Chapter 12 Overview of the Sympathetic Nervous System and Drugs Affecting Noradrenergic Transmission
The sympathetic (adrenergic) nervous system is the second major subdivision of the autonomic nervous system. This system acts in concert with the parasympathetic nervous system to regulate the heart, secretory glands, and vascular and non-vascular smooth muscle. Drugs that stimulate or block α- and β-adrenoceptors are very common in clinical practice today. Understanding the physiological responses mediated by adrenergic receptors aids in rationalising the pharmacological and adverse effects of drugs affecting noradrenergic transmission.
Key abbreviations
COMT catechol-O-methyltransferase
GPCR G-protein-coupled receptor
Key background
The sympathetic nervous system
IN the sympathetic nervous system the preganglionic fibres originate in the spinal cord from thoracic segment T1 to the lumbar segment at L2 level (Figure 11-2). The sympathetic ganglia lie on either side of the vertebral column in two chains, called the paravertebral sympathetic chains; hence the sympathetic preganglionic axons tend to be short. The only exception to the two-neuron arrangement is the adrenal medulla, which is supplied directly by a preganglionic neuron.
There are three naturally occurring catecholamines in the body: dopamine (DA), noradrenaline (NA) and adrenaline (A). Dopamine is the precursor for the synthesis of noradrenaline and adrenaline and has a major role as a neurotransmitter in certain areas of the central nervous system. (For information on CNS transmission, refer to various chapters in Unit 4.) Noradrenaline is released from sympathetic nerve terminals, while adrenaline is a circulating catecholamine released from the adrenal medulla. Nerves that contain noradrenaline or adrenaline are known as adrenergic neurons and are associated with adrenergic transmission. NA acts as the neurotransmitter between sympathetic postganglionic nerves and the organs they innervate (Figure 11-6).
The synthesis of these catecholamines is mediated by different enzymes located in the postganglionic nerve terminals and in the chromaffin cells of the adrenal medulla which are specialised cells that secrete both NA and A. Clinically, an abnormally high secretion of A and NA, which causes palpitations, excessive sweating, hypertension, headaches and skin pallor, occurs in phaeochromocytoma, a rare chromaffin cell tumour. The synthesis of NA begins with the amino acid tyrosine, which we obtain from dietary proteins. Tyrosine is taken up by adrenergic neurons and enzymatically converted into dihydroxyphenylalanine (DOPA), which in turn is metabolised (decarboxylated) to DA. DA is then taken up into neuronal storage vesicles, or granules, where it is metabolised into the neurotransmitter NA by the enzyme DA β-hydroxylase (Figure 12-1). Unlike adrenergic neurons, in the adrenal medulla the enzyme phenylethanolamine N-methyltransferase converts NA to A. On stimulation, both A and NA are released from the adrenal medulla and carried by the systemic circulation to all parts of the body.
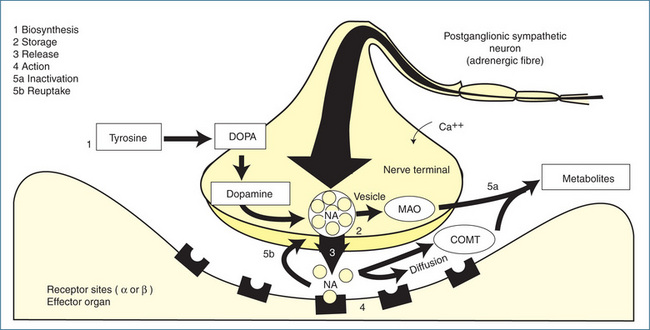
Figure 12-1 Adrenergic transmission at a neuroeffector junction. 1 Biosynthesis and storage of noradrenaline (NA): tyrosine is taken up by adrenergic neurons and metabolised to dopamine, which is taken up by the storage vesicles. Inside the vesicles dopamine is converted to noradrenaline. 2 Release: an action potential arriving at the nerve terminal causes the vesicle to attach itself to the membrane and release NA, which then diffuses across the synaptic cleft and combines with the receptors on the effector cell. 3 Action: the interaction of NA with the receptors results in a response. 4 The action of NA is terminated by reuptake of NA into the nerve terminal (5a), enzymatic degradation by monoamine oxidase (MAO) and catechol-O-methyltransferase (COMT) and by diffusion away from the receptors (5b).
In a manner similar to that of cholinergic transmission, the arrival of an action potential at the nerve terminal of the postganglionic neuron causes an influx of calcium ions, fusion of vesicles with the cell membrane, and release of stored NA into the junctional cleft. NA then diffuses across the cleft to the receptor sites on the postjunctional membrane of neuroeffector cells (smooth muscle, cardiac muscle or glands) (Figure 12-1). However, the uptake and degradation of catecholamines differs substantially from that of ACh. Once NA has bound to an adrenoceptor, its action must be rapidly terminated to prevent prolongation of its effects, which could lead to a loss of regulatory control of visceral function. The inactivation of NA occurs by reuptake of the NA into nerve terminals, enzymatic degradation and diffusion. Catecholamines are metabolised by the enzymes monoamine oxidase (MAO) and catechol-O-methyltransferase (COMT). Two types of MAO have been identified, MAO-A and MAO-B, and these are discussed in Chapter 18. Free NA within the cytoplasm of the nerve terminal is metabolised by MAO, which is bound to the surface membrane of intraneuronal mitochondria. COMT, which is located in both neuronal and non-neuronal tissues, metabolises both NA and the metabolites produced by the action of MAO.
The reuptake of NA plays a more significant role than enzymatic transformation in catecholamine inactivation. The reuptake processes, called uptake 1 (neuronal) and uptake 2 (extraneuronal), function collectively to accumulate catecholamines against a concentration gradient. NA is removed from the junctional sites by these active transport processes and is returned to the sympathetic nerve terminal and storage vesicles. In this way, an adequate supply of NA is provided by reuptake as well as by synthesis. Finally, a small portion of NA released at the synaptic cleft diffuses away, enters the systemic circulation and is metabolised elsewhere in the body (Figure 12-1).
Adrenoceptors
The adrenoceptors that are stimulated by the endogenous catecholamines NA and A consist of two subtypes: alpha (α) and beta (β). α-Adrenoceptors are then further divided into α1 and α2 subtypes, and β-adrenoceptors into the three subtypes β1, β2 and β3.
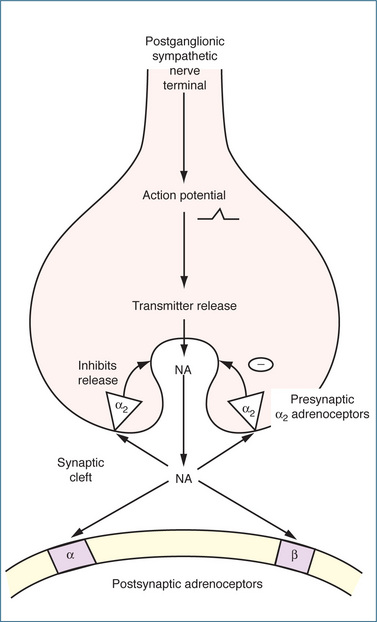
The α-adrenoceptors are differentiated primarily by neuronal location: α1-adrenoceptors are located on the postsynaptic membrane, while α2-adrenoceptors are found on presynaptic nerve terminals, postsynaptically on vascular and smooth muscle cells, and on sites remote from the nerve terminals, such as platelets and leucocytes. These inhibitory presynaptic α2 autoreceptors are pharmacologically distinct from the postsynaptic α1-adrenoceptors and control the amount of NA released through a negative feedback mechanism. When the concentration of NA in the synaptic cleft reaches a high level, it stimulates the α2-adrenoceptors, which prevent the further release of NA (Figure 12-2). This feedback prevents excessive and prolonged stimulation of postsynaptic α1-adrenoceptors on effector organs such as the eye, arterioles, veins, male sex organs and bladder neck (Figure 12-3). In contrast, the α1-adrenoceptors are generally located close to the nerve terminal so that the action of released noradrenaline is almost immediate.
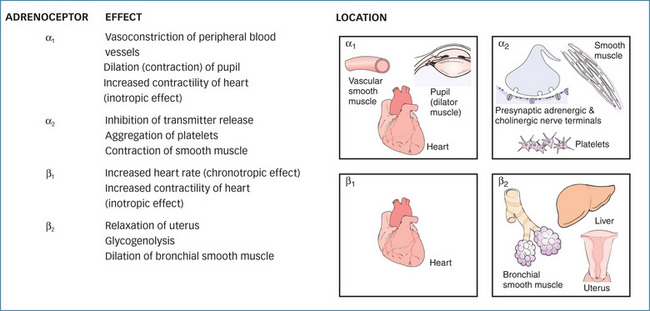
Important responses mediated by α-adrenoceptor stimulation in humans include:
Beta-adrenoceptors show wide tissue distribution (Table 11-1) and are subdivided into three types:

(See Clinical Interest Box 12-1.)
Clinical interest Box 12-1 β3-Adrenoceptors
The human β3-adrenoceptor (β3-AR) was cloned in 1989 and shown to mediate lipolysis and thermogenesis in adipose tissue. More recently β3-adrenoceptors have been demonstrated in other tissues including brain, heart, prostate, urinary bladder detrusor and GIT. Their widespread distribution has increased the potential for them to be targeted for the design of selective β3 agonists. Like the other adrenoceptors, β3-AR is a G-proteincoupled receptor and stimulation of the receptor leads to sequential activation of adenylyl cyclase and protein kinase A, which culminates in activation of various transcription factors that increase both the thermogenic response and lipolysis. Adrenaline and noradrenaline are both ligands for β3-AR. At present some β3-AR agonists are in clinical trials for the treatment of overactive bladder and the management of major depressive disorder because of the role of β3-ARs in anxiolytic and antidepressant-like effects.
In rodent models β3-AR agonists also exert potent anti-diabetic effects, which occur independently of the anti-obesity effects of the drugs. The mechanism of the anti-diabetic effect has not been elucidated but may result from improved insulin sensitivity. Although the potential role of β3-AR agonists for the treatment of human obesity was greeted with much enthusiasm, differences between the roles of white and brown adipocytes in humans and rodents and poor selectivity of the drugs for human β3-AR led to disappointing results in clinical trials. It appears that β3-AR is no longer seen as a viable target for the treatment of obesity (Ursino et al 2009).
NA acts principally on α1-, β1- and β3-adrenoceptors, with negligible activity at β2-adrenoceptors. In contrast, adrenaline acts on all α- and β-adrenoceptor subtypes, with significantly greater effects on α-adrenoceptors at higher doses. Isoprenaline, a synthetic catechol amine, acts only on β-adrenoceptors.
Important responses mediated by β-adrenoceptor stimulation include:
The effects of both α- and β-adrenoceptor stimulation when they are interrelated often results in a summation of action. Most arteries and veins contain both α- and β-adrenoceptors, and a change in blood pressure will depend on the degree of vasoconstriction in the skin and splanchnic area and the extent of vasodilation in skeletal muscle blood vessels, along with changes in heart rate.
Adrenergic drugs
Specific drugs that stimulate or block α- and β-adrenoceptors are available and many of these drugs are discussed in other chapters in the context of the relevant pharmacology. With the exception of central α2-adrenoceptors, which are discussed in Chapter 23, these agents all act at peripheral autonomic sites. The extent of either sympathetic innervation or the presence of adrenoceptors on various organs will determine the magnitude of response to an individual adrenergic drug.
Drugs affecting noradrenergic transmission include:
Direct-acting adrenoceptor agonists
During circulatory shock, the autonomic nervous system plays an essential compensatory role in an attempt to restore normal haemodynamics; therefore many drugs that target the sympathetic nervous system are used to manage this condition. The direct-acting adrenoceptor agonists (also called sympathomimetic drugs) directly stimulate α- and β-adrenoceptors, mimicking the effects of sym pathetic stimulation, such as increasing cardiac output, vasoconstriction of arterioles and veins, regulation of body temperature, bronchial dilation and a variety of other effects. (For additional information on the effects of the sympathetic nervous system, see Chapter 11.) Although there are other agents, the five drugs that are widely used for circulatory shock are the
Refer to Drug Monograph 12-1 for a description of the characteristics of adrenaline. The use of isoprenaline in shock is limited by β2-adrenoceptor mediated vasodilation that may worsen the hypotension (Table 12-1).
Drug monograph 12-1 Adrenaline
MECHANISM OF ACTION Adrenaline stimulates α- and β-adrenoceptors, which are GPCRs that link to various G-proteins and their respective signal transduction systems (see Chapter 5). The response elicited following activation of the adrenoceptors results from the generation of secondary messengers and subsequent changes in the activity of various ion channels. The primary action of adrenaline is on the β-adrenoceptors of the heart, smooth muscle of the bronchi and the blood vessels. At low doses adrenaline has predominantly β-adrenoceptor actions, but with increasing doses an increase in α-adrenoceptor activity is observed.
Cardiac effects. Adrenaline produces a significant increase in myocardial contraction (positive inotropic effect) as a result of increased influx of calcium into cardiac fibres. The strong myocardial contractions result in more complete emptying of the ventricles and an increase in cardiac work, oxygen consumption and cardiac output. If the heart is in ventricular fibrillation, adrenaline increases fibrillation vigour and frequently promotes successful defibrillation. In these situations the drug may be injected repeatedly; however, adrenaline cannot be used repeatedly to improve the function of a failing heart (congestive heart failure) because it increases oxygen consumption by cardiac muscle. This positive inotropic effect provides the rationale for the use of adrenaline in cardiac arrest.
A significant increase in cardiac rate (positive chronotropic effect) occurs as a result of the increased rate of membrane depolarisation in the pacemaker cells in the sinus node during diastole. Action potential threshold is reached sooner, pacemaker cells fire more often and heart rate increases. Adrenaline may also produce spontaneous firing of Purkinje fibres, which may cause them to exhibit pacemaker activity. This effect can cause ventricular extrasystoles and increase the susceptibility of ventricular muscle to fibrillation. An improvement in atrioventricular conduction (positive dromotropic effect) may also occur in conduction abnormalities.
Vascular effects. Vascular effects of adrenaline depend on the dose and the vascular bed affected. Low doses of adrenaline decrease total peripheral vascular resistance and lower blood pressure. In large doses adrenaline activates α receptors in the peripheral vascular system, which increases resistance and raises blood pressure. The dominant net response is often vasodilation; for example, during situations of high sympathetic demand, the release of adrenaline from the adrenal medulla constricts blood vessels in the skin and splanchnic areas but dilates those of skeletal muscles, thus shunting blood to the areas needed for ‘fight-or-flight’ type responses.
Renal artery constriction and resistance occurs with adrenaline, and renal blood flow may be substantially reduced. Direct action on β1-adrenoceptors on juxtaglomerular cells increases the secretion of renin.
Central nervous system effects. Adrenaline in therapeutic doses is not a CNS stimulant. Signs of restlessness, tremors and anxiety may be secondary to the effects of adrenaline on skeletal muscle, the cardiovascular system and changes in metabolism. Beneficial cerebral effects from adrenaline in persons with hypotension are thought to be the result of increased systemic pressure with a resultant improvement in cerebral blood flow.
Smooth muscle effects. Generally, adrenaline relaxes smooth muscle of the gastrointestinal tract. The stomach is relaxed and the amplitude and tone of intestinal peristalsis are reduced. In theory, this may retard gastrointestinal emptying and propulsion of food; however, this effect is rare in humans with therapeutic doses of catecholamines. In the urinary bladder, adrenaline causes trigone and sphincter constriction via α-adrenoceptor stimulation and detrusor relaxation (β-adrenoceptor agonist activity), which may cause a delay in the desire to void and hence urine retention. Adrenaline inhibits uterine contraction during the last months of pregnancy, and the β2-adrenoceptor agonists such as salbutamol are used to prevent premature labour.
Respiratory effects. Adrenaline is a powerful bronchodilator and relieves respiratory distress due to allergens such as bee venom. In asthma it is likely that beneficial effects also occur through the effect of adrenaline on mast cells and on bronchial mucosa (see Chapter 28).
Metabolic effects. Adrenaline inhibits insulin secretion and decreases the uptake of glucose by peripheral tissues, thus raising blood glucose levels. It stimulates lipolysis in adipose tissue, which results in an increase in free fatty acids in blood; thus in response to high sympathetic drive there is an abundant supply of fuel and energy. Adrenaline also has a calorigenic effect, primarily as a result of increased metabolism, which increases oxygen consumption (Figure 12-4).
Indications
Pharmacokinetics
Adrenaline should not be given orally because it is rapidly metabolised in the mucosa of the gastrointestinal tract and liver by COMT and MAO. Absorption after IM or SC injection can be slow because of local vasoconstriction. Adrenaline has a rapid onset of action of 3–5 minutes after inhalation. In severe anaphylaxis, asthma or cardiac arrest, doses may need to be repeated every 5–20 minutes, depending on the dosage used and the person’s response.
Drug interactions
If indicated adrenaline is not withheld because of concerns of drug interactions. The following effects can occur when adrenaline is given with the drugs listed:
Adverse reactions
Adverse reactions include increased nervousness, restlessness, insomnia, tachycardia, tremors, sweating, hypertension, nausea, vomiting, pallor and weakness. With inhalation devices, adverse reactions include bronchial irritation and coughing (with high doses), dry mouth and throat, headaches and flushing of face and skin. High doses may cause ventricular arrhythmias.
Warnings and contraindications
Adrenaline is used with caution in persons with diabetes mellitus, closed-angle glaucoma, hypertension, ischaemic heart disease, hyperthyroidism and Parkinson’s disease. When used in endartery areas, such as fingers, toes or penis, the reduced blood supply to the area may result in ischaemia and gangrene. Avoid use in persons with known hypersensitivity to adrenaline (or sympathomimetics), organic brain damage, coronary insufficiency or shock.
Noradrenaline
Noradrenaline has a high affinity for α-adrenoceptors and, because the blood vessels of the skin and mucous membrane contain α-adrenoceptors, NA produces vaso constriction in these tissues. The blood vessels (both arteriolar and venous beds) in the visceral organs, including the kidneys, also contain predominantly α-adrenoceptors. Consequently, NA causes vasoconstriction and a reduced blood flow in the kidneys and other visceral organs. Although NA activates β1-adrenoceptors on the heart, effecting changes in diastolic and systolic pressure, peripheral vascular resistance coupled with compensatory vagal reflexes results in no change in heart rate and cardiac output. Stimulation of α- and β1-adrenoceptors with NA is dose-related. At low doses (<0.002 mg/min), β1-adrenoceptors are stimulated; at doses higher than 0.004 mg/min stimulation of α-adrenoceptors increases total peripheral resistance. Titration of the dose in steps of 0.002–0.004 mg/min is based on haemodynamic response.
The main therapeutic effect of NA results from peripheral arteriolar vasoconstriction in all vascular beds. Both systolic and diastolic pressures are elevated, causing a rise in mean arterial pressure. Of importance during shock is constriction of the venous capacitance vessels, which reduces splanchnic and renal blood flow. This is brought about by severe restriction of tissue perfusion in these regions. In persistent hypotension after blood volume deficit has been corrected, NA helps to raise the blood pressure to an optimal level and establishes a more adequate circulation. Noradrenaline is selectively used for restoring blood pressure in acute hypotensive states such as sympathectomy, myo cardial infarction, phaeochromocytomectomy and blood transfusion reaction. It is also used as adjunct therapy in cardiac arrest.
Noradrenaline has limited therapeutic value and is administered only by intravenous infusion. Oral NA is rapidly metabolised in the mucosa of the GIT and liver by COMT and MAO. Subcutaneous NA is poorly absorbed owing to local vasoconstriction at the site of injection. Onset of action is rapid by intravenous infusion and distribution is mainly to the heart, spleen and glandular tissues. The half-life is short and ranges from about 30 seconds to 3 minutes. The drug is extensively metabolised in the liver and other tissues but the most significant clearance occurs by uptake into sympathetic nerves and other extraneuronal tissues. Most of the dose is excreted in urine as metabolites.
The drug interactions of NA are similar to those of adrenaline and include interactions with halogenated inhalational anaesthetics, β-blocking agents, digitalis glycosides, tricyclic antidepressants, MAO inhibitors, cocaine and oxytocics. Adverse reactions include anxiety, dizziness, pallor, tremor, insomnia, headache, palpitations and, infrequently, hypertension and bradycardia.
Noradrenaline should be used with caution in patients with atherosclerosis, mesenteric and peripheral vascular thrombosis or other occlusive vascular diseases, metabolic acidosis, hypoxia or hyperthyroidism. It should also be avoided in persons with hypertension, hypersensitivity to sodium metabisulfite (the preservative in the solution), hypovolaemia, myocardial infarction and ventricular arrhythmias. Importantly, NA solution should be protected from light and not used if brown colouration is present.
Isoprenaline
Isoprenaline, a synthetic catecholamine, is a non-selective β-adrenoceptor agonist—it stimulates β1- and β2-adrenoceptors. The β1-adrenoceptor activity produces an increase in the force of myocardial contraction and heart rate. The β2-adrenoceptor response of the smooth muscle of the bronchi, skeletal muscle, GIT and blood vessels of the splanchnic bed is relaxation. This drug also stimulates insulin secretion through β-adrenoceptor activation of pancreatic islet cells, and causes the release of free fatty acids from adipocytes.
Haemodynamically, the β1 activity of the heart increases cardiac output and venous return to the heart. However, peripheral vascular resistance is reduced and in normal individuals this may cause a significant drop in blood pressure with excessive dosage. Isoprenaline is used to increase automaticity and AV nodal conduction in Stokes–Adams syndrome and serious episodes of heart block. It may also be used as adjunctive therapy in treatment of hypovolaemic states, septic shock, congestive heart failure and cardiogenic shock.
Oral absorption of isoprenaline is erratic and this route is no longer recommended. Following IV administration, the plasma concentration of isoprenaline declines in a biphasic manner. The initial phase corresponds to rapid uptake in smooth muscle and cardiac tissue (around 5 minutes), while the second phase, which reflects widespread metabolism, lasts for more than 2.5 hours. Isoprenaline is metabolised by COMT in the GIT, liver and lungs and is excreted in the urine, predominantly as unchanged drug (60%).
Drug interactions with isoprenaline include β-blockers, which antagonise the therapeutic effect of isoprenaline (may precipitate asthma), and entacapone, which inhibits metabolism of isoprenaline and hence dose reduction of isoprenaline is necessary. Also avoid concurrent administration of isoprenaline with other sympathomimetic amines, as additive effects may occur and cardiotoxicity may result.
The range of adverse reactions for isoprenaline is similar to that for adrenaline. Use isoprenaline with caution in the elderly and in persons with diabetes mellitus, hyperthyroidism and ischaemic heart disease. Isoprenaline is contraindicated in the presence of tachycardia, ventricular arrhythmias and myocardial infarction and in persons with known hypersensitivity to isoprenaline.
Dopamine
Dopamine is a catecholamine and is the immediate precursor of NA (see Figure 12-1). It acts both directly on various receptors and indirectly by releasing NA. Dopamine stimulates dopaminergic receptors, β1-adreno ceptors and, in high doses, α1- and α2-adrenoceptors. Its actions are dose-dependent and very complex.
Unlike NA, in low doses (0.5–2 mcg/kg/min), DA acts mainly on dopaminergic (D1) receptors to cause vasodilation of the renal and mesenteric arteries. Renal vasodilation increases renal blood flow, usually with greater urine and sodium excretion.
In low to moderate doses (usually 2–10 mcg/kg/min), DA acts directly on the β1-adrenoceptors on the myocardium and indirectly by releasing NA from myocardial storage sites. These actions increase myocardial contractility and stroke volume, thereby increasing cardiac output. Systolic blood pressure and pulse pressure may rise, with either no effect or a slight elevation in diastolic blood pressure. Nevertheless, total peripheral resistance is usually unchanged. Coronary blood flow and myocardial oxygen consumption increase, while heart rate increases only slightly at low doses.
With higher doses of DA (10 mcg/kg/min or more), α-adrenoceptors are stimulated, increasing peripheral resistance. As a consequence, higher doses may reduce urinary output, eliminating the benefit of D1-mediated renal vasodilation.
Unlike NA, DA aids perfusion of vital splanchnic organs. The combination of cardiac and vascular effects has led to successful use of DA in the treatment of circulatory shock and refractory heart failure. DA is used to correct haemodynamic imbalances associated with shock syndrome caused by myocardial infarction, trauma, endotoxin septicaemia, open heart surgery, renal failure and chronic cardiac decompensation (as in congestive heart failure).
Dopamine is administered by IV infusion. The drug has a rapid onset of action (2–5 minutes) and a short duration of action (5–10 minutes). It is widely distributed throughout the body and is actively taken up into sympathetic nerves but does not cross the blood–brain barrier and therefore does not act on central dopaminergic receptors. Dopamine is rapidly metabolised to inactive metabolites by COMT and MAO in the liver, kidney and plasma; these metabolites are excreted in the urine.
Adverse reactions include headaches, nausea, vomiting, angina, respiratory difficulties, decreased blood pressure and, less frequently, hypertension, irregular or ectopic heart beats, tachycardia and palpitations. For drug interactions, warnings and contraindications, see discussion of NA (above).
Dobutamine
Dobutamine is a synthetic catecholamine used primarily in cardiogenic shock and it acts directly on heart muscle to increase the force of myocardial contraction. This response is attributed to the direct stimulation of cardiac β1-adrenoceptors. At the same time, dobutamine produces comparatively little increase in heart rate or peripheral vascular resistance. By enhancing stroke volume, this agent is an effective positive inotropic drug. Because of its minimal influence on heart rate and blood pressure (both major determinants of myocardial oxygen demand), it is valuable for use in individuals with low cardiac output. Dobutamine does not have any effect on DA receptors and does not cause vasodilation in the kidney.
Dobutamine is administered IV in the short-term management of patients requiring inotropic support, as in those with congestive heart failure, cardiogenic shock due to myocardial infarction or after cardiac surgery. Its beneficial effects include a progressive increase in cardiac output and a decrease in pulmonary capillary wedge pressure, thereby improving ventricular contraction. The onset of action of dobutamine is within 1–2 minutes and it has a duration of action of approximately 10 minutes. Its plasma half-life is less than 3 minutes, as it is rapidly metabolised by liver COMT to form methyldobutamine, which is conjugated with glucuronic acid and excreted in the urine. The glucuronide metabolite has no significant cardiovascular activity.
Drug interactions are similar to those of adrenaline with regard to α- and β-blockers, general anaesthetics and oxytocin. Additive vasodilatory effects also occur with the coadministration of nitroprusside. In combination with milrinone there is an increased potential for tachycardia and arrhythmias. Adverse reactions include nausea, headache, respiratory distress, angina, palpitations, tachycardia, hypertension and, commonly, ventricular ectopic beats. Hence dobutamine is contraindicated in persons with atrial fibrillation, ventricular arrhythmias and phaeochromocytoma. Table 12-1 provides comparative information on the endogenous and synthetic catecholamines.
In addition to the catecholamines other direct-acting α-adrenoceptor agonists include the topical ocular drugs naphazoline, tetrahydrozoline and phenylephrine (α1-adrenoceptor agonist), which are used for ocular congestion and are freely available as OTC products (refer to Chapter 31).
Mixed-acting adrenoceptor agonists
The prototypical mixed-acting adrenoceptor agonist is ephedrine (Drug Monograph 12-2). Ephedrine is a racemic drug and pseudoephedrine, the stereoisomer of ephedrine (l-ephedrine), has a similar pharmacological profile. Pseudoephedrine is most commonly used as an oral or intranasal decongestant as its actions on α-adrenoceptors in vascular smooth muscle result in vasoconstriction of dilated nasal blood vessels thereby relieving nasal congestion. Oral pseudoephedrine (commonly a component of OTC cough and cold medicines) is contraindicated in persons with a history of cardiovascular disease including coronary artery disease, hypertension and cardiac arrhythmias. Adverse effects include CNS stimulation, which adds to its potential for misuse, excitability, insomnia and tremor.
Drug monograph 12-2 Ephedrine (Sulfate)
Mechanism of action
Ephedrine has both a direct and an indirect sympathomimetic action. It acts indirectly by stimulating the release of noradrenaline from presynaptic nerve terminals and also acts directly on both α- and β-adrenoceptors. Like adrenaline and noradrenaline, ephedrine has positive inotropic and chronotropic activities, but it is a less effective vasoconstrictor.
Indications
Parenteral ephedrine has been used in hypotensive patients who do not respond to fluid replacement and as a vasopressor agent in hypotensive states secondary to spinal anaesthesia. Ephedrine has been used to produce bronchodilation in bronchial asthma and reversible bronchospasm, but generally more β2-selective drugs are used.
Pharmacokinetics
Absorption of the drug is rapid after IM or SC administration. Onset of action occurs within 10–20 minutes of IM administration. The duration of the pressor effects and cardiac responses after parenteral administration of ephedrine is 1 hour and the half-life of the drug is 3–11 hours. Most of the drug (55%–75%) is excreted unchanged in urine with the remainder metabolised in the liver.
Adverse reactions
These include a range of cardiac effects (e.g. palpitations, angina, bradycardia, tachycardia, hypotension and hypertension, arrhythmias), GIT effects (e.g. nausea, vomiting) and CNS effects (e.g. nervousness, insomnia, fear, irritability, confusion, delirium and euphoria).
These are similar to those for adrenaline, although ephedrine is not available in aerosol form, so coughing and local irritation are not reported. In addition, ephedrine may cause mood changes and hallucinations.
Dosage and administration
For vasopressor effects the adult ephedrine dose IM or SC is 10–50 mg, repeated if necessary. If an immediate effect is desired it may be administered IV at a dose of 10–25 mg, which may be repeated every 5–10 minutes if required. In children the dose recommended is 3 mg/kg/day via the IV or SC route, administered as 4–6 divided doses.
Indirect-acting adrenoceptor agonists
Indirect-acting adrenoceptor agonists can trigger the release of NA and adrenaline from their storage sites in the adrenal medulla and sympathetic neurons; these neurotransmitters then activate α- and β-adrenoceptors e.g. amphetamine (Chapter 19) and tyramine. Additionally, some of these drugs also stimulate adrenoceptors directly, e.g. metaraminol. Others block the uptake of NA into sympathetic neurons, e.g. cocaine, or inhibit MAO/COMT, e.g. entacapone (refer to Chapter 20).
Metaraminol
Metaraminol is a vasopressor agent with both direct (primarily) and indirect effects on the sympathetic system. It acts indirectly by causing the release of NA, and directly via an action on β- and α1-adrenoceptors, although it has predominantly more α-adrenoceptor activity. In general it is less potent than NA.
Metaraminol has a positive inotropic effect, constricts blood vessels, increases peripheral resistance, elevates both systolic and diastolic blood pressure and improves cardiac contractility. Adverse reactions are dose-related. Overdosage may cause severe hypertension, sinus arrhythmias, myocardial infarction and cardiac arrest.
Metaraminol is used for the treatment of acute hypotensive states occurring with spinal anaesthesia and as an adjunct in the treatment of hypotension due to haemorrhage, cardiogenic shock or septicaemia. This drug is infrequently used in anaesthesia and intensive care.
Adrenoceptor antagonists
α-Adrenoceptor antagonists
α-Adrenoceptor antagonists compete with catecholamines for binding at α-adrenoceptors and inhibit sympathetic stimulation. The main groups of drugs are:
α1-Adrenoceptor antagonists
The principal uses of prazosin (Drug Monograph 12-3), terazosin and tamsulosin are for the treatment of hypertension and for symptomatic relief of urinary obstruction in benign prostatic hypertrophy. Selective blockade of postsynaptic α1-adrenoceptors results in a decrease in peripheral vascular resistance because of inhibition of catecholamine-induced vasoconstriction. Only a minor increase in heart rate occurs because these drugs have negligible α2-adrenoceptor activity.
Prazosin was the first of the α1-adrenoceptor selective antagonists developed in the 1970s. From January 2009 to December 2009 >550,000 pre scriptions for prazosin were written in Australia. Blockade of α1-adrenoceptors in arteri oles and veins leads to a decrease in peripheral vascular resistance, reducing venous return to the heart. Unlike other vasodilator drugs, prazosin does not produce a reflex tachycardia.
Pharmacokinetics
Prazosin is well absorbed after oral administration, with bioavailability in the order of 50%–80%. Peak concentration occurs about 1–3 hours after an oral dose. Prazosin is highly bound (∼95%) in plasma to α1-acid glycoprotein. The plasma half-life ranges from 2.5 to 3.5 hours and more than 90% of the drug is metabolised in the liver. Both the metabolites and unchanged drug (<5%) are excreted in urine.
An increase in plasma half-life (about 7 hours) and bioavailability (2–3-fold) occurs in persons with conges tive cardiac failure. Some of the metabolites of prazosin also have antihypertensive activity, which may contrib ute to the effect of the drug.
Drug interactions
The first dose of prazosin may cause hypotension. The risk of this first-dose phenom enon may be increased by β-blockers, diuretics and calcium channel blockers.
Adverse reactions
Common adverse reactions include postural hypotension, dizziness, headaches, drowsi ness, fatigue, nasal congestion and urinary urgency.
Warnings and contraindications
Care should be exercised in persons with pre-existing renal disease (which may exacerbate the first-dose effect), liver disease (which may necessitate a dosage reduc tion) and the elderly (who are often more likely to suffer orthostatic hypotension). Prazosin is contraindicated in heart failure associated with mechanical obstruction such as aortic stenosis and in persons with known sensitivity to prazosin.
Terazosin and tamsulosin are used to treat benign prostatic hyperplasia. This condition is characterised by urinary frequency, nocturia and a weak urine stream in older men. The symptoms are due to a combination of mechanical pressure on the urethra resulting from prostate hyperplasia and α1-adrenoceptor mediated increased smooth muscle tone in the prostate and neck of the bladder. The duration of action of terazosin is approximately 18 hours while the half-life of tamsulosin is 5–10 hours, which allows once daily dosing for both drugs. Tamsulosin is extensively metabolised by CYP3A and CYP2D6 while <10% of terazosin is excreted as unchanged drug in urine.
Non-selective α1- and α2-adrenoceptor antagonists
These agents include phenoxybenzamine, phentolamine, carvedilol and labetalol.
Phenoxybenzamine is a long-acting, irreversible α1- and α2-adrenoceptor antagonist that abolishes or decreases the receptiveness of α-adrenoceptors to adrenergic stimuli. At higher doses it also antagonises the actions of acetylcholine, histamine and 5-hydroxytryptamine (5-HT, serotonin) because it covalently binds to the various receptors. This covalent interaction with α-adrenoceptors results in a long duration of action and a progressive decrease in peripheral vascular resistance. A reflex increase in heart rate occurs that may be exacerbated by blockade of presynaptic α2-adrenoceptors, which results in release of NA, which in turn causes tachycardia. Phenoxybenzamine is used in the management of phaeochromocytoma and the preparation of patients with this condition for surgery.
Oral absorption of the drug is variable and the onset of action is 1–2 hours. The clinical effect of the drug can persist for 3–4 days and this most probably relates to turnover time of the receptor. The half-life is in the order of 24 hours, with metabolism in the liver and excretion via urine and faeces.
Avoid concurrent use of phenoxybenzamine with other sympathomimetics, such as adrenaline, as unopposed stimulation of β2-adrenoceptors will exacerbate the hypotension and reflex tachycardia. Adverse reactions include dizziness (postural hypotension), miosis, tachycardia, nasal congestion, confusion, dry mouth, headache and inhibition of ejaculation. Use with caution in persons with heart failure, coronary artery disease, respiratory infections or renal impairment. The drug is contraindicated when hypotension is undesirable, e.g. after cerebrovascular accident and myocardial infarction.
Phentolamine competitively blocks α2- (presynaptic) and α1- (postsynaptic) adrenoceptors equally. The action occurs at both arterial and venous vessels. This direct relaxation of vascular smooth muscle lowers total peripheral resistance, inducing a marked reflex tachycardia. This drug is used to prevent or control hypertensive episodes in the individual with phaeochromocytoma, especially preoperatively and during surgery.
Phentolamine is administered IM and IV. Its half-life is approximately 19 minutes after IV administration but the haemodynamic response may persist for up to 12 hours. About 13% of the drug is excreted in urine unchanged. Drug interactions, adverse effects and warnings and contraindications are similar to those of phenoxybenzamine.
Non-selective α1- and β-adrenoceptor antagonists
Carvedilol (See Drug Monograph 12-4.)
Drug monograph 12-4 Carvedilol
Multiple clinical trials (COMET, COPERNICUS, CAPRICORN) have shown that carvedilol reduces mortality and morbidity in patients with mild-to-severe congestive heart failure. Additionally, in combination with standard drug therapy carvedilol reduces mortality in the setting of mycardial infarction.
Mechanism of action
Carvedilol is a unique cardiovascular drug with a wide range of therapeutic benefits. Its predominant haemodynamic effects are derived from blockade of β1-, β2- and α1-adrenoceptors. A racemic drug, both the R(+) and S(–) enantiomers possess similar α1-blocking activity, but only S(–) carvedilol exhibits β-blocking activity while the drug has little or no affinity for α2-adrenoceptors nor intrinsic sympathomimetic activity (ISA, refer to following section). Carvedilol is a potent antihypertensive and the reduction in blood pressure primarily due to blockade of β1-, β2- and α1-adrenoceptors is not associated with a reflex tachycardia. Total peripheral resistance decreases due to blockade of α1-adrenoceptors; this vasodilatory effect reduces afterload and offsets the negative inotropic effect of cardiac β-blockade. Consequently cardiac output and stroke volume are maintained.
Carvedilol and some of its metabolites possess antioxidant properties protecting the vascular system from reactive oxygen species. In addition this drug has antiproliferative and antiatherogenic actions, anti-ischaemic actions, antihypertrophic actions and anti-arrhythmic actions. A further additional benefit arises from improved insulin sensitivity (Cheng et al 2001).
Pharmacokinetics
Carvedilol is rapidly and completely absorbed after oral administration and undergoes extensive first-pass metabolism, which accounts for its bioavailability of 20%–25%. Peak plasma concentration occurs within 1–2 hours. As the S(–) enantiomer is cleared by hepatic metabolism more rapidly than the R(+) enantiomer the plasma concentration of R(+) carvedilol is 2–3 times higher than that of S(–) carvedilol. More than 95% of the drug is bound to plasma proteins (predominantly albumin) and the volume of distribution is ∼100–140 L. Carvedilol is metabolised in the liver by CYP2D6 and CYP2C9 with a half-life of 4–7 hours. The predominant route of excretion is biliary with <2% of the dose excreted as unchanged drug in urine. No dosage adjustment is necessary in patients with renal disease; however, in patients with hepatic disease bioavailability is increased.
Drug interactions and adverse reactions
See following section for general information on drug interactions and adverse reactions with β-blockers. A combination of carvedilol and rifampicin may result in decreased plasma concentration of carvedilol due to induction of metabolism (CYP2C9) by rifampicin. An increase in the dose of carvedilol may be necessary; alternatively consider use of a renally cleared β-blocker.
Warnings and contraindications
β-Blockers are in general contraindicated in shock and individuals with asthma, diabetes, hyperthyroidism, phaeochromocytoma, myasthenia gravis or bradycardia (45–50 beats/minute).
Dosage and administration
For the treatment of hypertension the usual adult dose is 12.5 mg daily for 2 days and then 25 mg once daily increased at intervals of >2 weeks up to a maximum once daily dose of 50 mg or in two divided doses. For individuals with heart failure the commencing dose is half the usual adult dose, increasing at intervals of >2 weeks to a maximum of 25 mg twice daily.
Labetalol acts on both α1- and β-adrenoceptors and competitively antagonises the action of catecho lamines. It is a complex drug that selectively blocks α1-, β1- and β2-adrenoceptors but also partially stimulates β2-adrenoceptors and inhibits the neuronal uptake of NA (similar to the action of cocaine). Blockade of α1-adrenoceptors leads to a fall in peripheral vascular resistance, while blockade of β1-adrenoceptors prevents the reflex sympathetic stimulation of the heart. Labetalol is indicated for the treatment of hypertension.
Rapid absorption occurs after oral administration, and peak plasma concentration occurs within 20–90 minutes. Bioavailability is highly variable (11%–86%), due primarily to extensive presystemic metabolism. Labetalol is extensively metabolised to glucuronide conjugates that are excreted in urine (55%–60%) and faeces (12%–27%).
Drug interactions, adverse effects, warnings, contraindications, dosage and administration are discussed in the following section in the context of the predominant β-blocking activity of labetalol.
β-adrenoceptor antagonists
β-Adrenoceptor antagonists, commonly referred to as β-blockers, competitively block the actions of catecholamines (Figure 12-5). The main group is the β1-selective blockers that are frequently referred to as cardioselective blockers because these agents block β1-adrenoceptors on the heart. At high doses, however, β1-adrenoceptor selectivity diminishes and the adverse effects of β2-adrenoceptor blockade then need to be considered. Drugs that block both types of adrenoceptors, β1- and β2-, are referred to as non-selective β-adrenoceptor antagonists. The use of all of these drugs is contraindicated in people with asthma because of inhibition of bronchodilation mediated by β2-adrenoceptors.
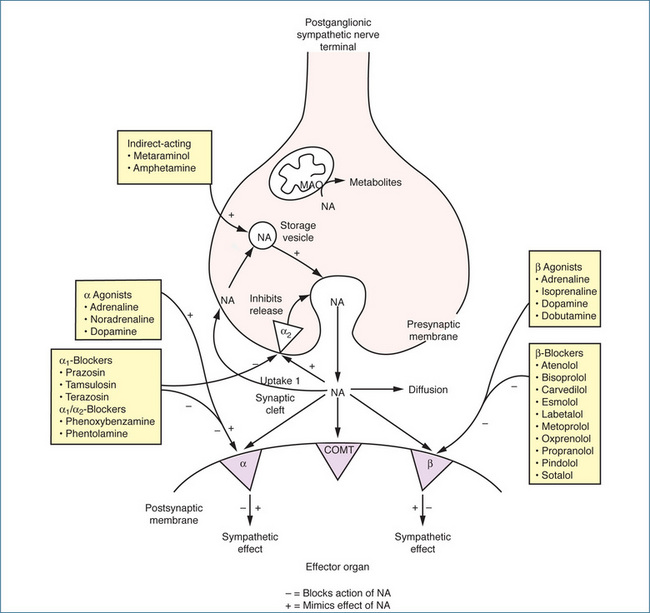
A further differentiation of β-blockers relates to intrinsic sympathomimetic activity (ISA). ISA was initially believed to be advantageous when compared with agents that exhibited β-blocking effects only. It was suggested that fewer serious adverse effects would occur with such agents but, clinically, the significance of this property has not been proved. ISA causes partial stimulation of the β-adrenoceptor, although this effect is less than that of a pure agonist. For example, if a person has a slow heart rate at rest, the partial agonists may help to increase the heart rate, but if the person has a rapid heart rate or tachycardia from exercise, these agents may help to slow the heart rate, primarily due to the predominant β-blocking effect. Table 12-2 classifies adrenergic blocking drugs by receptor activity. The proto type of the β-adrenoceptor antagonists is propranolol and it is the drug against which all others are compared.
Table 12-2 Classification of β-adrenoceptor antagonists
| TYPE | DRUGS |
| Selective β1-adrenoceptor antagonists | Atenolol, betaxolol,† bisoprolol, esmolol, metoprolol |
| Non-selective β1- and β2-adrenoceptor antagonists | Carvedilol,* labetalol,* levobunolol,† propranolol, sotalol, timolol† |
| Non-selective β1- and β2-adrenoceptor antagonists with ISA activity | Oxprenolol, pindolol |
† Available as eye-drops only.
Mechanism of action
β-Adrenoceptor antagonists competitively block β-adrenoceptor sites located on the heart, smooth muscle of the bronchi and blood vessels, kidney, pancreas, uterus, brain and liver. Cardiac muscle contains principally β1-adrenoceptors, while smooth muscle sites contain primarily β2-adrenoceptors.
Cardiovascular effects: Pharmacologically, blockade of β1-adrenoceptors on the heart decreases rate, conduction velocity, myocardial contractility and cardiac output. The antianginal effects produced by β-blockers are primarily a result of the reduction in myocardial oxygen requirements because of the diminished heart rate and myocardial contractility. Their antihypertensive actions result from decreased cardiac output (without a reflex increase in peripheral vascular resistance), diminished sympathetic outflow from the vasomotor centre in the brain to the peripheral blood vessels and reduced renin release by the kidney. Antiarrhythmic activity is associated with depression of sinus node function, slowing of conduction in the atria and the atrioventricular (AV) node and an increased refractory period of the AV node. Sotalol also prolongs the action potential duration and is used specifically as an anti-arrhythmic drug.
Central nervous system effects: Adverse effects of β-blockers include fatigue, insomnia, nightmares and depression. Although many studies have investigated an association between lipophilicity and CNS effects no clear correlation has been established. Studies have also identified that β-blockers decrease melatonin release via inhibition of central β1-adrenoceptors. Lower nocturnal melatonin concentration may contribute to the sleep disturbances.
Metabolic effects: Catecholamines are involved in the regulation of lipid and carbohydrate metabolism and, in response to hypoglycaemia, promote glycogen breakdown and mobilisation of glucose. Blockade of β-adrenoceptors prevents an adequate response to hypoglycaemia in people with insulin-dependent diabetes and may also mask the symptoms. Non-selective β-blockers raise plasma triglyceride levels and lower high-density lipoprotein levels, raising concern that this may be undesirable in people with hypertension.
Indications
β-Blocking drugs are used to treat angina pectoris, hypertension, Fallot’s tetralogy, tremors and tachycardia associated with anxiety and hyperthyroidism; to pre vent or treat cardiac arrhythmias, myocardial infarction (acute and in the long term), vascular headaches, phaeochromocytoma and glaucoma (topical eye-drops); and as an adjunct to conventional therapy for heart failure (the only approved drugs in this setting are bisoprolol, carvedilol and metoprolol).
Pharmacokinetics
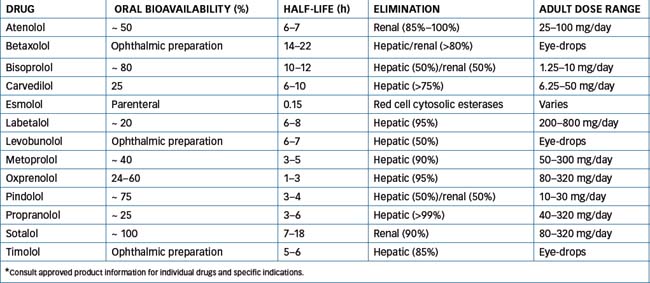
For the pharmacokinetics and usual adult dose range of β-blockers, see Table 12-3. These drugs are either metabolised in the liver or excreted as unchanged drug by the kidneys. This allows the use of different agents in pre-existing conditions of hepatic or renal impairment; for example, a drug such as metoprolol is metabolised by the liver, which is more suitable for use in persons with renal impairment, whereas atenolol is more suitable in a person with hepatic disease because it is predominantly cleared by the kidneys. When these agents are discontinued they should be withdrawn slowly to avoid inducing a potentially serious withdrawal syndrome. (See Clinical Interest Box 12-2 for information on withdrawal of a β-blocking agent.)
Clinical interest Box 12-2 Withdrawal of a β-blocking drug
Abrupt cessation of β-blockers can cause a rebound phe nomenon that exacerbates hypertension, angina or ven tricu lar arrhythmias and may precipitate a myocardial infarction. It is recommended that the dose of a β-blocking drug be halved every 2–3 days reducing the dose over 8–14 days (Therapeutic Guidelines: Cardiovascular, version 5, 2008). The person should be advised to avoid vigorous physical exercise or activity during this time to decrease the risk of a myocardial infarction or cardiac dysrhythmia. If withdrawal signs occur (angina or chest pain, sweating, rebound hypertension, dysrhythmias, tremors, tachycardia or respiratory distress), these may be controlled by temporary reinstitution of the drug.
Drug interactions and adverse reactions
See Drug Interactions 12-1 for the drug interactions of β-blockers.
Drug interactions 12-1 β-Adrenoceptor antagonists
| Drug | Possible effects and management |
| Adrenaline | Severe hypertension and bradycardia may occur. Use with extreme caution and monitor closely |
| Antidiabetic agents, oral hypoglycaemic agents, insulin | May mask symptoms of and prolong hypoglycaemia. Symptoms of hypoglycaemia such as increased heart rate and lowered blood pressure may be blocked, making monitoring difficult. Monitoring of blood glucose levels and dosage adjustments of the hypoglycaemic agent may be necessary |
| Digoxin | May have an additive effect, increasing atrioventricular conduction time. Monitor heart rate and use with caution |
| Calcium channel blockers (diltiazem and verapamil) | Enhanced cardiac-depressant effects, further decreasing rate, contractility and conduction |
| Clonidine | Combination may produce severe adverse reactions. Each drug is associated with withdrawal symptoms such as rebound hypertension. Avoid combination |
| MAO inhibitors | Combination may result in hypotension and bradycardia. Use with caution and monitor closely |
| NSAIDs | Antihypertensive effect of β-blockers may be reduced. Monitor blood pressure and avoid concurrent use |
Common adverse effects of β-blockers include insomnia, nightmares, depression, nausea, diarrhoea, dizziness, fatigue, hypotension, heart failure, heart block, bradycardia, cold hands and feet, bronchospasm and shortness of breath. Use β-blockers with caution in persons with liver or renal function impairment, heart failure, diabetes, hyperlipidaemia, peripheral vascular disease, hyperthyroidism, myasthenia gravis or phaeochromocytoma. β-blockers are contraindicated in persons with drug hyper sensitivity, cardiogenic shock, heart block, bradycardia, severe hypotension and asthma and chronic obstructive airways disease.
Key points
 The sympathetic nervous system is responsible for major physiological changes in the body in response to high demand or stressful situations. Drugs that affect this system are either adrenoceptor agonists (sympathomimetic) drugs (i.e. they mimic the effects of sympathetic nerve stimulation) or adrenoceptor antagonists (sympatholytic) drugs (i.e. drugs that compete at receptor sites to inhibit adrenergic sympathetic stimulation). These agents may be direct-acting or indirectacting drugs and affect α- and/or β-adrenoceptors. Adrenaline is an important drug (a direct-acting catecholamine) that stimulates α-, β1- and β2- adrenoceptors. It is commonly used in the treatment of asthma, emergency treatment of anaphylactic shock and cardiac arrest, treatment of local haemostasis and in management of simple open-angle glaucoma. Noradrenaline has a high affinity for α-adrenoceptors and is thus a potent peripheral arteriolar vasoconstrictor. It raises both systolic and diastolic pressure. Dobutamine is used for patients with low cardiac output because it directly stimulates the β1-adrenoceptors of the heart. Dopamine’s effects are dose-related and in the lower dose range this agent causes vasodilation of the renal and mesenteric arteries. Both these agents have been used for the treatment of circulatory shock. The adrenoceptor antagonists (sympatholytics) are classified by their receptor activity, i.e. α- and/or β-adrenoceptor antagonist effects. The main groups of α-adrenoceptor antagonists are the α1-selective antagonists such as prazosin, terazosin and tamsulosin, the nonselective α1- and α2-adrenoceptor antagonists such as phenoxybenzamine and phentolamine, and the nonselective α1- and β-adrenoceptor antagonists carvedilol and labetalol.
The sympathetic nervous system is responsible for major physiological changes in the body in response to high demand or stressful situations. Drugs that affect this system are either adrenoceptor agonists (sympathomimetic) drugs (i.e. they mimic the effects of sympathetic nerve stimulation) or adrenoceptor antagonists (sympatholytic) drugs (i.e. drugs that compete at receptor sites to inhibit adrenergic sympathetic stimulation). These agents may be direct-acting or indirectacting drugs and affect α- and/or β-adrenoceptors. Adrenaline is an important drug (a direct-acting catecholamine) that stimulates α-, β1- and β2- adrenoceptors. It is commonly used in the treatment of asthma, emergency treatment of anaphylactic shock and cardiac arrest, treatment of local haemostasis and in management of simple open-angle glaucoma. Noradrenaline has a high affinity for α-adrenoceptors and is thus a potent peripheral arteriolar vasoconstrictor. It raises both systolic and diastolic pressure. Dobutamine is used for patients with low cardiac output because it directly stimulates the β1-adrenoceptors of the heart. Dopamine’s effects are dose-related and in the lower dose range this agent causes vasodilation of the renal and mesenteric arteries. Both these agents have been used for the treatment of circulatory shock. The adrenoceptor antagonists (sympatholytics) are classified by their receptor activity, i.e. α- and/or β-adrenoceptor antagonist effects. The main groups of α-adrenoceptor antagonists are the α1-selective antagonists such as prazosin, terazosin and tamsulosin, the nonselective α1- and α2-adrenoceptor antagonists such as phenoxybenzamine and phentolamine, and the nonselective α1- and β-adrenoceptor antagonists carvedilol and labetalol.Review exercises
References and further reading
Australian Medicines Handbook 2010. Adelaide: AMH, 2010.
Benowitz N.L., Pentel P., Leatherman J. Drug use in the critically ill. Ch 13. In Speight T.M., Holford N.H.G., editors: Avery’s Drug Treatment, 4th edn, Auckland: Adis International, 1997.
Cardiovascular Expert Group. Therapeutic Guidelines: Cardiovascular, version 5, 2008. Melbourne: Therapeutic Guidelines Limited; 2008.
Cheng J., Kamiya K., Kodama I. Carvedilol: molecular and cellular basis for its multifaceted therapeutic potential. Cardiovascular Drug Reviews. 2001;19:152-171.
Gonzalez J.P., Clissold S.P. Ocular levobunolol: a review of its pharmacodynamic and pharmacokinetic properties and therapeutic efficacy. Drugs. 1987;34:648-661.
Rang H.P., Dale M.M., Ritter J.M., Flower R.J. Pharmacology, 6th edn. Edinburgh: Churchill Livingstone; 2007. [ch 11]
Ursino M.G., Vasina V., Raschi E., et al. The β3-adrenoceptor as a therapeutic target: current perspectives. Pharmacological Research. 2009;59:221-234.
Westfall T.C., Westfall D.P. Adrenergic agonists and antagonists. In Brunton L.L., Lazo J.S., Parker K.L., editors: Goodman & Gilman’s The Pharmacological Basis of Therapeutics, 11th edn, New York: McGraw Hill, 2006. [ch 10]