Chapter 10 Adverse Drug Reactions and Drug Interactions
The use of medications to alleviate ailments and to combat diseases has been common for centuries. Along with the development of therapeutic remedies came knowledge of the poisonous qualities of the ingredients. In modern times this evolved into the concept of adverse drug reactions and knowledge that in some instances these are dose-related (type A adverse drug reactions) but under other circumstances are unpredictable (type B adverse drug reactions). With the increasing use of multiple drug therapies, drug interactions are now a cause for concern because a drug interaction may result in loss of effi cacy (decreased effect) or the development of toxicity (enhanced effect). Many studies have confi rmed that adverse drug reactions and drug interactions are major clinical problems, accounting for a signifi cant number of hospital admissions, extended hospital stays and substantial costs to the health-care system. It is important that health-care professionals be aware of the adverse reaction and drug interaction profi les of drugs and be ever vigilant for the occurrence of adverse outcomes.
Key abbreviations
HERBS and medicinal products have been used for treating ailments and diseases for centuries. Ebers Papyrus (1550 BC) lists more than 700 remedies and describes in detail the procedures for preparing and administering them. Some of the individuals treated would certainly have suffered an adverse reaction, especially with the use of lizard’s blood, domestic animal excreta, tortoise bile, cat uterus and perhaps with the more reasonable ingredients of castor oil, squill and opium.
The relationship between a remedy and a poison was entrenched in the early teachings of the Chinese and Mayans. People learnt to avoid medicinal substances that were poisonous. The ancient Greeks later described the concept of a medicine and a poison in scientific terms, and in 1758 William Withering described both the therapeutic benefits and the adverse effects of digitalis. Throughout the centuries, the use of medicinal products has gone hand in hand with reports of adverse drug reactions (ADRs).
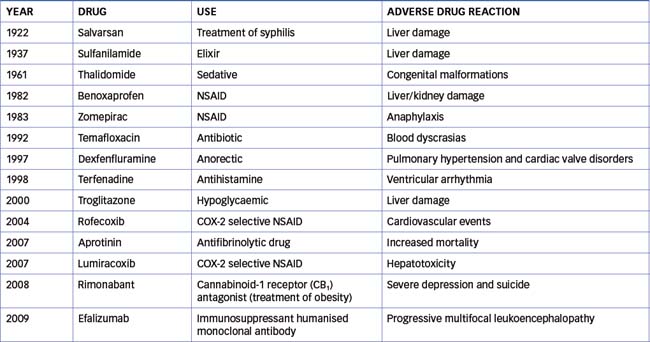
Public concern about ADRs arose in the late 19th century because of the number of sudden deaths associated with the use of chloroform. This led to the development of investigative committees such as the Food and Drug Administration (FDA) in the USA, which establishes the safety of new drugs before marketing. Despite regulatory frameworks, there have been many notable incidences of ADRs that have resulted in withdrawal of the offending drug (Table 10-1). Public interest in the safety of drugs has increased as a result of better communication between consumers and health-care professionals.
Definitions
An adverse drug reaction (ADR) has been defined as ‘any response to a drug which is noxious, unintended, and which occurs at doses normally (and appropriately) used in man for the prophylaxis, diagnosis, or therapy of disease’ (World Health Organization 1984). This definition has been in use for the last 26 years and at various times has been modified slightly, because the word ‘noxious’ is perhaps not correct in the context of the definition. An alternative definition has been proposed: ‘an appreciably harmful or unpleasant reaction, resulting from an intervention related to the use of a medicinal product, which predicts hazard from future administration and warrants prevention or specific treatment, or alteration of the dosage regimen, or withdrawal of the product’ (Edwards & Aronson 2000). Clinical responses to an ADR include modifying the dose, discontinuing the drug, hospitalising the patient or providing supportive measures. This definition does not encompass the situations of drug overdose, drug withdrawal, drug abuse or error in administration. The latter is included within the definition of an adverse drug event.
An adverse drug event (ADE) is defined as an ‘injury resulting from medical intervention related to a drug’ (Bates et al 1995): simply stated, an adverse event occurs while a person is taking a drug, but it is not necessarily ‘due to the drug’. Examples of ADEs include under- or overmedication resulting from misuse or malfunction of infusion pumps or devices; aspiration pneumonia resulting from drug overdose; and errors in ordering, dispensing or administration. These incidents are usually investigated and may be attributed to simple mistakes such as picking up the wrong syringe, giving treatment to the wrong body site or giving the wrong treatment. Under WHO guidelines, these types of incidents are not classed as ADRs.
The term ‘side effect’ is often used by health-care professionals and often appears in drug advertisements and consumer information. Often this term is interpreted by individuals to mean that the adverse reaction is insignificant (non-deleterious) or medically trivial and in general acceptable. In fact a side effect occurs via a mechanism different from the pharmacological effect and may be dose-related or not dose-related. An example of a side effect is the dose-related anticholinergic effect of tricyclic antidepressants as this is not associated with the therapeutic effect. The term ‘adverse effect’ is preferable to side effect, and generally relates to an unwanted effect that occurs via a different mechanism to the pharmacological effect and may or may not be dose-related. As an example, anaphylaxis with penicillin is both an adverse effect and an adverse drug reaction. The difference is that an adverse effect is ‘seen from the point of view of the drug, whereas an adverse reaction is seen from the point of view of the patient’ (Edwards & Aronson 2000). Throughout the literature, however, despite a difference in their definitions, in the disciplines of pharmacology, pharmacy and clinical toxicology the terms adverse effect and adverse drug reaction are used interchangeably.
Incidence of adverse drug reactions
Over the period 2005–2006 in Australia the total expenditure on medications was $11,501 million, of which $10,551 million was on prescription pharmaceuticals (Australian Institute of Health and Welfare 2008). In most developed countries the elderly take on average 4–5 prescription drugs and 2 OTC drugs at any one time. Taking multiple types of drugs contributes to the incidence of ADRs. In addition to prescribed medications and those bought over the counter, Australians have embraced the use of complementary and alternative medicines. The trend has been strongest among women, and includes the use of herbal medicines, aromatherapy oils and ginseng. The estimated annual retail sales of complementary medicines in Australia are estimated to be in the range of $800 million to $1 billion.
ADRs occur in people of all ages and are twice as common in women. They are a major cause of morbidity and mortality, especially in the elderly. A survey conducted in a major New South Wales teaching hospital found that 30% of elderly patients were taking 6–10 types of medications and 13% took more than 10 types each day (Nair 1999). A review of Australian studies published over the period 1988–1996 found that 2.4%–3.6% of all hospital admissions were reported to be drug-related, and 6%–7% of emergency admissions, 12% of all admissions to medical wards and 15%–22% of all emergency admissions among the elderly were drug-related. Between 32% and 69% of drug-related admissions were reported as definitely or possibly preventable (Roughead et al 1998). The drugs most commonly implicated were antihypertensives, anticoagulants, cardiovascular drugs, cytotoxics and nonsteroidal anti-inflammatory drugs (NSAIDs). Cytotoxic and anticoagulant agents were also the drugs most often involved in serious ADRs in general practice settings in France (Lacoste-Roussillon et al 2001). Similarly the drugs most commonly implicated in fatal ADRs in Sweden are antithrombotic drugs, NSAIDs, antidepressants and cardiovascular drugs (Wester et al 2007).
Little attention has been paid to the incidence of ADRs in neonates, infants, children and adolescents. Before release of a new drug, few if any studies are undertaken in children because of questions of ethics, responsibility, cost and regulations. This often leads prescribers to estimate dosage and hence increases the risk of ADRs. A recent analysis of prospective paediatric studies from the UK, USA and Spain reported that the incidence of ADRs in hospitalised children was 9.53%. The overall rate of hospital admissions due to ADRs was 2.09%, of which 39.3% involved life-threatening reactions. In the outpatient setting the overall incidence of ADRs in children was 1.46% (Impicciatore et al 2001). These data clearly show ADRs as a significant health issue in children.
Classification of adverse drug reactions
The current classification system is not ideal, and not every ADR may fit perfectly into one of the categories. It is generally accepted that there are two main categories of ADR, type A (augmented) and type B (bizarre), and two subordinate categories, type C (chronic) and type D (delayed). A further two classes include end-of-use or withdrawal effects (type E) and unexpected failure of therapy (type F).
TYPE A (AUGMENTED, DOSE-RELATED) ADRs are characterised by:
Factors predisposing to type A reactions include the dose, pharmaceutical variation in drug formulation, pharmacokinetic variation (e.g. renal failure), pharmacodynamic variation (e.g. altered fluid and electrolyte balance) and drug–drug interactions (e.g. inhibition of metabolism of one drug by another concomitantly administered drug). Examples include:
TYPE B (BIZARRE, NON-DOSE-RELATED) ADRs are characterised by:
These reactions are less common but often cause death. Factors contributing to type B reactions include pharmaceutical variation, receptor abnormalities, unmasking of a biological deficiency (e.g. glucose-6-phosphate dehydrogenase deficiency), abnormalities in drug metabolism (e.g. slow acetylators of the antituberculosis drug isoniazid), drug allergy (see next section) and drug–drug interactions (e.g. rare incidence of hepatitis) (Pirmohamed et al 1998). Examples include interstitial nephritis with the use of NSAIDs and eosinophilia with the use of anticonvulsants such as carbamazepine and phenytoin.
TYPE C (CHRONIC, DOSE-RELATED AND TIME-RELATED) ADRs are characterised by occurrence as a consequence of longterm use. Examples of reactions in this category include:
TYPE D (DELAYED, TIME-RELATED) ADRs are characterised by the appearance of delayed effects. These may be acceptable if the benefit of drug therapy outweighs the risk, as in the case of irreversible infertility in young persons receiving cytotoxic drugs for malignancies. In general, however, they are considered unacceptable. Examples include carcinogenesis (e.g. the association of lymphoma with immunosuppressive drugs) and teratogenesis.
TYPE E (END-OF-USE, WITHDRAWAL) ADRs are fortunately uncommon and are related to withdrawal of a drug. They include opiate withdrawal syndrome and myocardial ischaemia after abrupt cessation of β-blockers.
TYPE F (FAILURE, UNEXPECTED FAILURE OF THERAPY) ADRs are increasingly common and are often caused by a drug interaction (e.g. inadequate dose of the oral contraceptive when a drug that induces the metabolism of oestrogen is administered concomitantly).
Drug allergy
A drug allergy, or hypersensitivity, is a type B ADR. Drug allergies are characterised by:
The diagnosis of a drug allergy is often difficult to establish because there are no reliable laboratory tests that can identify the relevant drug, and in some cases the symptoms can imitate infectious disease symptoms. The situation may be easier if the drug administered is notorious for producing an allergic reaction (e.g. penicillin), but it is difficult if the drug used is seldom reported to produce an allergic reaction.
Some drugs can produce a pseudoallergic reaction, i.e. one resembling an allergic reaction but for which there is no immunological basis. Usually these occur as a result of mast cell degranulation and subsequent release of histamine. Clinically they resemble the type I hypersensitivity reaction but they do not involve drug-specific IGE. An example of a pseudoallergic reaction is the release of histamine that occurs with opiates (e.g morphine), vancomycin and radiocontrast media.
Allergic reactions to drugs generally follow the type I–IV classification (see Chapter 47). Table 10-2 lists the types of reactions, the main clinical manifestation and examples of drugs commonly implicated.
Table 10-2 Allergic drug reactions
| Type/reaction | Clinical manifestations | Examples of drugs |
| I Immediate hypersensitivity | Urticaria, anaphylaxis, angio-oedema, bronchospasm | Penicillins, streptomycin, local anaesthetics, neuromuscular blocking drugs, radiological contrast media |
| II Antibody-dependent cytotoxic | Cytopenia, vasculitis, haemolytic anaemia | Quinine, quinidine, rifampicin, metronidazole |
| III Complex-mediated | Serum sickness, vasculitis, interstitial nephritis | Anticonvulsants, antibiotics, hydralazine, diuretics |
| IV Cell-mediated or delayed hypersensitivity | Contact sensitivity | Local anaesthetic creams, antihistamine creams |
Immune-modulating drugs and adverse drug reactions
The use of drugs that modulate the immune system is common for the treatment of diseases such as cancer, rheumatoid arthritis, multiple sclerosis, inflammatory bowel disease and lupus. Although beneficial in many clinical settings the use of immune-modulating drugs is also associated with a significant number of ADRs (Table 10.3). Suppression of the immune response increases the risk of infection and cancer and paradoxically some of these drugs can also induce hypersensitivity (type B reactions) and/or autoimmune syndromes (Uetrecht 2009).
Table 10-3 Commonly used immune-modulating drugs and associated adrs
| Immune-modulating Drug | Therapeutic effect | Adverse Drug Reactions |
| Corticosteroids | Immune suppression; decreased expression of nitric oxide synthase; decreased expression of intracellular adhesion molecules | Increased risk of infections such as Pneumocystis carinii and systemic fungal infections |
| Cytotoxic drugs | Inhibition of cell proliferation | Increased risk of infections (e.g. herpes zoster, cytomegalolvirus) and life-threatening infections; increased risk of malignancies |
| Calcineurin inhibitors | Immune suppression | Increased risk of lymphomas and nonmelanoma skin cancers |
| Protein biological agents e.g. antibodies, interferons, antiadhesion molecules | Immune-modulating | Formation of autoantibodies; increased risk of infection and malignancies e.g. lymphomas (infliximab); progressive multifocal leukoencephalopathy (rituximab) |
| Sphingosine agonists | Immune suppression | Increased risk of infections |
Risk factors for developing an adverse drug reaction
Risk factors for ADRs are specific to both the person and the drug.
Factors relating to the person include:
Factors specific to the drug include:
Pharmacogenomics and adverse drug reactions
The role of genetics in predisposing individuals to ADRs has been known since the late 1950s through the discovery of enzyme deficiencies such as pseudocholinesterase and the link to succinylcholine apnoea. This easily recognised and diagnosed enzyme deficiency led many to conclude that we would be able to identify using genetic tests those individuals that were more likely to develop ADRs to some drugs. This concept although attractive has been replaced by the realisation that pharmacogenomics will aid in the prediction and prevention of only a limited number of ADRs, and as such the uptake of pharmacogenetic testing has been rather variable and limited (Alfirevic & Pirmohamed 2008). The era of pharmacogenomics and the prescribing of medications tailored to individual genotypes is on the horizon but genotyping would remove only one of the factors contributing to the risk of ADRs, leaving age, a history of atopy, polypharmacy and inappropriate prescribing to deal with. Examples of pharmacogenetic tests for the prevention of ADRs include:
Drug–drug interactions
A drug–drug interaction (commonly shortened to ‘drug interaction’ or DDI) occurs when the drug’s pharmacological effect is altered by another drug: that is, an increased therapeutic and/or adverse effect or a decreased therapeutic and/or adverse effect. Moreover, drug interactions often are unanticipated and go unrecognised and the clinical and economic importance is frequently underestimated.
Frequency of drug interactions
The exact frequency of drug interactions is unknown, although anecdotal evidence suggests that they are relatively common and result in a significant number of hospital admissions. The possibility of a drug interaction exists whenever two or more medications are prescribed to an individual, and the likelihood of an interaction will grow as the number of medications used increases. For a person taking two drugs the estimated incidence of a DDI is 5.6% while a person taking six drugs has an estimated incidence of a DDI of 56%. One of the problems in identifying a DDI is that the physiological/biochemical changes resulting from a DDI may be masked by, or confused with, the clinical signs and symptoms of the illness or other comorbidities. Additionally, it may be difficult to identify which ‘drugs’ are involved in a DDI especially if the prescriber is unaware of other medications the patient is taking, for example OTC drugs and/or complementary/alterative medications.
Individuals at greatest risk of a drug interaction are:
Drug interactions are of greatest concern with drugs that have a narrow therapeutic index. Even a small change in the concentration of the drug available at the target site (e.g. receptor, enzyme) can lead to a major alteration in response. For example:
Interactions involving drugs with a wide therapeutic index (e.g. penicillin antibiotics, β-adrenoceptor antagonists) cause fewer problems. Knowledge of the mechanisms of drug interactions is essential to enable health professionals to prevent interactions occurring (wherever possible) and to systematically analyse potentially new drug interactions. Indeed, analysis of known and potential interactions is critical in the planning of a therapeutic regimen.
Although drug interactions may have deleterious effects, they may also be used to advantage. For example, antimicrobial drugs with different mechanisms of action are commonly used in combination for increased effectiveness in treating bacterial infections. Similarly, combinations of drugs are commonly used in cancer chemotherapy and in the treatment of tuberculosis.
Classification of drug interactions
Drug interactions are broadly classified, according to their pharmacological mechanism, into either pharmacodynamic or pharmacokinetic interactions.
Pharmacodynamic drug interactions
Pharmacodynamic drug interactions may be ‘direct’ or ‘indirect’. Direct pharmacodynamic interactions involve effects at a common target, additively (and possibly potentiation) or antagonism due to actions at different sites in an organ. An example of antagonism at a common receptor site is the concurrent use of a β2-adrenoceptor agonist (used in the treatment of asthma, e.g. salbutamol) and a non-selective β-adrenoceptor antagonist (used in the treatment of hypertension, e.g. propranolol). Both drugs have opposing effects at the same receptor (i.e. the β2-adrenoceptor). Unintentional interactions of this type should not occur because they are so obvious from the known pharmacology of the drugs.
Examples of direct pharmacodynamic interactions involving drugs with different mechanisms of action include the following:
An indirect pharmacodynamic interaction occurs when the pharmacological effects of one drug alter the response to another drug, even though the two effects are not themselves directly related. Common examples include the following:
Pharmacokinetic drug interactions
The plasma concentration of a drug may be altered by interactions occurring during absorption, distribution, metabolism and excretion:
Coadministered drugs may also decrease the extent of drug absorption. Whereas changes in the rate of absorption generally affect only the time for onset of action, changes in extent of absorption can alter response. For example, cholestyramine is a bile acidbinding resin used in the treatment of hypercholesterolaemia. Unfortunately, cholestyramine also binds other drugs, reducing the amount of drug that is absorbed. As cholestyramine reduces the absorption of corticosteroids, digoxin, thyroxine and warfarin (and probably other drugs), these drugs should be administered either several hours before or after the cholestyramine dose.
Induction results in increased enzyme activity and drug metabolism, hence the steady-state blood concentration will decrease with the possibility of therapeutic failure (see Chapters 6 and 8)
Drugs known to cause induction are generally nonselective in their effects on CYP enzymes. Examples include:
Inhibitory drug interactions are relatively common, and inhibition of metabolism increases steady-state blood concentration and the likelihood of drug toxicity. Some drugs, notably the H2 antagonist cimetidine, inhibit the activity of most CYP enzymes (although UGT is unaffected). Conversely, probenecid inhibits most UGT enzymes (without affecting CYP). Most inhibitory interactions are relatively selective for one or a limited number of drug-metabolising enzymes, as they most commonly arise from competition for metabolism at the enzyme active site. It is generally not correct to refer to a drug as ‘an inhibitor of drug metabolism’. Rather, a drug will normally selectively inhibit the metabolism of other drugs for a limited number of enzymes, and this specificity of interaction is used to predict and interpret metabolic drug interactions. Some selective inhibitors of CYP enzymes are shown in Table 10-4. As an example, fluoxetine causes interactions with many drugs metabolised by CYP2D6 (e.g. other antidepressants and perhexiline), which generally requires a reduction of the dose. The clearances of drugs metabolised by CYP3A4 (see Clinical Interest Box 10-1) are similarly decreased by the commonly used antibiotic erythromycin, again generally requiring a dose reduction. Examples of CYP metabolic drug interactions include:
Table 10-4 Inhibitors of CYP enzymes
| CYP | Inhibitors |
| CYP1A2 | Fluvoxamine, propranolol |
| CYP2C8 | Gemfibrozil, trimethoprim |
| CYP2C9 | Amiodarone, fluconazole, gemfibrozil, sulfonamide antibiotics |
| CYP2C19 | Fluvoxamine, moclobemide |
| CYP2D6 | Fluoxetine, paroxetine, perphenazine, quinidine, quinine, thioridazine |
| CYP3A4 | Clarithromycin, diltiazem, erythromycin, indinavir, itraconazole, ketoconazole, nelfinavir, ritonavir, roxithromycin, verapamil |
Clinical interest Box 10-1 Colchicine and CYP3a4 inhibitors
Colchicine is used in the treatment of acute gouty arthritis (see Chapter 47) and has a narrow therapeutic index and dose-limiting severe adverse effects that include nausea, vomiting, diarrhoea and abdominal pain. Metabolised by CYP3A4, colchicine is subject to multiple DDIs involving drugs that inhibit CYP3A4 and hence increase the plasma concentration of colchicine and risk of ADRs. Four cases of colchicine toxicity have been reported to the TGA, three of which were fatal. One of the fatal cases involved the concomitant use of clarithromycin (a CYP3A4 inhibitor) as part of therapy for Helicobacter pylori eradication. The patient developed multi-organ failure and massive myelosuppression.
Source: Australian Adverse Drug Reactions Bulletin, Volume 27, No. 5, October 2008.
Up-to-date information on CYP DDIs can be found at the Indiana University, Division of Clinical Pharmacology website (http://medicine.iupui.edu/clinpharm/ddis/).
Metabolic drug interactions also occur with other drug-metabolising enzymes. Probenecid is a ‘universal’ inhibitor of drug glucuronidation, and there is evidence to suggest that rifampicin, phenobarbitone, phenytoin and carbamazepine may induce numerous UGT enzymes. For example, fluconazole appears to inhibit only UGT2B7 (which metabolises morphine and zidovudine). In the case of morphine, a rise in the plasma concentration may result in respiratory depression.
A potentially fatal interaction occurs when azathioprine and allopurinol are coadministered. Allopurinol is an inhibitor of the enzyme xanthine oxidase, and is used in the treatment of gout and gouty arthritis. Azathioprine (used mainly in the treatment of cancer) is converted to an active metabolite, 6-mercaptopurine, which is subsequently cleared by xanthine oxidase. Coadministration of azathioprine and allopurinol leads to accumulation of 6-mercaptopurine, resulting in potentially life-threatening bone marrow suppression.
Metabolic drug interactions involving nutrients and herbal medicines
Although there is wide appreciation of drug interactions, interactions between nutrients and/or food components and herbal remedies are often not considered and, in fact, may be discounted. Chemicals present in food may alter the activity of drug-metabolising enzymes. Notable in this regard are chemicals present in grapefruit (but not orange) juice that inhibit the activity of CYP3A4 present in the gastrointestinal tract. (CYP3A4 is localised in both liver and small bowel.) The enzyme present in the small bowel appears to contribute significantly to the first-pass metabolism of numerous CYP3A4 substrates. Thus, the bioavailability of a number of drugs, such as cyclosporin, felodipine, midazolam, triazolam and verapamil, increases significantly when they are taken with grapefruit juice, which enhances the potential for toxicity (see Clinical Interest Box 10-2).
Clinical interest Box 10-2 Grapefruit juice–drug interactions
Food–drug interactions are potentially an everyday occurrence. Many scientific findings have been serendipitous and perhaps none more so than an unexpected observation during a clinical study that the grapefruit juice that was used to mask the taste of ethanol led to an increase in the oral bioavailability of the drug being studied. Subsequent research indicated that the effect was due to the inhibition of CYP3A4 activity in the intestine, which resulted in a reduction of presystemic metabolism and subsequent increased bioavailability. This interaction is most clinically relevant with certain calcium channel blockers (e.g. verapamil), saquinavir, cyclosporin, midazolam and triazolam, and may be important with cisapride.
Although the active components in grapefruit juice respon sible for the inhibition of enzyme activity have not yet been fully identified, the effect is observed with a single glass of juice and can persist for 24 hours: ‘Since grocers do not take a drug history, physicians, pharmacists and other health profes sionals should educate patients about consumption of grapefruit juice with medications’ (Bailey et al 1998).
Currently there is intense interest in the effects of herbal medicines on drug metabolism and the consequences of herb–drug interactions, which are based on the same pharmacokinetic and pharmacodynamic mechanisms as DDIs. Herbal medicines are used widely, particularly by women, given the perception that ‘natural’ products are a safe and effective alternative to pharmaceuticals. As plant products, herbal medicines typically contain hundreds of different chemicals; it is not surprising that some of these will alter the activity of drug-metabolising enzymes. In the USA the seven top-selling herbal medicines in descending order are ginkgo, St John’s wort, ginseng, kava, saw palmetto, garlic and echinacea (Bressler 2005).
Important in terms of drug interaction is St John’s wort, which is taken to treat the symptoms of depression. St John’s wort contains chemicals called hyperforins, which mimic the effects of rifampicin as an inducer of CYP enzymes. Consumption for two weeks significantly induces the activity of both hepatic and intestinal CYP3A4 and the intestinal transporter P-glycoprotein (see Chapter 6). Thus the clearances of amitriptyline, carbamazepine, cyclosporin, HIV protease inhibitors, warfarin and several other drugs have been shown to be increased in subjects taking St John’s wort, with risk of therapeutic failure. Similarly, the plasma concentrations of alprazolam and midazolam have been shown to be reduced in healthy volunteers taking St John’s wort. Furthermore, oral contraceptives contain ethinyloestradiol, which is metabolised by CYP3A4. Concomitant administration of St John’s wort increases the metabolism of ethinyloestradiol and unplanned pregnancy has been reported as an issue in women who use oral contraceptive steroids and St John’s wort. Not surprisingly given its use in depression, interactions with ‘synthetic’ antidepressants have been reported. The combination of St John’s wort and the serotonin re-uptake inhibitors (sertraline, paroxetine, nefazadone and venlafaxine) may result in headaches, changes in mental state, tremors, autonomic instability, GI upset, myalgias and motor restlessness (symptoms similar to central serotonin excess). These symptoms may be explained by inhibition of serotonin reuptake in the brain by St John’s wort.
There is evidence to suggest that multiple herbal products interact (pharmacodynamically or pharmacokinetically) with ‘pharmaceutical’ drugs, and studies investigating the mechanisms involved and quantifying the magnitudes of any interactions are ongoing. Interactions between herbal medicines and conventional drugs are no longer just a theoretical possibility, and it is likely that their incidence is more common than anticipated initially. It is essential that a patient’s exposure to herbal medicines be determined when assessing the potential for drug interactions (refer to Appendix 3).
Strategies for limiting adverse drug reactions and drug interactions
Hospital admissions for ADRs are a significant and expensive health problem worldwide. The evidence from numerous studies indicates that ADRs may be impossible to avoid completely, but given the high predictability of many ADRs, there is significant room for improvement in reducing their incidence. Strategies for reducing/limiting ADRs include:
There is an overwhelming amount of information published on drug interactions, and sources such as the Australian Medicines Handbook, CARM (see Clinical Interest Box 10-3) and the website of Professor D Flockhart, School of Medicine, Indiana University, are excellent. Continued education of health professionals is essential, as new drugs enter the marketplace on an annual basis. Specific strategies for limiting drug interactions (in addition to those listed above) include:
Clinical interest Box 10-3 Centre for adverse reactions monitoring

Pharmacovigilance activities in New Zealand include the reporting of adverse drug reactions. Based in Dunedin, the Centre for Adverse Reactions Monitoring (CARM) collects and evaluates adverse reaction reports from health professionals. These are reported for medicines, herbal products and dietary supplements, and are held in a database of over 50,000 reports. Individual patients who suffer from lifethreatening adverse reactions have CARM warnings or danger alerts recorded against their National Health Index (NHI) numbers; the information is accessible through hospital systems and contributes to the safe use of medicines.
Anonymised data from various pharmacovigilance sources in New Zealand are fed into the database of the World Health Organization’s International Drug Monitoring Programme based in Uppsala, Sweden. CARM also provides feedback to the Medicines Adverse Reactions Committee which in turn makes recommendations to Medsafe, the regulatory medicines authority in New Zealand. Although the activities of CARM are focused on spontaneous reports, the Intensive Medicines Monitoring Programme (IMMP) monitors new medicines using a method called prescription event monitoring. IMMP operates in close collaboration with the spontaneous reporting programme and together they supplement the adverse reactions database.
Adapted from: http://carm.otago.ac.nz/CARM.asp [27 January 2010].
Our knowledge of adverse drug reactions and drug interactions continues to advance but our ability to apply this knowledge to patient populations seems to lag behind. A multidisciplinary approach involving the consumers, the prescribers and the carers is essential if we are to minimise both the personal and societal impact of ADRs and DDIs.
Key points

 An adverse drug reaction is defined as any response to a drug that is noxious and unintended, and that occurs at doses normally (and appropriately) used for the prophylaxis, diagnosis or therapy of disease. Public interest in the safety of drugs has increased as a result of better communication between patients and health-care professionals. Adverse drug reactions occur in people of all ages and are twice as common in women. They are a major cause of morbidity and mortality, especially in the elderly. There are two main categories of adverse drug reactions, type A (predictable) and type B (unpredictable), and subordinate categories, type C (chronic use), type D (delayed reactions), type E (end-of-use or withdrawal effects) and type F (unexpected failure of therapy). The type B adverse drug reactions include immunological reactions such as drug allergy and hypersensitivity. The diagnosis of a drug allergy can often be difficult to establish because there are no reliable laboratory tests that can identify the relevant drug, and the symptoms can sometimes imitate infectious disease symptoms (e.g. fever). Risk factors for developing an ADR include age, gender, presence of concurrent disease, genetics, a history of prior drug reaction, the drug dose and the duration and frequency of drug use. Drug interactions are broadly classified according to their pharmacological mechanisms: that is, pharmacodynamic or pharmacokinetic. Direct pharmacodynamic interactions involve effects at a common target, additively (and possibly potentiation) or antagonism due to actions at different sites in an organ. A pharmacodynamic interaction occurs when the pharmacological effects of one drug alter the response to another drug, even though the two types of effects are not themselves directly related.
An adverse drug reaction is defined as any response to a drug that is noxious and unintended, and that occurs at doses normally (and appropriately) used for the prophylaxis, diagnosis or therapy of disease. Public interest in the safety of drugs has increased as a result of better communication between patients and health-care professionals. Adverse drug reactions occur in people of all ages and are twice as common in women. They are a major cause of morbidity and mortality, especially in the elderly. There are two main categories of adverse drug reactions, type A (predictable) and type B (unpredictable), and subordinate categories, type C (chronic use), type D (delayed reactions), type E (end-of-use or withdrawal effects) and type F (unexpected failure of therapy). The type B adverse drug reactions include immunological reactions such as drug allergy and hypersensitivity. The diagnosis of a drug allergy can often be difficult to establish because there are no reliable laboratory tests that can identify the relevant drug, and the symptoms can sometimes imitate infectious disease symptoms (e.g. fever). Risk factors for developing an ADR include age, gender, presence of concurrent disease, genetics, a history of prior drug reaction, the drug dose and the duration and frequency of drug use. Drug interactions are broadly classified according to their pharmacological mechanisms: that is, pharmacodynamic or pharmacokinetic. Direct pharmacodynamic interactions involve effects at a common target, additively (and possibly potentiation) or antagonism due to actions at different sites in an organ. A pharmacodynamic interaction occurs when the pharmacological effects of one drug alter the response to another drug, even though the two types of effects are not themselves directly related.Review exercises
References and further reading
Alfirevic A., Pirmohamed M. Adverse drug reactions and pharmacogenomics: recent advances. Personalized Medicine. 2008;5:11-23.
Australian Institute of Health and Welfare. Australia’s Health 2008. Cat. no. AUS 99. Canberra: AIHW, 2008.
Bailey D.G., Arnold M.O., Spence J.D. Grapefruit juice–drug interactions. British Journal of Clinical Pharmacology. 1998;46:101-110.
Bates D.W., Cullen D.J., Laird N., Petersen L.A., Small S.D., et al. Incidence of adverse drug events and potential adverse drug events: implications for prevention. Journal of the American Medical Association. 1995;274:29-34.
Bressler R. Herb–drug interactions: interactions between kava and prescription medications. Geriatrics. 2005;60:24-65.
deShazo R.D., Kemp S.F. Allergic reactions to drugs and biologic agents. Journal of the American Medical Association. 1997;278:1895-1906.
Edwards I.R., Aronson J.K. Adverse drug reactions: definitions, diagnosis, and management. Lancet. 2000;356:1255-1259.
Flockhart DA. Drug Interactions: Cytochrome P450 Drug Interaction Table. Indiana University School of Medicine. 2009. Available: http://medicine.iupui.edu/clinpharm/ddis/table.asp [27 January 2010].
Hansten P.D. Drug interaction management. Pharmacy World Science. 2003;25:94-97.
Impicciatore P., Choonara I., Clarkson A., Provasi D., Pandolfini C., Bonati M. Incidence of adverse drug reactions in paediatric in/out-patients: a systematic review and metaanalysis of prospective studies. British Journal of Clinical Pharmacology. 2001;52:77-83.
Izzo A.A. Herb–drug interactions: an overview of the clinical evidence. Fundamental and Clinical Pharmacology. 2004;19:1-16.
Lacoste-Roussillon C., Pouyanne P., Haramburu F., Miremont G., Begaud B. Incidence of serious adverse drug reactions in general practice: a prospective study. Clinical Pharmacology and Therapeutics. 2001;69:458-462.
Nair B. Older people and medications: what is the right prescription? [editorial]. Australian Prescriber. 1999;22:130-131.
Pirmohamed M., Breckenridge A.M., Kitteringham N.R., Park B.K. Adverse drug reactions. British Medical Journal. 1998;316:1295-1298.
Riedl M., Casillas A.M. Adverse drug reactions: types and treatment options. American Family Physician. 2003;68:1781-1790.
Roughead E.E., Gilbert A.L., Primrose J.G., Sansom L.N. Drugrelated hospital admissions: a review of Australian studies published 1988–1996. Medical Journal of Australia. 1998;168(8):405-408.
Uetrecht J. Immune-mediated adverse drug reactions. Chemical Research Toxicology. 2009;22:24-34.
Wester K., Jonsson A.K., Spigset O., Druid H., Hagg S. Incidence of fatal adverse drug reactions: a population based study. British Journal of Clinical Pharmacology. 2007;65:573-579.
World Health Organization. Collaborating Centers for International Drug Monitoring. Geneva: WHO, 1984 (WHO publication DEM/NC/84:153.(E)).