Chapter 17 Antiepileptic drugs
Epilepsy is a common neurological illness involving recurrent epileptic seizures that may affect parts or the whole of the cerebral hemispheres and cause muscle twitching and impaired consciousness. It affects one in every 200 adults in Western societies; mild seizures in children are much more common. This chapter discusses classifications of the types of epilepsy and the various antiepileptic drugs available to treat this disorder. Clinical aspects of therapy of epilepsies, including choice of drug, compliance, therapeutic drug monitoring, lifestyle aspects and drug use in particular patient groups are considered.
Key abbreviations
Key background: epilepsy
EPILEPSY is a group of chronic neurological disorders characterised by sporadic, recurrent episodes of convulsive seizures resulting from occasional excessive disorderly discharges in neuronal pathways across the cerebral cortex. The seizures can lead to loss of consciousness, muscle jerking, sensory disturbances and abnormal behaviour. Although nearly 70% of seizures do not have an identifiable cause (primary, or idiopathic, epilepsy), around 30% have an underlying cause (secondary epilepsy) that may be treatable, e.g. head injury, cerebrovascular infarct or haemorrhage, infection, brain tumour, drug toxicity or a metabolic imbalance. It is estimated that about 2% of people will suffer seizures at some stage in their lives. The aim of therapy is to avoid factors that tend to trigger attacks (see Clinical Interest Box 17-1) and to find the drug or drugs that will effectively control the seizures and restore physiological homeostasis with a minimum of undesirable side effects or drug interactions.
Clinical interest Box 17-1 Triggers of epileptic seizures
Secondary, or organic, epilepsy frequently follows head injury or a focal lesion in the CNS, such as an infection or tumour, or birth damage or an endocrine disorder. These lesions may set off the high-frequency discharges in brain neuronal pathways that lead to the seizure.
Idiopathic epilepsy has no known organic cause, but many factors are likely to act as triggers to an attack: hyperventilation, trauma, lack of sleep, poor nutrition, fever, stress, bright lights—especially flashing lights of a TV set or strobe lights such as in a discotheque—or changes in blood levels of hormones, fluids or electrolytes.
A wide range of drugs has been implicated as potentially able to cause convulsions or lower the seizure threshold, including many common drugs (aspirin, some sedating antihistamines, anticholinesterases, antidepressants, antipsychotics, antibiotics, local and general anaesthetics, vaccines, neuroleptic agents, oral contraceptives, narcotic analgesics, bronchodilators), social drugs (alcohol, caffeine, cocaine, cannabis) and even some antiepileptic drugs themselves (clonazepam, sodium valproate). See ‘Drugs which may cause seizures’ Table in Australian Medicines Handbook, 2010.
Classification of seizures
The choice of appropriate antiepileptic (anticonvulsant) drugs for treating individual patients depends on accurate diagnosis and classification of the seizure type. A full medical history, laboratory tests, a neurological examination and electroencephalogram (EEG; see Figure 17-1) are necessary for classification. Computed tomography (CAT) scans and magnetic resonance imaging may also be used to detect anatomical defects or to locate small focal brain lesions. Identifying specific seizure types is critical to the development of a treatment plan.
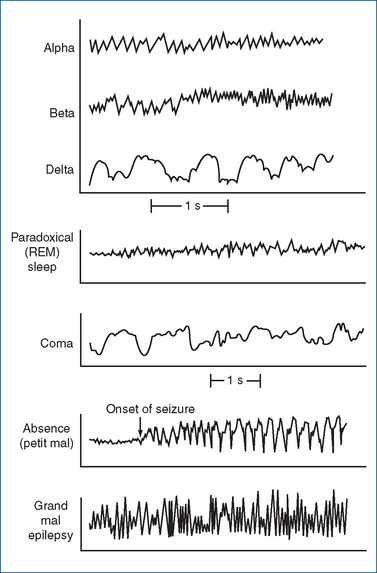
Figure 17-1 The electroencephalogram during sleep and in epilepsy.
Alpha waves: awake, eyes closed (8–13 cycles/second).
Beta waves: mental activity (14–30 cycles/second).
Delta waves: deep sleep (1–5 cycles/second).
REM sleep: EEG pattern similar to that for alpha waves when awake.
Epilepsy—generalised absence (petit mal) seizure: EEG tracing shows spikes and waves (3 cycles/second).
Epilepsy—generalised tonic–clonicseizure (grand mal): tracing shows spikes of clonic phase.
Adapted from: Tortora & Grabowski 2000; Vander et al 1998; Rang et al 2007.
The terminology currently used with epileptic seizures is shown in Clinical Interest Box 17-2, on the International Classification of Seizures. Mixed seizures are seen in some individuals who have more than one type of seizure disorder. In practice, many health-care providers still use the former common terms: grand mal, jacksonian, psychomotor and petit mal epilepsy.
Clinical interest Box 17-2 International classification of seizures
Partial seizures
Simple (no impairment of consciousness)
Complex (impaired consciousness)
Partial seizures
Partial simple motor (jacksonian) epilepsy is described as a type of focal seizure; it is associated with irritation of a specific part of the brain. A single body part such as a finger or an extremity may jerk, and such movements may end spontaneously or spread over the whole musculature. Consciousness may not be lost unless the seizure develops into a generalised convulsion.
Partial complex (psychomotor) seizures are characterised by brief alterations in consciousness, unusual stereotyped movements (such as chewing or swallowing movements) repeated over and over, changes in temperament, confusion and feelings of unreality. These seizures may spread and evolve to generalised grand mal seizures, and are likely to be resistant to therapy with drugs.
Generalised seizures
Generalised absence1seizures, simple or complex (petit mal), are most often seen in childhood and consist of temporary lapses in consciousness that last a few seconds. Generally, children appear to stare into space or daydream, are inattentive and may exhibit a few rhythmic movements of the eyes (slight blinking), head or hands, but they do not convulse. They may have many attacks in a single day. The EEG records a 3/second spike-wave pattern (see Figure 17-1). Sometimes an attack of generalised absence seizures is followed by a generalised tonic–clonic seizure. When the child reaches adulthood, other types of seizures may occur.
Myoclonic seizures are characterised by bilaterally symmetrical muscle jerks, often with loss of consciousness.
Tonic–clonic generalised (grand mal) epilepsy is the type most commonly seen. Such attacks may be characterised by an aura and a sudden loss of consciousness and motor control. The aura is specific to the individual; it may consist of numbness, visual disturbance or a particular form of dizziness that warns the person of an approaching seizure. The person falls forcefully due to continuous tonic spasm (stiffening, increased muscle tone), which may be followed by a series of clonic (rapid, synchronous jerking) muscular contractions. The eyes roll upwards, the arms flex and the legs extend. The force of the muscular contractions causes air to be forced out of the lungs, which accounts for the cry that the person may make on falling. Respiration is suspended temporarily, the skin becomes sweaty and cyanotic, saliva flows, incontinence may occur and the person may froth at the mouth and bite the tongue if it gets caught between the teeth. No pain is felt, as the person is deeply unconscious. When the seizure subsides, the individual regains partial consciousness, may complain of aching and then tends to fall into a deep sleep.
Status epilepticus is a clinical emergency. It is the state of recurrent seizures for more than 30 minutes without an intervening period of consciousness. A 10%–20% mortality rate results from anoxia in this state. The major cause of status epilepticus is non-compliance with an antiepileptic drug regimen; other causes include cerebral infarction, central nervous system (CNS) tumour or infection, trauma or low blood concentration of calcium or glucose.
Relation of age to seizures
A relationship exists between age and onset of an epileptic seizure state. Most people with epilepsy have their initial seizure before the age of 20; however, seizures may have an onset at any age in life. Idiopathic (of no defined aetiology, or genetic in origin or cause) seizures are often diagnosed between the ages of 5 and 20. Onset before or after this age period is often from non-idiopathic (identifiable) causes and is termed ‘symptomatic’ (acquired, organic) epilepsy.
Neonates
Neonatal seizures occur in children younger than 1 month. Among the more common causes of neonatal seizures are congenital defects or malformation of the brain, infections (meningitis, encephalitis, abscess) within the CNS, hypoxia (in utero or during delivery), premature birth and defects in metabolism.
Infants
In infants younger than 2 years, the seizure types most frequently described are sudden, brief contractions of the head, neck and trunk, in runs lasting several minutes. The infantile spasm (West syndrome) is not classified as a partial or generalised type of epileptic seizure itself. Among the more common causes of infant seizures are those reported above for neonatal seizures, plus infection, exposure to toxins (in utero, caused by maternal exposure to or use, misuse or abuse of drugs), maternal exposure to X-rays and postnatal trauma. Infantile spasms may lead to atonic epileptic seizures seen in later development (ages 2–5 years).
Children
In children 2–5 years of age the seizure types often diagnosed include generalised tonic–clonic seizures and atonic seizures. The causes are similar to those mentioned above for newborns and infants, with the addition of chronic diseases involving the CNS. The parents of the child may wrongly believe the child has a behavioural disorder rather than a treatable seizure disorder. Febrile convulsions are frequently associated with a fever from a source outside the CNS.
In children 6 years and over the seizure types that emerge most commonly on diagnosis are absence seizures and generalised tonic–clonic seizures, which may be idiopathic in origin. Sometimes the convulsive seizure is associated with a brain tumour, vascular disease, brain infection, head trauma (accident or sport), fever, growth of scar tissue, presence of a toxin or a poison or drug withdrawal. (See Clinical Interest Box 17-3 for more information on paediatric implications.)
Clinical interest Box 17-3 Paediatric implications
Problems related to phenytoin use in children
Young adults
Within the 16–25-year age group, generalised seizures may be idiopathic in origin. The partial seizure and generalised seizures may result from the use of alcohol; social or recreational drug use; drug abuse, misuse or withdrawal; or head injury. Patients who had been previously stabilised on antiepileptic therapies may require monitoring and review of treatment as pharmacokinetic parameters change; teenagers often become embarrassed about taking medications for chronic conditions and compliance with drug therapy may drop.
Elderly
People over 60 years of age are at greater risk of seizure episodes. Common causes of seizures in the elderly include trauma, brain tumours, vascular disease, embolic stroke and Alzheimer’s disease. In this population, osteoporosis and cerebrovascular disease are common and therefore seizures may lead to fractures, intracranial bleeding, neurological deficit, cognitive impairment and severe limitation in daily functioning. (See Clinical Interest Box 17-4 for more information on geriatric implications.)
Clinical interest Box 17-4 Geriatric implications
Antiepileptic therapy
Clinical aspects
While secondary seizures usually respond to correction of the underlying condition and perhaps short-term use of antiepileptic agents, primary recurrent seizures require long-term antiepileptic drug therapy. The main goal of drug therapy is to control or prevent the recurrence of the seizure disorder while ensuring that unwanted effects of the treatment do not handicap the person more than further seizures would. Lifestyle aspects, including issues related to emergency management of seizures, sleep patterns, employment, driving and other hazardous activities, use of social drugs, sport, relationships and pregnancy, also need to be discussed with patients.
Monotherapy
If possible, epilepsy is controlled with one antiepileptic drug (monotherapy) introduced slowly; if maximum tolerated doses of one drug are not effective, another drug is substituted. Only if seizure control cannot be achieved with any safe drug is a second drug added to the regimen. About 70% of patients can be well controlled with one drug; half of the remainder may require two to three drugs, and the rest may remain refractory to treatment or require surgery.
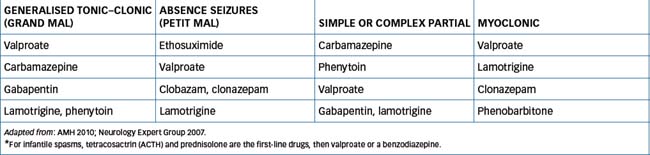
Choice of antiepileptic drug
As stated earlier, different types of seizure may respond to particular antiepileptic agents, hence the importance of accurate diagnosis of seizure type. The currently recommended drugs are listed in Table 17-1; other second-line agents may be tried if the first-line drugs are not successful in controlling seizures.
Compliance
Good compliance with chronic, life-long therapy is often difficult. Lack of compliance can lead to drug withdrawal symptoms, including lack of seizure control and onset of convulsions. Therapeutic monitoring is usually carried out regularly (see below), partly to facilitate adjustment of doses and also to check compliance and toxicity. Compliance is improved if the patient (and family, teachers and carers) understands the condition and the importance of regular therapy (see Clinical Interest Box 17-5).
Clinical interest Box 17-5 Epilepsy support groups
Epilepsy Action Australia (http://www.epilepsy.org.au/) has affiliated groups with branches in major cities. Its mission is the enhancement of the quality of life of people living with epilepsy, and it provides education and support services to children and adults with epilepsy or other seizure disorders across Australia, so that all Australians affected by seizures will have the opportunity to live confident lives.
Its team of nurses and educators is equipped with a rich base of technical expertise, built up during five decades of services to the community. They run workshops, educational sessions, epilepsy awareness programs, clinics, parent support groups, referral services, family camps, forums and continuing education for health professionals. Library facilities offer information on specific issues; loans of books, videos, periodicals, clippings collections and education kits; and publications including newsletters and brochures.
Publications are available on topics such as epilepsy medications, managing epilepsy, driving and epilepsy, employing people with epilepsy and sudden unexplained deaths in epilepsy.
Special situations
Infancy
Febrile seizures in infancy occur commonly with mild infections and fevers. While distressing to parents, these do not indicate that the child will develop epilepsy. Paracetamol reduces fever symptoms in children but does not prevent febrile convulsions; in susceptible infants, phenobarbitone or sodium valproate prevents recurrences.
Epilepsy in women
In some women, seizure frequency increases during menstruation. Antiepileptic drugs may reduce the effectiveness of the oral contraceptive pill, leading to breakthrough bleeding, pill failure and pregnancy (see Drug Interactions 17-1).
Clinical interest Box 17-6 Eclampsia—toxaemia of pregnancy
Eclampsia is a possible serious complication of pregnancy in which dangerous seizures occur with a high maternal and fetal mortality.
It is always preceded by pre-eclampsia, a condition characterised by elevated blood pressure, oedema of the extremities (hands, feet and ankles) and proteinurea. This occurs in about 5% of all pregnancies; regular antenatal care with monitoring of maternal blood pressure and weight gain helps warn of impending pre-eclampsia.
Pregnancy-induced hypertension is associated with primigravidae (first-time mothers), diabetes, multiple pregnancy and underlying renal or hypertensive disease.
The treatment plan for pre-eclampsia is to control the elevated blood pressure, prevent seizures, maintain renal function and generally provide optimal conditions for the fetus. This approach is primarily symptomatic because the only real cure for this syndrome is delivery of the baby.
The usual antiepileptic drugs (diazepam, pheno barbitone or phenytoin) are administered parenterally to prevent convulsions and for sedation. Magnesium sulfate has useful CNS-depressant effects as well as reducing neuromuscular transmission and hence muscular contractions; however, decreased muscle tone and respiratory depression may be seen in the neonate.
The mother should be monitored for up to 2 days after delivery, as seizures may still occur in the immediate postpartum period.
Drug interactions 17-1 Antiepileptics
| Drug | Possible effects and management |
| Barbiturates, benzodiazepines, phenytoin | Can cause raised plasma concentrations of other AEDs and of many other drugs, and increase toxicity; dosages of these drugs may need to be lowered |
| Barbiturates, carbamazepine, phenytoin and sodium valproate | Can induce drug-metabolising enzymes and cause lowered plasma concentrations of other AEDs and even of themselves (and of other drugs, including hormones, cardiovascular drugs and antimicrobial agents) and reduce seizure control; drug dosages may need to be raised |
| Barbiturates, carbamazepine, phenytoin and topiramate | Induce hepatic enzymes and increase metabolism of many hormonal contraceptives, excluding levonorgestrel IUD and medroxyprogesterone depot which are preferred hormonal contraceptives |
| Drugs that lower the convulsive threshold (including antidepressants, antipsychotics and anticholinesterases)—usually contraindicated in epilepsy | Potential danger; AED requirements are altered |
| Drugs that inhibit CYP3A4, including cimetidine, –conazole antifungals, protease inhibitors, grapefruit juice and quinolone antibiotics | May inhibit metabolism of some benzodiazepines, carbamazepine and tiagabine, and prolong their effects; reduce dose of AED |
| Drugs that induce CYP3A4, including corticosteroids, rifampicin, some antivirals and St John’s wort | May increase metabolism of some benzodiazepines, carbamazepine and tiagabine, and reduce their effects; increase dose of AED or use another drug |
| Sodium valproate | May reduce platelet aggregation and prolong bleeding time; monitor effects if giving with other drugs that affect bleeding times |
No antiepileptic drug (AED) is completely safe to use in pregnancy; many are potentially teratogenic or can affect cognitive development of the child (see pregnancy safety categories in Drugs at a Glance at the end of this chapter). All the ‘old’ AEDs are implicated, especially high-dose sodium valproate. Fetal abnormalities are 2–3 times more likely in babies whose mothers took AEDs during pregnancy; hence treatment of epileptic women of childbearing age must always include consideration of these risks. Increasing the intake of folic acid (5 mg/day) in women taking an AED before conception and for the first 3 months thereafter may decrease the risk of spina bifida in the fetus. Overall, seizure control is of the highest priority, as seizures during pregnancy pose a greater risk to mother and fetus than do AEDs. (See Lander [2008] and Clinical Interest Box 17-6 for seizure prevention in eclampsia.)
Although breastfeeding is not usually contraindicated, CNS-depressant drugs may pass into breast milk, so the infant should be monitored for drowsiness or feeding difficulties.
Treatment of children or elderly patients
See Clinical Interest Boxes 17-3 and 17-4, respectively, for paediatric and geriatric implications.
Status epilepticus

Generalised convulsive status epilepticus is a medical emergency, with significant morbidity and mortality. First-line drugs are fast-acting benzodiazepines (diazepam, midazolam), then a long-acting AED such as IV phenytoin, phenobarbitone, sodium valproate or levetiracetam. If these are ineffective, general anaesthetics are required.
Maintenance therapy
After a drug regimen is found that successfully controls seizures without significant adverse effects, it is continued until the patient has been seizure-free for 2–3 years. Plasma drug levels are occasionally monitored to check compliance (non-compliance being the commonest cause of failure of seizure control). Carers should watch for delayed adverse effects such as gum hypertrophy, poor school performance or liver failure.
Therapeutic monitoring
Epilepsy is not a stable condition and seizures may occur at irregular intervals; hence it is difficult to control clinically. As with other conditions in which relapses and remissions occur, therapeutic monitoring can be useful in optimising drug therapy. Plasma levels of AEDs are often monitored, especially for carbamazepine, phenobarbitone and phenytoin (but are not useful for benzodiazepines, vigabatrine or valproate). This is helpful for establishing baseline data, predicting toxicity, detecting interactions that affect blood levels and checking compliance.
Published therapeutic plasma ranges of various antiepileptic agents are used as a guide to therapy. This allows the prescriber to adjust dosages according to the individual’s requirement, to reach the therapeutic range (see Figure 17-2) or to achieve seizure control without adverse effects. While the time needed to reach a steady-state drug level in plasma is usually five times the elimination half-life of the drug, dosage requirements of individual patients are unpredictable, so monitoring of plasma concentrations can help keep patients within the target range.
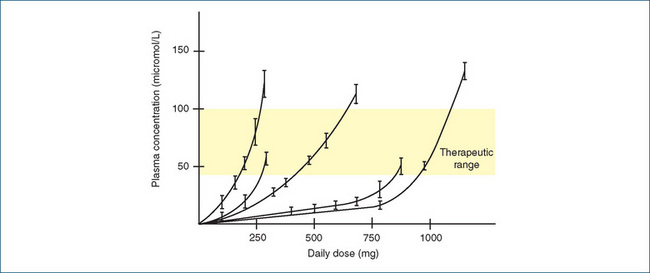
Figure 17-2 Non-linear relation between daily dose of phenytoin and steady-state plasma concentration in five individual human subjects. Although the therapeutic range is quite broad (40–100 micromol/L), the daily dose required varies greatly between individuals, and for any indi vidual the dose has to be adjusted in small increments to keep within the acceptable plasma concentration range. Note: 1 mmol is equal to about 250 mg phenytoin. Adapted from: Rang et al 2007 with permission; data redrawn from Richens and Dunlop 1975.
Parenteral use of antiepileptics
Antiepileptic drugs are administered parenterally (usually IV or IM) in acute conditions involving seizures, such as eclampsia, status epilepticus, severe recurrent seizures, tetanus, seizure during neurosurgery and in toxicity due to convulsant drugs. Phenobarbitone, phenytoin and the benzodiazepines diazepam and lorazepam are given by injection; there is variable absorption of diazepam after IM injection depending on the muscle mass injected. If it is impossible to administer the drugs by injection because of severe convulsions, the rectal route may be used.
Discontinuing antiepileptic therapy
A diagnosis of epilepsy no longer implies a lifetime of drug therapy: studies have indicated that AEDs may be safely withdrawn from up to 70% of patients who are seizurefree for at least two years. In long-term studies, seizures recurred in 12%–36% of the patients who were monitored for more than 20 years after complete drug withdrawal. There are some risk factors that help in predicting which patients may have seizure recurrence after drug withdrawal. These include:
Withdrawal from phenytoin or valproate is associated with a higher rate of recurrence than for other drugs.
Antiepileptic medications should be tapered down slowly (in a non-emergency situation) to avoid the potential for inducing seizures and status epilepticus. If the patient is taking more than one AED, each drug is withdrawn separately and slowly over several months.
Drugs that may cause seizures
A wide variety of drugs have been noted to reduce the seizure threshold and are thus potentially dangerous in people predisposed to or with epilepsy. Groups of drugs known to have this effect include anticholinesterases, antipsychotics, some antihistamines, interferons, monoamine oxidase inhibitors, quinolone antibiotics and some other antimicrobials, many neurological drugs, selective serotonin reuptake inhibitors and other serotonergic drugs, and tricyclic antidepressants, plus many individual drugs. Metabolites of drugs can also cause seizures, e.g. norpethidine. Such drugs should be used cautiously if at all in conjunction with antiepileptic drugs. (See also ‘Triggers of epileptic seizures’ in Clinical Interest Box 17-1 and a useful list of drugs that may cause seizures given in an Appendix of the Australian Medicines Handbook.)
Use of antiepileptics in neuropathic pain
Antiepileptic agents are sometimes used in conditions other than epilepsy, notably in pain syndromes such as neuropathic pain (see Chapter 15) that do not respond to the usual analgesic drugs. In particular, carbamazepine is used in trigeminal neuralgia, sodium valproate in migraine headache, some of the newer antiepileptics such as gabapentin and pregabalin in painful diabetic neuropathy and post-herpetic neuralgia, and lamotrigine in spinal cord injury pain. However, understanding of the mechanism of efficacy of antiepileptic drugs in these conditions is still limited (see Besson et al [2008]).
Antiepileptic drugs
The ideal antiepileptic drug
Although there is no ideal antiepileptic drug, the following characteristics would be desirable:
The major drugs used in the treatment of partial seizures and generalised tonic–clonic seizures are sodium valproate, phenytoin, carbamazepine, benzodiazepines (especially clonazepam) and the barbiturate phenobarbitone. Newer AEDs include gabapentin, lamotrigine, vigabatrin, tiaga bine, topiramate and levetiracetam. Typical AEDs will be discussed briefly, with more detailed drug monographs on topiramate and phenytoin as examples from different groups.
As no antiepileptic agent is ideal, there is considerable current research into new mechanisms of action of AEDs. It is very expensive to carry out large-scale, high-powered clinical trials, particularly in a chronic condition like epilepsy in which the manifestations (seizures) occur occasionally and randomly, so there is little level-one evidence on the efficacy and safety of the newer AEDs from randomised controlled clinical trials.
Mechanisms of action
The aim of using a drug to prevent seizures is to decrease the likelihood of excessive neuronal transmission in CNS pathways; however, most CNS depressants are too sedating to be clinically useful in epilepsy. For those that are effective AEDs, the exact modes and sites of action are complex and incompletely understood. A common mechanism of action relates to stabilisation of the nerve cell membrane by altering cation transport, especially that of sodium, potassium or calcium.
Drugs enhancing GABA inhibition
Neuronal activity is reduced by drugs that enhance gamma (γ)-aminobutyric acid (GABA)-mediated inhibition, e.g. by facilitating GABA-mediated opening of chloride channels; by inhibiting GABA-transaminase, the enzyme that inactivates GABA; or by inhibiting the GABA reuptake processes. The benzodiazepines such as clonazepam, barbiturates (phenobarbitone) and some newer drugs (vigabatrin, tiagabine and topiramate) act by these mechanisms. Some GABA-ergic drugs are now being trialled in anxiety, affective disorders and pain conditions.
Drugs inhibiting sodium channel function
Drugs that inhibit sodium channel function block repetitive depolarisation of neurons; these drugs appear to block preferentially the excitation of cells that are firing repetitively. For example, phenytoin blocks sodium channels and possibly also calcium influx, thus stabilising cell membrane excitability and reducing the spread of seizure discharge. Carbamazepine also inactivates sodium channels, which alters neuronal excitability and decreases synaptic transmission. Other examples of drugs acting by this mechanism are sodium valproate and lamotrigine.
Drugs inhibiting calcium channel function
Compounds may also reduce CNS neuronal excitation by blocking the excitatory amino acid transmitter glutamate.2 Three types of glutamate receptors are of interest: NMDA (N-methyl-D-aspartate) receptors, kainate receptors and AMPA (alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid) receptors (see Table 14-1). All are ionotropic receptors involving calcium channels. Antagonists of these receptors are being assessed for clinical efficacy in neurodegenerative conditions and in epilepsy, anxiety, hyperalgesia and psychosis.
Miscellaneous drugs
This group includes ethosuximide, gabapentin and some drugs that are more commonly used in other clinical conditions but have useful membrane-stabilising actions (acetazolamide, sulthiame, adrenocorticotrophic hormone [ACTH]); their mechanisms of action vary. The precise mechanism of action of levetiracetam is unknown.
Adverse drug reactions
Adverse drug reactions are common with AEDs; in particular, CNS depression is likely (see Table 17-2). While each drug has its own adverse-effect profile, common reactions include excessive sedation, ataxia and confusion; depression of the cardiovascular and respiratory centres; and adverse cognitive effects such as impaired memory and learning, which can impair progress of children in school. Paradoxical reactions (excitation rather than depression) sometimes occur with benzodiazepines and barbiturates, especially in children and the elderly. Adverse effects on the gastrointestinal tract and haema tological system are also possible.
Table 17-2 Central nervous system adverse effects of some antiepileptic drugs
| Drug | Behavioural alterations | Cognitive effects |
| Phenobarbitone | Confusion, physical dependence, altered mood; may cause a paradoxical effect, especially in children or the elderly (e.g. increased activity or excitement, irritability, altered sleep patterns, increased tiredness) | Impaired judgement, short-term memory impairment, decreased attention span |
| Carbamazepine | Drowsiness, anorexia, increased irritability, insomnia, behavioural changes (especially in children), depression | Less than phenytoin, phenobarbitone or primidone |
| Clonazepam | Drowsiness, dizziness, ataxia, impaired speech and vision, hysteria; tolerance; dependence and withdrawal symptoms after cessation; paradoxical reactions (excitement, insomnia, agitation) | Anterograde amnesia, memory impairment, confusion, impaired concentration |
| Lamotrigine | Dizziness, ataxia, somnolence, hyperkinesia | |
| Phenytoin | Insomnia/sedation, fatigue, increased clumsiness, confusion, mood alterations, agitation, vertigo | Decreased attention span, decreased ability to solve problems, impaired learning |
| Sodium valproate | Sedation, ataxia, depression, increased appetite and weight, tremor; hyperactivity and aggression in children | Stupor (associated with excess dosage or polytherapy) |
Anticonvulsant hypersensitivity syndrome is a rare but potentially serious reaction related to the CYP450 metabolites of barbiturates, carbamazepine and analogues and phenytoin. It can occur after 1–4 weeks treatment and involves fever, rash that can develop into Stevens–Johnson syndrome and impairment of systemic organs.
Drug interactions
Drug interactions with AEDs are common, variable and unpredictable; they need to be anticipated and monitored whenever adding or withdrawing an antiepileptic drug. Typical examples are shown in Drug Interactions 17-1. The possibilities for variable plasma drug concentrations and responses can make treatment confusing; a good general rule is ‘see an antiepileptic, think drug interactions’. Regular monitoring may be required until the patient is stabilised on an effective drug regimen. (As it is difficult to generalise drug interaction effects with AEDs, a reference text should be consulted for details of adverse drug interactions with individual antiepileptic agents; see especially Australian Medicines Handbook Appendix and Diaz et al [2008].)
Antiepileptics that enhance GABA inhibition
Benzodiazepines
The benzodiazepines used as AEDs are those with long half-lives, such as clonazepam, diazepam, nitrazepam and clobazam. These drugs are discussed in detail in Chapter 16 as the standard sedative–hypnotic and antianxiety agents (see Drug Monograph 16-1 for details on diazepam). Their mechanism of action is to occupy specific benzodiazepine-binding sites in the GABA receptor and hence facilitate GABA-mediated inhibition of neural activity and suppress the propagation of seizure activity produced by foci in the cortex, thalamus and limbic areas. Dependence, tolerance and withdrawal reactions are common problems.
Clonazepam is a long-acting benzodiazepine used to treat absence seizures, myoclonic seizure disorders and status epilepticus. It has been used alone but more often it is prescribed as an adjunct to other AEDs to establish seizure control. Clonazepam is given orally or by slow IV injection. Diazepam can be given orally or IV, or rectally when IV injection is not possible, e.g. in prolonged convulsions in children. Diazepam has a relatively rapid onset but short duration of antiepileptic action, as it redistributes rapidly out of the CNS, then has a long elimination half-life.
Drug interactions with other CNS depressants (including alcohol, which is contraindicated) are common. Severe withdrawal reactions and an increase in seizures may follow abrupt withdrawal of benzodiazepines. Dosage is usually individualised for each patient and increased as necessary. Elderly or debilitated persons and patients taking other CNS-depressant-type medications usually receive a smaller dose with a slower dosage increase.
Barbiturates
The mechanism of action for barbiturates is non-selective depression of the CNS via facilitation of chloride entry into cells at GABAA receptors (acting at a different site from those where GABA or benzodiazepines bind), hence enhancement of inhibitory systems that use GABA as a neurotransmitter. Barbiturates can also decrease excitatory neurotransmitter effects. There is a selective depressant action on the motor cortex even in small doses, which explains their use as AEDs. Depression of the ascending reticular formation decreases cortical stimuli, reducing wakefulness and alertness. High doses of barbiturates can induce anaesthesia.
Barbiturates, especially phenobarbitone, have been used for many years for the treatment of generalised tonic–clonic and partial seizures and for neonatal febrile convulsions and status epilepticus. Primidone, while strictly speaking not a barbiturate, has two active metabolites, phenobarbitone and phenylethyl-malonamide, which contribute to antiepileptic activity. Primidone has been used for control of generalised tonic–clonic (grand mal) and complex seizures but is less well tolerated than phenobarbitone alone and so is dropping out of use.
Adverse drug reactions and interactions
Barbiturates have a much lower therapeutic index (safety margin) than do the benzodiazepines; they were the classic ‘sleeping pills’ with which many people committed suicide or died after an inadvertent overdose. Large doses, especially when administered intravenously, depress the respiratory and vasomotor centres. Elderly or debilitated patients are especially sensitive to the CNS-depressant effects and can exhibit confusion, disorientation and mental depression. Paradoxical CNS stimulation can occur in children and some elderly people. Physical dependence on the drugs is common. Phenobarbitone parenteral solutions are highly alkaline and can cause local tissue necrosis.
Drug interactions are frequent, especially with other CNS depressants, including alcohol. The barbiturates are also the main group of drugs that induce hepatic drugmetabolising enzymes, thus enhancing the metabolism and inactivation of many other drugs, including anticoagulants, anticonvulsants, oral contraceptives and corticosteroids.
Vigabatrin
Vigabatrin also enhances GABA-mediated inhibition but by a very different mechanism: it is an irreversible inhibitor of GABA transaminase, the enzyme that inactivates GABA, and thereby allows a build-up of the neurotransmitter in synapses. It is a relatively new drug, indicated for adjunctive (add-on) treatment, especially of complex partial seizures, focal epilepsy and infantile spasms. It has a specific adverse effect on vision: it can cause an irreversible visual field constriction in 20%–40% of patients taking the drug, so visual fields should be tested before starting therapy, then every 3–6 months. Vigabatrin should only be prescribed when other treatments have proved unsuccessful.
Tiagabine and topiramate
Tiagabine is a GABA reuptake inhibitor and is indicated as adjunctive therapy in patients with partial seizures. Drug interactions may occur when tiagabine is given in combination with other AEDs such as carba mazepine, phenytoin, primidone or phenobarbitone. It has been reported that tiagabine clearance is increased by nearly 60% when combined with these AEDs; therefore tiagabine dosage increase may be necessary. Adverse reactions occur most commonly in the CNS and gastrointestinal tract.
Topiramate is another new AED (see Drug Monograph 17-1). Although the exact mechanism of action for topiramate is unknown, it has four useful properties:
Drug monograph 17-1 Topiramate
Topiramate is a relatively new AED that stabilises neuronal membranes both by blocking sodium channels, thus reducing the frequency of action potentials, and by enhancing inhibitory neuronal activity at kainate-type GABAA receptors. It is considered safer than some of the older AEDs; however, the newer drugs are currently considerably more expensive. Its mechanism of action in migraine is unknown.
Indications
Topiramate is indicated for monotherapy in newly diagnosed epilepsy, and in conversion to monotherapy, and as add-on therapy of partial onset seizures and primary generalised tonic–clonic seizures. It is also used for prophylaxis of migraine headaches in adults.
Pharmacokinetics
Topiramate is admin istered orally and is well and rapidly absorbed. It is distributed to the total body water, with low protein binding, reaching peak plasma concentration by 2–3 hours. It is not extensively metabolised unless metabolism has been enhanced by enzyme-inducers, and metabolites are inactive. It is mainly cleared by the kidneys, with a long half-life of approximately 21 hours; steady state is not reached for several days. Topiramate is not a potent inducer of drug-metabolising enzymes.
Drug interactions
As with all AEDs, there are potential additive effects with other CNS depressants. The metabolism of topiramate may be increased by drugs that induce drug-metabolising enzymes, including other AEDs such as carbamazepine and phenytoin, necessitating dose increase. Topiramate may increase the concentration of phenytoin in plasma.
Adverse reactions
The most common adverse effects are due to CNS depression and include cognitive impairment, ataxia and speech disorders; patients need to be warned against driving or operating machinery. Psychiatric disorders also occur, including confusion, mood disturbances and depression and amnesia. Other possible adverse effects include fatigue, diarrhoea, weight loss, reduced sweating and hyperthermia, nephrolithiasis, myopia and secondary angle-closure glaucoma, and metabolic acidosis.
Warnings and contraindications
Precautions are required before prescribing to patients predisposed to the adverse effects, especially renal stone formation, psychiatric disturbances, metabolic acidosis or glaucoma. In patients with reduced renal function, topiramate’s half-life may be even longer. The drug is classified B3 with respect to pregnancy safety; however, the risk to a baby of maternal seizures is much greater than the risk of malformations. Prophylactic dosing with folic acid 1 month before and 3 months after conception is advised.
Dosage and administration
Topiramate is available as tablets and as ‘sprinkle capsules’, which may be opened and the contents sprinkled on soft food before swallowing without chewing. The usual starting dose in adults is 25 mg/day, taken at night, gradually increasing to 100 mg/day; dosage in children is 0.5–1 mg/kg/day, increasing to 3–6 mg/kg/day to maximum 500 mg daily.
Topiramate is used as adjunctive therapy for partial and generalised seizures in adults. The main adverse reactions are CNS-depressant effects.
Antiepileptics that inhibit sodium channel functions
Phenytoin
The prototype hydantoin drug is phenytoin (diphenylhydantoin; Drug Monograph 17-2), which was developed from a search for an AED that would cause less sedation than the barbiturates. Phenytoin is recommended for the treatment of all types of epilepsy except absence seizures. It blocks voltage-dependent sodium channels, decreasing the propagation of seizures. It is particularly interesting from the pharmacokinetic point of view, as it has non-linear pharmacokinetic parameters, which often make clinical use of phenytoin difficult (see Figure 17-2 and Clinical Interest Box 17-7).
Indications
Phenytoin acts by blocking voltage- and usedependent sodium channels; it is more effective for generalised and partial seizures than for petit mal seizures. It is also frequently prescribed in combination with phenobarbitone and may be prescribed for patients to prevent seizures after surgery on the brain, after head trauma and for status epilepticus.
Pharmacokinetics
With most drugs, as the dose is increased the plasma drug concentration increases in a linear, arithmetic (direct) relationship; however, because of saturable metabolism, the non-linear pharmaco kinetics of phenytoin means that the dose–plasma concentration relationship is not linear, and a small rise in dose may cause an unexpectedly large rise in plasma drug levels (see Figure 17-2). Oral absorption of phenytoin is slow and variable (poor in neonates); it is highly bound to plasma albumin. The time to peak serum level is 1.5–3 hours and half-life varies with dose and serum level, ranging from 7 to 42 hours, with an average of about 24 hours. Steady-state levels are achieved after 7–10 days. It is inac tivated in the liver and metabolites excreted in the bile and in urine.
Drug interactions
There are many important drug interactions with phenytoin (see Drug Interactions 17-1 and Australian Medicines Handbook Appendix B). In particular, many drugs (including chloramphenicol, cimetidine, disulfiram, isoniazid, amiodarone, oral anticoagulants, allopurinol, omeprazole, imipramine, azole antifungals, sulfonamides) may inhibit the metabolism of phenytoin and hence prolong the halflife, leading to neurotoxic effects.
Adverse reactions
There are many dose-related neurotoxic effects (drowsiness, dizziness, confusion) at plasma concentrations >80 micromol/L (20 mg/L), also idiosyncratic reactions such as hirsutism, gingival hyperplasia with bleeding, sensitive gum tissue or overgrowth of gum tissue, acne and facial coarsening. Signs of overdose or toxicity include blurred or double vision, slurred speech, clumsiness, dizziness, confusion and hallucinations. In addition, signs of toxicity with intravenous phenytoin include cardiovascular collapse, CNS depression, ischaemia of distal extremities and hypotension. The rate of IV administration (25–50 mg/min) is critical, as severe cardiotoxic reactions and fatal outcomes have been reported with faster infusions.
Warnings and contraindications
Use with caution in pregnancy (category D) and in persons with drug allergies, diabetes mellitus, cardiac arrhythmias or liver or renal impairment. Women relying on oestrogen-containing contraceptives may require higher doses or should use nonhormonal contraception. Regular dental care is important to detect gum problems. Avoid use in persons with hydantoin hypersensitivity and use caution with similar compounds such as phenobarbitone and carbamazepine.
Dosage and administration
The usual adult dosage is 200–500 mg daily; however, careful monitoring and titration of dose is required. It should be noted for dosage calculations that 100 mg phenytoin sodium contains approximately 92 mg phenytoin. Phenytoin has very low water solubility and is provided in specially formulated IV solutions, which should not be mixed with other drugs or glucose solutions.
Clinical interest Box 17-7 Titrating phenytoin doses
As explained in the caption to Figure 17-2, for each individual patient the dose of phenytoin may need to be adjusted (titrated) frequently and precisely to keep the plasma concentration within the therapeutic range, quoted as 40–80 micromol/L (10–20 mg/L). This is done through regular monitoring of drug level in samples of the patient’s plasma and with the help of various formulations of phenytoin, which allow small changes in dose. As the half-life of phenytoin is long, about 24 hours in adults, it can take 5–10 days to reach steady state. Free phenytoin levels may need to be measured at steady state in patients with impaired protein binding (e.g. infants, or renal failure, hypoalbuminaemia, pregnancy).
The average (adult) dose is about 300 mg/day, with a range of 200–500 mg/day; however, owing to the non-linear pharmacokinetics, a small increase in dose, say from about 375 to 500 mg, may more than double the plasma concentration.
In Australia, phenytoin is available in several dose forms, including:
Fosphenytoin is a phenytoin analogue, a prodrug that is rapidly converted to phenytoin in the body. It has been formulated to overcome the problems with parenteral phenytoin (not very soluble and the injectable form is very alkaline and can cause venous irritation). Fosphenytoin is less alkaline and can be given IM for status epilepticus and to treat and prevent seizures associated with neurosurgery or head trauma.
Carbamazepine
Carbamazepine also blocks sodium channels, decreasing the propagation of seizures. The drug’s effects are somewhat similar to those of phenytoin. Carbamazepine is indicated in the treatment of generalised tonic–clonic seizures, partial complex seizures and psychomotor seizures and for mixed seizure patterns. It is also indicated in the treatment of neuropathic pain, such as that associated with trigeminal neuralgia, and for bipolar disorder and mania.
Pharmacokinetics
Oral absorption is slow and onset of action may range from hours to days, depending on the individual. Due to autoinduction of metabolism (i.e. it induces higher levels of the enzymes that metabolise it) it may take a month to reach a stable therapeutic serum level. Carbamazepine is metabolised in the liver (it has one active metabolite) and excreted primarily by the kidneys.
Adverse reactions
These include CNS depression, possible severe hypersensitivity reactions including skin reactions and depressed white cell counts and antidiuretic hormone-like effects.
Drug interactions
Again, there are many clinically significant drug interactions with carbamazepine. It enhances the metabolism and thus decreases the effectiveness of many drugs, including anticoagulants (warfarin), other AEDs and carbamazepine itself, corticosteroids and oral contraceptives. Plasma concentration should be monitored whenever any of these medications is added or discontinued in persons receiving carbamazepine, as dosage adjustment may be necessary. The half-life of carbamazepine is prolonged by drugs that inhibit CYP3A4 enzymes and by grapefruit juice.
Oxcarbazepine
Oxcarbazepine, an analogue of carbamazepine, has been developed to overcome some of the problems of the latter. Oxcarbazepine is less toxic and has fewer drug interactions; it is useful in adults and children with partial and generalised seizures uncontrolled by other drugs and has been used as both adjunctive and monotherapy. Hyponatraemia (low sodium concentrations) can develop, so it is recommended that plasma sodium concentration be monitored.
Sodium valproate
The mechanism by which sodium valproate exerts its antiepileptic effects has not been fully established. It may enhance brain levels of GABA and also block sodium, potassium and/or calcium channels. By competitive inhibition it may prevent the reuptake of GABA by glial cells and axonal terminals.
Indications
As can be seen from Table 17-1, sodium valproate is one of the most generally useful AEDs. It is indicated for use as sole or adjunctive therapy in the treatment of absence seizures, including petit mal seizures, and in patients with multiple seizure types, including partial (simple and complex), generalised, myoclonic or atonic seizures and also in bipolar disorder and migraine.
Pharmacokinetics
Sodium valproate is converted in the stomach to valproic acid, which is rapidly absorbed from the gastrointestinal tract; food delays absorption. Valproate has variable onset time and half-life (6–16 hours), depending on the formulation administered.
Adverse effects and drug interactions
These include drowsiness, tremors, mild gastric distress, hair thinning, weight gain, irregular menstruation, skin reactions and hepatotoxicity (especially in infants) or pancreatitis. Drug interactions occur, particularly with CNS depressants (alcohol, general anaesthetics, barbiturates), anticoagulants and aspirin (increased risk of bleeding), and with combinations of AEDs because of drug metabolism interactions (levels should be monitored to check toxicity or compliance). Valproate causes an increased risk of congenital malformations, including spina bifida, and is in Pregnancy Safety Category D.
Lamotrigine
Lamotrigine is believed to stabilise seizures by blocking sodium channels and thus inhibiting the release of excitatory neurotransmitters (glutamate and aspartate). It is indicated as adjunctive therapy for the treatment of partial seizures and generalised epilepsy. It has a long half-life (30 hours) that may be reduced by enzymeinducing drugs and female sex hormones but increased by sodium valproate.
Early clinical experience with lamotrigine has shown that there is a high risk of severe potentially life-threatening skin reactions, including toxic epidermal necrolysis, in particular with high dosage or when drug interactions prolong the half-life. Any rashes or skin reactions should be carefully evaluated and recommended doses not exceeded.
Lacosamide
Lacosamide has recently been approved in Australia for adjunctive therapy in partial seizures; it enhances slow inactivation of voltage-gated sodium channels and also binds to a protein (CRMP2) involved in neuronal differentiation, outgrowth and epileptogenesis. In clinical trials in patients poorly controlled on at least two other anticonvulsants who had added 200- or 400-mg oral doses of lacosamide, seizure frequency decreased by 35%–40%. Adverse reactions were dizziness, altered vision, headache and vomiting. As the drug is very new, there is little experience yet with its use in children or pregnant or lactating women.
Other antiepileptics
Ethosuximide
This is the only surviving member of the succinimide group of AEDs. These agents produce a variety of effects; by decreasing calcium conductance in the motor cortex they increase the seizure threshold and reduce the EEG spike-and-wave pattern of absence seizures. Ethosuximide has a long half-life, allowing once-daily administration. Common adverse reactions are disturbances in CNS and gastrointestinal functions. Other AEDs may increase the metabolism of ethosuximide, decreasing its effectiveness, or may change the pattern of seizures.
Gabapentin
Gabapentin is an AED that was designed as a GABA analogue but unexpectedly appears not to mimic the actions of GABA; the mechanism for its antiepileptic action is not yet established; however, it raises brain GABA levels and inhibits glutamate synthesis. It is indicated for the treatment of partial seizures with or without secondary generalisation and also for neuropathic pain, such as in diabetic neuropathy and post-herpetic neuralgia. Absorption is reduced with high doses and by antacids.
Pregabalin
Pregabalin is another GABA analogue that appears not to act via GABA-ergic mechanisms; it binds to calcium channels and also inhibits release of some excitatory transmitters. It is indicated for partial seizures and neuropathic pain. It is excreted almost 100% unchanged, so dosage is adjusted depending on renal clearance, and there are few drug interactions. Main adverse effects are CNS depression, weight gain and oedema.
Levetiracetam
Levetiracetam is a relatively new AED, indicated as adjunctive therapy for patients whose partial seizures are not well controlled with other drugs. Its mechanism of action is as yet unknown. Common adverse effects include somnolence, headache and altered behaviours, but long-term safety has not yet been established. There are few significant drug interactions.
Acetazolamide and sulthiame
Acetazolamide is a carbonic anhydrase inhibitor usually prescribed for the treatment of open-angle glaucoma. Its membrane-stabilising activity may be due to inhibition of carbonic anhydrase in the CNS, resulting in an increase in carbon dioxide that retards neuronal activity. Systemic metabolic acidosis may also play a part in its action. It is occasionally used in combination with other AEDs.
Sulthiame, another older AED, is also a carbonic anhydrase inhibitor. It is indicated mainly for childhood epilepsy and for temporal lobe and myoclonic seizures.
Miscellaneous and new drugs
Magnesium sulfate
Magnesium sulfate has a depressant effect on the CNS and reduces striated muscle contractions. It is used to treat toxaemia of pregnancy (see Clinical Interest Box 17-6).
Tetracosactrin and prednisolone
The most effective treatment for infantile myoclonic spasms is IM tetracosactrin (also known as corticotrophin or ACTH) or oral prednisolone. The mechanism of action is not understood but it is thought that the hormones may act as central neurotransmitters or neuromodulators. They improve the associated psychomotor retardation more effectively than do the usual AEDs.
New antiepileptic drugs
Since there is no ideal AED, the search is always on for new, safer drugs; recent discoveries include rufinamide,

carisbamate, eslicarbazepine and retigabine. Rufinamide shows promise in effective, well-tolerated treatment of Lennox–Gastaut syndrome, a serious paediatric epilepsy syndrome for which there are few treatment options.
Key points


 Epilepsy is characterised by sporadic recurrent episodes of convulsive seizures and is classified by extent (generalised/partial) and signs exhibited (loss of consciousness; muscle tone and twitching). The seizures may be idiopathic, triggered by external events or internal changes, or secondary to head injury or focal brain damage. Antiepileptic drug therapy to control seizures may be lifelong; choice of drug is determined by type of seizure, likely adverse drug reactions, other drugs that may interact and individual aspects such as pregnancy and compliance. Therapeutic monitoring is regularly carried out for some AEDs by measuring drug concentration in plasma samples; this helps check whether levels are in the therapeutic range and monitors compliance. While all CNS-depressant drugs may reduce seizure incidence, most are too sedating to be useful. The major drugs used to treat seizures act by enhancing GABAmediated inhibition of neural transmission or inhibit neurotransmission by blocking sodium channel functions.
Epilepsy is characterised by sporadic recurrent episodes of convulsive seizures and is classified by extent (generalised/partial) and signs exhibited (loss of consciousness; muscle tone and twitching). The seizures may be idiopathic, triggered by external events or internal changes, or secondary to head injury or focal brain damage. Antiepileptic drug therapy to control seizures may be lifelong; choice of drug is determined by type of seizure, likely adverse drug reactions, other drugs that may interact and individual aspects such as pregnancy and compliance. Therapeutic monitoring is regularly carried out for some AEDs by measuring drug concentration in plasma samples; this helps check whether levels are in the therapeutic range and monitors compliance. While all CNS-depressant drugs may reduce seizure incidence, most are too sedating to be useful. The major drugs used to treat seizures act by enhancing GABAmediated inhibition of neural transmission or inhibit neurotransmission by blocking sodium channel functions.Review exercises
References and further reading
Ashton H., Young A.H. GABA-ergic drugs: exit stage left, enter stage right. Journal of Psychopharmacology. 2003;17(2):174-178.
Australian Medicines Handbook 2008. Adelaide: AMH, 2010.
Besson M., Piguet V., Dayer P., Desmeules J. New approaches to the pharmacotherapy of neuropathic pain. Expert Reviews in Clinical Pharmacology. 2008;1(5):683-693.
Buchanan N. Medications which may lower seizure threshold. Australian Prescriber. 2001;24(1):8-9.
Camfield P., Camfield C. When is it safe to discontinue AED treatment? Epilepsia. 2008;49(Suppl 9):25-28.
Caswell A., editor. MIMS Annual June 2005. Sydney: CMP Medica Australia, 2005.
Diaz R.A., Sancho J., Serratosa J. Antiepileptic drug interactions. Neurologist. 2008;14(6 Suppl 1):S55-S65.
Herkes G.K. Epilepsy. Medical Journal of Australia. 2001;174:534-539.
Kilpatrick C.J. Withdrawal of anti-epileptic drugs in seizurefree adults. Australian Prescriber. 2004;27(5):114-117.
Lander C. Antiepileptic drugs in pregnancy and lactation. Australian Prescriber June. 2008;31(3):70-72.
LaRoche S.M., Helmers S.L. The new anti-epileptic drugs: scientific review. Journal of the American Medical Association. 2004;291(5):605-614.
Miller L.C., Drislane F.W. Treatment of status epilepticus. Expert Reviews in Neurotherapy. 2008;8(12):1817-1827.
Morrell M.J., Flynn K.L., editors. Women with Epilepsy: A Handbook of Health and Treatment Issues. Cambridge: Cambridge University Press, 2003.
Moshe S.L. Mechanisms of action of anticonvulsant agents. Neurology. 2000;55(5 Suppl. 1):S54-S58. S32-S40
Neurology Expert Group. Therapeutic Guidelines: Neurology, version 3. Melbourne: Therapeutic Guidelines Limited; 2007.
Rang H.P., Dale M.M., Ritter J.M., Flower R.J. Rang & Dale’s Pharmacology, 6th edn. Edinburgh: Churchill Livingstone; 2007.
Richens A., Dunlop A. Serum-phenytoin levels in management of epilepsy. Lancet. 1975;2:247-248.
Smith R.L. Withdrawing anti-epileptic drugs from seizure-free children. Australian Prescriber. 2006;29(1):18-20.
Tortora G.J., Grabowski S.R. Principles of Anatomy and Physiology, 9th edn. New York: HarperCollins; 2000. [ch 14]
Vander A., Sherman J., Luciano D. Human Physiology: The Mechanisms of Body Function, 7th edn. Boston: McGraw–Hill; 1998. [ch 13]
Epilepsy Action Australia: www.epilepsy.org.au/
New Zealand Medicines and Medical Devices Safety Authority: www.medsafe.govt.nz
More weblinks at: http://evolve.elsevier.com/AU/Bryant/pharmacology