Chapter 22 Overview of the Heart and Drugs Affecting Cardiac Function
Cardiovascular disease is a major health problem in Australia and New Zealand (Clinical Interest Box 22-1) and annually accounts for approximately 40% of all deaths. Indigenous Australians are three times more likely to die from cardiovascular disease than non- Indigenous Australians. As life expectancy increases, it is anticipated that more people will be diagnosed with acute and chronic cardio vascular conditions, and they will rely increasingly on care provided by health professionals. An understanding of the anatomy and physiol ogy of the heart and vascular system is essential to understanding the action and use of drugs in the treatment of hypertension, cardiac failure, angina and thromboembolic disorders. Numerous drugs affect the heart both directly and indirectly and include the autonomic transmitter adrenaline and related drugs (refer to Chapter 12), antiarrhythmic drugs, the cardiac glycoside digoxin and the calcium channel blockers (refer to Chapter 23). Digoxin has a narrow therapeutic index and there is a need to be aware of its proarrhythmogenic tendency and its range of adverse effects. The drugs used to treat arrhythmias are very potent, with the potential to cause sudden cardiac death. Careful drug selection, along with close monitoring of a person’s clinical condition, is crucial to achieving the goal of safe and effective anti-arrhythmic therapy.
Clinical interest Box 22-1 New zealand health information
 Population health surveys and record linkage studies are managed by Health and Disability Intelligence. The findings are integrated into the New Zealand Health Monitor, which compiles social statistics and operates under strict ethical standards. The surveys are conducted nationwide at different intervals, as shown below:
Population health surveys and record linkage studies are managed by Health and Disability Intelligence. The findings are integrated into the New Zealand Health Monitor, which compiles social statistics and operates under strict ethical standards. The surveys are conducted nationwide at different intervals, as shown below:
| Survey | Frequency (Years) | Next survey |
| NZ Health Survey | 3 | 2009/10 |
| NZ Adult Nutrition Survey | 10 | 2018 |
| NZ Child Nutrition Survey | 10 | 2012 |
| NZ Tobacco Use Survey 2 out of every | 3 | 2011 |
| NZ Alcohol and Drug Use Survey | 3 | 2011 |
| NZ Mental Health and Wellbeing Survey | 10 | 2012 |
Key findings on chronic disease from the 2006/07 survey include:
Adapted from: Ministry of Health, A Portrait of Health: Key results of the 2006/07 New Zealand Health Survey. Wellington: Ministry of Health; 2008 (www.moh.govt.nz).
Key abbreviations
Key background
The heart
THE heart, which lies in the mediastinum slightly to the left of the midline of the thoracic cavity, is a hollow muscular organ that consists of four chambers—the upper right and left atria and the lower right and left ventricles. It is surrounded and protected by the pericardium; the outer fibrous pericardium prevents overstretching and anchors the heart in the mediastinum. The serous (inner) pericardium has two layers and between them is secreted the thin slippery pericardial fluid, which reduces friction between the layers as the heart moves. The heart wall consists of the external smooth epicardium, the middle layer of myocardium (or muscle tissue) and the inner endocardium, which lines the chambers of the heart and the valves. The valves open, enabling blood to flow in the forward direction, and close, preventing backflow into the chambers. The tricuspid valve (three cusps) lies between the right atrium and right ventricle, the mitral (bicuspid) valve between the left atrium and left ventricle, the semilunar valves (three cusps) between the right ventricle and the pulmonary artery (pulmonary valve) and between the left ventricle and the aorta (aortic valve) (Figure 22-1). Movement of the cusps (or leaflets) can be detected by echocardiography, and the sounds they produce during closure can be heard with a stethoscope. Regurgitant (leaky) or stenotic (narrowed) valves make characteristic sounds that are called murmurs.
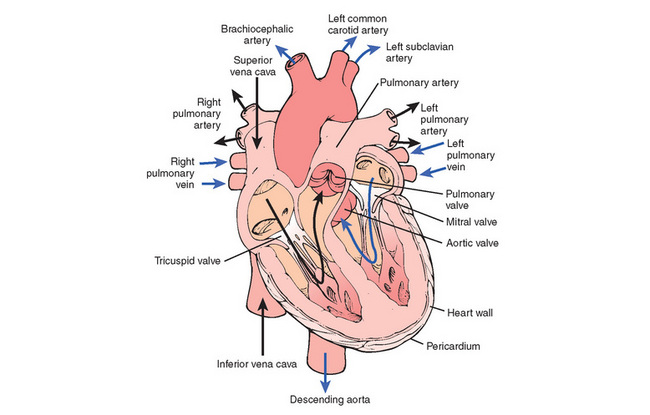
The pumping action of the heart depends on the ability of the cardiac muscle to contract. The myocardium is composed of interconnected branching muscle fibres, or cells, that form the walls of the atria and the ventricles. The thickness of the myocardium of the atria and ventricles varies. The atria tend to be thinner because they act more as delivery containers while the ventricles tend to be thicker because they forcibly contract and pump blood against a resistance. The resistance of the pulmonary bed is low so the wall of the right ventricle is not as thick as that of the left ventricle, which has to pump blood to all parts of the body against total systemic vascular resistance. Contractility of the heart is energy dependent and the heart derives most of its energy from oxidative metabolism of fatty acids and lactate, which occurs in mitochondria and cardiac muscle cells.
Coronary vascular supply of the heart
The entire blood supply to the myocardium is provided by the right and left coronary arteries, which arise from the base of the aorta (Figure 22-2). The right atrium and ventricle are supplied with blood from the right coronary artery. The left coronary artery divides into the anterior (descending) branch and the circumflex branch and supplies blood to the left atrium and ventricle. These main coronary vessels continue to divide, forming numerous branches, resulting in a profuse network of coronary vessels. The major arterial vessels supplying the heart are located on the external surface of the ventricles. Branches penetrate the myocardium towards the endocardial (inner) surface. Venous coronary blood drains via the coronary sinus into the right atrium. Coronary perfusion occurs as a result of the high pressure of blood in the aorta and occurs primarily when the ventricles have relaxed and the coronary vessels are no longer compressed. Ventricular contraction compresses the coronary vascular bed but increases coronary outflow. Increased oxygen delivery to the myocardium is supported almost exclusively by the increased coronary blood flow.
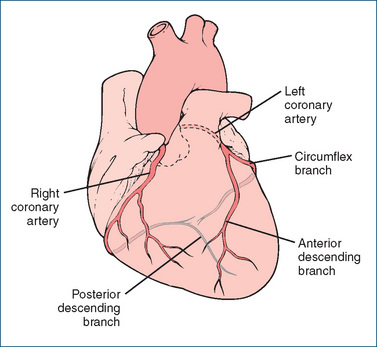
Figure 22-2 Coronary blood supply to the heart. Dark shaded vessels are those located on the external surface of the ventricles; light shaded vessels show penetration of arterial branches towards the endocardial surface.
When the demand for oxygen and nutrients by body tissues increases, cardiac output must increase. At the same time, the heart muscle itself must be supplied with enough oxygen and nutrients to replace the energy it expends. In other words, a balance must be maintained between energy expenditure and energy restoration. The increase in heart rate increases the metabolic needs of the heart and, normally, coronary dilation occurs in an attempt to meet the higher metabolic demand and to overcome restricted blood inflow. Whenever the delivery of oxygen to the myocardium is inadequate to meet the increased oxygen consumption by the heart, myocardial ischaemia occurs. In many instances atheroma formation is one of the major causes of ischaemia, which manifests in the signs and symptoms of angina (Chapter 23).
Control by the autonomic nervous system
Although the cardiac conduction system possesses the inherent ability for spontaneous rhythmic initiation of the cardiac impulse, the autonomic nervous system has an important role in the regulation of the rate, rhythm and force of myocardial contraction of the heart. Postganglionic fibres of the sympathetic nervous system, which release noradrenaline (NA), innervate the sinoatrial (SA) node, atria and ventricles. Action of NA on β1 receptors located in both nodes and atrial/ventricular muscles increases heart rate, automaticity, conduction velocity and force of contraction. Circulating adrenaline from the adrenal medulla also elicits cardiac responses e.g. tachycardia. Clinically, high doses of administered adrenaline may exert a direct effect on the electrophysiological properties of cardiac tissue, causing cardiac dysrhythmias.
Vagal nerve fibres of the parasympathetic branch, which release acetylcholine, are found primarily in the SA and atrioventricular (AV) nodes and atrial muscle. Acetylcholine acts on muscarinic (M2) receptors of the SA node to decrease heart rate, on the AV node to decrease conduction velocity and, to a limited extent, on the M2 receptors on cardiac myocytes to reduce cardiac contractility. Control by the vagus nerve ensures that the heart rate is slowed to about 75 beats/minute. In the absence of any regulation from the parasympathetic nervous system the heart would contract at about 90–100 beats/minute, which is the normal automatic firing rate of the SA node. Normally the heart rate is under the continuous influence of both parasympathetic and sympathetic nervous systems; the resting heart rate is the result of their opposing influences and at rest the firing rate of the sympathetic cardiac nerves is less than that of the vagus nerve.
Cardiac natriuretic peptides
The natriuretic peptides include atrial natriuretic peptide (ANP), B-type natriuretic peptide (BNP) and C-type natriuretic peptide. These endogenous neurohormones increase sodium and water excretion by the kidney and, with the exception of the efferent arterioles of the kidney, they relax vascular smooth muscle. Additionally, natriuretic peptides increase vascular permeability and inhibit the actions and/or release of endothelin, antidiuretic hormone and mediators of the renin–angiotensin–aldosterone system. ANP is located in secretory granules of atrial cells and is released by stretching of the atria. In contrast, BNP and C-type NP are located in the ventricles and vascular smooth muscle, respectively. Stretching of the ventricles results in the release of BNP, and the circulating concentration of BNP correlates with severity of heart failure.
The cardiac conduction system
Contraction of the heart depends on the regularity of events occurring in the cardiac cycle. Each cycle consists of a period of relaxation, diastole, followed by a period of contraction, systole. The rhythm and rate of the cardiac cycle are regulated by the conduction system, specialised cardiac cells that have the ability to initiate and transmit the electrical impulses needed to stimulate contraction of the cardiac muscle.
The conduction system (Figure 22-3) comprises:
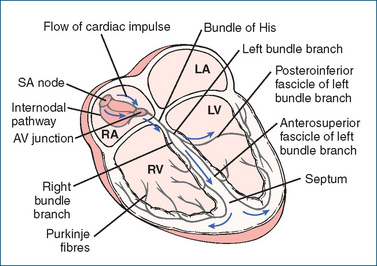
Figure 22-3 Conduction system of the heart. The cardiac impulse is initiated at the SA node and is transmitted through the internodal pathways to the two atria, resulting in atrial contraction. At the AV node, the electrical impulse is delayed. Conduction then speeds up at the bundle of His, with the impulse travelling through the right bundle branch and the left bundle branch and continuing through the posteroinferior fascicle and anterosuperior fascicle of the latter bundle branch. Finally, the arrival of impulses at the Purkinje fibres results in their distribution to all parts of both ventricles where, on excitation, ventricular contraction is produced. RA = right atrium; RV = right ventricle; LA = left atrium; LV = left ventricle; SA = sinoatrial.
In the normal heart, the SA node, located in the right atrium, initiates the heartbeat. The impulses generated are then conducted through the interatrial and internodal pathways to both atria, producing atrial contraction. Having travelled through the atria the impulses arrive at the AV node, which links the conducting pathways of the atria to the ventricles. Electrical conduction is delayed at the AV node, allowing time for the atria to contract fully and the ventricles to finish filling before ventricular contraction. At the bundle of His, conduction speeds up and the impulses travel through the right and left bundle branches, then through the posteroinferior and anterosuperior fascicles of the left bundle branch. The transmission of impulses at the Purkinje fibres, which consist of tiny fibrils that spread around the ventricles and connect directly with the myocardial cells, is very rapid. Finally, the synchronised depolarisation of both ventricles produces ventricular contraction, resulting in the ejection of blood through both the pulmonary artery and the aorta by the ventricles. The coordinated pumping action of the heart is initiated and regulated by the specialised fibres of the conduction system. The individual fibres of this system possess three basic electrophysiological properties: automaticity, conductivity and refractoriness.
Automaticity
The specialised fibres of the conduction system have the inherent ability to spontaneously initiate an electrical impulse. The cells that possess this property of automaticity are called pacemaker cells. They are found in specialised conducting tissues such as the SA and AV nodes and the His–Purkinje system.
Normally, impulses are spontaneously and regularly initiated at the pacemaker cells of the SA node. During the resting phase the membrane of the cell depolarises spontaneously and gradually, until it reaches the threshold and generates an action potential (see later section, ‘Electrical excitation’). Thus the membrane of pacemaker cells is never truly at rest. The slow depolarisation of the membrane in the resting state is called spontaneous diastolic depolarisation, or phase 4 depolarisation, and defines automaticity. In the SA and AV nodes this property is attributed to changes in potassium and calcium currents and the pacemaker current. The resting potential of automatic pacemaker cells differs from that of contractile myocardial cells. After full repolarisation, the membrane of myocardial cells maintains a steady resting potential until an external stimulus causes it to depolarise. Automaticity is thus a property of fibres of the conduction system that normally controls heart rhythm — it is not a feature of ‘working’ muscle (atria and ventricles). However, in some circumstances (e.g. cardiac disease, use of certain drugs), myocardial cells have the potential to exhibit spontaneous depolarisation. This is often referred to as an ‘early after-depolarisation’, which occurs because of a shift closer to the threshold for an action potential resulting from an abnormal interaction of the calcium current and the repolarising potassium current. If an early afterdepolarisation is sufficiently large it may trigger an extrasystole, often referred to as a premature ventricular contraction. If a run of extrasystoles occurs this may result in ventricular tachycardia (120–150 beats/minute), which has the potential to degenerate into ventricular fibrillation.
The spontaneous excitation of pacemaker cells establishes the normal rhythm of the heart. The regularity of such pacemaking activity is termed rhythmicity. Under normal circumstances, only one functional pacemaker, the SA node, predominates because it has the highest frequency of depolarisation. The normal rate of impulse formation is about 72 beats/min. If the SA node substantially slows its rate of impulse formation, then the AV node becomes the primary pacemaker of the heart and will drive the heart at about 40 beats/min.
Conductivity
Conductivity refers to the ability of a cell to transmit an action potential along its plasma membrane. The property of conductivity therefore exists not only in the cells of the conduction system but also in the cardiac musculature. The speed with which electrical activity is spread within the SA node is quite slow—about 0.05 m/s. The impulse then spreads out rapidly over the atrial musculature at a rate of about 1 m/s. When the impulse reaches the AV node, there is a delay of about 0.01 seconds, then atrial systole occurs, allowing the atria to contract fully and the ventricles to fill. The impulse then spreads rapidly at about 2–4 m/s, along the right and left bundle branches and Purkinje fibres. This rapid activation of contractile elements evokes a synchronous contraction of the ventricles. The conduction velocity is determined by the threshold size of the resting potential of the cell membrane and by membrane responsiveness.
Refractoriness
Cardiac tissue is non-responsive to stimulation during the initial phase of systole (contraction). This is known as refractoriness, and it determines how closely together two action potentials can occur. Throughout most of the repolarisation phase, the cell cannot respond to a stimulus. The effective refractory period represents that period in the cardiac cycle during which a stimulus, no matter how strong, fails to produce an action potential. After the effective refractory period and as repolarisation nears completion, a relative refractory period occurs. This is defined as that period during which a propagated action potential can be elicited, provided that the stimulus is stronger than normally required in diastole. When this happens, the fibre is stimulated to contract prematurely, giving rise to an ectopic (extra) beat. Drugs such as digoxin, caffeine and nicotine can trigger ectopic activity.
Myocardial contraction
Throughout the past decade, our understanding of the fundamental mechanisms governing contraction of cardiac muscle in both normal and disease states has improved tremendously. Yet some aspects of this complicated process are still unknown. Cardiac cells are electrically coupled to each other through gap junctions that allow the action potential to propagate from cell to cell. Cardiac muscle contraction begins with a rapid change in the resting membrane potential of the cell. This electrical current spreads to the interior of the cell, where it causes the release of calcium ions from the sarcoplasmic reticulum. The calcium ions then initiate the chemical events of contraction. The overall process for controlling cardiac muscle contraction, called excitation–contraction coupling, involves electrical excitation, mechanical activation and contractile mechanisms.
Electrical excitation
Cardiac muscle contraction begins with an action potential initiated by the SA node. The action potential, the difference in conductance, which produces rapid changes in concentrations of sodium, potassium and calcium ions, occurs in the membrane of the myocardial cell. The resting state of a muscle cell in the ventricle is created by the difference in electrical charge across the sarcolemma. In this case, the inside of the cell is negative with respect to the outside, which is positively charged. Because the sarcolemma separates these opposite charges, the membrane is in effect polarised. At rest, the extracellular environment is rich in sodium ions (Na+) and the intracellular environment in potassium ions (K+), with a rich calcium ion (Ca2+) concentration in the region of the sarcolemma and where it invaginates on the sarcotubule (Figure 22-6).
The cardiac action potential is divided into two stages, depolarisation and repolarisation. These stages are further subdivided into five phases, 0–4. The resting potential of a myocardial cell is called phase 4; in this phase the membrane is polarised with a charge of around −90 millivolts (mV). At this voltage the interior of the cell is negative with respect to the exterior and the membrane is relatively impermeable to ions. Any stimulus that changes the resting membrane potential to a critical value, called the threshold, can generate an action potential. See Figure 22-4 for the stages of an action potential.
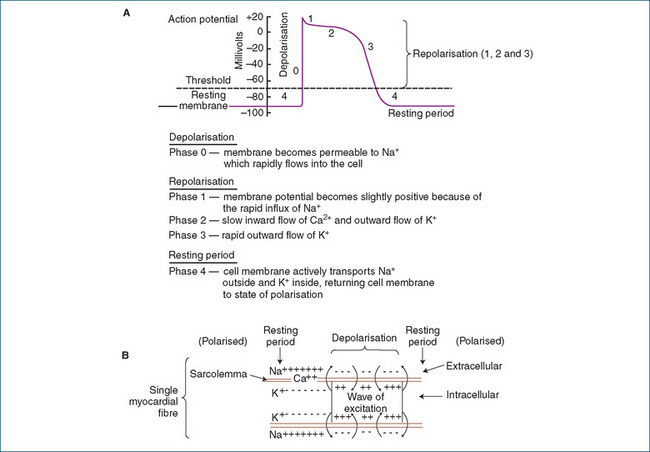
Figure 22-4 A Action potential of a single myocardial cell. B Ion movements across the myocardial cell membrane during an action potential.
In the cells of the SA and AV nodes, the action potential consists of only phases 0, 3 and 4 (Figure 22-5). The principal distinguishing feature of the pacemaker fibre resides in phase 4 or the pacemaker potential. A slow spontaneous depolarisation occurs that requires no external stimulus and is termed diastolic depolarisation. This is responsible for automaticity. Unlike the fast sodium channels of the myocardium, depolarisation (or phase 0) is achieved predominantly by the slower current carried by calcium ions (to a minor extent by sodium ions) through the slow calcium channels of nodal cells. Thus, phase 0 results in a slower conduction velocity in nodal cells than in myocardial cells. Calcium channel blockers inhibit these slow channels. Repolarisation is more gradual and involves only phase 3. The membrane then finally returns to phase 4.
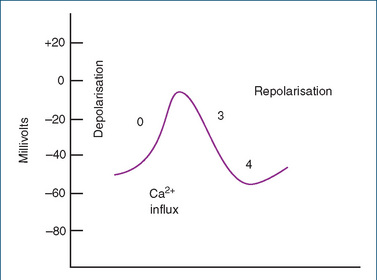
Figure 22-5 Three-phase action potential of a slow-channel fibre, the SA node. Unlike in the case of the fast fibres of myocardial cells, the depolarisation (phase 0) is attributed primarily to Ca2+ influx through slow calcium channels of the cell membrane. Repolarisation involves only phase 3, which is followed by phase 4.
Mechanical activation
Each individual cardiac muscle cell contains a nucleus in the middle and a plasma membrane (cell membrane), the sarcolemma (Figure 22-6A). By joining end to end, the cells form a long fibre, with each cell contacting its neighbour through a thickening of the sarcolemma called the intercalated disc. These discs contain desmosomes, which hold the fibres together, and gap junctions, which provide sites of low electrical resistance, permitting the spread of muscle action potentials throughout the cardiac muscle.
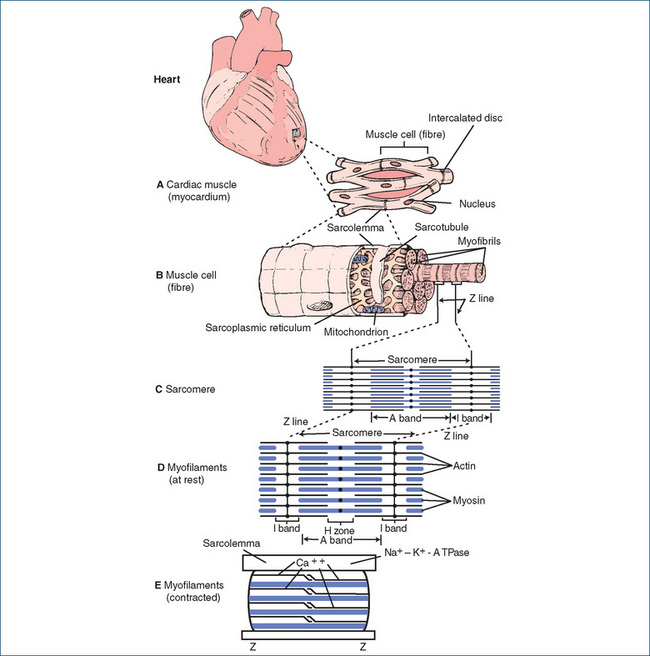
Figure 22-6 Structure of heart and cardiac muscle cell fibres. The enlargement of the square illustrates a portion of the cardiac muscle (myocardium) (A), which is composed of myocardial cells. Each cell contains a centrally located nucleus and a limiting plasma membrane (sarcolemma), which forms the intercalated disc at the termination of each cell. An individual muscle cell (fibre) (B) consists of multiple parallel myofibrils. Each myofibril is arranged longitudinally in a series of light and dark repeating units. Each unit is called a sarcomere. At the Z line, the sarcolemma invaginates to form the transverse sarcotubules, or T system. An extensive network, called the sarcoplasmic reticulum, encircles groups of myofibrils and makes contact with the sarcotubules. The sarcoplasmic reticulum contains a high concentration of calcium ions. The mitochondria appear in long chains between the myofibrils. The sarcomere (C) is the unit of muscle contraction. It is composed of two types of bands, the A band and the I band. The Z line divides the latter. Myofilaments (D) of the sarcomere include the thin filament, actin, and the thick filament, myosin. The dark appearance of the A band is caused by the myosin and the lighter appearance of the I band by the actin. When contracted (E), the sarcomere shortens so that the thick filaments approach the Z line and the width of the H zone between the thin filaments narrows. Calcium ions are required for contraction.
Each individual muscle fibre (cell) comprises a group of multiple parallel myofibrils, the end unit of which is the myofilament. The myofibrils are arranged end to end in a series of repeating units called sarcomeres (Figure 22-6B, C). At the point of separation of the sarcomeres, known as the Z line, the sarcolemma of the muscle fibre interlocks (invaginates) at its end with the sarcomere to form the transverse sarcotubule, or T system, which penetrates deeply into the cell. An extensive network of internal membranes, the sarcoplasmic reticulum, encircles groups of myofibrils and makes contact with the sarcotubules.
The sarcomere, which is the basic unit of contraction in the heart, lies between two successive Z lines and consists of two contractile proteins, actin and myosin. Examination by light microscopy reveals the most characteristic feature of the muscle fibre, alternating light (I) bands and dark (A) bands (Figure 22-6C). The darkness of the A bands results from the thicker myosin filaments, while the lightness of the I bands reflects the thinner actin filaments. In the middle of the darker A band is a less dense portion called the H zone; the myosin filaments run the entire length of the A band, passing through the H zone. The lighter I band, in contrast, is divided by the darker-appearing Z line, where actin filaments from neighbouring sarcomeres join each other; actin filaments run through the whole I band and terminate at the edge of the H zone, which accounts for the lighter appearance of this zone (Figure 22-6D, E). Cross-bridges, which are small projections that extend from the sides of the myosin filament, appear along the entire length of the filament. The interaction between these cross-bridges of myosin and the active sites of actin produces contraction by sliding the A bands and I bands with respect to each other. Myosin contains the ATPase needed to hydrolyse ATP, which is required to provide the energy for contraction. ATP is synthesised in the mitochondria, which are normally abundant in cardiac muscle. Actin is involved with calcium ion activity. These two filaments combine to help effect cardiac contraction.
Contraction is initiated when the impulse reaches the myocardial cell and travels along the sarcolemma of the muscle fibre. As the depolarisation wave spreads along the sarcotubules, calcium enters through ‘L-type’ (longlasting and large) voltage-sensitive calcium channels, causing a secondary release of calcium from the sarcoplasmic reticulum. Hence the plateau, which is phase 2 of the action potential, is maintained through this slow inward calcium current. Calcium ion movement is the chief component that couples electrical excitation of the sarcolemma with muscle activation of the myofilaments in the sarcomere. Normally, interaction between actin and myosin is prevented by tropomyosin, which is bound to the actin filament. Binding of calcium ions to troponin C, a component of the troponin complex, results in a conformational change that moves tropomyosin out of the way and allows binding of the myosin cross-bridges to the actin filaments. These changes initiate the contractile mechanism.
The contractile mechanism
Activation of the actin filaments by calcium ions allows formation of the myosin cross-bridges. This interaction pulls the actin along the immobile myosin filaments towards the centre of the A band, shortening the sarcomere and producing muscle contraction. In this process, the lengths of individual filaments remain unchanged. The I band narrows as the thick filaments approach the Z line, and the H zone narrows between the ends of the thin filaments when they meet at the centre of the sarcomere (Figure 22-6D, E). The greater the quantity of calcium ions delivered to troponin, the greater the rate and numbers of interactions between actin and myosin. As a result of this response, the development of tension and contractility is increased.
When magnesium is present, ATP is cleaved by myosin ATPase. This reaction provides the energy necessary for the actin filaments to move along the myosin and produce muscle contraction. Muscle relaxation depends on removing calcium ions from the sarcomere, thereby allowing the actin–myosin filaments of the sarcomere to return to their resting positions. This is achieved by a calcium ATPase (located in the walls of the sarcoplasmic reticulum), which actively returns some calcium ions to the sarcoplasmic reticulum while the remainder are removed from the cell by a Na+–Ca2+ exchange protein that exchanges three sodium ions for every calcium ion.
The electrocardiogram
An electrocardiogram (ECG) is a graphic representation of electrical currents produced by the heart. It is a useful tool in determining abnormalities of cardiac rhythm, the heart’s response to exercise and the effectiveness of certain drugs. An electrode is placed on each limb and a single electrode placed independently in six different positions on the chest. Combinations of limb and chest leads provide 12 different recordings that, when compared, provide information on the functioning of the heart.
Electrical activity typified by three distinct waves on the ECG (P, QRS and T) always precedes mechanical contraction. The P wave represents atrial depolarisation and follows the firing of the SA node. Immediately after, a wave of electrical activity moves through atrial muscle, the muscle contracts and blood flows from the atria into the ventricles. After the P wave, a short pause or interval (P–R interval) occurs while the electrical activity is transmitted to the AV node, conduction tissue and ventricles. The second wave, the QRS complex, represents ventricular depolarisation and the ventricles contract shortly after it begins. Repolarisation, or recovery, of the ventricles is indicated by the third and smaller T wave. Rarely a U wave may be seen, which is thought to represent repolarisation of papillary muscle. Atrial recovery or repolarisation does not show on the ECG because it is hidden in the QRS complex (Figure 22-7).
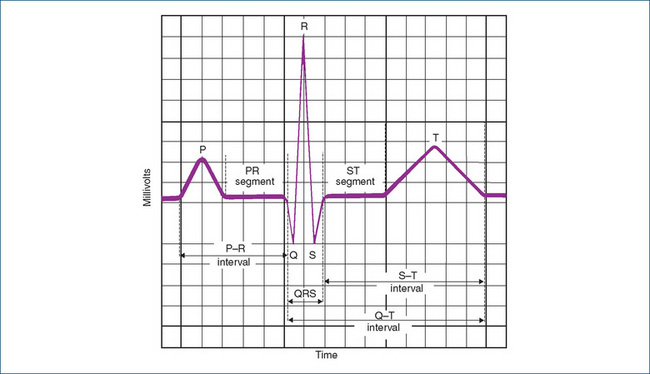
Figure 22-7 Graphic representation of the normal electrocardiogram. Vertical lines represent time, each square represents 0.04 s and every five squares (set off by heavy black lines) represents 0.20 s. The normal P–R interval is less than 0.20 s; the average is 0.16 s. The average P wave lasts 0.08 s, the QRS complex is 0.08 s, the ST segment is 0.12 s, the T wave is 0.16 s, and the Q–T interval is 0.32–0.40 s if the heart rate is 65–95 beats/min. Each horizontal line represents voltage; every five squares equals 0.5 mV.
Cardiac output
The primary function of the heart is the supply of oxygenated blood to the rest of the body, both during periods of rest and during increased physical activity. When the body’s requirement for oxygen increases, heart rate and cardiac output increase to meet the demand. Cardiac output (CO) is a function of both the stroke volume (SV) and heart rate (HR); that is:
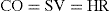
SV of the heart depends on the volume of blood remaining in the heart at the end of diastole and the volume that remains after ventricular contraction. For example, in a healthy resting adult, if SV was about 70 mL and HR 72 beats/min, CO would equal 5040 mL/min.
The factors that regulate SV include the degree of stretch of heart fibres before contraction (preload), the force of contraction of the ventricles and the pressure that must be overcome before the ventricles can eject the blood (afterload). The greater the preload, the greater is the stretch and the greater the contraction. This relation means that the longer the muscle fibres are at the end of diastole, the more forceful the contraction will be during systole. This mechanism applies only when the muscle fibre is lengthened within physiological limits and is known as the Frank–Starling relation (or the Frank–Starling law of the heart). This relation ensures that outputs from the right and left ventricles are the same.
If a diseased heart is dilated and the fibres are stretched to a critical point beyond their limit of extensibility, the forces of contraction and CO are both diminished and ineffective. If the right ventricle fails, blood pools in systemic vessels, causing peripheral oedema, while failure of the left ventricle results in pulmonary oedema because of the backing up of blood in the lungs. Thus the functional significance of the Frank–Starling relation is that effective CO can be brought about only by adequate relaxation and refilling of cardiac chambers after each myocardial contraction.
 Drugs affecting cardiac function
Drugs affecting cardiac function
Numerous drugs affect the heart and vascular system and provide the mainstay for treating diseases such as heart failure (see Clinical Interest Box 22-2), dysrhythmias, hypertension, ischaemic heart disease and shock and hypotensive states. (The authors acknowledge that the prefix ‘a’ means ‘without’, and the only arrhythmia is asystole. The correct term is ‘dysrhythmia’, the prefix ‘dys’ meaning ‘difficulty with’. However, as the terms ‘arrhythmia’ and ‘anti-arrhythmic drugs’ dominate the
Clinical interest Box 22-2 Heart failure
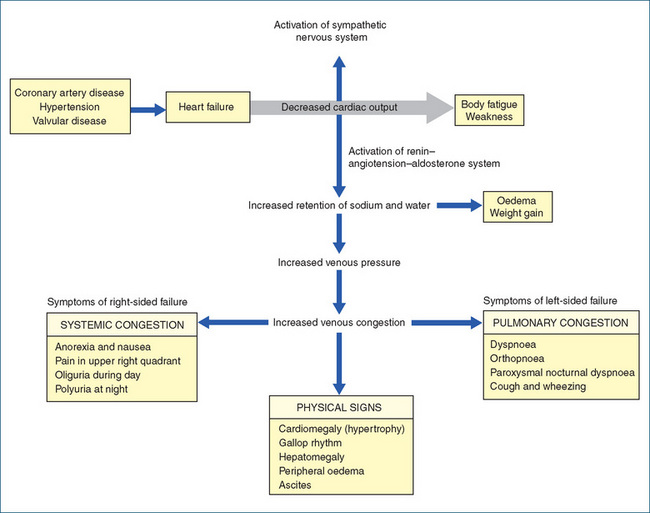
In general, heart failure is a disease of the elderly population, with most hospital admissions occurring in people over the age of 65 years. The prevalence increases from ∼10% in persons aged 65 years to >50% in persons 85 years and older. Risk factors predisposing to heart failure include high blood pressure and coronary heart disease. Heart failure is a complex problem and the symptoms (fatigue, shortness of breath and congestion) are related to inadequate cardiac output (and hence inadequate tissue perfusion) during exertion and to the retention of fluid (Figure 8Figure 22-8). As a consequence of inadequate performance of the myocardium, compensatory mechanisms are activated, and incomplete emptying of the heart during ventricular systole eventually allows blood to accumulate, causing dilation or enlargement of the heart. In the left atrium, this can lead to pulmonary congestion; in the right atrium, systemic congestion, including ascites, may occur. During the interim, the heart attempts to pump blood out to the systemic circulation, but instead the increased fluid in the left ventricle produces stretching of the myocardial fibres and dilation of the ventricles. The ventricles start to fail and cardiac output is reduced.
Mechanisms to compensate, involving adrenergic (sympathetic) stimulation, occur as the body attempts to maintain an adequate cardiac output. The increased heart rate and peripheral vascular resistance also elevate the heart’s demand for oxygen, further contributing to myocardial dysfunction. The decrease in cardiac output leads to decreased tissue perfusion and, following activation of the renin–angiotensin–aldosterone system, the kidneys respond by retaining more sodium and water. The increase in circulatory blood volume increases the demands on the heart.
The short-term goals of therapy are the relief of symptoms and improvement in the quality of life. Long-term management is aimed at retarding disease progression and prolonging survival. Non-pharmacological approaches include modifying risk factors (diet, smoking and alcohol intake), encouraging exercise, often through rehabilitation programs, and providing home support.
Although digoxin was previously the mainstay of therapy for heart failure, the agents more commonly used include the angiotensin-converting enzyme (ACE) inhibitors, diuretics, β-blockers, aldosterone antagonists and angiotensin receptor antagonists (used in persons who experience adverse effects from ACE inhibitors). Studies have shown that β-blockers may have favourable effects in some cases of heart failure but, because of adverse effects on left ventricular function, these drugs are started in low doses and titrated upwards. Digoxin provides valuable therapy in people with chronic heart failure accompanied by atrial fibrillation (National Heart Foundation of Australia and the Cardiac Society of Australia and New Zealand 2006).
literature, they have been retained.) Many of these drugs exert a direct effect on the heart or vasculature, while others indirectly affect cardiac function as a consequence of actions on vascular tissue.
Drugs acting directly on the heart include:
Drugs with a positive inotropic effect increase the force of myocardial contraction (e.g. digoxin, dobutamine, adrenaline and isoprenaline), whereas drugs with a negative inotropic effect decrease the force of myocardial contraction (e.g. propranolol).
Drugs with a positive chronotropic effect accelerate the HR by increasing the rate of impulse formation in the SA node (e.g. adrenaline). A drug with a negative chronotropic effect has the opposite effect and slows the HR by decreasing impulse formation (e.g. digoxin).
A drug with a positive dromotropic effect increases conduction velocity through specialised conducting tissues (e.g. phenytoin), while a drug with a negative dromotropic effect delays conduction (e.g. verapamil).
Drugs in the digitalis group are among the oldest drugs known to affect both cardiac contractility and rhythm. They increase the force of contraction (positive inotropism) and alter the electrophysiological properties of the heart by slowing the HR (negative chronotropism) and slowing conduction velocity (negative dromotropism).
Drugs that increase myocardial contraction
Digoxin
The use of digitalis demonstrates a herbal remedy (it was called ‘housewife’s recipe’) that was used for hundreds of years by ‘common’ people (farmers and housewives) for dropsy (fluid accumulation). More than 400 years ago, Dr Leonhard Fuchs recommended that physicians use it ‘to scatter the dropsy, to relieve swelling of the liver, and even to bring on menstrual flow’ (Silverman 1942). Dr Fuchs was a botanist–physician, and at that time the medical profession paid little attention to a ‘mere flower picker’.
In the mid-1700s, a female patient shared an old family recipe for curing dropsy with Dr William Withering, which he then used for his dropsy patients. After studying digitalis for 10 years, he published his conclusions in An Account of the Foxglove. This remarkable publication stressed instructions that are still valid today—for example, the necessity of individualising dosage according to response. Digitalis was listed in the London Pharmacopoeia in 1722.
The term digitalis glycoside refers specifically to cardiac glycosides derived from the Digitalis species and includes digoxin and digitoxin. The mechanisms of action of the cardiac glycosides (digoxin, digitoxin and oubain) are fundamentally the same, with minor differences occurring among the pharmacokinetic parameters of the individual agents. Digoxin is the only cardiac glycoside used clinically and was previously considered first-line therapy for heart failure. Its use in that setting has declined in the face of more effective drugs (See Drug Monograph 22-1).
The main effects produced by digoxin on the heart are: increased contractile force, decreased conduction through the AV node, decreased heart rate and rhythm disturbances.
MECHANISM OF ACTION Digoxin inhibits the active transport of sodium and potassium across the myocardial cell membrane by inhibiting the action of the membrane-bound enzyme Na+–K+- ATPase. Normally, this enzyme hydrolyses ATP to provide the energy for the Na+–K+ pump that expels intracellular sodium and transports potassium into the cardiac cell during repolarisation. Digoxin binds specifically to Na+–K+-ATPase and inhibits its action (Figure 22-9). Intracellular sodium accumulates, which inhibits the extrusion of calcium ions, and more calcium is taken up by the sarcoplasmic reticulum. Free calcium ions are essential for linking the electrical excitation of the cell membrane to the mechanical contraction of the myocardial cell, a mechanism known as excitation–contraction coupling. The increased availability of calcium ions released from the sarcoplasmic reticulum increases the coupling of actin and myosin, which results in more forceful myocardial contraction with a concomitant increase in cardiac output. Inhibition of Na+–K+-ATPase activity is proposed to be the mechanism by which the cardiac glycosides increase myocardial contraction without causing increased oxygen consumption.
Digoxin decreases heart rate and slows conduction velocity by altering the electrophysiological properties of cardiac tissues. At therapeutic plasma concentration, digoxin decreases automaticity and increases the resting membrane potential of atrial tissue and the AV node. These actions occur as a result of augmentation of vagal activity (slowing of heart rate) by a direct effect on the central vagal nuclei, which modifies the excitability of efferent vagal fibres, and by a decrease in the sensitivity of the SA and AV nodes to catecholamines and sympathetic impulses. With increasing plasma concentration of digoxin, severe bradycardia and heart block can occur. On the other hand, toxic concentrations of digoxin can increase sympathetic nervous system activity and directly increase automaticity. This increases the rate of spontaneous depolarisation and is one of the mechanisms responsible for digitalis-induced ectopic pacemakers. Toxic doses of digitalis can significantly increase impulse formation in latent or potential pacemaker tissue, causing arrhythmias.
Digoxin decreases AV conduction velocity by a direct action, which increases the effective refractory period of the AV node, and by augmenting vagal activity. The effect of digoxin on the refractory period varies in different parts of the heart. A prolonged refractory period occurs because of decreased conduction velocity and an increase in the effective refractory period of the AV conduction system, which is very sensitive to digoxin. This action is partly direct and partly caused by increased vagal tone. Toxic doses of digoxin can prolong the refractory period and depress conduction in the AV conduction system until complete heart block occurs.
INDICATIONS Digoxin is used for cardiac arrhythmias, especially atrial fibrillation, atrial flutter and paroxysmal atrial tachycardia and heart failure. During atrial fibrillation, several hundred impulses originate from the atria, but only a few of them are transmitted through the AV junction. Digoxin slows the ventricular rate because it increases the refractory period of the AV junction and slows conduction at this site, thereby reducing the possibility of inducing ventricular tachycardia.
PHARMACOKINETICS The absorption of digoxin is influenced by both formulation and the activity of the intestinal efflux transporter P-gp (reduces absorption). Digoxin is 60%–80% absorbed from tablets, and 70%–85% from the oral liquid. Absorption is enhanced by coadministration of the P-gp inhibitor quinidine, which increases the digoxin plasma concentration (Igel et al 2007). Digoxin is not hepatically metabolised; ∼20% of the dose is eliminated by biliary excretion and the remainder is renally excreted as unchanged drug (about 70%–80%) in urine. Digoxin is both filtered and secreted and renal elimination is impaired (∼16% reduction) by coadminstered drugs that inhibit digoxin efflux via the renal P-gp transporter (Fenner et al 2009). In the presence of normal renal function the plasma half-life of digoxin is 20–50 hours, thus permitting once-daily dosing, and steady-state plasma concentration is reached after about 5 days. In situations of impaired renal function the half-life can increase to 3–5 days necessitating dosage adjustment. Digoxin is widely distributed to all body tissues and the concentration of digoxin in tissues such as the heart, liver and skeletal muscle tends to be higher than that in plasma.
DRUG INTERACTIONS There are multiple drug interactions with digoxin and relevant drug information sources should be consulted. Drug interactions 22-1 lists examples of interactions that decrease absorption and increase or decrease digoxin plasma concentration.
ADVERSE REACTIONS Adverse reactions include anorexia and gastrointestinal disturbances such as nausea, vomiting and diarrhoea. Central nervous system effects, such as visual disturbances, confusion, nightmares, agitation and drowsiness are less frequent, as are arrhythmias. The arrhythmias seen with digitalis toxicity are premature ventricular beats, paroxysmal atrial tachycardia with AV block, progressing AV block and ventricular arrhythmias such as ventricular tachycardia or fibrillation. Loss of appetite, nausea, vomiting and abdominal distress may indicate digoxin toxicity. Hypokalaemia and hypomagnesaemia increase the risk of digoxin toxicity, whereas hypocalcaemia may reduce the effectiveness of digoxin.
WARNINGS AND CONTRAINDICATIONS Use with caution in people with renal impairment (decreased elimination), hypothyroidism (increased sensitivity), hyperthyroidism (digoxin resistance), electrolyte abnormalities (e.g. hypokalaemia, hypomagnesaemia or hypercalcaemia) or acute myocardial infarction. Digoxin is contraindicated in people with digoxin hypersensitivity, Wolff–Parkinson–White syndrome, constrictive pericarditis, cor pulmonale, complete heart block, ventricular arrhythmias or obstructive cardiomyopathy.
DOSAGE AND ADMINISTRATION Digoxin has a narrow therapeutic range and people can display toxic effects when the drug is in the therapeutic range. Dosage should be individualised depending on the underlying condition (i.e. heart failure or atrial fibrillation/flutter) and on assessment of renal function, clinical response and plasma drug concentration monitoring. Elderly people may have age-related renal or hepatic impairment and a decreased volume of distribution for digoxin; thus lower doses are necessary to avoid toxicity. Use of a loading dose is not usually required for heart failure but is used for the treatment of arrhythmias. Oral dosing is most commonly used while IV administration is infrequently used.
For situations of atrial fibrillation, an initial loading dose (oral/IV) of 250–500 mcg is given, with further dosing every 4–6 hours to a maximum of 1.5 mg. In the elderly or in the presence of chronic renal failure, an initial loading dose of 125–250 mcg is given, with further dosing every 4–6 hours to a maximum of 500 mcg. The usual adult oral maintenance dose is 125–250 mcg once daily, or 62.5–125 mcg in the elderly. The paediatric (infant up to 2 years) loading dose is 30–40 mcg/kg in three or four divided doses, and an oral maintenance dose of 5–10 mcg/kg daily in one or two divided doses.
Drug interactions 22-1 Digoxin
| Drug | Possible effects | Management |
| Amiodarone | Marked increase in plasma concentration of digoxin, with increased risk of toxicity. Additive effect on slowing cardiac conduction |
Reduce dosage of digoxin and monitor plasma digoxin concentration and clinical status, especially bradyarrhythmias |
| Antacids, antidiarrhoeals (kaolin–pectin type), bile acidbinding resins, oral neomycin, macrolides and sucralfate | Decreased absorption of digoxin and reduced efficacy | Separate administration of these drugs; consult relevant sources for further information |
| Calcium channel-blocking drugs (verapamil and diltiazem) | Increased plasma digoxin concentra tion, enhanced negative effect on atrioventricular conduction and heart rate | Monitor plasma digoxin concentration and anticipate need to reduce dose |
| Potassium-depleting drugs, such as amphotericin B (parenteral), corticosteroids, or loop or thiazide diuretics | The potential for inducing hypokalaemia with these drugs, if used concurrently with digoxin, increases the possibility of digoxin toxicity | Ensure adequate intake of potassium, monitor potassium levels closely and watch for clinical signs and symptoms of hypokalaemia |
| Quinine | Possibly an increase in plasma concentration of digoxin | Monitor plasma concentration and clinical response closely; dosage reduction may be necessary |
| Spironolactone | Increased plasma digoxin concentration | Monitor plasma digoxin concentration and anticipate need to reduce dose |
| St John’s wort | Possibly decreases plasma digoxin concentration and clinical effect | Avoid combination |
| Suxamethonium | Risk of dangerous arrhythmias (e.g. bradyarrhythmia) | Avoid or potentially serious drug interaction may occur |
Sources: AMH 2010; Speight & Holford 1997
Digoxin toxicity
Almost every type of arrhythmia can be produced by digoxin. The type of arrhythmia produced varies with age and other factors. Premature ventricular contractions and bigeminal rhythm (two beats and a pause) are common signs of digoxin toxicity in adults, whereas children tend to develop ectopic nodal or atrial beats. Other digoxininduced arrhythmias are caused by depression of the SA and AV nodes of the heart. This results in various conduction disturbances (first- or second-degree heart block or complete heart block). Digoxin can also cause increased myocardial automaticity, producing extrasystoles or tachycardia.
Health-care professionals need to be aware of the predisposing factors for digoxin toxicity. The presence of any of these factors indicates the need for close observation for signs and symptoms of toxicity:
Treatment of digoxin poisoning
The antidote for life-threatening digoxin poisoning is an ovine digoxin-specific immune antigen-binding frag ment (Fab). These fragments, which are derived from antidigoxin antibodies, bind the digoxin molecules, preventing them from interacting at their site of action. The digoxin– fragment complex accumulates in blood and is excreted by the kidneys. As more tissue digoxin is released into the blood to maintain equilibrium, it is bound by the antigen fragments and removed, which results in lower levels of digoxin in tissues, thereby reversing its effects.
After IV administration the onset of action is rapid and initial signs of improvement in digoxin toxicity may be seen within 15–30 minutes. The half-life of digoxin immune Fab appears to be in the order of 15–20 hours, but data on use in humans are limited.
Close monitoring is necessary, as withdrawal of digoxin can result in a decrease in cardiac output, congestive heart failure and hypokalaemia. An increase in ventricular rate may be seen in people with atrial fibrillation. Safety of digoxin immune Fab has not been completely defined because of its limited use. There are no known contraindications to use, but caution should be exercised in people with kidney function impairment; a history of allergies, particularly to sheep proteins; and in those previously treated with digoxin immune Fab.
The adult dose varies according to the amount of digoxin that is required to be complexed. One vial of antibody binds about 0.5 mg digoxin. The dose required can be calculated from the number of tablets ingested or from the plasma digoxin concentration. The full product information should be consulted for calculation of the dosage of digoxin antibodies, and the shelf expiration date of the product checked before use.
Formulae for digoxin immune fab (ovine) (digibind)
body load (mg) = [digoxin concentration (ng/mL) × 5 L/kg2 × body weight (kg)] ÷ 1000
Therapeutic drug monitoring of plasma digoxin concentration
Digoxin has a narrow therapeutic range of 0.5–2 mcg/L (0.6–2.6 nanomol/L). In situations of chronic heart failure, studies suggest that a plasma concentration of 0.5–1 mcg/L (0.6–1.3 nanomol/L) should be the target range. Although adverse effects are in general related to plasma digoxin concentration, often the plasma concentration does not clearly delineate patients with toxic levels from those with non-toxic levels. It has been reported that 38% of individuals with actual digoxin toxicity had digoxin plasma concentrations of 2 mcg/L while some with hypokalaemia exhibited toxic signs with plasma levels of 1.5 mcg/L (Kradjan 1995). Plasma digoxin concentration should be used as a guide in conjunction with clinical observations.
Criteria for determining plasma digoxin concentration include:
The time that a blood sample is drawn for determination of plasma digoxin concentration is critical. Blood should be taken at least 6–8 hours after the last oral dose or immediately before the next dose (trough concentration).
Phosphodiesterase inhibitor
Milrinone is a selective inhibitor of phosphodiesterase, the enzyme that metabolises cAMP in cardiac and vas cular tissue (Figure 22-9). Inhibition of the breakdown of cAMP results in elevated levels of cAMP within those tissues. This then results in increased calcium influx and uptake by the sarcoplasmic reticulum, causing improvement in myocardial contractility and vasodilation without increasing myocardial oxygen consumption and HR. Milrinone is a positive inotrope and vasodilator with very little chronotropic activity.
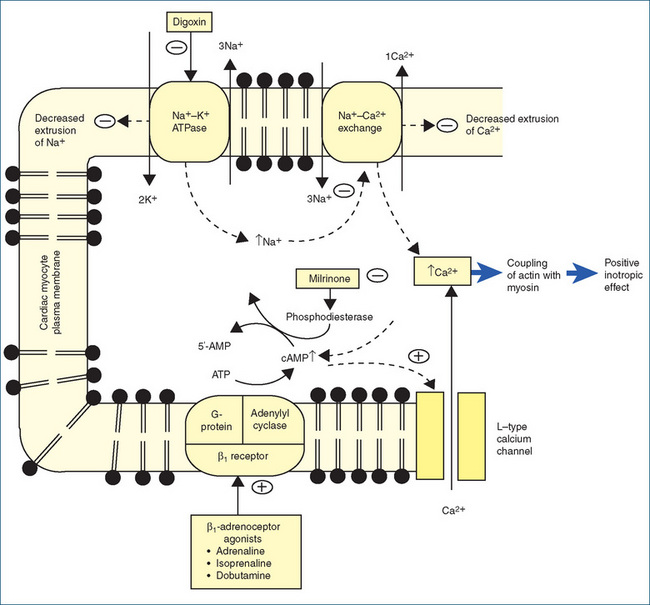
Figure 22-9 Schematic representation of cardiac myocyte indicating sites of action of digoxin, milrinone and β-adrenoceptor agonists. − = inhibitory effect; + = positive effect.
It is indicated for short-term treatment (about 48 hours) of severe heart failure refractory to other drugs, and for low-cardiac-output states (e.g. following cardiac surgery). It is principally used in coronary and intensive care units, and prolonged use is associated with increased mortality.
Administered intravenously, milrinone has a half-life of 2.5 hours and duration of action of 3–6 hours. It is excreted by the kidneys, and a reduction in dose is necessary in people with severe renal impairment. Common adverse reactions include ventricular arrhythmias, angina and hypotension.
Arrhythmias and anti-arrhythmic drugs
Arrhythmias
A cardiac arrhythmia is defined as any deviation from the normal rhythm of the heartbeat (Table 22-1). Disorders of cardiac rhythm arise because of abnormality in spontaneous initiation of an impulse, i.e. in automaticity; or abnormality in impulse conduction, i.e. in conductivity. In some circumstances a combination of both processes occurs.
Table 22-1 Examples of arrhythmias
| TYPE | DESCRIPTION | POSSIBLE CAUSES |
| Bradyarrhythmias | ||
| Atrioventricular block | Intermittent or absent conduction between atria and ventricles. Commonly occurs at AV node or within bundle branch system | Drug induced (e.g. β-blockers, digoxin, diltiazem, verapamil), infection, myocardial infarction |
| Sick sinus syndrome | Associated with SA node dysfunction, usually SA block or inadequate SA node conduction. Characterised by severe sinus bradycardia and symptoms of weakness, dizziness, lethargy and syncope. Treatment usually requires a pacemaker | Various cardiomyopathies, inflammatory myocardial disease, myocardial ischaemia, digoxin toxicity |
| Tachyarrhythmias | ||
| Atrial tachyarrhythmias | ||
| Atrial fibrillation | Common sustained arrhythmia characterised by disordered electrical activity in atria resulting in a fast, irregular ventricular response. High risk of stroke and heart failure | Acute myocardial infarction, can be idiopathic, cardiac surgery, mitral stenosis, rheumatic disease, elderly age |
| Atrial flutter | Atrial tachycardia characterised by contraction rate 230–380/min with ventricular contractions in 1:2, 1:3, 1:4 or variable ratio | Cause often unknown, heart disease, AV node dysfunction, chronic hypertension, overactive thyroid |
| Supraventricular tachycardia | Atrial tachycardia and arrhythmias arising from AV junction | Hypoxia, electrolyte and acid–base abnormalities, enlarged atria, digoxin toxicity |
| Ventricular tachyarrhythmias | ||
| Premature ventricular ectopics | Ectopics | Alcohol, caffeine, stress |
| Ventricular tachycardia | >100 beats/min | Acute myocardial infarction |
| Torsades de pointes | Characterised by a prolonged ventricular action potential (prolonged QT interval). Can be congenital (mutations in the cardiac sodium or potassium channels) or acquired | Acquired: electrolyte disturbances or from a number of drugs and drug classes e.g. antiarrhythmic drugs (e.g. amiodarone, disopyramide, sotalol), antipsychotics, antimicrobial drugs, arsenic trioxide, methadone, tacrolimus, tricyclic antidepressants |
Abnormality in automaticity
A disturbance in automaticity can alter the heart’s rate, rhythm or site of origin of impulse formation. When the rate of pacemaker activity is affected, a decrease in automaticity of the SA node produces sinus bradycardia (an abnormal condition in which the myocardium contracts steadily but at less than 60 contractions per minute). An increase in automaticity of the SA node results in sinus tachycardia (an abnormal condition in which the myocardium contracts regularly but at more than 100 beats/ minute). A shift in the site of origin of impulse formation can generate an abnormal pacemaker or an ectopic focus, resulting in activation of a part of the heart other than the SA node. This is called an ectopic pacemaker, and it may discharge at either a regular or an irregular rhythm. It occurs because the cardiac fibres depolarise more frequently than the SA node. Abnormal automaticity can develop in cells that usually do not initiate impulses (e.g. atrial or ventricular cells). Clinical disorders such as hypoxia or ischaemia can cause impulse disturbances in automaticity and in conductivity, and both manifestations are responsible for ectopic beats. Ectopic beats are classified as escape beats, premature beats or extrasystoles, and ectopic tachyarrhythmia.
Abnormality in conductivity
Altered conduction of the cardiac impulse probably accounts for more arrhythmias than changes in automaticity. A disturbance in conductivity may be caused by a delay or block of impulse conduction or by the re-entry phenomenon. In abnormal circumstances, conduction of an atrial impulse to the ventricles can be delayed or blocked in the AV node or structures beyond this region in the conduction pathway. In first-degree AV block, the impulses from the SA node pass through to the ventricles very slowly; this is shown by a prolonged P–R interval on the ECG. In second-degree block, some atrial beats fail to pass into the ventricles through the AV node. In third-degree block, or complete heart block, no impulses reach the ventricle, in which case the Purkinje fibres initiate their own spontaneous depolarisation at a very slow rate. This results in independent ventricular and atrial rhythms referred to as ventricular ‘escape’.
Re-entry phenomenon
The re-entry phenomenon is the mechanism responsible for initiating ectopic beats. For example, when an impulse travels down the Purkinje fibre, it normally spreads along two branches, and when it enters the connecting branch impulses are extinguished at the point of collision in the centre (Figure 22-10A). At the same time, other impulses that begin laterally from the Purkinje fibres activate ventricular muscle tissue. In an abnormal situation, the impulse descending from the central Purkinje fibre travels down one branch normally but encounters a block in the other branch due to ischaemia or injury (Figure 22-10B). This is a unidirectional block, because the impulse can pass in one direction only. In the injured branch, where the impulse is blocked in the forward direction at the site of injury, a retrograde (reverse) impulse from the ventricular tissue re-enters the depressed region from the other direction, provided the pathway proximal to the block is no longer refractory. When the effective refractory period of the blocked area is over, re-entry of the impulse from the ventricular muscle into this site causes the impulse to circulate or recycle repetitively through the loop, resulting in a circus-type movement that produces arrhythmia.
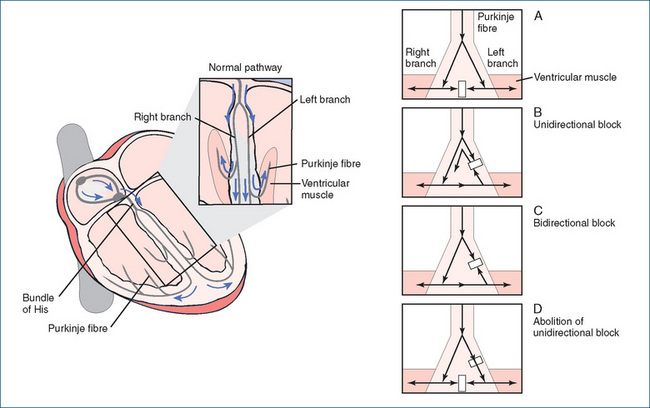
Figure 22-10 Re-entry phenomenon. Illustration of a branched Purkinje fibre that activates ventricular muscle.
Drugs that decrease or slow conduction velocity can convert unidirectional block to a two-way or bidirectional block (Figure 22-10C). As the impulses travelling in the antegrade (forward) direction and those moving in a retrograde (reverse) direction are blocked at the injured site, the re-entry pathway is interrupted, abolishing the ectopic beats. In Figure 22-10D, the conditions required for preventing re-entry by another mechanism are also illustrated.
Anti-arrhythmic drugs
Anti-arrhythmic drugs are used for the treatment and prevention of disorders of cardiac rhythm. These drugs were classified into categories based on their fundamental effects on cardiac electrophysiology by Vaughan Williams in 1970. This grouping is of value in predicting the drug’s therapeutic efficacy, although not all drugs belonging to a particular class necessarily possess identical actions. The currently available antiarrhythmic drugs are grouped into four classes (Table 22-2) according to their mechanisms of action (Figure 22-11) but there are a number of drugs that are not classified in the Vaughan Williams system.
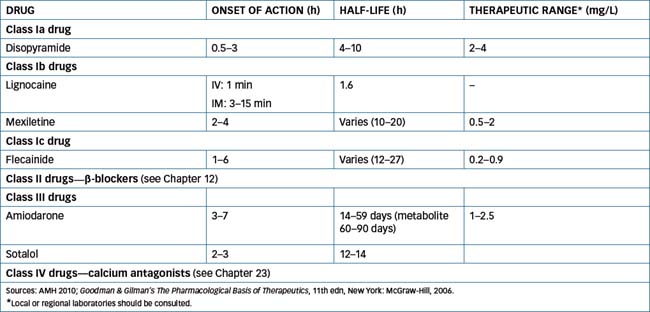
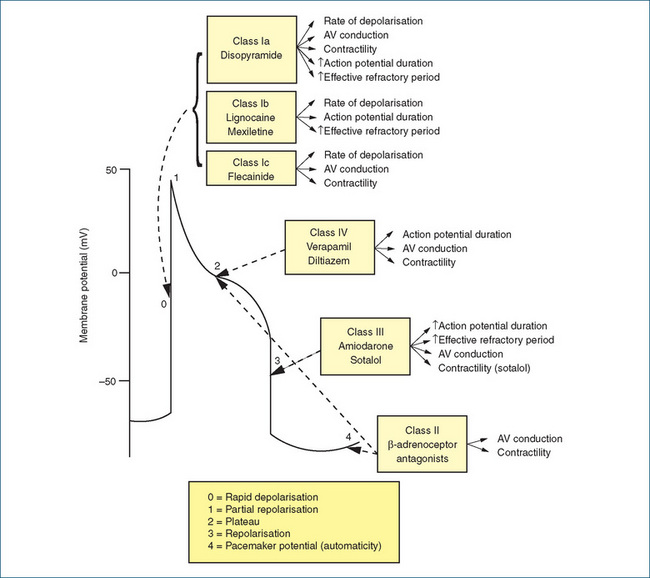
Figure 22-11 Phases of the cardiac action potential and the effects produced by the various classes of antiarrhythmic drugs.
Classification of anti-arrhythmic drugs
Drugs not classified under this scheme include adenosine, atropine (Chapter 11), adrenaline (Chapter 12) and digoxin.
The rationale for use of anti-arrhythmic drugs includes restoration of haemodynamic stability, prevention of life-threatening arrhythmias, prevention of sudden cardiac death, controlling ventricular rate and preventing thromboembolism in atrial fibrillation. Despite their use for the treatment of arrhythmias, these drugs all possess proarrhythmogenic potential, and can worsen the arrhythmia and cause sudden death. Use of these drugs requires careful consideration of other treatment options and, following institution of therapy, careful monitoring of the clinical condition of the patient.
Class Ia drugs
The use of class Ia drugs such as quinidine has declined because of evidence of increased mortality with chronic use (Ninio 2000).
Disopyramide prevents the movement of sodium and potassium across cell membranes. This inhibition of cation exchange results in a decrease in the rate of diastolic depolarisation from the resting potential during phase 4 and an increase in the threshold potential (the voltage shifts towards 0 mV). This results in decreased impulse conduction and delayed repolarisation in the atria, ventricles and Purkinje fibres. By decreasing impulse generation at ectopic sites, disopyramide suppresses or abolishes arrhythmias. Abnormal or ectopic pacemaker tissue appears to be more sensitive to disopyramide than the SA node, thus permitting the SA node to re-establish control over impulse formation in the heart.
The most significant action of disopyramide is its ability to prolong the effective refractory period of atrial and ventricular fibres. A delay in completion of repolarisation probably exerts an important anti-arrhythmic action. The tissue remains refractory for a period after full restoration of the resting membrane potential. This property is believed to influence the conversion of unidirectional block to bidirectional block, thereby abolishing the re-entry type of arrhythmia (Figure 22-10C).
Disopyramide exerts an anticholinergic effect, resulting in inhibition of vagal action on the SA node and AV junction. This effect permits the sinus node to accelerate and can often provoke a dangerous sinus tachycardia. The latter is the reason why a drug that slows AV conduction may be administered with disopyramide when it is used in the treatment of atrial flutter or atrial fibrillation. Unlike quinidine, disopyramide does not possess β-adrenergic receptor antagonist properties.
Disopyramide is well absorbed and has a weakly active metabolite with both antiarrhythmic and anticholinergic effects. The therapeutic plasma concentration range for disopyramide is 2–4 mg/L (see Table 22-2).
The following drug interactions may occur with disopyramide:
Common adverse reactions explained by the anticholinergic effects include blurred vision, constipation, urinary retention and dry mouth. In addition, disopyramide can cause hypersensitivity reactions, severe disturbances of cardiac rhythm and exacerbation of heart failure. Disopyramide should be used with caution in people with diabetes mellitus, glaucoma (closed-angle), hypokalaemia, myasthenia gravis, enlarged prostate or renal impairment. Avoid use in people with disopyramide hypersensitivity, AV block, cardiogenic shock, cardiac conduction abnormality, cardiomyopathy or heart failure.
Dosage and administration are individualised according to response and tolerance, to a maximum of 800 mg per day. The usual adult oral loading dose is 200–300 mg, with a maintenance dose of 100–150 mg every 6 hours (400–600 mg daily). A dose of 100 mg is administered every 8 hours in people with mild renal impairment, every 12 hours in people with moderate impairment and once daily in people with severe renal impairment.
Class Ib drugs
The class Ib drugs lignocaine and mexiletine differ from class Ia drugs because in general they do not affect conduction velocity. Lignocaine and mexiletine are structurally very similar and are useful for acute ventricular arrhythmias. A high incidence of adverse effects has limited the usefulness of both lignocaine and mexiletine. Like the class Ia drugs, these drugs can worsen arrhythmias.
Lignocaine, an agent used extensively as a local and topical anaesthetic agent, is also an anti-arrhythmic agent, infrequently used for ventricular arrhythmias occurring after cardiac surgery or an acute myocardial infarction. Lignocaine appears to act primarily on the sodium channel, blocking both the activated and inactivated sodium channels, although its greater effect is in depolarised or ischaemic tissues. These effects are indicative of the efficacy of lignocaine for suppressing arrhythmias associated with depolarisation (such as ischaemia and digoxin-induced toxicity) and its lack of effectiveness in arrhythmias that occur in normal polarised tissues (atrial fibrillation and atrial flutter). Lignocaine has few electrophysiological effects in normal cardiac tissue. Lignocaine does not inhibit vagal activity, nor does it influence cardiac output and arterial pressure. In addition, it does not depress myocardial contractility, thereby reducing the potential for development of heart failure. Because it exerts a limited effect, if any, on the SA node and atrial myocardium, the drug has no use in the treatment of supraventricular tachycardia. The major use of lignocaine is in the treatment of severe ventricular arrhythmias.
Lignocaine is only administered intravenously and is metabolised by the liver to the active metabolites, glycine xylidide (GX) and monoethyl glycine xylidide; after a 24-hour infusion, GX also contributes to the therapeutic and toxic effects (see Table 22-2). Concurrent drug administration with β-blockers, fluvoxamine or cimetidine can inhibit metabolism of lignocaine and increase the risk of toxicity. Additive toxicity may also occur with other antiarrhythmic drugs e.g. mexiletine.
Adverse reactions include dizziness, anorexia, nausea, vomiting, tinnitus, tremor and visual disturbances. Of a more serious nature are bradycardia, convulsions, respiratory depression and cardiac arrest. Use lignocaine with caution in people with liver or kidney function impairment, as lignocaine and its active metabolites can accumulate. Avoid use in people with lignocaine hypersensitivity, complete heart block, sinus bradycardia or Stokes–Adams syndrome.
The lignocaine adult dose of 1 mg/kg is given IV over 1–2 minutes and is repeated after 5 minutes if necessary. Children receive the same 1 mg/kg dose initially, but repeated doses should not exceed a total dose of 3 mg/kg. By IV infusion, the maintenance dose is usually 10–50 mcg/kg/min.
Mexiletine is a structural analogue of lignocaine that was chemically modified to reduce hepatic first-pass metabolism, and hence it is administered orally (see Table 22-2 for pharmacokinetics). Recovery of sodium channel blockade is so rapid mexiletine is of no use for atrial fibrillation or flutter. Ventricular arrhythmias that respond to parenteral lignocaine are usually responsive to mexiletine.
Drug interactions occur with mexiletine and other antiarrhythmic drugs, resulting in additive depressant effects on conduction of the AV node and myocardium. Mexiletine inhibits the metabolism of theophylline and increases the risk of theophylline toxicity.
Adverse reactions are similar to those for lignocaine. In addition, paraesthesia of fingers and toes, rash, tremors and indigestion can occur. Rarely, pulmonary fibrosis, Stevens–Johnson syndrome (erythema multiforme presenting with macules, papules and vesicles on the mucous membranes of the lips, mouth, genitalia and conjunctiva), hepatotoxicity and cardiac arrest have been reported. Use mexiletine with caution in people with severe heart failure, an acute myocardial infarction, convulsive disorders, hypotension, sinus node or intraventricular conduction dysfunction or liver function impairment. Avoid use of mexiletine in people with drug hypersensitivity, AV block or cardiac shock.
The mexiletine adult oral loading dose is usually 400 mg, with an oral maintenance dose of 100–250 mg three times daily.
Class Ic drugs
The class Ic drug available in Australia is flecainide, which is used to treat atrial fibrillation and flutter and serious ventricular arrhythmias. The potential for proarrhythmic effect is of special concern, especially in people with poor left ventricular function or sustained ventricular arrhythmias. The class Ic drugs can also aggravate congestive heart failure.
Flecainide is a sodium channel-blocking agent used to treat ventricular arrhythmias; it has minimal effects on repolarisation and no anticholinergic properties. It suppresses premature ventricular contractions, and in high doses can exacerbate arrhythmias in people with a pre-existing ventricular tachyarrhythmia or in people with a previous myocardial infarction.
Flecainide is well absorbed after oral administration. It is both hepatically metabolised by CYP2D6 and renally excreted (∼45% as unchanged drug). The therapeutic range for flecainide is 0.2–0.9 mg/L (see Table 22-2). The administration of flecainide with other anti-arrhythmic drugs (digoxin, β-blockers, verapamil) can result in enhanced adverse cardiac effects. In people with diureticinduced hypokalaemia, there is an increased risk of arrhythmias.
Adverse reactions include blurred vision, dizziness, headaches, constipation, nausea, weakness, chest pain, irregular heartbeats and arrhythmias. Use flecainide with caution in people with heart failure, hypokalaemia or hyperkalaemia and renal impairment. Flecainide is contraindicated post-myocardial infarction, in people with heart block or in situations of cardiogenic shock.
The flecainide adult oral dose is 50–100 mg every 12 hours, increasing by 50 mg every four days to a maximum of 400 mg daily.
Class II drugs
The class II drugs include atenolol, esmolol and metoprolol. All three drugs are β-adrenoceptor antagonists that are used to control cardiac arrhythmias caused by excessive sympathetic nerve activity. They reduce the rate of the SA node and slow conduction in the atria and AV node and increase the functional refractory period. These drugs are the only class of anti-arrhythmics to show a reduction in mortality post-myocardial infarction.
Drugs such as atenolol are used to treat atrial tachyarrhythmias and ventricular arrhythmias, whereas esmolol is indicated for short-term treatment of supraventricular tachycardia induced by atrial fibrillation or atrial flutter (these drugs are discussed extensively in Chapter 12).
Class III drugs
The electrophysiological properties of amiodarone (see Drug Monograph 22-2) and sotalol (Chapter 12) differ markedly from those of the other classes. Drugs in this group prolong the effective refractory period by prolonging the action potential duration.
Drug monograph 22-2 Amiodarone
MECHANISM OF ACTION Amiodarone increases the refractory period in all cardiac tissues through a direct effect on the tissues. It decreases automaticity, prolongs AV conduction and decreases the automaticity of fibres in the Purkinje system. It can block potassium, sodium (class I effect) and calcium channels (class IV effect) and β receptors (class II effect). It has the potential to cause a variety of complex effects on the heart and has serious adverse effects. Its main active metabolite desethylamiodarone (DEA) produces increasing depression of the rate of depolarisation during phase 0 and hence the changing electrophysiological effects observed with chronic dosing may reflect accumulation of both amiodarone and DEA.
INDICATIONS Amiodarone is used for the prevention and treatment of serious atrial and ventricular arrhythmias and for the management of acute tachyarrhythmias.
PHARMACOKINETICS The pharmacokinetics of amiodarone are subject to large interindividual variability in bioavailability, plasma concentration and elimination half-life. Additionally, there are pharmacokinetic differences between single dose and chronic administration. Amiodarone is a structural analogue of thyroid hormone and is highly lipophilic. It is poorly absorbed and has a bioavailability that ranges from 20% to 86%. It is highly protein bound (>99%), widely distributed in the body (e.g. in adipose tissues, liver and lung) and reaches steady-state plasma concentration after several weeks. Its onset of action varies from several days to weeks, even if loading doses are administered. It has a biphasic elimination half-life: the initial half-life is 2.5–10 days and the terminal half-life is 26–107 days. It has one active metabolite, DEA, which has a terminal half-life of about 60 days (see Table 22-2).
DRUG INTERACTIONS Amiodarone inhibits CYP1A2, CYP2C9, CYP2D6 and CYP3A4 and is subject to multiple drug interactions. Examples include:
ADVERSE REACTIONS These include dizziness, bitter taste, headache, flushing, nausea, vomiting, constipation, ataxia, weight loss, tremors, paraesthesiae of fingers and toes, photosensitivity, blue–grey skin discolouration, pulmonary fibrosis or pneumonitis, cough, fever, allergic reaction and blurred vision.
WARNINGS AND CONTRAINDICATIONS Use amiodarone with caution in people with heart failure and liver or thyroid function impairment. Avoid use in people with amiodarone hypersensitivity, second- or third-degree AV block and bradycardia.
DOSAGE AND ADMINISTRATION The usual adult dose for chronic atrial or ventricular tachyarrhythmias is 200–400 mg 8 hourly for 1 week, then 200–400 mg twice daily for 1 week. The maintenance dose is 100–400 mg (or less if indicated) once daily. For children, the oral maintenance dose is 4 mg/kg once daily.
Sotalol is a β-adrenoceptor antagonist that also blocks cardiac potassium channels, prolonging the action potential duration and increasing the effective refractory period in atrial and ventricular tissue and at the AV node. It is indicated for treatment and prevention of atrial and serious ventricular arrhythmias. Sotalol is predominantly cleared renally (around 90%) and therefore accumulates in people with renal impairment (Table 2Table 22-2). Additive depressant effects occur with other anti-arrhythmic drugs, including verapamil and diltiazem, producing bradyarrhythmia, AV block and an increased risk of heart failure. Increased risk of arrhythmias also occurs in the presence of diuretic-induced hypokalaemia.
Common adverse effects include hypotension, dyspnoea, fatigue, dizziness, impotence, nausea, vomiting and diarrhoea. Similar to the other classes of antiarrhythmic drugs, sotalol is proarrhythmogenic, potentially producing new or worsening arrhythmias. Care should be exercised in people with heart failure, airways disease, diabetes or peripheral vascular disease. Sotalol is contraindicated in the presence of heart block, sinus bradycardia, severe heart failure and hypotensive states. Sotalol is available as both an IV and an oral formulation.
Class IV drugs
Drugs in this class are used for the treatment of supraventricular tachycardia and for control of ventricular rate in atrial fibrillation and atrial flutter. The Class IV drugs include verapamil, which is reviewed in Chapter 23.
Unclassified anti-arrhythmic agents
This group includes digoxin (discussed previously), adenosine and magnesium sulfate. Adenosine is produced endogenously and acts on G-protein-coupled A1 receptors on the AV node. By activating acetylcholine sensitive potassium currents in the atria and AV node it shortens the action potential duration and slows normal automaticity. It is indicated for the acute treatment of supraventricular tachycardia. Administered by intravenous bolus, it has an immediate onset and its action is terminated rapidly (20–30 s) by uptake into red blood cells and vascular endothelial cells. It is metabolised to inosine (which is processed to uric acid) and adenosine monophosphate (AMP). Caffeine and theophylline can antagonise the effect of adenosine, whereas dipyridamole increases the effects of adenosine because it inhibits metabolism of adenosine and blocks adenosine reuptake.
Adverse reactions include dyspnoea, flushing, cough, dizziness, tingling in arms, nausea, headache and transient
arrhythmias such as premature ventricular contractions, sinus bradycardia, sinus tachycardia, skipped beats and chest pain/pressure. These all resolve rapidly because of the short duration of action of adenosine. Use adenosine with caution in people with asthma as it may cause bronchospasm. Avoid use in people with adenosine hypersensitivity, AV block or sick sinus syndrome.
The usual dose is 3 mg administered rapidly in an IV bolus over 1–2 s, followed by 6 mg as a second dose if the arrhythmia has not resolved. If the arrhythmia is still present 1–2 min after the second injection, a third dose of 12 mg may be administered.
Magnesium sulfate blocks calcium entry by inhibiting L-type calcium channels during phase 3 of the action potential, an effect that potentially could shorten the QT interval. Additionally it also blocks the outward movement of potassium via potassium channels, which could potentially prolong the QT interval. These counterbalancing cellular actions may terminate torsades de pointes independently of the QT interval. Magnesium sulfate also decreases early after-depolarisations contributing to an anti-arrhythmic effect. It is cleared renally and hypermagnesaemia is an issue in situations of renal impairment. Adverse effects relate to hypermagnesaemia and include loss of deep tendon reflexes and respiratory depression resulting from neuromuscular blockade. The latter is potentiated in combination with aminoglycosides and the combination should be used with extreme caution and with monitoring of respiratory function.
Key points


 The heart comprises four chambers, two upper atria and two lower ventricles, with the left atrium and ventricle separated from the right by the septum, and the atria from the ventricles by the atrioventricular (tricuspid and mitral) valves. The myocardium is composed of cardiac muscle tissue that comprises sarcomeres, the basic contractile unit of the heart. The sympathetic and parasympathetic nervous systems increase the rate and force of contraction and decrease heart rate, respectively. Postganglionic fibres of the sympathetic nervous system, which release noradrenaline (NA) and innervate the SA node, atria and ventricles. Action of NA on β1 receptors located in both nodes and atrial and ventricular muscles increases heart rate, automaticity, conduction velocity and force of contraction. Vagal nerve fibres of the parasympathetic branch, which release acetylcholine, are found primarily in the SA and AV nodes and atrial muscle. Acetylcholine acts on muscarinic (M2) receptors of the SA node to decrease heart rate, on the AV node to decrease conduction velocity and, to a limited extent, on the M2 receptors on cardiac myocytes to reduce cardiac contractility. The natriuretic peptides include atrial natriuretic peptide (ANP), B-type natriuretic peptide (BNP) and the C-type natriuretic peptide. These endogenous neurohormones increase sodium and water excretion by the kidney and, with the exception of the efferent arterioles of the kidney, they relax vascular smooth muscle. Stretching of the ventricles results in the release of BNP and the circulating concentration of BNP correlates with the severity of heart failure. The cardiac conduction system comprises the SA and AV nodes, internodal pathways, the bundle of His, right and left bundle branches and the Purkinje fibres. The cardiac conduction system exhibits automaticity, conductivity and refractoriness, attributes required for the initiation and transmission of electrical impulses necessary for myocardial contraction. The overall process of cardiac muscle contraction involves electrical excitation, mechanical activation and contractile mechanisms. The cardiac action potential is divided into two stages, depolarisation and repolarisation, which are further subdivided into five phases (0–4) on the basis of ion movement. Electrocardiograms are graphic representations of electrical currents produced by the heart, and consist of a P wave (atrial depolarisation), the QRS complex (ventricular depolarisation) and the T wave (ventricular repolarisation). Factors regulating stroke volume include the degree of stretch of heart fibres before contraction (preload), the force of contraction of the ventricles and the pressure that must be overcome before the ventricles can eject the blood (afterload). The Frank–Starling law of the heart defines the relationship between the force of ejection and the length of cardiac muscle fibres. Numerous drugs that affect the heart and vascular system provide the mainstay for the treatment of diseases such as heart failure, arrhythmias, hypertension and ischaemic heart disease. Many drugs exert a direct effect on the heart or vasculature; others indirectly affect cardiac function as a consequence of actions on vascular tissue. Drugs acting directly on the heart include catecholamines (Chapter 12), cardiac glycosides (typified by digoxin) and anti-arrhythmic drugs. Digoxin decreases heart rate and slows conduction. It is used in the treatment of cardiac arrhythmias, especially atrial flutter and fibrillation, and heart failure. Digoxin has a narrow therapeutic index, can exacerbate or worsen arrhythmias and is subject to many serious drug interactions. Several factors, such as hypokalaemia, hypercalcaemia and hypomagnesaemia, may predispose to digoxin toxicity. Cardiac rhythm disorders are usually the result of an abnormality in the electrophysiology of the cells in the cardiac conduction system or cardiac muscle cells. The anti-arrhythmic drugs are subdivided into four classes (I–IV) according to their mechanism of action. Class Ia, Ib and Ic drugs suppress automaticity. Class Ia includes disopyramide, which decreases conduction velocity and prolongs the action potential.
The heart comprises four chambers, two upper atria and two lower ventricles, with the left atrium and ventricle separated from the right by the septum, and the atria from the ventricles by the atrioventricular (tricuspid and mitral) valves. The myocardium is composed of cardiac muscle tissue that comprises sarcomeres, the basic contractile unit of the heart. The sympathetic and parasympathetic nervous systems increase the rate and force of contraction and decrease heart rate, respectively. Postganglionic fibres of the sympathetic nervous system, which release noradrenaline (NA) and innervate the SA node, atria and ventricles. Action of NA on β1 receptors located in both nodes and atrial and ventricular muscles increases heart rate, automaticity, conduction velocity and force of contraction. Vagal nerve fibres of the parasympathetic branch, which release acetylcholine, are found primarily in the SA and AV nodes and atrial muscle. Acetylcholine acts on muscarinic (M2) receptors of the SA node to decrease heart rate, on the AV node to decrease conduction velocity and, to a limited extent, on the M2 receptors on cardiac myocytes to reduce cardiac contractility. The natriuretic peptides include atrial natriuretic peptide (ANP), B-type natriuretic peptide (BNP) and the C-type natriuretic peptide. These endogenous neurohormones increase sodium and water excretion by the kidney and, with the exception of the efferent arterioles of the kidney, they relax vascular smooth muscle. Stretching of the ventricles results in the release of BNP and the circulating concentration of BNP correlates with the severity of heart failure. The cardiac conduction system comprises the SA and AV nodes, internodal pathways, the bundle of His, right and left bundle branches and the Purkinje fibres. The cardiac conduction system exhibits automaticity, conductivity and refractoriness, attributes required for the initiation and transmission of electrical impulses necessary for myocardial contraction. The overall process of cardiac muscle contraction involves electrical excitation, mechanical activation and contractile mechanisms. The cardiac action potential is divided into two stages, depolarisation and repolarisation, which are further subdivided into five phases (0–4) on the basis of ion movement. Electrocardiograms are graphic representations of electrical currents produced by the heart, and consist of a P wave (atrial depolarisation), the QRS complex (ventricular depolarisation) and the T wave (ventricular repolarisation). Factors regulating stroke volume include the degree of stretch of heart fibres before contraction (preload), the force of contraction of the ventricles and the pressure that must be overcome before the ventricles can eject the blood (afterload). The Frank–Starling law of the heart defines the relationship between the force of ejection and the length of cardiac muscle fibres. Numerous drugs that affect the heart and vascular system provide the mainstay for the treatment of diseases such as heart failure, arrhythmias, hypertension and ischaemic heart disease. Many drugs exert a direct effect on the heart or vasculature; others indirectly affect cardiac function as a consequence of actions on vascular tissue. Drugs acting directly on the heart include catecholamines (Chapter 12), cardiac glycosides (typified by digoxin) and anti-arrhythmic drugs. Digoxin decreases heart rate and slows conduction. It is used in the treatment of cardiac arrhythmias, especially atrial flutter and fibrillation, and heart failure. Digoxin has a narrow therapeutic index, can exacerbate or worsen arrhythmias and is subject to many serious drug interactions. Several factors, such as hypokalaemia, hypercalcaemia and hypomagnesaemia, may predispose to digoxin toxicity. Cardiac rhythm disorders are usually the result of an abnormality in the electrophysiology of the cells in the cardiac conduction system or cardiac muscle cells. The anti-arrhythmic drugs are subdivided into four classes (I–IV) according to their mechanism of action. Class Ia, Ib and Ic drugs suppress automaticity. Class Ia includes disopyramide, which decreases conduction velocity and prolongs the action potential.Review exercises
Australian Medicines Handbook 2010. Adelaide: AMH, 2010.
Boulpaep E.L. Organization of the cardiovascular system. In Boron W.F., Boulpaep E.L., editors: Medical Physiology, Updated edition., Philadelphia: Elsevier Saunders, 2005. [ch 17]
Expert Group. Therapeutic Guidelines: Cardiovascular, version 5. Melbourne: Therapeutic Guidelines Ltd; 2008.
Fenner K.S., Troutmen M.D., Kempshall S., et al. Drug–drug interactions mediated through P-glycoprotein: clinical relevance and in vitro-in vivo correlation using digoxin as a probe drug. Clinical Pharmacology and Therapeutics. 2009;85:173-181.
Igel S., Drescher S., Murdter T., et al. Increased absorption of digoxin from human jejunum due to inhibition of intestinal transporter-mediated efflux. Clinical Pharmacokinetics. 2007;46:777-785.
Kradjan W.A. Congestive heart failure. In Young L.Y., Koda-Kimble M.A., editors: Applied Therapeutics: the Clinical Use of Drugs, 6th edn., Vancouver: Applied Therapeutics Inc, 1995.
Krum H. Recent advances in the management of chronic heart failure. Australian and New Zealand Journal of Medicine. 2000;30:475-482.
Lederer W.J. Cardiac electrophysiology and the electrocardiogram. In Boron W.F., Boulpaep E.L., editors: Medical Physiology, Updated edition., Philadelphia: Elsevier Saunders, 2005. [ch 20]
National Heart Foundation of Australia and the Cardiac Society of Australia and New Zealand (Chronic Heart Failure Guidelines Expert Writing Panel). Guidelines for the prevention, detection and management of chronic heart failure in Australia, 2006. Available: http://www.heartfoundation.org.au/SiteCollectionDocuments/CHF%202006%20Guidelines%20NHFA-CSANZ%20WEB.pdf [27 August 2009].
Ninio D.M. Contemporary management of atrial fibrillation. Australian Prescriber. 2000;23:100-102.
Quinn D.I., Day R.O. Guide to clinically more important drug interactions. In Speight T.M., Holford N.H.G., editors: Avery’s Drug Treatment., 4th edn., Auckland: Adis International, 1997. [Appendix B]
Rang H.P., Dale M.M., Ritter J.M., Flower R.J. Pharmacology, 6th edn. Edinburgh: Churchill Livingstone; 2007. [ch 18]
Rocco T.P., Fang J.C. Pharmacotherapy of congestive heart failure. In Brunton L.L., Lazo J.S., Parker K.L., editors: Goodman & Gilman’s The Pharmacological Basis of Therapeutics, 11th edn., New York: McGraw-Hill, 2006. [ch 33]
Roden D.M. Antiarrhythmic drugs. In Brunton L.L., Lazo J.S., Parker K.L., editors: Goodman & Gilman’s The Pharmacological Basis of Therapeutics, 11th edn., New York: McGraw-Hill, 2006. [ch 34]
Silverman M. Magic in a Bottle. New York: Macmillan; 1942.
Speight T., Holford N., editors. Avery’s Drug Treament, 4th edn., Auckland: Adis Inernational, 1997.