontinuance of the therapy in the presence of precipitating factors results in a rapid reappearance of tetany.
Raised values of magnesium or hypermagnesemia have been reported in uncontrolled diabetes mellitus, adrenocortical insufficiency, hypothyroidism, advanced renal failure, and acute renal failure.
Pathologic Calcification
Pathologic calcification implies the abnormal deposition of calcium salts together with smaller amounts of iron, magnesium, and other mineral salts. Pathologic calcification is commonly classified as:
It is not always possible to make a clear distinction between these various forms.
Dystrophic Calcification
In the dystrophic form of calcification, calcium salts are deposited in dead or degenerating tissues. This is the most frequent type of pathologic calcification and is found in a wide variety of tissues. Areas of tuberculous necrosis, blood vessels in arteriosclerosis, scars and areas of fatty degeneration are commonly recognized as sites of dystrophic calcification by the general pathologist. This type of calcification is not dependent upon an increase in the amount of circulating blood calcium, but appears to be related to a change in the local condition of the tissues. A local alkalinity in comparison with adjacent undamaged tissues appears to be an important factor in initiating the precipitation of calcium in degenerating or nonvital tissues.
In the mouth, areas of dystrophic calcification may frequently be found in the gingiva, tongue or cheek. Such areas are also found in the benign fibromas of the mouth and adjacent structures (Fig. 15-2). One of the most common intraoral dystrophic calcifications is found in the pulp of teeth, and this has been discussed in Chapter 13 on Regressive Alterations of the Teeth. Boyle described the pulp calcifications as calcific degeneration of the pulp tissue. They are usually found in the teeth of older persons, although they also may be seen in young people. They may occur in the wall of blood vessels or in the perineural connective tissue of the pulp, or they may be rather diffusely scattered both in the pulp chamber and in the root canal. They appear as fine fibrillar calcifications which may coalesce to form large masses of calcific material.
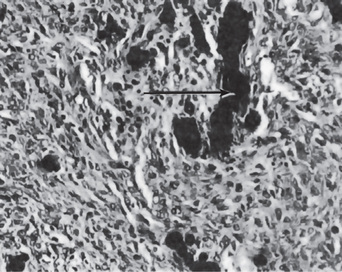
Hill classified calcific degenerations of the pulp into two types. The first, a nodular type, is a result of calcification of hyalinized connective tissue. Such calcification is usually perivascular or perineural and is often associated with increased fibrosis. The calcium deposits are most frequently found in the coronal portion of the pulp chamber and increase in size by accretion and deposition of calcium along the collagenous fibrils. The second type of calcification of the pulp is that found in and around necrotic cells and corpora amylacea. It occurs in a multicentric manner and is most frequently found in the radicular portion of the pulp canal. This type of calcification always shows a nidus in the center and increases in size by concrescence which is obvious on histologic examination.
Many of the deposits of calcareous material are found in degenerative processes of the pulp as well as in pulps which are the seat of inflammatory processes. In these cases the calcifications probably have the same relation to body health as calcifications within arterial walls in arteriosclerosis. This type of calcification probably does not cause pulpal inflammation, and there is no justification for considering it a source of dental infection. The other types of pulp stones or pulp nodules (denticles) are discussed in Chapter 13 on Regressive Alterations of the Teeth.
Metastatic Calcification
In metastatic calcification, calcium salts are precipitated in previously undamaged tissues. This precipitation is due to an excess of blood calcium and occurs particularly in such diseases as hyperparathyroidism, which depletes the bone calcium and causes a high level of blood calcium. Metastatic calcifications also occur in hypervitaminosis D. In this type of calcification, the deposits of calcium occur mainly in the kidneys, lungs, gastric mucosa, and media of blood vessels. Since any degenerating or necrotic tissue will also be calcified when there is an increase in blood calcium levels, the differentiation between metastatic calcification and dystrophic calcification becomes extremely difficult.
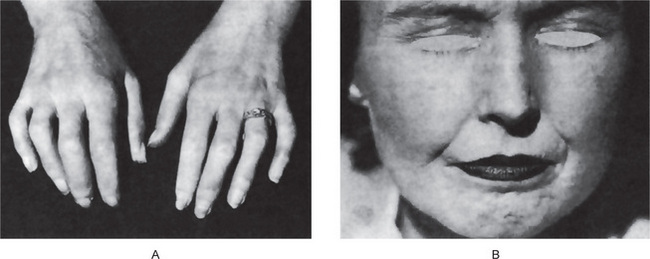
Calcinosis
Calcinosis is the presence of calcifications in or under the skin. There are two forms of calcinosis: calcinosis circumscripta, which, as the name suggests, is a circumscribed form, and calcinosis universalis, which is a generalized form. Calcinosis universalis is often associated with scleroderma and sometimes dermatomyositis. These different forms of calcinosis have been discussed by Johnson (Fig. 15-3).
Sodium
The sodium found in the body is mainly associated with chloride and as NaCl and NaHCO3. The sodium ion content of the normal (70 kg) adult male ranges from 83–97 gm. Over one-third of this amount is in the skeleton, of which 65–75% is unexchangeable. Most of the remaining sodium is extracellular and accounts for 90% of the basic ions of both extracellular fluid and plasma. Enamel ash contains about 0.3%. The question of whether the sodium of the dental tissues is associated with the inorganic or organic fractions or with small quantities of tissue fluid present in the teeth remains unanswered.
Requirements and Excretion
The minimal requirement of salt is thought to be about 0.5 gm. The lower limit of salt intake is not really known. The estimate of 0.5 gm was reached based on the salt intake of breastfed infants. Breast milk contains 0.4 gm NaCl per liter. Interestingly, cow’s milk contains 1.7 gm NaCl per liter. The maximal intake without accumulating edema fluid is 35–40 gm per day. In the United States, the average dietary intake of sodium is 10–15 gm per day. The normal blood level is 160 mg/dl of whole blood, or 340 mg/dl of plasma (147.8 mEq/liter of plasma).
Under conditions of profuse sweating, 1 gm of salt should be ingested for each liter of water in excess of 4 liters. Sweat may contain 2–3 gm of salt per liter in hot environments if the person has not been acclimatized; after acclimatization, 0.5 gm of salt per liter is found.
The kidney is the principal organ for the excretion of water and salt. Abnormal losses of either sodium or chloride must be balanced by the kidney. When the diet is low in salt, or when there is profuse sweating, practically no sodium or chloride is found in the urine. The regulatory mechanism controlling the reabsorption of sodium and chloride by the renal tubules is controlled in part by the adrenal glands. An inadequate intake or excessive loss of sodium stimulates the adrenal cortex to secrete aldosterone, a steroid hormone which acts directly on the renal tubules to increase reabsorption and to conserve sodium. The adrenal glands also control, to a smaller degree, the salt content of sweat.
Function
Sodium ions play an important role in the maintenance of the acid-base equilibrium as well as of osmotic pressure, which depends largely on total base. In fact, the bulk of basic metabolic energy expenditure is concerned exclusively with the maintenance of proper intracellular sodium concentration, i.e. the sodium pump. When tissues are depleted of potassium, sodium may substitute for it, regulating the contraction of the heart. Sodium also helps in maintaining the neuromuscular excitability, viscosity of blood, and fluid balance.
Deoxycorticosterone, cortisone, and hydrocortisone act to increase the tubular reabsorption of glomerular filtrate of sodium and to decrease tubular reabsorption of filtrate potassium. It should be realized, therefore, that care must be taken in using these drugs to avoid edema resulting from excess sodium retention. Potassium loss must also be anticipated and provided for by increased potassium ingestion. For every 24 hours approximately 25,000 mmol of sodium are filtered by the kidneys. However, due to tubular reabsorption, less than 1% of this sodium appears in the urine. Two types of clinical conditions exist in sodium metabolism—hypernatremia and hyponatremia. Specific conditions where hypernatremia occurs are simple dehydration, diabetes insipidus, excess sodium intake, and steroid therapy. Hyponatremia occurs as a result of diuretic medication, excessive sweating, kidney diseases, congestive heart failure, and in cases of increased gastrointestinal loss as in case of diarrhea.
Many of the features of Addison’s disease are referable to salt depletion. In this disease extracellular water and sodium are rapidly excreted by the kidneys, resulting in a fall of plasma sodium concentration and an increase in the serum potassium level. Water migrates intercellularly, and the result is a deprivation of both sodium and water. Studies with radioactive sodium (Na22) have shown that the sodium of bone, which constitutes about 30% of the body sodium, is located on the surface of the apatite crystal lattice.
Deficiency
Sodium deficiency in man probably never occurs in an uncomplicated form, but it may be present as a sodium and chloride deficiency. When diets very low in salt are used for long periods of time, gradual weakness, excessive fatigue, lassitude, apathy, anorexia, a sense of exhaustion, nausea, muscle cramps, and peripheral vascular collapse may ensue.
Potassium
Potassium is the major intracellular cation. It is widely distributed in the body fluid (whole blood and plasma) and tissues such as nerve, muscle as well as in cells. Most of the potassium of the body is intracellular. It is the predominant base in the cells. Radioactive potassium (K42) studies have indicated that there is a constant exchange of potassium between its intracellular and extracellular phases, although it is clear from the studies of Peters and Van Slyke that potassium is prevented from diffusing freely out of cells by a membrane or by some other restraining factor or factors in the cellular or extracellular fluids.
Requirements and Excretion
An average amount of 4 gm of potassium is present in the diet. The requirement for potassium is greatest during periods of rapid growth. As soon as potassium is absorbed, it enters the cells. About 90% of the excreted potassium is eliminated in the urine. The amount of potassium excretion increases, where there is an excessive dietary intake of sodium. The average normal human body contains 3.6 moles of potassium. Urinary excretion is influenced by aldosterone, which controls the active tubular secretion of potassium. The normal blood plasma level is about 4 mEq/liter of plasma. Potassium is also excreted in the gastrointestinal tract, saliva, and gastric, bile, pancreatic, and intestinal juices.
Function
The major functions of potassium and sodium are carried out in coordination with each other and are common. It influences the muscular activity, is involved in the acidbase balance, has a role in cardiac function, and is involved in neuromuscular irritability and the nerve conduction process.
Deficiency
Primary dietary deficiency of potassium has not been observed, but depletion secondary to some pathologic condition has been encountered. It may occur in gastrointestinal disorders, in which there may be a loss of potassium through diarrhea and vomiting. It may also occur in general malnutritional states. It develops as a result of the administration of diuretics or ion-exchange resins. Excessive doses of cortisone or hydrocortisone may result in potassium depletion, and potassium deficiency is common in diabetic acidosis during insulin therapy.
Death in potassium deficiency may result from cardiac or respiratory failure or from paralytic ileus. The signs of potassium deficiency are primarily those of decreased muscular irritability, muscular weakness, reduced or absent reflexes, mental confusion, paralysis, disturbances in conductivity and contractility of heart muscle, and alterations in the gastrointestinal tract.
Hyperkalemia
Hyperkalemia, which may result from extensive tissue breakdown, adrenal insufficiency, advanced dehydration or administration of excessive amounts of potassium, will produce such signs and symptoms as mental confusion, numbness and tingling of the extremities, pallor, cold skin, weakness, disturbances in cardiac rhythm, and peripheral collapse, as noted by Darrow. Clinically, most cases of hyperkalemia are due to kidney failure with decreased excretion of potassium or due to the sudden release of potassium from the intracellular compartment which may happen in a variety of diseases. The effects of potassium deficiency or of excess potassium on the oral structures per se have not been reported.
Chlorine
The metabolism of chlorine, together with that of sodium and potassium, is closely related to the water balance and the acid-base equilibrium of the body. The average intake is about 6–9 gm per day. Chlorine is taken in diet as sodium chloride. The absorption of chlorine takes place in small intestines. The mechanism of chloride uptake is unclear, but it appears to depend on an exchange process with the bicarbonate, whilst the accompanying sodium exchange for a hydrogen ion. Quantitatively, chlorine and sodium are the most important mineral constituents of the extracellular fluids. Chlorine is excreted primarily through the kidney. It is one of the so-called threshold substances which are reabsorbed into the circulation after passing through the glomeruli to maintain normal body fluid concentrations. The normal blood plasma concentration of chlorine is 550–650 mg/dl as sodium chloride. Chlorine is important in the production of HCl in the gastric juice and is also important in chloride shift.
Chloride activates salivary amylase. Little else is known of the function of chlorine in the animal organism. Rats placed on synthetic diet low in chlorides failed to grow normally. The only histologic lesions reported were in the kidney. The role of chloride per se in sodium deficiency in man is not clear. Large quantities of chloride ions may be lost in pyloric obstruction with gastric tetany, leading to signs of hyperexcitability and convulsions. These may be prevented by the administration of chloride ions. No oral manifestations of chloride deficiency have been reported.
Trace Elements
A large number of elements have been shown to occur in a wide range of animal tissues and fluids in such minute quantities that they are usually described as ‘traces.’ Demonstration of a physiologic role for many of these elements has lagged far behind their mere detection in the living organism. It has been shown that both barium and strontium are essential for growth and especially for calcification of the bones and teeth of rats and guinea pigs. Mertz has reported silicon, vanadium, nickel, and arsenic to be essential in various animal species. However, no imbalances in humans have been reported.
Iodine
Iodine in small amounts is widely distributed in living matter. Sea foods are the best natural source and useful amounts may be present in vegetables and milk. The food color erythrosine is very rich in iodine. Normal whole blood contains an average of 8–12 μg/dl (range, 3–30 μg); protein-bound iodine varies from 3–8 μg/dl. The level of protein-bound iodine is increased during pregnancy and in hyperthyroidism and decreased in hypothyroidism. Iodine is essential for the formation of thyroid hormone. No other function for iodine in the nutrition of higher animals is known.
Iodine deficiency in man results in goiter. Iodine deficiency in experimental animals does not lead to colloid goiter. On the other hand, addition of iodine to the salt or water supply of endemic goiter areas has been successful in acting as a prophylactic in colloid goiter. About one-third of the total body iodine is found in the thyroid. The precise mechanism of conversion of thyroid-concentrated iodine to colloid is unclear. However, thyroxin formation is intimately related to tyrosine metabolism.
The effects of the thyroid gland on oral structures will be considered in the section dealing with the endocrine glands. The ovaries also contain a high concentration of iodine.
Copper
Iron and copper have been inextricably involved in the development of all forms of life since the earth’s atmosphere dramatically changed from a reducing to an oxidizing environment. Copper deficiency in experimental animals leads to anemia. Adult humans contain 100–150 mg of copper, out of which approximately 65 mg is found in muscles, 23 mg in bones, and 18 mg in liver. Fetal liver contains approximately 10 times more copper than adult liver.
Requirement and Absorption
Copper requirements for infants and children are 0.05 mg/kg body weight per day, whereas adult requirement is approximately 2.5 mg/day. Ordinary diets consumed daily contain about 2.5–5.0 mg of copper. Acute copper deficiency in human beings has not been demonstrated.
The value of copper supplements, with and without iron, in the treatment of anemias of infancy and childhood and of secondary anemias of adults has been extensively studied. Copper is necessary for normal erythropoiesis as well as for iron absorption. Copper deficiency produces microcytic hypochromic anemia, due to impairment of erythropoiesis and decrease in erythrocyte survival time, which cannot be corrected by administration of iron. Iron absorption is mediated by ceruloplasmin, which acts as a ferroxidase. Other metalloenzymes which require copper are cytochrome c oxidase, superoxide dismutase, tyrosinase, and lysyl oxidase. Human copper deficiency diseases of importance are hepatolenticular degeneration (Wilson’s disease) and Menkes’ syndrome (steelyor kinky-hair syndrome).
Iron
Iron is one of the most essential trace elements in the body. In spite of the fact that iron is the fourth most abundant element in the earth’s crust, iron deficiency is one of the most important prevalent nutritional deficiencies in India. The total iron content in a human of 70 kg body weight varies approximately from 2.3–3.8 gm. The average iron content of adult males is about 3.8 gm and of females about 2.3 gm. There are two broad categories that are used to describe iron in the body. They are essential (or functional) iron and storage iron. Essential iron is involved in the normal metabolism of cells whereas storage iron is present in two major compounds— ferritin and hemosiderin.
Requirement and Absorption
The requirement of iron varies according to age, gender, weight, and state of health. An adult male requires approximately 10 mg/day and adult female 20 mg/day. Pregnancy and lactation demand more: pregnant women require 10 mg/day and lactating mothers 25–30 mg/day. Children require 10–15 mg/day. Iron is absorbed in the upper portion of the duodenum, either as ferrous or as ferric salts, depending on the species studied. Absorption depends on the amount of the element that the organism has stored. If the tissues are depleted, iron is absorbed rapidly; if sufficient quantities are present, absorption is slight. Since little excretion of iron takes place either by the alimentary canal or by the kidneys, this element has been called a ‘oneway substance’. Normally, the loss of iron from the body of a man is limited to 1 mg/day.
Few studies have been reported on the histopathologic changes occurring in the tissues of human beings or experimental animals with iron deficiency anemias. Iron deficiency in the human being, particularly in women and children, however, is more common than has been realized. Changes in the resulting anemia include formation of an esophageal web in the Plummer-Vinson syndrome, spooning of the nails (koilonychia), normoblastic arrest in the bone marrow and microcytosis, anisocytosis, and hypochromia of the erythrocytes in the peripheral blood. Sore tongue, similar to that found in nicotinic acid and riboflavin deficiencies, has been described in the iron deficiency anemias. These anemias respond well to iron therapy. It is imperative to determine iron levels in all patients with anemias, since there are disorders such as thalassemias that may be present and misdiagnosed as iron deficiency.
Iron overload can occur in a number of conditions. Idiopathic hemochromatosis results in excessive iron absorption and is characterized by micronodular cirrhosis with marked brown pigmentation, diabetes mellitus, and skin pigmentation called ‘Bronze diabetes’. Hemoglobinopathies such as sideroblastic anemia and thalassemia can also cause iron overload. Bantu siderosis, a form of iron overload resulting from ingestion of home made beer fermented in iron pots, has been extensively described.
Zinc
The role of zinc as an essential nutrient is known for more than 100 years. Zinc is obtained from liver, milk and dairy products, eggs, unmilled cereals, legumes, pulses, oil seeds, and leafy vegetables. An average man has about 1.4–2.3 gm of zinc in the body. The zinc is distributed in highest concentration in skin and prostate where it is about 70–80 mg/100 gm followed by bone and teeth where zinc concentration varies between 10–15 mg/100 gm. The concentration of zinc in enamel and dentin is about 0.02%, which is higher than in many other hard tissues of the body. Bone, nails, and hair have a slightly lower concentrations.
Only a small percentage of dietary zinc is absorbed from duodenum and ileum. A low molecular weight zinc binding factor secreted by the pancreas, forms complex with zinc and helps in its absorption. High amounts of dietary calcium and phosphates interfere with zinc absorption. In a normal healthy adult, approximately 9.0 mg of zinc is excreted through feces and urine and only about 0.5 mg retained in the body. Adult men and women require about 15–20 mg as the recommended daily dose is about 0.3 mg/kg body weight.
Among many functions of zinc, the most important is its role in enzyme action as it forms an integral part of several enzymes in the body. Important zinc containing enzymes are superoxide dismutase, carbonic anhydrase, and leucine aminopeptidase. Zn++ has been claimed to stimulate the release of vitamin A from the liver into the blood and thus increases its plasma level and its utilization in rhodopsin synthesis. Protamine zinc insulin and globin zinc insulin contain Zn++ for its functioning. Zinc content of pancreas also has been found to diminish in diabetes mellitus. Zinc is also necessary for the healing of wounds as zinc has been found to accumulate in granulation tissues and zinc deficiency delays wound healing.
In 1961, Prasad and his associates reported a symptom complex of dwarfism and hypogonadism in male Iranians which stemmed from a deficiency of zinc in the diet. This deficiency was thought to occur from zinc binding with phytates present in bread. Subsequent studies in Egypt by Prasad and his coworkers confirmed this impression. The zinc-deficient subjects appeared much younger than their stated age, lacked facial, axillary and pubic hair, had atrophic testes and small external genitalia and were retarded in bone age. The zinc content of the plasma, red blood cells and hair was consistently lower than in normal ethnically identical controls. Radioisotope studies demonstrated a significantly increased plasma zinc turnover and a decreased excretion of Zn65 in the urine and stools of the dwarfs, indicative of zinc retention and conservation. A low plasma level of alkaline phosphatase, a zinc-containing enzyme, was also found in these patients.
Acrodermatitis enteropathica, a specific multiorgan disorder resulting from zinc deficiency, has been described. It is an autosomal recessive disorder in which the primary defect is in zinc absorption. Its symptoms include diarrhea and a wide range of mucocutaneous problems including vesicles, eczematoid and hyperkeratotic plaques, alopecia, stomatitis, and glossitis. In leukemias, zinc content is almost reduced to 10% of the normal amount. Zinc in leukocytes probably has immunologic function. Serum zinc levels are decreased in cirrhosis of liver and lower plasma levels of zinc has been noted in acute viral hepatitis which returns to normal level with recovery. Zinc deficiency in humans results in a number of disorders involving taste, keratogenesis, bone growth, wound healing, and reproduction.
Manganese
Manganese is an essential oligo element widely distributed in the crust of the earth. The total amount of manganese distributed in our body is in the range of 10–18 mg and is found in highest concentration in the kidney and liver. Manganese is obtained in diet principally from cereals, vegetables, fruits, nuts and tea. Blood manganese is usually about 4–20 μg/100 ml. They are mainly in RBCs in combination with several porphyrins and are transported in the plasma in combination with a β1 globulin called transmanganin.
Manganese acts as a ‘cofactor’ or as an activator of many enzymes like arginase, isocitrate dehydrogenase (ICD), lipoprotein lipase, cholinesterase, and many others. Manganese may be associated with mitochondrial respiratory chain enzymes and act as a cofactor of all hydrolases and decarboxylases. Manganese has also been shown to have a role in animal reproduction and plays a part in the synthesis of mucopolysaccharides in the cartilaginous matrices of long bones. Deficiencies in animals produce alterations of bones, ataxia, and infertility.
Manganese excess in primates produces damage to the extrapyramidal system, with depletion of dopamine and serotonin in the caudate nucleus. In man, there is a transient period of psychosis followed by irreversible Parkinsonism. Increased susceptibility to manganese poisoning has been reported in anemic adults and in newborn and premature children, which appears to be due to increased intestinal absorption of manganese.
Cobalt
Cobalt is an important constituent and an integral part of vitamin B12. The main source of cobalt is from animal origin and the normal diet contains about 5–8 μg of cobalt. This is more than the recommended daily allowance of 1–3 μg of vitamin B12 containing about 0.0045–0.09 μg of cobalt. It has a principal role in the formation of cobamide enzyme (adenosyl coenzyme) and is also required to maintain normal bone marrow function and maturation of RBCs.
A deficiency of cobalt results in decreased vitamin B12 supply resulting in nutritional macrocytic anemia. Excessive availability of cobalt results in polycythemia. The polycythemic effect is a result of inhibition of certain respiratory enzymes namely cytochrome oxidase, and succinate dehydrogenase leading to relative anoxia. The element does not seem to have a storage depot in the animal organism. Vitamin B12 contains about 4.5% cobalt. If this is the only cobalt required by man, the amount must be infinitesimal, since 1–2 μg of vitamin B by injection each day will adequately treat pernicious anemia.
Chromium
The nutritional importance of trace quantities of Cr3+ for mammals has been conclusively established since this element was first identified as necessary dietary ingredient for normal glucose metabolism in rats in 1959 by Shwartz and Mertz. The level of chromium in normal healthy adult is 6–20 μg/100 ml. Significant amounts of chromium is obtained in the diet by cooking foods in stainless steel utensil. Mertz has suggested that chromium may facilitate insulin binding to cell membranes via a ‘chromium bridge’. This appears to be accomplished by the participation of Cr3+ in a ternary complex with insulin and insulin receptor sites that expedites the initial attachment of insulin to these sites. Inorganic Cr3+ is poorly absorbed and has only limited biological activity compared with naturally occurring organic chromium complex that has been termed the glucose tolerance factor (GTF). While it may potentiate insulin action, chromium is not thought to be a hypoglycemic agent per se.
Chromium deficiency has been described in cases of malnutrition and total parenteral alimentation. The total body content of chromium is less than 6 mg. It appears to have a role in carbohydrate and lipid metabolism. Results of supplementation studies using physiological quantities of Cr3+ indicate that chromium depletion is one etiologic factor that has to be considered in a variety of disorders of carbohydrate metabolism, ranging from infants suffering from protein calorie malnutrition to elderly people with impaired glucose intolerance. Chromium metabolism appears to be significantly disturbed in diabetes mellitus, and may be one etiologic factor in some gestational and maturity onset diabetes mellitus.
Selenium
The importance of trace element selenium was first reported when it was described to prevent liver cell necrosis. Current evidence suggests selenium to be an essential trace element for all species including humans. Biological forms of selenium occur as selenium analogs of selenium containing amino acids namely selenomethionine, selenocysteine, and selenocystine, at a mean concentration of less than 0.2 μg/gm. It is found in higher concentrations in the liver, nails, and kidneys. Major source of selenium for food is the plant material; and selenium is absorbed mainly from the duodenum, particularly in the form of methionine analog. The total body selenium has been estimated to be approximately 4–10 mg. Selenium levels in the blood and tissues are very much influenced by dietary selenium. Selenium concentration in the blood is about 0.22 μg/ml. In selenium deficient areas of China, blood levels may be as low as 0.009 μg/ml in selenium concentration.
The metabolic role of selenium is played by the prosthetic group of selenium enzyme glutathione peroxidase which is present in cytoplasm and mitochondria. This results in the reduction of hydroperoxide, thus functioning as part of the multicomponent antioxidant defense system within the cell. It is supplementary to vitamin E and acts as the primary antioxidant by scavenging reactive oxygen species and free radical intermediates of polyunsaturated lipid peroxidation. Selenium has a sparing effect on vitamin E and reduces the vitamin E requirement; conversely vitamin E appears to reduce the selenium requirements by preventing loss of selenium from the body or by maintaining it in an active form. Rotruck and coworkers have reported that glutathione peroxidase is a selenoenzyme that is responsible for eliminating potentially harmful peroxides and free radicals. Burk and coworkers have described selenium deficiency in humans with protein energy malnutrition.
Specific features of selenium deficiency include liver cell necrosis, exudative diathesis, pancreatic degeneration, muscular dystrophies, and myopathies. Numerous reports of selenium toxicity in humans are also available. Clinical manifestations include chronic dermatitis, loss of hair, brittle nails, and an early indication of selenium toxicity is a garlicky breath caused by exhalation of dimethyl selenide. The likely cause is occupational exposure in electronics, glass, and paint industries.
Fluoride
Fluoride is one of the most interesting trace elements and man’s intake of fluoride comes from the food and water ingested every day. Tea, salmon, sardine, and mackerel are among other sources of fluoride in addition to it being derived from water. Fluoride is essential in human nutrition and one part of fluorine in one million parts of drinking water (1 ppm) seems to serve the daily requirement of fluorine in human adults and children. Several studies from India indicate that many foods from the continent contain appreciably higher concentrations of fluoride than are found in western foods. For example, samples of rice contained 0.4–14 ppm fluoride, table salt contained 0.88–17.6 ppm fluoride, and various spices contained 0.9–14.4 ppm fluoride.
If 1 ppm of fluoride is added to the drinking water, about 1–2 mg of fluoride will be added to the diet daily. Daily dietary fluoride should not exceed 3 mg as it is a toxic element. Balance studies in man have shown that when the quantities of fluoride ingested do not exceed 4–5 mg daily, little is retained by the body. This finding indicates the safety of the preventive dentistry programs based on the addition of fluoride to drinking water in concentrations of approximately 1 ppm. Dietary fluorides are absorbed by diffusion from the intestine and approximately 20 μg of fluoride is present in blood mostly in the ionized form. Fluorides are mainly excreted in urine. One must remember; however, that the rates of absorption and excretion as well as the rate of retention are related to the nature of the diet; e.g. intake of calcium above certain minimal levels will reduce the absorption of dietary fluoride.
Reports of the effect on animals of diets low in fluoride have been conflicting. With the exception of McClendon’s work, there have been no published findings which indicate that fluoride is essential to animal growth, development, and reproduction. McClendon, using hydroponic techniques for preparing fluoride free foods, showed that a few rats raised on such foods evidenced severe caries that they were unable to eat and eventually starved. It is a well known fact that fluorine is present in the human tooth in trace quantities and helps in tooth development and hardening of surface enamel, thereby resisting acid dissolution. Cariostatic effects of fluorine are due to its entry into the apatite salts of dental enamel. Fluorine is also present in human bones in trace amounts. Catalytic amounts of fluorine are required for the conversion of the phosphates of calcium to apatite salts of bones and teeth, which is the basis of the role of fluorine in teeth and bone development.
The effects of fluoride as a prophylactic in dental caries are reviewed in Chapter 9 on Dental Caries. We should mention here; however, some of the work done on toxic fluorosis. Although fluoride normally accumulates slowly in bones as the person ages, it accumulates rapidly if ingested in abnormally high quantities.
Excess of fluoride in drinking water or diet is harmful and is considered to be the main cause of the crippling disease known as fluorosis. Chronic fluoride intoxication, such as that described in cryolite workers in Denmark, is characterized by widespread calcification of tendons and muscle sheaths, by extensive arthritic changes in the spine, producing rigidity, and by osteosclerosis of the bones. These workers probably inhaled 20–80 mg F/daily for 10–20 years. The afflicted individuals could not perform simple daily tasks. Hypermineralization due to osteofluorosis has been identified in workmen employed in various industries with fluoride exposure and also in residents of India associated with excessive fluoride concentrations in the drinking water. The progressive development of crippling fluorosis initially results in the osteosclerosis of pelvic vertebra, later in other bones, and finally in all bones with ligamentous calcification and exostoses. The mechanism of osteofluorosis is not well understood; but fluoride in certain doses stimulates osteoblasts and alters osteoclastic and osteoblastic activity differentially.
Due to increased fluoride levels, collagen synthesis is found to be adversely affected due to reduced proline uptake. The formation of deficient collagen fibers with abnormal biochemical sites provides an impulsion for pathological calcification which occurs during fluoride intoxication.
Disturbances in Protein Metabolism
Proteins are complex biologic compounds of high molecular weight containing nitrogen, hydrogen, oxygen, carbon, and small amounts of sulfur. As the third principal group of organic compounds, they are much more complex in structure and have a larger range of functions than carbohydrates or lipids. All living tissues, whether plant or animal, contain proteins. The fundamental difference between protein metabolism of plants and that of animals is the ability of plants to synthesize proteins from the nitrogen and sulfur of soil and from the carbon, oxygen, and hydrogen of air. Animals must ingest, breakdown, absorb, and rearrange dietary proteins to form tissue proteins. The chemical process of digestion, which is essentially hydrolytic, is common to all heterotrophic organisms. Substances of high molecular weight—proteins, nucleic acids, and carbohydrates are hydrolyzed to yield smaller molecules which are absorbed and assimilated. A normal adult has about 12–18% protein. Nitrogen balance studies are used to determine the lowest protein intake that will support homeostasis.
Protein Requirements
The accepted figure of 1 gm of protein for each kilogram of body weight is designed to give a factor of safety to cover individual differences in requirement. Protein is required in increased quantity in the last half of pregnancy and during lactation, and in even greater amounts in infancy, childhood, and adolescence. Proteins constitute the most important group of foodstuffs. In addition to contributing to cells and intercellular materials, proteins and their constituent amino acids are of importance in the formation of hormones, enzymes, plasma proteins, antibodies, and numerous other physiologically active substances.
Complete proteins contain sufficient amounts of the essential amino acids for normal metabolic reactions and these are usually found in foods of animal origin. Incomplete proteins are those that have insufficient quantities of one or more essential amino acids and among few are the corn protein which is low in lysine and legume protein that is low in methionine. Complementary proteins are proteins that, when ingested singly, are incomplete but, when combined, provide sufficient essential amino acids.
Comparatively little is known of the processes by which digested protein is recombined to form body proteins. Buildup of body protein is particularly active during growth, late pregnancy, and lactation. There is apparently a constant flux of tissue breakdown and tissue formation, producing a dynamic equilibrium. Proteins have an important bearing on the pre-eruptive and post-eruptive effects on teeth. They form an integral component of cells necessary for the normal development of the tooth and specifically for the formation of the matrix of hard tissues of teeth. The chemical nature of protein foods can neutralize the acids produced by oral bacteria.
Protein Energy (Calorie) Malnutrition
Protein energy malnutrition (PEM) is a spectrum of diseases with kwashiorkor whose essential feature is deficiency of protein at one end; and nutritional marasmus, which is total inanition of infant due to severe and prolonged restriction of all food at the other end. In the middle of the spectrum is marasmic kwashiorkor in which there are clinical features of both disorders. Some children adapt to prolonged energy and protein shortage by nutritional dwarfism. The most prevalent of all the varieties is mild to moderate PEM or the underweight child. PEM, in its various forms, has a higher incidence in India, south east Asia, parts of Africa, the Middle East, the Caribbean Islands, and in South and Central America.
In some developing countries, marasmus is of greater clinical importance than kwashiorkor. The factors which predispose to marasmus are a rapid succession of pregnancies, and early and often abrupt weaning, followed by artificial feeding of infants in inadequate amounts. The two constant features of marasmus are retarded growth and wasting of subcutaneous tissues, giving the child an aged appearance. Marasmus is usually associated with energy deficiency and occurs in many pathologic states besides simple starvation. Protein deficiency is common in prolonged febrile illness, in massive burns and large chronic ulcers, in ‘stress’, hyperthyroidism and other hypermetabolic states, in conditions interfering with digestion and absorption and in metabolic diseases which interfere with utilization. The other clinical findings in protein deficiency include loss of weight and of subcutaneous fat, wasting of muscles, pigment changes in the skin with hair loss, hypotension, weakness, and edema. Anemia is common. A decrease in serum proteins, hemoconcentration, and a decrease in blood volume are other frequent findings.
In kwashiorkor, some amino acid or protein deficiency arises typically after prolonged breastfeeding and the child is weaned on to a low protein family diet. This combined protein energy deficiency in children in many parts of the world, due to insufficient supply of amino acids, leads to inadequate protein synthesis, reduced synthesis of enzymes, and plasma proteins and impaired development of organs. The child’s weight is usually well below standard for age but the deficit may be masked by edema often due to hypoalbuminemia. Impaired synthesis of digestive enzymes may be partially responsible for diarrhea which is so commonly present and which leads to loss of potassium and magnesium in the stools. The child is prone to infections due to subnormal levels of immune responsiveness.
The oral lesions, when apparent, include a bright reddening of the tongue with a loss of papillae, bilateral angular cheilosis, fissuring of the lips, and a loss of circumoral pigmentation. In addition, the mouths of kwashiorkor patients have been described by Van Wyk as being dry, dirty, caries-free, and easily traumatized, with the epithelium readily becoming detached from the underlying tissues, leaving a raw, bleeding surface. In oral cytologic smears from his patients, he described a perinuclear vacuolization or halo around the nucleus in a remarkable number of the epithelial cells present and interpreted this as a sign of epithelial atrophy.
King has pointed out that about half of the world’s population lives in areas where the lack of milk, meat, poultry, fish, eggs, and so on, leads to early retardation of growth. Typically, children so retarded have edema, episodes of diarrhea, skin pigmentation, liver enlargement, alopecia, and poor resistance to infection, especially of the lungs and intestinal tract. The death rate may reach 25 times than that considered normal for the age group. Those that survive show permanent physical stunting. This stunted condition is so general that it is often mistaken for a genetic phenomenon. In some areas 50% of the children die before school age. It is significant that most of the children exhibit a normal growth rate up to weaning time.
Frandsen and his coworkers, Chawla and Glickman, Di Orio and coworkers, Navia, Aponte-Merced and Navia, Menaker and Navia, and Navia and coworkers have studied the effect of protein and protein energy deprivation on salivary glands and teeth, and their supporting structures in experimental animals. Overall growth and growth of the jaws were decreased. Eruption was delayed, and incisor and molar growth was retarded. Radicular osteocementum was decreased. The enamel of affected incisors exhibited increased acid solubility. Increased dental caries was also reported. The gingiva and periodontal membranes exhibited varying degrees of degeneration. Salivary volume was decreased as were the DNA, RNA, and protein concentrations of affected animals. The severity of these changes was dependent on the degree of protein deprivation.
Some of the genetic disorders, where dietary modifications become important include, phenylketonuria (PKU), which is an inherited enzyme defect in which individuals cannot metabolize the phenylalanine found in nearly all proteins. Patients with this condition are prescribed a diet which is protein restricted, just enough to meet growth and maintenance needs. Gout is yet another disorder of protein metabolism which is characterized by excessive uric acid production leading to the formation of urate crystals deposited in joints. The treatment often includes restriction of protein to limit purine and uric acid production.
Protein needs increase during fever, after severe injury and surgery, intestinal malabsorption, increased protein loss from the kidneys, or diminished protein synthesis by the liver. Dietary protein must be restricted when the kidneys can no longer remove nitrogenous wastes from the body or in severe liver disease when the nitrogenous by-products of protein catabolism can no longer be synthesized.
Individual Amino Acids
The inadequacy of zinc as a sole source of protein in rat nutrition brought out the importance of the variations in amino acid content of different proteins and led to the work of Rose and his collaborators and others on the essential and nonessential amino acids. The essential amino acids are histidine, isoleucine, leucine, lysine, methionine, phenylalanine, threonine, tryptophan, and valine. This list of nine essential amino acids must not be accepted as final; however, more may have to be added and some may eventually be dropped. The original concepts of ‘essential’ and ‘nonessential’ must be modified, since the determination of essentiality depends not only on the species studied, but also on the experimental criteria used (e.g. nitrogen balance, growth), the age of the animal used and the presence or absence of vitamins in the diet. For example, arginine is nonessential in the adult. However, infants are incapable of producing sufficient amounts of arginine for normal physiologic functions. Therefore arginine is considered essential in infants. It is unlikely; however, that a deficiency of a single essential amino acid occurs in humans.
Amyloidosis
An abnormal proteinaceous substance that is deposited between cells in tissues and organs of the body in a variety of clinical disorders is referred to as an amyloid. Using routine stains, it is seen as intercellular pink translucent material by light microscopy. Despite its morphologic uniformity, it is quite clear that amyloid is a complex material with at least two distinct forms: type A and type B. By electron microscopy, X-ray crystallography and infrared spectroscopy, amyloid appears to be made up largely of nonbranching fibrils with a characteristic ‘β-pleated sheet confirmation’, which is unique among mammalian fibrillar proteins. Two major classes of amyloid identified include amyloid light chain (AL), composed of immunoglobulin light chain, and amyloid associated (AA), made up of nonimmunoglobulin protein.
Type A (secondary) amyloid is a fibrillar protein of unknown origin that is seen in prolonged inflammatory diseases, genetic diseases, and syndromes such as familial Mediterranean fever. Type B (primary) amyloid is thought to be of immune origin because of its sequence homology with the NH2 terminal end of immunoglobulin light chains. Type B amyloid is commonly seen in patients with multiple myeloma and macroglobulinemia. Clinically asymptomatic patients found to have type B amyloid serum and urine immunoglobulin abnormalities. A third type of amyloid (type C) includes amyloid of aging, localized nonspecific amyloid, and amyloid adjacent to APUD (amine precursor uptake and decarboxylase) tumors, i.e. pheochromocytoma (Fig. 15-4). A review of the association of amyloid with a variety of diseases in men and animals has been published by Rigdon. The most common diseases predisposing to amyloidosis are the collagen diseases, particularly rheumatoid arthritis, chronic infections such as tuberculosis and osteomyelitis, regional enteritis, ulcerative colitis, and certain malignant diseases, particularly multiple myeloma, Hodgkin’s disease, and renal cell carcinoma. Since modern surgery and medicine have largely eliminated chronic suppurative disease, rheumatoid arthritis and myeloma are now the chief predisposing causes for amyloidosis. Excellent reviews of amyloidosis were written by Franklin and by Kyle and Bayrd.
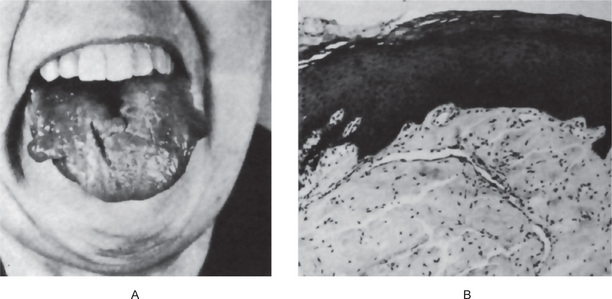
Figure 15-4 Amyloidosis.
Amyloid tumors of the tongue (A). The photomicrograph (B) illustrates a section through such a nodule Courtesy of Dr Boynton H Booth.
While any organ may be involved, those most commonly affected are the kidneys, heart, gastrointestinal tract, liver, and spleen. Amyloidosis is also seen with considerable frequency in the respiratory tract, skin, eye, adrenals, and nerves, and may involve bone. A primary localized cutaneous amyloidosis is also recognized. Amyloidosis itself is generally considered to be an irreversible disease.
Amyloid deposition in the tongue, resulting in macroglossia, and gingiva is also reported to be commonly seen. Because of the reported frequency of amyloid in gingival tissues, it has often been suggested that the gingival biopsy may be used conveniently for the diagnosis of amyloidosis. However, the results have been quite varied, some investigators reporting a high incidence of positive biopsies, while others have found so few positive results that the technique has been considered of little value. This subject has been reviewed by Lovett and his associates. Ulmansky, and Stanback and Peagler have discussed the oral manifestations of primary amyloidosis, and van der Waals and coworkers have reported on the significance of amyloidosis in oral surgery.
The amyloidosis may or may not be apparent on macroscopic examination. However, when the cut surface of the suspected organ is painted with iodine and sulfuric acid, a peculiar mahogany brown staining of the amyloid deposit is revealed. If large amounts of amyloid are accumulated the affected organ is frequently enlarged and the tissue appears gray with a waxy firm consistency. Histologically, the deposition always begins between the cells and eventually surround and destroy the trapped native cells. The amyloid in the microscopic sections of involved tissue appears as a hyaline, homogeneous material, often perivascular in distribution especially in the immune-associated form. It is best demonstrated by special stains such as Congo red and crystal violet or by the thioflavin-T fluorescent technique. Under polarized light the Congo red-stained amyloid shows green birefringence. This reaction is shared by all forms of amyloid and is due to the crossed β-pleated configuration of amyloid fibrils. AA and AL amyloid can be distinguished in histologic sections. AA protein loses affinity for Congo red after incubation of tissue sections with potassium permanganate, whereas AL proteins do not. In suspected cases of immunocyte associated amyloidosis, serum and urinary protein electrophoresis and immunoelectrophoresis should be performed.
Porphyria
Porphyria is a term which has been generally used to connote one of the inborn errors of porphyrin metabolism, characterized by overproduction of uroporphyrin and related substances. Not all cases of porphyria; however, represent a constitutional disturbance, since porphyria may appear as a sequel to some infections or intoxications. The classification of the porphyrias remains unsettled, although the most basic classification defines two types:
• Erythropoietic porphyria, characterized by early photosensitivity, splenomegaly, and excessive abnormal porphyrin formation in developing erythrocytes. Two subclasses, uroporphyria (congenital porphyria) and protoporphyria, have been described based on their respective porphyrin precursor type.
• Hepatic porphyria, also a multisystem disorder, which has four subclasses; acute intermittent porphyria, porphyria variegata, porphyria cutanea tarda, and hereditary coproporphyria.
Heritable enzymatic effects have been identified in uroporphyria, acute intermittent porphyria and porphyria cutanea tarda. An excellent review of the porphyrias has been published by Elder and coworkers.
Erythropoietic Uroporphyria: (Congenital porphyria)
This disease, the most important of the group, is transmitted as a nonsex-linked recessive character, both genders being equally affected. The first sign of erythropoietic porphyria is usually the excretion of red urine containing much uroporphyrin. This may be noted at birth or only during the first year of life. Photosensitivity is frequently absent in the neonatal period, but may become apparent during the first year of life as exposure to sunlight increases. A vesicular or bullous eruption appears on the face, back of the hands, and other exposed parts of the body (Fig. 15-5). The vesicles contain a serous fluid which usually exhibits red fluorescence. Ruptured vesicles heal slowly and leave depressed, pigmented scars. Occasionally the cutaneous manifestations may be relatively mild, resulting in little scarring. There is an interesting oral finding. The deciduous and permanent teeth may show a red or brownish discoloration, although this is not invariably present. Under ultraviolet light; however, the teeth always exhibit red fluorescence. Deposition of porphyrin in the developing teeth and bones is believed to be due to its physical affinity for calcium phosphate. The presence of porphyrin in the deciduous teeth indicates that the metabolic disorder may have been present during fetal life.
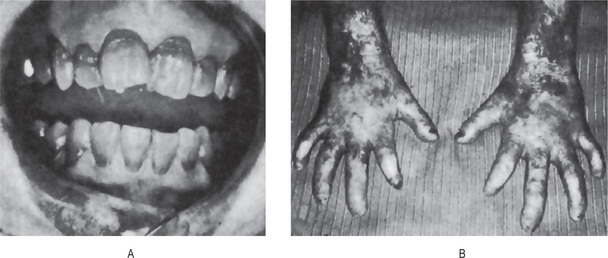
Lysosomal Storage Diseases
Lysosomal storage diseases are a heritable group of heterogeneous disorders characterized by the accumulation of undigested macromolecules intralysosomally, resulting in an increase in the size and number of these organelles, and ultimately in cellular dysfunction and clinical abnormalities. Lysosomal storage diseases are generally classified by the accumulated substrate and they include sphingolipidoses, glycoproteinoses, mucolipidoses, mucopolysaccharidoses (MPSs), and others. The concept of lysosomal storage disorders is now being extended to include deficiencies in lysosomal enzymes, noncatalytic role of lysosomal proteins, and abnormalities of lysosomal function.
The pathologic manifestations of this error in metabolism mainly depends on the nature and quantity of the accumulating material as well as the organs affected. For example, neurons which are incapable of cell division and cell turnover are particularly vulnerable to intracellular accumulations. Similarly, cells of the mononuclear phagocyte system are especially rich in lysosomes and are frequently affected by lysosomal storage disorders. Recent advances in molecular genetics have shifted the focus from conventional theories, holding gene products and genes themselves responsible for these disorders. The defective genes in most of these genetic disorders have been isolated, characterized and their specific mutations identified. At the gene level, genetic heterogeneity is complex despite similarities in phenotypes, biochemistry, and enzymology.
Disturbances in Carbohydrate Metabolism
Mucopolysaccharidoses
Mucopolysaccharidoses (MPS) result from abnormal degradation of glycosaminoglycans such as dermatan sulfate, keratan sulfate, heparan sulfate, and chondroitin sulfate resulting in organ accumulation and eventual dysfunction. Glycosaminoglycans or mucopolysaccharides are normally a component of the cornea, cartilage, bone, connective tissue, and the reticuloendothelial system and are therefore target organs for excessive storage. The catabolic enzymes involved in the breakdown of glycosaminoglycans or mucopolysaccharides are deficient. Ten known enzyme deficiencies give rise to six distinct MPS.
The stepwise degradation of the glycosaminoglycans requires four glycosidases, five sulfatases, and one nonhydrolytic transferase. The mode of transmission is autosomal recessive except for MPS II, which is X-linked. A variety of mutations are described, and the correlation of genotype with disease severity is beginning to emerge from mutation analysis.
In general, MPS are progressive disorders, characterized by the involvement of multiple organs, including the brain, liver, spleen, heart, and blood vessels; many are associated with coarse facial features, clouding of the cornea, and mental retardation. Diagnosis can often be made by examination of urine, which reveals increased concentration of glycosaminoglycan fragments (Table 15-1).
Table 15-1
The genetic mucopolysaccharidoses*
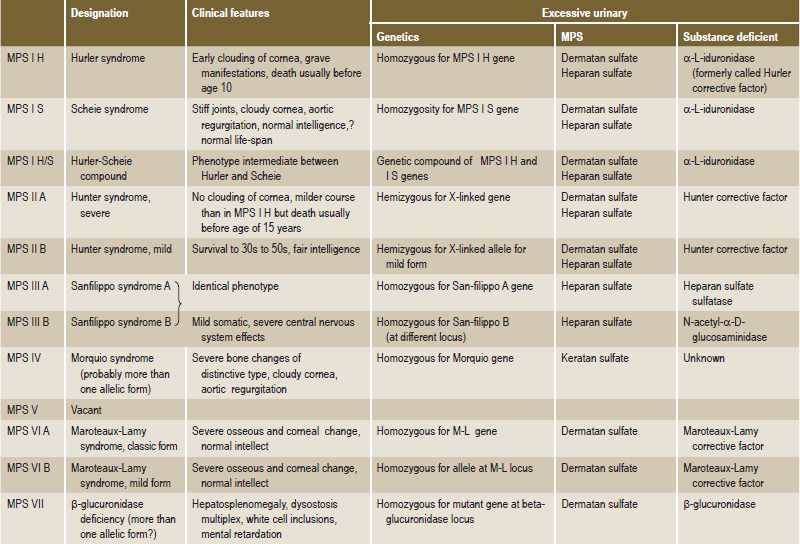
*By permission of Dr Victor A McKusick. From McKusick VA. Heritable Disorders of Connective Tissue (4th ed). CV Mosby, St Louis, 1972.
Clinical Presentation
MPS type I includes Hurler, Hurler-Scheie, and Scheie syndromes.
MPS type I H (Hurler syndrome).
MPS type I S (Scheie syndrome)
Biochemical findings are identical to type I Hurler syndrome, but the clinical features are less severe.
Glycosaminoglycan fragments are generated by alternative pathways and are excreted in the urine. Simple enzyme assays are available for the diagnosis of MPS from fibroblast, leukocyte, or serum samples. Because heterozygous individuals are identified on the basis of enzyme activity, the diagnosis can be difficult. However, it is becoming more definitive as specific mutations are identified. Prenatal diagnosis is made by means of amniocentesis or chorionic villus biopsy.
Hurler Syndrome: (Mucopolysaccharidosis I, MPS IH, gargoylism)
A chromosomal abnormality occurs in chromosome arm 4p16.3.
Hurler syndrome is a disturbance of mucopolysaccharide metabolism exhibiting a variety of classic clinical features. It is characterized by an elevated mucopolysaccharide excretion level in the urine. The disease, in which there is an excessive intracellular accumulation of both chondroitin sulfate B and heparan sulfate in those tissues and organs where they are normally found, is inherited as an autosomal recessive trait.
Clinical Features
The disease usually becomes apparent within the first two years of life, progresses during early childhood and adolescence and terminates in death usually before puberty. The head appears large and the facial characteristics are quite typical, consisting of a prominent forehead, broad saddle nose and wide nostrils, hypertelorism, puffy eyelids with coarse bushy eyebrows, thick lips, large tongue, open mouth, and nasal congestion with noisy breathing. Progressive corneal clouding is a classic manifestation of the disease as is hepatosplenomegaly, resulting in a protuberant abdomen. A short neck and spinal abnormalities are typical, while flexion contractures result in the ‘claw hand’. These dwarfed individuals are mentally retarded.
Oral Manifestations
The oral manifestations of the Hurler syndrome have been reviewed by Gardner. These consist of a shortening and broadening of the mandible with prominent gonions, a wide intergonial distance and a greater than normal distance around the arch from ramus to ramus accounting, at least in part, for the typical spacing of the teeth. Localized areas of bone destruction in the jaws may be found which appear to represent hyperplastic dental follicles with large pools of metachromatic material, probably mucopolysaccharide. The teeth themselves are frequently described as being small, widely spaced, and mishappen. However, many investigators have been unable to demonstrate abnormalities of the teeth, except for some delay in the time of eruption.
Gingival hyperplasia has been repeatedly described in patients with Hurler syndrome, although it is not a constant feature of the disease. In some patients the gingiva appears normal, while in others the gingiva appears enlarged as a result of local factors such as poor oral hygiene or mouth breathing. In occasional patients, the gingival tissues appear to be involved in a manner similar to fibromatosis gingivae. Finally, the tongue is also characteristically enlarged.
Histologic Features
There is excessive accumulation of intracellular mucopolysaccharide in many tissues and organs throughout the body including the liver, spleen, reticuloendothelial system, nervous system, cartilage, bone, and heart. Abnormal deposits are also found in many sites, with involved fibroblasts assuming the appearance of clear or gargoyle cells.
Gardner has reported the demonstration of these ‘Hurler cells’ or ‘gargoyle cells’ in the gingival tissues of affected patients. The Hurler cells are relatively large, with metachromatically staining cytoplasm which is either agranular or finely granular, often with crescent-shaped nuclei. These cells are not identified with hematoxylin and eosin but become evident with toluidine blue or with Alcian blue/aldehyde fuchsin stains. Some difficulty may be encountered in differentiating these from mast cells.
Lipoid Proteinosis: (Hyalinosis cutis et mucosae, Urbach-Wiethe disease)
Lipoid proteinosis is a rare, autosomal recessive disorder typified by generalized thickening of skin, mucosae and certain viscera. Lipoid proteinosis was first described by a Viennese dermatologist and otorhinolaryngologist, Urbach and Wiethe, in 1929.
Clinical Features
Classical features include beaded eyelid papules and laryngeal infiltration leading to hoarseness of voice. One of the characteristic features of the disease is the inability of infants to cry at birth and the hoarseness of the voice also present from birth. These features are due to the yellowish-white plaques in the epiglottis, aryepiglottic folds, and interarytenoid region. On laryngoscopic examination, the cords are seen to be thickened and nodular. On rare occasion, dyspnea may be so severe as to necessitate stripping the nodules from the cords or laryngectomy. The exact pathogenesis of this disease is not known but has been postulated to be the result of either a lysosomal storage disorder involving multiple enzyme defects or from a disturbance in collagen synthesis, as evidenced by a decrease in the ratio of type I to type III collagen associated with a decrease in mRNA for type I procollagen. There is also an increase in mRNA for type IV procollagen resulting in underproduction of fibrous collagens and an overproduction of basement membrane collagens, which tend to deposit in the skin and various organs, which form the hallmark of the disease.
Oral Manifestations
The oral cavity is usually severely affected in this disease with much of the oral mucous membrane developing the characteristic yellowish-white papular plaques which become increasingly more prevalent and prominent from childhood into adult life. The lips become thickened and nodular while the tongue becomes thickened, enlarged, very firm on palpation, and sometimes bound to the floor of the mouth (Fig. 15-6). Recurrent painful parotitis may occur as a result of involvement of the buccal mucosa, with stenosis of the parotid duct opening. Congenital absence of teeth and severe enamel hypoplasia have also been reported. The oral manifestations of the disease have been discussed by Gorlin, Williams, Hofer, and Bergenholtz.
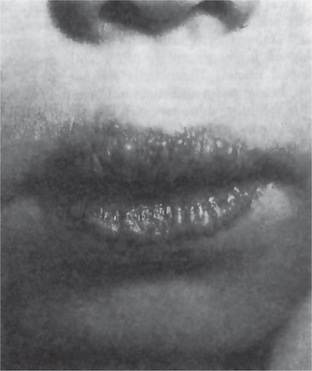
Other mucocutaneous changes may include thickening of the tongue and frenulum, blisters, warty skin papules, scarring, alopecia, nail dystrophy, and dental anomalies. Extracutaneous features may include epilepsy and neuropsychiatric abnormalities, sometimes in association with calcification in the temporal lobes or hippocampi.
Lipoid proteinosis occurs worldwide, but is more common in certain areas such as the Northern Cape province of South Africa. Lipoid proteinosis is mapped to 1q21 and identified the extracellular matrix protein 1 gene (ECM1) as the LP gene.
Histologic Features
Lipoid proteinosis is characterized by deposition of PAS-positive, diastase-resistant material at the level of the basement membrane (resulting in its thickening at the dermoepidermal junction), papillary dermis, surrounding blood vessels, and around adnexal epithelia especially sweat glands. There is widespread deposition of hyaline (glycoprotein) material and disruption/reduplication of basement membrane. Ultrastructural examination reveals concentric rings of excess basement membrane surrounding blood vessels, and irregular reduplication of lamina densa at dermoepidermal junction resulting in onion-skin appearance. Biochemically, this material is characterized by decrease in type I collagen with overproduction of type IV or basement-membrane collagen. The hyaline deposits in the biopsies examined consist of a carbohydrate-protein complex containing hyaluronic acid and probably chondroitin sulfate, plus large amounts of lipids.
Hereditary Fructose Intolerance
Over 25 years ago Chambers and Pratt reported an unusual case of a young woman who repeatedly became nauseated and vomited after the ingestion of fruit or cane sugar. Numerous reports have since appeared in the literature, and over two dozen families have been diagnosed as having hereditary fructose intolerance.
Clinical Features
The disease is transmitted as an autosomal recessive trait and is manifested by hypoglycemia and vomiting after the ingestion of fructose-containing foods. It results from a deficiency in fructose 1-phosphate aldolase. Affected individuals rapidly acquire an intense aversion to all sweets and fruits.
Oral Manifestations
Newbrun and coworkers reported on the dietary habits and dental health of 17 affected individuals. Subjects with hereditary fructose intolerance had a total sucrose intake of less than 5% of that of controls. Caries scores (DMFS) were less than 10% of those of controls. This study confirms the previous observations of Cornblath and coworkers, Levin and colleagues, and Marthaler and Froesch.
Disturbances in Lipid Metabolism
Lipid metabolism is concerned with the assimilation, utilization, replacement, and synthesis of the various fatty acids of the cell. All living cells contain fatty acids, largely in the form of esters with glycerol, cholesterol or other alcohols, or combined with phosphoric acids, nitrogenous bases or carbohydrates.
Disturbances of lipid metabolism are rare, but they do occur. These disturbances have been classified as ‘lipoid storage diseases’, xanthomatoses, lipid granulomas, and so on. Several disease entities have been identified on the basis of the particular lipid involved.
Gaucher’s Disease
Gaucher’s disease is a common lysosomal storage disease, characterized by the deposition of glucocerebroside in cells of the macrophage-monocyte system. Deficiency of a specific lysosomal hydrolase, glucocerebrosidase, which cleaves glucocerebroside to ceramide is held responsible for this disorder. Three clinical subtypes exist and are delineated by the absence or presence of neurologic involvement and its progression.
Clinical Features
Gaucher’s disease has been divided into three clinical forms:
Type I. Chronic nonneuronopathic form often presents in childhood with hepatosplenomegaly, pancytopenia, and skeletal disease, although striking clinical variability occurs in disease severity. This is the most common variety (99%) and has a striking predilection for occurrence amongst individuals of Ashkenazi Jewish descent.
Most patients have radiographic evidence of skeletal involvement, including an Erlenmeyer flask deformity of the distal femur, which is an early skeletal change.
Type II. Infantile or acute neuronopathic form causes rapidly progressive neurovisceral involvement and results in death at infancy.
Type III. Juvenile or Norrbottnian form (intermediate between type I and type II). The patients are juveniles presenting with systemic involvement. Progressive central nervous system involvement usually begins in teens or twenties.
All three subtypes are inherited as autosomal recessive traits. When bone is involved, the bone marrow shows diffuse changes. Numerous large, foamy, slightly granular cells with small, round pyknotic nuclei, which are the Gaucher’s cells, group together and replace the normal marrow structure. These skeletal manifestations have been discussed by Amstutz and Carey. Gaucher’s cell accumulations are also found in the spleen, the lymph nodes, and the liver. An excellent review of both the infantile and adult forms of Gaucher’s disease was published by Levin. In all three types of Gaucher’s disease, sternal puncture or examination of the biopsies of the spleen or liver will reveal the typical Gaucher’s cell. This is a round pale cell, measuring between 20 and 80μ in diameter, containing a small eccentric nucleus and a wrinkled or ‘crumpled silk’ cytoplasm.
Treatment and Prognosis
The prognosis of the malignant infantile form is very poor, the disease resulting in death usually within the first year. The less virulent form may persist until the sixth decade of life, when the patients usually die of some intercurrent infection. Brady and coworkers have reported that the administration of purified glucocerebrosidase to affected patients results in a dramatic decrease in hepatic accumulations of glucocerebroside. Although this therapeutic regime is experimental and fraught with potential side effects, it nevertheless holds promise for future treatment of the disease. Enzyme replacement therapy with recombinant enzymes for Gaucher’s disease is now available, which is very effective but extremely expensive.
Niemann-Pick Disease
In 1914, German pediatrician Albert Niemann described a young child with brain and nervous system impairment. Later, in the 1920s, Luddwick Pick studied tissues after the death of such children and provided evidence of a new disorder, distinct from those storage disorders previously described. It is the least common of the genetic disturbances of lipid metabolism. It is inherited as an autosomal recessive trait. Niemann-Pick disease results from lysosomal accumulation of sphingomyelin resulting from inherited deficiency of sphingomyelinase.
Niemann-Pick disease can be classified as: Type A is the acute infantile; Type B is a less common, chronic, non-neurological form, while Type C is a biochemically (intracellular cholesterol esterification) and genetically distinct form of the disease. The mutant gene is localized to 18q11–12.
Niemann-Pick disease type A is a severe infantile form with extensive neurologic involvement, marked visceral accumulation of sphingomyelin and progressive wasting resulting in early death within first three years of life. Niemann-Pick disease type B has a more variable course, with the first symptoms occurring in early childhood and many persons surviving into adulthood. In this type of patients, organomegaly is seen commonly but no nervous system involvement is noticed. Like Gaucher’s disease, it is more common in Ashkenazi Jews and has pathognomonic cell, the Niemann-Pick cell. Niemann-Pick cells are foamy, lipid-laden cells distributed throughout the reticuloendothelial system. Unlike Gaucher’s cells, they are positive for cholesterol and only weakly positive for alkaline phosphatase. They are most easily distinguished from Gaucher’s cells by phase or electron microscopy. Epstein and coworkers have developed a procedure for detecting Niemann-Pick disease in utero by measuring sphingomyelinase activity in cultured amniotic cells. The clinical findings in this disease have been discussed by Gildenhorn and Amromin as well as by Knudson.
Histologic Features
Histologically, the affected cells become extremely enlarged secondary to the distention of lysosomes due to the accumulation of sphingomyelin and cholesterol. The cells show foamy cytoplasm due to numerous vacuoles which stain for fat.
Treatment
Enzyme replacement therapy in Niemann-Pick disease is currently being explored. Current treatment is symptomatic and consists mainly of antibiotic therapy for infections resulting from pulmonary involvement. Organ transplantation, e.g. the liver, has been proposed. To date the prognosis is poor, and the vast majority of patients die of the disease.
Letterer-Siwe Disease
Letterer-Siwe disease is an acute, often fulminating histiocytic disorder which invariably occurs in infants, usually before the age of three years.
Clinical Features
The initial manifestation of this disease is often a skin rash involving the trunk, scalp, and extremities. This rash may be erythematous, purpuric, or ecchymotic, sometimes with ulceration. The patients will also commonly have a persistent, low-grade spiking fever with malaise and irritability. Splenomegaly, hepatomegaly, and lymphadenopathy are early manifestations as well as nodular or diffuse involvement of visceral organs, particularly the lungs and gastrointestinal tract, later in the course of the disease. Diffuse involvement of the skeletal system also usually occurs later in the disease.
Oral Manifestations
The oral lesions may consist of ulcerative lesions, although gingival hyperplasia has also been described. Furthermore, diffuse destruction of bone of the maxilla and mandible may occur, causing loosening and premature loss of teeth. In some cases the disease has such a rapid course that significant oral involvement does not occur.
Histologic Features
The microscopic appearance of the lesions is very similar to that seen in Hand-Schüller-Christian disease where there is basically a histiocytic proliferation with or without eosinophils. However, these histiocytes do not contain significant amounts of cholesterol so that foam cells are not a feature of the disease, nor is fibrosis encountered. In some cases cytologically altered histiocytes are present in sufficient numbers to resemble a histiocytic lymphoma.