Chapter 11 Overview of the Autonomic Nervous System and Drugs Affecting Cholinergic Transmission
The peripheral nervous system is subdivided functionally and anatomically into two divisions: the autonomic nervous system and the somatic nervous system (Chapter 13). The autonomic nervous system, which comprises the parasympathetic, sympathetic and enteric nervous systems, is responsible for regulation of the internal viscera such as the heart, blood vessels, digestive organs, kidneys and reproductive organs.
The parasympathetic component of the autonomic nervous system uses principally acetylcholine as the neurotransmitter. The action of acetylcholine on muscarinic receptors leads to responses primarily in the gastrointestinal and respiratory tracts, bladder, heart, eye and glands. This chapter reviews clinically relevant drugs that mimic, intensify or block the action of acetylcholine on muscarinic receptors in the parasympathetic system.
Key abbreviations
Key background
The autonomic nervous system
THE two principal divisions of the nervous system are the central nervous system (see Chapter 14) and the peripheral nervous system (PNS). The PNS is divided further on a functional and anatomical basis into two subdivisions: the autonomic and the somatic nervous systems. The afferent (incoming) fibres of both systems carry sensory information to the central nervous system, which is integrated at various levels within the brain. The information that flows out from the central nervous system is conducted along efferent (outgoing motor) neurons of either the autonomic efferent system or the somatic efferent system. These systems innervate various organs and tissues (commonly called effectors) that produce a physiological response when stimulated by the appropriate nerves (Figure 11-1). Innervation of skeletal muscle is principally coordinated by the somatic nervous system, discussed in Chapter 13.
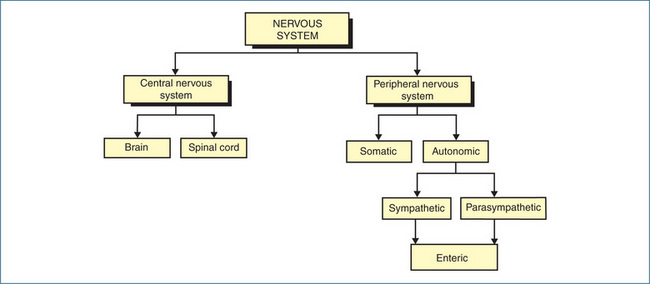
The autonomic nervous system (ANS) primarily maintains the internal environment of the body at an optimal level (homeostasis) and cannot function independently of the central nervous system. The activities regulated by the ANS are not under direct conscious control and include the contraction and relaxation of smooth muscle, regulation of heartbeat and glandular secretions (Figure 11-2). The simplest means by which these types of activities are adjusted are autonomic (visceral) reflexes.
The first component of the reflex arc is the receptor, which detects changes such as a rise or fall in temperature or in pressure in blood vessels, or distension in the viscera. Information from the receptor is then transmitted via a sensory (afferent) neuron to the central nervous system, the site of integration. The preganglionic, autonomic efferent (motor) neuron then conveys nerve impulses from the central nervous system to ganglia and onwards to the effector, which produces the appropriate alteration of activity of muscles and glands.
The information carried to the central nervous system (sensory input) and instructions sent from the central nervous system (motor output) constitute a feedback control mechanism. Information fed back to the central nervous system from a receptor is modulated so that nerve impulses may vary in frequency and pattern according to the degree of activity required of the effector. The control of visceral function is involuntary, so the feedback mechanism must include all the components of a control system essential to performing the reflex act, the sole purpose of which is to prevent extreme changes in function that may create a disturbance in the internal environment.
Several centres in the central nervous system integrate all ANS activities. There is evidence that the hypothalamus, in particular, performs such integrating activities. It contains centres that regulate body temperature, water balance and carbohydrate and fat metabolism, and also integrates information concerned with emotional behaviour, the waking state and sleep. The medulla oblongata integrates the control of blood pressure, respiration and cardiac function; a series of ‘vital centres’, including the vasomotor centre, respiratory centre and cardiac centre, respectively, coordinate these activities. The midbrain, limbic system, cerebellum and cerebral cortex are all involved in the control of physiological functions regulated by the ANS. An understanding of autonomic reflexes is integral to appreciating the actions of autonomic drugs, especially those that evoke significant compensatory ‘reflex’ responses.
The ANS is organised into three subdivisions (Figure 11-1):
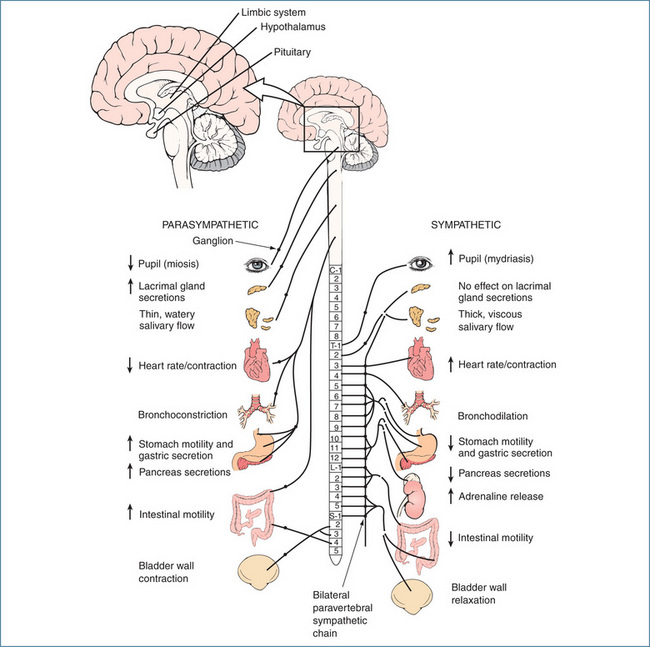
Most organs receive dual innervation from the parasympathetic and sympathetic systems. In many instances the systems produce opposite effects but they may also produce the same effect, e.g. in salivary glands stimulation from both systems produces secretion. The noticeable exceptions are the lacrimal (tear) glands of the eye, which receive only parasympathetic fibres. In contrast, the arrector pili muscles attached to hair follicles in the skin, adipocytes (fat cells), kidneys and blood vessels are innervated solely by sympathetic fibres. With regard to the human airway the situation is even more complex. The dominant neural control is exerted through parasympathetic cholinergic nerves that mediate bronchoconstriction. In contrast, sympathetic innervation of the airways is sparse and few noradrenergic fibres have been demonstrated in human airways. However, both α- and β-adrenoceptors are present in airways and these are activated by circulating catecholamines, predominantly adrenaline (refer to Chapter 28). In general, the opposing actions of the two systems balance one another; for example, a rise in parasympathetic activity is accompanied by a fall in sympathetic input (Table 11-1).
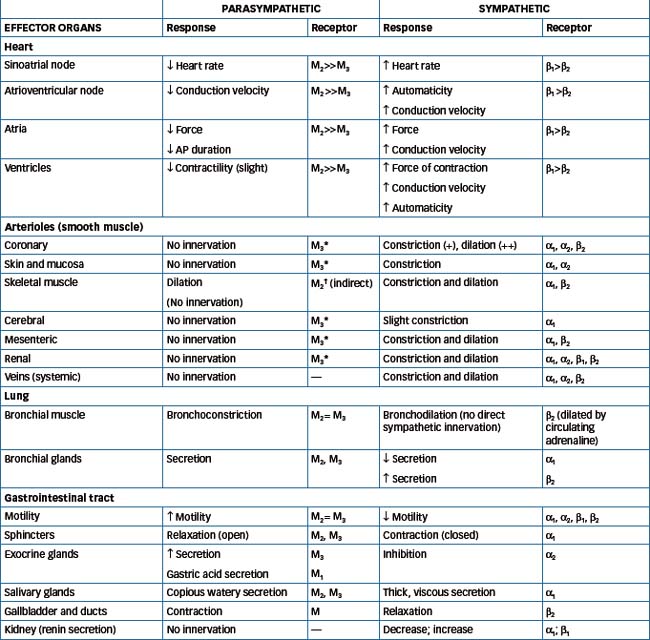
Physiological differences between the subdivisions of the ANS
The parasympathetic system functions mainly to conserve energy and restore body resources. This includes reducing heart rate, increasing gastrointestinal activity and secretion of digestive enzymes associated with increased digestion and absorption. In contrast, the sympathetic system dominates the body during emergency and stress situations and is often called the ‘fight-or-flight’ system. The sympathetic response to physical or emotional stress involves expenditure of energy and includes an increase in the blood sugar concentration, heart activity and blood pressure (Table 11-1). The concept of these two extreme situations (rest and flight) in humans is outdated, as in everyday life the ANS functions continually and the balance of sympathetic and parasympathetic control depends on the needs of a particular organ at any given time.
Unlike the parasympathetic and sympathetic systems, the enteric nervous system consists of a collection of neural plexuses (networks) in the walls of the gastrointestinal tract that can function independently of the central nervous system. Incoming nerves from the parasympathetic and sympathetic systems serve primarily to modulate the intrinsic activity of the enteric neural network, which controls motility, secretion and the microcirculation of the gastrointestinal tract.
Anatomical differences between the subdivisions of the ANS
The parasympathetic and sympathetic efferent pathways consist of two neurons (nerve cells) and an autonomic ganglion, which is a collection of neuronal cell bodies. The first neuron is known as the preganglionic neuron and extends from the cell body in the central nervous system to the autonomic ganglion. The second neuron is called the postganglionic neuron and extends from the autonomic ganglion to the effector organ, gland or cell. The parasympathetic preganglionic fibres emerge with the cranial nerves (III, VII, IX and X) and at the sacral spinal levels from about S2 through S4 (Figure 11-2). The 10th cranial nerve (X), or vagus nerve, has extensive branches that supply fibres to the heart, lungs and almost all the abdominal organs. The parasympathetic ganglia are located close to the effectors that produce the physiological response, and consequently the preganglionic axon tends to be long.
The sympathetic system is also called the thoracolumbar system because its preganglionic fibres originate in the spinal cord from thoracic segment T1 to the lumbar segment at L2 level (Figure 11-2). The sympathetic ganglia lie on either side of the vertebral column in two chains, called the paravertebral sympathetic chains; hence the sympathetic preganglionic axons tend to be short. The only exception to the two-neuron arrangement is the adrenal medulla, which is supplied directly by a preganglionic neuron.
Action potential generation and neurochemical transmission
Communication throughout the nervous system occurs through a highly integrated network of neurons and neuroglia (support cells). Neurons are the cells that transmit electrical impulses (also called action potentials), and they have three basic parts (Figure 11-3):
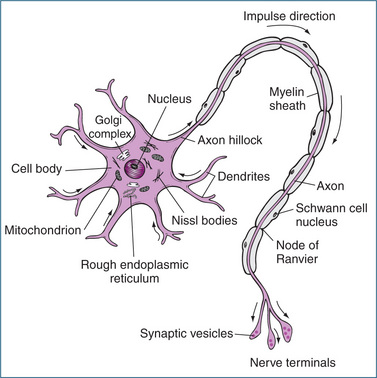
Most axons are covered with a whitish lipid–protein coating called the myelin sheath. The myelin sheath is formed by Schwann cells that wrap themselves around the axon, forming as many as 100 concentric circles. Myelin protects the neuron, insulates it from other neurons and aids the speed of conduction of electrical impulses. Gaps (nodes of Ranvier) occur at regular intervals in the myelin sheath and speed conduction, as the electrical impulse is forced to jump from one node to the next. Unmyelinated fibres also exist but impulses tend to be conducted very slowly (around 1 m/s or less). In the ANS, the preganglionic neuron is myelinated and the postganglionic neuron is unmyelinated.
Action potential generation
Neurons communicate with each other via generation of an electrical signal or action potential. Although the human body is electrically neutral, having the same number of positive and negative charges, the inside (cytoplasmic side) of the membrane of a resting neuron is negatively charged with respect to the outside. This potential or voltage difference is called the resting membrane potential and is in the order of −40 to −90 mV, typically −70 mV. Neurons exhibiting a membrane potential are referred to as polarised. The principal ions involved in generating the membrane potential are potassium and sodium. Movement of sodium into the neuron and movement of potassium out of the neuron occur principally via voltage-gated sodium and potassium channels. Under normal (resting) conditions, a slow inward leakage of sodium ions occurs. These are pumped out of the neuron by a sodium–potassium pump that expels three sodium ions for each two potassium ions imported. This pump maintains the resting membrane potential at around −70 mV (Figure 11-4).
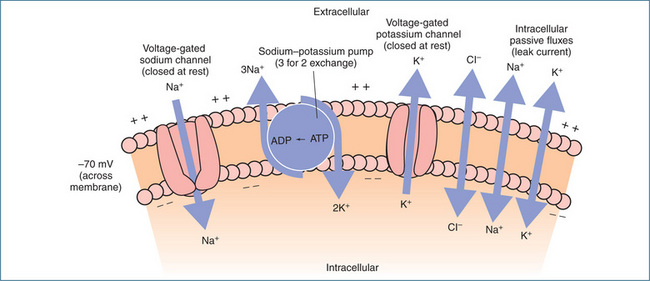
Figure 11-4 Primary determinants of the resting membrane potential. At rest the small build-up of positive ions along the outside of the membrane and negative ions along the inside of the membrane gives rise to the resting membrane potential of about −70 mV.
An action potential (Figure 11-5) occurs when:
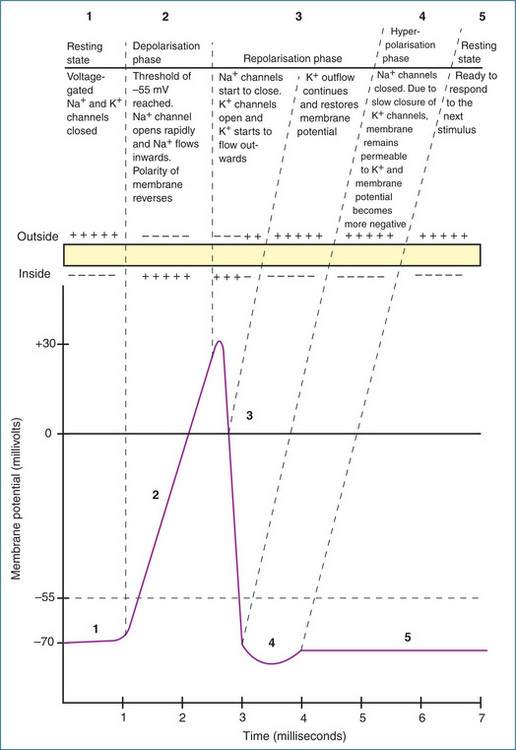
After an action potential is initiated it self-propagates along the full length of the axon in one direction only, away from the cell body towards the nerve terminals. Nerve fibres conduct electrical impulses only along the axon but communication throughout the neuronal network relies completely on the conversion of these impulses into chemical signals at synapses or effector junctions.
Neurochemical transmission
The passage of a nerve impulse from one neuron to another neuron (e.g. at autonomic ganglia) or from a neuron to an effector via a chemical signal is called neurochemical transmission. When the action potential reaches the presynaptic nerve terminal, the electrical signal is converted to a chemical signal by release of a neurotransmitter, which acts as a chemical messenger enabling nerve cells to communicate signals to the structures they innervate. The site at which communication between neurons occurs is called a synapse. Communication between a neuron and an effector occurs at a neuroeffector junction. In the parasympathetic and sympathetic nervous systems, synapses occur at ganglia, which are the sites of synapses between the preganglionic and postganglionic neurons and between the postganglionic neuron and the effector tissue or organ. The presence of a specific chemical at these synapses determines the type of information a neuron can receive and the range of responses it can yield in return. Receptors on the postsynaptic membrane bind the transmitter, which initiates a postsynaptic response that can be either excitatory or inhibitory. There are many specific neurotransmitters and these will be discussed in the context of the relevant pharmacology in the appropriate chapters.
Acetylcholine and cholinergic transmission
There are multiple neurotransmitters in the ANS (see Clinical Interest Box 11-1) but the two about which most is known are acetylcholine (ACh) and noradrenaline. Nerves that release ACh are called cholinergic neurons and are involved in cholinergic transmission (Figure 11-6). ACh is the neurotransmitter released from:
Clinical interest box 11-1 Neurotransmitters
To date, over 50 different chemicals have been identified as neurotransmitters or as putative neurotransmitters. These include acetylcholine, adrenaline, noradrenaline, dopamine, 5-hydroxytryptamine (5-HT, also commonly called serotonin), ATP, histamine, γ-aminobutyric acid, glycine, aspartate, glutamate, nitric oxide and numerous peptides such as neuropeptide Y, calcitonin gene-related peptide, gonadotrophin-releasing hormone, substance P, vasoactive intestinal peptide (VIP) and the endorphins and enkephalins. Some neurons synthesise and release only one type of neurotransmitter but equally many synthesise and release multiple transmitters. The pathology of many diseases has been linked to neurotransmitter–receptor dysfunction, including Parkinson’s disease, myasthenia gravis, Alzheimer’s disease, depression and schizophrenia.
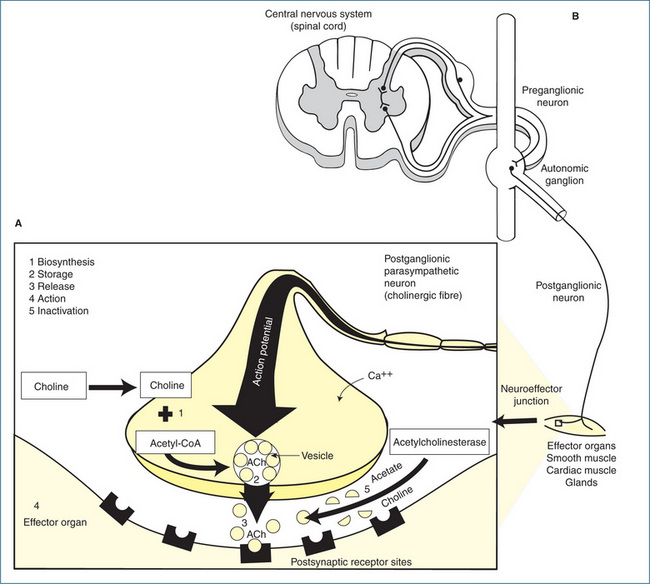
Figure 11-6 Chemical neurotransmitters and receptor sites in the autonomic nervous system. A = adrenaline; ACh = acetylcholine; NA = noradrenaline; NN = neuronal nicotinic receptor; M = muscarinic receptor.
For correct transmission across synapses to occur the neurotransmitter must first be synthesised, stored and then released, so that it can bind to and activate receptors and finally be inactivated. Many autonomic drugs affect one of these individual events, so it is essential to understand the basic mechanisms involved in neurotransmission.
ACh is synthesised in the cytoplasm of the nerve terminal from free choline and acetyl coenzyme A (acetyl-CoA) via the action of the enzyme choline acetyltransferase. Once synthesised, the ACh is packaged into synaptic vesicles or granules, which are located in the nerve terminal (Figure 11-7). The arrival of an action potential at the nerve ending facilitates the entry of calcium, which induces the synaptic vesicles containing ACh to attach to specific docking sites on the synaptic membrane and release the neurotransmitter molecules (via a process called exocytosis) into the synaptic cleft. The whole process of vesicle docking, cycling and exocytosis is under the control of various trafficking proteins. Once free, ACh diffuses across the synaptic cleft and attaches to specialised postsynaptic receptors on the membrane of the next neuron or effector. The binding of ACh to the receptor increases the permeability of the postsynaptic membrane to sodium and potassium ions, causing a depolarising action that results in excitation or inhibition of neural, muscular or glandular activity (Figure 11-7).
Figure 11-7 A Cholinergic transmission at a neuroeffector junction. 1 Biosynthesis of acetylcholine (ACh): choline is taken up by the axon terminal and ACh is synthesised from choline and acetyl-CoA. 2 Storage: after synthesis, ACh is stored in the vesicle until the arrival of a nerve impulse. 3 Release: an action potential arriving at the nerve terminal causes the vesicle to attach itself to the membrane and release ACh, which then diffuses across the synaptic cleft and combines with the receptors on the effector cell. 4 Action: the interaction of ACh with the receptors results in a response. 5 Inactivation of ACh: at the synaptic cleft, ACh is hydrolysed by the enzyme acetylcholinesterase. B Schematic representation of the relation between a neuron in the CNS, a preganglionic neuron and an effector organ innervated by a postganglionic parasympathetic neuron.
Acetylcholine receptors and acetylcholinesterase
Sir Henry Dale, investigating the pharmacological properties of ACh in 1914, distinguished two actions that were reproduced by the alkaloids, muscarine and nicotine. As the effects of muscarine mimicked the parasympathetic nervous system, he termed the receptors ‘muscarinic’, while those in autonomic ganglia and at the skeletal neuromuscular junction were termed ‘nicotinic’ (N). In the periphery, muscarinic (M) receptors, which are all G protein-coupled receptors (GPCRs, refer to Chapter 5) are located in smooth muscle, cardiac muscle and glands while, in the CNS, M receptors are involved in motor control, memory and cardiovascular and temperature regulation.
Five distinct subtypes of M receptors have been identified, of which three are relevant pharmacologically. These subtypes are classified broadly as the neural type (M1), the cardiac and presynaptic type (M2) and the glandular or smooth muscle type (M3) (Table 11-1). Nicotinic receptors are classed as either neuronal (NN) or muscle (NM) type. The neuronal nicotinic receptors (NN) are in the ganglia of both the parasympathetic and sympathetic systems and the adrenal medulla.
To ensure that the action of released ACh is brief (1–2 milliseconds), after acetylcholine has exerted its effect on the postsynaptic receptors the excess amount is inactivated rapidly by the enzyme acetylcholinesterase (AChE). This enzyme, which is bound to the basement membrane of the nerve terminal, is a target for drugs, insecticides and nerve gases (refer to Chapter 13). As a result of the action of AChE, ACh is metabolised to choline and acetate, which have no transmitter action and are recycled for synthesis of ACh (Figure 11-7).
Drugs acting at muscarinic receptors
Muscarinic receptor agonists, also referred to as parasympathomimetic drugs (e.g. bethanechol), bind to muscarinic receptors and mimic the action of acetylcholine on the parasympathetic nervous system. Muscarinic receptor antagonists, also referred to as parasympatholytic or anticholinergic drugs (e.g. atropine), block competitively the action of ACh on muscarinic receptors and, hence, they block the effect that would result from stimulation of the parasympathetic nervous system.
Acetylcholine has two major actions on the nervous system:
The first action resembles the effects of nicotine, such as tachycardia, elevated blood pressure and peripheral vasoconstriction, and is referred to as the nicotinic effect of ACh. The second action of ACh at the post-ganglionic nerve endings is like that of muscarine (an alkaloid obtained from the toadstool Amanita muscaria) and is referred to as the muscarinic effect of ACh. Refer to Figure 11-6 to review nicotinic and muscarinic receptor sites.
Although ACh is important physiologically, it has no therapeutic value because it lacks selectivity, binding to both nicotinic and muscarinic receptors, and its duration of action is exceedingly brief (∼ 100 microseconds) due to rapid hydrolysis by AChE. The lack of tissue selectivity, which is due to the widespread distribution of muscarinic receptors, is common to many of the drugs discussed in this chapter.
Muscarinic receptor agonists
These drugs are divided into two groups:
Drug monograph 11-1 Bethanechol
Bethanechol acts on muscarinic receptors on the detrusor muscle of the urinary bladder and smooth muscle of the gastrointestinal tract. In the bladder, the resulting contraction of the smooth muscle is sufficiently strong to initiate micturition and empty the bladder. In the gastrointestinal tract, the drug stimulates gastric motility, increases gastric tone and often restores impaired peristaltic activity of the oesophagus, stomach and intestine. It also promotes defecation. Unlike acetylcholine, bethanechol is not degraded by acetylcholinesterase and its effects therefore are more prolonged than those of acetylcholine. Therapeutic doses in normal human subjects have little effect on heart rate, blood pressure or the peripheral circulation.
Indications
More effective drugs have generally replaced bethanechol; it is available for treating postoperative and postpartum non-obstructive urinary retention (although not recommended as the effect is inconsistent) and for neurogenic atony of the urinary bladder associated with retention.
Pharmacokinetics
Despite being poorly absorbed from the gastrointestinal tract, bethanechol chloride is effective orally. It does not penetrate the blood–brain barrier in therapeutic doses but it is distributed to areas of low blood flow. Onset of action is within 30–90 minutes of oral administration, peak effect occurs within 90 minutes and duration of action is up to 6 hours, depending on the dose administered. When the drug is administered subcutaneously, the onset of action is within 5–15 minutes, peak effect occurs within 15–30 minutes and the duration of action is about 2 hours. Routes of metabolism and excretion are unknown.
Drug interactions
The following effects can occur when bethanechol is given with the drugs listed:
Warnings and contraindications
Use is contraindicated in people with known bethanechol hypersensitivity, Parkinson’s disease, asthma (causes bronchoconstriction), epilepsy, hypotension, severe bradycardia, coronary artery disease, gastrointestinal obstruction, hyperthyroidism (may precipitate atrial fibrillation) and peptic ulcer. No data are available regarding excretion in breast milk. Bethanechol should be avoided during pregnancy because of excitatory effects on bladder smooth muscle.
Bethanechol is a polar (charged) quaternary ammonium compound, which is poorly absorbed orally. Its actions are similar to, although longer-acting than, those of the physiological mediator ACh, and its effect is blocked by atropine. The adverse effects of this drug, which include bradycardia, hypotension, sweating, salivation, vomiting, diarrhoea and intestinal cramps, are a conse quence of parasympathetic stimulation (Table 11-1).
Muscarinic receptor antagonists
Muscarinic receptor antagonists are often referred to as parasympatholytic, antimuscarinic or anticholinergic drugs because they competitively block the action of ACh at muscarinic receptors. (See Figure 11-6 for muscarinic receptor sites.) These drugs are categorised as:
The best-known muscarinic antagonists are atropine and hyoscine. Atropa belladonna (deadly nightshade) contains mainly atropine, whereas Hyoscyamus niger (henbane) and Datura stramonium (jimsonweed) contain hyoscine. Atropine (see Drug Monograph 11-2) is the prototype muscarinic antagonist and its use for more than half a century is testimony to its therapeutic effectiveness. With the exception of some degree of selectivity for the heart and GI tract, all the muscarinic receptor antagonists produce peripheral effects similar to those observed with atropine. To avoid the widespread unwanted effects of muscarinic receptor antagonism more selective drugs have been developed. These include the M1 selective antagonist pirenzipine (not available in Australia and New Zealand), which inhibits gastric acid secretion but has little effect elsewhere in the body, and oxybutynin, tolterodine and the M3-selective drugs darifenacin and solifenacin, which are all used to treat urinary incontinence (refer to Chapter 25). Muscarinic receptor antagonists and their main uses are shown in Table 11-3.
Atropine has very little effect on the actions of acetyl choline at nicotinic receptor sites but can produce a wide range of pharmacological effects because of the wide spread distribution of muscarinic receptors in the body. The main effects are summarised below.
Eyes
The pupil is dilated (mydriasis) and relaxation of the ciliary muscle causes failure of accommodation (cycloplegia) impairing near vision. Pupil dilation may reduce outflow of aqueous humour, causing a rise in intraocular pressure, a hazardous situation for people with narrow-angle glaucoma. These effects in the eye occur with local and systemic administration of atropine, although the usual single therapeutic dose of atropine given orally or parenterally has little effect on the eye. Following pupil dilatation photophobia occurs, and the usual reflexes to light and accommodation disappear.
Skin and mucous membranes
Low doses of atropine inhibit secretion from lacrimal, bronchial, salivary and sweat glands. This produces the characteristic drying of the mucous membranes of the mouth, nose, pharynx and bronchi, and causes the skin to become hot and dry.
Respiratory system
Atropine relaxes the smooth muscle of the bronchial tract but is less effective than adren aline as a bronchodilator and is not used for asthma.
Cardiovascular system
When very low doses of atropine are administered, the heart rate is temporarily slowed because of a central action that augments vagal activity (paradoxical bradycardia). Larger doses block the effect of vagal stimulation on the sinoatrial node and atrioventricular junction, which leads to an increased heart rate. In therapeutic doses, atropine has little or no effect on blood pressure because most vascular beds lack significant cholinergic innervation.
Gastrointestinal tract
The effect of atropine on the secretions of the pancreas and intestinal glands is not therapeutically significant but atropine (in larger doses) incompletely inhibits GI motility. Gastric acid secretion is reduced slightly.
Urinary tract
Atropine slightly relaxes smooth muscle of the urinary tract, and therapeutic doses decrease the tone of the fundus of the urinary bladder. It also causes constriction of the internal sphincter, which can produce urinary retention, particularly in elderly men with prostatic enlargement.
Central nervous system
Atropine has prominent effects on the CNS and in large doses causes excitement, agitation, irritability, hallucinations, delirium and finally stupor and coma. These effects are due to blockade of central muscarinic receptors. A rise in temperature is sometimes seen, especially in infants and young children, probably as a result of suppression of sweating. Because they reduce cholinergic transmission, muscarinic receptor antagonists are used to treat the extrapyramidal effects (tremor, involuntary movements and rigidity) associated with both Parkinson’s disease and antipsychotic drug use.
Pharmacokinetics
Atropine is readily absorbed after oral and parenteral administration; it is also absorbed from mucous membranes. After intramuscular administration, peak plasma concentration is reached within 30 minutes. The duration of action is 4–6 hours but ocular effects can last longer. Approximately 50% of the drug is bound to plasma proteins and it readily crosses the placental barrier and the blood–brain barrier. Atropine is metabolised primarily in the liver but approximately 30%–50% is excreted unchanged in the urine.
Drug interactions and adverse reactions
The anticholinergic effect of drugs such as tricyclic antidepressants (e.g. amitriptyline, nortriptyline, clomipramine, dothiepin), some antihistamines (e.g. promethazine) and the phenothiazines (e.g. chlorpromazine) may be additive with atropine, increasing the therapeutic and adverse effects, including central delirium. Avoid a combination of drugs with anticholinergic effects and muscarinic receptor antagonists if possible. The reduction in gastric motility caused by atropine can also impair the absorption of other drugs. For adverse reactions see Table 11-2.
Warnings and contraindications
Avoid use in people with atropine hypersensitivity or known hypersensitivity to other muscarinic antagonists. Atropine is contraindicated in myasthenia gravis, severe cardiac disease, GI obstructive disease, narrow-angle glaucoma, acute haemorrhage, prostatic hypertrophy, urinary retention, pyloric obstruction, ulcerative colitis, toxaemia of pregnancy and febrile conditions, and in debilitated patients with intestinal atony or paralytic ileus. Caution should be exercised in any situation where there is a higher likelihood of adverse effects, e.g. Down syndrome, the elderly, people with autonomic neuropathy and hepatic and renal disease.
Dosage and administration
Atropine is used in a variety of circumstances including before surgery, to treat anticholinesterase poisoning, to treat bradycardia during resuscitation and for asystole. Current drug information sources or if applicable a Poisons Information Centre should be consulted before administration. As a premedication to prevent excessive salivation and respiratory tract secretions in adults during anaesthesia, 0.3–0.6 mg may be given IM about 1 hour before anaesthesia or IV immediately before induction.
Hyoscine hydrobromide
Similar to atropine, hyoscine is a muscarinic receptor antagonist. The peripheral effects of hyoscine are similar to those of atropine but, due to greater permeation of the blood–brain barrier, it has marked effects on the central nervous system. At therapeutic doses, it depresses the CNS and causes drowsiness, euphoria, memory loss, relaxation and sleep. It does not increase blood pressure or respiration.
Because of its depressant action on vestibular function, it is used for motion sickness, to prevent nausea and vomiting and as an adjunct medication with general anaesthesia to reduce respiratory tract secretions. Its pharmacokinetics are similar to those of atropine. For adverse reactions see Table 11-2.
Table 11-2 Drugs affecting the parasympathetic nervous system: adverse reactions
| Drug | Adverse reactions |
| Muscarinic agonist | |
| Bethanechol | Abdominal pains or upset, increased salivation and sweating, nausea or vomiting; flushed skin; blurred or disturbed vision; unsteadiness; headache and diarrhoea |
| Muscarinic antagonists | |
| Atropine and hyoscine | Inhibition of sweating; constipation; dry mouth, throat and skin; blurred vision; urinary retention; headache; photophobia; drowsiness; weakness; nausea or vomiting; urticaria; dermatitis and eye pain from raised intraocular pressure. In addition, euphoria, amnesia and insomnia are reported more often with hyoscine |
| Glycopyrrolate (synthetic antispasmodic) | Abdominal distension; headache; dizziness; constipation; nausea; vomiting; sedation; dry mouth, nose, throat and skin; blurred or disturbed vision; dysuria; weakness; hypotension and decreased sexual ability |
For travel sickness the adult oral dose of hyoscine hydrobromide is 0.3–0.6 mg 30 minutes before travel and repeated 3–4 hours later if required, to a maximum dosage of 1.2 mg/day. The dosage for children varies: age 2–7 years, a maximum of 0.15–0.3 mg/daily; over 7 years of age, a maximum of 0.6 mg/daily. The elderly are more sensitive to this drug at the usual adult dosage; sensitivity can manifest as confusion, blurred vision and ataxia.
Synthetic cogeners and semisynthetic derivatives
The usefulness of atropine is limited by the fact that it is a complex drug and because it produces effects in a range of organs or tissues simultaneously, owing to the widespread distribution of muscarinic receptors. When it is administered for its antispasmodic effects, it also produces prolonged effects in the eye, causing dilated pupils and blurred vision. It also causes dry mouth and possibly tachycardia. Atropine does have some desirable effects, and a large number of drugs have been synthesised in an effort to take advantage of the antispasmodic effect of atropine without its other effects (see Table 11-3).
Table 11-3 Muscarinic antagonists: clinical use and route of administration
| Drug | Clinical use | Route of administration |
| Atropine | Mydriatic, cycloplegic, antisecretory | IV, IM, SC, topical (eye-drops) |
| Benztropine | Parkinson’s disease, drug-induced extrapyramidal disorders | Oral, IV, IM |
| Benzhexol | Parkinson’s disease, drug-induced extrapyramidal disorders | Oral |
| Biperiden | Parkinson’s disease, drug-induced extrapyramidal disorders | Oral |
| Cyclopentolate | Mydriatic, cycloplegic | Topical (eye-drops) |
| Glycopyrrolate | Antisecretory | IM, IV |
| Homatropine | Mydriatic, cycloplegic | Topical (eye-drops) |
| Hyoscine butylbromide | Antispasmodic | Oral, IM, IV |
| Ipratropium bromide (Ch 28) | Bronchodilator | Inhalational |
| Mebeverine* | Antispasmodic | Oral |
| Orphenadrine | Parkinson’s disease, drug-induced extrapyramidal disorders | Oral |
| Oxybutynin† | (Ch 25) Antispasmodic (urge incontinence) | Oral |
| Propantheline bromide | Antispasmodic (urge incontinence) | Oral |
| Tiotropium (Ch 28) | Bronchodilator | Inhalational |
| Tolterodine (Ch 25) | Antispasmodic (urge incontinence) | Oral |
| Tropicamide | Mydriatic, cycloplegic | Topical (eye-drops) |
* Has multiple mechanisms of action and is not solely a muscarinic antagonist.
† Has calcium channel-blocking activity at high doses.
Many products are marketed as antispasmodic and anticholinergic agents but their formulations are either modifications of a belladonna alkaloid or include one or more of the natural alkaloids as their active ingredients. The pharmacological properties are therefore similar to atropine. One of the more commonly used systemic agents is glycopyrrolate.
Glycopyrrolate
Glycopyrrolate is a synthetic muscarinic receptor antagonist with effects similar to those of atropine. Unlike atropine, it is unable to easily cross lipid membranes (such as the blood–brain barrier) and hence has minimal CNS effects. It is also less likely to produce pupillary or ocular effects. Glycopyrrolate is indicated as an antimuscarinic drug to reduce salivary, tracheobronchial and pharyngeal secretions preoperatively, to prevent bradycardia induced during anaesthesia and to prevent or reduce the peripheral effects of AChE inhibitors (neostigmine or pyridostigmine).
Following an IV dose, the onset of action occurs within about 1 minute, and following an IM dose, about 15–30 minutes. Vagal blocking action lasts 2–3 hours and the antisialagogue effect (inhibition of the flow of saliva) can last up to 7 hours. Glycopyrrolate is predominantly excreted by the kidneys as unchanged drug.

To reduce excessive salivation and respiratory tract secretions in adults during anaesthesia, the dose is 0.2–0.4 mg IV or IM, or 0.004–0.005 mg/kg of body weight to a maximum of 0.4 mg given 30–60 minutes before anaesthesia. For children aged 1 month to 12 years, 0.004–0.008 mg/kg of body weight to a maximum of 0.2 mg IV or IM is given 30–60 minutes before anaesthesia.
Ganglion-blocking drugs
The major neurotransmitter at all autonomic ganglia is ACh. Ganglion-blocking drugs block the action of ACh at autonomic ganglia by competing with ACh at the synapse. This results in reduced impulse transmission from preganglionic to postganglionic neurons in both the sympathetic and parasympathetic systems. Because of the profound physiological effects (hypotension, loss of cardiovascular reflexes) elicited by ganglion-blocking drugs, they are now clinically obsolete.
Key points
 The principal divisions of the nervous system are the central nervous system (CNS) and the peripheral nervous system (PNS), which has two subdivisions—the autonomic nervous system (ANS) and the somatic nervous system (SNS). The ANS regulates the function of smooth muscle, cardiac muscle and glandular secretions, which are not under direct conscious control. The ANS is organised in three subdivisions: the parasympathetic, sympathetic and enteric nervous systems. The parasympathetic system functions to conserve energy, while activity of the sympathetic system increases in stress situations. The enteric nervous system receives parasympathetic and sympathetic inputs but can function independently of the CNS to modulate motor and secretory functions of the gastrointestinal tract. Parasympathetic and sympathetic efferent (motor) pathways consist of preganglionic neurons that extend from cell bodies in the CNS to the autonomic ganglia, and postganglionic neurons that extend from the ganglia to various effector organs. A neuron is composed of a cell body, dendrites and axon. Neurons communicate with each other or with effectors via generation of an electrical signal or action potential. Neurotransmitters are stored in presynaptic vesicles, released by exocytosis and have their actions terminated by metabolism, reuptake into nerve terminals and diffusion. ACh is the transmitter between pre- and postganglionic neurons in both the parasympathetic and sympathetic systems, and between postganglionic parasympathetic nerves and effector organs. The cholinergic receptor sites that are stimulated by acetylcholine are either nicotinic (NN, NM) or muscarinic (M1, M2, M3). Nicotinic receptors appear in the ganglia of the parasympathetic and sympathetic systems, adrenal medulla and skeletal muscle (somatic motor system), whereas muscarinic receptors are located at postganglionic sites in smooth muscle, cardiac muscle and glands. Muscarinic agonists are also referred to as parasympathomimetic drugs (e.g. bethanechol) and they mimic the action of ACh on the parasympathetic nervous system. Muscarinic antagonists are also referred to as parasympatholytic or anticholinergic drugs (e.g. atropine) and they block the action of ACh and hence the effect that would result from parasympathetic nervous system stimulation. Muscarinic agonists such as bethanechol are used systemically for non-obstructive urinary retention and for atony of the urinary bladder associated with retention. Pilocarpine is used for the treatment of glaucoma. Adverse reactions to bethanechol include abdominal pain or upset, increased salivation and sweating, nausea or vomiting, flushed skin, blurred or disturbed vision, unsteadiness, headache and diarrhoea. Muscarinic antagonists such as atropine and synthetic substitutes are used clinically as antispasmodics, mydriatics and cycloplegics. They are also used as preanaesthetic drugs to decrease respiratory secretions. Adverse effects of atropine include: constipation; dry mouth, throat and skin; blurred vision; urinary retention and headache (Table 11-2).
The principal divisions of the nervous system are the central nervous system (CNS) and the peripheral nervous system (PNS), which has two subdivisions—the autonomic nervous system (ANS) and the somatic nervous system (SNS). The ANS regulates the function of smooth muscle, cardiac muscle and glandular secretions, which are not under direct conscious control. The ANS is organised in three subdivisions: the parasympathetic, sympathetic and enteric nervous systems. The parasympathetic system functions to conserve energy, while activity of the sympathetic system increases in stress situations. The enteric nervous system receives parasympathetic and sympathetic inputs but can function independently of the CNS to modulate motor and secretory functions of the gastrointestinal tract. Parasympathetic and sympathetic efferent (motor) pathways consist of preganglionic neurons that extend from cell bodies in the CNS to the autonomic ganglia, and postganglionic neurons that extend from the ganglia to various effector organs. A neuron is composed of a cell body, dendrites and axon. Neurons communicate with each other or with effectors via generation of an electrical signal or action potential. Neurotransmitters are stored in presynaptic vesicles, released by exocytosis and have their actions terminated by metabolism, reuptake into nerve terminals and diffusion. ACh is the transmitter between pre- and postganglionic neurons in both the parasympathetic and sympathetic systems, and between postganglionic parasympathetic nerves and effector organs. The cholinergic receptor sites that are stimulated by acetylcholine are either nicotinic (NN, NM) or muscarinic (M1, M2, M3). Nicotinic receptors appear in the ganglia of the parasympathetic and sympathetic systems, adrenal medulla and skeletal muscle (somatic motor system), whereas muscarinic receptors are located at postganglionic sites in smooth muscle, cardiac muscle and glands. Muscarinic agonists are also referred to as parasympathomimetic drugs (e.g. bethanechol) and they mimic the action of ACh on the parasympathetic nervous system. Muscarinic antagonists are also referred to as parasympatholytic or anticholinergic drugs (e.g. atropine) and they block the action of ACh and hence the effect that would result from parasympathetic nervous system stimulation. Muscarinic agonists such as bethanechol are used systemically for non-obstructive urinary retention and for atony of the urinary bladder associated with retention. Pilocarpine is used for the treatment of glaucoma. Adverse reactions to bethanechol include abdominal pain or upset, increased salivation and sweating, nausea or vomiting, flushed skin, blurred or disturbed vision, unsteadiness, headache and diarrhoea. Muscarinic antagonists such as atropine and synthetic substitutes are used clinically as antispasmodics, mydriatics and cycloplegics. They are also used as preanaesthetic drugs to decrease respiratory secretions. Adverse effects of atropine include: constipation; dry mouth, throat and skin; blurred vision; urinary retention and headache (Table 11-2).Review exercises
References and further reading
Australian Medicines Handbook 2010. Adelaide: AMH, 2010.
Boron W.F., Boulpaep E.L. Medical Physiology, Updated edition. Philadelphia: Elsevier; 2005.
Brown J.H., Taylor P. Muscarinic receptor agonists and antagonists. In Brunton L.L., Lazo J.S., Parker K.L., editors: Goodman & Gilman’s The Pharmacological Basis of Therapeutics, 11th edn, New York: McGraw-Hill, 2006. [ch 7]
Dollery C., editor. Therapeutic Drugs, 1. Churchill Livingstone, London, 1991.
Eglen R.M., Choppin A., Dillon M.P., Hegde S. Muscarinic receptor ligands and their therapeutic potential. Current Opinion in Chemical Biology. 1999;3:426-432.
Eglen R.M., Watson N. Selective muscarinic receptor agonists and antagonists. Pharmacology and Toxicology. 1996;78:59-68.
Pendry Y.D. Neural control of airway smooth muscle. Pharmacology and Therapeutics. 1993;57:171-202.
Rang H.P., Dale M.M., Ritter J.M., Flower R.J. Pharmacology, 6th edn. Edinburgh: Churchill Livingstone; 2007. [ch 10]
Westfall T.C., Westfall D.P. The autonomic and somatic motor nervous system. In Brunton L.L., Lazo J.S., Parker K.L., editors: Goodman & Gilman’s The Pharmacological Basis of Therapeutics, 11th edn, New York: McGraw-Hill, 2006. [ch 6]