Chapter 15 Analgesics
Pain is a distressing and incapacitating symptom experienced by most people at some stage. Many chemical mediators are potentially involved. Pain varies, and can be classified depending on its aetiology and duration. Fortunately, the main analgesics currently available—opioids such as morphine and non-steroidal anti-inflammatory drugs such as aspirin and paracetamol—are safe and effective when properly selected and administered, based on individual patient needs and responses and on individual drug pharmacokinetics.
Key abbreviations
AMI acute myocardial infarction
COX cyclo-oxygenase glu glutamate
5-HT 5-hydroxytryptamine (serotonin)
NSAID non-steroidal anti-inflammatory drug
Key background
PAIN has been defined by the International Association for the Study of Pain as ‘an unpleasant sensory and emotional experience associated with actual or potential tissue damage, or described in terms of such damage’. This definition emphasises the dual aspects of pain (sensory and emotional); only the person suffering the hurt can tell how much pain is being experienced. Pain is an important protective mechanism, warning of potential injury from the environment or from inside the body. Understanding pain and the actions of analgesics (pain-relieving drugs) in treating it requires first an understanding of how pain is generated.
Physiology of pain
Pain and suffering
Because of its highly subjective nature, pain is difficult to study objectively. It can be viewed as having two components: the physical component, or the sensation of pain (nociception), which involves peripheral and central nerve pathways; and the psychological component, or the emotional response to pain, which involves factors such as a person’s anxiety level, previous pain experiences, age, sex and culture. Suffering, a broader term, may include physical pain, emotional fears (fear of the unknown, fear of dying, fear of dying alone, lack of social supports, loss of independence or integrity), social conflicts (unresolved conflicts with family and friends) and spiritual despair. Such problems are often best addressed by interdisciplinary teams in hospices and pain management programs.
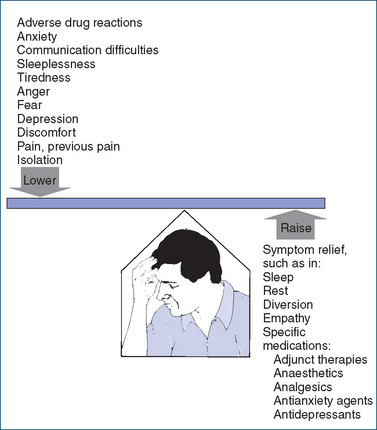
People have a relatively constant pain threshold under normal circumstances; for example, heat applied to the skin at an intensity of 45°–48°C will initiate the sensation of pain in almost all people. By contrast, pain tolerance—the point beyond which pain becomes unbearable—varies widely among individuals and in a single person under different circumstances. Figure 15-1 shows some factors affecting pain perception and tolerance.
Detection and transmission of pain
Tissue injury in the periphery is detected by nociceptors (pain receptors) and the signals are transmitted to the spinal cord via A-delta (δ) fibres (mediating sharp, transient fast pain) or C-fibres (mediating burning, aching, slow, visceral pain) (see Figure 15-2A). These primary (first-order) afferent fibres terminate in the dorsal horn of the spinal cord, where voltage-gated calcium channels are opened and the transmitter glutamate is released to cross the synaptic gap and activate NMDA receptors on the second-order neurons. These neurons cross over and continue upwards in the anterolateral spinothalamic tracts (Figure 15-2B, right-hand side) to the thalamus where they synapse with third-order neurons which connect to specific areas of the limbic system and cerebral cortex, where the messages are perceived as pain.
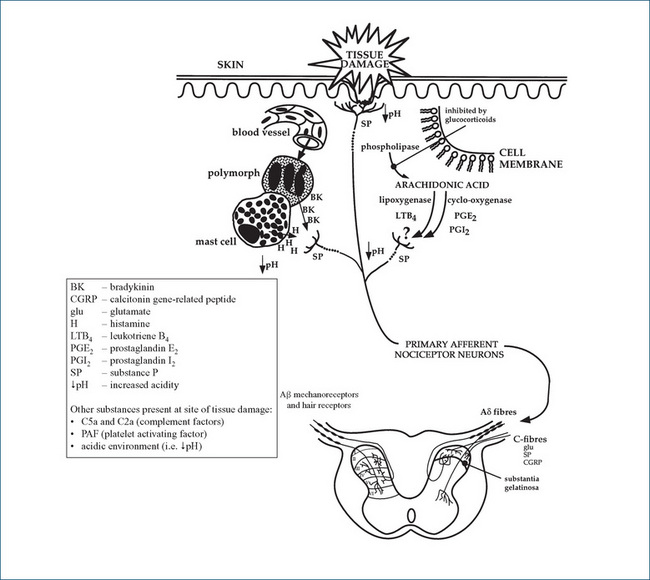
Figure 15-2A Reproduced with permission from Analgesic Expert Group.
Diagram of peripheral factors involved in nociception (Figure 1). In: Therapeutic Guidelines: Analgesic. Version 5. Melbourne: Therapeutic Guidelines Ltd; 2007. p 7.
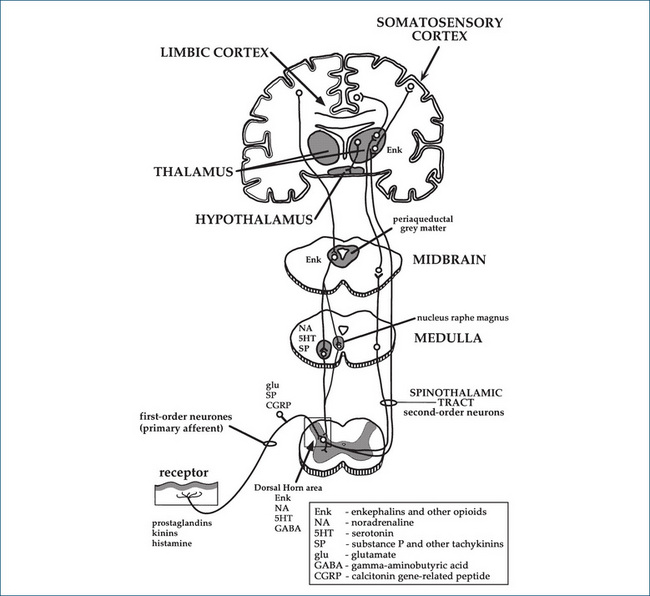
Figure 15-2B Reproduced with permission from Analgesic Expert Group.
Diagram of nerve pathways and some of the neurotransmitters involved in nociception (Figure 2). In: Therapeutic Guidelines: Analgesic. Version 5. Melbourne: Therapeutic Guidelines Ltd; 2007. p 9.
The gate control theory
Several theories of pain transmission and pain relief have been proposed. The gate control theory, put forward by Melzack and Wall in 1965, proposes that a mechanism in the dorsal horn of the spinal cord (the spinal ‘gate’) can modify the transmission of painful sensations from the peripheral nerve fibres to the thalamus and cortex of the brain. The gate is influenced by descending inhibition from the brain: efferent anti-nociceptive (analgesic) pathways from the cortex descend via the periaqueductal grey matter down the spinal cord. (Descending pathways that ‘gate’ impulse transmission in the dorsal horn area are shown on the left-hand side of Figure 15-2B.) In the dorsal horn areas they synapse with short interneurones, via releasing noradrenaline and 5-HT as transmitters. The interneurones modify afferent impulses by release of the inhibitory transmitter GABA, thus reducing transmission of incoming pain sensation between the first-order and second-order neurons.
Hyperalgesia
Facilitation in the dorsal horn area results in greatly increased sensitivity (hyperalgesia, or ‘opened gate’), which spreads beyond the injured area. Substance P, glutamate and nitric oxide are thought to be involved as transmitters. Conversely, techniques of afferent stimulation to relieve pain, such as transcutaneous electrical nerve stimulation (TENS), acupuncture and rubbing or ‘itching’ the skin, are thought to act through inhibitory circuits within the dorsal horn to diminish nociceptive transmission through the C-fibres, thus decreasing pain (‘closing the gate’).
Mediators of pain
Many chemicals are potentially involved in the transmission or relief of pain, especially glutamate, GABA, endogenous opioids, 5-HT and noradrenaline (see Figure 15-2B). Modulation of these chemical mediators is the basis for many methods of pain relief, including opioids, NSAIDs, local anaesthetics, γ-aminobutyric acid (GABA) agonists, N-methyl-D-aspartate (NMDA) antagonists, tachykinin antagonists, cannabinoids, calcium-channel blockers, α2-adrenergic agonists and non-drug techniques such as TENS, acupuncture and the Lamaze (psychoprophylaxis) technique.
Endogenous opioids
There are high concentrations of receptors for the body’s natural opioid pain-relievers, the endorphins and enkephalins, in many areas of the CNS, particularly in the periaqueductal grey matter of the midbrain, in the limbic system and at interneurons in the dorsal horn areas. These areas are known to be involved in pain transmission or perception. The enkephalins (pentapeptides), endorphins (larger polypeptides: the name implies ‘endogenous morphines’) and dynorphin are thought to be the body’s natural pain-relieving chemicals and to act by enhancing inhibitory effects at opioid receptors (see later discussion). One main group of analgesic drugs, the opioids, such as morphine and codeine, cause their effects by mimicking the actions of endorphins and enkephalins on opioid receptors. Endorphin release in the body is higher after acupuncture and TENS, and both effects may be reversed by the use of naloxone, an opioid antagonist. It has been proposed that the analgesic response associated with the use of a placebo may result from an increased release of endorphins in the body.
Prostaglandins
Damage to tissue may directly activate sensory nerves and also sets in train the process of inflammation (see Figure 15-2A), in which a large number of inflammatory mediators are released (so many in fact that this has been referred to as the ‘inflammatory soup’). Of particular importance in the context of pain mechanisms is arachidonic acid, a compound produced from damaged cell membranes that is metabolised by the cyclo-oxygenase enzyme system to tissue hormones called prostaglandins (PGs), which lower the threshold of nociceptors to other mediators. (These mechanisms are discussed in greater detail in Chapter 47.) The mechanism of action of the second main group of analgesic agents, the non-opioids (and the non-steroidal anti-inflammatory drugs [NSAIDs]), is inhibition of the production of PGs.
Tachykinins
Another group of modulators in the nociceptive pathway is the tachykinins (fast-acting polypeptides), including substance P and neurokinins A and B. Tachykinins are involved in inflammatory and neurogenic pain (see below). Competitive antagonists at neurokinin receptors are being developed and studied as potential new analgesic agents.
Pain classification
Pain can be classified in various ways, e.g. on the basis of its time course as acute or chronic (see Table 15-1). Pain may also be classified on the basis of its origin, i.e. as nociceptive, neurogenic or psychogenic.
Table 15–1 Comparisons between acute and chronic pain
| Acute pain | Chronic pain | |
| Onset | Usually sudden | Long duration (>3 months) |
| Characteristics | Generally sharp, localised, may radiate | Dull, aching, persistent, diffuse |
| Physiological responses | Raised blood pressure, respiratory and heart rates; sweating, pallor, dilated pupils; increased muscle tension | Often absent: normal blood pressure, respiratory and heart rates, and pupil size |
| Emotional/behavioural responses | Increased anxiety and restlessness; focuses on pain, rubs affected part; cries, grimaces, moans | Person may be angry, depressed, withdrawn, expressionless and exhausted; physical inactivity or sleep; no report of pain unless questioned |
| Therapeutic goals | Cure of cause; relief of pain; prevent transition to chronic pain; sedation often desirable | Restore functions; tolerance of some pain; improve quality of life; sedation not usually wanted |
| Drug administration | Usually opioids | Paracetamol, NSAIDs, opioids and/or adjuvants |
| Timing | Start as soon as possible; assess regularly; patient-controlled analgesia is useful | Regular preventive schedule |
| Dose | Standard dosages are often adequate; dose reviewed frequently | Individualise according to patient response |
| Route | Parenteral (IV or SC) | Oral or transdermal |
Acute and chronic pain
Acute pain, a state in which a person experiences the sensation of severe discomfort, has a sudden onset, often with a protective function and an obvious cause; it is expected to last only a short time, and usually subsides with treatment. Examples of acute pain include the pain of a broken limb, myocardial infarction, burn, appendicitis and kidney stones. Responses to acute pain include behavioural responses (withdrawal, protection, crying out) and autonomic effects (pallor, sweating, tremor, tachycardia).
If acute pain continues, there can be many changes, including physical changes (loss of fitness, fatigue, changes in weight and muscle mass), psychological changes (uncertainty, grieving, hyperalgesia) and social changes (adoption of a ‘sick role’, dependence, isolation, loss of employment).
Chronic pain, such as that accompanying cancer or rheumatoid arthritis, is a persistent or recurring pain that continues for more than 3 months or after completion of healing, and may be difficult to treat effectively. A person with chronic severe pain experiences an adaptation process (see Table 15-1); patients are likely to become trapped within a chronic pain disability cycle in which ineffective treatments increase anxiety and contribute to the pain persisting. The primary goal of treatment then becomes not total relief from pain, but minimisation of pain-related disabilities, avoidance of unnecessary investigations and ineffective therapies and improving quality of life. The principles of the ‘analgesic ladder’ should be followed. For chronic non-cancer pain, opioids are reserved for pain unresponsive to other treatments and should be reviewed regularly. (Treatment of neuropathic pain is discussed below, and cancer pain and palliative care in Chapter 42.)
Nociceptive pain
Nociceptive pain is ‘physiological’ pain; it arises from stimulation of superficial or visceral (deep) nociceptors by noxious stimuli such as tissue injury or inflammation.
Somatic nociceptive pain originates especially in the skin, mucosal surfaces, bones and joints, pleura and peritoneum; it is usually well localised. It is described as being sharp, shooting, throbbing, burning, stinging or cutting. Examples are the pain from a skin ulcer, arthritis, bony metastases of cancer or minor surgery. Somatic pain responds best to treatment with NSAIDs.
Visceral nociceptive pain originates in the walls of visceral organs, such as the liver and pancreas, and large muscle masses. It is described as being deep, aching, diffuse, cramping and nagging, and may be associated with nausea and vomiting or sweating. The pain may be referred to another area of the body (which has sensory nerves running to the same segment of the spinal cord), such as the pain of a myocardial infarction that may be felt initially in the arm or shoulder. Examples include pain from bowel obstruction, abdominal tumours, ischaemic muscle or major surgery. Visceral pain usually responds well to opioid analgesics.
Muscle spasm nociceptive pain originates in skeletal or smooth muscle, is mediated by PGs and is worse on movement or when smooth muscle is stretched (colic). Biliary colic, bowel obstruction, spinal cord damage and some types of acute low-back pain exemplify muscle spasm pain. Muscle relaxants and NSAIDs are usually the analgesics of choice.
Neuropathic pain
Neuropathic pain arises from a primary lesion, alteration or dysfunction in the peripheral or central nervous system pathways. It may be due to nerve compression, for example by a prolapsed intervertebral disc, or to inflammation, trauma or degeneration. This pain has been described as burning, shooting and/or tingling, and is often associated with paraesthesia, hyperalgesia and allodynia (pain due to a stimulus that does not usually cause pain, e.g. pressure from clothing). This type of pain, caused for example by post-herpetic neuralgia, limb amputation, trigeminal neuralgia, diabetic neuropathy or cancer, may be accompanied by sympathetic nervous system dysfunction.
Neuropathic pain responds less well to opioid analgesics and often requires the addition of adjunct medication to the patient’s drug regimen, such as:
Psychogenic pain
Psychogenic pain has psychological, psychiatric or psychosocial causes as its primary aetiology. Anxiety, depression and fear of dying have been known to cause severe pain. It is a CNS syndrome and, although no obvious somatic source may be evident, can be real and distressing to the sufferer and may lead to anger and depression. In such patients, drug therapy alone does not usually bring relief; a multimodal approach with psychotherapy is indicated.
Specific pain syndromes
Other more specific types of pain are treated whenever possible with specific therapies known as directed analgesia. Tension headaches, for example, usually respond to over-the-counter analgesics such as aspirin and paracetamol, sinus headaches to NSAIDs plus a decongestant, trigeminal neuralgia to carbamazepine, pain from osteoporotic fractures may be helped by the osteoblastic actions of calcitonin, while migraine headaches may require more specific vasoactive drugs such as sumatriptan or ergotamine (see Chapter 20). Dental pain and toothache usually require treatment of the underlying dental or oral disease; since most dental pain is caused by inflammation, NSAIDs are the best choice for analgesics.
Cancer pain relief requires a multimodal approach of palliative care (see Chapters 41 and 42), possibly involving analgesics and anaesthetics, other cancer therapies (radiotherapy, hormones, surgery, chemotherapy), neurosurgical techniques, physical therapies (splints, electrotherapy, occupational therapy), complementary and alternative medicine methods and psychological support for patients and their carers.
Breakthrough pain
When pain occurs between doses of regular analgesics in patients with severe chronic pain, this is referred to as ‘breakthrough pain’ or incident pain. It is usually managed with extra doses of short-acting oral (morphine liquid) or transmucosal (fentanyl lozenge) opioids (see section on breakthrough pain in Chapter 42).
Pain management
Assessment of pain
Pain measurement in the laboratory
To test the activity of analgesic drugs, there must be some standard (mild) pain stimulus against which analgesic activity can be measured. The traditional method was to place white mice gently onto a heated metal surface (a ‘hot plate’ at 55°C), and measure the time taken for the mouse to show some evidence of pain, such as lifting or licking its paws or trying to hop off the plate. Groups of mice previously administered test drugs might show longer times before pain elicited a response. This method obviously raises ethical issues—should animals have to suffer pain for us (and them) to have access to better analgesic drugs?
A newer method with fewer ethical problems is that of the ‘third molar model’, in which (human) patients undergoing a standard dental procedure, extraction of third molars (wisdom teeth), are recruited into trials in which various new analgesics or new routes of administration can be compared (see Clinical Interest Box 15-1).
Clinical interest box 15-1 A model for pain?
The ‘third molar model’ is commonly used as a standard method whereby analgesic activity can be assessed in a typical clinical situation, without causing unnecessary pain to the subjects. Wisdom tooth extraction is a common dental procedure carried out using standard operative procedures in otherwise healthy young people.
This situation allows prospective clinical trials to be carried out to investigate the onset, depth and duration of analgesic activity of drugs such as local anaesthetics and analgesics. As there are other possible sequelae from the procedure, including infection, anxiety, swelling, impaired sleep and difficulty opening the mouth, other drugs such as antibiotics, sedatives and anti-inflammatory agents can also be tested. Since patients may return for subsequent extraction of other wisdom teeth, cross-over studies may be possible, in which the patient acts as his/her own control.
In a recent clinical trial using this pain model, a novel intranasal formulation of morphine was trialled against standard analgesics (oral morphine and IV morphine) and against placebo. Intranasal morphine (15 mg) was shown to be statistically similar to IV 7.5 mg morphine. Study medications were well tolerated, with no serious adverse events; the intranasal route was shown to be an effective non-invasive method for administering morphine for treatment of postoperative pain. After: Christensen et al 2008.
Measurement of clinical pain
As ‘pain is what the patient says hurts’, it is important to assess from the patient’s experience the time course, type, site and extent of pain and its associations and effects. A Pain Assessment Chart or the ‘PQRST’ approach helps the patient describe the pain:
With respect to locating the pain, a body chart such as that shown in Figure 15-3 (I Location) can be helpful. For an estimate of the severity of the pain, scales such as those in Figure 15-4 help patients indicate the intensity and distress levels of the pain. For children, a pictorial scale can be used, with faces to ‘show how much it hurts’ (Figure 15-4 III).

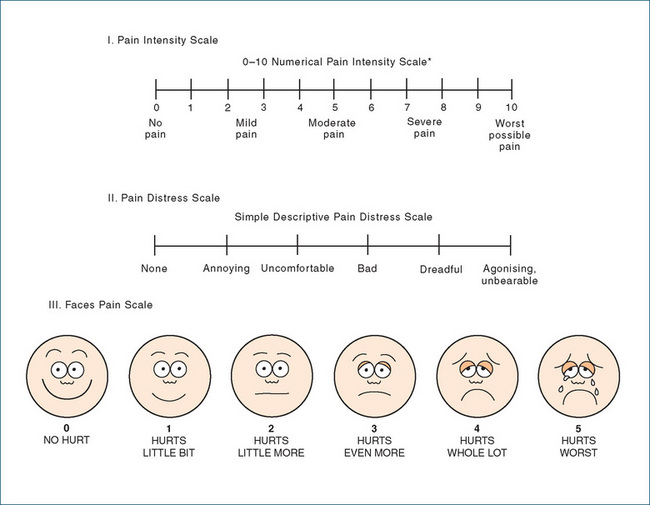
Figure 15-4 Scales for rating the intensity and distress of pain. In the Faces Pain Scale used for paediatric patients, the gradation in ‘hurt’ or ‘pain’ is explained to the child, with increasing pain shown from left to right; the child is asked to point to the face that shows how much she/he hurts now.
Adapted from: Salerno 1999; Carr et al 1992; Wong et al 2001; reproduced with permission.
Physical examination, with attention to tender spots, patient’s responses to movement and stretch and non-verbal behaviours, helps accurate diagnosis. Regular reassessment of pain is essential, to monitor both the disease process and analgesic therapy, and to assess whether other therapy is required.
General principles
Some important principles in pain management are summarised below and in Clinical Interest Box 15-2.
Clinical interest box 15-2 World health organization guidelines on analgesic use
The World Health Organization guidelines on clinical use of analgesics in cancer pain management, published over 20 years ago, are that analgesics should be prescribed:
The ‘analgesic ladder’ was devised to legitimise the appropriate prescribing of ‘strong’ opioids for patients with moderate–severe cancer pain.
It has since been criticised for being non-specific, for the lack of strong evidence for its effectiveness and for not specifying nonpharmacological remedies.
In the ensuing 20 years the guidelines have been evaluated and updated; they are still valid in promoting the appropriate prescribing of readily available analgesics for individual patients.
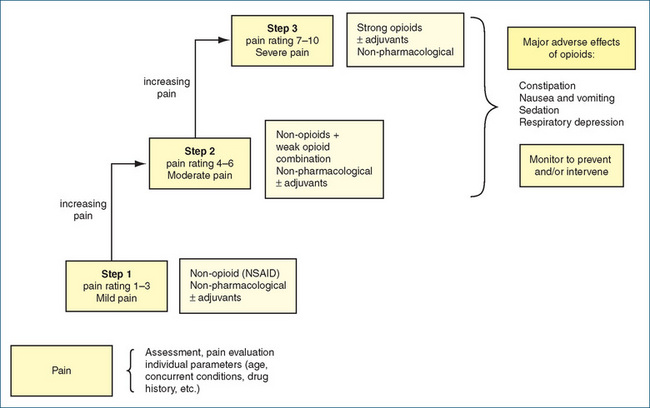
Figure 15-5 Flowchart for the ‘stepwise’ pharmacological management of pain. Adjuvants may include antidepressants, anti-inflammatories, antianxiety agents and local anaesthetics; non-pharmacological techniques include physiotherapy, acupuncture, counterirritants, psychotherapeutic methods and complementary and alternative therapies. NSAID = non-steroidal anti-inflammatory drugs.
Adapted from: Salerno & Willens 1996; Therapeutic Guidelines: Analgesic 2007.
Avoid under-treatment of pain
Even though effective pain management techniques are available, the wide application of such approaches has been slow and many patients still suffer pain. Some of the reasons for under-treatment of pain are summarised in Clinical Interest Box 15-3. Despite health-care providers being legally as well as morally responsible for pain relief, undertreatment and improper use of analgesics continue to be major problems in both acute and chronic pain settings.
Clinical interest box 15-3 Fears or myths about pain and pain management
Many mistaken ideas contribute to the mismanagement of pain:
Endpoints of treatment
Pain assessment charts and scales are useful for monitoring pain intensity during treatment and to assess need for ongoing analgesia. Doses are titrated depending on clinical responses and adverse effects; after opioids, depth of respiratory depression is correlated with depth of sedation. The aim is to maintain comfort for the patient, avoiding peaks and troughs of pain relief and relapses.
Routes of administration
If it is possible to deliver an analgesic drug directly to the site of pain or to the sensory nerve pathway, this will localise the effects, minimise the dose required and reduce the time to onset of action. Examples are the epidural administration of local anaesthetics and opioids, intraarticular administration of corticosteroids and topical administration of local anaesthetics and NSAIDs. Generally, however, analgesics must be administered systemically to circulate to the required site of action, whether in the painful tissues or in the CNS.
Oral route
The oral route is preferred as being the simplest and most acceptable and has the advantage of minimising the risk of IV drug-related problems. Opioid drugs may undergo significant hepatic metabolism after oral administration (first-pass effect), so higher doses are required than for parenteral administration; however, as the metabolites may be pharmacologically active, they contribute to the analgesic effects. Sustained-release preparations (e.g morphine sulfate controlled-release tablets, Drug Mono graph 15-1) help prolong the half-life of morphine from 3–4 hours to 12–24 hours, and are useful for stable, chronic pain.
Continuous infusion of opioids
Continuous opioid infusions by SC or IV routes may be used when there is intractable vomiting; for severe pain that is not relieved by oral, rectal or intermittent parenteral opioid dosing; or for pain management in the postoperative period.
Opioids may be infused by a microdrip infusion set and pump or by a patient-controlled analgesia (PCA) infusion pump unit. PCA is commonly ordered in a hospital or hospice setting, usually after surgery or for chronic cancer pain. It is a microprocessor-controlled injector programmed to deliver a predetermined IV opioid dose when the patient triggers the pump mechanism. This dose is based on the prescriber’s order of a specific analgesic dose and a lock-out interval (5–20 minutes), which protects the patient from overdosing. The unit may record all patient dosing attempts so that the prescriber can evaluate the individual’s need for the analgesic.
Other routes for systemic absorption
The rectal route is useful in patients who cannot swallow or who are vomiting, and for slower absorption; absorption from the rectal route is variable, and paracetamol suppositories are no longer recommended. Transdermal administration is effective for chronic administration of lipid-soluble drugs: fentanyl patches are available for patients who cannot tolerate oral morphine (see Drug Monograph 15-2). Additional analgesics may be prescribed to cover rising levels of pain (breakthrough pain).
Aspirin has analgesic, antipyretic, anti-inflammatory and antiplatelet effects. Its commonest formulation is in tablets containing 300 mg aspirin.
Indications
Aspirin is indicated for the treatment of pain and fever (in those over 18 years), headaches, rheumatic fever, rheumatoid arthritis and osteoarthritis, and in prevention of AMI, reinfarction or stroke. The advantages of aspirin over paracetamol include its anti-inflammatory effects and its effectiveness in preventing AMI and thrombi.
Pharmacokinetics
Aspirin taken orally is rapidly absorbed, partly from the stomach (as the drug is itself acidic), and also from the intestine. Peak serum levels of acetylsalicylic acid are reached within 20–40 minutes.
Rapid metabolism by tissue and blood esterases occurs, hydrolysing aspirin to acetic acid and salicylate; the peak plasma salicylate level is reached in 2–4 hours. Salicylate is distributed throughout most body tissues and fluids, including synovial fluid and cerebrospinal fluid, and is 50%–90% bound to plasma proteins. Salicylate then undergoes hepatic metabolism to inactive metabolites. The plasma salicylate level required for analgesic and antipyretic effects is about 28 mg/L, whereas 100 mg/L is required for anti-inflammatory effects. Toxic effects (salicylism) are seen above about 200 mg/L.
Common adverse effects
Common adverse effects include gastrointestinal irritation or discomfort and nausea or vomiting (see earlier section on adverse drug reactions). Taking aspirin with a full glass of water helps reduce these effects. Toxic reactions due to salicylate poisoning (salicylism) include tinnitus, vertigo, complicated effects on acid–base balance (both respiratory alkalosis and metabolic acidosis) and Reye’s syndrome in young people. See Clinical Interest Box 15-8 for clinical management of aspirin overdose.
Drug interactions
There are many potential drug interactions, in particular with other drugs affecting platelet function or the blood-clotting process; drugs eliminated mainly by renal excretion; drugs used to treat heart failure (as aspirin can impair renal function); antihypertensive agents (as aspirin can raise blood pressure and impair renal perfusion); other NSAIDs (increased risk of gastric ulceration or bleeding); corticosteroids (may decrease salicylate concentration and clinical effect); drugs affecting blood glucose concentration; probenecid (uricosuric effect may be reduced); valproate (concentration and effects of valproate may be enhanced, as may effects of aspirin on blood parameters). In most cases, low-dose aspirin (e.g. 100 mg daily) is safe with other drugs.
Warnings and contraindications
Precautions with aspirin use are required in heart failure and hypertension, renal impairment, severe liver disease, during surgery and in people predisposed to bleeding, peptic ulcer or asthma, or if previous serious adverse effects with salicylates have occurred, and during late pregnancy. It should not be used in children under 18 years, especially those with fever, because of the possibility of rare but potentially fatal Reye’s syndrome (severe liver damage and encephalopathy).
Dosage and administration
Aspirin products are available in tablet, effervescent tablet, capsule, enteric-coated, extendedrelease tablet and oral powder dosage forms. Aspirin should be taken after food, to minimise irritant effects in the gastrointestinal tract (this also has the effect of delaying absorption). The usual adult dose is 1–2 regular-strength tablets (300 mg) every 4–6 hours or as needed. Higher doses are required for anti-inflammatory effects.
For prophylactic antiplatelet effects, much lower doses are sufficient, e.g. 75–150 mg/day (¼ to ½ standard tablet).
Nitrous oxide and other gaseous and volatile general anaesthetics are administered by inhalation. IM and SC injection routes are common for opioid analgesics, the latter having a slower onset of action because the tissues are less vascular.
Intravenous injection is obviously the fastest route for rapid pain control, as it avoids the absorption phase. Relatively poor lipid solubility delays the onset of analgesia when morphine is administered by epidural or intrathecal injection. The risk of inducing respiratory depression is greater by the intrathecal route than by epidural administration, so patients must be monitored for at least 24 hours after administration by the intrathecal route.
Analgesic use in special groups
Pregnancy, labour and delivery
During pregnancy, the analgesic of choice for mild to moderate pain is paracetamol or codeine. In late pregnancy, NSAIDs should be avoided because of increased risk of bleeding (especially after aspirin); adverse effects on the fetal cardiovascular, respiratory and renal systems; and prolongation of gestation and labour (see also discussion in Chapter 38 on drugs in pregnancy and the perinatal period).
Labour and lactation
During labour, most women experience pain. Ideally, the analgesic used will provide pain relief without any interference with labour and without increasing the risk or danger to the mother or fetus. While there is no ideal analgesic available for use during childbirth, inhaled nitrous oxide is commonly used. For more severe pain, epidural administration of combined local anaesthetic and opioid is effective and allows the mother to remain conscious even through caesarean section. Morphine is a potent analgesic when used during labour, but has been associated with greater neonatal respiratory depression than pethidine and has a slower onset of action. Both drugs cross the placenta to enter fetal circulation. Naloxone, an opioid antagonist, should always be available to treat the mother or neonate if excessive CNS or respiratory depression occurs.
If an opioid analgesic or methadone is administered to a woman who is breastfeeding a baby, the feeding should be immediately before the next dose of drug, to minimise the quantity of drug passed on to the infant in breast milk.
Opioid dependence
A concern with the use of opioids during pregnancy (particularly in an addicted woman) is that these agents may lead to physical drug dependence in the fetus, causing severe withdrawal reactions in the neonate after birth. Pregnant women dependent on an opioid and/or enrolled in methadone maintenance programs may present with fetal distress syndrome in utero and often deliver an underweight baby at birth. Such infants are usually lethargic, with difficulty breathing, high-pitched cry and poor feeding and sleeping patterns; the infant will require small doses of morphine postnatally to prevent potentially fatal opiate withdrawal effects and may require specialcare nursing for weeks while being weaned off the opioids (see Clincal Interest Box 21-7).
Children
Children are often untreated or inadequately treated for pain, due to an incorrect belief that children do not ‘feel pain’ in the same way that adults do, which results in needless suffering. Assessing pain in young children is more difficult than in an older child or adult. At a minimum, it should be based on knowledge of the procedure or event that caused the pain, as well as the child’s non-verbal behaviour. Even when children can verbalise their feelings they may be somewhat reluctant to report pain, often fearing that the consequences (diagnostic test, examination or injection) will be more painful than the pain they are experiencing.
General rules for analgesic use in children
Infants and neonates
Medicating a child under 2 years who cannot verbally report pain is justified if the child displays increased irritability, restlessness, crying, anorexia and decreased activity. The approach to a child should be individualised, based on the child’s age and stage of development and the various assessment tools available, such as figure drawings to identify the area that hurts and scales that rate pain intensity (Figure 15-4 III).
Painful procedures often need to be carried out on neonates: injections, heel pricks or venepuncture for blood sampling or placement of a peripheral venous or arterial line. Non-pharmacological methods of pain relief include feeding, calming and warming. A simple pharmacological technique that can easily be implemented by nurses is oral sucrose. For example, 0.2 mL of Syrup BP (66.7% sucrose solution) dropped onto the front of the tongue from a syringe has been shown to reduce stress from painful procedures, as evidenced by reduced crying time and grimacing behaviours.
Elderly persons
Analgesic use in the elderly usually requires careful adjustments in dosage and dosing interval according to the person’s liver and kidney functions, therapeutic responses and development of undesirable adverse effects (increased pain, confusion or respiratory depression). The elderly often have enhanced drug responses and may not tolerate adverse effects as well as younger patients. Elderly patients may have multiple medical problems and several medications prescribed for them (polypharmacy).
Elderly people often report pain differently from younger persons because of the belief that pain is a part of old age, because they do not want to cause difficulties to their carer or because they deny their discomfort as a cultural and ethnic issue. In such instances, non-verbal communication and behaviours should be carefully assessed, such as increased irritability, loss of appetite, decreased activity, crying easily or tightly gripping an object. Cognitive impairment, dementia and confusion may make pain assessment in these people difficult; methods are available for assessing pain in patients with dementia.
The elderly may have impaired circulatory function, which results in slower absorption of drugs administered by the IM and SC routes. Administering additional doses in such a situation may result in unpredictable or increased drug absorption, which increases the potential for adverse reactions.
Specific analgesics that are inappropriate for use in the elderly because of high risk of toxicity include dextropropoxyphene and pethidine; safer analgesics are available. All NSAIDs are relatively dangerous in the elderly because of gastrointestinal, renal and cardiovascular adverse effects; those with long half-lives, such as naproxen, piroxicam and tenoxicam, must be avoided.
Opioid-dependent persons
Managing acute pain in patients with an opioid-dependence disorder is difficult, due to tolerance and/or blockade of opioid receptors by antagonists or partial agonists used in treatment. Pain and the addictive disorder must be care fully assessed, and prescribers need to be alert to drug-seeking behaviours. Non-opioid analgesics and non-pharmacological methods are used whenever possible (see Roberts [2008]).
Analgesic drugs
Opioid analgesics
The prototype opioid analgesic, and still the ‘gold standard’ most commonly used clinically, is morphine, so most discussion here will be based on the actions and clinical uses of morphine (see Drug Monograph 15-1 and Clinical Interest Box 15-4). Other opioids will be mentioned briefly, highlighting the main aspects in which they differ from morphine.
Drug monograph 15-1 Morphine sulfate controlled-release tablets
Morphine is a strong analgesic with central actions on pain perception, used in moderate to severe acute and chronic pain; it mimics the actions of enkephalins and endorphins at opioid receptors.
Indications
Morphine is indicated for the treatment of opioidresponsive severe pain, such as after trauma or surgery or for cancer pain. It may be given to suppress an unproductive nagging cough and to patients with lung cancer to treat pain aggravated by coughing; the sedative and euphoriant actions are also useful. Morphine increases GIT tone and decreases peristalsis and glandular secretions; these actions are useful in treating diarrhoea.
Pharmacokinetics
Morphine may be administered by many routes—PO, IM, IV, SC, epidural, intrathecal and rectal. It is rapidly absorbed and is subject to an extensive first-pass effect in the liver, leading to poor bioavailability (about 40% when taken orally), so the oral dose may need to be 2–6 times the parenteral dose (see Table 15-3). The main metabolites are morphine-6-glucuronide (M6G, an active metabolite) and morphine-3-glucuronide (M3G).
Morphine is distributed widely in most body tissues but only a small fraction crosses the blood–brain barrier. Metabolites are excreted primarily via the kidneys, with 7%–10% undergoing enterohepatic circulation, which extends the half-life. The mean elimination half-life is 2–3 hours but this is increased with slowrelease preparations (tablets, capsules, granules, oral suspension), such that the peak morphine concentrations during chronic use occur 4–8 hours after dosing and therapeutic effects may extend for 16–24 hours.
Adverse reactions
The most serious adverse reactions reported are constipation, nausea and vomiting, itch, urinary retention, sedation, circulatory and respiratory depression and miosis (pin-point pupils). People who overdose on opioids are likely to die from cessation of respiration (see Clinical Interest Box 15-5). Tolerance occurs to analgesia as well as to depressant effects (but not to constipation), requiring higher dosages. Constipation requires treatment with a laxative; a diet high in fibre helps prevent constipation. Respiratory depression, dependence and withdrawal reactions are not usually problems when opioids are used clinically for relief of severe pain.
Warnings and contraindications
Avoid the use of opioids in patients with known opioid drug hypersensitivity, acute respiratory depression, acute alcoholism or head injury. Use opioids with caution in patients with acute asthma, chronic obstructive pulmonary disease (COPD) or any respiratory impairment or a history of drug abuse, and in patients with elevated intracranial pressure (may rise), biliary colic or pancreatitis (may cause spasm of biliary tract muscle and sphincter), acute abdominal conditions or severe inflammatory bowel disease (risk of obscuring the diagnosis, or risk of toxic megacolon). Doses need to be reduced in patients with renal or liver impairment and in the elderly and children. Administration during pregnancy may result in dependence in the infant; use during labour may cause respiratory depression in the infant, treated with naloxone.
Clinical interest box 15-4 Opium, opiates, opioids and narcotics
A note on terminology: opium is the dried extract of the seed capsules of the opium poppy, Papaver somniferum (meaning ‘the poppy bringing sleep’). Opium contains many pharmacologically active alkaloids (nitrogenous compounds), including morphine, codeine and papaverine. The term ‘opiate’ strictly refers only to opium derivatives, whereas ‘opioid’ means any opium-like compound and includes endogenous painrelieving transmitters as well as synthetic drugs that mimic the actions of the opiates.
The medicinal effects of opium have been known in many cultures for over 6000 years. Opium was almost literally ‘the panacea for all ills’, as it is effective in treating pain, diarrhoea, cough and sleeplessness—what more could a sufferer ask for? One of the Latin synonyms for opium preparations was ‘laudanum’, meaning praiseworthy. A doctor’s bag could have contained many opium preparations, such as Tincture of Opium, Aromatic Chalk with Opium Mixture, Compound Aspirin and Opium Tablets, Gall and Opium Ointment, Ipecacuanha and Opium Powder (Dover’s Powder), Opiate Squill Linctus and Opium and Atropine Suppositories.
Opium preparations were standardised in terms of their morphine content (see Chapter 4, ‘Standardisation of drugs’). It is now considered preferable to administer pure forms of single drugs (e.g. morphine) rather than crude extracts (e.g. opium) that contain not only varying amounts of several active ingredients but also unknown amounts of contaminants.
Pharmacologists too have known (and experimented with) morphine and similar drugs for many decades, naming and classifying the morphine receptors, probably without stopping to wonder why evolution had endowed the human CNS with receptors for a poppy extract! It was not until 1975, when Hughes and Kosterlitz in Aberdeen, Scotland, succeeded in isolating from mammalian brain two pentapeptides that competed with and mimicked the actions of morphine, that the body’s natural analgesic compounds, the enkephalins, were discovered. Since then, the receptors mediating pain relief have been referred to as opioid receptors rather than morphine receptors.
The term ‘narcotic’ has also suffered misuse and confusion: literally, it means a compound causing numbness or stupor; hence ‘narcotic analgesics’ was the group name for the morphine-like drugs, which cause sedation and pain relief. (Morphine was named after Morpheus, the Greek god of sleep and dreams.) The term ‘narcotic’ was later extended to refer to all drugs causing addiction and likely to be abused, and thence to all illicit drugs—so it now includes stimulants like cocaine as well as sedatives like morphine. It is probably best avoided in the medical context.
Because of their addictive potential, opium and opioids are tightly controlled worldwide. Most opioids (except low-dose codeine, pholcodine, dextropropoxyphene, diphenoxylate and tramadol preparations) are ‘Controlled Drugs’ (Schedule 8) in Australia and New Zealand, requiring strict controls on storage and supply.
Mechanism of action of opioids
The mechanism of the analgesic action of opioids is still not totally clear despite decades of intensive study. At the spinal cord level, morphine stimulates opioid receptors and thus inhibits release of substance P from dorsal horn neurons. (Substance P is a neurokinin present especially in nociceptive primary afferent neurons—see Figures 15-2A and B—and is involved in mediating pain, inflammation, smooth muscle contraction, a slow excitatory response and stimulation of many exocrine glands.)
At supraspinal levels, opioids act to ‘close the gate’ in the dorsal horn, thus inhibiting afferent transmission. Opioids are also capable of altering perception and emotional responses to pain because opioid receptors are widely distributed in the CNS, especially in the limbic system, thalamus, hypothalamus and midbrain. Pain perception is altered, thus patients have reported they could still feel the pain, but it no longer worried them.
Opioid receptors
The endogenous opioid peptides involved in nociception and sensory pathways have been described earlier (‘Mediators of pain’, ‘Endogenous opioids’). High-affinity binding sites for the enkephalins and endorphins are located in the membranes of central neurons (also in peripheral tissue, especially in the gut), and are responsive to various opioid agonists. On the basis of their actions at these opioid receptors, drugs may be classed as opioid agonists (natural or synthetic agents that have a full morphine-like effect), antagonists or partial agonists (e.g. buprenorphine, having a less than maximal effect at mu (μ) receptors).
Opioid receptors are G-protein-coupled transmembrane receptors, activation of which inhibits adenylate cyclase and reduces cyclic adenosine monophosphate (cAMP) levels. G-protein coupling also promotes opening of potassium channels and inhibits opening of calcium channels, which reduces neuronal excitability and inhibits release of excitatory (pain) transmitters, leading to inhibitory effects at the cellular level. Effects that appear to be excitatory are probably actually due to suppression of firing of inhibitory neurons. Tolerance to opioid effects is thought to be due to both a gradual loss of inhibitory functions and an increase in excitatory signalling. Withdrawal effects may be due to a rebound increase in cAMP formation which has been activated by chronic administration of opioid via delta opioid receptors.
Opioid receptor subtypes
Subtypes of opioid receptors have been classified according to their responses to the actions of different agonists and antagonists (just as there are several subtypes of noradrenaline receptors). The primary opioid receptors concentrated in the CNS are named by the Greek letters μ (m; mu), κ (k; kappa) and δ (d; delta). (A new member, the ‘opioid receptor-like 1 receptor’, discovered in the Human Genome Project, is being studied as a potential target for new agents involved in analgesia, anxiety and drug addiction.) Analgesia and constipation have been associated with all three receptors, while euphoria (feeling good) is associated mainly with actions at μ-receptors and dysphoria (feeling bad) at κ-receptors What were formerly thought to be specific sigma (σ) opioid receptors are now considered general ‘psychotomimetic receptors’, primarily associated with unwanted effects such as dysphoria, hallucinations and confusion.
Agonists and antagonists
The agonist analgesics (e.g. morphine, pethidine) activate both the μ and κ receptors, while the partial agonist agents such as buprenorphine activate one type of receptor (agonist effect) and have minimal effects on other receptors (partial agonist or antagonist effect). The partial agonist drugs may induce the undesirable effects associated with σ-receptor activity. Pure antagonists (naloxone, naltrexone) antagonise all opioid receptors. A summary of opioid receptor responses is shown in Table 15-2; the situation is complicated by the fact that some drugs show varying effects in different tissues or species.
Table 15–2 Opioid receptor responses
| RECEPTOR | DRUG EXAMPLES | RESPONSE |
| mu (μ) | Strong agonist: morphine, fentanyl, methadone, hydromorphone, β-endorphin Partial agonist: buprenorphine Weak agonist: pethidine |
Supraspinal analgesia, euphoria, respiratory depression, sedation, constipation, miosis, drug dependence |
| Antagonist: naloxone, nalorphine, naltrexone | Reverses opioid effects, induces acute withdrawal in opioid dependency | |
| kappa (κ) | Agonist: morphine, β-endorphin, dynorphin Little or no activity: methadone, pethidine |
Spinal and peripheral analgesia, sedation, miosis, dysphoria, respiratory depression |
| Antagonist: naloxone, naltrexone | Reverses opioid effects, induces acute withdrawal in opioid dependency | |
| delta (δ) | Agonist: enkephalins, β-endorphin | Spinal analgesia, respiratory depression, constipation |
| Antagonist: naltrexone |
Pharmacological effects of opioids
Considering the widespread distribution of opioid receptors in both peripheral and central tissues, it is not surprising that opioids have a broad spectrum of actions. (Aspects of opioid actions relevant to drug dependence and social pharmacology are discussed in Chapter 21.)
Central effects
Effects of opioids in the CNS include:
Peripheral effects
Effects of opioids in the PNS include:
Adverse drug reactions
As described above, the most serious adverse effects of opioids are respiratory depression, excessive sedation, dysphoria, constipation, nausea and vomiting, tolerance and dependence. The cause of death from acute toxicity after an overdose of an opioid such as heroin is usually respiratory failure (see Clinical Interest Box 15-5).
Clinical interest box 15-5 A case of iatrogenic opioid overdose
A 29-year-old woman with bacterial tonsillitis was treated with IM procaine penicillin, oral phenoxymethylpenicillin and ibuprofen. Some hours later the patient was still unwell, with severe headache, nausea and vomiting; the GP administered IM morphine (30 mg) and metoclopramide. After admission to the local hospital, a further 30 mg of morphine was given IM and prochlorperazine. When the patient was checked 2 hours later, it was found that she had succumbed to respiratory depression and was unable to be resuscitated.
Death was attributed to respiratory depression, caused by excess doses of morphine and exacerbated by narrowed airways and CNS depressants. The recommended IM morphine dose for a 29-year-old woman naive to morphine was 7.5–12.5 mg, repeated after 2 hours if necessary. Appropriate dosing and monitoring of the patient could have averted this death.
The usual treatment for an oral opioid overdose includes aspiration or gastric lavage and activated charcoal. For respiratory depression, a patent airway is established, respiration is assisted with oxygen and IV naloxone is administered to reverse the opioid-induced respiratory depression and sedation.
Based on: Dowden (2009) ‘Medicinal Mishap’; see Drug Monograph 21-1 for additional relevant information.
Tolerance to opioid analgesics
Drug tolerance is defined as the gradual decrease in the effectiveness of a drug given repeatedly over a period of time; if tolerance develops, higher doses are required to achieve the same effect. Morphine and the opioids provide the classic example of this. If this is not recognised and higher doses are not given, patients may be under-treated with opioid analgesics. The dose of an opioid may therefore be gradually increased to large amounts (doses potentially fatal in ‘opioid-naïve’ individuals) to control pain in patients with cancer, without producing the adverse effects of respiratory depression or excessive sedation. Tolerance develops to the analgesic effects and to the sedation, nausea and vomiting that opioids cause. Unfortunately, tolerance does not develop to the accompanying constipation, confusion, nightmares and hallucinations, so these adverse reactions may become more of a problem as doses are increased. A change to another opioid (e.g. fentanyl) sometimes minimises the adverse CNS effects.
Pharmacokinetic aspects
Opioids generally are not well absorbed after oral administration and have a low and variable bioavailability due to extensive first-pass metabolism in the liver.2 Even after parenteral administration there is variability in plasma concentrations, metabolism and rates of elimination, so doses need to be individualised.
People with liver damage may accumulate active drug and are very sensitive to the depressant effects of opioids. The toxic pethidine metabolite, norpethidine, may cause convulsions. Because methadone is not metabolised to glucuronides, it may be a safer alternative in liver disease. Renal disease can extend the half-lives of opioids that are excreted in an active form and lead to respiratory depression, especially from methadone, M6G and norpethidine.
Morphine is not highly protein-bound (35%) and is relatively hydrophilic, so it crosses only slowly into the CNS. (By comparison, fentanyl and its analogues are highly lipophilic and so have rapid onset and short duration of action and can be administered transdermally.)
In the elderly and in infants under 1 year, doses need to be reduced because of increased CNS sensitivity and decreased clearance. In patients with hypovolaemia (e.g. from burns or trauma), IM medications are poorly absorbed.
Equianalgesic dosing
Some patients experience intolerable adverse effects from a particular opioid agent, and need to be switched to a different analgesic. Doses of other opioids are compared to those of morphine and quoted in terms of equianalgesic dose to standard morphine 10 mg IM/SC or 30 mg orally; comparative doses are shown in Table 15-3. Switching requires careful assessment of pain levels, adverse effects and tolerance that has developed. It is recommended that the initial dose of the new agent should be only half of that indicated by comparing doses, as tolerance developed to the previous agent may not fully extend to the new agent. (Note that switching from morphine to methadone is complicated and expert advice should be sought; see Australian Medicines Handbook.)
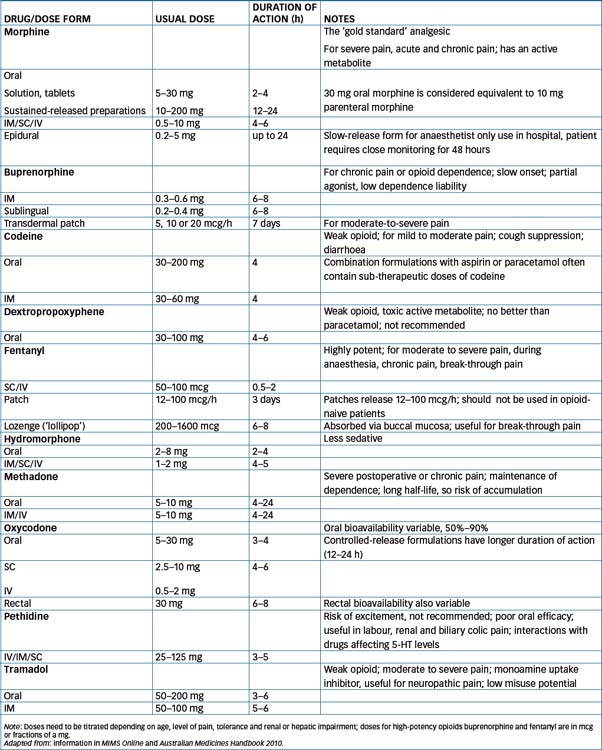
Drug interactions with opioids
Some clinically important drug interactions that may occur when morphine or other opioids are given with concomitant drugs are listed in Drug Interactions 15-1.
Drug interactions 15-1 Opioids
| Drug | Possible effects and management |
| Alcohol or other CNS depressants (other opioids, anaesthetics, sedatives, psychotropics) | May result in enhanced CNS depression, respiratory depression and hypotension; reduce dosage and monitor closely |
| Buprenorphine (partial agonist) | May result in additive effect of respiratory depression if given concurrently with low doses of μ- or κ-receptor agonists; avoid concurrent usage. Partial agonists given with an opioid agonist may reduce the analgesic effects of the full agonist or precipitate withdrawal symptoms |
| Monoamine oxidase inhibitors (phenelzine, tranylcypromine; moclobemide and selegiline) | MAOIs intensify the effects of opioids (especially pethidine and tramadol) and may cause serotonin syndrome; caution should be taken and dosages of opioids reduced |
| Opioid antagonists (naltrexone, naloxone) | Will produce withdrawal symptoms in patients dependent on opioid medications; avoid concurrent administration |
| Diltiazem, erythromycin and fluconazole | May inhibit metabolism and increase concentration of alfentanil, thus exacerbating respiratory depression; dose may need to be decreased |
| Rifampicin | May enhance metabolism and decrease concentration of morphine, codeine and alfentanil, thus reducing their effects; effects should be monitored and dose may need to be increased or another analgesic substituted |
| Many drugs (including anticonvulsants, antivirals, antifungals, rifampicin and St John’s wort) | May enhance metabolism and decrease concentration of methadone, thus reducing its effects; effects should be monitored and dose may need to be increased |
Opioid receptor agonists
Morphine
Morphine is the prototype opioid analgesic; all new analgesics are compared with morphine for potency and for therapeutic effects or adverse reactions (see Clinical Interest Box 15-1), particularly in the palliative care situation (Clinical Interest Box 15-6).
Clinical interest box 15-6 Brompton cocktail
This traditional oral mixture for pain relief dates back to the 19th century when pulmonary tuberculosis (TB) was rife, particularly among poor and malnourished populations in overcrowded cities, and antibiotics to treat Mycobacterium tuberculosis infection had not yet been discovered. Chronic infection caused granulomatous reactions and necrosis in various tissues, most commonly in the lungs and airways, but also in the bones and marrow, bowel, liver, lymph nodes, meninges and genital tracts.
The disease could have a silent course or manifest as fever, weakness, dyspnoea and destruction of lung tissue, with productive cough, bloodstained sputum and haemoptysis (coughing up of blood). The only treatment was removal of the patient to fresh air in a sanatorium and to trust the immune system to overcome the infection. In many 19th century operas and novels, the heroine afflicted with ‘consumption’ (pulmonary TB) dies tragically but musically—think of Mimi in ‘La Boheme’, Violetta in ‘La Traviata’, Fantine in ‘Les Miserables’, Beth in ‘Little Women’ and (later) Satine in the movie ‘Moulin Rouge’.
The Brompton Chest Hospital in London developed a powerful analgesic mixture to relieve the pain of pulmonary tuberculosis. The mixture became known as Brompton’s Cocktail, also known as Haustus E or Mist. Euphoriens (euphoriant mixture). A typical recipe combined three effective euphoriant analgesic drugs: opium, cocaine and gin (alcohol).
Many hospital pharmacies still have their own formula for a dose of Brompton’s Cocktail, such as:
with the gin or brandy replaced by alcohol, syrups and flavourings. It is used nowadays to relieve severe terminal cancer pain; the CNS-depressant effects help cloud the patient’s consciousness.
Morphine is available in many dosage forms, including injection, oral mixture, tablets; controlled-release capsules, granules and tablets, and slow-release epidural injection. Because the controlled-release oral preparations are very commonly used clinically (and interesting pharmacokinetically), they are considered in detail in Drug Monograph 15-1; note that controlled-release preparations are not suitable for treatment of acute pain.
Codeine
Codeine (see Drug Monograph 28-5) is absorbed well after either oral or parenteral administration. Codeine is the 3-methyl ether of morphine and is actually a prodrug, being rapidly metabolised in most people to morphine (by CYP2D6). In the 6%–10% of the (Caucasian) population who lack the enzyme to metabolise codeine, it has no analgesic effect, whereas rapid metabolisers may reach toxic concentrations of morphine.
Oral administration is used for analgesic, antitussive and antidiarrhoeal effects. Codeine may also be injected SC or IM for treatment of mild to moderate pain. Constipation is a frequent adverse effect and may require treatment or may limit the clinical usefulness of this drug.
Codeine is often combined with a non-opioid analgesic such as aspirin or paracetamol in compound analgesic tablets to provide stronger relief than the NSAID alone can achieve. In Australia, tablets containing <10 mg codeine are available over the counter (Schedule 2 or 3 [Pharmacy Medicines, Pharmacist Only Medicines, respectively]), but at higher doses (30 mg) must be prescribed (S4: Prescription Only or S8: Controlled Drugs, respectively).
Fentanyl
Fentanyl is a very potent opioid with a short duration of action and a good adverse-effect profile, so it has become popular for use as a component of anaesthesia in day-surgery procedures (see Chapter 14) and in break-through pain in cancer therapy. Fentanyl is formulated for IM or slow IV injection, as a lozenge (‘lollipop’) for absorption via the oral mucosa and, in combination with bupivacaine or ropivacaine, for epidural administration for postoperative or obstetric analgesia.
Fentanyl is also available in a patch dosage form to provide continuous opioid administration through the skin, with doses of 2.5–10 mg, indicated for severe pain associated with malignant neoplasia in patients experiencing problems with other opioids. The transdermal patch has a slow onset of action, so other shorter-acting analgesics should be administered as required when therapy is initiated. Heat and fever increase release of active drug. Fentanyl has a long duration of action (up to 72 hours), so adverse effects are not easily reversed. Common adverse effects include rash and itching; constipation is less of a problem than after oral morphine or pethidine. Even after 3 days’ wearing, patches retain about 50% of fentanyl activity, so must be disposed of carefully, as the contents of patches may be retrieved and abused by addicts. It is recommended that used patches be folded so that the adhesive side of the patch adheres to itself, wrapped and disposed of carefully. Unused patches should be returned to a pharmacy.
Related drugs are sufentanil, alfentanil and remifentanil. These are used as adjunct opioid analgesics in general anaesthesia, postoperatively and in intensive care situations.
Methadone
Methadone (see Drug Monograph 21-2) is an effective analgesic with properties similar to those of morphine, with the exception of its extended half-life and better oral bioavailability. The duration of action for methadone is usually listed at 4–6 hours, but with repeated oral dosing it may extend to 72 hours (perhaps even longer in the elderly and in patients with renal dysfunction). To control pain, methadone is administered once or twice daily, based on the individual’s response. Accumulation can occur and steady-state concentrations may not be reached for several days. Cardiac arrhythmias may occur with high doses.
Because of its extended half-life, which reduces the need for frequent dosing, methadone is approved for use in opioid detoxification and maintenance treatment programs in individuals who are physiologically dependent on heroin, opium or other opioids. Oral administration in liquid form is preferred for detoxification and required for maintenance programs, as this removes the need for injections.
Pholcodine
Pholcodine (see Drug Monograph 28-5), an opioid compound chemically similar to the opium derivative papaverine, is interesting because it has virtually no analgesic effects but retains many morphine-like effects, including suppressing cough and respiration and causing mild sedation, nausea and vomiting, dependence and constipation. It is used mainly as a cough suppressant, as are dextromethorphan and dihydrocodeine.
Tramadol
Tramadol is a relatively new centrally acting synthetic analgesic that is not chemically related to the opioids. It appears to bind to the μ-opioid receptors and also inhibits the reuptake of noradrenaline and 5-HT; hence it is sometimes referred to as an opioid–SSRI (selective serotonin reuptake inhibitor) analgesic. It is indicated for the treatment of moderate to severe pain and in neuropathic pain, but is less effective and more expensive than morphine; however, it may have a lesser potential for respiratory depression and drug dependency. Common adverse reactions include nausea, dizziness, hypertension and seizures. Tramadol is a prodrug, requiring enzymatic activation by CYP2D6, hence there are many drug–drug interactions, including with other drugs inducing or inhibiting CYP2D6 and with other drugs affecting 5-HT levels, contributing to serotonin syndrome.3
Pethidine
Pethidine (known as meperidine in the USA) is an effective analgesic for short-term use but is unsuitable for oral administration because of low bioavailability. It is less apt than morphine to release histamine or to raise biliary tract pressure; it is thus often prescribed for patients with acute asthma, biliary colic or pancreatitis. A metabolite (norpethidine) produced in the liver is neurotoxic and can accumulate, so pethidine is used only short-term, e.g. postoperatively, or in acute pain such as for obstetric analgesia. Concurrent use of MAO inhibitors may result in severe, unpredictable life-threatening reactions, including serotonin syndrome. Pethidine is often requested by illicit drug users (who may very effectively mimic the signs and symptoms of severe pain), and prescribers are warned of the danger of this drug-seeking behaviour.
Hydromorphone
Hydromorphone is a semisynthetic opioid with a faster onset but a shorter duration of action than morphine. It is prescribed for its analgesic and antitussive effects and is administered as tablets, oral liquid or injection.
Oxycodone
Oxycodone is a potent synthetic opioid about 10 times more potent than codeine; it is available in many different strength tablets, and as capsules and oral liquid. It is well absorbed through the rectal mucosa, making the suppository dosage form (30 mg) useful as a night-time analgesic and in patients unable to swallow. (Note: Suppositories should not be cut (divided up) to reduce the dosage, as the pieces may not contain an even distribution of the drug.) Doses may need to be reduced in renal impairment.
Dextropropoxyphene
Dextropropoxyphene is a synthetic analgesic, structurally related to methadone, indicated for the treatment of mild to moderate pain. It has significant dysphoric effects, accumulation and cardiotoxicity can occur and it has no marked advantages over safer analgesics such as codeine, aspirin or paracetamol, so its use is not recommended.
Heroin
The case is often put for the legalisation of heroin for treatment of intractable pain because of its analgesic and euphoric effects. Some advocates believe it is more potent, faster-acting and produces a more prolonged analgesic and euphoric effect than other analgesics. Pharmacologically, however, heroin is a prodrug: when it is administered, it is rapidly converted in the liver to morphine and morphine metabolites, and its analgesic effects are due to the morphine produced. Due to its greater lipid solubility, heroin crosses the blood–brain barrier faster than does morphine, inducing a greater ‘rush’; hence it is preferred by opioid-dependent persons; however, it has a shorter duration of action.
Heroin is a popular illegal drug of abuse (Schedule 9), so an additional fear associated with legalising it is that there may be an increased risk for drug diversion, pharmacy burglaries and crime. As heroin offers few (if any) advantages over the already marketed opioids, it is Australian policy that legalisation and clinical use of heroin are not essential to optimal treatment of pain.
Partial agonists
Partial agonists produce less than maximal effects at a receptor; for example, buprenorphine is a partial agonist at μ-receptors and antagonist at κ-receptors (see Table 15-3). Generally these drugs are less effective analgesics and have a lower dependency potential and less severe withdrawal symptoms than full opioid agonist medications. However, their use as analgesics is not recommended, as they may precipitate pain or withdrawal reactions in patients taking other opioids, their actions may not be reversible with an antagonist (naloxone) and patients taking a partial agonist may not respond fully if a full agonist needs to be given subsequently.
Buprenorphine
Buprenorphine is available as tablets, injection or patches, and is indicated for relief of moderate-to-severe pain and for treatment of opioid dependence. However, as it has a prolonged onset of action it is not suitable for acute pain and, as a partial agonist, it may precipitate withdrawal in patients dependent on other opioids, and its effects are not readily reversed by naloxone.
Opioid antagonists
The search for pure morphine antagonists produced naloxone and naltrexone, opioid antagonists that competitively displace opioid analgesics from their receptor sites, thus reversing their effects. Antagonism of endogenous opioids (enkephalins and endorphins) released during inflammatory reactions and acupuncture can lead to a state of hyperalgesia (exacerbated pain).
Antagonists block subjective and objective opioid effects and can precipitate withdrawal symptoms in people physically dependent on opioids. Naloxone and naltrexone are used to reverse the adverse or overdosage effects of opioid agonists (morphine, codeine, fentanyl, heroin, methadone etc). Respiratory difficulties in newborn babies, caused by opioids given to the mother for pain relief during childbirth, can also be treated. Respiratory depression induced by non-opioids (e.g. barbiturates), CNS depression or respiratory disease will usually not respond to opioid antagonist drug therapy.
In an opioid analgesic overdosage, the antagonist drugs will reverse the respiratory depression, sedation, pupillary miosis (constriction) and euphoric effects. The drugs are believed to bind to all three receptor sites, but their greatest affinity is for μ-receptors.
Naloxone, a short-acting antagonist administered parenterally, is used mainly for treatment of overdose or for reversal of opioid depressant effects (see Clinical Interest Box 15-5). The serum half-life of naloxone is approximately 0.5–1 hour, whereas that of morphine is 1.5–2 hours, hence frequent doses of naloxone may be necessary to prevent the person slipping back into the overdose state. Naltrexone, a long-acting antagonist given orally, is used mainly for treatment of alcohol or opioid dependence and for rapid opioid detoxification (see Chapter 21 and Drug Monograph 21-1). Adverse reactions include nausea, dizziness, nervousness, headache and fatigue.
Reversal of opioid-induced constipation
A new derivative, methylnaltrexone, is useful in treating opioid-induced constipation, as it is a relatively selective antagonist for peripheral opioid receptors. Injected SC, it causes a bowel motion within 4 hours in most patients, with little change in pain scores. At present its use is reserved for palliative care patients for whom laxatives have been ineffective.
Alvimopan (not yet available in Australia) has been designed as a peripherally-acting μ-opioid receptor antagonist to accelerate gastrointestinal tract recovery following bowel surgery. It is a charged molecule with relatively high molecular weight and low lipid solubility, hence has very low oral bioavailability into the systemic circulation or CNS. Clinical trials show efficacy in improving GIT symptoms after bowel surgery or chronic use of opioids for pain with no evidence of impaired analgesia. It is hoped that faster recovery will mean reduction in length of hospitalisation of such patients.
Non-opioid analgesics—the NSAIDs
Pharmacological actions and clinical uses
The second main group of analgesic drugs is the non-opioid or non-narcotic analgesics, typified by aspirin (see Drug Monograph 15-2). As these drugs also have significant anti-inflammatory and antipyretic (antifever) actions but do not possess the steroidal chemical structure of endogenous and exogenous anti-inflammatory corticosteroids, they are also known as non-steroidal anti-inflammatory drugs (NSAIDs) or antipyretic analgesics.4 The drugs also have antiplatelet actions, which inhibit platelet aggregation and thus decrease the risk of thrombosis. The main drugs in this classification include aspirin and paracetamol, ibuprofen, indomethacin, diclofenac and the newer specific cyclo-oxygenase-2 (COX-2) inhibitors such as celecoxib. Only aspirin and paracetamol will be considered as non-opioid analgesics in this chapter; the NSAIDs are covered in Chapter 47, with other drugs used for inflammatory conditions.
Aspirin and paracetamol are effective for mild to moderate pain (Step 1 in the stepwise management of pain, Figure 15-5), and are often combined with opioid analgesics to enhance pain control in patients with moderate to severe pain (Step 2). These agents are some of the most commonly used of all drugs (see OTC use in Chapter 3 and Clinical Interest Box 15-7): they are used for the treatment of mild to moderate pain, fever, and inflammation caused by rheumatoid arthritis, osteoarthritis and various other acute and chronic musculoskeletal and soft tissue inflammations (Chapter 47). No particular non-opioid has been shown to be generally more effective as an analgesic than others. The drugs are also used to treat bone pain associated with metastatic cancer, usually in combination with an opioid analgesic. Aspirin is used in very low doses to prevent thrombosis and reduce the risk of stroke or heart attacks (see Chapter 26).
Clinical interest box 15-7 Willow bark, salicylates and melaleuca
The history of the development of analgesics is an interesting one in the realm of pharmacognosy (the study of properties of drugs obtained from plants and other natural sources). Extracts of bark from trees of the willow family (Salicaceae) were used medicinally in ancient times, but later fell into disuse. In the 18th century in England, cinchona bark, which was imported for use as an antimalarial, analgesic, antipyretic and anti-inflammatory agent, was very expensive, so local plants were tested as cheaper substitutes.
The Rev. Edward Stone, noting that many of his parishioners suffered from ‘the ague’ (fevers, shivering, rigor and rheumatism), trusted that the cure for their ills would be provided nearby. As willows flourish in damp areas, he tested extracts of willow bark, with great success. Salicin, the bitter glycoside of the Salix species, was extracted in 1827 and found to contain saligenin, an active ingredient from which salicylic acid was prepared. Sodium salicylate was first used in 1875, and acetylsalicylic acid was introduced into medicine in 1899 as aspirin. The trade name Aspro was popularised by the Nicholas drug company in Australia. Many other derivatives of salicylic acid have been synthesised and trialled; some of those still in use are methyl salicylate (present in oil of wintergreen), salicylamide and choline salicylate.
Despite its many serious adverse effects, aspirin remains the standard antipyretic analgesic worldwide, due to its long history of availability and usage. It has been stated that, if aspirin had been discovered after the development of laws regulating drug development and clinical trials, it would never have got past phase 1 trials.
In Australian indigenous bush medicine, several traditional remedies are available to relieve the symptoms of headaches. Often, inhalations of volatile oils obtained from the leaves of such plants as small-leaf clematis (Clematis microphylla), stinkwood (Ziera spp.) or Melaleuca species are said to provide relief; their leaves may be crushed and the vapour inhaled, or leaves bound around the head to relieve pain.
Mechanism of action: Inhibition of prostaglandin formation
Despite salicylates having been used for thousands of years for the relief of pain, fever and inflammation, their mechanism of action was discovered only relatively recently (Vane 1971; see Figure 4-2). Aspirin and the other NSAIDs peripherally inhibit the synthesis and release of prostaglandins (PGs) by inhibiting the arachidonate cyclooxygenase (COX) isoenzymes (see Figure 47-5). As shown in Figure 15-2A, the products of arachidonic acid, such as PGs, play important roles in both pain and inflammation.
NSAIDs reduce the production at the site of injury of PGs that sensitise nociceptors to the algesic actions of bradykinin and other mediators of pain. Both COX-1 and COX-2 isoenzymes catalyse synthesis of PGs involved in pain, and thus COX inhibition accounts for most of the analgesic effects and also the adverse effects of the non-opioid analgesics. The analgesic effects of NSAIDs are particularly useful in relief of pain of inflammatory origin. (Selective COX-2 inhibitors are preferred for anti-inflammatory effects and for reduced gastrointestinal adverse effects).
The analgesic action is peripheral, and non-opioids do not cause tolerance or dependence, or modify psychological reactions to pain. They may, however, also have a central analgesic action in the spinal cord. The antipyretic action is due to inhibition of PG synthesis in the hypothalamus, the temperature-regulating centre of the body.
Adverse drug reactions
Common adverse drug reactions to non-opioid analgesics include gastrointestinal tract disorders (dyspepsia, nausea and vomiting, diarrhoea/constipation and gastritis) due to reduced synthesis of mucoprotective PGs by systemically absorbed NSAIDs; hence NSAIDs are contraindicated in peptic ulcer disease. Other adverse effects include renal damage (due to inhibition of vasodilator PGs—particularly a problem in elderly patients on long-acting NSAIDs), asthma attacks, skin reactions (rashes, urticaria), sodium retention and consequent heart failure and hypertension in predisposed individuals. Individual NSAIDs may cause specific adverse effects, e.g. salicylates generally can cause tinnitus, impaired haemostasis and acid–base imbalances (Clinical Interest Box 15-8), while large overdose of paracetamol can cause fatal acute liver damage if not promptly treated (see Clinical Interest Box 15-9).
Clinical interest box 15-8 Managing aspirin overdose
The effects of severe overdose of aspirin can be dramatic and potentially fatal, especially in children, causing ketosis and metabolic acidosis. Treatment includes gastric lavage and forced alkaline diuresis, and close monitoring of vital functions is necessary. Fluid, electrolyte and acid–base imbalances must be corrected and hyperthermia, hyperglycaemia or hypoglycaemia treated.
Plasma salicylate concentration is monitored until it is lowered to a non-toxic level. If large amounts of aspirin have been consumed and the salicylate concentration 2 hours after ingestion is 500 mg/L, this indicates a serious toxicity, whereas a plasma concentration of 800 mg/L is potentially fatal. If large amounts of delayed-release formulations have been taken, plasma salicylate concentration is not reliable for estimating degree of toxicity.
Exchange transfusion, haemodialysis or peritoneal dialysis may be necessary in cases of severe salicylate overdose. It is important to note that there is no specific antidote for aspirin poisoning, as there is for paracetamol (acetylcysteine).
Clinical interest box 15-9 Managing paracetamol overdose
Patients suspected of taking an overdose of paracetamol are notoriously unreliable as to the amount taken and the time of ingestion, and may appear well for 1–2 days after a potentially fatal overdose. As there is a specific antidote (acetylcysteine, which replaces depleted glutathione, see Figure 15-6), it is important that any patient with possible paracetamol overdose be tested for paracetamol plasma levels. Toxicity is probable if the patient has ingested more than 150 mg paracetamol per kg body weight (about 20 standard 500 mg paracetamol tablets for an average adult) or 10 g total paracetamol.
Symptoms
Treatment
Aspirin and other salicylates
Aspirin
Aspirin, the prototype NSAID (Drug Monograph 15-2), is readily available, both over the counter and by prescription for some combination products. After absorption, administered salicylates are rapidly metabolised in the plasma to salicylic acid, which is the active form of salicylate drugs.5 Aspirin is indicated for prophylaxis of thrombosis, and hence prevention of stroke and acute myocardial infarction (AMI), and treatment of mild-to-moderate pain, fever, and inflammation.
Topical salicylates
Salicylates can also be absorbed after topical application to inflamed soft tissues; e.g. methyl salicylate (oil of wintergreen) is used in creams, ointments and liniments for relief of inflammatory pain such as from sporting injuries and arthritic conditions. Salicylic acid itself is too strong an irritant to be taken orally but is used topically in many dermatological preparations for its antimicrobial and keratolytic effects (see Chapter 48).
Antiplatelet action of aspirin
In addition to its analgesic, anti-inflammatory and antipyretic actions, aspirin decreases the risk of arterial thrombosis in blood vessels by selectively inhibiting the formation in platelets of thromboxanes, which mediate vasoconstriction and platelet aggregation. As this antiplatelet action ‘kicks in’ at very low concentrations of aspirin, it has been suggested that aspirin should really be classified as an antiplatelet drug that has analgesic and other actions at higher doses. (Note that aspirin does not prevent or break down fibrin clots such as in venous thromboembolism.)
This effect of aspirin is qualitatively different from that of the other NSAIDs, as it causes irreversible inhibition of platelet cyclo-oxygenase by acetylation of the active site of the enzyme (see Clinical Interest Box 26-3 and Figure 26-4) for the lifetime of the platelet (a few days)—hence chronic aspirin dosage should be ceased 7 days before surgery if possible to reduce the risk of bleeding. Aspirin also appears to be effective prophylactically in decreasing the risk of bowel cancer. The benefits in reduced cardiovascular events must always be weighed against the risks of enhanced gastrointestinal or intracranial bleeding due to reduced platelet functions.
Paracetamol
Paracetamol (known as acetaminophen in the USA; see Drug Monograph 3-1) is an effective antipyretic analgesic, producing these effects by inhibiting PG synthesis in the CNS. Although its anti-inflammatory effects are minimal, paracetamol does appear to inhibit COX in some tissues in some species, so its exact mechanisms of action are not clear. There is recent evidence that paracetamol also acts as a prodrug, with one of its metabolites active at cannabinoid receptors in the CNS, thus mimicking the analgesic actions of marijuana.
In normal doses, it is a safer OTC analgesic than aspirin, for the following reasons:
Paracetamol is an effective analgesic provided that adequate regular doses are maintained; typical adult dosage is two tablets to start, then one tablet (500 mg) every 3–6 hours, with maximum daily dose 4 g (8 standard tablets). The dose is lower for small elderly patients.
Paracetamol is recommended particularly for treatment of mild pain and fevers in children. Paracetamol is indicated for treatment of a wide range of conditions causing mild-to-moderate pain (especially of non-inflammatory origin), fever, migraine and tension headache, and some forms of arthritis (osteoarthritis; see Day and Graham [2005]). Aspirin or other NSAIDs are preferred in moderate-to-severe arthritis when there is a major inflammatory component, as in rheumatoid arthritis (see Chapter 47).
Pharmacokinetics of paracetamol
Paracetamol taken orally is rapidly absorbed, reaching peak serum levels in 15–60 minutes; its elimination half-life is 1–3 hours. It is metabolised in the liver (see Figure 15-6), and glucuronide and sulfate metabolites are excreted by the kidneys. The usual maximum daily paracetamol dose for adults is 4 g/day (8 × 500 mg tablets); for children, the single dose is 15 mg/kg orally, with a maximum of 60–90 mg/kg/day. Paracetamol is available in infant, paediatric and adult strengths, as tablets, capsules, chewable tablets, elixirs, suppositories (not recommended) and as an injection. Extended-release tablets are also now available, containing 655 mg paracetamol in a formulation that maintains therapeutic ally active levels of drug for up to 8 hours after oral administration. The dose is two tablets every 6–8 hours, with a maximum of six tablets in any 24 hours; as with most sustained-release preparations, the tablets must not be crushed.
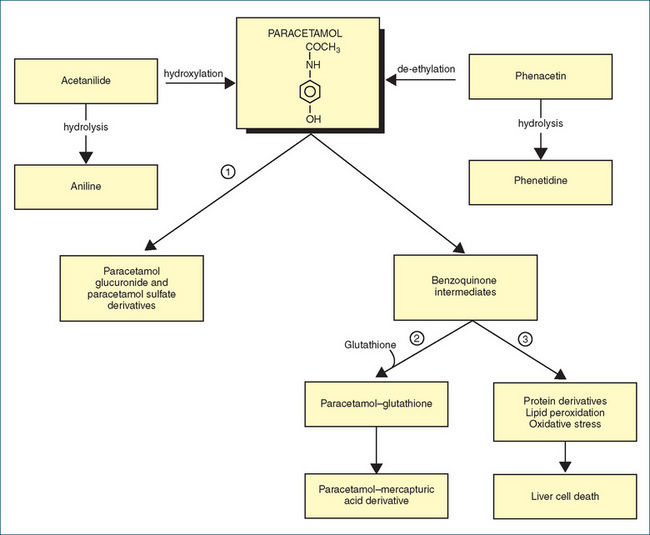
Figure 15-6 Metabolic pathways involving paracetamol. In normal doses, paracetamol is conjugated (pathway 1) to non-toxic glucuronide and sulfate derivatives. In higher doses, pathway 1 becomes saturated and a benzoquinone intermediate (BQI) is produced. Combination of the BQI with glutathione (GSH, a gamma-glutamyl-cysteinyl-glycine tripeptide involved in amino acid transport in cells) via pathway 2 produces mercapturic acid metabolites. In large overdoses, GSH reserves are used up and BQIs are diverted via pathway 3, in which toxic derivatives cause potentially lethal reactions in liver cells. Paracetamol overdose is treated with acetylcysteine, a precursor of the natural compound GSH; this replenishes GSH supplies and avoids formation of toxic BQI metabolites.
Adverse drug reactions
Adverse effects are rare with paracetamol in normal therapeutic doses, although in some instances nausea and rash have occurred. Overdose can cause serious damage to the liver and kidneys and hypoglycaemia. The hepatotoxic effects of very high levels of paracetamol are due to build-up of a toxic metabolite (Figure 15-6). This can occur after ingestion of 20 or more tablets (10 g), and an overdose is potentially fatal. Inadvertent overdose can also occur, for example by exceeding the recommended daily dose in attempts to manage increasing pain or by additive effects of paracetamol present in more than one OTC prepara tion. (See Clinical Interest Box 15-9 for management of paracetamol overdose and Drug Monograph 28-1 for use of acetylcysteine via a different formulation as a mucolytic agent in respiratory disorders.)
Compound analgesics
Simple analgesics (500 mg paracetamol, 300–500 mg aspirin) are sometimes formulated with other drugs (such as other NSAIDs, antihistamines, mild opioids, caffeine, decongestants, antiemetics or antacids) to combine the pharmacological actions of two or more drugs. Examples are Disprin Forte6 (500 mg aspirin + 9.5 mg codeine) or Panadeine Forte (500 mg paracetamol + 30 mg codeine). Codeine is also formulated with other NSAIDs such as ibuprofen.
Such combinations, however, suffer the disadvantages of all fixed-dose combinations: it is impossible to titrate the dose of either individual drug; the drug with the longer half-life may accumulate; and combinations may be more expensive than taking the individual drugs at the appropriate intervals and dosages. Codeine and caffeine have been thought to enhance the analgesic effects of aspirin; however, while codeine (60 mg) may have additive analgesic actions, caffeine provides no adjunctive analgesia. In the past, APC combinations of aspirin, phenacetin and caffeine caused disastrous ‘epidemics’ of renal damage in people who became dependent on them for pain relief (Clinical Interest Box 15-10).
Clinical interest box 15-10 A cup of tea, an APC and analgesic nephropathy
A ‘classic’ Australian compound analgesic formulation, APC powders or tablets, contained standard doses of aspirin, phenacetin and caffeine. Many people suffering repetitive strain injuries, headache or muscular or soft tissue pain followed the common advice, ‘Have a cup of tea, an APC and a good lie-down’, and took the compound analgesics regularly in high doses for relief of pain and inflammation (aspirin and phenacetin) and for mild CNS stimulation (caffeine). It was a common practice for women doing boring manual jobs in factories to order a box of APC powders or tablets every day with their lunch order.
Unfortunately, the aspirin and phenacetin components caused gastric upsets and pain, leading to more doses of APC being taken; the caffeine withdrawal as drug levels decreased caused a rebound headache and consequent dependence on the preparation; and the chronic use of NSAID analgesics, especially phenacetin, caused chronic nephritis, renal tubular necrosis and eventually chronic renal failure requiring regular dialysis or kidney transplantation. Compound analgesics have been blamed for the high incidence of analgesic nephropathy common in Australia for much of the 20th century, and requirements for renal dialysis or transplantation. (Phenacetin has long been withdrawn from use.)
There is little evidence that combinations (even buffered aspirin or aspirin–antacid preparations) offer any advantage over individual drugs. There are, however, at least three situations in which NSAID–other drug combinations are clinically useful:
NSAIDs and polypharmacy
Aspirin or paracetamol is often included in products that contain more than one ingredient, such as pain relievers, cough and cold remedies, sedatives and medicines for allergy, menstrual or premenstrual problems. Taking more than one of these products at the same time, or with a prescribed NSAID, can lead to overdose and potential toxicity. In particular, elderly patients may be unaware that they are taking several NSAIDs concurrently,7 for example for headache, fever, arthritis, cough and cold and prophylaxis of AMI or stroke. The pharmacological and toxic effects are additive, and elderly patients with impaired renal function are particularly at risk of adverse drug reactions and interactions, especially from NSAIDs with long half-lives.
Other pharmacological analgesics
GABA analogues
New analgesics unrelated to opioids or NSAIDs are pregabalin and gabapentin. Pregabalin (an analogue of the neurotransmitter GABA, and related to the anticonvulsant drug gabapentin) reduces the release of various transmitters via interference with the calcium channels in nerve terminals. It is particularly useful in neuropathic pain such as occurs in diabetic neuropathy and post-herpetic neuralgia, but patient response is variable. Adverse reactions are mainly due to effects on CNS transmitters and include dizziness, sedation and incoordination. It is also useful as adjunctive therapy in partial seizures. The related gabapentin, a new anticonvulsant drug (see Chapter 17), has also been shown to be effective in the relief of neuropathic pain.
Capsaicin
Capsaicin, an alkaloid found in chilli peppers, is formulated into topical creams that are indicated for the treatment of neuralgias, arthritic pain and pain associated with cystitis and HIV infection. It is said to activate capsaicin receptors; on application it causes an initial release and then a depletion of substance P from nerve fibres, which eventually results in a decrease in pain transmission.
Other drugs useful for their analgesic effects
Clinical interest box 15-11 Herbal remedies for pain
The most important herbal remedies for pain are, of course, morphine and codeine in opium extracts from the poppy Papaver somniferum and salicin and salicylates from the bark of the willow tree Salix alba and from the herb meadowsweet (Filipendula ulmaria). Many other plants also produce herbal extracts that have analgesic properties, including the following, which have been clinically tested and proven:
Adapted from: Braun & Cohen (2007); this excellent evidence-based resource contains details of herbal remedies’ chemical components, pharmacological activities, clinical uses, adverse drug reactions and interactions and practice points, with references to scientific studies on the plants’ properties.
New pharmacological approaches to pain management include research on drugs that enhance the inhibitory effects of adenosine on nociceptors, mimic the actions of analgesic neuropeptides (e.g. nociceptin/orphanin FQ, a novel peptide discovered in brain extracts as the natural ligand of the opioid receptor-like 1-receptor, identified during the elucidation of the human genome) or inhibit the enzymes that inactivate enkephalins or endorphins.
Adjuvants to analgesics
Adjuvant (co-analgesic) medications are used in combination with opioid or NSAID analgesics to enhance pain relief or to treat symptoms that exacerbate pain. In some instances they are used alone to treat specifically identified pain. Adjuvant analgesic medications include a variety of drugs whose main clinical indications are in other conditions, such as anticonvulsants, antidepressants, antihistamines, corticosteroids, antiarrhythmics, psychostimulants, clonidine and capsaicin, as follows:
Non-pharmacological analgesic techniques
Non-pharmacological analgesic methods include:
Key points
 Pain is a major worldwide health problem, with sensory and emotional components contributing to suffering that disables and distresses people. Physiological theories of pain involve nociceptors, ascending afferent pathways of sensory nerves and spinothalamic tracts and descending efferent pathways from the cortex, which modulate dorsal-horn ‘gating’ mechanisms. Many neurotransmitters and other chemical factors are involved in pain sensation; analgesic drugs in particular affect endorphins and prostaglandins as mediators. Pain is often inadequately or inappropriately treated because of attitudes, fears and biases of health professionals, patients and family. Clinical principles of pain management emphasise the importance of adequate regular doses of appropriate analgesics, stepping up the analgesic ladder as necessary, with adjunctive care. Many effective pharmacological and non-drug methods are available for the treatment and/or prevention of pain. Assessment tools, pain scales and clinical guidelines are the basis for an effective pain management program. The main group of strong analgesic drugs is the opioids, which are agonists centrally on opioid receptors. Opioid antagonists are used to treat adverse effects, overdoses or dependence.
Pain is a major worldwide health problem, with sensory and emotional components contributing to suffering that disables and distresses people. Physiological theories of pain involve nociceptors, ascending afferent pathways of sensory nerves and spinothalamic tracts and descending efferent pathways from the cortex, which modulate dorsal-horn ‘gating’ mechanisms. Many neurotransmitters and other chemical factors are involved in pain sensation; analgesic drugs in particular affect endorphins and prostaglandins as mediators. Pain is often inadequately or inappropriately treated because of attitudes, fears and biases of health professionals, patients and family. Clinical principles of pain management emphasise the importance of adequate regular doses of appropriate analgesics, stepping up the analgesic ladder as necessary, with adjunctive care. Many effective pharmacological and non-drug methods are available for the treatment and/or prevention of pain. Assessment tools, pain scales and clinical guidelines are the basis for an effective pain management program. The main group of strong analgesic drugs is the opioids, which are agonists centrally on opioid receptors. Opioid antagonists are used to treat adverse effects, overdoses or dependence.Review exercises
References and further reading
Analgesic Expert Group. Therapeutic Guidelines: Analgesic, version 5. Melbourne: Therapeutic Guidelines; 2007.
Australian Medicines Handbook 2010. Adelaide: AMH, 2010.
Beggs S. Paediatric analgesia. Australian Prescriber. 2008;31(3):63-65.
Benedict D.G. Walking the tightrope: chronic pain and substance abuse. Journal for Nurse Practitioners. 2008;4(8):604-609.
Besson M., Piguet V., Dayer P., Desmeules J. New approaches to the pharmacotherapy of neuropathic pain. Expert Reviews in Clinical Pharmacology. 2008;1(5):683-693.
Braun L., Cohen M. Herbs and Natural Supplements: An Evidence-Based Guide, 2nd edn. Sydney: Elsevier Mosby; 2007.
Carr DB, Jacox AK, Chapman CR et al. Acute Pain Management: Operative or Medical Procedures and Trauma. Clinical Practice Guideline. AHCPR Pub. no. 92-0032. Rockville, MD: Agency for Health Care Policy and Research, Public Health Service, US Department of Health and Human Services, 1992.
Christensen K.S., Cohen A.E., Mermelstein F.H., et al. The analgesic efficacy and safety of a novel intranasal morphine formulation (morphine plus chitosan), immediate release oral morphine, intravenous morphine, and placebo in a postsurgical dental pain model. Anesthesia and Analgesia. 2008;107(6):2018-2024.
Day R.O., Graham G.G. Paracetamol should be first-line therapy in osteoarthritis. Medical Journal of Australia. 2005;182(4):198-199.
Dowden J.S. Medicinal mishap: monitor morphine. Australian Prescriber. 2009;32(1):13.
Editorial. The World Health Organization three-step analgesic ladder comes of age. Palliative Medicine. 2004;18:175-176.
Hicks C.L., von Baeyer C.L., Spafford P., van Korlaar I., Goodenough B. The Faces Pain Scale—revised: toward a common metric in pediatric pain measurement. Pain. 2001;93:173-183.
Hunt S.P., Koltzenberg M. The Neurobiology of Pain. Oxford: Oxford University Press; 2005.
Hurley R.W., Adams M.C. Sex, gender, and pain: an overview of a complex field. Anesthesia and Analgesia. 2008;107(1):309-317.
Lin J.G., Chen W.L. Acupuncture analgesia: a review of its mechanisms of actions. American Journal of Chinese Medicine. 2008;36(4):635-645.
McCaffery M., Pasero C. Pain: Clinical Manual. St Louis: Mosby; 1999.
McMahon S.B., editor. Wall and Melzack’s Textbook of Pain, 5th edn, Philadelphia: Elsevier/Churchill Livingstone, 2005.
Melzack R. The tragedy of needless pain. Scientific American. 1990;262(2):19-25.
Melzack R., Wall P.D. Pain mechanisms: a new theory. Science. 1965;150:971-979.
Meunier J.C. Utilizing functional genomics to identify new pain treatments: the example of nociceptin. American Journal of Pharmacogenomics. 2003;3(2):117-130.
Murtagh J.E. Managing painful paediatric procedures. Australian Prescriber. 2006;29:94-96.
Palliative Care Expert Group. Therapeutic Guidelines: Palliative Care, version 2. Melbourne: Therapeutic Guidelines; 2005.
Pasternak G.W. Multiple opiate receptors: deja vu all over again. Neuropharmacology. 2004;47(Suppl 1):312-323.
Roberts L.J. Managing acute pain in patients with an opioid abuse or dependence disorder. Australian Prescriber. 2008;31(5):133-135.
Salerno E. Pharmacology for Health Professionals. St Louis: Mosby; 1999. [chs 3, 8]
Salerno E., Willens J.S. Pain Management Handbook. St Louis: Mosby; 1996.
Speight T.M., Holford N.H.G., editors. Avery’s Drug Treatment, 4th edn, Auckland: Adis, 1997.
Spencer J.W., Jacobs J.J. Complementary and Alternative Medicine: An Evidence-Based Approach. St Louis: Mosby; 1999.
Stillman M. Clinical approach to patients with neuropathic pain. Cleveland Clinic Journal of Medicine. 2006;73(8):726-739.
Surratt C.K., Adams W.R. G protein-coupled receptor structural motifs: relevance to the opioid receptors. Current Topics in Medicinal Chemistry. 2005;5(3):315-324.
Vane J.R. Inhibition of prostaglandin synthesis as a mechanism of action for aspirin-like drugs. Nature New Biology. 1971;231:232-239.
Vane J.R. The fight against rheumatism: from willow-bark to COX-1 sparing drugs. Journal of Physiology and Pharmacology. 2000;51(4):573-586. Part 1
Wong D., Hockenberry-Eaton M., Wilson D., et al. Wong’s Essentials of Pediatric Nursing, 6th edn. St Louis: Mosby; 2001.