Chapter 23 Drugs affecting vascular smooth muscle
The focus of this chapter is the vasodilator drugs that produce vasodilation by relaxing smooth muscle in the blood vessel walls by either a direct or indirect action. Some drugs act primarily on veins or arterioles, while others dilate both types of blood vessels. The principal uses of these drugs, which include the organic nitrates, calcium channel blockers, potassium channel activators, angiotensin-converting enzyme inhibitors and angiotensin-receptor antagonists, are in the treatment of angina, hypertension and heart failure. These drugs are frequently prescribed, either alone or in combination therapy, so it is important that health-care professionals are knowledgeable about all of these drug classes.
Key abbreviations
ACE angiotensin-converting enzyme
Key background
Vascular smooth muscle
THE vascular system comprises the arteries and arterioles and venules and veins that carry blood away from and back to the heart, respectively. Arterioles and capillaries are the main resistance vessels and regulate afterload, while the venules and veins are capacitance vessels, contributing to preload of the ventricles. The arterial wall consists of three layers: the inner, tunica intima; the middle, tunica media; and the outer, tunica adventitia. The middle (thickest) layer is composed of elastic and smooth muscle fibres, and the outer of elastic and collagen fibres. Actin and myosin are present in smooth muscle but the striations are not visible (unlike skeletal and cardiac muscle) and the relationship between actin and myosin is less highly organised. The smooth muscle is arranged in a circular layer, and stimulation by the sympathetic nervous system causes contraction of the smooth muscle, which narrows the lumen of the vessel (vasoconstriction). In contrast, a diminution in sympathetic stimulation results in relaxation of the smooth muscle (vasodilation). The elastic properties of the arteries enable distension when the ventricles eject a volume of blood, and the elastic recoil aids in the forward propulsion of the blood. Arteries branch to form arterioles and capillaries, the main resistance vessels, which play a key role in blood pressure regulation. Capillaries are the smallest of the arterial vessels and connect the arterioles to the venules. The combined resistance of the systemic blood vessels, but principally the arterioles, capillaries and venules, is referred to as systemic vascular resistance (SVR) or total peripheral resistance (TPR).
Venules are the conduits through which blood flows from the capillaries to the veins. Veins consist of the same three layers as arteries but these differ in terms of their relative thickness, with the tunica adventitia forming the thickest layer. Unlike arteries, veins have a system of valves that ensure blood flows in a forward direction towards the heart. About 60% of the blood volume is contained within the systemic veins and venules; hence they are referred to as capacitance vessels.
Smooth muscle action potentials
Blood vessel smooth muscle depends primarily on the presence of calcium ions to initiate and sustain contraction. It is believed that the onset of depolarisation (phase 0) in smooth muscle is caused mainly by calcium ions rather than by sodium ions. There are three mechanisms that can lead to an increase in calcium ions, which then triggers smooth muscle contraction: (1) calcium entry through voltage-gated L-type calcium channels, (2) calcium release from the smooth endoplasmic reticulum of the smooth muscle cell and (3) calcium entry through either ligandgated channels or channels activated by G-protein-coupled receptors. In smooth muscle the action potential generally has a slower upstroke and longer duration than that observed in skeletal muscle action potentials. The upstroke or depolarising phase reflects the opening of voltage-gated L-type calcium channels and influx of calcium ions that causes more voltage-gated calcium channels to open. The slow rise of the action potential is due to the slowness of opening of the calcium channels in contrast to the more rapid opening of sodium channels in skeletal and cardiac muscle. The repolarisation phase is also relatively slow because of a combination of the slow inactivation of L-type calcium channels and the slow activation of voltage-gated potassium channels and calcium-activated potassium channels.
The rise in free calcium ion concentration is considered to be the primary event in triggering smooth muscle contraction via the calcium–calmodulin complex that activates myosin light chain kinase (refer Chapter 22), increasing smooth muscle tone and causing vasoconstriction. Activation of smooth muscle can reduce the calibre of small vessels markedly, as is apparent from the ‘spasm’ that may occur in coronary vessels. Calcium channelblocking drugs are capable of blocking the slow calcium ion influx in smooth muscle of blood vessels, thereby producing relaxation. Modulation of calcium concentration forms the basis for the actions of a range of drugs that affect the vascular system. Drugs causing vasoconstriction by acting on α-adrenoceptors (e.g. α-adrenoceptor agonists such as adrenaline) and vasodilation (α1-adrenoceptor antagonists such as prazosin) are discussed in Chapter 12.
This chapter describes the drugs that directly and indirectly affect vascular smooth muscle contraction. Emphasis is on vasodilator drugs used for the treatment of a variety of disorders, including hypertension, angina, shock, cardiac failure and peripheral vascular conditions. These drugs produce vasodilation by relaxing smooth muscle in the blood vessel walls. The main groups of drugs to be discussed are:
Angina
The term angina pectoris refers to temporary interference with blood flow that reduces oxygen and nutrient supply to heart muscle, resulting in intermittent myocardial ischaemia, typically characterised by pain and can be of a number of types.
Angina may occur at rest or can be precipitated by exertion or excitement. When coronary blood flow is inadequate, hypoxia causes an accumulation of painproducing substances such as lactic acid (an anaerobic metabolite) and other chemicals such as potassium ions, kinins and adenosine. Stimulation of cardiac sensory nerve endings, which transmit impulses to the central nervous system, results in the typical anginal pain response. Coronary atherosclerosis or vasomotor spasm of the coronary vessels may cause inadequate oxygenation. Other causes of anginal pain may be pulmonary hypertension and valvular heart disease. Individuals with severe anaemia, even with minimal coronary artery disease, may suffer from anginal attacks because of inadequate oxygen supply.
Drug therapy of angina is aimed at either relaxing coronary artery smooth muscle, thus improving perfusion, or reducing the metabolic demand of the heart, or both. An ideal antianginal drug:
Currently no one drug meets all these criteria and the drugs now available provide only temporary relief (see Clinical Interest Box 23-1).
Clinical interest Box 23-1 Management of angina
A 2009 report from Access Economics estimated that there would be 10,000 deaths in Australia in 2009 from heart attack and that together heart attack and unstable angina would ‘cost the community $17.9 billion, of which $1.8 billion would be in direct health care costs’. Guidelines for the management of acute coronary syndromes were published jointly by the National Heart Foundation of Australia and the Cardiac Society of Australia and New Zealand in 2006. These evidence-based guidelines provide recommendations on the management of acute chest pain, management of patients with ST-segmentelevation myocardial infarction, management of patients with non-ST-segment-elevation acute coronary syndromes and longterm management of patients after control of myocardial ischaemia.
Therapies recommended (in the absence of contra indications) for acute management of chest pain include aspirin (300 mg), oxygen, glyceryl trinitrate and intravenous morphine as required.
Recommendations for long-term management include, where appropriate, the use of aspirin, clopidogrel (stent implantation), β-blockers, angiotensin-converting enzyme inhibitors and statin therapy. Additionally, lifestyle education, cardiac rehabilitation programs and chest pain action plans now form part of the longterm management strategy. Check regularly for updates and amendments (http://www.heartfoundation.org.au).
Direct-acting vasodilator drugs

Organic nitrates
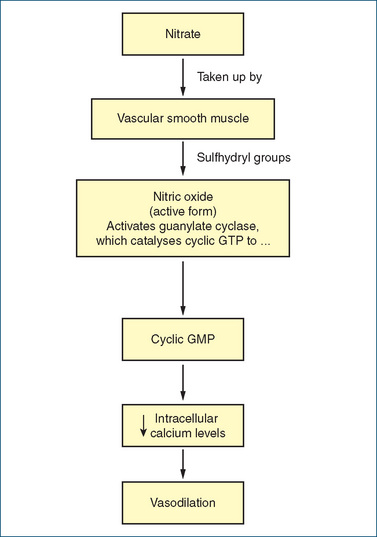
The nitrates, glyceryl trinitrate (also called GTN or nitroglycerine), isosorbide dinitrate and isosorbide mononitrate, are very effective drugs for the treatment of angina pectoris because of their dilating effects on veins and arteries (Drug Monograph 23-1; Figure 23-1). The resulting pooling of blood in the veins (capacitance blood vessels) decreases the amount of blood returned to the heart (preload), which reduces left ventricular end-diastolic volume. This decrease in blood return helps reduce the myocardial oxygen demand (chest pain caused by angina pectoris largely results from an inadequate supply of oxygen to the heart).
Drug monograph 23-1 Glyceryl trinitrate
Glyceryl trinitrate (GTN) is the key drug in the nitrate category. It is available as a sublingual tablet, sublingual spray, transdermal patch or IV infusion. The other drugs in this category include isosorbide dinitrate, which is available as a sublingual or oral tablet, and isosorbide mononitrate, available as a slow-release formulation.
Mechanism of action
The precise mechanism by which GTN activates vascular soluble guanylyl cyclase remains unclear. However, the common theme is release of the free radical nitric oxide (NO), which may in part arise from metabolism of GTN by mitochondrial aldehyde dehydrogenase. The release of NO activates guanylate cyclase in vascular smooth muscle, thereby increasing formation of cyclic guanosine monophosphate (cGMP). This in turn leads to changes in the degree of phosphorylation of smooth muscle proteins. Ultimately, dephosphorylation of the myosin light chain leads to relaxation. The biochemical steps for the metabolism of nitrates to the active NO and the ultimate therapeutic effect of vasodilation are illustrated in Figure 23-1.
Glyceryl trinitrate at low doses causes venodilation, with little effect on arterial resistance vessels. This causes a reduction in preload and stroke volume. With higher doses, dilation of arteries occurs, resulting in a reduction in arterial pressure which, coupled with venous pooling when standing, often results in postural hypotension and dizziness. As both cardiac output and arterial pressure are reduced, the oxygen demand by the myocardium is also reduced. Nitrates also dilate normal coronary and coronary collateral vessels. The resultant increased coronary perfusion and hence oxygen delivery ensures more efficient distribution of blood to ischaemic areas of the myocardium.
Indications
Glyceryl trinitrate is used to prevent or treat stable angina, and to treat unstable angina and heart failure associated with acute myocardial infarction.
Pharmacokinetics
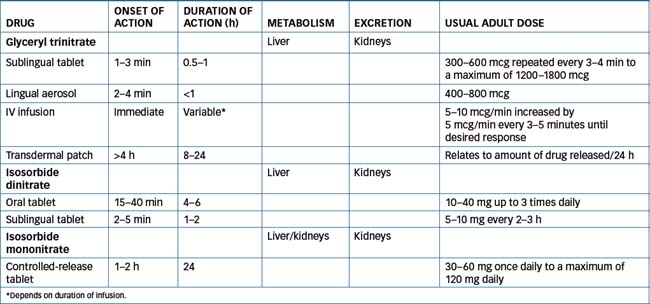
(Table 23-1) Glyceryl trinitrate is rapidly metabolised by the liver to dinitrates that have about 10% of the biological activity of the parent drug.
Drug interactions
The concurrent use of nitrates with alcohol, antihypertensives, other drugs causing hypotension and vasodilators (including sildenafil, tadalafil and vardenafil) may result in enhanced orthostatic hypotensive effects.
Adverse reactions
These include dizziness, headaches, nausea or vomiting, agitation, facial flushing, increased pulse rate, dry mouth, rash, prolonged headaches and blurred vision.
Warnings and contraindications
Nitrates are contraindicated in persons with cardiomyopathy, hypotension, hypovolaemia, aortic or mitral stenosis, severe anaemia, raised intracranial pressure and glaucoma, and with concurrent use of sildenafil.
Dosage and administration
See Table 23-1. Sublingual tablets and the sublingual spray are used during an acute attack or during an episode of angina. The sublingual tablet is placed under the tongue or in the buccal pouch allowing it to dissolve fully, or spray the aerosol under the tongue. Patients should not swallow, eat, drink or smoke while the tablet is in the mouth. If necessary, the sublingual tablet dosage (300–600 mcg) is repeated at 5-minute intervals to a maximum of 2–3 tablets (1200–1800 mcg). A maximum of two sublingual sprays 5 minutes apart is used. After use of sublingual preparations, a transient headache lasting 15–20 minutes and flushing may occur.
The sublingual tablets should be stored in a tightly closed dark container and the bottle dated when first opened. The container should not be left opened or exposed to heat or moisture. To maintain potency the unused tablets should be discarded 3 months after opening the container.
The lingual spray should not be inhaled and the mouth should be closed immediately after delivery of the dose. If using a pump delivery system the pump may need to be primed to ensure an even spray.
Oral sustained-release tablets should not be crushed or chewed but swallowed whole with a full glass of water.
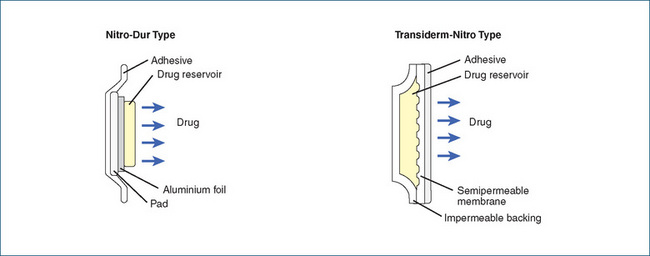
Transdermal GTN is a patch system that contains a drug reservoir from which the drug is slowly released (passive diffusion). The drug is absorbed through the skin and transported by blood to the site of action to produce its beneficial effects. This system is applied daily to a hairless skin area, usually on the chest (preferred site), shoulder or inside upper arm. The site should be changed to avoid skin irritation but avoid applying the patch to extremities, especially below the knee or elbow.
Transdermal systems with different release rates are available (releasing 0.2–0.6 mg/h), and each has a different mechanism for drug delivery. The systems should not be considered to be interchangeable. (See Figure 23-2 for an illustration of transdermal systems.) As tolerance develops to glyceryl trinitrate, it is recommended that the patch be applied for 12–14 hours and removed for 10–12 hours each day. This drug-free interval helps maintain the efficacy of the product.
Calcium channel blockers
Mechanism of action
The calcium channel blockers, while having diverse chemical structures, all block the inward movement of calcium through the slow channels of the cell membranes of cardiac and smooth muscle cells. This activity, however, varies according to the tissue/cells: cardiac muscle, or myocardium; the cardiac conduction system (SA and AV nodes); and vascular smooth muscle. These drugs are used to treat angina, supraventricular tachyarrhythmias (verapamil), hypertension and cerebral vasospasm after subarachnoid haemorrhage (nimodipine).
Clinical interest Box 23-2 Nitrate tolerance
Tolerance to nitrates was first reported in 1888 and at that time it was perceived as a major clinical problem. Despite a century of use the problem persists and only recently has some light been shed on the mechanism of nitrate tolerance. It remains a complex problem as the organic nitrates induce both tolerance and endothelial dysfunction in healthy individuals and in persons with coronary artery disease. Multiple mechanisms have been investigated and nitrate tolerance is thought to be due to a combination of nitrateinduced stimulation of superoxide and/or peroxynitrite production by vascular mitochondria, which contributes to endothelial dysfunction. Production of these reactive species in turn inhibits mitochondrial aldehyde dehydrogenase (ALDH-2), which is responsible for converting GTN to nitrite prior to further metabolism to NO. Hence, inhibition of ALDH-2 impairs the metabolism of GTN and the formation of the bioactive metabolite NO that causes the vasodilation. However, impaired bioactivation of GTN does not explain the associated endothelial dysfunction and increased sensitivity to vasoconstrictors. ALDH-2 activity is reduced in nitrate tolerant individuals and this does provide some support for the proposed mechanism. It remains to be seen whether agents that maintain ALDH-2 activity prevent nitrate tolerance (Munzel et al 2005). Tolerance is not a uniform problem as some individuals appear to develop only partial tolerance. Strategies to reduce nitrate tolerance with the patches include ensuring nitrate-free intervals of 10–12 hours/day. However, nocturnal angina during the nitrate-free interval can be problematic in some patients.
Effects on myocardium
Calcium channel blockers decrease the force of myocardial contraction by blocking the inward flow of calcium ions through the slow channels of the cell membrane during phase 2 (plateau phase) of the action potential (see Figure 22-5). The diminished entry of calcium ions into the cells fails to trigger the release of large amounts of calcium from the sarcoplasmic reticulum within the cell. This free calcium is needed for excitation–contraction coupling, an event that activates contraction by allowing cross-bridges to form between the actin and myosin filaments of muscle. The force of contraction by the heart is determined by the number of actin–myosin cross-bridges formed within the sarcomere. Decreasing the amount of calcium ions released from the sarcoplasmic reticulum results in fewer actin–myosin cross-bridges being formed, thus decreasing the force of contraction and resulting in a negative inotropic effect.
Effects on SA node and AV junction
In these tissues, calcium channel blockers decrease automaticity in the SA node and decrease conduction in the AV junction. Depolarisation (phase 0) of the action potential is normally generated by the inward calcium ion current through the slow channels. These drugs block the inward calcium ion current across the cell membrane of the SA node, decreasing the rate of depolarisation and depressing automaticity. The result is a decrease in heart rate (negative chronotropic effect). Similarly, decreasing calcium ion influx across the cell membrane of the AV junction slows AV conduction (negative dromotropic effect) and prolongs AV refractory time. When AV conduction is prolonged, fewer atrial impulses reach the ventricles, thus slowing the rate of ventricular contractions.
Effects on vascular smooth muscle

The effect of calcium channel blockers on smooth muscle of the coronary and peripheral vessels has a significant influence on cardiovascular haemodynamics. Coronary artery dilation occurs, which lowers coronary resistance and improves blood flow through collateral vessels, as well as improving oxygen delivery to ischaemic areas of the heart.
These agents also inhibit the contraction of smooth muscle of the peripheral arterioles. This results in widespread reduction in resistance to blood flow through the body (determined by the tone of the vascular musculature and the diameter of the blood vessels) and blood pressure. The haemodynamic change reduces afterload, which also decreases oxygen demand of the heart.
The calcium channel blockers include the:
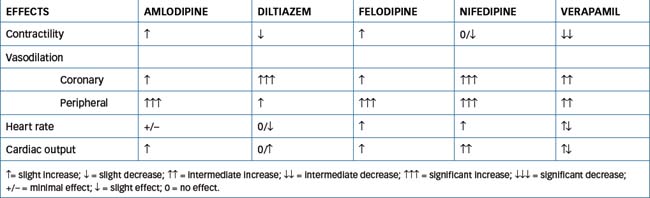
Verapamil was the first calcium channel blocker released and is the key drug for this category. It has greater effects on the heart, reducing AV conduction and blocking the SA node, resulting in a decrease in heart rate and contractility. It is considered a moderate peripheral vasodilator. Diltiazem has similar pharmacological effects on vascular tissue but has less effect on the heart than verapamil. These agents dilate coronary arteries and arterioles, inhibit coronary artery spasm and dilate peripheral arterioles, reducing TPR (afterload), thus lowering arterial blood pressure at rest and during exercise.
The dihydropyridine drugs, exemplified by nifedipine, have minimal effect on cardiac tissue at therapeutic doses. They act principally on vascular smooth muscle, reducing peripheral vascular resistance. In some circumstances the reflex sympathetic response to vasodilation results in tachycardia, which may be deleterious. Table 23-2 provides a comparison of the effects of the different types of calcium channel blockers.
Pharmacokinetics
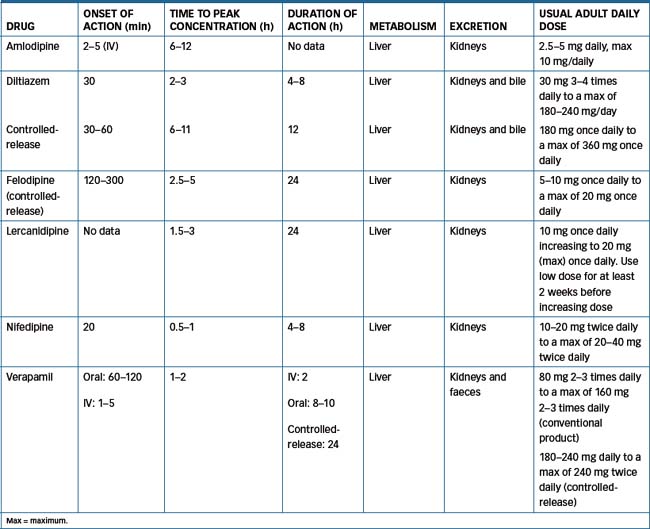
See Table 23-3 for pharmacokinetics of the calcium channel-blocking agents. These drugs are all metabolised to a varying degree by CYP3A4. Diltiazem is metabolised to a major metabolite, desacetyldiltiazem, which may be responsible for up to 50% of its coronary dilation effect. Norverapamil, the active metabolite of verapamil, accounts for about 20% of the antihypertensive effect of verapamil. Nifedipine has no known active metabolite, while the other agents have metabolites that have varying therapeutic effects. Nimodipine is highly lipophilic and crosses the blood–brain barrier, having a greater effect on cerebral arteries than other arteries in the body.
Drug interactions
Due to the common involvement of CYP3A4 drug interactions are extensive and vary for each of the calcium channel-blocking drugs. Relevant drug information sources should always be consulted. Examples include:
Adverse reactions
These include headache, nausea, hypotension, dizziness, skin flushing or rash, oedema of the ankles and feet, dry mouth and tachycardia. Gingival hyperplasia is a rare adverse effect reported with amlodipine, diltiazem, felodipine, verapamil and, most often, nifedipine. It starts as an inflammation of the gums, usually in the first 9 months of therapy. When the drug is discontinued, this effect usually improves within 1–4 weeks. Good dental hygiene, along with professional teeth cleaning, is necessary to reduce the potential for this adverse effect.
Warnings and contraindications
Calcium channel blockers should be used with caution in persons with severe bradycardia, congestive heart failure (caution with felodipine, nifedipine and nimodipine, as they have a slight negative inotropic effect), hypotension, acute myocardial infarction or liver or kidney impairment. Avoid use in persons with hypersensitivity to calcium channel blockers, cardiac shock, severe bradycardia or congestive heart failure (use extreme caution with diltiazem and verapamil).
The elderly are more susceptible to these agents and the adverse effects of increased weakness, dizziness, fainting episodes and falls. In the presence of hepatic impairment treatment should be started at a lower dose for amlodipine and felodipine because a reduced hepatic metabolism may result in increased plasma concentration. Plasma half-life increases in the elderly with amlodipine, diltiazem, felodipine and verapamil. These agents should not be discontinued abruptly, as severe rebound angina attacks may result (gradual drug withdrawal is recommended).
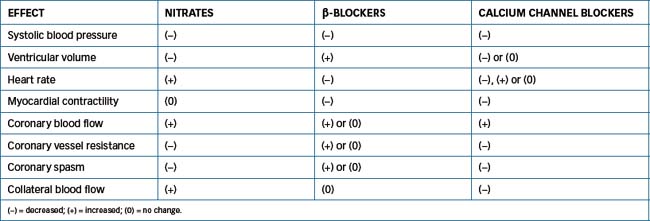
All the antianginal drugs discussed in preceding sections (nitrates, calcium channel blockers and β-blockers [Chapter 12]) provide symptomatic relief in persons with angina. These drugs may be used alone or in combination because they improve the balance between myocardial oxygen supply and demand. A comparison of the effects of these drugs on cardiovascular parameters is shown in Table 23-4.
Potassium channel activators
Potassium channel activators include nicorandil, which may be used as an alternative to long-acting nitrates to reduce the frequency of anginal attacks, and diazoxide and minoxidil, which have limited use in treating hypertension. These drugs relax smooth muscle by acting on ATP-sensitive potassium channels. Normally, intracellular ATP closes the channel, causing the smooth muscle cells to depolarise. Drugs that activate the potassium channel antagonise the action of ATP, preventing closure of the channel. This results in hyperpolarisation and relaxation of the vascular smooth muscle.
Nicorandil
In addition to its action as an activator of potassium channels, which leads to arterial vasodilation and a reduction in afterload, nicorandil relaxes the venous vascular system. This is due to an increase in cGMP brought about by the nitrate moiety of the drug (a similar effect to that of glyceryl trinitrate). Nicorandil is used in chronic stable angina because of evidence that it exerts a direct effect on normal and stenotic coronary arteries.
After oral administration nicorandil is absorbed rapidly, with bioavailability in the order of 75%, indicating no extensive first-pass metabolism. Maximal plasma concentration is reached in 30–60 minutes and the drug has a rapid elimination phase, with a half-life of about 60 minutes. Nicorandil is metabolised by denitration, with the metabolites excreted in the urine within 24 hours.
Common adverse reactions include nausea, flushing, headache, dizziness, palpitations and myalgia. At high doses, hypotension may be problematic. Precaution should be exercised in persons with severe hepatic impairment, as lower doses may be required. Nicorandil is contraindicated in individuals with hypotension or left ventricular failure.
The initial dose is 5 mg orally twice daily (less for patients prone to headache or adverse reactions), increasing after 1 week to 10–20 mg twice daily.
Diazoxide
The antihypertensive action of diazoxide results from potassium channel activation and hence relaxation of smooth muscles in the peripheral arterioles, which causes a decrease in peripheral resistance. As blood pressure falls, a reflex increase in heart rate and cardiac output occurs, with resultant maintenance of coronary and cerebral blood flow. This cardiovascular reflex mechanism also inhibits the development of orthostatic hypotension. Concurrent use with other antihypertensives or peripheral vasodilators may result in additive effects.
Diazoxide is administered intravenously to reduce blood pressure promptly in hypertensive emergencies such as malignant hypertension and hypertensive crisis. Intravenous diazoxide is ineffective in reducing elevated blood pressure in persons with MAO-induced hypertension or phaeochromocytoma. Because of its adverse effects, the drug is not used orally for treatment of chronic hypertension. Administered intravenously (intermittently or by infusion) the onset of action is 1 minute, the peak effect occurs within 2–5 minutes, the half-life is about 28 hours, and the duration of effect is 2–12 hours. Diazoxide is metabolised by the liver and excreted by the kidneys.
Adverse reactions include hyperglycaemia, tachycardia, anorexia, headache, flushing, dizziness, constipation, abdominal cramps, changes in taste perception and oedema (sodium and water retention). With rapid IV injection severe adverse reactions including angina, bradycardia, hypotension, cerebral ischaemia and confusion may occur. Use diazoxide with caution in persons with gout, diabetes and heart failure. Avoid use in persons with diazoxide hypersensitivity, coronary or cerebral insufficiency or aortic dissection.
The adult dose is 1–3 mg/kg up to a maximum of 150 mg IV, repeated after 5–15 minutes if necessary. Intravenous infusion at a rate of 7.5–30 mg/min is the preferred route and dosage, to minimise the risk of a precipitous fall in blood pressure.
Capsules of diazoxide are available through the Special Access Scheme for the treatment of intractable hypoglycaemia. The oral route is not normally used for the treatment of hypertension because of poor tolerance of oral diazoxide.
Minoxidil
Minoxidil is an orally effective direct-acting peripheral vasodilator. It reduces blood pressure by decreasing peripheral vascular resistance in the arteriolar vessels, with little effect on veins. It does not cause orthostatic hypotension. It is a potent vasodilator and also causes a reflex increase in cardiac output, induces sodium retention, promotes development of oedema, and increases plasma renin activity. Minoxidil is reserved for severe hypertension unresponsive to traditional agents, i.e. severe hypertension associated with chronic renal failure. Concomitant administration of a β-adrenergic-blocking agent such as propranolol is necessary to prevent severe reflex tachycardia. Administration of a diuretic agent is also essential to counteract sodium and water retention. Its more commonly recognised topical use is for male-pattern baldness in both men and women.
The onset of action of minoxidil is 30 minutes, with the peak effect occurring in 2–3 hours (after a single dose). Although the plasma half-life is about 4 hours, the duration of its hypotensive action may exceed 24 hours. With daily administration steady-state plasma concentration is achieved after 3–7 days. It is metabolised by the liver and excreted by the kidneys.
Adverse reactions occur with oral dosing and include: nausea; vomiting; tachycardia; anorexia; headaches; exces sive hair growth (hypertrichosis), usually on face, arms and back; red flushing of skin; oedema; angina and peri carditis. Use with caution in persons with heart failure, angina, phaeochromocytoma, cerebrovascular disease and postmyocardial infarction. Avoid use in persons with minoxidil hypersensitivity or pulmonary hypertension secondary to mitral stenosis, and in pregnant or lactating women.
For children 12 years and over and adults, the initial dose is 5 mg orally daily as a single dose. For adults this can be increased every 3 days by 5–10 mg until a maintenance dose of 10–40 mg daily in one or two divided doses is achieved. For children up to 12 years of age the dose is 0.2 mg/kg (maximum 5 mg/day). The maximal daily dose for adults is 100 mg, and for children 50 mg.
Miscellaneous vasodilators
The drugs discussed below include hydralazine and sodium nitroprusside. Both agents are used during hypertensive emergencies because they are rapid directacting vasodilators that produce an immediate fall in arterial pressure.
Hydralazine
Hydralazine hydrochloride is believed to produce its hypotensive effects by direct relaxation of vascular smooth muscle, particularly the arterioles, with little effect on veins, leading to reduction in peripheral resistance. Consequently, renal blood flow is increased, providing an advantage to patients with renal failure. Hydralazine also maintains cerebral blood flow and causes sodium and water retention. The resulting hypotension is thought to stimulate the baroreceptor reflex, causing an increase in heart rate and cardiac output. Unfortunately, this response offsets the antihypertensive effects of the drug. Tolerance to the antihypertensive action may be counteracted by combination with other antihypertensive drugs. Hydralazine also increases plasma renin activity. It is used for the treatment of hypertensive emergencies and in combination with a β-blocker and a diuretic in persons with hypertension refractory to other drugs.
An oral dose of hydralazine has an onset of action of 45 minutes; with IV administration the onset is within 10–20 minutes. The peak effect is within 1 hour (orally) or 15–30 minutes (IV). The plasma half-life is 3–7 hours and duration of action is 3–8 hours. It is metabolised principally by acetylation in the liver, with the metabolites excreted by the kidneys. Identification of a ‘slow-acetylator’ phenotype (see Chapter 7) in both Caucasians (about 50%) and Asians (about 20%), which results in significant increases in the plasma concentration of the drug and hence the risk of toxicity, has limited the usefulness of this drug. Concurrent drug administration with monoamine oxidase inhibitors or other antihypertensives may result in severe hypotension.
Adverse reactions include diarrhoea, nausea, vomiting, tachycardia, anorexia, headache, facial flushing, stuffy nose, oedema, angina, rash, peripheral neuritis and a systemic lupus erythematosus (SLE)-like syndrome. The SLE-like syndrome may include myalgia, arthralgia, arthritis, weakness, fever and skin changes. Use with caution in patients with angina, cerebral artery disease and renal and hepatic impairment. The drug is contraindicated in persons with hydralazine hypersensitivity, aortic dissection, severe tachycardia and heart failure, and SLE.
The adult oral dose is 25 mg twice daily increasing as required to a maintenance dose of 50–200 mg daily. Doses above 100 mg daily increase the risk of the SLE-type adverse reaction, and the acetylator phenotype should be determined in advance. Parenterally, the adult dose is 5–10 mg IV slowly over 20 minutes, repeated if necessary.
Sodium nitroprusside
Sodium nitroprusside (nitroferricyanide) is a potent and rapid direct-acting vasodilator agent that greatly reduces arterial blood pressure. It relaxes both arterial and venous smooth muscles. The decrease in systemic resistance causes a reduction in preload and afterload, improving cardiac output. It is indicated for rapid reduction of blood pressure in hypertensive emergencies and for controlled hypotension during surgery.
Sodium nitroprusside is useful only for short-term treatment, as it must be given intravenously. Its onset of action and peak effect occur almost immediately (within minutes) after administration by IV infusion. The half-life of nitroprusside is 2 minutes; the half-life of thiocyanate, a toxic metabolite, is 3 days. The duration of effect is 1–10 minutes after discontinuation of the infusion. Metabolism is by erythrocytes (to cyanide) and the liver (cyanide to thiocyanate). The drug is excreted by the kidneys.
The hypotensive effect of sodium nitroprusside is exacerbated by other antihypertensive drugs, volatile anaesthetics and other negative inotropes. Adverse reactions include dizziness, excessive sweating, headaches, anxiety, abdominal cramps, tachycardia, hypothyroidism, flushing, rash and muscle twitching. Ataxia, blurred vision, headache, nausea, vomiting, tinnitus, shortness of breath, delirium and unconsciousness may occur with thiocyanate toxicity. Hypotension, metabolic acidosis, pink colouration, very shallow breathing pattern, decreased reflexes, coma and widely dilated pupils may be observed with cyanide toxicity. Use with caution in persons with hypothyroidism, hypothermia or lung disease. Avoid use in persons with nitroprusside hypersensitivity, cerebrovascular or coronary artery disease, liver disease, kidney disease or metabolic-induced vitamin B12 deficiency.
For hypertensive emergency, the initial dose is 0.3 mcg/kg/min, which is slowly increased as necessary. The maximum dose is 10 mcg/kg/min.
Solution should be freshly prepared using 5% glucose intravenous infusion (no other solution should be used) and protected from light by wrapping the container in the supplied opaque sleeve, aluminium foil or other opaque material. The prepared solution should be discarded within a 24-hour period. A freshly prepared solution has a faint brown tinge; discard if it is highly coloured (e.g. blue, green or dark red).
Management of sodium nitroprusside overdose
Nitroprusside contains five cyanide groups. When the drug is administered IV, one molecule of nitroprusside reacts with one molecule of haemoglobin to form one molecule of cyanmethaemoglobin, four cyanide ions and nitric oxide, the active substance. Nitric oxide then activates the enzyme guanylate cyclase to produce cGMP, and vasodilation. The free cyanide ions react with thiosulfates, catalysed by the mitochondrial enzyme rhodanase, forming the final metabolite thiocyanate, which is excreted by the kidneys. Processing of cyanide ions from sodium nitroprusside to thiocyanate can proceed normally at a rate of about 2 mcg/ kg/min. Infusion rates greater than this can lead to accumulation of cyanide ions. In persons with normal renal function the half-life of thiocyanate is 3 days, but this may double or triple in persons with renal failure.
Cyanide toxicity may manifest as tachycardia, sweating, hyperventilation, metabolic acidosis, hypotension, arrhythmias and death. In contrast, thiocyanate toxicity causes nausea, dyspnoea, blurred vision, confusion, psychosis and tinnitus. If nitroprusside overdose is suspected, discontinue sodium nitroprusside and administer sodium nitrite (3% solution) at a dose of 4–6 mg/kg IV over 2–4 minutes. After administration of sodium nitrite, administer sodium thiosulfate (150–200 mg/kg) IV to convert the cyanide to thiocyanate. The nitrite/thiosulfate regimen can be repeated using 2–3 mg/kg sodium nitrite and 75–100 mg/kg sodium thiosulfate after 2 hours (eMIMS 2009).
Thiocyanate is less toxic and rarely a problem, but if thiocyanate toxicity occurs, use haemodialysis. Be aware, however, that haemodialysis does not remove cyanide.
Peripheral vascular disease

Peripheral vascular disease, which results in coolness or numbness of the extremities, intermittent claudication and leg ulcers, is a common problem in the elderly. The primary risk factors include hyperlipidaemia, diabetes, obesity, high blood pressure and smoking. The use of various direct-acting vasodilators for peripheral occlusive arterial disease has generally been very disappointing. Oxpentifylline and hydroxyethylrutosides are used for symptomatic relief but there is a lack of convincing evidence for efficacy of either drug. If no benefit is seen after a short trial of use, the drugs should be stopped (see Drug Monograph 23-2).
Drug monograph 23-2 Oxpentifylline
Oxpentifylline is a xanthine derivative that improves haemorrhagic disorders in the microcirculation, which involves the flow of blood through the fine vessels (arterioles, capillaries and venules). Although the mecha nism of action of oxpentifylline is not completely understood, current evidence shows the drug possesses several properties that improve microcirculatory blood flow to ischaemic tissues. (It improves red blood cell flexibility and reduces blood viscosity by decreasing fibrinogen concentrations and inhibiting aggregation of red blood cells and platelets.) The result is increased microcirculatory blood flow and oxygenation of tissues.
Indications
Oxpentifylline is indicated for the treat ment of intermittent claudication caused by occlusive arterial disease of the limbs.
Pharmacokinetics
Oxpentifylline is administered orally as a controlled-release tablet. The drug is almost completely absorbed and undergoes extensive first-pass metabolism. The plasma half-life of the parent drug ranges from 0.4 to 0.8 hours and for the metabolites from 1 to 1.6 hours. Peak plasma concentration occurs in 2–4 hours and the onset of action with chronic dosing is between 2 and 4 weeks. It is metabolised in the liver and excreted by the kidneys.
Drug interactions
Concurrent use with anti platelet and thrombolytic medications may prolong prothrombin time and bleeding. Use with sympatho mimetics and xanthines may result in an increase in central nervous system stimulation.
Warnings and contraindications
Avoid use in persons with bleeding problems (e.g. peptic ulcers), cerebrovascular or coronary artery disease or acute myocardial infarction. In severe renal impairment dosage reduction may be required.
Indirect-acting vasodilator drugs
The main groups of drugs in this category include centrally acting drugs that inhibit vasoconstriction through mediation by the sympathetic nervous system and inhibitors of the renin–angiotensin–aldosterone system (RAAS).
Centrally acting adrenergic inhibitors
The centrally acting adrenergic inhibitors clonidine, methyldopa and moxonidine are effective antihypertensive drugs, but are not considered first-line drugs because of both their adverse effects profile and lack of clinical evidence of significant cardiovascular benefit.
Clonidine
Clonidine is a centrally acting α2 agonist. It reduces systolic and diastolic blood pressures by stimulating central α2 receptors, which decreases sympathetic outflow from the brain to the blood vessels and heart. Blood pressure is lowered as a result of decreased cardiac output, heart rate and peripheral vascular resistance. The effect on cardiac output is the result of a reduction in both heart rate and stroke volume, which can lead to bradycardia.
The decreased sympathetic outflow to the kidneys reduces renal vascular resistance, preserving renal blood flow. In some persons, renin activity may be suppressed. With continued clonidine use, a diuretic is used to correct fluid retention.
Clonidine is marketed for the treatment of hypertension and menopausal flushing but is also used for the diagnosis of phaeochromocytoma, for attention-deficit hyperactivity disorder and for managing the symptoms of opioid withdrawal. At a low dose (25 mcg), clonidine is also used for migraine or recurrent vascular headache prophylaxis in adults unresponsive to other drug therapies.
Clonidine is well absorbed after oral administration, with bioavailability approaching 100%. Oral clonidine has an onset of action within 0.5–1 hour, a peak effect in 2–4 hours and a duration of action of up to 8 hours. Clonidine is excreted predominantly as unchanged drug in urine (60%), with the remainder excreted as hydroxylated metabolites.
Clonidine is subject to a number of drug interactions including:
Adverse reactions include dry mouth, headaches, constipation, weakness, postural hypotension, impotency or decreased sexual drive, insomnia, anxiety, anorexia, nausea, vomiting and pruritus.
Use with caution in the elderly and in persons with impaired AV node or sinus node function, coronary insufficiency, depression or a history of depression, Raynaud’s syndrome or a recent myocardial infarction. Clonidine is contraindicated in sick sinus syndrome and heart block.
The adult dose for the treatment of hypertension is 0.05–0.1 mg twice daily initially, increased every 2–3 days by 0.1 or 0.2 mg, to a maximum of 0.6 mg daily. For maintenance, the dose is 0.15–0.3 mg daily in two divided doses.
Methyldopa
Although the exact hypotensive mechanism of methyldopa is unknown, the theory is that a metabolite of methyldopa (α-methylnoradrenaline) stimulates the central α2 receptors, which results in a reduction in noradrenaline (sympathetic) outflow to the heart, kidneys and peripheral vasculature.
Methyldopa’s peak effect occurs in 4–6 hours after a single dose or in 48–72 hours with multiple dosing. The duration of action is 12–24 hours (after a single oral dose), 1–2 days (after multiple oral doses) or 10–16 hours (after IV administration). Methyldopa is metabolised centrally to α-methylnoradrenaline within adrenergic nerve endings and in the liver to a sulfate conjugate (30–60%). Excretion is primarily by the kidneys. Methyldopa is often used to treat hypertension in pregnant women but in nongestational hypertension its usefulness is limited by CNS and hepatic adverse effects.
Methyldopa is subject to a number of drug interactions including:
Adverse reactions include drowsiness, dry mouth, headaches, oedema of the feet and legs, fever, postural hypotension, impotency, insomnia, depression, anxiety and nightmares. Use with caution in persons with sulfite sensitivity or kidney impairment. Avoid use in persons with methyldopa hypersensitivity, hepatitis, cirrhosis, haemolytic anaemia or phaeochromocytoma. Note that methyldopa is often the antihypertensive of choice during early pregnancy due to its pregnancy safety rating of A (most newer drugs have a C or D rating).
The initial adult oral dose is 125–250 mg twice daily for 2 days, adjusted by 250–500 mg daily at 2-day intervals as necessary. The maintenance dose is 250–2000 mg/day, divided into 2–4 individual doses.
Moxonidine
Unlike clonidine and methyldopa moxonidine has minimal actions on α2 adrenoceptors. Moxonidine acts on central imidazoline I1 receptors present in the rostral ventral, medulla decreasing sympathetic tone. Adrenaline, noradrenaline and renin levels fall resulting in a decrease in blood pressure and systemic vascular resistance. Heart rate, cardiac output and stroke volume are unaffected. The drug is well absorbed and bioavailability is ∼90%. Moxonidine is largely excreted as unchanged drug (∼60%) and the half-life is ∼2.5 hours. Renal dysfunction decreases clearance and increases half-life. A large clinical trial (MOXCON) of a controlled release formulation was terminated early because of evidence of increased mortality and morbidity, questioning the safety of central sympathetic inhibition in heart failure patients (Cohn et al 2003). Hence moxonidine is contraindicated in heart failure and may exacerbate symptoms of coronary heart disease. Adverse effects are fewer in comparison with clonidine and include somnolence, weakness, dizziness, hypotension and bradycardia. Experience with this drug is limited. The initial dose is 0.2 mg once daily, increasing to 0.4 mg (as two divided doses after 2 weeks) to a maximum of 0.6 mg daily as two divided doses.
Management of hypertension
Non-pharmacological measures are the first-line approach for managing hypertension (Clinical Interest Box 23-3) and include weight and alcohol reduction, smoking cessation, limiting dietary sodium intake and embarking on a program of regular physical activity of at least 30 minutes of moderate-intensity exercise daily on most days of the week. When instituting drug therapy the lowest dose of the chosen drug is used, adding a second drug from a different drug class if necessary. The overriding goal of drug therapy is to lower the blood pressure, with minimal adverse effects. Long-acting drugs that allow once-daily dosing are preferable, as they aid adherence.
Clinical interest Box 23-3 Hypertension
Hypertension is defined as an elevated systolic blood pressure, diastolic blood pressure or both. In clinical practice elevated systolic blood pressure is a greater predictor of cardiovascular risk than elevated diastolic pressure. Worldwide definitions of hypertension vary and a suggested classification has been developed following a review of systems in the USA and Europe (National Heart Foundation of Australia 2008).
| Category | Systolic (mmHg) | Diastolic (mmHg) |
| Normal | <120 | <80 |
| High–normal | 120−139 | 80−89 |
| Grade 1 (mild hypertension) | 140−159 | 90−99 |
| Grade 2 (moderate hypertension) | 160−179 | 100−109 |
| Grade 3 (severe hypertension) | ≥180 | ≥110 |
| Isolated systolic hypertension | ≥140 | <90 |
This classification has also stratified people based on blood pressure levels, the presence of risk factors and the degree of targetorgan damage secondary to hypertension. The major risk factors in hypertensive patients include cigarette smoking, diabetes mellitus, raised total or LDL cholesterol or reduced HDL cholesterol, age (>55 years male, >65 years female), family history of heart disease, male gender (increased risk at any age compared to females), obesity, excessive alcohol intake and a sedentary lifestyle. Psychosocial risk factors include depression, social isolation and lack of quality support. Those populations most at risk include people of Aboriginal, Torres Strait Islander, Maori or Pacific Islander origin and those in lower socioeconomic groups. The targetorgan damage or cardiovascular disease in hypertensive patients includes stroke or transient ischaemic attacks (TIA), kidney disease, retinopathy and various cardiac diseases such as angina, heart failure, left ventricular hypertrophy and prior myocardial infarction.
‘Hypertension should be managed within a comprehensive management plan to reduce BP, reduce overall cardiovascular risk and minimise end-organ damage’ (National Heart Foundation of Australia 2008).
Effective monotherapy regimens include:
Effective drug combinations include:
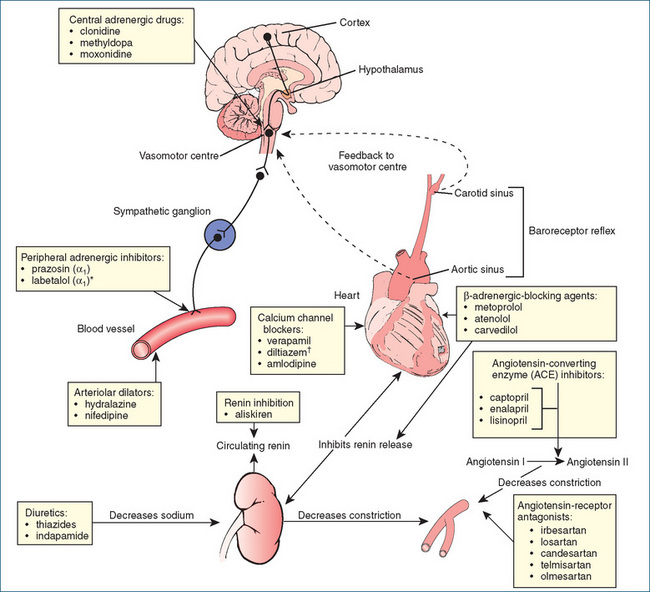
Figure 23-3 summarises the physiological factors controlling blood pressure (the sympathetic nervous system and the RAAS) and indicates the sites of action of currently used oral antihypertensive drugs.
The renin–angiotensin–aldosterone (RAAS) system
The kidneys are by far the most important organs in the body for long-term regulation of blood pressure and, normally, excessive fluid retention is controlled by negative feedback mechanisms that operate to restore normal fluid and electrolyte balance. The RAAS regulates blood pressure by increasing or decreasing blood volume through modulation of renal function (Figure 23-4). Abnormal activation of the RAAS plays a key role in the development and pathophysiology of hypertension and cardiovascular disease and correlates directly with the incidence and extent of end-organ damage. The major effector molecules of the RAAS are renin, angiotensin II and the mineralocorticoid aldosterone. Additionally, it is now recognised that aldosterone is a proinflammatory molecule that plays a major role in the progression of ischaemic heart disease and that a raised aldosterone-to-renin ratio is common in patients with hypertension.
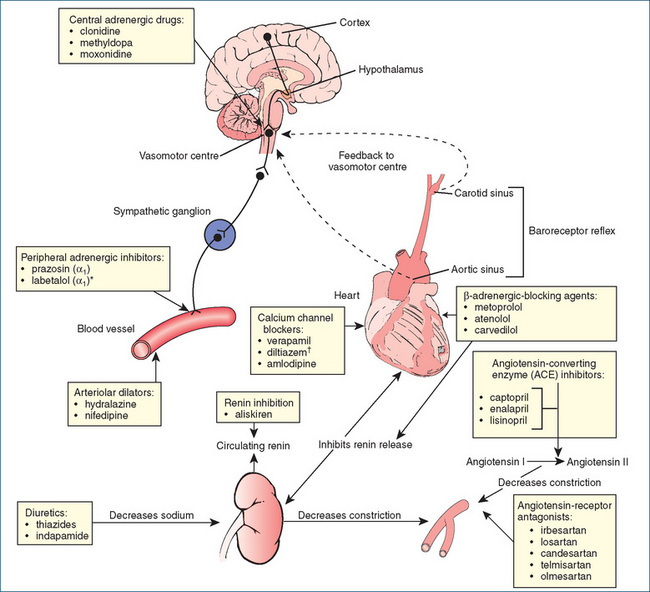
Renin is an enzyme secreted from the juxtaglomerular cells located in the afferent arteriolar walls of the nephron. When blood flow through the kidneys is reduced, renal arterial pressure is reduced, which causes the release of renin into the circulation. Renin catalyses the cleavage of angiotensinogen (a plasma globulin) to form angiotensin I (a weak vasoconstrictor). Subsequently, in endothelial cells (primarily in the lung), angiotensin I is converted by angiotensin-converting enzyme (ACE) to angiotensin II.
Angiotensin II, acting via AT1 receptors, is one of the most potent vasoconstrictors known. It is particularly effective in constricting arterioles, which increases peripheral resistance and raises blood pressure. In addition, angiotensin II acts on the adrenal cortex to stimulate the secretion of aldosterone, which promotes reabsorption of sodium by the kidneys and the excretion of potassium. The increased sodium elevates the osmotic pressure in the plasma, causing a release of antidiuretic hormone from the hypothalamus, leading to increased reabsorption of water from the renal tubules, which adds further to the rise in blood pressure. Angiotensin II itself also acts on the kidney tubules to promote reabsorption of water. Furthermore, activation of AT1 receptors produces adverse effects on the cardiovascular system, including the promotion of vascular and cardiac hypertrophy, inflam mation and fibrosis.
In addition to vascular effects angiotensin II reduces insulin sensitivity and impairs insulin secretion. Not surprisingly there is a close association between the metabolic syndrome, the risk of type II diabetes and the RAAS system, as blockade of the RAAS results in an antidiabetogenic effect improving measures of diabetic nephropathy.
Inhibition of the RAAS is now a common therapeutic strategy used to intervene in the pathogenesis of cardiovascular and renal dysfunction. The key drug groups are those that:
Renin inhibitors
Development of an inhibitor of renin, the rate-limiting step in the activation of the RAAS has been the goal of a number of pharmaceutical companies for years. Aliskiren is the first of the orally effective direct renin inhibitors and was registered in Australia in April 2009. Renin cleaves angiotensinogen to angiotensin I and this is inhibited by aliskiren, which binds to the active site of renin thereby blocking its action. The plasma concentration of renin increases but its actions are suppressed, resulting in a reduction in activity and hence a consequential reduction in angiotensin II formation. In clinical trials aliskiren was as efficacious as an ARB in patients with mild–moderate hypertension. Its potential place as first-line antihypertensive therapy remains to be established.
Aliskiren is poorly absorbed and has a bioavailability of 2.6%. Peak plasma concentration is reached within 1–3 hours, the half-life is in the order of 24–40 hours and steady state is achieved in approximately a week. Although a substrate for CYP3A4, hepatic metabolism is minimal and aliskiren is primarily eliminated in bile as the parent drug. Less than 1% of the orally administered drug is excreted in urine. The most common adverse effects are fatigue, dizziness, diarrhoea, nasopharyngitis and back pain (Sanoski 2009).
Angiotensin-converting enzyme (ACE) inhibitors
ACE inhibitors competitively block the angiotensinconverting enzyme necessary for the conversion of angiotensin I to angiotensin II. Angiotensin II is a powerful vasoconstrictor that raises blood pressure and also causes aldosterone release, resulting in sodium and water retention (Figure 23-4). Inhibition of ACE results in:
Large clinical studies (HOPE, EUROPA, PEACE, QUIET) have now established that ACE inhibitors reduce cardiovascular mortality and morbidity in patients with coronary artery disease and reduce the incidence of stroke and the need for revascularisation procedures compared to placebo. Further studies (IDNT, DETAIL) have demonstrated the beneficial effects of ACE inhibitors in delaying progression of renal impairment in patients with early diabetic nephropathy. The ELITE and ValHeFT studies showed a reduction in cardiovascular mortality and morbidity in patients with congestive heart failure.
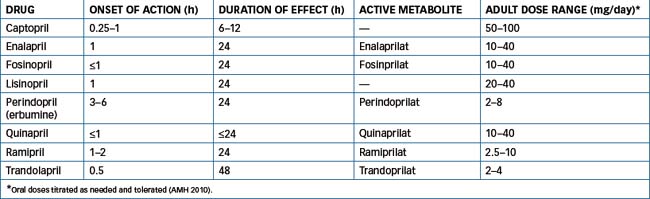
Captopril is the prototype drug of this class, which now also includes enalapril, fosinopril, lisinopril, perindopril, quinapril, ramipril and trandolapril.
Indications
ACE inhibitors are indicated for the treatment of hypertension, heart failure, diabetic nephropathy (type 1 diabetes) and left ventricular dysfunction, and after myocardial infarction. All drugs of this class demonstrate similar antihypertensive efficacies, and adverse reactions do not differ significantly among the individual ACE inhibitors. With the exception of captopril, most of these drugs maintain an antihypertensive effect for up to 24 hours, allowing once-daily dosing. Some of these drugs are available in combination with either the diuretic hydrochlorothiazide (enalapril, fosinopril, quinapril) or indapamide (perindopril) or with a calcium channel blocker (enalapril, ramipril, trandolapril).
Pharmacokinetics
(Refer to Table 23-5.) With the exception of captopril and lisinopril the remainder of the ACE inhibitors are esterprodrugs that following cleavage by esterases form the active metabolite (listed in Table 23-5).
Drug interactions
A consequence of ACE inhibition is potassium retention, which may lead to hyperkalaemia. Combination of ACE inhibitors with other drugs that cause potassium retention should be avoided or the plasma potassium concentra tion should be monitored closely. Other drug interactions include:
Clinical interest Box 23-4 The triple ‘whammy’
The ‘triple whammy’, which described the adverse renal effects of a combination of an ACE inhibitor/angiotensinreceptor antagonist, diuretics and NSAIDs, was first reported in 2000 (Thomas 2000). The mechanism of the nephrotoxic ‘triple whammy’ is complex. The first thing to appreciate is that in healthy volume-replete individuals prostaglandins play a very limited (if any) role in the regulation of renal haemodynamics. However, in elderly patients or in those with heart failure, chronic renal failure, renal artery stenosis or volume depletion (from diuretic use) plasma volume is decreased. This results in activation of the RAAS, increasing the concentration of angiotensin II, which controls efferent arteriolar tone. Consequently, in those individuals renal prostaglandins then play an important role in maintaining renal vasodilation and hence renal blood flow.
NSAIDs inhibit the production of vasodilatory prostaglandins and interfere with the secretion of frusemide in the proximal tubule, thereby impairing frusemide-induced natriuresis and increased renal blood flow. In addition, both ACE inhibitors and NSAIDs lower glomerular hydrostatic pressure and hence reduce the glomerular filtration rate. These three drugs in combination are truly a ‘triple whammy’ and adverse renal outcomes of this combination constituted a significant number of the reports to ADRAC in 2002. Of the 129 reports to ADRAC of acute renal failure (predominantly in elderly patients, median age 76 years), 28 implicated the ‘triple whammy’ combination. Of the ADRAC cases of renal failure with the ‘triple whammy’ a fatal outcome was reported in 10% of the cases.
Adverse reactions
These include headaches, diarrhoea, loss of taste, weakness, nausea, dizziness, hypotension, rash, fever and joint pain. The cough associated with ACE inhibitor use occurs in a significant number of people and is thought to be due to the inhibition by the ACE inhibitor of the enzyme that degrades bradykinin (see Clinical Interest Box 23-5).
Clinical interest Box 23-5 Ace inhibitor cough
A well-known adverse effect of ACE inhibitors is a dry persistent cough that has been reported to occur in 5%–35% of individuals prescribed ACE inhibitors. Interestingly, this cough occurs more often in women, non-smokers and Chinese persons. The cough is not dose-dependent, can occur within hours of the first dose, may take weeks to months to develop and normally resolves within 1–4 weeks of cessation of therapy. In a small number of individuals the cough may persist for 3 months after stopping the ACE inhibitor.
In general, it has been considered that the cough occurs as a consequence of inhibition of ACE, which is normally responsible for the metabolism of bradykinin (a vasodilator peptide) to inactive fragments. Inhibition of ACE results in accumulation of bradykinin and substance P, protussive mediators that sensitise airway nerves that produce a cough as a result of a tickling or scratching sensation in the throat (Dicpinigaitis 2006).
Warnings and contraindications
Use with caution in persons with SLE, scleroderma, bone marrow depression, cerebrovascular insufficiency, coronary insufficiency, type 1 diabetes or liver function impairment (the last of these warnings is for all ACE inhibitors except for lisinopril and quinapril). Avoid use in persons with ACE inhibitor hypersensitivity, angiooedema or hyperkalaemia and in renal artery stenosis, transplant or impairment. ACE inhibitors should be avoided in pregnancy because of their potential to produce a range of abnormalities.
Angiotensin-receptor (AT1) antagonists (ARBs)
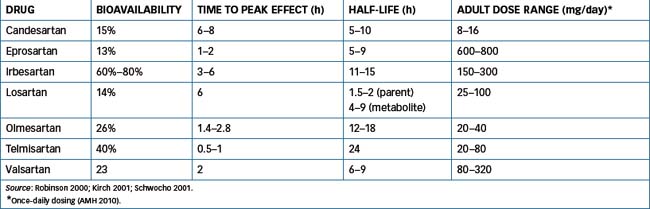
Although ACE inhibition clearly has beneficial effects in patients with hypertension, ischaemic heart disease and congestive heart failure, approximately 10% of patients have adverse effects that limit the usefulness of ACE inhibitors. This led scientists to investigate the potential of drugs to inhibit angiotensin II. After the discovery of two subtypes of angiotensin II receptors, it was found that stimulation of the type 1 receptor (AT1) mediated all the actions of angiotensin II. This meant a more precise target was available for blocking the vasoconstrictor effects of angiotensin II, rather than the broader effects (and possibly adverse effects) resulting from inhibition of ACE. The AT1 receptor quickly became the target for the development of new antihypertensive drugs, the non-peptide angiotensin receptor (AT1) antagonists (blockers). These agents, commonly called ‘sartans’, include candesartan, eprosartan, irbesartan, losartan, olmesartan, valsartan and telmisartan (see Table 23-6). They block the AT1 receptor thus inhibiting angiotensin II-mediated vasoconstriction and aldosterone release but they have very little effect on plasma potassium concentration. In addition, they do not inhibit the breakdown of the cough-producing bradykinin. Losartan was the first AT1 receptor antagonist synthesised and is considered the key drug in this category (see Drug Monograph 23-3). With the exception of losartan the ARBs are available as combination products with hydrochlorothiazide while valsartan is also available as a combination product with amlodipine.
Table 23-6 Pharmacokinetics and adult dosing of angiotensin receptor (AT1) antagonists in hypertension
Losartan is an orally active non-peptide ARB with 10,000-fold greater selectivity for AT1 receptors than AT2 receptors.
Pharmacokinetics
(Table 23-6) Losartan is completely absorbed following oral administration, with peak concentration occurring within 1 hour, while that of the active carboxylic acid metabolite occurs in 3–4 hours. The drug undergoes substantial first-pass metabolism in the liver and is metabolised by hepatic CYP2C9 and CYP3A4 (see Chapter 6) to at least six metabolites. The active carboxylic acid metabolite is 10–40 times more potent than the parent drug. The half-life of losartan is 2 hours and, for the carboxylic acid metabolite, between 6 and 9 hours. The duration of action is at least 24 hours, with metabolism in the liver and excretion in bile (around 60%) and urine (35%).
Drug interactions
When losartan is adminis tered with a diuretic, potentiation of the hypotensive effect may result. Usually, a lower losartan dose is necessary, with close monitoring. Rifampicin induces the activity of both CYP2C9 and CYP3A4 and hence metabolism of losartan may be increased, resulting in decreased plasma concentration and loss of BP control. Dose increase may be necessary. Other drug interactions are similar to those of the ACE inhibitors, e.g. NSAIDs, thiazide and loop diuretics.
Adverse reactions
These include hyperkalaemia (common in the presence of renal disease), headache, tiredness, back or muscle pain, diarrhoea, nasal congestion and dizziness. Decreases in haemoglobin and haematocrit may occur and this has also been observed with the other sartans. Dry cough and insomnia are considered rare effects.
Warnings and contraindications
Use losartan with caution in persons with kidney impairment. Use extreme caution if administering losartan to volume- or sodium-depleted individuals, as severe hypotension may result. Correct the deficiency first or start with a smaller drug dose. Avoid use in persons with losartan hypersensitivity, liver impairment or stenosis of the renal arteries, and in women contemplating pregnancy. A lower dosage may be required in persons with hepatic impairment.
Indications
ARBs are indicated for the treatment of hypertension and/ or heart failure in patients unable to tolerate ACE inhibitors. These drugs reverse endothelial dysfunction and atherosclerosis and reduce the risk of cardiovascular events. Additionally, ARBs reduce end-organ damage of the kidney, brain and heart and decrease mortality of patients with congestive heart failure.
Pharmacokinetics
(Refer to Table 23-6.) Some of the ARBs are metabolised to varying degrees by CYPs and UGTs and the resulting metabolites in general (with the exception of losartan and the prodrug candesartan cilexetil) have a lower affinity for the AT1 receptor. Valsartan and candesartan are minimally metabolised and are excreted primarily as unchanged drug. In contrast irbesartan is extensively metabolised with <2% excreted in urine as unchanged drug. The ratio of hepatic to renal clearance varies for each individual sartan, e.g. 35%–50% of olmesartan is excreted as unchanged drug in urine. In general the sartans are well absorbed and bioavailability varies from 13% (eprosartan) to 60%–80% (irbesartan).
Drug interactions
Similar to the ACE inhibitors, drug interactions with the sartans include those drugs that cause potassium retention, and the combination should be avoided or the plasma potassium concentration monitored closely. Additional interactions occur with loop diuretics, NSAIDs and lithium (refer to the information with ACE inhibitors).
Adverse reactions
As expected adverse reactions include hyperkalaemia, dizziness and headache (refer to Drug Monograph 23-3 for further information).
Aldosterone receptor antagonists
In addition to a role in sodium and water homeostasis aldosterone contributes to the progression of cardiovascular and renal disease by increasing magnesium and potassium excretion, impairing endothelial and baroreceptor function, reducing vascular compliance and promoting myocardial fibrosis. Aldosterone binds to mineralocorticoid receptors (see Chapter 35) that upregulate the absorption of sodium ions and water through epithelial sodium channels and potassium excretion by epithelial cells in the distal nephron. Spironolactone, the prototypical aldosterone receptor antagonist, was launched in 1960 as a potassiumsparing diuretic (refer to Chapter 25 for pharmacokinetic details) for the treatment of primary aldosteronism, oedematous conditions, essential hypertension and hypokalaemia. However, further advances in our understanding of the RAAS led to the development of ACE inhibitors and ARBs that were devoid of the adverse effects of spironolactone.
Structurally spironolactone is similar to progesterone and non-selectively binds to mineralocorticoid receptors and to progesterone and androgen receptors. Use of spironolactone for extended periods of time results in profound hyperkalaemia (resulting from antagonism of aldosterone) and major endocrine effects including loss of libido, menstrual irregularities, gynaecomastia and impotence. This non-selectivity limits the use of spironolactone to primary hyperaldosteronism, refractory oedema and hirsutism in females. However, in recognising the deleterious actions of aldosterone on the CV system, this led to a trial of spironolactone in patients with severe heart failure, RALES (Randomised Aldactone Evaluation Study), in 1999. Patients randomised to receive spironolactone had a 35% reduction in hospitalisation and a 30% reduction in mortality. However, hyperkalaemia and progestogenic and antiandrogenic adverse effects limit its usefulness in this setting.
Further investigations led to the development of eplerenone, which is a competitive antagonist with greater mineralocorticoid receptor selectivity and reduced progestogenic and antiandrogenic actions. In 2005 the Eplerenone Post-Acute Myocardial Infarction Heart Failure Efficacy and Survival Study (EPHESUS) showed that eplerenone reduced all-cause mortality by 15% in patients post-myocardial infarction and with clinical signs of heart failure, which is now its indication for use (see Drug Monograph 23-4).
Drug monograph 23-4 eplerenone
Eplerenone is approved for the treatment of heart failure and left ventricular impairment post-myocardial infarction. Due to its mineralocorticoid receptor selectivity, eplerenone is expected to provide clinical benefits exceeding those of spironolactone
Pharmacokinetics
The absolute oral bioavailability of eplerenone is unknown but absorption is not affected by food. Peak plasma concentration occurs within 1.5 hours post-dose. Eplerenone is extensively metabolised by CYP3A4 to the major metabolites 6 β-hydroxy eplerenone (32%), 6 β, 21-hydroxy eplerenone (21%) and 21-hydroxy eplerenone (8%). Approximately 67% of the dose is excreted in urine and 32% in faeces predominantly as metabolites (<2.5% is excreted as unchanged drug). The plasma half-life is ∼3 hours.
Drug interactions
The aldosterone antagonists cause hyperkalaemia, particularly in the presence of renal impairment or when coadministered with other drugs that cause potassium retention or with potassium supplements. The plasma concentration of eplerenone is increased and the risk of hyperkalaemia exacerbated when administered with amiodarone, diltiazem, erythromycin, fluconazole, ketoconazole, saquinavir or verapamil. Combination with NSAIDs increases the risk of hyperkalaemia and worsening of renal function.
Adverse reactions
Common adverse effects include hyperkalaemia, hypotension, dizziness and renal dysfunction.
Warnings and contraindications
Eplerenone is contraindicated in existing situations of hyperkalaemia (potassium >5.5 mmol/L), diabetes and/or proteinuria and severe hepatic impairment. Plasma potassium concentration should be checked regularly and the eplerenone dose reduced or ceased if hyperkalaemia occurs. Data are limited and current drug information sources should be consulted.
Key points


 Vasodilator drugs are used for the treatment of a range of disorders, including hypertension, angina, shock, cardiac failure and peripheral vascular conditions. Vasodilator drugs work by relaxing smooth muscle in the blood vessel walls. Some drugs act primarily on veins or arterioles, while others dilate both types of blood vessels. The main groups of direct-acting vasodilator drugs are the organic nitrates, calcium channel blockers and potassium channel activators. The indirect-acting vasodilators include the centrally acting adrenergic inhibitors, renin inhibitors, ACE inhibitors and angiotensin receptor (AT1) antagonists. The organic nitrates produce vasodilation through the production of nitric oxide and an increase in cGMP. At low doses, glyceryl trinitrate causes venodilation, reducing preload and stroke volume. Higher doses dilate arteries and the overall effect is a reduction in myocardial work and oxygen demand, hence their beneficial effect in angina. Calcium channel-blocking drugs block the inward movement of calcium ions through the slow channels of the cell membrane of cardiac and vascular smooth muscle cells. These drugs decrease the force of myocardial contraction (negative inotropic effect), decrease automaticity in the SA node and decrease conduction in the AV junction (negative chronotropic and negative dromotropic effects, respectively), and inhibit calcium ion influx in smooth muscle cells (reduction in peripheral vascular resistance). Not all calcium channel blockers have equivalent therapeutic effects. Nifedipine is a potent peripheral vasodilator with minimal cardiac effects; therefore it is an effective antihypertensive agent. Nimodipine is indicated for the treatment of subarachnoid haemorrhage while the other calcium channel-blocking agents are used for their cardiovascular effects (antianginal, anti-arrhythmic and antihypertensive). Drugs such as diazoxide, nicorandil and minoxidil relax smooth muscle by activating ATP-dependent potassium channels. They activate the potassium channel, antagonising the action of ATP and preventing closure of the channel. This results in hyper-polarisation and relaxation of the vascular smooth muscle. Diazoxide and minoxidil have limited use in the treatment of hypertension, while nicorandil is used for the treatment of angina. The main indirect-acting vasodilator drugs are the centrally acting drugs that inhibit sympathetic outflow and inhibitors of the renin–angiotensin–aldosterone system. The centrally acting agents, clonidine and methyldopa, are effective antihypertensives, especially when combined with a diuretic. When given as the sole agent, clonidine and methyldopa usually cause sodium and water retention. Aliskiren is the first of the orally effective direct renin inhibitors. Renin cleaves angiotensinogen to angiotensin I and this is inhibited by aliskiren, which binds to the active site of renin, thereby blocking its action. The plasma concentration of renin increases but its actions are suppressed resulting in a reduction in activity and hence a consequential reduction in angiotensin II formation. ACE inhibitors such as captopril competitively block the angiotensin-converting enzyme necessary for the conversion of angiotensin I to angiotensin II. Angiotensin II is a powerful vasoconstrictor that raises blood pressure and also causes aldosterone release, resulting in sodium and water retention. ACE inhibitors are indicated for the treatment of hypertension, heart failure, diabetic nephropathy, left ventricular dysfunction and after myocardial infarction. All drugs of this class demonstrate similar antihypertensive efficacy, and adverse reactions do not differ significantly among the individual ACE inhibitors. Angiotensin receptor (AT1) antagonists inhibit the action of angiotensin II, thereby preventing vasoconstriction, and are indicated for the treatment of hypertension. In addition to a role in sodium and water homeostasis, aldosterone contributes to the progression of cardiovascular and renal disease. Spironolactone, the prototypical aldosterone receptor antagonist, was launched in 1960 as a potassiumsparing diuretic for the treatment of primary aldosteronism, oedematous conditions, essential hypertension and hypokalaemia. Although beneficial in a setting of heart failure, hyperkalaemia and progestogenic and antiandrogenic adverse effects limit spironolactone’s usefulness. Further investigations led to the development of eplerenone, which is a competitive aldosterone antagonist with greater mineralocorticoid receptor selectivity and reduced progestogenic and antiandrogenic actions. Five drug classes are used to treat hypertension. These include diuretics, β-blockers, ACE inhibitors, angiotensin receptor (AT1) antagonists and calcium channel blockers.
Vasodilator drugs are used for the treatment of a range of disorders, including hypertension, angina, shock, cardiac failure and peripheral vascular conditions. Vasodilator drugs work by relaxing smooth muscle in the blood vessel walls. Some drugs act primarily on veins or arterioles, while others dilate both types of blood vessels. The main groups of direct-acting vasodilator drugs are the organic nitrates, calcium channel blockers and potassium channel activators. The indirect-acting vasodilators include the centrally acting adrenergic inhibitors, renin inhibitors, ACE inhibitors and angiotensin receptor (AT1) antagonists. The organic nitrates produce vasodilation through the production of nitric oxide and an increase in cGMP. At low doses, glyceryl trinitrate causes venodilation, reducing preload and stroke volume. Higher doses dilate arteries and the overall effect is a reduction in myocardial work and oxygen demand, hence their beneficial effect in angina. Calcium channel-blocking drugs block the inward movement of calcium ions through the slow channels of the cell membrane of cardiac and vascular smooth muscle cells. These drugs decrease the force of myocardial contraction (negative inotropic effect), decrease automaticity in the SA node and decrease conduction in the AV junction (negative chronotropic and negative dromotropic effects, respectively), and inhibit calcium ion influx in smooth muscle cells (reduction in peripheral vascular resistance). Not all calcium channel blockers have equivalent therapeutic effects. Nifedipine is a potent peripheral vasodilator with minimal cardiac effects; therefore it is an effective antihypertensive agent. Nimodipine is indicated for the treatment of subarachnoid haemorrhage while the other calcium channel-blocking agents are used for their cardiovascular effects (antianginal, anti-arrhythmic and antihypertensive). Drugs such as diazoxide, nicorandil and minoxidil relax smooth muscle by activating ATP-dependent potassium channels. They activate the potassium channel, antagonising the action of ATP and preventing closure of the channel. This results in hyper-polarisation and relaxation of the vascular smooth muscle. Diazoxide and minoxidil have limited use in the treatment of hypertension, while nicorandil is used for the treatment of angina. The main indirect-acting vasodilator drugs are the centrally acting drugs that inhibit sympathetic outflow and inhibitors of the renin–angiotensin–aldosterone system. The centrally acting agents, clonidine and methyldopa, are effective antihypertensives, especially when combined with a diuretic. When given as the sole agent, clonidine and methyldopa usually cause sodium and water retention. Aliskiren is the first of the orally effective direct renin inhibitors. Renin cleaves angiotensinogen to angiotensin I and this is inhibited by aliskiren, which binds to the active site of renin, thereby blocking its action. The plasma concentration of renin increases but its actions are suppressed resulting in a reduction in activity and hence a consequential reduction in angiotensin II formation. ACE inhibitors such as captopril competitively block the angiotensin-converting enzyme necessary for the conversion of angiotensin I to angiotensin II. Angiotensin II is a powerful vasoconstrictor that raises blood pressure and also causes aldosterone release, resulting in sodium and water retention. ACE inhibitors are indicated for the treatment of hypertension, heart failure, diabetic nephropathy, left ventricular dysfunction and after myocardial infarction. All drugs of this class demonstrate similar antihypertensive efficacy, and adverse reactions do not differ significantly among the individual ACE inhibitors. Angiotensin receptor (AT1) antagonists inhibit the action of angiotensin II, thereby preventing vasoconstriction, and are indicated for the treatment of hypertension. In addition to a role in sodium and water homeostasis, aldosterone contributes to the progression of cardiovascular and renal disease. Spironolactone, the prototypical aldosterone receptor antagonist, was launched in 1960 as a potassiumsparing diuretic for the treatment of primary aldosteronism, oedematous conditions, essential hypertension and hypokalaemia. Although beneficial in a setting of heart failure, hyperkalaemia and progestogenic and antiandrogenic adverse effects limit spironolactone’s usefulness. Further investigations led to the development of eplerenone, which is a competitive aldosterone antagonist with greater mineralocorticoid receptor selectivity and reduced progestogenic and antiandrogenic actions. Five drug classes are used to treat hypertension. These include diuretics, β-blockers, ACE inhibitors, angiotensin receptor (AT1) antagonists and calcium channel blockers.Review exercises
Australian Medicines Handbook 2010. Adelaide: AMH, 2010.
Boron W.F., Boulpaep E.L. Medical Physiology. In Updated Version. Philadelphia: Elsevier; 2005. [ch 9]
Cohen J.N., Pfeffer M.A., Rouleau J., et al. Adverse mortality effect of central sympathetic inhibition with sustained-release moxonidine in patients with heart failure (MOXCON). European Journal of Heart Failure. 2003;5:659-667.
Cook C.S., Berry L.M., Bible R.H., et al. Pharmacokinetics and metabolism of [14C] eplerenone after oral administration to humans. Drug Metabolism and Disposition. 2003;31:1448-1455.
Davis K.L., Nappi J.M. The cardiovascular effects of eplerenone, a selective aldosterone-receptor antagonist. Clinical Therapeutics. 2003;25:2647-2668.
Dicpinigaitis P.V. Angiotensin-converting enzyme inhibitorinduced cough. Chest. 2006;129(Suppl 1):169S-173S.
Hoffman B.B. Therapy of hypertension. In Brunton L.L., Lazo J.S., Parker K.L., editors: Goodman & Gilman’s The Pharmacological Basis of Therapeutics, 11th edn, New York: McGraw-Hill, 2006. [ch 32]
Kirch W., Horn B., Schweizer J. Comparison of angiotensin II receptor antagonists. European Journal of Clinical Investigation. 2001;31:698-706.
Michel T. Treatment of myocardial ischaemia. In Brunton L.L., Lazo J.S., Parker K.L., editors: Goodman & Gilman’s The Pharmacological Basis of Therapeutics, 11th edn, New York: McGraw-Hill, 2006. [ch 31]
Munzel T., Daiber A., Mulsch A. Explaining the phenomenon of nitrate tolerance. Circulation Research. 2005;97:618-628.
National Heart Foundation of Australia. Guidelines for the management of acute coronary syndromes 2006. Available: http://www.heartfoundation.org.au.
National Heart Foundation of Australia and the Cardiac Society of Australia and New Zealand. Guidelines for the management of acute coronary syndromes 2006. Medical Journal of Australia 2006; 184: S1-S30.
National Heart Foundation of Australia (National Blood pressure and Vascular Disease Advisory Committee). Guide to management of hypertension 2008. Available: http://www.heartfoundation.org.au.
Quinn D.I., Day R.O. Guide to clinically more important drug interactions. Ch 1 Appendix B. In Speight T.M., Holford N.H.G., editors: Avery’s Drug Treatment, 4th edn, Auckland: Adis International, 1997.
Robinson M. Angiotensin receptor antagonists for hypertension. Australian Pharmacy Trade. 2001;14 June:14-18.
Sanoski C.A. Aliskiren: an oral direct renin inhibitor for the treatment of hypertension. Pharmacotherapy. 2009;29:193-212.
Schwocho L.R., Masonson H.N. Pharmacokinetics of CS-866, a new angiotensin II receptor blocker, in healthy subjects. Journal of Clinical Pharmacology. 2001;41:515-527.
Sica D.A., Gehr T.W.B., Gosh S. Clinical pharmacokinetics of losartan. Clinical Pharmacokinetics. 2005;44:797-814.
Thomas M.C. Diuretics, ACE inhibitors and NSAIDs—the triple whammy. Medical Journal of Australia. 2000;172:184-185.
Werner C., Baumhakel M., Teo K.K., et al. RAS blockade with ARB and ACE inhibitors: current perspective on rationale and patient selection. Clinical Research Cardiology. 2008;97:418-431.
Westfall T.C., Westfall D.P. Adrenergic agonists and antagonists. In Brunton L.L., Lazo J.S., Parker K.L., editors: Goodman & Gilman’s The Pharmacological Basis of Therapeutics, 11th edn, New York: McGraw-Hill, 2006. [ch 10]