Correlation of physical signs and neurological syndromes and disease
Upper motor neurone lesions
In neurology a clinical diagnosis is made by defining the deficit that is present, deciding on its anatomical level and then considering the likely causes. It is important to be able to distinguish upper motor neurone signs from lower motor neurone signs (see Figure 34.1 on page 437; see also List 35.3 on page 464). The former occur when a lesion has interrupted a neural pathway at a level above the anterior horn cell: for example, motor pathways in the cerebral cortex, internal capsule, cerebral peduncles, brainstem or spinal cord. When this occurs there is greater weakness of abductors and extensors in the upper limb, and flexors and abductors in the lower limb, as the normal function of this pathway is to mediate voluntary contraction of the antigravity muscles. All muscles, however, are usually weaker than normal. Muscle wasting is slight or absent, probably because there is no loss of trophic factors normally released from the lower motor neurone. The disuse that results from severe weakness may, however, cause some atrophy.
Upper motor neurone signs occur when the lesion is in the brain or spinal cord above the level of the lower motor neurone (see List 35.1).
Spasticity occurs because of destruction of the corticoreticulospinal tract, resulting in stretch reflex hyperactivity.
Monoplegia is paralysis affecting only one limb, when there is a motor cortex or partial internal capsule lesion. Hemiplegia affects one side of the body due to a lesion affecting projection of pathways from the contralateral motor cortex.
Paraplegia affects both legs, while quadriplegia affects all four limbs and is the result of spinal cord trauma or, less often, a brainstem lesion (e.g. basilar artery thrombosis).
CAUSES OF HEMIPLEGIA (UPPER MOTOR NEURONE LESION)
Vascular disease (stroke or TIA)
Thrombosis, embolism or haemorrhage occur in specific vascular territories (see Figure 35.1).1,2 The deficit usually progresses over a period of minutes but continuing deterioration may occur over hours or days as a result of oedema or haemorrhage around or into the infarct. When symptoms and signs last for less than 24 hours, the episode is called a transient ischaemic attack (TIA). Lesions in the territory of the internal carotid artery result in hemiplegia on the opposite side of the body if a large area of the internal capsule or hemisphere is involved. Homonymous hemianopia, hemianaesthesia and dysphasia may occur (see Table 35.1). Stenosis of the internal carotid artery in the neck may be associated with a bruit.3
TABLE 35.1
Intracerebral thrombosis or embolism: clinical features
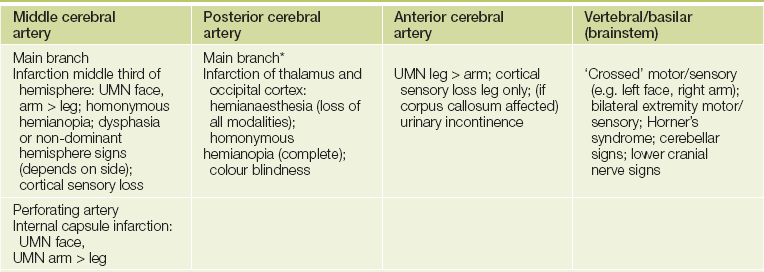
UMN = upper motor neurone lesion.
*Effects are variable because of anastomoses with distal middle cerebral artery branches and supply from posterior communicating artery, but one would examine particularly for occipital and temporal lobe dysfunction.
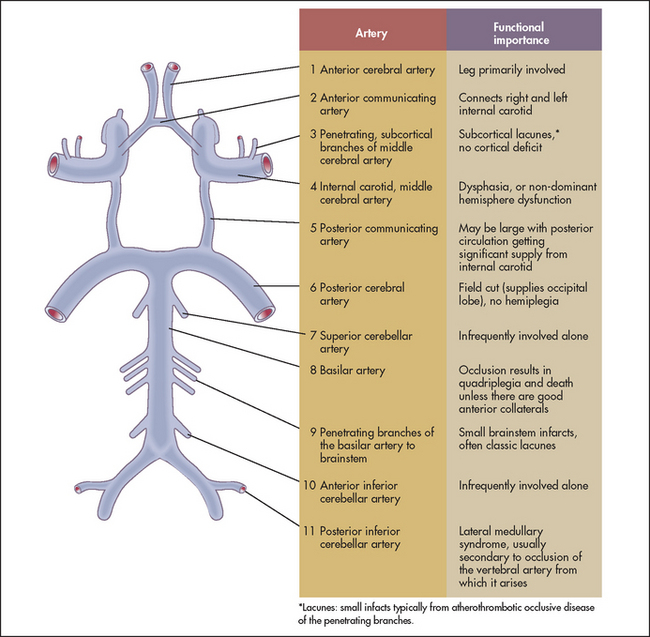
Figure 35.1 Anatomy of the circle of Willis, showing the functional importance of the arterial blood supply (Adapted from Weiner HL, Levitt LP. Neurology for the house officer, 6th edn. Baltimore: Williams & Wilkins, 2000.) Williams & Wilkins
Haemorrhagic strokes often involve the internal capsule and putamen (causing a contralateral hemiparesis and often sensory loss) or the thalamus (causing a contralateral hemianaesthesia).2
Lesions in the territory of the vertebrobasilar arterymay produce cranial nerve palsies, cerebellar signs, Horner’s syndrome and sensory loss, as well as upper motor neurone signs (often bilateral because of the close proximity of structures in the brainstem). For example, a lesion in the midbrain may be associated with a third nerve paralysis and upper motor neurone signs on the opposite side. Hemianaesthesia and homonymous hemianopia may occur if the posterior cerebral arteries are affected. An important syndrome to recognise is the lateral medullary syndrome (see List 35.2). Atheroma in the ascending aorta is increasingly recognised as a source of cerebral emboli.
Compressive and infiltrative lesions
Tumours tend to occur in the lobes of the brain, and focal signs will depend on the tumour site. Signs localised to the parietal, temporal, occipital or frontal lobe suggest this disease process. There may, however, be false localising signs in the presence of raised intracranial pressure: for example, a unilateral or bilateral sixth nerve palsy (because of the nerve’s long intracranial path). Papilloedema is usually associated if there is raised intracranial pressure.
Lower motor neurone lesions
Lower motor neurone lesions interrupt the spinal reflex arc and therefore cause muscle wasting, reduced or absent reflexes and sometimes fasciculations. This results from a lesion of the spinal motor neurones, motor root or peripheral nerve (see List 35.3).
Motor neurone disease
This disease of unknown aetiology results in pathological changes in the anterior horn cells, the motor nuclei of the medulla and the descending tracts. It therefore causes a combination of upper motor neurone and lower motor neurone signs, although one type may predominate.
Importantly, fasciculations are almost always present. The muscle stretch reflexes are usually present (often increased) until late in the course of the disease, and there are rarely any objective sensory changes (15–20% of patients report sensory symptoms).
Peripheral neuropathy
Distal parts of the nerves are usually involved first because of their distance from the cell bodies, causing a distal loss of sensation or motor function, or both, in the limbs. A typical sensory change is a symmetrical glove and stocking loss to all modalities (see Figure 35.2). This is unlike the pattern found with individual nerve or nerve root disease, which should be suspected if sensory loss is asymmetrical or confined to one limb. Peripheral muscle weakness may be present due to motor nerve involvement. Occasionally, motor neuropathy may occur without sensory change. In the latter case, reflexes are reduced but may not be absent in the distal parts of the limbs (see List 35.4).
Guillain-Barré syndrome (acute inflammatory polyradiculoneuropathy)
This disease, thought to have an immune basis, may begin 7–10 days after an infective illness. It results in flaccid proximal and distal muscle paralysis, which typically ascends from the lower to the upper limbs. Wasting is rare. The reflexes are reduced or absent. The cranial nerves can be affected; occasionally disease is confined to these. Sensory loss is minimal or absent. Unlike transverse myelitis, the sphincters are little affected. Weakness of the respiratory muscles can be fatal but the disease is usually self-limiting. HIV infection can cause a similar syndrome. It is important that the patient’s respiratory function be assessed regularly by measuring the forced expiratory volume in the first second (FEV1) and the forced vital capacity (FVC). A decline in these can lead to respiratory failure and death.
Multiple sclerosis
This disease with unknown cause is characterised by scattered areas of inflammation in the central nervous system (CNS). A careful history is necessary as the diagnosis of multiple sclerosis (MS) depends on the occurrence of at least two neurological episodes separated in time and place within the CNS; see List 35.5.
The signs can be very variable. Look particularly for signs of spastic paraparesis and posterior column sensory loss as well as cerebellar signs. Examine the cranial nerves. Look carefully for loss of visual acuity, optic atrophy, papillitis and scotomata (usually central). Internuclear ophthalmoplegia is an important sign and is almost diagnostic in a young adult. Internuclear ophthalmoplegia is weakness of adduction in one eye as a result of damage to the ipsilateral medial longitudinal fasciculus; there may be nystagmus in the abducting eye. Bilateral internuclear ophthalmoplegia is almost always caused by MS.
Other cranial nerves may rarely be affected (III, IV, V, VI, VII, pseudobulbar palsy) by lesions within the brainstem. Charcot’s triad for MS consists of nystagmus, intention tremor and scanning speech, but occurs in only 10% of patients.
Look for Lhermitte’sa sign (an electric shock-like sensation in the limbs or trunk following neck flexion). This can also be caused by other disorders of the cervical spine, such as subacute combined degeneration of the cord, cervical spondylosis, cervical cord tumour, foramen magnum tumours, nitrous oxide abuse and from mantle irradiation.
Thickened peripheral nerves
If there is evidence of a peripheral nerve lesion, peripheral neuropathy or a mononeuritis multiplex (see List 35.6), palpate for thickened nerves. The median nerve at the wrist, the ulnar nerve at the elbow, the greater auricular nerve in the neck and the common peroneal nerve at the head of the fibula are the most easily accessible. If nerves are thickened, consider the following diagnoses:
• chronic inflammatory demyelinating polyradiculoneuropathy
• hereditary motor and sensory neuropathy (autosomal dominant; see List 35.10 on page 472)
• other (e.g. sarcoidosis, diabetes mellitus, neurofibromatosis).
Spinal cord compression
It is important to remember that a spinal cord lesion causes lower motor neurone signs at the level of the lesion and upper motor neurone signs below that level (see List 35.7). Do not forget the spinal cord’s anatomy and vascular supply (see Figure 35.3). Examine any suspected case as follows.
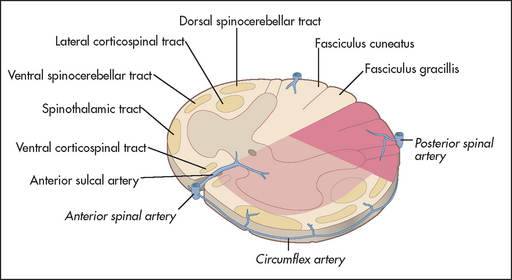
Figure 35.3 Anatomy and vascular supply of the spinal cord Note: Anterior spinal artery occlusion spares posterior column function.
After carefully examining the lower limbs (see above) determine the level of any sensory impairment (see Table 35.2). Then examine the back for signs of a local lesion. Look for deformity, scars and neurofibromas. Palpate for vertebral tenderness and auscultate down the spine for bruits. Next examine the upper limbs and cranial nerves to determine the upper level, if this is not already obvious.
TABLE 35.2
Important patterns of abnormal sensation
| Sign | Location of lesion |
| Total unilateral loss of all forms of sensation | Thalamus or upper brainstem (extensive lesion) |
| Pain and temperature loss on one side of the face and opposite side of the body | Medulla involving descending nucleus of the spinal tract of the fifth nerve and ascending spinothalamic tract (lateral medullary lesion) (see Figure 35.9) |
| Bilateral loss of all forms of sensation below a definite level | Spinal cord lesion (if only pain and temperature affected: anterior cord lesion) |
| Unilateral loss of pain and temperature below a definite level | Partial unilateral spinal cord lesion on opposite side (Brown-Sequard syndrome; see Figure 35.8) |
| Loss of pain and temperature over several segments but normal sensation above and below | Intrinsic spinal cord lesion near its centre anteriorly (involves the crossing fibres)—e.g. syringomyelia, intrinsic cord tumour (note: more posterior lesions cause proprioceptive loss) |
| Loss of sensation over many segments with sacral sparing | Intrinsic cord compression more likely |
| Saddle sensory loss (lowest sacral segments) | Cauda equina lesion (touch preserved in conus medullaris lesions) |
| Loss of position and vibration sense only | Posterior column lesion |
| Glove and stocking loss (hands and feet) | Peripheral neuropathy |
| Loss of all forms of sensation over a well-defined body part only | Posterior root lesion (purely sensory) or peripheral nerve (often motor abnormality associated) |
Important spinal cord syndromes
BROWN-SÉQUARD SYNDROMEb
Clinical features are shown in Figure 35.8. These signs result from hemisection of the cord.

Figure 35.8 Brown-Séquard syndrome Loss of pain and temperature on the opposite side to the lesion, with loss of vibration and proprioception on the same side as the lesion. (Courtesy of Glenn McCulloch)
• Motor changes: (1) upper motor neurone signs below the hemisection on the same side as the lesion; (2) lower motor neurone signs at the level of the hemisection on the same side.
• Sensory changes: (1) pain and temperature loss on the opposite side to the lesion—note that the upper level of sensory loss is usually a few segments below the level of the lesion; (2) vibration and proprioception loss occur on the same side; (3) detection of light touch is often normal.
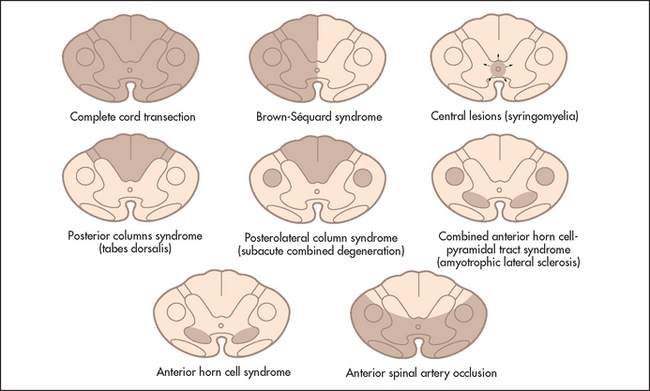
Figure 35.4 Spinal cord syndromes (Adapted from Brazis PW. Localisation in clinical neurology, Philadelphia: Lippincott, Williams & Wilkins, 2001.) Lippincott, Williams & Wilkins
• Causes: (1) multiple sclerosis; (2) angioma; (3) trauma; (4) myelitis; (5) post-radiation myelopathy.

Figure 35.6 Pattern of sensory loss with intrinsic spinal cord disease For example, central tumour or less commonly with extrinsic compression of the spinal cord—sacrum is spared. (Courtesy of Glenn McCulloch)
SUBACUTE COMBINED DEGENERATION OF THE CORD (VITAMIN B12 DEFICIENCY)
Clinical features are: (1) posterior column loss symmetrically (vibration and joint position sense), causing an ataxic gait; and (2) upper motor neurone signs in the lower limbs symmetrically with absent ankle reflexes; knee reflexes may be absent or, more often, exaggerated. There may also be (3) peripheral sensory neuropathy (less common and mild); (4) optic atrophy; and (5) dementia.
DISSOCIATED SENSORY LOSS
This usually indicates spinal cord disease but may occur with a peripheral neuropathy.
• Causes of spinothalamic loss only: (1) syringomyelia; (2) Brown-Séquard syndrome (contralateral leg); (3) anterior spinal artery thrombosis; (4) lateral medullary syndrome (contralateral to the other signs; see Figure 35.9); (5) small fibre peripheral neuropathy (e.g. diabetes mellitus, amyloid).

• Causes of dorsal column loss only: (1) subacute combined degeneration; (2) Brown-Séquard syndrome (ipsilateral leg); (3) spinocerebellar degeneration (e.g. Friedreich’sc ataxia); (4) multiple sclerosis; (5) tabes dorsalis; (6) peripheral neuropathy (e.g. diabetes mellitus, hypothyroidism); (7) sensory neuronopathy (a dorsal root ganglionopathy that may be caused by carcinoma, diabetes mellitus or Sjögren’s syndrome).
SYRINGOMYELIA (A CENTRAL CAVITY IN THE SPINAL CORD)
• Clinical triad: (1) loss of pain and temperature over the neck, shoulders and arms (a ‘cape’ distribution); (2) amyotrophy (atrophy and areflexia) of the arms; and (3) upper motor neurone signs in the lower limbs.
There may also be thoracic scoliosis due to asymmetrical weakness of the paravertebral muscles.
AN EXTENSOR PLANTAR RESPONSE PLUS ABSENT KNEE AND ANKLE JERKS
• Causes: (1) subacute combined degeneration of the cord (B12 deficiency); (2) conus medullaris lesion; (3) combination of an upper motor neurone lesion with cauda equina compression or peripheral neuropathy, such as a stroke in a diabetic; (4) syphilis (taboparesis); (5) Friedreich’s ataxia; (6) motor neurone disease; (7) human T-cell lymphotropic virus (HTLV-I) infection.
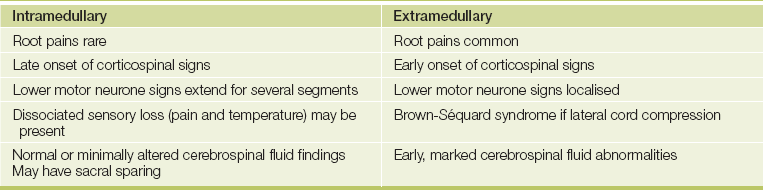
A summary of the features that differentiate intramedullary from extramedullary cord lesions is presented inTable 35.3.
Myopathy
Muscle weakness can be due to individual peripheral nerve lesions, mononeuritis multiplex, peripheral neuropathy or spinal cord disease. Each of these has a characteristic pattern. Primary disease of muscle (myopathy) causes weakness without sensory loss. The motor weakness is similar to that of the lower motor neurone type. There are two major patterns: proximal myopathy and distal myopathy.
Proximal myopathy is the more common form. On examination there is proximal muscle wasting and weakness (see Lists 35.8 and 35.9 and Figures 35.10 and 35.11). Reflexes involving these muscles may be reduced. This can be caused by genetic (e.g. muscular dystrophy) or acquired disease. Distal myopathy also occurs and is always genetic, although peripheral neuropathy is a much more common cause of distal muscle weakness. If the distal limbs are affected, consider hereditary motor and sensory neuropathy (see List 35.10). Motor neurone disease also causes weakness without any sensory loss.
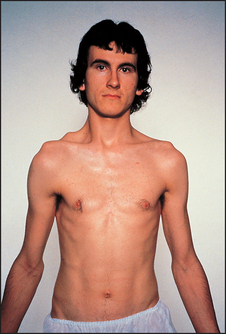
Figure 35.10 Fascioscapular muscular dystrophy (From Mir MA. Atlas of clinical diagnosis, 2nd edn. Edinburgh: Saunders, 2003.)
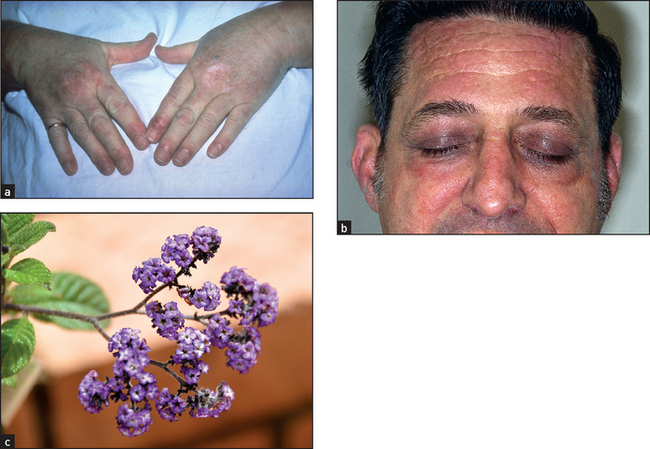
Figure 35.11 Dermatomyositis (a) Gottren’s sign in dermatomyositis—heliotrope (lilac-coloured) flat-topped papules, which occur over the knuckles, but may also be seen over the elbows or knees and may ulcerate. (b) Dermatomyositis may also cause a heliotrope rash on the face (especially on the eyelids, upper cheeks and forehead), periorbital oedema, erythema, maculopapular eruptions and scaling dermatitis. Dermatomyositis and the closely related condition polymyositis are idiopathic myopathies. Up to 10% of adult patients with dermatomyositis may have an underlying malignancy. (c) Heliotrope in flower. ((a) and (b) From McDonald FS, ed. Mayo Clinic images in internal medicine, with permission. © Mayo Clinic Scientific Press and CRC Press.)
Dystrophia myotonica
If this disease (which is inherited as an autosomal dominant condition) is suspected because of an inability on the part of the patient to let go when shaking hands (myotonia), or because general inspection reveals the characteristic appearance (see Figure 35.12), examine as follows.
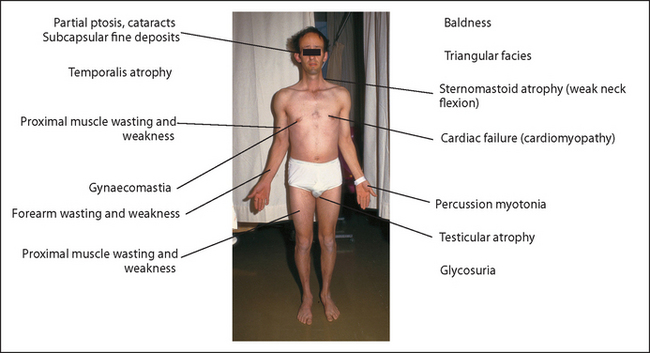
Observe the face for frontal baldness (the patient may be wearing a wig), the expressionless triangular facies, atrophy of the temporalis muscle and partial ptosis. Thick spectacles, a traditional sign of this disease, are not often seen now because of lens replacement surgery. The eyes should still be examined, as these patients can develop characteristic iridescent cataracts and subcapsular fine deposits.
Look at the neck for atrophy of the sternocleidomastoid muscles and then test neck flexion (neck flexion is weak, while extension is normal).
Go to the upper limbs. Shake hands and test for percussion myotonia. Tapping over the thenar eminence causes contraction and then slow relaxation of abductor pollicis brevis. Examine the arms for signs of wasting and weakness distally (forearms are usually affected first) and proximally. There are no sensory changes.
Go to the chest and look for gynaecomastia (uncommon). Examine the cardiovascular system for cardiomyopathy. Next palpate the testes for atrophy. Examine the lower limbs. The tibial nerves are affected first. Always ask to test the urine for sugar (diabetes mellitus is associated with this disease).
Note: Muscle myotonia can also occur in the hereditary diseases myotonia congenita (autosomal dominant or recessive) and hereditary paramyotonia (autosomal dominant cold-induced myotonia).
Myasthenia gravis
Myasthenia gravis is an autoimmune disease of the neuromuscular junction. There are circulating antibodies against acetylcholine receptors. It differs from the proximal myopathies in that muscle power decreases with use. There is little muscle wasting and no sensory change.
About 65% of patients present with ocular symptoms including diplopia and drooping eyelids. Patients (or their students, if the patient is a university lecturer) may report that their speech becomes unintelligible during prolonged speaking.
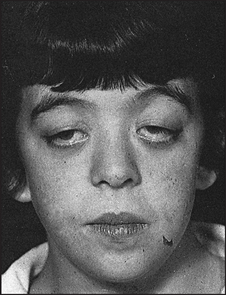
It is necessary to test for muscle fatigue. Test the oculomotor muscles by asking the patient to sustain an upward gaze by looking up at the ceiling for 1 minute, and watch for progressive ptosis (see Figure 35.13). Test for the peek sign for orbicularis oculi weakness. Ask the patient to close the eyes; if positive, within 30 seconds the lid margin will begin to separate, showing the sclera. This test strongly increases the likelihood of myasthenia (LR+ 30.0, LR– 0.88).4 Weakness of facial muscles is also common but less often reported by the patient. The transverse smile sign may present. Weakness of the levator muscles of the mouth makes an attempt at prolonged smiling look more like a grimace. To elicit this sign, tell the patient a series of mildly amusing anecdotes and watch the face carefully.
Then test the proximal limb girdle muscles—ask the patient to hold the arms above the head. You can repeatedly press the abducted arms down until they weaken. Power will decrease with repeated muscle contraction.
Look for a thymectomy scar (over the sternum)—thymectomy is often undertaken as treatment for generalised myasthenia.
The cerebellum
If the patient complains of clumsiness or problems with coordination of movement, a cerebellar examination is indicated (see Figure 35.14). Signs of cerebellar disease occur on the same side as the lesion in the brain. This is because most cerebellar fibres cross twice in the brainstem, both on entry to and exit from the cerebellum. Proceed as follows with the examination.
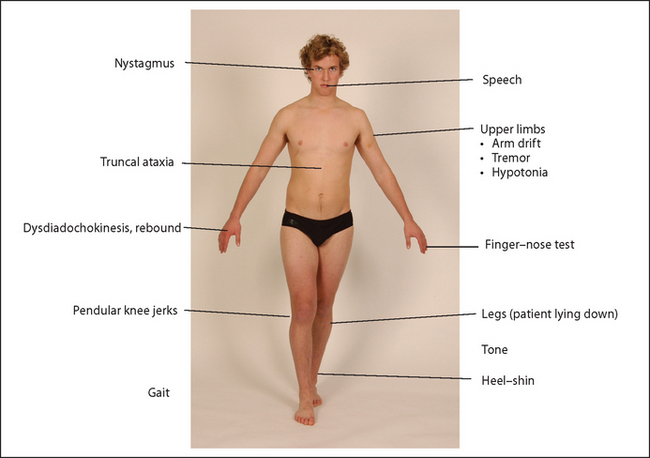
Look first for nystagmus—usually jerky horizontal nystagmus with an increased amplitude on looking towards the side of the lesion. The direction of fast movement is the side of the lesion. Assess speech next. Ask the patient to say ‘British Constitution’ or ‘West Register Street’ (see Figure 35.15). Cerebellar speech is jerky, explosive and loud, with an irregular separation of syllables.

Go to the upper limbs. Ask the patient to extend the arms and look for upward arm drift due to hypotonia of the agonist muscles. Test tone. Hypotoniad is due to loss of a facilitatory influence on the spinal motor neurones.
Next perform the finger–nose test. The patient touches the nose, then rotates the finger and touches your finger. Note any intention tremor (tremor that increases as the target is approached—this is due to loss of cerebellar connections in the brainstem) and past-pointing (the patient overshoots the target). Test rapidly alternating movements: the patient taps alternately the palm and back of one hand on the other hand or thigh. Inability to perform this movement smoothly is called dysdiadochokinesis. Now test rebound: ask the patient to lift the arms quickly from the sides, then stop (incoordination of antagonist and agonist action causes the patient to be unable to stop the arms). Before testing, always demonstrate these movements for the patient’s benefit.
Go on to examine the legs. Again, test tone here. Then perform the heel–shin test looking for accuracy of fine movement when the patient slides the heel down the shin slowly on each side for several cycles. Then ask the patient to lift the big toe up to touch your finger, and look for intention tremor and past-pointing. Ask the patient then to tap each heel on the other shin.
Test for truncal ataxia by asking the patient to fold the arms and sit up. While the patient is sitting, ask him or her to put the legs over the side of the bed and test for pendular knee jerks (the lower leg continues to swing a number of times before coming to rest—this is evidence of hypotonia).
Test gait (the patient will stagger towards the affected side if there is a unilateral cerebellar hemisphere lesion). If there is an obvious unilateral cerebellar problem, examine the cranial nerves for evidence of a cerebellopontine angle tumour (fifth, seventh and eighth nerves affected) or the lateral medullary syndrome, and auscultate over the cerebellum.
Always look in the fundi for papilloedema. Next examine for peripheral signs of malignant disease and for vascular disease (carotid or vertebral bruits). Examine the base of the skull for scars from previous neurosurgery.
If there is evidence of a midline lesion, such as truncal ataxia, abnormal heel–toe walking or abnormal speech, consider either a midline tumour or a paraneoplastic syndrome (see List 35.11). If there is bilateral disease, look for signs of multiple sclerosis, Friedreich’s ataxia (pes cavus is the most helpful initial clue; see List 35.12) and hypothyroidism (rare). Alcoholic cerebellar degeneration (which affects the anterior lobe of the cerebellar vermis) classically spares the arms. If there are, in addition, upper motor neurone signs, consider the causes in List 35.13.
Remember that there are important reciprocal connections between the cerebellum and the parietal and frontal lobes. These explain the problems that cerebellar abnormalities can cause with functions other than coordination. Loss of verbal fluency, grammatical problems with speech and difficulty with memory and planning can all sometimes be features of cerebellar disease.
Parkinson’s disease
This is a common extrapyramidal disease of middle to old age (1% of people older than 65) where there is degeneration of the substantia nigra and its pathways. This results in dopamine deficiency and a relative excess of cholinergic transmission in the caudate nucleus and putamen, which causes excessive supraspinal excitatory drive. There may be a history of insidious and asymmetrical onset. Non-specific symptoms (sleep abnormalities, constipation, depression and dementia) may precede or accompany the classic tremor.
Examine as follows.5
INSPECTION
Note the lack of facial expression, which leads to a mask-like facies. The posture is characteristically flexed and there are few spontaneous movements.
GAIT AND MOVEMENTS
Ask the patient to rise from a chair, walk, turn quickly, stop and start.
The characteristic gait is described as shuffling—there are small steps, and the patient hardly raises the feet from the ground. There is often difficulty in initiating walking, but once it begins the patient hurries (festination) and has difficulty stopping. The Parkinsonian patient seems always to be trying to catch up with the centre of gravity. There is a lack of the normal arm swing. Walking heel-to-toe will be difficult.
Testing for propulsion or retropulsion (propulsion involves pushing the patient from behind and retropulsion pushing from in front) is of uncertain value and must be done with some caution because the patient may be unable to stop and may fall over. You can stand behind the patient and pull him or her backwards, but should stand braced to catch the patient.
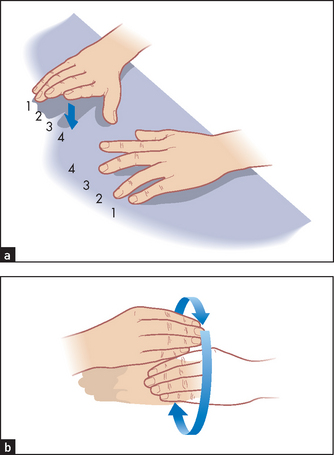
Bradykinesia (a decrease in the speed and amplitude of complex movements) may be the result of a lesion in the nigrostriatal pathway (a dopaminergic pathway), which affects connections between the caudate nucleus, the putamen and the motor cortex, causing abnormal movement programming and abnormal recruitment of single motor units. Two simple tests (see Figure 35.16) for this are finger tappingand twiddling. Ask the patient to tap the fingers in turn onto a surface repeatedly, quickly and with both hands at once. Twiddling is rotating the hands around each other in front of the body. These movements are slow and clumsy in Parkinsonian patients but obviously depend on motor and cerebellar function as well. Difficulty getting out of a chair can be another sign of bradykinesia and patients often have difficulty turning over in bed.
Kinesia paradoxica is the striking ability of a patient to perform rapid movements (especially if startled) but not slow ones; for example, the patient may be able to run down the stairs in response to a fire alarm but be unable to stop at the bottom—this is not a recommended test.
TREMOR
Have the patient return to bed. Look for a resting tremor, which is often asymmetrical. The characteristic movement is described as pill-rolling. Movement of the fingers at the metacarpophalangeal joints is combined with the movements of the thumb. Various attending movements may also occur at the wrist. On finger–nose testing the resting tremor decreases, but a faster action tremor may supervene.
Tremor can be facilitated by getting the patient to perform ‘serial 7s’—take 7 from 100, then 7 from the answer and so forth (mental stimulation)—or to move the contralateral limb (e.g. by rapidly opposing the contralateral thumb and fingers). Other types of tremor are summarised in List 35.14.
TONE
Test tone at both wrists. The characteristic increase in tone is called cogwheel or plastic (lead pipe) rigidity. Tone is increased with an interrupted nature, the muscles giving way with a series of jerks. If hypertonia is not obvious, obtain reinforcement by asking the patient to turn the head from side to side or to wave the contralateral arm. Cogwheel rigidity occurs because the exaggerated stretch reflex is interrupted by tremor.
Remember, the signs are often asymmetrical early in the course of Parkinson’s disease.
FACE
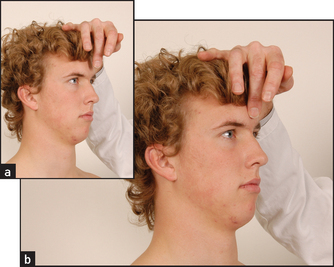
There may be titubation (tremor) of the head, absence of blinking, dribbling of saliva and lack of facial expression. Test the glabellar tap (reflex): keeping your finger out of the patient’s line of vision, tap the middle of the patient’s forehead (glabella) with your middle finger (see Figure 35.17). This sign is positive when the patient continues to blink as long as you tap. Normal people blink only a couple of times and then stop. The glabellar reflex is a primitive reflex that is also frequently present in frontal lobe disease.
Assess speech, which is typically monotonous, soft and faint, lacking intonation. Sometimes palilalia is present; this is repetition of the end of a word (the opposite of stuttering).
Now test ocular movements, particularly for weakness of upward gaze. Isolated failure of upward gaze is a feature of Parkinson’s disease. There is a separate group of patients with marked rigidity and paralysis of gaze who should be diagnosed as having progressive supranuclear palsy rather than Parkinson’s disease. These people develop loss of downward gaze first, then loss of upward gaze and finally loss of horizontal gaze.
Feel the brow for greasiness (seborrhoea) or sweatiness, due to associated autonomic dysfunction. Orthostatic hypotension may also be present for the same reason.
The palmomental reflex is commonly present in these patients and tends to be more prominent in those with severe akinesia. Dementia develops in 30% of patients.
WRITING
Ask the patient to write his or her name and address. Micrographia (small writing) is characteristic. The patient may also be unable to do this because of the development of dementia, a late manifestation. Test the higher centres if appropriate. See Good signs guide 35.1.
Other extrapyramidal movement disorders (dyskinesia)
CHOREA
Here there is a lesion of the corpus striatum, which causes non-repetitive, abrupt, involuntary jerky movements. These may be unilateral or generalised. Often the patient attempts to disguise this by completing the involuntary movements with a voluntary one. In this disease, dopaminergic pathways dominate over cholinergic transmissions.
Chorea can usefully be distinguished from hemiballismus, athetosis and pseudoathetosis. Hemiballismus is due to a subthalamic lesion on the side opposite the movement disorder. It causes unilateral wild throwing movements of the proximal joints. There may be skin excoriation due to limb trauma. These movements may persist during sleep. Athetosis or dystonia is due to a lesion of the outer segment of the putamen and causes slow sinuous writhing distal movements that are present at rest. Pseudoathetosis is a description given to athetoid movements in the fingers in patients with severe proprioceptive loss (these are especially prominent when the eyes are shut).
If the patient has chorea or if chorea is suspected, proceed as follows. First shake hands. There may be tremor and dystonia superimposed on lack of sustained hand grip (‘milkmaid’s grip’). Ask the patient to hold out the hands, then look for a choreic (dystonic) posture. This typically involves finger and thumb hyperextension and wrist flexion.
Go to the face and look at the eyes for exophthalmos (thyrotoxicosis), Kayser-Fleischer rings (Wilson’s disease) and conjunctival injection (polycythaemia). Ask the patient to poke out the tongue and note frequent retraction of the tongue (serpentine movements). Look for skin rashes (e.g. systemic lupus erythematosus, vasculitis). If the patient is a young girl, examine the heart for signs of rheumatic fever (Sydenham’se chorea).
Test the reflexes. The abdominal reflexes are usually brisk, but tendon reflexes are reduced and may be pendular (due to hypotonia).
Assess the higher centres for dementia (Huntington’sf chorea).
Causes of chorea are shown in List 35.16.
DYSTONIA
The patient manifests an involuntary abnormal posture with excessive co-contraction of antagonist muscles. Dystonia may be focal (e.g. spasmodic torticollis), segmental or generalised. Other forms of movement disorder may be present (e.g. myoclonic dystonia). The acute onset of dystonia is seen most commonly as a side effect of various drugs (e.g. levodopa, phenothiazines, metoclopramide).
The unconscious patient
The rapid and efficient examination of the unconscious patient is important. The word COMA provides a mnemonic for four major groups of causes of unconsciousness:
C O2 narcosis (respiratory failure: uncommon)
O verdose (e.g. tranquillisers, alcohol, salicylates, carbon monoxide, antidepressants)
M etabolic (e.g. hypoglycaemia, diabetic ketoacidosis, uraemia, hypothyroidism, hepatic coma, hypercalcaemia, adrenal failure)
A poplexy (e.g. head injury, cerebrovascular accident [infarction or haemorrhage], subdural or extradural haematoma, meningitis, encephalitis, epilepsy).
Coma occurs when the reticular formation is damaged by a lesion or metabolic abnormality, or when the cortex is diffusely damaged.
GENERAL INSPECTION
Airway and breathing
Look to see if the patient is breathing, as indicated by chest wall movement. If not, urgent attention is required, including clearing the airway and providing ventilation. Note particularly the pattern of breathing (see Table 9.4 on page 134). Important signs to look for are Cheyne-Stokes respiration (the rate and depth of respiration waxes and wanes often to the point of ceasing altogether for periods, which may indicate diencephalic injury, but is not specific), irregular ataxic breathing (Biot’s breathing, from an advanced brainstem lesion) and deep rapid respiration (e.g. Kussmaul breathing, secondary to a metabolic acidosis, as in diabetes mellitus).
Circulation
Look for signs of shock, dehydration and cyanosis. A typical cherry-red colour occurs rarely in cases of carbon monoxide poisoning. Take the pulse rate and blood pressure.
Posture
Look for signs of trauma. Note any neck hyperextension (from meningism in children or cerebellar tonsillar herniation).
1. A decerebrate or extensor posture, which may be held spontaneously or occur in response to stimuli, and which suggests severe midbrain disease. The arms are held extended and internally rotated and the legs are extended.
2. A decorticate or flexor posture, which suggests a lesion above the brainstem. It can be unilateral or bilateral. There is flexion and internal rotation of the arms and extension of the legs.
Involuntary movements
Recurrent or continuous convulsions, which may be focal or generalised, suggest status epilepticus. Myoclonic jerks can occur after hypoxic injury and as a result of metabolic encephalopathy. Remember that complex partial seizure status epilepticus can cause a reduced level of consciousness without convulsive movements.
LEVEL OF CONSCIOUSNESS
Tickle the patient’s nose with cottonwool and watch for facial movements. This is less likely to harm the patient than the traditional method of firmly pressing the knuckles over the patient’s sternum to cause pain.
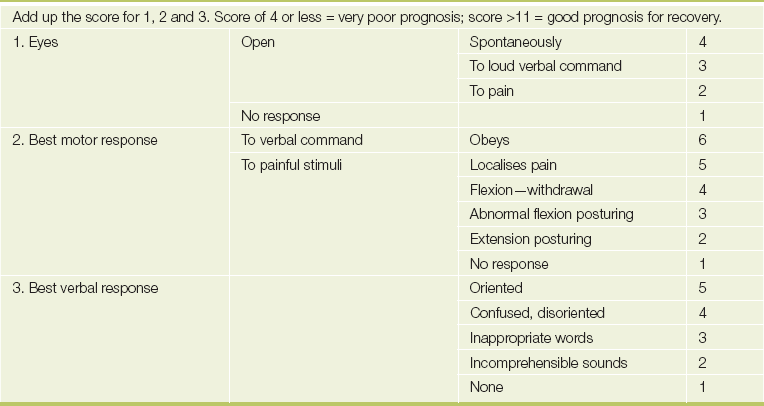
Determine the level of consciousness. Coma is unconsciousness with a reduced response to external stimuli. Coma in which the patient responds semi-purposefully is considered light. In deep coma there is no response to any stimuli and no reflexes are present (it is usually due to a brainstem or pontine lesion, although drug overdose, such as with barbiturates, can be responsible). Stupor is unconsciousness, but the patient can be aroused. Purposeful movements occur in response to painful stimuli. Drowsiness resembles normal sleepiness. The patient can be fairly easily roused to normal wakefulness, but when left alone falls asleep again. The Glasgow Coma Scale (see Table 35.4) is used to assess the depth of coma more accurately: record the subscores and total scores.
NECK
If there is no evidence of neck trauma, assess for neck stiffness and Kernig’s sign (for meningitis or subarachnoid haemorrhage).
HEAD AND FACE
Inspect and palpate for head injuries, including Battle’sg sign (bruising behind the ear indicating a fracture of the base of the skull). Look for facial asymmetry (i.e. facial weakness). The paralysed side of the face will be sucked in and out with respiration. A painful stimulus (e.g. pressing the supraorbital notch) may produce grimacing and make facial asymmetry more obvious. Note jaundice (e.g. hepatic coma) or manifestations of myxoedema.
Eyes
Inspect the pupils. Very small pupils (but reactive to light) occur in pontine lesions and with narcotic overdoses. One small pupil occurs in Horner’s syndrome (e.g. as part of the lateral medullary syndrome or in hypothalamus injury; see Figure 35.18). Two midpoint non-reactive pupils suggest midbrain disease, anoxia or drugs (anticholinergics). One dilated pupil suggests a subdural haematoma, raised intracranial pressure (unilateral tentorial herniation) or a subarachnoid haemorrhage from a posterior communicating artery aneurysm. Widely dilated pupils may occur when increased intracranial pressure and coning cause secondary brainstem haemorrhage, or with anticholinergic drugs.
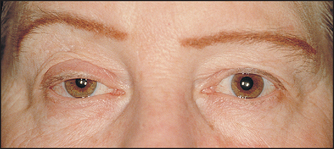
Figure 35.18 Horner’s syndrome Note ipsilateral right-sided ptosis and pupillary miosis (constriction). (Yanoff M, Duker J. Ophthalmology, 3rd edn. Mosby, 2008.)
Conjunctival haemorrhage suggests skull fracture. Look in the fundi for papilloedema, diabetic or hypertensive retinopathy, or subhyaloid haemorrhage. The locked-in syndrome is rare; in a lower brainstem lesion patients are awake but can only control their eye movements (see page 570).
Look at the position of the eyes. Particular cranial nerve palsies may cause deviation of an eye in various directions. The sixth nerve is particularly vulnerable to damage because of its long intracranial course. Deviation of both eyes to one side in the unconscious patient may be due to a destructive lesion in a cerebral hemisphere, which causes fixed deviation towards the side of the lesion. An irritative (epileptic) focus causes the direction of gaze to be away from the lesion. Upward or downward eye deviation suggests a brainstem problem. Trapping of the globe or extraocular muscles by fracture may also lead to an abnormal eye position or to an abnormal eye movement.
Perform doll’s eye testing by lifting the patient’s eyelids and rolling the head from side to side. When vestibular reflexes are intact (i.e. an intact brainstem), the eyes maintain their fixation as if looking at an object in the distance, but change their position relative to the head. This is the normal ‘doll’s eye phenomenon’. Brainstem lesions or drugs affecting the brainstem cause the eyes to move with the head, so that fixation is not maintained.
Ears and nostrils
Look for any bleeding or drainage of cerebrospinal fluid (the latter indicating a skull fracture). A watery discharge can be simply tested for glucose. The presence of glucose confirms that it is cerebrospinal fluid.
Tongue and mouth
Trauma may indicate a previous seizure, and corrosion around the mouth may indicate ingestion of a corrosive poison. Gum hyperplasia suggests that the patient may be taking phenytoin for epilepsy. Smell the breath for evidence of alcohol poisoning, diabetic ketosis, hepatic coma or uraemia. Remember that alcohol ingestion may be associated with head injury. Test the gag reflex; its absence may indicate brainstem disease or deep coma, but is not a specific sign. Bite marks on the tongue suggest that an epileptic seizure may have been the cause of unconsciousness.
UPPER AND LOWER LIMBS
Look for injection marks (drug addiction, diabetes mellitus). Test tone in the normal way and by picking up the arm and letting it fall. Compare each side, assessing for evidence of hemiplegia. In coma and acute cerebral hemiplegia, the muscle stretch reflexes may be normal or reduced at first on the paralysed side. Later the muscle stretch reflexes become increased and the cutaneous reflexes are absent.
Test for pain sensation by placing a pen over a distal finger or toe just below the nail bed. Press firmly and note if there is arm or leg withdrawal. Test all limbs. There will be no response to pain if sensation is absent or if the coma is deep. If sensation is intact but the limb is paralysed, there may be grimacing with movement of the other limbs.
The presence of grimacing or purposeful movements is important. Segmental reflexes alone can cause the limb to move in response to pain.
URINE
Note whether there is incontinence. Test the urine for glucose and ketones (diabetic ketoacidosis), protein (uraemia) and blood (trauma).
BLOOD GLUCOSE
Always prick the patient’s finger, place a drop of blood on an impregnated test strip and test for hypoglycaemia or hyperglycaemia. If this cannot be done immediately, give the patient a bolus of intravenous glucose anyway (which will not usually harm the patient in diabetic ketoacidosis, but will save the life of a patient with hypoglycaemia). If there is any suspicion of Wernicke’s encephalopathy, thiamine must be given as well.
TEMPERATURE
Hypothermia (e.g. exposure or hypothyroidism) or fever (e.g. meningitis) must be looked for.
COMA SCALE
It is most useful to score the depth of coma, as changes in the level of consciousness can then be judged more objectively (see Table 35.4).
References
1. Goldstein, LB, Matchar, DB. Clinical assessment of stroke. (The Rational Clinical Examination. JAMA. 1994; 271:1114–1120. [Discusses the limitations of physical examination in identifying the lesion.].
2. Runchey, S, McGee, S. Does this patient have a hemorrhagic stroke? Clinical findings distinguishing hemorrhagic stroke from ischemic stroke. JAMA. 2010; 303(22):2280–2286. [Nothing works well enough to avoid neuroimaging, but a history of headache, seizures and vomiting and finding neck stiffness and hypertension increase the probability of a subarachnoid haemorrhage. Finding a carotid bruit decreases the likelihood.].
3. Sauve, JS, Laupacis, A, Ostbyte, T, et al. Does this patient have a clinically important carotid bruit? (The Rational Clinical Examination. JAMA. 1993; 270:2843–2846. [Describes how to interpret the findings of a carotid bruit. Distinguishing high grade from moderate symptomatic carotid stenosis based on the bruit itself is difficult and the absence of a bruit does not mean the absence of a significant carotid stenosis.].
4. Scherer, K, Bedlack, RS, Simel, DL. Does this patient have myasthenia gravis. JAMA. 2005; 293:1906–1914. [Speech failure when speaking over a prolonged period and the peek test increased the likelihood of myasthenia; their absence was unhelpful.].
5. Rao, G, Fisch, L, Srinivason, S, et al. Does this patient have Parkinson disease. JAMA. 2003; 289:347–353. [Rigidity, the glabella tap and walking (heel–toe) seem to be useful signs of Parkinsonism.].
aJacques Jean Lhermitte (1877–1939), French neurologist and neuropsychiatrist.
bCharles Edouard Brown-Séquard (1817–1894) succeeded Claude Bernard at the College de France. He was the son of an American sea captain (pirate) and a French woman. He was born in Mauritius at a time when it was under British rule. With this background he roved around the world, working in Paris, Mauritius, London and New York. His syndrome usually arose from failed murder attempts. Traditionally Mauritian cane cutters, when trying to murder someone, used a very long thin knife that was slipped between the ribs from behind, to cut the aorta or penetrate the heart. Only such a knife could have caused a cord hemitransection.
cNicholaus Friedreich (1825–1882), German physician, described this in 1863. He was professor of pathology at Heidelberg. Pes cavus is also called Friedreich’s foot.
dThe concepts of hypotonia, rebound and pendular jerks in cerebellar disease stem from Gordon Holmes’ 1917 description of signs in acute unilateral cerebellar disease. They may well not exist in other cerebellar problems. Students will, however, still be expected to know how to test for these signs.
eThomas Sydenham (1624–89). He was a captain in Cromwell’s army and became the most famous English physician of his time, providing clinical descriptions of gout (from which he suffered), fevers, hysteria and venereal disease. He was called the Father of English Clinical Medicine.
fGeorge Huntington (1850–1916), American general practitioner. He described this disease in his only clinical paper in 1872, when he was 22.
gWilliam Battle (1855–1936), surgeon, St Thomas’s Hospital, London.