Chapter 67 NURSING MANAGEMENT: respiratory failure and acute respiratory distress syndrome
1. Compare the pathophysiological mechanisms that result in hypoxaemic and hypercapnic respiratory failure.
2. Differentiate between early and late clinical manifestations of acute respiratory failure.
3. Describe the nursing and collaborative management of the patient with hypoxaemic or hypercapnic respiratory failure.
4. Relate the pathophysiological mechanisms that result in acute respiratory distress syndrome (ARDS) to the clinical manifestations.
5. Describe the nursing and collaborative management of the patient with ARDS.
6. Identify complications that may result from acute respiratory failure or ARDS and the measures to prevent or reverse these complications.
acute respiratory distress syndrome (ARDS),
hypercapnic respiratory failure,
Acute respiratory failure
The major function of the pulmonary system is gas exchange, which involves the transfer of oxygen (O2) and carbon dioxide (CO2) between the atmosphere and the blood, which then transports these gases to and from the body cells. This involves three essential steps: ventilation (movement of gas into the lungs), diffusion (movement of gas between the lungs and the blood) and perfusion (transfer of blood to the tissues; see Fig 67-1). These processes are vital to the homeostatic balance of O2 and CO2 in the body. The regulation of O2 and CO2 is complex and involves multiple levels of control, including mechanical, chemical and neural controls, which are intimately coordinated to match metabolic demand.1 During periods of increased metabolism, such as when a person is exercising, chemoreceptors detect the increase in CO2 production and neural controls at the respiratory centres in the pons and medulla initiate an increase in ventilation. This occurs to adequately oxygenate the exercising muscles and remove accumulated CO2.
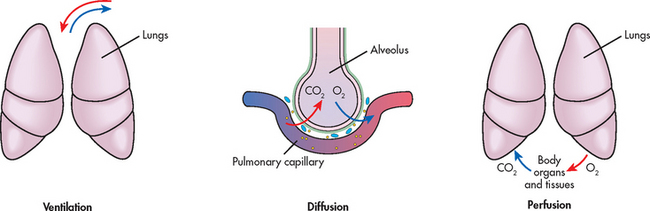
Figure 67-1 Three essential processes are involved in gas transport: ventilation, diffusion and perfusion. Ventilation occurs when air moves in and out of the lungs. Gases diffuse from the alveoli to the capillaries until the partial pressures are equal. Perfusion is the process of blood transport to and from the lungs. Alterations to any of these processes can impair gas exchange.
When disease affects the pulmonary system, changes may occur to ventilation, diffusion and perfusion. In patients with respiratory failure, the exchange of O2 and CO2 is inadequate. This means that insufficient O2 is transferred to the blood or insufficient CO2 is removed from the lungs, or both.1 Clinical states that interfere with adequate O2 transfer result in hypoxaemia, manifest as a decrease in the partial pressure of arterial O2 (PaO2) with a concomitant decrease in arterial O2 saturation (SaO2). Insufficient CO2 removal results in hypercapnia, an increase in CO2 in the blood, which causes an increase in the partial pressure of arterial CO2 (PaCO2).1
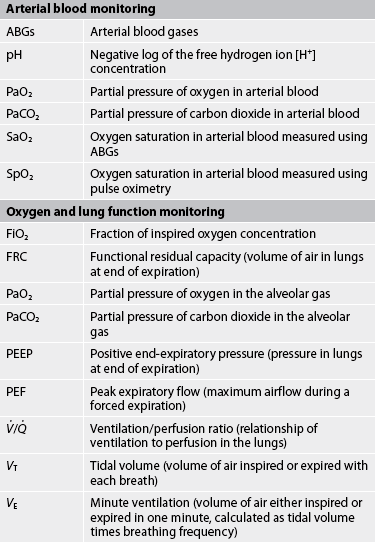
In clinical practice, arterial blood gases (ABGs) can be used to assess the changes in pH, PaO2, PaCO2 and SaO2. More routinely, pulse oximetry is used to indirectly assess the arterial O2 saturation (SpO2). Data should be interpreted within the context of the clinical assessment findings, as well as the patient’s baseline observations—for example, an individual with chronic lung disease may have a baseline PaCO2 higher than what is considered the ‘normal’ range. This needs to be considered when making clinical decisions.
Respiratory failure is not a single disease entity; rather, it is a condition that occurs as a result of one or more diseases involving the lungs or other body systems (see Tables 67-1 and 67-2). Respiratory failure can be classified as acute or chronic, depending on the acuity of the presenting signs and symptoms. Acute respiratory failure is diagnosed when the onset is sudden and gas exchange is sufficiently impaired to be life-threatening. Chronic respiratory failure arises when respiratory function declines gradually and gas exchange is impaired.2,3 The usual cause of chronic respiratory failure is chronic bronchitis and emphysema, commonly referred to as chronic obstructive pulmonary disease (COPD; see Ch 28).4,5 Both acute and chronic respiratory failure can be classified as either hypoxaemic or hypercapnic based on the gas exchange derangement.
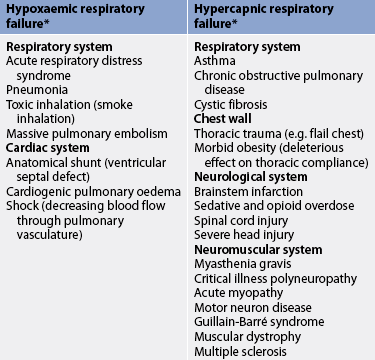
Hypoxaemic respiratory failure, also called type I respiratory failure, is a failure of oxygenation because the primary problem is inadequate O2 transfer between the alveoli and the pulmonary capillary bed.2 Although no universal definition exists, hypoxaemic respiratory failure is commonly defined as a PaO2 of 60 mmHg (8 kPa) or less despite the patient receiving an inspired O2 concentration of 60% or greater. This definition incorporates two important concepts: (1) the PaO2 is at a level that indicates inadequate O2 saturation of haemoglobin (<90%); and (2) this PaO2 level exists despite the administration of supplemental O2 at a percentage (60%) that is about three times that in the atmosphere (21%). Disorders that interfere with O2 transfer into the blood include pneumonia, pulmonary oedema, pulmonary emboli and alveolar injury related to inhalation of toxic gases (e.g. smoke inhalation). In addition, disorders that result in low cardiac output—for example, left heart failure (congestive heart failure) and shock—can also cause hypoxaemic respiratory failure.1,3
Hypercapnic respiratory failure, also called type II respiratory failure, is ventilatory failure because the primary problem is insufficient CO2 removal. Hypercapnic respiratory failure is defined as a PaCO2 above normal (>50 mmHg [>7 kPa]) with respiratory acidosis (arterial pH <7.30).3 This criteria incorporates three important concepts: (1) the PaCO2 is higher than normal (>45 mmHg [6 kPa]); (2) there is evidence of an inability to compensate for the increased PaCO2; and (3) the pH is at a level where a further decrease may lead to severe acid–base imbalance. (See Ch 16 for a discussion of acid–base balance.) Disorders that can lead to hypercapnic respiratory failure include chronic respiratory diseases that affect CO2 removal (COPD, severe asthma), drug overdose with central nervous system depressant effects, neuromuscular diseases (e.g. myasthenia gravis) and trauma that affects ventilation.
It is important to note that many patients experience both hypoxaemic and hypercapnic respiratory failure. This occurs when the ventilation is reduced leading to ventilatory insufficiency, which elevates PaCO2 and simultaneously decreases PaO2.2
The PaO2 and PaCO2 thresholds in hypoxaemic and hypercapnic respiratory failure represent a level where significant respiratory compromise will ensue. In normal healthy lungs, substantial respiratory adjustments occur when either the PaO2 is less than 60 mmHg (8 kPa) or the PaCO2 is greater than 50 mmHg (6.5 kPa) (see Fig 67-2). Therefore, in a patient with acute respiratory failure, the changes in gas exchange are significant and the patient often requires critical care management.
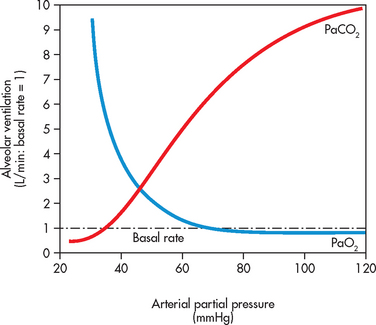
Figure 67-2 Change in arterial O2 and CO2 partial pressure against alveolar ventilation rate. As PaO2 declines, there is no change in alveolar ventilation until the level is below 60 mmHg. Thereafter, there is a rapid increase in ventilation in response to the hypoxaemia. This is sensed at the carotid chemoreceptors, which triggers the respiratory centres to increase ventilation. CO2 sensitivity changes alveolar ventilation over a smaller range. When PaCO2 increases above 50 mmHg, there is a substantial increase in ventilation. This occurs as ventilation is predominant driven by changes in CO2; therefore, it is a more sensitive driver of breathing.
AETIOLOGY AND PATHOPHYSIOLOGY
Hypoxaemic respiratory failure
Common diseases and conditions that cause hypoxaemic respiratory failure are listed in Table 67-1. Four physiological mechanisms may cause hypoxaemia and subsequent hypoxaemic respiratory failure: (1) mismatch between ventilation (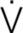 ) and perfusion (
) and perfusion (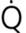 ), commonly referred to as / mismatch; (2) shunt; (3) diffusion limitation; and (4) hypoventilation. The most common causes are / mismatch and shunt.1–3
), commonly referred to as / mismatch; (2) shunt; (3) diffusion limitation; and (4) hypoventilation. The most common causes are / mismatch and shunt.1–3
Ventilation–perfusion (/) mismatch
In the normal lung, the volume of blood perfusing the lungs each minute (5–6 L/min) usually exceeds the amount of gas that reaches the alveoli each minute (4–5 L/min). In a perfectly matched system, each portion of the lung would receive about 1 mL of air for eaCh 1 mL of blood flow. This match of ventilation and perfusion would result in a / ratio of 1:1 (i.e. 1 mL of air per 1 mL of blood), which is expressed as / = 1. In this scenario, ventilation is ideally matched with perfusion. However, this is not the case. Although this example implies that ventilation and perfusion are ideally matched in all areas of the lung, this situation does not normally exist. In reality, regional mismatch throughout the lung fields is normal and dependent on many factors, such as gravity, posture, pulmonary vascular tone and alveolar space. At the lung apex, / ratios are usually greater than 1, whereby there is greater ventilation compared to perfusion. The opposite is true at the base of the lungs, where / ratios are less than 1 because there is relatively less ventilation compared with the perfusion. However, differences across the lungs even out when the entire lung is considered, giving a close overall match (see Fig 67-3).
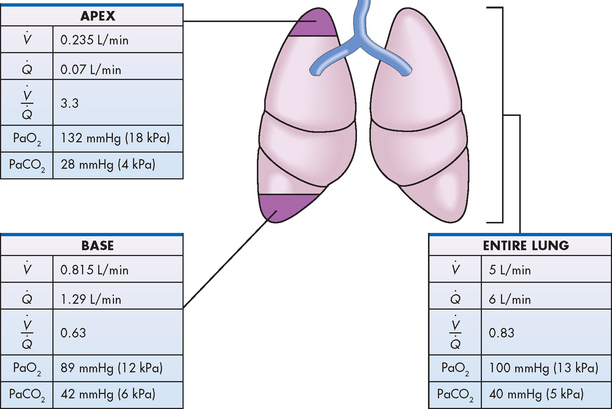
Figure 67-3 Regional / differences in the normal lung. At the lung apex, the / ratio is 3.3 as there is relatively greater ventilation compared to perfusion. The opposite occurs at the base with greater perfusion compared with ventilation, resulting in a lower / ratio, 0.63. This difference causes the PaO2 to be higher at the apex of the lung and lower at the base. Values for PaCO2 are the opposite (i.e. lower at the apex and higher at the base). When viewed across the entire lung, the average / ratio is approximately 0.83, as perfusion is slightly greater than ventilation. This is advantageous, as increases in ventilation do not require concomitant increases in basal perfusion rates.
Many diseases and conditions alter the / ratio across the lungs and cause / mismatch (see Fig 67-4). These conditions and diseases can alter either the ventilation or the perfusion, or both. The most common are those in which increased secretions are present in the airways, such as in COPD, in the alveoli (e.g. pneumonia) or when bronchospasm is present (e.g. asthma). / mismatch may also result from alveolar collapse (atelectasis) or as a result of pain; this is especially valid in patients with severe postoperative pain. Unrelieved or inadequately relieved pain diminishes chest and abdominal wall movement, compromising ventilation as patients tend to inhale smaller volumes leading to hypoventilation. Additionally, pain increases muscle and motor neuron tension (producing generalised muscle rigidity), causes systemic vasoconstriction and activation of the stress response, and increases O2 consumption and CO2 production.6 All of these conditions result in limited airflow to alveoli (ventilation) but have no effect on blood flow (perfusion) to the gas-exchange units. The consequence is / mismatch. In contrast, perfusion inequalities may result from a pulmonary embolus. The embolus limits blood flow but has no effect on airflow to the alveoli; however, this will also result in a / mismatch (see Fig 67-4).
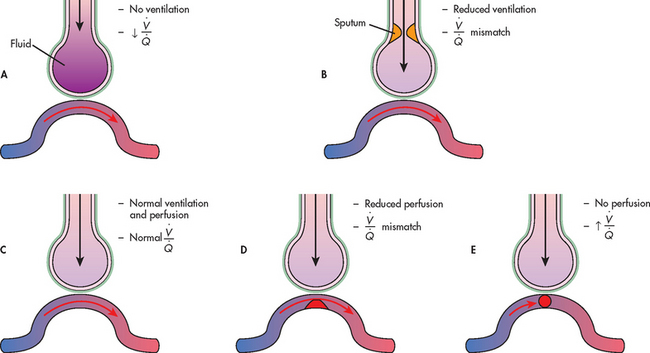
Figure 67-4 Range of ventilation to perfusion (/) relationships. A, Absolute shunt: no ventilation due to fluid filling the alveoli. B, / mismatch: ventilation partially compromised by secretions in the airway. C, Normal lung unit, with a physiologically normal /. D, / mismatch: perfusion partially compromised by clot (emboli) obstructing pulmonary capillary blood flow. E, Dead space: no perfusion due to total obstruction of the pulmonary artery.
Shunt
Shunt occurs when blood exits the heart without having participated in gas exchange. A shunt can be viewed as an extreme / mismatch (see Fig 67-4). For instance, if one entire lung was not ventilated but received all the pulmonary blood flow, this would result in a shunt. There are two types of shunt: anatomical and intrapulmonary. An anatomical shunt occurs when blood passes through an anatomical channel in the heart (e.g. ventricular septal defect) and therefore a significant amount of blood does not pass through the lungs. An intrapulmonary shunt occurs when blood flows through the pulmonary capillaries without any gas exchange occurring. An intrapulmonary shunt is seen in conditions in which the alveoli fill with fluid (e.g. acute respiratory distress syndrome [ARDS], pneumonia, pulmonary oedema). O2 therapy alone may be ineffective in increasing the PaO2 if hypoxaemia is due to shunt because: (1) blood passes from the right to the left side of the heart without passing through the lungs (anatomical shunt); or (2) the alveoli are filled with fluid, which prevents gas exchange (intrapulmonary shunt). Patients with a shunt are usually more hypoxaemic than patients with / mismatches, and they may require tracheal intubation, mechanical ventilation and a high concentration of inspired oxygen to significantly improve gas exchange.
Diffusion limitation
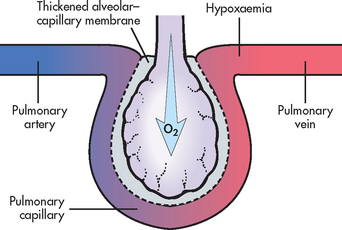
Diffusion limitation occurs when gas exchange across the alveolar–capillary membrane is compromised by a process that thickens or destroys the membrane (see Fig 67-5). Diffusion limitation can be worsened by conditions that affect the pulmonary vascular bed, such as severe emphysema or recurrent pulmonary emboli. Some diseases cause the alveolar–capillary membrane to become thicker (meaning the membrane becomes fibrotic), which slows diffusion of gases. These diseases include ARDS, pulmonary fibrosis and interstitial lung disease.
Alveolar hypoventilation
Alveolar hypoventilation is a generalised decrease in ventilation that results in an increase in the PaCO2 and a consequent decrease in PaO2. Alveolar hypoventilation may be the result of restrictive lung disease (see Ch 27), central nervous system (CNS) disease, chest wall dysfunction or neuromuscular disease. Alveolar hypoventilation is primarily a mechanism of hypercapnic respiratory failure; however, it can also cause hypoxaemia.
Interrelationship of mechanisms
Frequently, hypoxaemic respiratory failure is caused by a combination of two or more of the following: / mismatch, shunting, diffusion limitation and hypoventilation. For example, the patient with acute respiratory failure secondary to pneumonia may have a combination of / mismatch and shunt because the inflammation, oedema and hypersecretion of exudate within the bronchioles and terminal respiratory units obstruct the airways (/ mismatch) and fill the alveoli with exudate (shunt). In addition, the shunt may be increased because of improper positioning (affected lung down) and endogenous vasodilator mediators (such as prostaglandins and nitric oxide), as is the case with healthcare-acquired pneumonia.7 The patient with ARDS may have a combination of shunt and / mismatch because some alveoli are completely filled with fluid from oedema (shunt) and others are only partially filled with fluid (/ mismatch). The important aspect is that usually multiple factors cause the hypoxaemia and if severe can be very difficult to treat. Therefore, these patients need to be admitted to an intensive care unit for monitoring and respiratory interventions, such as tracheal intubation, if respiratory insufficiency is pronounced.
Hypercapnic respiratory failure
Hypercapnic respiratory failure results from an imbalance between ventilatory supply and ventilatory demand. Ventilatory capacity or supply is the maximum ventilation (maximal gas flow in and out of the lungs) that the patient can sustain without developing respiratory muscle fatigue. The ventilator capacity is dependent on the oxygen consumption requirements, CO2 production and deadspace. In contrast, ventilatory demand is the amount of ventilation needed to keep the PaCO2 within normal limits (35–45 mmHg [4–6 kPa]). The total amount of ventilatory supply is related to the central respiratory drive from the respiratory centres in the brainstem, ventilator musculature and the mechanics of breathing. Normally, ventilatory supply far exceeds ventilatory demand. As a consequence, individuals with normal lung function can engage in activities that increase metabolic rate, such as strenuous exercise, which greatly increases CO2 production, without causing an elevation in PaCO2. Conversely, patients with pre-existing lung disease, such as severe emphysema, do not have this advantage and cannot effectively increase lung ventilation in response to metabolic demands or exercise. Considerable dysfunction is typically present before ventilatory demand exceeds ventilatory supply. Usually, at this stage the patient’s condition is quite severe and therefore their capacity to cope with increased ventilatory requirements is greatly limited.
When ventilatory demand does exceed ventilatory supply, the PaCO2 can no longer be sustained within normal limits and hypercapnia occurs. With the presence of hypercapnia there is substantial lung dysfunction. Hypercapnic respiratory failure is sometimes called ventilatory failure because the primary problem is the inability of the respiratory system to excrete sufficient CO2 to maintain a normal PaCO2. One of the most typical presentations of hypercapnic respiratory failure is in patients with chronic respiratory insufficiency who experience an acute exacerbation of their condition, which precipitates the acute hypercapnic ventilatory failure.
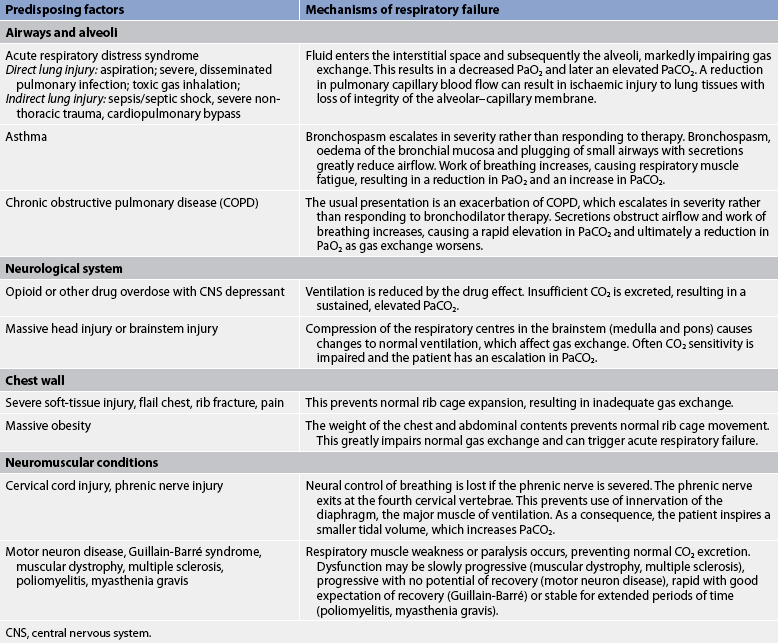
Many different diseases can cause a limitation in ventilatory supply (see Tables 67-1 and 67-2). These diseases can be grouped into four categories: (1) abnormalities of the airways and alveoli; (2) abnormalities of the CNS; (3) abnormalities of the chest wall; and (4) neuromuscular conditions.
• Airways and alveoli. Patients with asthma, emphysema, chronic bronchitis (COPD) and cystic fibrosis are at high risk of hypercapnic respiratory failure because the underlying pathophysiology of these conditions results in airflow obstruction and air trapping.
• Central nervous system. A variety of problems may suppress the drive to breathe. A common example is an overdose of an opioid or other respiratory depressant drug. A brainstem infarction or severe head injury may also interfere with the normal function of the respiratory centre in the medulla. Patients with these conditions are at risk of respiratory failure because the respiratory centre in the medulla cannot alter the respiratory rate in response to an elevation in PaCO2. CNS dysfunction may also include spinal cord injuries that limit innervation to the respiratory muscles, with injuries above C5 usually causing some degree of respiratory compromise. (Spinal cord injury is discussed in Ch 60.)
• Chest wall. A variety of conditions may prevent normal movement of the chest wall and hence limit lung expansion. In patients with a flail chest, fractures prevent the rib cage from expanding normally because of pain, mechanical restriction and muscle spasm. In patients with morbid obesity (body mass index [BMI] equal to or greater than 40 kg/m2), the weight of the chest and abdominal contents may limit full lung expansion. Patients with these conditions are at risk of respiratory failure because these dysfunctions limit lung expansion or diaphragmatic movement and, consequently, gas exchange.
• Neuromuscular conditions. Various types of neuromuscular diseases may result in respiratory muscle weakness or paralysis (see Table 67-1). For example, patients with Guillain-Barré syndrome, muscular dystrophy or multiple sclerosis are at risk of respiratory failure because the respiratory muscles are weakened or paralysed as a consequence of the underlying neuromuscular condition. Therefore, they are unable to maintain normal PaCO2 levels.
In summary, respiratory failure may occur in these three categories (CNS, chest wall, neuromuscular conditions) despite the presence of normal lungs. Respiratory failure occurs because the medulla, chest wall, peripheral nerves or respiratory muscles are not functioning normally. The patient may have no damage to lung tissue but may have a tidal volume that is insufficient to expel CO2 from the lungs.
Tissue oxygen needs
It is important to remember that even though PaO2 and PaCO2 determine the pathophysiological origin of the respiratory failure, the major threat is the inability to meet the O2 demands of the tissues (hypoxaemia). This inability may occur as a result of inadequate tissue O2 delivery or because the tissues are unable to use the O2 delivered to them. It may also occur as a result of the stress response and dramatic increases in tissue O2 consumption.1 Tissue O2 delivery is determined by the amount of O2 carried on haemoglobin (oxyhaemoglobin), as well as cardiac output (heart rate × stroke volume). Thus, respiratory failure places the patient at greater risk if there are pre-existing cardiac problems or anaemia. Failure of O2 utilisation most commonly occurs as a result of septic shock. In this situation, adequate O2 may be delivered to the tissues but an abnormally high amount of O2 returns in the venous blood, indicating that O2 uptake at the tissue level is impaired. (Shock is discussed in Ch 66.)
CLINICAL MANIFESTATIONS
As mentioned, respiratory failure may develop acutely or be chronic. A sudden decrease in PaO2 or a rapid rise in PaCO2, without adequate compensation, implies a serious condition, which can develop into a life-threatening clinical emergency. An example is an asthmatic patient who develops severe bronchospasm with a marked decrease in airflow, resulting in a respiratory arrest. In contrast, a gradual change in PaCO2 and, to a lesser extent, PaO2 is better tolerated because compensation can occur. An example is the patient with COPD who develops a progressive increase in PaCO2 over several days following the onset of a respiratory tract infection. Because the change occurred over several days, there is usually adequate renal compensation of the respiratory acidosis (e.g. retention of bicarbonate), which minimises the decrease in arterial pH. The patient has compensated for the respiratory acidosis (discussed in detail in Ch 16).1,8
Manifestations of respiratory failure are related to the extent of change in PaO2 or PaCO2, or both, the rapidity of change (acute versus chronic) and the patient’s ability to compensate these changes. When the patient’s compensatory mechanisms are inadequate or fail, respiratory failure occurs. As clinical manifestations are variable, it is important to monitor trends in respiratory signs and symptoms (such as dyspnoea, respiratory rate, thoracic expansion), ABGs and pulse oximetry to evaluate the extent of change. Frequently, the initial indication of respiratory failure is a change in the patient’s mental status, as the cerebral cortex is sensitive to variations in oxygenation and acid–base balance. Mental status changes often occur early and restlessness, confusion, agitation and combative behaviour in a patient suggest inadequate delivery of cerebral O2 and should be fully investigated.
The nurse may detect manifestations of respiratory failure that are specific (arise from the respiratory system) or non-specific (arise from other body systems; see Table 67-3). An understanding of the significance of these manifestations is critical to the ability to detect the onset of respiratory failure and the effectiveness of treatment.

Tachycardia and mild hypertension can also be early signs of respiratory failure. Such changes may indicate an attempt by the cardiovascular system to compensate for decreased O2 delivery. A severe morning headache may suggest that hypercapnia may have been present overnight, increasing cerebral blood flow by vasodilation and causing the headache. Rapid, shallow breaths suggest that the tidal volume may be inadequate to remove CO2 from the lungs. Cyanosis is an unreliable indicator of hypoxaemia and is a late sign of respiratory failure because it does not occur until hypoxia is severe (typically a PaO2 <45 mmHg [<6 kPa]).9
Consequences of hypoxaemia
Hypoxaemia occurs when the O2 concentration in arterial blood is less than the normal value. This usually leads to hypoxia, in which there is low cellular/tissue oxygenation. It should be noted that hypoxaemia is not the sole cause of hypoxia; patients can experience hypoxia in the presence of anaemia, despite an adequate PaO2. Hypoxaemia/hypoxia occurs when the PaO2 has fallen sufficiently to cause signs and symptoms of inadequate oxygenation (see Table 67-3). If hypoxia or hypoxaemia is severe, there may be a shift from aerobic to anaerobic metabolism in some body regions. For example, renal tubular cells are very sensitive to hypoxia and will undergo anaerobic metabolism and die without adequate oxygenation.10 Anaerobic metabolism uses more fuel (e.g. glucose) to produce less energy (ATP) and is therefore less efficient in ATP production than aerobic metabolism pathways. Additionally, waste products of anaerobic metabolism, such as lactic acid, are more difficult to remove from the body than CO2 because lactic acid has to be buffered with sodium bicarbonate. When the body does not have adequate amounts of sodium bicarbonate to buffer the lactic acid, metabolic acidosis results and cell death may occur.
Hypoxaemia and metabolic acidosis have deleterious adverse effects on the vital organs, especially the heart and brain. Cardiac compensation for a decreased arterial O2 level causes an increase in heart rate and cardiac output. As the PaO2 decreases and acidosis increases, the myocardium (heart muscle) may become dysfunctional and cardiac output may decrease. In addition, angina and arrhythmias may occur. All of these consequences result in a further decrease in oxygen delivery. Permanent brain damage may occur because of O2 deprivation. Renal function may also be impaired, and sodium retention, oedema formation, acute tubular necrosis and uraemia may occur. Gastrointestinal (GI) system alterations include tissue ischaemia, increased permeability of the intestinal wall and possible translocation of bacteria from the GI tract into the circulation.
Specific clinical manifestations
The patient in acute respiratory failure may have several clinical findings indicating distress. One of the most common clinical findings is tachypnoea. Patients often present with rapid breathing indicative of hypoxaemia or hypercapnia. Patients may increase the respiratory rate in an effort to exhale accumulated CO2 or increase O2 content. However, this breathing pattern requires a substantial amount of work (termed the work of breathing) and predisposes the patient to respiratory muscle fatigue. A change from a rapid rate to a slower rate in a patient in acute respiratory distress suggests extreme fatigue and the possibility of an impending respiratory arrest.
The position that the patient assumes is an indication of the effort associated with breathing. The patient may be able to lie down (mild distress), be able to lie down but prefer to sit (moderate distress) or be unable to breathe unless sitting upright (severe distress). A common position is to sit with the arms propped on the bedside table. This position, called the tripod position, helps decrease the work of breathing because propping the arms increases the anterior–posterior diameter of the chest and changes the pressure in the thorax. Pursed-lip breathing may also be observed. This strategy causes an increase in SaO2 because it slows the breathing rate, allows more time for expiration and increases intrathoracic pressure, which prevents the small bronchioles from collapsing, thus facilitating gas exchange. (Pursed-lip breathing is discussed in Ch 25.) Another assessment parameter is the number of pillows the patient requires to breathe comfortably when resting in bed. This is termed orthopnoea and may be documented as one-, two-, three- or four-pillow orthopnoea. While this is a crude differentiation, it provides evidence about the magnitude of dyspnoea and possible respiratory compromise, and it is easy to explain and understand.
The patient who is experiencing dyspnoea is working hard to breathe and may be able to speak only a few words at a time between breaths. The patient’s ability to speak without pausing to breathe is an indication of the severity of dyspnoea. The patient may speak in sentences (mild or no distress), phrases (moderate distress) or words only (severe distress). The number of words is also a clue (e.g. how many words can the patient say without pausing to breathe?). There may also be earlier onset of fatigue with walking. An additional assessment parameter is how far the patient is able to walk without stopping to rest. In fact, assessment criteria for patients with lung disease who require pulmonary surgery include the 6-minute walk test, which measures the distance a patient can walk in 6 minutes.11
There may be a change in the inspiratory (I) to expiratory (E) (I:E) ratio. Normally, the I:E ratio is 1:2, which means that expiration is twice as long as inspiration. In patients in respiratory distress, the ratio may increase to 1:3 or 1:4. This change signifies airflow obstruction and that more time is required to empty the lungs.
The nurse may observe retraction (i.e. inward movement) of the intercostal spaces or the supraclavicular area and use of the accessory muscles during inspiration or expiration. Use of the accessory muscles signifies moderate distress. Paradoxical breathing indicates severe distress. Normally, the thorax and abdomen move outwards on inspiration and inwards on exhalation. During paradoxical breathing, the abdomen and chest move in the opposite manner—outwards during exhalation and inwards during inspiration. Paradoxical breathing results from maximal use of the accessory muscles of respiration. The patient may also be diaphoretic from the work associated with breathing.
Auscultation should be performed in order to assess the patient’s baseline breath sounds, as well as any changes from the baseline. The nurse should note the presence and location of any adventitious breath sounds. Crackles may indicate pulmonary oedema or emphysema. Absent or diminished breath sounds may indicate atelectasis, pleural effusion or pneumothorax. The presence of bronchial breath sounds over the lung periphery often results from lung consolidation that is seen with pneumonia. A pleural friction rub may also be heard in the presence of pneumonia that has involved the pleura.
A thorough nursing assessment may result in early detection of manifestations associated with respiratory insufficiency, allowing therapy to be instituted before the patient experiences respiratory failure. Patients with end-stage (severe) chronic lung disease are likely to have elevated PaCO2 or low PaO2 values and crackles as their ‘normal’ baseline observations. It is especially important to monitor specific and non-specific signs of respiratory failure in patients with COPD because a small change can cause significant exacerbation of the baseline condition, which is often tenuous (see Table 67-3). Any deterioration in mental status, such as agitation, combative behaviour, confusion or decreased level of consciousness, should be reported and acted on immediately, because this change may indicate the onset of rapid deterioration in clinical status and the need for mechanical ventilation.
DIAGNOSTIC STUDIES
After physical assessment, the most common diagnostic study used to determine respiratory failure is ABG analysis. ABGs are used to determine the levels of pH, PaCO2, bicarbonate (HCO3−), PaO2, SaO2 and base excess (BE). Collectively, the pH, PaCO2, and HCO3− provide information about the type and magnitude of acid–base disturbance. A cannula may be inserted into a peripheral artery (usually the radial artery) for monitoring systemic blood pressure and obtaining blood routinely for ABG analyses. Pulse oximetry is frequently used for continuous monitoring of oxygenation status but provides no reliable information about ventilation. In acute respiratory failure, ABG results are necessary to determine both ventilation (PaCO2) status and oxygenation (PaO2) status, as well as information related to acid–base balance.
Other diagnostic studies that may be done include a chest X-ray, full blood count, serum electrolyte levels, urinalysis and electrocardiogram (ECG). Cultures of the sputum and blood are obtained in patients with suspected infection. If pulmonary embolus is suspected, a / lung scan or pulmonary angiography may be done. For patients in severe respiratory failure requiring endotracheal intubation, end-tidal CO2 monitoring may be used to assess tube placement within the trachea immediately following intubation. End-tidal CO2 is the partial pressure of CO2 at the end of expiration and is a non-invasive measurement of alveolar ventilation and pulmonary blood flow.12 End-tidal CO2 monitoring may also be used throughout mechanical ventilation to assess trends in lung function as determined by expired CO2.
In severe respiratory failure, a pulmonary artery catheter may be inserted to measure cardiac and pulmonary pressures and cardiac output, as well as mixed venous oxygen saturation or gases. This information is helpful in determining the adequacy of tissue perfusion and the patient’s response to treatment measures. Pulmonary artery systolic, diastolic and wedge pressures are monitored to determine whether the accumulation of fluid in the lungs is the result of cardiac or pulmonary problems. These parameters are also monitored to determine the response of the lungs and heart to hypoxaemia and the patient’s response to therapy. In addition, pulmonary arterial pressure monitoring can provide feedback on the physiological effects of mechanical ventilation on haemodynamic status. However, it is important to acknowledge that the use of pulmonary artery catheters is controversial in critically ill patients. Often, there are no useful clinical indicators associated with the catheters and the decision to insert a pulmonary artery catheter is usually based on the doctor’s preference, as clinical trials do not support their use.13
 NURSING AND COLLABORATIVE MANAGEMENT: ACUTE RESPIRATORY FAILURE
NURSING AND COLLABORATIVE MANAGEMENT: ACUTE RESPIRATORY FAILURE
Because many different problems cause respiratory failure, specific care of these patients is varied. This section discusses general assessment and multidisciplinary care measures that apply to patients with acute respiratory failure. In acute care settings the whole healthcare team is generally involved in the patient’s care and there is often an overlap in function between nurses and other members of the multidisciplinary team.
Nursing assessment
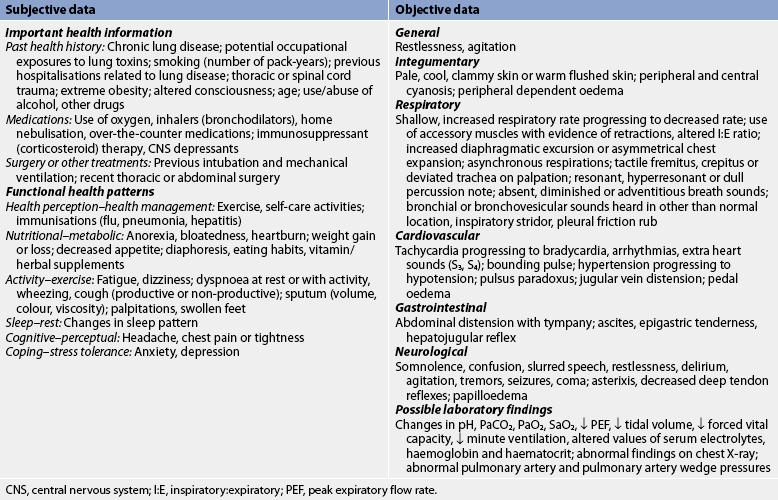
Subjective and objective data that should be obtained from the patient with acute respiratory failure are presented in Table 67-4.
Nursing diagnoses
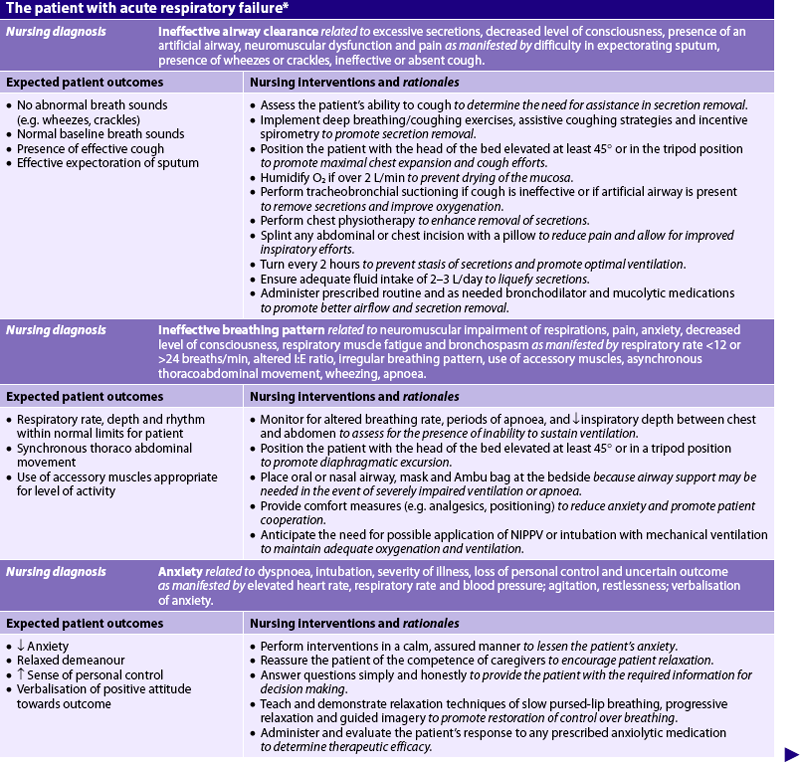
Nursing diagnoses for the patient with acute respiratory failure include, but are not limited to, those presented in NCP 67-1.
NURSING CARE PLAN 67-1 The patient with acute respiratory failure*
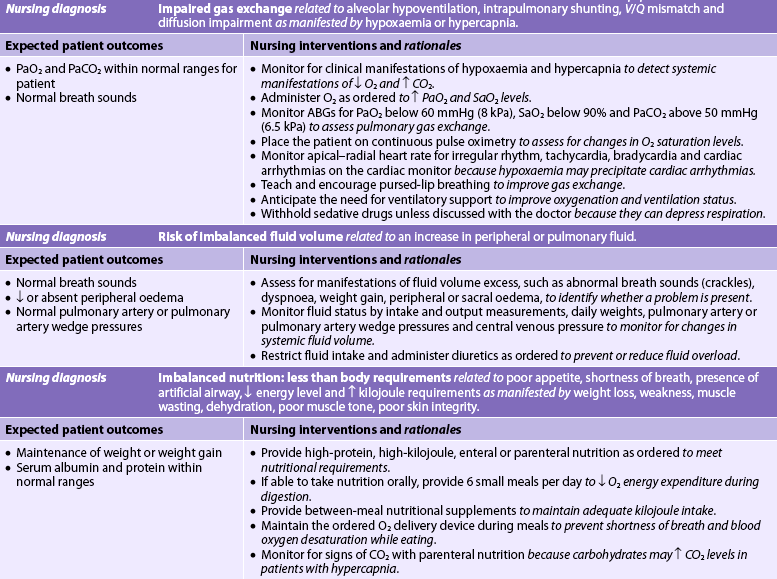
ABGs, arterial blood gases; I:E, inspiratory:expiratory; NIPPV, non-invasive positive pressure ventilation; /, ventilation–perfusion.
*The nursing care for the patient on mechanical ventilation is presented in NCP 65-1 and discussed in Chapter 65.
Planning
The overall goals are that the patient with acute respiratory failure will have: (1) ABG values within the patient’s baseline levels; (2) breath sounds within the patient’s baseline; (3) no dyspnoea, and breathing patterns within the patient’s baseline; and (4) effective cough and ability to clear secretions.
Prevention
As part of the plan of care for any patient who may be at risk of respiratory failure, prevention and early recognition of respiratory distress are important. Prevention involves a thorough physical assessment and history to identify the patient at risk of respiratory failure, followed by the initiation of appropriate nursing interventions. For example, a patient at risk of respiratory failure should receive appropriate patient teaching regarding coughing, deep breathing, incentive spirometry and ambulation as appropriate. Preventing atelectasis, pneumonia and complications of immobility, as well as optimising hydration and nutrition, can potentially decrease the risk of respiratory failure in the acutely or critically ill patient.
Respiratory therapy
The major goals of care for acute respiratory failure include maintaining adequate oxygenation and ventilation. This goal is accomplished by collaboration between nursing, medical and physiotherapy teams. The interventions used include O2 therapy, mobilisation of secretions and positive pressure ventilation (see Box 67-1).
BOX 67-1 Acute respiratory failure
MULTIDISCIPLINARY CARE
Oxygen therapy
Oxygen therapy is an essential first step to reverse hypoxaemia, especially caused by / mismatch. This is because not all gas-exchange units are affected. O2 therapy will increase the PaO2 in the blood leaving normal gas-exchange units, thus raising PaO2. This also increases the affinity of O2 for haemoglobin, thereby increasing O2 saturation. This well-oxygenated blood mixes with the poorly oxygenated blood from the alveolar regions that are underventilated, raising the overall PaO2 of blood leaving the lungs. Initially, supplemental O2 administered at 1–3 L/min by nasal cannula or 24–50% by simple face mask or Venturi mask should improve the PaO2 and SaO2. If not, the application of a non-rebreather O2 mask can provide higher concentrations of O2 delivery (up to 70%; see Fig 67-6). The optimal approach to hypoxaemia caused by a / mismatch is one that treats the cause. Hypoxaemia secondary to an intrapulmonary shunt is usually not responsive to high O2 concentrations and the patient will usually require positive pressure ventilation (PPV). PPV may be provided via an endotracheal tube (most frequently) or non-invasively by means of a tight-fitting mask.14 (Mechanical ventilation is discussed in detail in Ch 65.) PPV offers a means of providing O2 therapy and humidification, decreasing the work of breathing and reducing respiratory muscle fatigue. In addition, the positive pressure may assist in opening collapsed airways (alveolar recruitment) and decreasing shunt.
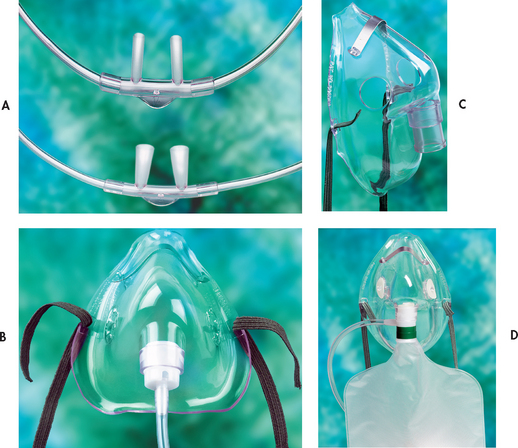
Figure 67-6 Delivery devices for supplemental O2. A, Nasal cannulas for delivery of 2 L/min O2 (24–44% concentration). B, Face mask (Hudson) for delivery of 5–10 L/min of O2 (40–60% concentration). C, Venturi mask for precise O2 concentration delivery between 24% and 50%. These masks provide high-flow O2. D, Non-rebreather mask for high concentration O2 delivery.
Source: Walsh TD. Palliative medicine. Philadelphia: Saunders; 2008.
The type of O2 delivery system chosen for the patient in acute respiratory failure should: (1) be tolerated by the patient, because anxiety caused by feelings of claustrophobia related to the face mask or dyspnoea may prompt the patient to remove the O2 device; and (2) maintain PaO2 at 55–60 mmHg (7.4–8 kPa) or more and SaO2 at 90% or more at the lowest O2 concentration possible. High O2 concentrations replace the nitrogen gas normally present in the alveoli, causing instability and atelectasis. In intubated patients, exposure to 60% or more O2 for longer than 48 hours poses a significant risk of O2 toxicity. In non-intubated patients, the risk is less clear. The effects of prolonged exposure to high levels of O2 include increased pulmonary microvascular permeability, decreased surfactant production and surfactant inactivation, and fibrotic changes in the alveoli.15
Additional risks of O2 therapy are specific to the patient with chronic hypercapnia, such as the patient with COPD (see Ch 28). Chronic hypercapnia may blunt the response of chemoreceptors in the medulla, a condition termed CO2 narcosis. In this situation, ventilation and breathing rate are stimulated by hypoxia (PaO2 less than 60 mmHg [8 kPa]). If the PaO2 is suddenly increased, the patient will no longer be hypoxic, will often have a decreased stimulus to breathe and may experience respiratory arrest. The patient with chronic hypercapnia should receive low concentration O2 through a nasal cannula at 1–2 L/min, or a Venturi mask at 24–28%. They should be closely monitored for changes in mental status, respiratory rate and ABG results until the PaO2 level has reached the patient’s baseline value, which may be less than the normal values.
Mobilisation of secretions
Retained pulmonary secretions may cause or exacerbate acute respiratory failure by blocking movement of O2 into the alveoli and pulmonary capillary blood and removing CO2 during the respiratory cycle. Secretions can be mobilised through effective coughing, adequate hydration and humidification, chest physiotherapy and tracheal suctioning.
Effective coughing and positioning
If secretions are obstructing the airway, the patient should be encouraged to cough. The patient with a neuromuscular weakness or exhaustion may not be able to generate sufficient airway pressures to produce an effective cough. Assisted coughing may be of benefit to these patients. Assisted coughing is performed by placing the palm of the hand or hands on the abdomen below the xiphoid process. As the patient ends a deep inspiration and begins the expiration, the hands should be moved forcefully downwards, increasing abdominal pressure and facilitating the cough. This measure helps increase expiratory flow and thereby facilitates secretion clearance.
Some patients may benefit from therapeutic cough techniques. Huff coughing is a series of coughs performed while saying the word ‘huff’. This technique prevents the glottis from closing during the cough. Patients with COPD generate higher flow rates with a huff cough than is possible with a normal cough. The huff cough is effective in clearing only the central airways but it may assist in moving secretions upwards. The staged cough also assists secretion mobilisation. To perform the staged cough, the patient sits in a chair, breathes three or four times in and out through the mouth, and coughs while bending forwards and pressing a pillow inwards against the diaphragm.
Positioning the patient either by elevating the head of the bed to at least 45° or by using a reclining chair or chair bed may help maximise thoracic expansion, thereby decreasing dyspnoea and improving secretion mobilisation. A sitting position improves pulmonary function and assists in venous pooling in dependent body areas, such as the lower extremities. When lungs are upright, ventilation and perfusion are best in the lung bases. Lateral or side-lying positioning may be used in patients with disease involving only one lung. This position, termed good lung down, allows for improved / matching in the affected lung. Pulmonary blood flow and ventilation are optimal in dependent lung areas. This positioning also allows for secretions to drain out of the affected lung to the point where they may be removed by suctioning. For example, in patients with significant right middle lobe pneumonia, optimal positioning would be to place them on their left side to maximise ventilation and perfusion in the ‘good’ lung and facilitate secretion removal from the affected lung (postural drainage). All patients should be side-lying if there is any possibility that the tongue will obstruct the airway or that aspiration may occur. An oral or nasal airway should be kept at the bedside for use if necessary.
Hydration and humidification
Thick and viscous secretions are difficult to remove and should be thinned. Adequate fluid intake (2–3 L per day) is necessary to keep secretions thin and easy to expel. If the patient is unable to take sufficient fluids orally, intravenous (IV) hydration will be necessary. Thorough assessment of the patient’s cardiac and renal status is paramount to determine whether they can tolerate the intravascular volume and avoid congestive heart failure and pulmonary oedema. Assessment for signs of fluid overload (e.g. crackles, dyspnoea and increased central venous pressure) at regular intervals is paramount. These considerations would also apply to the patient with renal dysfunction. An appropriate humidification device is an adjunct in secretion management. Aerosols of sterile normal saline, administered by a nebuliser, may be used to liquefy secretions. Oxygen may also be administered by aerosol mask to thin secretions and facilitate their removal. Aerosol therapy may induce bronchospasm and severe coughing, causing a decreased PaO2. As such, frequent assessment of patient tolerance to therapy is paramount.16 Mucolytic agents, such as nebulised acetylcysteine mixed with a bronchodilator, may be used to thin secretions but, as a side effect, they may also cause airway erythema and bronchospasm. They are therefore used only in specific situations (e.g. during bronchoscopy to remove thick, copious secretions).
Chest physiotherapy
Chest physiotherapy is indicated in patients who produce more than 30 mL of sputum per day or have evidence of severe atelectasis or pulmonary infiltrates. If tolerated, postural drainage, percussion and vibration to the affected lung segments may assist in moving secretions to the larger airways, where they may be removed by coughing or suctioning. Because positioning may affect oxygenation, patients may not tolerate head-down or lateral positioning because of extreme dyspnoea or hypoxaemia caused by / mismatch. (Chest physiotherapy is discussed in Ch 28.)
Airway suctioning
If the patient is unable to expectorate secretions, nasopharyngeal, oropharyngeal or nasotracheal suctioning (blind suctioning without a tracheal tube in place) is indicated. Suctioning through an artificial airway, such as an endotracheal or tracheostomy tube, may also be performed (see Chs 27 and 65).
Positive pressure ventilation
If intensive care measures fail to improve ventilation and oxygenation and the patient continues to exhibit manifestations of acute respiratory failure, ventilatory assistance may be initiated. PPV may be provided invasively through endotracheal or nasotracheal intubation or non-invasively through a nasal or face mask. Patients who require PPV are typically cared for in a critical care unit.
Non-invasive positive pressure ventilation (NIPPV) may be used as a treatment for patients with acute or chronic respiratory failure. During NIPPV a mask is placed over the patient’s nose or nose and mouth and the patient breathes spontaneously while PPV is delivered (see Fig 67-7). With NIPPV it is possible to decrease the work of breathing without the need for endotracheal intubation. Bilevel positive airway pressure (BiPAP ventilatory support system) is a form of NIPPV in which different positive pressure levels are set for inspiration and expiration (see Fig 67-7). Continuous positive airway pressure (CPAP) is another form of NIPPV in which a constant positive pressure is delivered to the airway during inspiration and expiration.14
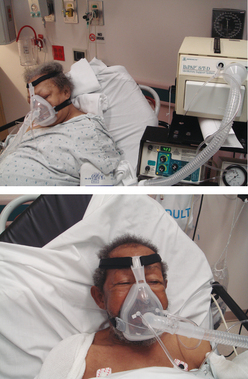
Figure 67-7 Non-invasive bilevel positive airway pressure ventilation. A mask is placed over the nose or the nose and mouth. Positive pressure from a mechanical ventilator assists the patient’s breathing efforts, decreasing the work of breathing.
Source: Reardon RF, Mason PE, Clinton JE. Basic airway management and decision-making. In: Roberts JR. Clinical procedures in emergency medicine. 5th edn. Philadelphia: Saunders; 2009.
NIPPV is most useful in managing chronic respiratory failure in patients with COPD,17,18 chest wall and neuromuscular disease (see Table 67-1). NIPPV has been used in patients with hypoxaemic respiratory failure (e.g. ARDS, cardiogenic pulmonary oedema) but with less success.19–21 NIPPV is not appropriate for the patient who has absent respirations, excessive secretions, decreased level of consciousness, high O2 requirements, facial trauma or haemodynamic instability. Clinical trials are continuing in an attempt to find the optimal use of NIPPV in patients with acute hypoxaemic and hypercapnic and mixed respiratory failures.
Drug therapy
Goals of drug therapy for patients in acute respiratory failure include relief of bronchospasm, reduction of airway inflammation and pulmonary congestion, treatment of pulmonary infection, and reduction of severe anxiety and restlessness.
Relief of bronchospasm
Alveolar ventilation will be increased with relief of bronchospasm. Short-acting bronchodilators, such as salbutamol, are frequently administered to reverse bronchospasm using either a hand-held nebuliser or a metered-dose inhaler with a spacer. In acute bronchospasm this drug may be given at 15–30 minute intervals until it can be determined that a response is occurring. If severe bronchospasm continues, IV salbutamol or adrenaline may be administered. The bronchodilator effects of all this medication can sometimes cause a worsening of arterial hypoxia by redistributing the inspired gas to areas of decreased perfusion. Due to the general β-receptor effects of these medications, patients are likely to experience an increased heart rate. In those with existing cardiac disease, chest pain or arrhythmias may be precipitated, so close assessment and pre-emptive care may be required.
Reduction of airway inflammation
Corticosteroids (e.g. methylprednisolone) may be used in conjunction with bronchodilator agents when bronchospasm and inflammation are present. When administered IV, corticosteroids have an immediate onset of action. Inhaled corticosteroids are not used for acute respiratory failure because they require 4–5 days before optimum therapeutic effects are seen. Evidence suggests that moderate-dose corticosteroids improve respiratory function; however, more clinical trials are required to ascertain the full effect.22,23
Reduction of pulmonary congestion
Pulmonary interstitial fluid can occur as a consequence of direct or indirect injury to the alveolar capillary membrane (e.g. ARDS) or from right- or left-sided heart failure, and therefore can be either cardiac or non-cardiac in origin. The result is decreased alveolar ventilation and hypoxaemia. IV diuretics (e.g. frusemide) are used to decrease the pulmonary congestion caused by heart failure. If atrial fibrillation is also present, calcium channel blockers (e.g. diltiazem) and β-adrenergic blockers (e.g. metoprolol) may be used to decrease heart rate and improve cardiac output (see Ch 34).
Treatment of pulmonary infections
Pulmonary infections (pneumonia, acute bronchitis) result in excessive mucus production, fever, increased oxygen consumption and inflamed, fluid-filled or collapsed alveoli. Alveoli that are fluid-filled or collapsed cannot participate in gas exchange. Pulmonary infections can either cause or exacerbate acute respiratory failure. IV antibiotics, such as cephalosporins (e.g. ceftriaxone), are frequently administered to treat bacterial infections. Chest X-rays are performed to determine the location and extent of a suspected infectious process. Sputum cultures are used to determine the type of organisms causing the infection and their sensitivity to antimicrobial medications.
Reduction of severe anxiety, pain and agitation
Anxiety, restlessness and agitation result from cerebral hypoxia. In addition, fear caused by the inability to breathe and a sense of loss of control may exacerbate anxiety. Anxiety, pain and agitation increase O2 consumption, which may worsen the degree of hypoxaemia. Anxiety, pain and agitation also increase CO2 production, affect ventilator management and increase morbidity.24–26 Several nursing strategies can assist the patient in reducing the level of anxiety and pain (see NCP 67-1).
Sedation and analgesia with drug therapy such as benzodiazepines (e.g. diazepam, midazolam) and opioids (e.g. morphine, fentanyl) may be used to decrease anxiety, agitation and pain. Continued agitation will increase the patient’s work of breathing, O2 consumption, CO2 production and risk of injury (e.g. accidental extubation). When receiving any sedative or analgesic agent, patients must be monitored closely for haemodynamic instability and respiratory depression.25,27 In the critical care setting, a combination of sedation and analgesia medications is commonly used for severely restless, anxious and agitated patients who may be experiencing pain and are in acute respiratory failure, although there is evidence to suggest that this prolongs time on the mechanical ventilator and weaning in critically ill patients with respiratory failure.28
Patients who breathe asynchronously with mechanical ventilation (‘fight the ventilator’) may benefit from addressing treatable causes of agitation, such as hypoxaemia, hypercapnia or pain. Patients who remain asynchronous during positive pressure ventilation may require neuromuscular blocking agents, such as vecuronium, to produce skeletal muscle relaxation (predominately the diaphragm) and synchrony with mechanical ventilation, although the use of neuromuscular blockade remains controversial in critically ill patients.29 Neuromuscular block may also decrease the patient’s risk of lung injury related to excessive inspiratory/intrathoracic pressures. In this way, the ventilator can then provide optimal respiratory support. Patients receiving neuromuscular block should receive adequate sedation and analgesia to the point of unconsciousness for comfort, to eliminate awareness and to avoid the terrifying experience of being awake and in pain while paralysed. Monitoring levels of sedation in patients receiving neuromuscular blockade is challenging. Non-invasive electroencephalogram-based technology is available that may help guide sedative and analgesic therapy in this population.30 Monitoring levels of pharmacological paralysis is commonly done through the use of a peripheral nerve stimulator. Clinical assessment is essential to determine the adequacy of sedation, analgesia and neuromuscular blockade in critically ill patients (see the Nursing research box overleaf).
Medical supportive therapy
Therapeutic goals and interventions to maximise O2 delivery and treat the underlying cause of the respiratory failure are essential to improving the patient’s oxygenation and ventilation status. The primary goal is to treat the underlying cause of the respiratory failure. Other goals include maintaining an adequate cardiac output and haemoglobin concentration.
Sedation management in critical care
NURSING RESEARCH
Citation
Bucknall TK, Manias E, Presneill JJ. A randomized trial of protocol-directed sedation management for mechanical ventilation in an Australian intensive care unit. Crit Care Med 2008; 36:1444–1450.
Purpose
To evaluate the effectiveness of protocol-directed ICU sedation for mechanical ventilation, where registered nurses routinely assess, titrate and wean mechanical ventilation and sedation/analgesia of patients.
Methods
Randomised controlled trial of 312 adult mechanically ventilated patients in a 24 bed ICU. Patients were randomly assigned to either sedation directed by formal guidelines (experimental: n = 153) or local practice (control: n = 159). Outcomes were length of time on mechanical ventilator, duration of ICU and hospital stay, mortality rate, tracheostomy requirement or self-extubation.
Results and conclusions
Overall rates of sedation, analgesia and neuromuscular blockage were similar between the groups. Interestingly, the length of mechanical ventilation was longer in the experimental group compared with the control group (79 vs 58 hours), although this was not statistically significant. All other outcome variables were equivalent and demonstrated no statistical significance. These results indicate that protocol-directed sedation does not influence mechanical ventilation variables in critically ill patients.
Implications for nursing practice
There has been evidence from the US and Europe which suggests that protocol-directed ICU sedation for mechanical ventilation provides superior outcomes compared with usual practice. However, this excellent Australian study showed no benefit to the use of protocol-directed ICU sedation in patients requiring mechanical ventilation. It is thought that the differences in nursing qualifications, staffing numbers and the Australian nursing responsibilities in managing mechanically-ventilated patients may account for the differences from the North American studies. Therefore, based on the findings of the current study, the standard care of mechanically-ventilated patients in Australian ICUs appears to provide adequate vigilance of sedation needs.
Treating the underlying cause
Interventions are directed towards reversing the disease process that resulted in the development of acute respiratory failure. Patients with hypoventilation can be diagnosed and treated rapidly. Patients with / mismatch, shunting or diffusion limitation are managed differently depending on the underlying cause. In all patient situations, monitoring treatment effects, including trends in ABGs and changes in respiratory status, is a continuous process.
Maintaining adequate cardiac output
Cardiac output is the product of heart rate and stroke volume. In most critically ill patients with low cardiac output, compensatory tachycardia occurs to increase cardiac output and intravenous filling increases intravascular volume, thereby increasing preload and stroke volume. This provides patients with adequate perfusion of organs. Blood pressure, in particular mean arterial pressure (MAP), and total peripheral resistance are important indicators of the adequacy of cardiac output and accordingly an adequate blood pressure is required for organ perfusion. Usually a systolic blood pressure of at least 90 mmHg or a MAP of 60 mmHg is adequate to maintain perfusion to the vital organs. If the systolic blood pressure is at least 90 mmHg or the MAP at least 60 mmHg, changes in mentation may be attributed to the level of O2 and CO2 rather than decreased cerebral perfusion. In some patients with hypertension, a systolic blood pressure of 90–100 mmHg may be inadequate to maintain systemic and cerebral perfusion. Hence, aiming for higher systemic arterial pressures and MAP may be part of the management strategy to prevent episodes of brain ischaemia. Decreased cardiac output is treated by administration of IV fluids or medications, or both. (See Ch 66 for a discussion on drugs used to treat decreased cardiac output and shock.) Cardiac output may also be decreased by changes in intrathoracic or intrapulmonary pressures from positive pressure ventilation.
Patients experiencing an exacerbation of COPD or asthma, as well as those receiving controlled positive pressure ventilation, are at risk of alveolar hyperinflation, increased right ventricular afterload and excessive intrathoracic pressures.31 These alterations in right-sided thoracic pressure dynamics may affect the pulmonary vasculature to the left side of the heart, which may result in dramatic haemodynamic compromise. In addition, blood return from the systemic circulation to the right side of the heart may be impaired, decreasing preload.31 Each of these physiological consequences can potentially lead to severe haemodynamic compromise. Consequently, blood pressure and other clinical indicators of adequate cardiac output and tissue perfusion should be monitored closely, with initiation or titration of mechanical ventilation.
Maintaining adequate haemoglobin concentration
Haemoglobin is the primary carrier for delivering O2 to the tissues. If a patient is anaemic, tissue O2 delivery will be compromised. A haemoglobin concentration of 90–100 g/L or greater typically ensures adequate O2 saturation of haemoglobin. The patient should be monitored for signs of blood loss and transfused with packed red blood cells if an adequate haemoglobin concentration cannot be maintained and the patient is symptomatic.
Nutritional therapy
Maintenance of protein and energy stores is especially important in patients who experience acute respiratory failure because nutritional depletion causes a loss of muscle mass, including the respiratory muscles, which may prolong recovery. The nurse and dietician can work together to determine the optimal method of feeding as well as the optimal energy and fluid requirements. (See Fig 65-1 for nutrition guidelines for patients in ICU.) During the acute manifestations of respiratory failure, the risk of aspiration typically limits adequate oral nutritional intake. Therefore, it is optimal if enteral nutrition is initiated within 24 hours in these patients. A multitude of specific nutritional supplements are available for this patient population. A high-carbohydrate diet may need to be avoided in the patient who retains CO2 because carbohydrates metabolise into CO2 and increase the CO2 load. However, research in this area remains controversial.32
Evaluation
The expected outcomes for the patient with acute respiratory failure are presented in NCP 67-1.
Gerontological considerations: respiratory failure
Residents of Australia and New Zealand have some of the highest average life expectancies in the world.33,34 Consequently, the elderly population represents a greater proportion of society than at any other time in history. This trend is also evident within patient populations in acute and critical care settings. Multiple factors contribute to an increased risk of respiratory failure in older adults. They are at higher risk of developing respiratory failure because of the reduction in ventilatory capacity that accompanies ageing, especially if other risk factors are present. Physiological ageing of the lung may produce alveolar dilation, larger air spaces and loss of surface area. Diminished elastic recoil within the airways, decreased chest wall compliance and decreased respiratory muscle strength are also evident. In older adults, the PaO2 falls further and the PaCO2 rises to a higher level before the respiratory system is stimulated to alter the rate and depth of breathing. This delayed response can contribute to the development of respiratory failure.35 Poor nutritional status and lower physiological reserve in cardiovascular, respiratory and autonomic nervous systems increase the risk of additional disease states, such as pneumonia and cardiac disease, which may compromise respiratory function and precipitate respiratory failure.36,37
The elderly are more vulnerable to delirium, healthcare-acquired infections and the effects of (multiple) medications while being managed in the critical care environment. Delirium has been identified as an independent risk factor for increased mortality and morbidity in the critically ill patient.38 It can complicate mechanical ventilation management and weaning from the ventilator. Delirium increases CO2 elimination and O2 consumption, increases the risk of unplanned extubation and device removal, and contributes to increased length of hospital stay and ventilator days.38
Assessment parameters must also be adjusted for a patient’s age—for example, blood pressure generally increases with age. Thus, determining the patient’s baseline vital signs and using these as a basis for comparison of physical assessment findings is most appropriate in evaluating changes in cardiopulmonary function in the older adult.
Acute lung injury/acute respiratory distress syndrome
Acute respiratory distress syndrome (ARDS) is characterised as acute lung injury (ALI) with a sudden and progressive form of acute respiratory failure in which the alveolar capillary membrane becomes damaged and more permeable to intravascular fluid (see Fig 67-8). The alveoli fill with fluid, resulting in severe dyspnoea, hypoxaemia refractory to supplemental O2, reduced lung compliance and diffuse pulmonary infiltrates.39–41 ARDS was recognised as early as 1967; however, it was not until 1994 that consensus on the definition of ARDS was accepted by the American-European Consensus Committee and the term ALI was introduced. ALI was defined as: ‘a syndrome of inflammation and increased permeability that is associated with a constellation of clinical, radiological, and physiological abnormalities that cannot be explained by, but may coexist with, left atrial or pulmonary capillary hypertension.’42 Practically, ARDS is a more severe form of ALI, with profound hypoxaemia as the defining characteristic. Therefore, apart from the clinical manifestation of hypoxaemia, ALI and ARDS will be discussed here synonymously as ARDS.
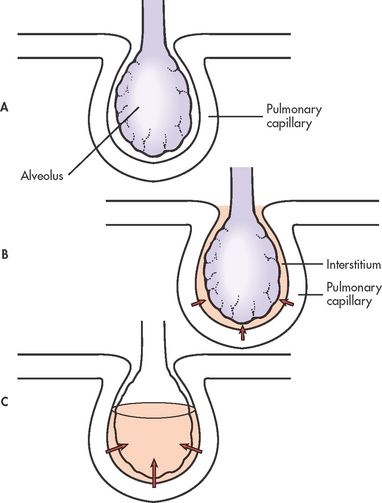
Figure 67-8 Stages of oedema formation in acute respiratory distress syndrome. A, Normal alveolus and pulmonary capillary. B, Interstitial oedema occurs with increased flow of fluid into the interstitial space. C, Alveolar oedema occurs when the fluid crosses the blood–gas barrier.
Previously, the incidence of ARDS was difficult to quantify as there was no global definition; however, since the advent of the consensus statement, many studies have estimated incidence per population. In the US, the incidence has been reported as high as 200,000 patients per year.43 In Australia, the incidence is estimated at 28 cases per 100,000 population per annum, and the overall mortality rate (at 28 days) is estimated to be 32% in patients with ALI and 34% in patients with ARDS.44 Morbidity is a significant problem in patients with ARDS. The most common complications are an extended hospitalisation period, muscle mass loss, muscle weakness and an increased susceptibility to healthcare-acquired infections.40,45 There has also been a reported increase in psychiatric conditions in patients recovering from ARDS.46
AETIOLOGY AND PATHOPHYSIOLOGY
There are a variety of conditions that predispose patients to the development of ARDS (see Table 67-5). The most common cause of ARDS is sepsis. Patients with multiple risk factors are three to four times more likely to develop ARDS.
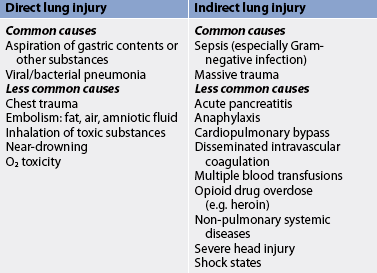
Direct lung injury may cause ARDS, or it may develop as a consequence of the systemic inflammatory response syndrome (SIRS; see Fig 66-1). SIRS may have an infectious or non-infectious aetiology and is characterised by widespread inflammation or clinical responses to inflammation following a variety of physiological insults, including severe trauma, gut ischaemia, lung injury and sepsis.40,41 ARDS may also develop as a consequence of multiple organ dysfunction syndrome (MODS). MODS results from single organ system dysfunction that progressively increases in severity and ultimately results in multisystem organ failure. (SIRS and MODS are discussed in Ch 66.)
An exact cause for the damage to the alveolar–capillary membrane is not known. However, the pathophysiological changes of ARDS are thought to be due to stimulation of the inflammatory and immune systems, which causes an attraction of neutrophils to the pulmonary interstitium (see Fig 67-8).1,47 The neutrophils cause a release of biochemical, humoral and cellular mediators (see Box 67-2) that produce changes in the lungs, including increased pulmonary capillary membrane permeability, destruction of epithelium and endothelium, formation of pulmonary microemboli and pulmonary artery vasoconstriction.40,41 (These mediators are discussed in Chs 12 and 13.)
The pathophysiological course of ARDS can be explained in phases: the exudative phase, the proliferative phase, the fibrotic phase and the recovery phase.
Exudative phase
The initial injury causes the cascade of inflammatory responses known as the exudative phase. This occurs approximately 1–7 days (usually 24–48 hours) after the initial direct lung injury or host insult. Neutrophils adhere to the pulmonary microcirculation, causing damage to the vascular endothelium and increased capillary permeability. In the earliest phase of injury, there is engorgement of the peribronchial and perivascular interstitial space, which produces interstitial oedema. Next, fluid from the interstitial space crosses the alveolar epithelium and enters the alveolar space. Intrapulmonary shunt develops because the alveoli fill with fluid and blood passing through them cannot be oxygenated (see Figs 67-4 and 67-9). Alveolar type I and type II cells (which produce surfactant) are damaged, leading to further fluid and protein accumulation and surfactant dysfunction. The function of surfactant is to maintain alveolar stability by decreasing alveolar surface tension and preventing alveolar collapse. Decreased synthesis of surfactant and inactivation of existing surfactant cause the alveoli to become unstable and collapse (atelectasis). Widespread atelectasis further decreases lung compliance, compromises gas exchange and contributes to hypoxaemia.41 These changes in the exudative phase are summarised in Figure 67-10.
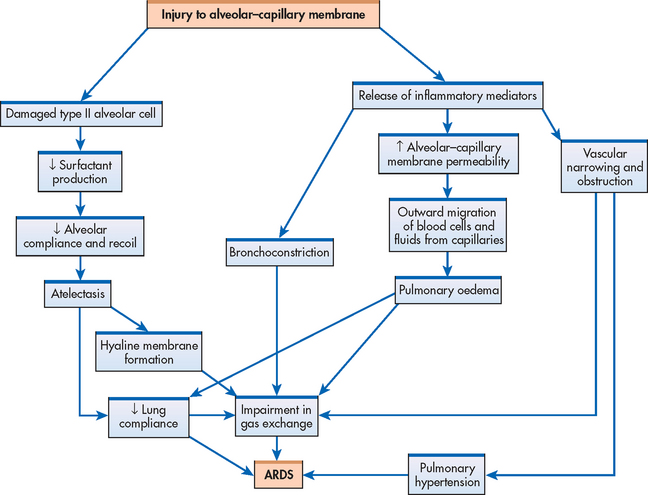
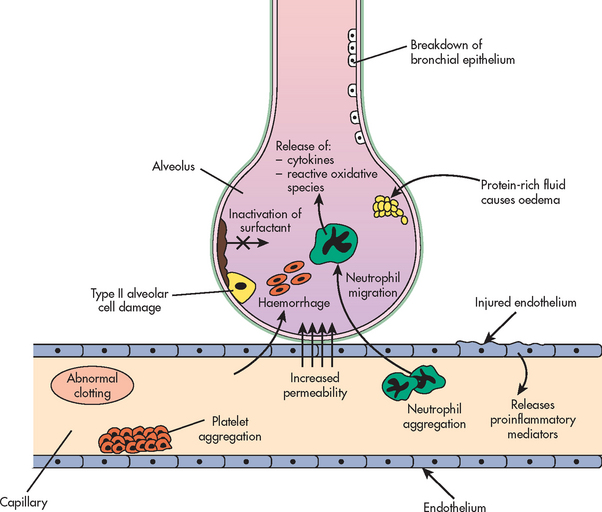
Figure 67-10 Summary of pathophysiological mechanisms during the exudative phase of the development of acute respiratory distress syndrome. Multiple events unfold, including a breakdown in the epithelium and endothelium leading to emigration of neutrophils into the alveoli. This cascade triggers the release of pro-inflammatory mediators, altering coagulation, surfactant and alveolar–capillary permeability.
The primary pathophysiological changes that characterise the exudative phase are interstitial and alveolar oedema (non-cardiogenic pulmonary oedema) and atelectasis. Severe / mismatch and shunting of pulmonary capillary blood result in hypoxaemia unresponsive to increasing concentrations of O2 (termed refractory hypoxaemia). Diffusion limitation, caused by disruption of the alveolar-capillary membrane, further contributes to the severity of the hypoxaemia. As the lungs become less compliant because of decreased surfactant, pulmonary oedema and atelectasis, the patient must generate higher airway pressures to inflate ‘stiff’ lungs. Reduced lung compliance greatly increases the patient’s work of breathing.
Severe hypoxaemia initially causes an increase in breathing rate and a decrease in tidal volume. This breathing pattern increases CO2 removal, producing respiratory alkalosis (pH >7.45). Cardiac output also increases in response to hypoxaemia, which is a compensatory effort to increase pulmonary blood flow. However, as atelectasis, pulmonary oedema and pulmonary shunt increase, compensation fails and hypoventilation, decreased cardiac output and decreased tissue O2 perfusion eventually occur, leading to respiratory acidosis (pH <7.45).
Proliferative phase
The proliferative phase begins 1–2 weeks after the initial lung injury. During this phase, there is an influx of neutrophils, monocytes and lymphocytes and fibroblast proliferation as part of the continuing inflammatory response. The injured lung has an immense regenerative capacity after acute injury. The proliferative phase is complete when the diseased lung becomes characterised by dense, fibrous tissue. Increased pulmonary vascular resistance and pulmonary hypertension may occur in this stage because fibroblasts and inflammatory cells destroy the pulmonary vasculature. Lung compliance continues to decrease as a result of interstitial fibrosis. Hypoxaemia worsens because of the thickened alveolar membrane, causing diffusion limitation and shunting. If the proliferative phase persists, widespread pulmonary fibrosis results. If the proliferative phase is arrested, the lesions resolve.
Fibrotic phase
The fibrotic phase occurs approximately 2–3 weeks after the initial lung injury. This phase is also called the chronic or late phase. During this time the lung is completely remodelled by sparsely collagenous and fibrous tissues. There is diffuse scarring and fibrosis, resulting in decreased lung compliance. In addition, the surface area for gas exchange is significantly reduced because the interstitium is fibrotic, and therefore hypoxaemia continues. Pulmonary hypertension results from pulmonary vascular destruction and fibrosis. The resolution of ARDS may take up to 12 months and is dependent on the severity of the condition.
CLINICAL PROGRESSION
Progression of ARDS varies among patients. Some survive the acute phase of lung injury, pulmonary oedema resolves and complete recovery occurs in a few days. The chance for survival is poor in patients with significant comorbidities. It is not known why injured lungs repair and recover in some patients and why ARDS progresses in others. Several factors seem to be important in determining the course of ARDS, including the nature of the initial injury, the extent and severity of coexisting diseases, and pulmonary complications.40,41
CLINICAL MANIFESTATIONS
The initial presentation of ARDS is often insidious. At the time of the initial injury, and for several hours to 1–2 days afterwards, the patient may not experience respiratory symptoms, or the patient may exhibit only dyspnoea, tachypnoea, cough and restlessness. Chest auscultation may be normal or reveal fine, scattered crackles. ABGs usually indicate respiratory alkalosis caused by hyperventilation and mild hypoxaemia. The chest X-ray may be normal or exhibit evidence of minimal scattered interstitial infiltrates.
As ARDS progresses, symptoms worsen due to increased fluid accumulation and decreased lung compliance. Respiratory discomfort becomes evident as the work of breathing increases. Tachypnoea, intercostal and suprasternal retractions may be present. Tachycardia, diaphoresis, changes in sensorium with decreased mentation, cyanosis and pallor may also be present. Chest auscultation usually reveals scattered-to-diffuse crackles. Chest X-ray demonstrates diffuse and extensive bilateral interstitial and alveolar infiltrates (see Fig 67-11). A pulmonary artery catheter may be inserted and reveal increased pulmonary artery pressure values. Pulmonary artery wedge pressure, however, does not increase in ARDS because the cause is non-cardiogenic (i.e. not related to cardiac function).
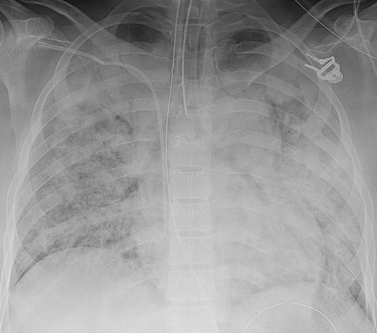
Figure 67-11 Chest X-ray of a patient with acute respiratory distress syndrome. The X-ray shows bilateral, diffuse consolidation (pulmonary infiltrates). In addition, a nasogastric tube, central venous catheter and endotracheal tube are visible.
Source: Imaging Consult. From Muller NL, Silva CIS. The teaching files: chest. Philadelphia: Saunders: 2010.
The cardinal sign of ARDS is the magnitude of hypoxaemia and a PaO2/FiO2 ratio below 200. In ALI the PaO2/FiO2 ratio is below 300. This ratio is present despite an increased FiO2 usually during mechanical ventilation via an endotracheal tube. This means that if patients have a normal PaO2 (80–100 mmHg [11–13 kPa]) but are receiving high concentrations of O2, then the PaO2/FiO2 ratio is likely to be below 200. ABGs may initially demonstrate a normal or decreased PaCO2 despite severe dyspnoea and hypoxia. Hypercapnia signifies that hypoventilation is occurring and the patient is no longer able to maintain the level of ventilation needed to provide optimum gas exchange.
As ARDS progresses it is associated with profound respiratory distress requiring endotracheal intubation and PPV. The chest X-ray often shows what is termed whiteout, because consolidation and coalescing infiltrates are widespread throughout the lungs, leaving few recognisable (dark) air spaces (see Fig 67-11). Pleural effusions may also develop. Severe hypoxaemia, hypercapnia and metabolic acidosis, with symptoms of target organ or tissue hypoxia (such as haematological disturbances and renal dysfunction), may ensue if prompt therapy is not instituted.48
In summary, ARDS is considered to be present if the patient has: (1) refractory hypoxaemia; (2) a chest X-ray with new bilateral interstitial or alveolar infiltrates; (3) a PaO2/FiO2 ratio below 200; and (4) a predisposing condition for ARDS within 48 hours of clinical manifestations.
COMPLICATIONS
Complications may develop as a result of ARDS itself or its treatment. Table 67-6 lists the common complications of ARDS. The major cause of death in ARDS is MODS, often accompanied by sepsis. The vital organs most commonly involved are the kidneys, liver and heart. The organ systems most often involved are the CNS, haematological system, renal system and gastrointestinal system.
TABLE 67-6 Complications associated with Ards
Healthcare-acquired pneumonia
A frequent complication of ARDS is healthcare-acquired pneumonia, occurring in as many as 68% of patients with ARDS. Risk factors include impaired host defences, contaminated medical equipment, invasive monitoring devices, aspiration of gastrointestinal contents and prolonged mechanical ventilation, as well as colonisation of the respiratory tract. Strategies to prevent healthcare-acquired pneumonia include infection control measures (e.g. strict hand-washing and sterile technique during endotracheal suctioning) and elevating the head of the bed 30–45° to prevent aspiration, unless contradicated.49 (Pneumonia is described in Ch 27.)
Barotrauma
Barotrauma may result from the rupture of overdistended alveoli during mechanical ventilation. The high-peak airway pressures that may be required to ventilate patients with ARDS predispose them to this complication. Barotrauma results in the presence of alveolar air in locations where it is not usually found. This can lead to pulmonary interstitial emphysema, pneumothorax, subcutaneous emphysema, pneumoperitoneum, pneumomediastinum and tension pneumothorax (see Ch 27). Barotrauma is minimised with the use of smaller tidal volumes, thereby minimising the risk of alveolar overdistension. This is discussed further in the section on mechanical ventilation below.
Physiological stress ulcers
Critically ill patients with acute respiratory failure are at high risk of gastric stress ulcers. Management strategies include correction of predisposing conditions, such as hypotension, shock and acidosis. Prophylactic management includes anti-ulcer agents (e.g. omeprazole, sucralfate). Early initiation of enteral nutrition also helps prevent mucosal damage.
NURSING AND COLLABORATIVE MANAGEMENT: ACUTE RESPIRATORY DISTRESS SYNDROME
Multidisciplinary care for acute respiratory failure is essential when managing patients with ARDS. The following section discusses additional collaborative measures for the patient with ARDS (see Box 67-3). These patients are usually cared for in intensive care units as they are commonly intubated requiring mechanical ventilation and intensive nursing care. The nursing care plan for acute respiratory failure (see NCP 67-1) is applicable to patients with ARDS.
Nursing assessment
Since ARDS causes acute respiratory failure, the subjective and objective data that should be obtained from the patient with ARDS are the same as those obtained for the patient with acute respiratory failure (see Table 67-4). Abnormal findings on physical examination are indications that ARDS has progressed beyond the initial stages.
Nursing diagnoses
Nursing diagnoses for the patient with ARDS may include, but are not limited to, those described for acute respiratory failure (see NCP 67-1).
Planning
With appropriate therapy, the overall goals for the patient with ARDS are a PaO2 of at least 60 mmHg (8 kPa) and adequate lung ventilation to maintain normal pH. Following recovery from ARDS the patient will have: (1) PaO2 within normal limits for their age or baseline values on room air (FiO2 of 0.21); (2) SaO2 greater than 90%; (3) a patent airway; and (4) clear lungs on auscultation.
Respiratory therapy
Oxygen administration
The primary goal of O2 therapy is to correct hypoxaemia. O2 administered via a simple face mask or nasal prongs is inadequate to treat the refractory hypoxaemia that is associated with ARDS. Masks with high-flow systems (Venturi masks) that have the ability to deliver different O2 concentrations are initially used to maximise O2 delivery. Pulse oximetry (SpO2) is used to continuously monitor the O2 saturation to assess the effectiveness of the O2 therapy. The general standard for O2 administration is to give the patient the lowest concentration that results in a PaO2 of 60 mmHg (8 kPa) or greater. Patients with ARDS commonly need tracheal intubation and mechanical ventilation support because the PaO2 cannot otherwise be maintained at acceptable levels.
Mechanical ventilation
Endotracheal intubation and mechanical ventilation provide additional respiratory support. However, even with these interventions it may be necessary to maintain the FiO2 at 60% or above to maintain the PaO2 at 60 mmHg (8 kPa) or greater. It has been conclusively shown that the use of smaller tidal volumes compared to traditional tidal volumes during mechanical ventilation of ARDS patients improves mortality.50 In this landmark study,50 ARDS patients who received low tidal volumes (6 mL/kg) compared with usual tidal volumes (12 mL/kg) had such significantly improved outcomes that the study was discontinued early because the evidence was so overwhelming. It was shown that mortality was reduced by 22% in the small tidal volume group compared to the traditional tidal volume group. This has extensively changed critical care ventilator practices for ARDS patients since the early 2000s.
What is less clear is the selection of positive end-expiratory pressure (PEEP) in these patients. The mechanism of action of PEEP is related to its ability to increase functional residual capacity (FRC) and ‘recruit’ (open up) collapsed alveoli because of increased residual pressure in the lungs. It appears that the best evidence suggests that a PEEP of 8–15 cm H2O is the most appropriate in ARDS patients.51,52 However, higher levels of PEEP may be used in patients where a greater level of alveolar recruitment is required.52 The implementation of appropriate PEEP levels may improve / in parts of the lung that collapse at low airway pressures, thus allowing the FiO2 to be lowered.
Positioning strategies
In the exudative phase of ARDS, fluid moves freely throughout the lung. Because of gravity, this fluid pools in dependent regions of the lung. As a consequence, some alveoli are fluid-filled (termed dependent areas), whereas others are air-filled (termed non-dependent areas). In addition, when the patient is supine the heart and mediastinal contents place more pressure on the lungs than in the prone position, which changes pleural pressure and predisposes to atelectasis. If the patient is turned from the supine to the prone position, air-filled, non-atelectic alveoli in the ventral (anterior) portion of the lung become dependent. Perfusion may be better matched to ventilation, causing less / mismatching. Therefore, many studies have examined the impact of prone positioning in ARDS patients, in an effort to improve lung recruitment.53–55 It is known that the prone position improves oxygenation in ARDS patients, especially those with refractory hypoxaemia.52 However, not all patients respond to prone positioning with an increase in PaO2 and there is no reliable way of predicting who will respond. Prone positioning is often reserved for patients with refractory hypoxaemia who do not respond to other strategies to increase PaO2. In addition, it has been recommended that the prone position be used only as short-term therapy in patients with high FiO2 requirements or elevated mechanical ventilation airway pressures.52,56
Medical supportive therapy
Maintenance of cardiac output and tissue perfusion
Patients on PPV, and especially with PEEP, frequently experience decreased cardiac output. One cause is decreased venous return, which results from the PEEP-induced increase in intrathoracic pressure. Cardiac output may also be decreased by impaired cardiac contractility and decreased preload. Continuous haemodynamic monitoring is essential to detect these changes and titrate therapy. An arterial catheter is inserted to permit continuous monitoring of blood pressure and sampling of blood for ABG analysis. If cardiac output falls, it may be necessary to administer crystalloid fluids or colloid solutions or to lower PEEP. Use of inotropic drugs to maintain adequate mean blood pressure values may also be necessary. Additionally, the insertion of a pulmonary artery catheter to monitor pulmonary artery pressures that reflect the left atrial pressure may be considered. It should be noted, however, that there is considerable controversy regarding the use of pulmonary artery catheters, with some studies showing no added benefit in terms of mortality and length of hospitalisation, and an increase in complication rates.57,58 Therefore, the use of these catheters is usually based on doctor preference.
The haemoglobin level is usually kept at levels of more than 90–100 g/L to ensure oxygen saturations of 90% or greater (when PaO2 is more than 60 mmHg [>8 kPa]). Packed red blood cells may be transfused to increase haemoglobin and thus the O2-carrying capacity of the blood.
Maintenance of nutrition and fluid balance
Maintenance of fluid balance is challenging in the patient with ARDS. The increasing pulmonary capillary permeability results in fluid sequestration in the lungs and causes pulmonary oedema. At the same time, the patient may have intravascular volume depletion and therefore be prone to hypotension and decreased cardiac output from mechanical ventilation and PEEP. Daily weights (if feasible) and intake and output are monitored to assess the patient’s fluid status. Conservative fluid administration of incremental volumes in the context of patient response should be adopted. The patient is usually placed on mild fluid restriction, and diuretics are used as necessary.
Other therapies
There has been much research on other therapy modalities for ARDS patients, primarily due to the high mortality rate. These include pharmacological therapies such as inhaled nitric oxide, corticosteroids and surfactant therapy, as well as the use of algorithms to guide care.52,59–61 The evidence for any one or combinations of these pharmacological therapies has not been substantiated as yet. In addition, extracorporeal membrane oxygenation (ECMO) may be used; this provides external gas exchange through the use of an extracorporeal bypass circuit. It is used in specialised critical care centres and appears to offer only an option for refractory hypoxaemia that is unresponsive to other therapies.52,62 Its efficacy has yet to be determined and further research is required.
Evaluation
The expected outcomes for the patient with ARDS are similar to those for the patient with acute respiratory failure and are presented in NCP 67-1.
CASE STUDY
Patient profile
Asad Jamali is a 55-year-old Arabic man who was admitted 72 hours ago to a general surgical unit after surgery for an acutely ischaemic bowel. The surgical procedure involved extensive abdominal surgery to repair a perforated colon, irrigate the abdominal cavity and provide haemostasis. During surgery his systolic blood pressure dropped to 70 mmHg. Seven units of packed red blood cells and 4 L of normal saline were administered intravenously to restore blood loss and circulating volume. He is receiving 50% O2 through a Venturi mask with humidification. He is being monitored with a cardiac monitor and pulse oximeter. He has a central intravenous catheter in place and is receiving 0.9% saline intravenously at 125 mL per hour. A urinary catheter is in place. His pulmonary status continues to worsen. He requires progressively higher fractions of inspired oxygen. Urgent endotracheal intubation and mechanical ventilation are necessary. Sedation and analgesia are initiated to achieve pain control and ventilator synchrony.
His lung function continues to worsen and his hypoxaemia is refractory to 100% FiO2 and high levels of PEEP. He experiences kidney and liver failure. There is little hope of weaning him from the ventilator. He has an advance directive that indicates he does not want to be kept alive by artificial means.
Objective data
Physical assessment
• General: sedated, paralysed, well-nourished man; head of bed elevated 45°; skin cool with moderate diaphoresis
• Respiratory: no accessory muscle use, retractions or paradoxical breathing; respiratory rate 18 breaths/min; SpO2 85%; fine crackles at lung bases
• Cardiovascular: blood pressure 100/60 mmHg; cardiac monitor shows sinus tachycardia at 120 beats/min, with equal apical–radial pulse; temperature 38°C rectally
• Gastrointestinal: surgical dressing dry and intact; colostomy draining serosanguineous drainage.
• Urological: urinary catheter draining concentrated urine, less than 30 mL per hour
CRITICAL THINKING QUESTIONS
1. How does the pathophysiology of ARDS predispose to the development of refractory hypoxaemia?
2. What clinical manifestations does this patient exhibit that support a diagnosis of ARDS?
3. What are the possible causes of ARDS in this patient?
4. What are the possible complications this patient is at risk of developing secondary to ARDS?
5. What respiratory care interventions might be implemented to improve this patient’s hypoxaemia?
6. Based on the assessment data presented, write one or more appropriate nursing diagnoses.
7. Given his progressive decline in cardiopulmonary function, what information should be given to this patient’s family?
1. What should the key components of an assessment tool be that will predict patients at risk of acute respiratory failure?
2. Determine the efficacy of prone positioning in patients with ARDS.
3. What are the effects of a nurse-implemented sedation protocol for patients requiring mechanical ventilation on the patient’s and family’s comfort and satisfaction with care?
1. Hypercapnic respiratory failure can be caused by:
2. An early sign of acute respiratory failure is:
3. The oxygen delivery system chosen for the patient in acute respiratory failure should:
4. The most common early clinical manifestations of ARDS that the nurse may observe are:
5. Maintenance of fluid balance in the patient with ARDS involves:
6. Which of the following interventions is designed to prevent or limit barotrauma in the patient with ARDS who is mechanically ventilated?
1 West JB. Pulmonary physiology and pathophysiology: an integrated, case-based approach. Philadelphia: Lippincott Williams & Wilkins, 2007.
2 Ali JS, Summer W, Levitzky M. Pulmonary pathophysiology: a clinical approach, 3rd edn. New York: McGraw-Hill, 2009.
3 Fauci AS, Braunwald E, Kasper DL, Hauser S, Longo DL, Jameson JL, Loscalzo J. Harrison’s manual of medicine, 17th edn. New York: McGraw-Hill, 2009.
4 Rabe KF, Hurd S, Anzueto A, Barnes PJ, Buist SA, Calverley P, et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: GOLD executive summary. Am J Respir Crit Care Med. 2007;176:532–555.
5 Budweiser S, Jorres RA, Pfeifer M. Treatment of respiratory failure in COPD. Int J Chron Obstruct Pulmon Dis. 2008;3:605–618.
6 Pinsky MR, Brochard L, Mancebo J, Hedenstierna G. Applied physiology in intensive care medicine, 2nd edn. Heidelberg: Springer, 2009.
7 Sogaard OS, Lemoh C, Spelman D, et al. A binational cohort study of ventilator-associated pneumonia in Denmark and Australia. Scand J Infect Dis. 2006;38(4):256–264.
8 Johnson KL. Diagnostic measures to evaluate oxygenation in critically ill adults: implications and limitations. AACN Clin Issues. 2004;15(4):506–524.
9 Considine J. The reliability of clinical indicators of oxygenation: a literature review. Contemporary Nurse. 2005;18:258–267.
10 Huang Y-C T. Monitoring oxygen delivery in the critically ill. Chest. 2005;128(5):555S–560S.
11 Jenkins S. 6-minute walk test in patients with COPD: clinical applications in pulmonary rehabilitation. Physiotherapy. 2007;93:175–182.
12 St John RE. End-tidal carbon dioxide monitoring. Crit Care Nurse. 2003;23:83–88.
13 Frazier SK, Skinner GJ. Pulmonary artery catheters: state of the controversy. J Cardiovasc Nurs. 2008;23:113–121.
14 Duke G, Bersten A. Non-invasive ventilation. In Bersten A, Soni N, Oh T, eds.: Oh’s intensive care manual, 6th edn., Oxford: Butterworth-Heinemann, 2008.
15 McHugh G, Freebairn R. Optimal oxygen therapy in the critically ill patient with respiratory failure. Curr Respir Med Rev. 2010;6:229–237.
16 Ricard J-D, Boyer R. Humidification during oxygen therapy and non-invasive ventilation: do we need some and how much? Intensive Care Med. 2009;35:963–965.
17 Schettino G, Altobelli N, Kacmarek RM. Noninvasive positive pressure ventilation reverses acute respiratory failure in select ‘do-not-intubate’ patients. Crit Care Med. 2005;33:1976–1982.
18 Burns KE, Adhikari NK, Keenan SP, Meade MO. Noninvasive positive pressure ventilation as a weaning strategy for intubated patients with respiratory failure. Cochrane Database Syst Rev. (4):2010. CD004127.
19 Keenan S, Sinuff T, Cook DJ, Hill NS. Does noninvasive positive pressure ventilation improve outcome in acute hypoxemic respiratory failure? A systematic review. Crit Care Med. 2004;32(12):2516–2523.
20 Guilherme S, Altobelli N, Kacmarek R. Non-invasive positive-pressure ventilation in acute respiratory failure outside of clinical trials: experiences at the Massachusetts General Hospital. Crit Care Med. 2008;36:441–447.
21 Hill NS, Brennan J, Garpestad E, Nava S. Non-invasive ventilation in acute respiratory failure. Crit Care Med. 2007;35:2402–2407.
22 Salpeter SR, Buckley NS. Systematic review of clinical outcomes in chronic obstructive pulmonary disease: beta-agonist use compared with anticholinergics and inhaled corticosteroids. Clin Rev Allergy Immunol. 2006;31(2–3):219–230.
23 Raoof S, Goulet K, Esan A, Hess DR, Sessler CN. Severe hypoxemic respiratory failure. Part 2: non-ventilatory strategies. Chest. 2010;137:1437–1448.
24 Hynes-Gay P, Leo M, Molino-Carmona S, et al. Optimizing sedation and analgesia in mechanically ventilated patients: an evidence-based approach. Dynamics. 2003;14(4):10–13.
25 Kress JP, Hall JB. Sedation in the mechanically ventilated patient. Crit Care Med. 2006;34:2541–2546.
26 Freely K, Gardner A. Sedation and analgesia management for mechanically ventilated adults: literature review, case study and recommendations for practice. Aus Crit Care. 2006;19:73–77.
27 Jacobi J, Fraser GL, Coursin DB. Clinical practice guidelines for the sustained use of sedatives and analgesics in the critically ill adult. Crit Care Med. 2002;30:119–141.
28 Arroliga A, Thompson BT, Ancukiewicz M, Gonzalez JP, Guntupalli KK, et al. Use of sedatives, opioids, and neuromuscular blocking agents in patients with acute lung injury and acute respiratory distress syndrome. Crit Care Med. 2008;36:1083–1088.
29 Vender JS, Szokol JW, Murphy GS, Nitsun M. Sedation, analgesia and neuromuscular blockade in sepsis: an evidence-based review. Crit Care Med. 2004;32:S554–S561.
30 Arbour R. Impact of bispectral index monitoring on sedation and outcomes in critically ill adults: a case series. Crit Care Nurs Clin North Am. 2006;18(2):227–241.
31 Pinsky MR. Cardiovascular issues in respiratory care. Chest. 2005;128:592S–597S.
32 Malone AM. The use of specialized enteral formulas in pulmonary disease. Nutrition Clin Pract. 2004;19:557–562.
33 Australian Institute of Health & Welfare (AIHW). Australia’s health, 2008. Catalogue no. AUS 99. Canberra: AIHW, 2008.
34 Estimated resident population of New Zealand. Available at www.stats.govt.nz/ accessed December 2010.
35 Miller MR. Structural and physiological age-associated changes in aging lungs. Sem Respir Crit Care Med. 2010;31:521–527.
36 Ely EW, Wheeler AP, Thompson BT, et al. Recovery rate and prognosis in older persons who develop acute lung injury and the acute respiratory distress syndrome. Ann Internal Med. 2002;136(1):25–36.
37 Sprung J, Gajic O, Warner DO. Age related alterations in respiratory function: anesthetic considerations. Can J Anaesthiol. 2006;53(12):1244–1257.
38 Thomason JW, Shintani A, Peterson JF, et al. Intensive care unit delirium is an independent predictor of longer hospital stay: a prospective analysis of 261 non-ventilated patients. Crit Care. 2005;9(4):335–336.
39 Bersten A. Acute respiratory distress syndrome. In Bersten A, Soni N, Oh T, eds.: Oh’s intensive care manual, 6th edn., Oxford: Butterworth-Heinemann, 2008.
40 Bernard GR. Acute respiratory distress syndrome, a historical perspective. Am J Respir Crit Care Med. 2005;172:798–806.
41 Ware LB. Pathophysiology of acute lung injury and the acute respiratory distress syndrome. Sem Respir Crit Care Med. 2006;27:337–349.
42 Bernard GR, Artigas A, Brigham KL, Carlet J, Falke K, Hudson L, et al. The American-European Consensus Conference on ARDS. Definitions, mechanisms, relevant outcomes, and clinical trial coordination. Am J Respir Crit Care Med. 1994;149:818–824. (Seminal text)
43 Rubenfeld GD. Epidemiology of acute lung injury. Crit Care Med. 2003;31:S276–S284.
44 Bersten AD, Edibam C, Hunt T, et al. Incidence and mortality of acute lung injury and the acute respiratory distress syndrome in three Australian states. Am J Respir Crit Care Med. 2002;165:443–448.
45 Herridge MS, Cheung AM, Tansey CM. One-year outcomes in survivors of the acute respiratory distress syndrome. New Eng J Med. 2003;348:683–693.
46 Davydow DS. Psychiatric morbidity in survivors of the acute respiratory distress syndrome: a systematic review. Psychosomatic Med. 2008;70:512–519.
47 Fudala R, Krupa A, Stankowska D, Allen TC, Kurdowska AK. Anti-interleukin-8 autoantibody: interleukin-8 immune complexes in acute lung injury/acute respiratory distress syndrome. Clin Science. 2008;144:403–412.
48 Bastarache JA, Ware LB, Bernard GR. The role of the coagulation cascade in the continuum of sepsis and acute lung injury and acute respiratory distress syndrome. Sem Respir Crit Care Med. 2006;27:365–376.
49 Johnson KL, Meyenburg T. Physiological rationale and current evidence for therapeutic positioning of critically ill patients. AACN Adv Crit Care. 2009;20:228–240.
50 The Acute Respiratory Distress Syndrome Network. Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. New Eng J Med. 2000;342:1301–1308.
51 MacIntyre N. Ventilatory management of ALI/ARDS. Sem Respir Crit Care Med. 2006;27(4):396–403.
52 Esan A, Hess DR, Raoof S, George L, Sessler CN. Severe hypoxemic respiratory failure part 1: ventilatory strategies. Chest. 2010;137:1203–1216.
53 Mancebo J, Fernandez R, Blanch L, et al. A multicenter trial of prolonged prone ventilation in severe acute respiratory distress syndrome. Am J Respir Crit Care Med. 2006;173:1233–1239.
54 Pelosi P, Brazzi L, Gattinoni L. Prone position in acute respiratory distress syndrome. Eur Respir J. 2002;20:1017–1028.
55 Romero CM, Cornejo RA, Galveza LR, Llanos OP, Tobar EA, et al. Extended prone position ventilation in severe acute respiratory distress syndrome: a pilot feasibility study. J Crit Care. 2009;24:81–88.
56 Girard TD, Bernard GR. Mechanical ventilation in ARDS: a state-of-the-art review. Chest. 2007;131:921–929.
57 The NHLBI ARDS Clinical Trials Network. Pulmonary-artery versus central venous catheter to guide treatment of acute lung injury. New Eng J Med. 2005;354:2213–2224.
58 Shah MR, Hasselblad V, Stevenson LW, Binanay C, O’Connor CM, Sopko G, Califf RM. Impact of the pulmonary artery catheter in critically ill patients: meta-analysis of randomized clinical trials. JAMA. 2005;294:1664–1670.
59 Deja M, Hommel M, Weber-Carstens S, Moss M, von Dossow V, et al. Evidence-based therapy of severe acute respiratory distress syndrome: an algorithm-guided approach. J Int Med Research. 2008;36:211–221.
60 Tang BMP, Craig JC, Eslick GD, Seppelt I, McLean AS. Use of corticosteroids in acute lung injury and acute respiratory distress syndrome: a systematic review and meta-analysis. Crit Care Med. 2009;37:1594–1603.
61 Lewis JF, Veldhuizen RAW. The future of surfactant therapy during ALI/ARDS. Sem Resp Crit Care Med. 2006;27:377–388.
62 Brown JK, Haft JW, Bartlett RH, Hirschl RB. Acute lung injury and acute respiratory distress syndrome: extracorporeal life support and liquid ventilation for severe acute respiratory distress syndrome in adults. Sem Respir Crit Care Med. 2006;27:416–425.
American Association for Respiratory Care. www.aarc.org
Ardsnet Group. www.ardsnet.com
Asian Pacific Society of Respirology. www.apsresp.org
Australian & New Zealand Society of Respiratory Science. www.anzsrs.org.au
Become an expert in spirometry. www.spirxpert.com
Lung Health Promotion Centre at The Alfred. www.lunghealth.org
The Australian Lung Foundation. www.lungfoundation.com.au
The Thoracic Society of Australia & New Zealand. www.thoracic.org.au
Timely Topics in Medicine: respiratory. www.ttmed.com/respiratory
